

ABSTRACT
The amino acid sequence of Dnmt2 is very similar to the catalytic domains of bacterial and eukaryotic DNA-(cytosine 5)-methyltransferases, but it efficiently catalyzes tRNA methylation, while its DNA methyltransferase activity is the subject of controversial reports with rates varying between zero and very weak. By using composite nucleic acid molecules as substrates, we surprisingly found that DNA fragments, when presented as covalent DNA-RNA hybrids in the structural context of a tRNA, can be more efficiently methylated than the corresponding natural tRNA substrate. Furthermore, by stepwise development of tRNAAsp, we showed that this natural Dnmt2 substrate could be engineered to employ RNAs that act like guide RNAs in vitro. The 5’-half of tRNAAsp was able to efficiently guide methylation toward a single stranded tRNA fragment as would result from tRNA cleavage by tRNA specific nucleases. In a more artificial setting, a composite system of guide RNAs could ultimately be engineered to enable the enzyme to perform cytidine methylation on single stranded DNA in vitro.
KEYWORDS: 5-methylcytosine, DNA, Dnmt2, enzyme kinetics, modification pathway crosstalk, RNA methylation, RNA modification, tRNA
Introduction
A central question that may join the fields of DNA and RNA modification concerns the action of such rare enzymes that act on nucleobases in both types of nucleic acid.1,2 Ever since the discovery of its robust tRNA methylation,3 Dnmt2 has been a paradigm in this respect, because its nearest neighbors in evolution all are DNA-cytosine C5-methyltransferases (MTases) and methylate DNA.4 While the deoxyribo-5-methylcytidine (5mC or dm5C) residues formed by these related Dnmt enzymes are classical epigenetic marks, the role of Dnmt2 and its product, ribo-5-methylcytidine (rm5C) at position 38 of different tRNAs, is ill understood at present, with different studies showing effects of this modification on tRNA fragmentation and stability,5,6 tRNA charging7 and fidelity in translation.8 The search for a Dnmt2 substrate had been exclusively focused on DNA until the discovery of tRNA methylation activity of Dnmt2 in the seminal paper by Goll.3 With the discovery of tRNAAsp as very active substrate the search for substrates took an unexpected turn, and reports on DNA methylation were now subject to highly controversial discussion.3,9-13 Meanwhile, a recurrent set of tRNAs other than tRNAAsp have been identified as Dnmt2 substrates in various organisms. These include tRNAVal and tRNAGly in mouse,6 tRNAGlu in Schizosaccharomyces pombe,14 tRNAGly and tRNAGlu in Dictyostelium discoideum,15 tRNAVal, and tRNAGly in Drosophila melanogaster 5,16 and tRNAGly in Arabidopsis thaliana.17 In Geobacter sulfurreducens, which so far represents the only prokaryote known to express a Dnmt2 protein, substrate specificity includes tRNAGlu but not tRNAAsp, and the discrimination hinges upon the presence of certain sequence elements in the variable loop.18
Since the revelation of Dnmt2 as an RNA methyltransferease, the performance of methods for selective and sensitive detection of nucleic acid methylation has significantly improved.19 One particularly important development was that bisulfite mapping of dm5C in DNA, an established method that has seen dozens of improvements over the decades, was finally adapted to rm5C detection in RNA.16 This approach 17,20-24 and a related transcriptome-wide approach based on azacytidine 25 have produced numerous candidates for rm5C in RNA, although most of these candidates were not attributed to Dnmt2 activity, but rather dependent on Trm4/NSUN2, a related bona fide RNA methyltransferases.6 Although these approaches allegedly identified several RNAs as Dnmt2 substrates, to date there is no published biochemical data showing any in vitro activity of Dnmt2 on substrates other than tRNA.4 This leaves open the possibility that additional factors influence or even determine substrate specificity of Dnmt2 in vivo or that search paradigms have not been correctly aimed.
Inspired by the ongoing discussion about whether Dnmt2 is capable of methylating DNA, or any other RNA in addition to tRNA, we have set out to determine if the biochemical potential for DNA methylation in vitro is present in the enzyme. We argued that composite nucleic acid molecules with varying contents of RNA versus DNA should allow gauging a preference of an enzyme for one or the other type of nucleic acid. We surprisingly found that Dnmt2 can efficiently methylate short DNA stretches when these are presented in the form of covalent chimeric molecules in the structural context of a tRNA. This activity is interpreted as a residual property originating from the evolutionary proximity of Dnmt2 to DNA methyltransferases.4,26 Furthermore, starting from these structures, artificial guide RNAs can be developed, which allow targeting of Dnmt2 activity to the methylation of short single stranded RNA or DNA oligomers in vitro.
Results and discussion
Dnmt2, but not Trm4, methylates single deoxycytidines at the defined target site
Since linear double-strand DNA is a poor in vitro Dnmt2 substrate at best,9 whereas tRNAAsp is efficiently methylated, we have constructed a series of tRNAAsp hybrids, in which RNA nucleotides were substituted with their corresponding deoxy-surrogates, starting with the single target nucleotide C38, which is methylated to rm5C in native tRNAAsp.3 Several hybrid constructs of increasing DNA content were assembled from synthetic fragments by splint ligation 27 and their properties as methylation substrates of recombinant human Dnmt2 28 were assessed in a tritium incorporation assay with 3H-SAM.29 As expected, the natural all-ribo tRNAAsp displayed robust methylation (Fig. 1).
Figure 1.
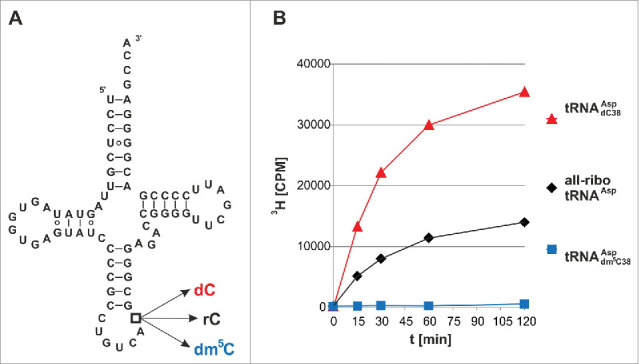
Tritium incorporation assay suggests that Dnmt2 methylates a tRNA containing a deoxycytidine at position 38. (A) Cloverleaf structure of unmodified human cytosolic tRNAAsp. Position 38 has been engineered to contain rC, dC, or dm5C in three hybrid tRNAs of corresponding name. (B) In vitro methylation measured by tritium incorporation from 3H-SAM. Standard deviations of three replicates are given in Fig. S1.
However, we surprisingly found that a chimeric tRNA with a single deoxyribose substitution at the target site C38 (tRNAAspdC38) was an even better substrate for hDnmt2 (Fig. 1a, b, Fig. S1). As a negative control, a dm5C38 hybrid containing synthetic dm5C at position 38 did not show any methyl group acceptor properties in the tritium incorporation assay (Fig. 1b, blue trace). To confirm the structure of the methylated nucleotide, both methylated substrates were re-isolated from the reaction mixture, hydrolyzed to nucleosides, and analyzed by LC-MS/MS. Using fragmentation patterns from synthetic rm5C and dm5C (Fig. 2a), we established a multiple reaction monitoring (MRM) fragmentation method with a limit of detection in the single digit femtomol range.30 While no methylated deoxycytidine was present in the substrates before the reaction, analysis of the material after the reaction revealed the presence of a new peak in the hybrid tRNAAspdC38 that corresponds to dm5C in both retention time and fragmentation pattern (Fig. 2b). Both RNAs contained trace amounts of rm5C, that was already present in the synthetic oligonucleotides used for ligation of the full length substrates,30 but a strong increase of rm5C in the all-ribo substrate was detected upon incubation with Dnmt2. These results clearly show methylation of dC38, and prove that Dnmt2 can accept and modify deoxynucleotides in its active site. To analyze, whether this is a particular feature of Dnmt2, or a generic features of m5C:MTases, we performed analogous experiments with yeast Trm4, an enzyme known to methylate cytidines in the variable loop of yeast tRNAs.31 While recombinant Trm4 showed robust tritium incorporation into an all-ribo tRNA (Fig. S2), substitution of the known target sites rC48 and rC49 with dC led to complete ablation of tritium incorporation, suggesting strongly that Trm4, in contrast to Dnmt2, is not capable of DNA methylation.
Figure 2.

LC-MS analysis confirms that Dnmt2 methylates a tRNA containing a deoxycytidine at position 38. (A) Fragmentation patterns and mass transitions used for scanning of rm5C and dm5C in LC-MS. (B) LC-MS analysis of all-ribo tRNAAsp and hybrid tRNAAspdC38 before and after in vitro methylation by human Dnmt2.
Dnmt2 methylates deoxycytidines faster than ribocytidines at its target site
An in-depth analysis of the methylation reaction showed that it followed Michaelis Menten kinetics (Fig. S3), yielding similar apparent Km values for all-ribo tRNAAsp and hybrid tRNAAspdC38 (4.2 μM vs. 4.0 μM), while the kcat value of the hybrid tRNAAspdC38 was increased by a factor of 2.3. This indicates similar binding properties combined with an accelerated turnover as a consequence of the lack of the 2’-hydroxylgroup at position 38. In the absence of a crystal structure of Dnmt2 29 complexed to its tRNA substrate, it is unclear if the 2’-hydroxylgroup is interacting directly with the enzyme,32 as an influence on base-flipping is possible as well.33
Yet more surprising observations were made in comparing hybrid tRNA with further increasing DNA content: introduction of a 3 nucleotide DNA stretch spanning from d37 to d39 did also improve methylation in comparison to all-ribo tRNAAsp (Upper curves in Fig. 3).
Figure 3.

Dnmt2 methylates hybrid tRNAs containing up to 10 deoxynucleotides. (A) Positions of DNA substitutions in the tRNA cloverleaf in hybrid tRNAs. (B) Methylation of hybrid tRNAs of increasing DNA content. Average values and standard deviations are given in Fig. S1.
Dnmt2 methylates DNA presented as covalent hybrid with RNA in the structural context of a tRNA
In chimeras with longer deoxynucleotide stretches, substrate quality decreased with the DNA content, but a hybrid tRNAAspd36-40, containing a stretch of 5 deoxynucleotides between positions 36 and 40, was still a substrate comparable to the all-ribo tRNAAsp. Chimeric tRNAAspd34-43 (10 deoxynucleotides) was the first to show clearly decreased, albeit still readily detectable activity (Fig. 3).
Exploring the limits of DNA substitution in tRNAAsp
The above data clearly show methylation of DNA, provided it is presented in a structure that can be assumed to resemble the canonical tRNA substrate structure to a reasonable degree. The data also implies a drop in methylation efficiency with increasing DNA content. These findings provoke numerous questions as to where and how much DNA could be incorporated into the tRNA substrate, and if, ultimately, this approach would reveal an all-DNA substrate.
In the investigation of further DNA-tRNA chimeras of increasing DNA content, the substrate quality was assessed in an endpoint assay after 120 minutes, foregoing a time-consuming determination of Michaelis-Menten parameters. Results, as given in Table 1a, were normalized to the all-ribo tRNA as a reference. Based on this quantitative data, a qualitative ranking in descending order of substrate quality was visualized in Fig. 4 to facilitate the following discussion. When the DNA stretch starting at position 34, was further extended toward, and into, the 5’-side of the T-stem, methylation efficiency remained at a moderate level of ∼25-40% of the all-ribo tRNA (Fig. 4e,f,g). To assess the general requirement for the 3D structure of a tRNA, an anticodon stem loop construct (ASL) of tRNAAsp was tested, but found to not be a substrate. Because this strongly suggested that typical elements of tRNA architecture were required for substrate recognition and methylation by Dnmt2, the possibility of extending the DNA stretch into the D- and T-stems was tested by chimeras featuring either an all-deoxy-T-domain or an all-deoxy-D-domain. However, the fact that replacement of either the entire D-loop or the entire T-loop led to undetectable levels of methylation, ablated the idea that an all-DNA substrate might be derived from the tRNA structure in its full length.
Table 1.
tRNA-derivatives constructed by ligation and/or hybridization. Numbering in column 2 corresponds to that used in Figs. 4 and 5.
| Name | tRNA derivative | Relative efficiency / % | N = | SD / % |
|---|---|---|---|---|
|
(a) |
See Fig. 4 for graphical representation |
|||
| all-ribo tRNAAsp | 4c, 5b | 100 | ||
| tRNAAspdC38 | 4a | 177 | 3 | 74 |
| tRNAAspd37-39 | 4b | 180 | 3 | 56 |
| tRNAAspd36-40 | 4d | 103 | 3 | 12 |
| tRNAAspd34-51 | 4e | 38 | 3 | 4 |
| tRNAAspd34-46 | 4f | 32 | 3 | 3 |
| tRNAAspd34-43 | 4g | 25 | 3 | 6 |
|
(b) |
See Fig. 5 for graphical representation |
|||
| nicked tRNAAspdC*dC38 | 5a | 145 | 3 | 20 |
| nicked tRNAAspdC*dC38-43 | 5c | 72 | 3 | 7 |
| nicked tRNAAsp d27-33 | 5d | 71 | 3 | 3 |
| nicked tRNAAspdC38 | 5e | 43 | 3 | 5 |
| nicked tRNAAspI | 5f | 43 | 3 | 3 |
| nicked tRNAAspdX-43-3’HX | 5g | 22 | 3 | 3 |
| nicked tRNAAspdX-53-5’HX | 5h | 13 | 3 | 1 |
| nicked all-ribo tRNAAsp | 5i | 11 | 3 | 0 |
| nicked tRNAAspdX-53 | 5j | 9 | 3 | 1 |
| See Fig. 6 | ||||
| RgD | 6A | 2 | 3 | 0,2 |
Figure 4.

Relative methylation efficiency of covalent chimeric tRNAsAsp compared to the all-ribo tRNAAsp (entry 4c in Table 1), which is set to 100 on the black scale bar on the right.
In vitro development of an RNA-guided system for DNA methylation (RgD)
Having delineated the limits of the DNA content in a covalent DNA-RNA chimera, we turned to the task, of developing a non-covalent RNA-DNA hybrid that might support DNA methyltransferase activity to Dnmt2. Using the previous chimera as a starting point, we tested the effects on methylation of several manipulations, designed to lead to guide-RNAs for Dnmt2 methylation of a ssDNA fragment. These manipulations are graphically summarized in the inset in Fig. 5, and comprise (i) the introduction of a nick in the anticodon at a previous junction of RNA and DNA domains, e.g. at position 34 of constructs 4e-4g (constructs 5e, 5i). Furthermore, (ii) the DNA content would be enlarged similarly to what was discussed above, including also an oligo-dC tail adjacent to the target C38 site to offer multiple potential methylation sites (5a, 5c, 5f-h, 5j). The generated bipartite system would then (iii) be stabilized by extending basepairs in the newly opened anticodon (construct 5g), and (iv) near the 3’-CCA end of the tRNA (construct 5h). The results in Table 1b and the activity scale in Fig. 5 show, that a nick between positions 33 and 34 as in substrates 5i and 5e, which results in an open anticodon loop, is still tolerated by the enzyme, although substrate activity of construct 5i is clearly reduced in comparison to the intact all-ribo tRNA (4c,5b). This result, although obtained rather in passing, is of biological significance. It shows that a hybrid of 2 tRNA-fragments (tRFs), which is essentially a nicked tRNA as would result from specific cleavage by a tRNase such as angiogenin,5 was still methylated. Therefore, the 5’-tRF half could be viewed as a “guide RNA” that steers the enzyme to an RNA target, in this case the 3’-tRF. We took the biochemical proof of concept of a tRF as guide RNA for methylation as an important prerequisite to engineer an artificial system for ssDNA methylation. To this end DNA residues were successively introduced into the target strand, and trends already observed were indeed recapitulated. For example, an exchange of rC38 against dC38 improved methylation (5i vs. 5e), or substitution of the 3’-side of the anticodon stem against DNA entailed moderate losses in methylation (5c, 5f-5h, 5j) as was previously the case with covalent chimeras (4e, 4f, and 4g).
Figure 5.
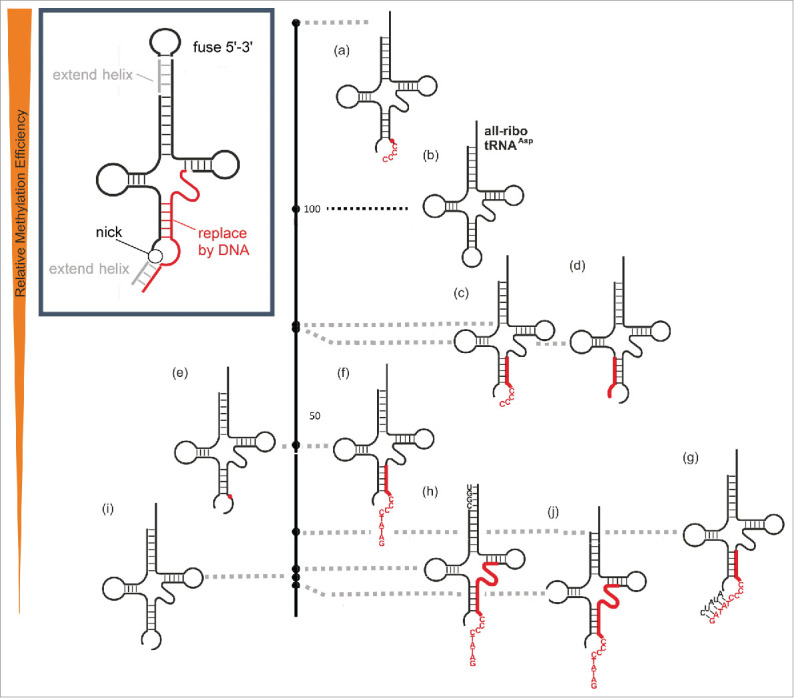
Relative methylation efficiency of 2-component hybrid-chimeric tRNAsAsp compared to the all-ribo tRNAAsp (entry 4c/5b in Table 1) which is set to 100 on the black scale bar in the middle. Deviations from the native tRNAAsp sequence are presented in capital letters, DNA is highlighted in red.
An extension of the DNA fragment on its 5’-end was tolerated, albeit with some loss of activity (compare 5c vs. 5f). In comparison to the covalent chimeras, the newly implemented exchange of the Asp anticodon sequence against an oligo-C stretch was also tolerated by the enzyme (5 a,c,f,g,h,j). Subsequent extension of the acceptor stem helix led to moderate improvement (5j vs. 5h), while the artificial introduction of an extra helix in the anticodon domain led to a drop in methylation (5f vs. 5g) pointing to a dangling DNA single strand as the better substrate. Indeed, a construct featuring a 5’-tRNA-half hybridized to an RNA-DNA chimera with an oligo-C segment was a good substrate (5a) and its DNA content could be extended to the 3’ (5c) and the 5’ (5f) and still retained appreciable activity. An extension of the 5’-tRNA-half, designed to form an additional helix, lowered activity (compare 5f vs. 5g), pointing to a dangling DNA single strand as the better substrate.
The next step was aimed at replacing the covalent RNA-DNA chimera with 2 separate oligonucleotides, one DNA and one RNA. The underlying rationale was that this would lead to a ternary system of 2 RNA strands, which would be assembled into a methylation competent complex upon hybridization with the substrate DNA strand, effectively acting as artificial guide RNAs in vitro. In case of the construct (5h), the hitherto covalent link between the DNA and the RNA part was simply omitted by using 2 separate oligomers, in essence replacing that covalent link with a nick between positions 51 and 52. This particular construct (Fig. 6A) was chosen for 2 reasons, namely (i) to have a maximum length of the DNA for hybridization onto/into the tRNA scaffold, and (ii) because of the stretch of 4 consecutive G residues at positions 51-54 in the T-stem, which was liable to induce potential misfolding by hybridization to C residues 26-29 if the nick was placed elsewhere. The corresponding 3 oligonucleotides needed to assemble the ternary tRNA structural mimic were submitted to a hybridization protocol, but a subsequent test for tritium incorporation showed methylation activity that was only ∼2 fold above the background signal. We therefore changed the assay conditions to include higher enzyme and SAM concentrations, and sampled at 15 and 120 minutes of incubation. Under these enhanced conditions however, a clear signal was measured, exceeding background by a factor of 2.8 after 15 minutes, and by a factor of 9.3 after 120 minutes in 3 replicates (Fig. 6B). Omission of the RNA fragments resulted in signal intensity of background level (not shown), confirming that the methylation depended on the presence of the 2 “guide” RNAs.
Figure 6.
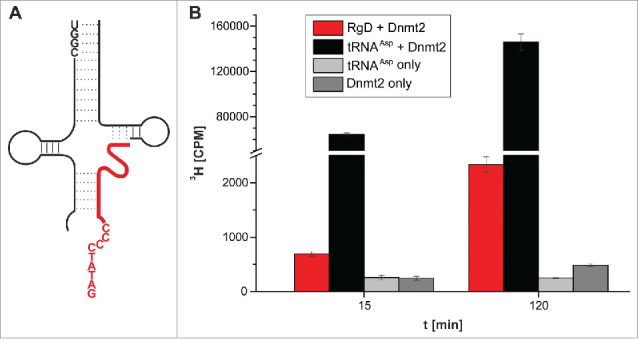
Methylation of an RNA-guided DNA oligonucleotide by Dnmt2. (A) Structure of the hybridized construct. DNA shown in red. (B) Average values and standard deviations of 3 tritium incorporation assays are shown.
To verify, that the methyl group of 3H-SAM was indeed incorporated into the DNA strand our customary LC-MS assay turned out to be inadequate, since the synthetic oligomers used to assemble the various substrates were discovered to already contain substantial amounts of non-radioactive m5C in both RNA and DNA,30 which increased the background to a level (compare Fig. 2) that precluded adequate quantification. Of note, the relative content of tritium in 3H-SAM is so low, that detection of 3H-by LC-MS would require quantities incompatible with biochemical handling and radiation safety. Instead, we resorted to thin-layer chromatography (TLC) for detection of tritium-containing dm5C. The reaction mixture was precipitated, digested to mononucleosides, and separated by TLC in 2 dimensions after addition of authentic standards.34 The corresponding spots were visualized on the TLC by UV light, scratched from the plate, and the tritium quantified by scintillation counting. As shown in Fig. 7, recovered radioactivity resided in the dm5C spot, showing that no formation of rm5C was catalyzed in the RNA part of the substrate complex.
Figure 7.
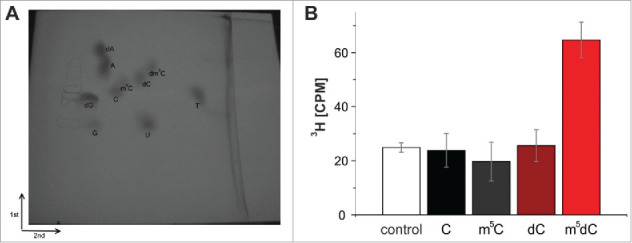
(A) Two-dimensional thin-layer chromatography of nucleosides on a 10 cm x 10 cm cellulose TLC plate. The starting point is marked by an X. (B) Methylation of an RNA-guided DNA oligonucleotide by Dnmt2. The oligonucleotides were hydrolyzed to nucleosides after the tritium incorporation assay, separated by 2D thin-layer chromatography and analyzed with the Cherenkov counter. Tritium could only be detected in m5dC. Average values and standard deviations of 3 experiments are shown. Control corresponds to background signal of the TLC plate. Note that an identical Fig. S4 with enhanced contrast can be found in the supplement.
Discussion
As an opening remark, 3 classes of new Dnmt2 substrates have been identified purely in vitro, and we are well aware that evidence for their biological significance is scarce in the absence of in vivo data. However, one of the new substrate classes has a known correspondent in tRNA biology, namely the all-RNA hybrid composed of 2 RNA strands in Fig. 5j. Such tRNAs with a nicked anticodon may result from incorrect tRNA splicing, the action of ribotoxins or other endonuclease enzymes like angiogenin, and the latter actually cleaves the Dnmt2 substrate tRNAAsp.5 Our results imply, that tRNAs may still be methylated after cleavage, but also that the 5’-tRNA half can, in principle act as a the equivalent of a guide RNA in trans, directing the methylation of Dnmt2 onto a single stranded short RNA such as a tRF. Given that tRFs were found associated to RISC,36 modifications can be expected to affect RNAi, and might constitute a pathway through which Dnmt2 influences RNAi.
Two other classes of newly defined Dnmt2 substrates are of interest in our quest to understand the origin and mechanism of Dnmt2 catalytic action. The definition of a number of covalent RNA-DNA chimeras with variegated substrate properties illustrates, that Dnmt2 still bears strong traits of a DNA methyltransferase. These findings are strongly supporting the result of multiple sequence alignments indicating that Dnmt2 has evolved from a DNA methyltransferase precursor and the DNA methylation activity might be considered an evolutionary “relict” in terms of biochemical catalysis.26
Although our unexpected results cannot prove that any DNA methylation by Dnmt2 does indeed occur in vivo, the fact that constructs featuring small DNA stretches near the target site (substrates 3a, 3b) actually improve methylation over the bona fide natural substrate tRNAAsp, is indeed very suggestive. Based on our data one may speculate that different full or partially single stranded all-DNA structures which present a target deoxycytidine in a loop region, might function as Dnmt2 substrates.
Should Dnmt2 indeed never methylate DNA in vivo, then our finding might be construed to signify that DNA at the target site of a methyltransferase is more easily amenable to catalysis than RNA in general, e.g., because it facilitates base flipping.37 However, this is not the case for the TRM4 enzyme, which does not accept a deoxyribonucleotide at its target position C49 at all (Fig S1), thus supporting once more that the DNA methylation activity of Dnmt2 is likely related to its evolutionary ancestry among DNA methyltransferases.26
The substrates 3a-4i all contain DNA covalently linked to RNA, and these chimeras are derived from a tRNA scaffold whose structure depends, among other features, on the presence of A-form helices and a number of hydrogen bridges mediated by 2’-OH groups.32 Both these features are gradually diminished upon increasing the DNA content and the resulting chimeras can reasonably be expected to gradually lose their structural resemblance to a proper tRNA, a view which receives support from the concomitantly fading methylation activity.
Our construction of an artificial guide RNA system that can efficiently direct methylation activity onto a non-tRNA oligoribonucleotide, and, less efficiently, onto a single stranded DNA strand, brings new concepts to the currently very lively field of nucleic acid modification. While guide RNAs are well established parts of several RNP complexes, e.g. effecting pseudouridinylation,38,39 2’O-methylation,40,41 or U-insertion,42,43 the fact that tRNA derived fragments may potentially turn any given modification enzyme into an RNA-guided modification RNP complex, can reasonably be expected to give impulse to the field. Of interest might be our attempts to simplify the tripartite DNA methylation system to a dual one by fusing the 2 guide RNA fragments. By applying again the splint ligation approach we obtain ∼60% ligation yield, independently of where within the tRNA structure the junction is situated.44 However, a series of resulting constructs featuring the former CCA-3’-end now forming a loop between the former 5’ and 3’-ends (Fig. 5 inset) were, surprisingly, completely inactive for reasons yet to be evaluated, but likely linked to particular structural features of this loop (data not shown). This structural impact is likely to somehow affect the local structure at the remote modification site, because the known data on the enzyme suggest that its interaction with the substrate tRNA is restricted to the anticodon stem loop structure.4,45
As a concluding remark, we are aware that the low efficiency of our artificial DNA methylation system certainly quenches hope that this very construct might exhibit any activity in vivo. However, the principle possibility that Dnmt2 might do so in a more appropriate structural context should certainly be considered. Since such context would be expected to contain both RNA and DNA, possibly covalently linked, a future search might be directed toward replication forks, transcription sites, DNA repair events, or retroviral replication. However, even viral, bacterial and eukaryotic RNA contain a certain amount of DNA.46
Material and methods
Splinted ligation of hybrid tRNAs
Splinted ligation was performed as described previously, by annealing 2 synthetic fragments (IBA Göttingen, Germany, see Table 2) of RNA or DNA:RNA hybrids, corresponding in sequence to tRNAAsp from human (or mouse, as the sequences are identical) onto a 52 nt long complementary oligodeoxynucleotide.47 Appropriate fragments (4 nmol) were 5’-phosphorylated by incubating in KL buffer supplemented with 5 mM ATP, 5 mM DTT and 0.75 u/μl T4 polynucleotide kinase (PNK, Fermentas, Germany) in a final volume of 150 μl in the thermomixer at 37°C for 1 h. To the phosphorylation reaction mixture an equimolar amount of the 5’-fragment and the DNA splint were added, as well as KL buffer, ATP (5 mM) and DTT (5 mM) leading to a final volume of 500 μl and a 8 μM concentration of each fragment. The RNA fragments were hybridized to the DNA splint by heating to 75°C in the thermomixer for 4 min and letting the reaction mixture cool down to room temperature for 15 min. Then T4 DNA ligase (1.5 u/μl; Fermentas) and T4 RNA ligase 2 (22 ng/μl) were added and the ligation was performed in the thermomixer at 16°C over night. Template DNA in constructs tRNAAspdC38 and all-ribo tRNAAsp was removed by addition of 1.5 u/μL DNase I (Fermentas), followed by 1 h of incubation at 37°C. The DNase digestion was omitted for hybrids containing DNA stretches of 3 nucleotides in length or more. All tRNA and tRNA hybrids were purified from ligation mixtures by denaturing PAGE, excised and eluted from the gel, and precipitated with ethanol. Concentrations were calculated from absorption at 254 nm, as determined on a Nanodrop ND-1000 spectrometer.
Table 2.
Oligonucleotides used in this study. Ribonucleotides are indicated by plain letters, deoxyribonucleotides are indicated as dC, dT, dA, dG, or dm5C.
| Construct name | 5‘ fragment | 3‘ fragment | ||
|---|---|---|---|---|
| all-ribo tRNAAsp | MH 565 | UCCUCGUUAGUAUAGUGGUGAGUAUCCCCGCCU | MH 566 | GUCACGCGGGAGACCGGGGUUCGAUUCCCCGACGGGGAGCCA |
| tRNAAspdC38 | MH 565 | UCCUCGUUAGUAUAGUGGUGAGUAUCCCCGCCU | MH 571 | GUCAdCGCGGGAGACCGGGGUUCGAUUCCCCGACGGGGAGCCA |
| tRNAAspd37-39 | MH 565 | UCCUCGUUAGUAUAGUGGUGAGUAUCCCCGCCU | MH 584 | GUCdAdCdGCGGGAGACCGGGGUUCGAUUCCCCGACGGGGAGCCA |
| tRNAAspd36-40 | MH 565 | UCCUCGUUAGUAUAGUGGUGAGUAUCCCCGCCU | MH 585 | GUdCdAdCdGdCGGGAGACCGGGGUUCGAUUCCCCGACGGGGAGCCA |
| tRNAAspd34-43 | MH 565 | UCCUCGUUAGUAUAGUGGUGAGUAUCCCCGCCU | MH 587 | dGdTdCdAdCdGdCdGdGdGAGACCGGGGUUCGAUUCCCCGACGGGGAGCCA |
| tRNAAspd34-46 | MH 565 | UCCUCGUUAGUAUAGUGGUGAGUAUCCCCGCCU | MH 623 | dGdTdCdAdCdGdCdGdGdGdAdGdACCGGGGUUCGAUUCCCCGACGGGGAGCCA |
| tRNAAspd34-51 | MH 565 | UCCUCGUUAGUAUAGUGGUGAGUAUCCCCGCCU | MH 642 | dGdTdCdAdCdGdCdGdGdGdAdGdAdCdCdGdGGGUUCGAUUCCCCGACGGGGAGCCA |
| tRNAAspd27-43 | MH 586 | UCCUCGUUAGUAUAGUGGUGAGUAUCdCdCdCdGdCdCdT | MH 587 | dGdTdCdAdCdGdCdGdGdGAGACCGGGGUUCGAUUCCCCGACGGGGAGCCA |
| tRNAAspd27-33 | MH 586 | UCCUCGUUAGUAUAGUGGUGAGUAUCdCdCdCdGdCdCdT | MH 566 | GUCACGCGGGAGACCGGGGUUCGAUUCCCCGACGGGGAGCCA |
| tRNAAspd48-49 | MH 565 | UCCUCGUUAGUAUAGUGGUGAGUAUCCCCGCCU | MH 621 | GUCACGCGGGAGAdCdCGGGGUUCGAUUCCCCGACGGGGAGCCA |
| tRNAAspdm5C38 | MH 565 | UCCUCGUUAGUAUAGUGGUGAGUAUCCCCGCCU | MH 569 | GUCAdm5CGCGGGAGACCGGGGUUCGAUUCCCCGACGGGGAGCCA |
| Splint | MH 570 | dGdGdAdAdTdCdGdAdAdCdCdCdCdGdGdTdCdTdCdCdCdGdCdGdTdGdAdCdAdGdGdCdG dGdGdGdAdTdAdCdTdCdAdCdCdAdCdTdAdTdAdC | ||
| nicked tRNAAsp dC*dC38 | MH 565 | UCCUCGUUAGUAUAGUGGUGAGUAUCCCCGCCU | MH 619 | dCdCdCdCdCGCGGGAGACCGGGGUUCGAUUCCCCGACGGGGAGCCA |
| nicked tRNAAspdC*dC38-43 | MH 565 | UCCUCGUUAGUAUAGUGGUGAGUAUCCCCGCCU | MH 638 | dCdCdCdCdCdGdCdGdGdGAGACCGGGGUUCGAUUCCCCGACGGGGAGCCA |
| nicked tRNAAspdX-43 | MH 565 | UCCUCGUUAGUAUAGUGGUGAGUAUCCCCGCCU | MH 643 | dGdAdTdAdTdCdCdCdCdCdGdCdGdGdGAGACCGGGGUUCGAUUCCCCGACGGGGAGCCA |
| nicked tRNAAspdX-43-3’HX | MH 646 | UCCUCGUUAGUAUAGUGGUGAGUAUCCCCGCCUAUAUC | MH 643 | dGdAdTdAdTdCdCdCdCdCdGdCdGdGdGAGACCGGGGUUCGAUUCCCCGACGGGGAGCCA |
| nicked tRNAAspdC38 | MH 565 | UCCUCGUUAGUAUAGUGGUGAGUAUCCCCGCCU | MH 571 | GUCAdCGCGGGAGACCGGGGUUCGAUUCCCCGACGGGGAGCCA |
| nicked tRNAAsp d27-33 | MH 586 | UCCUCGUUAGUAUAGUGGUGAGUAUCdCdCdCdGdCdCdT | MH 566 | GUCACGCGGGAGACCGGGGUUCGAUUCCCCGACGGGGAGCCA |
| nicked tRNAAspdX-53 | MH 565 | UCCUCGUUAGUAUAGUGGUGAGUAUCCCCGCCU | MH 647 | dGdAdTdAdTdCdCdCdCdCdGdCdGdGdGdAdGdAdCdCdGdGGGUUCGAUUCCCCGACGGGGAGCCA |
| nicked all-ribo tRNAAsp | MH 565 | UCCUCGUUAGUAUAGUGGUGAGUAUCCCCGCCU | MH 566 | GUCACGCGGGAGACCGGGGUUCGAUUCCCCGACGGGGAGCCA |
| nicked tRNAAspdX-53-5’HX | MH 648 | UGGCUCCUCGUUAGUAUAGUGGUGAGUAUCCCCGCCU | MH 647 | dGdAdTdAdTdCdCdCdCdCdGdCdGdGdGdAdGdAdCdCdGdGGGUUCGAUUCCCCGACGGGGAGCCA |
| RgD | MH 648 | UGGCUCCUCGUUAGUAUAGUGGUGAGUAUCCCCGCCU | MH 649 | GGUUCGAUUCCCCGACGGGGAGCCA |
| MH 650 (middle) | dGdAdTdAdTdCdCdCdCdCdGdCdGdGdGdAdGdAdCdCdGdG | |||
Protein preparation
Cloning, expression and purification by IMAC of the human DNMT2-His6 fusion protein was performed as described before.9,28 Expression was done in E. coli (DE3) Rosetta2 pLysS cells. The protein expression was induced at OD(600 nm) = 0.6 with 1 mM IPTG and the cells were harvested 3 h after induction.
Tritium incorporation assay of in vitro methylation
In vitro methylation was done essentially as described.27 The in vitro methylation assay measures the transfer of a tritiated methyl group from its donor [3H]-S-adenosyl-methionine (3H-SAM , 1 μCi/μL, 80 Ci/ mmol, from Hartmann Analytics, Braunschweig, Germany) onto the target tRNA, which is catalyzed by m5C-methyltransferases. The tRNA is precipitated on small filters and the radioactive signal counted in a scintillation counter. The standard reaction volume was 40 μl. Unless stated otherwise, 120 pmol tRNA were diluted in water and heated to 65°C for 2 min. tRNA-MT-assay buffer and DTT were added immediately to final concentrations of 100 mM Tris-HCl pH 8.0; 100 mM NH4OAc; 0.1 mM EDTA, 10 mM MgCl2 and 10 mM DTT. 3H-SAM-Stock solution (10x; containing cold and 3H-SAM) was added to a final concentration of 0.9 μM SAM and 1 μCi per sample. The enzyme was added to final concentrations of 1 μM and mixed well by pipetting. At various time points 8 μL aliquots were spotted onto small Whatman filters and precipitated in 5 % ice-cold TCA, followed by 2 washes at room temperature for 20 min and 10 min, respectively in 5 % TCA. Then the filters were swirled in EtOH. After drying, the Whatman filters were transferred into scintillation vials and 3 ml of Ultima Gold MV liquid scintillation cocktail (PerkinElmer, Waltham, USA) were added into each vial. The incorporated tritium was measured by liquid scintillation counting with a Wallac 1409 liquid scintillation counter (PerkinElmer, Waltham, USA). Measuring duration was 60 sec per sample. 1 μL of the 10x SAM-stock solution was spotted in duplicate as a standard for specific activity. Initial reaction rates were extracted from methylation kinetics (Fig. S2) by linear regression after background correction. From the initial rates, Km and Vmax values were obtained by a non-linear 2 component least square fit, and kcat was calculated from Vmax.
HPLC -MS analysis
0.5 μg of either Dnmt2 treated or untreated tRNA was dissolved in 20 mM NH4OAc pH 5.3 and digested to nucleosides as described before.48 Additionally the commercial oligomer MH569 (Table 1) containing deoxy-5-methyl-cytidine was digested and used as a reference sample for MS fragmentation experiments. For ribo-5-methyl-cytidine a commercial nucleoside was used (Sigma-Aldrich, Missouri, US).
The digested RNA was analyzed on an Agilent 1260 series equipped with a diode array detector (DAD) and Triple Quadrupol mass spectrometer Agilent 6460. A Synergy Fusion RP column (4 μm particle size, 80 Å pore size, 250 mm length, 2 mm inner diameter) from Phenomenex (Aschaffenburg, Germany) was used at 35 °C. The solvents consisted of 5 mM ammonium acetate buffer adjusted to pH 5.3 using acetic acid (solvent A) and pure acetonitrile (solvent B). The elution started with 100% solvent A followed by a linear gradient to 20% solvent B at 10 min. Initial conditions were regenerated by rinsing with 100% solvent A for 7 minutes. The flow rate was 0.5 mL/min.
The effluent from the column was first measured photometrical at 254 nm by the DAD followed by the mass spectrometer equipped with an electrospray ion source (Agilent Jet Stream). ESI parameters were as follows: gas temperature 300°C, Gas flow 5 L/min, Nebulizer pressure 35 psi, Sheath gas temperature 350°C, Sheath gas flow 12 L/min, capillary voltage 3500 V. The MS was operated in positive ion mode monitoring multiple fragmentation reactions (MRM mode) at previously optimized conditions. The transitions and retention times used for identification of nucleosides can be found in Table 3.
Table 3.
Mass transitions for MRM and retention times in LC-MS analysis.
| Nucleoside | Precursor ion [m/z] | Product ion [m/z] | Retention time [min] |
|---|---|---|---|
| ribocytidine (C) | 244 | 112 | 2.8 |
| ribouridine | 245.2 | 113 | 3.7 |
| deoxycytidine (dC) | 228 | 112 | 4.0 |
| ribomethylcytidine (rm5C) | 258.1 | 126.1 | 5.1 |
| Deoxymethylcytidine (dm5C) | 242 | 126.1 | 6.0 |
| adenosine | 268 | 136 | 7.5 |
Thin layer chromatography
For the RgD Experiments the concentration of nucleic acid and enzyme was doubled and the ratio of hot to cold SAM was increased (2.3 μCi per sample). The total reaction volume was 50 μl. At two time points after reaction start (15 and 120 minutes), aliquots were spotted on a whatman filter paper and measured with the Cherenkov counter. The remaining portion of the sample was subjected to a nucleic acid precipitation with LiClO4 in acetone to purify the nucleic acids. The precipitation with perchlorate and acetone was chosen over standard EtOH precipitation, as it is more suited to precipitate small oligonucleotides. The pellet was then resuspended in H2O and further purified with a MicroSpin G50 size-exclusion column (GE Healthcare, Solingen, Germany). The purified oligonucleotides were enzymatically hydrolyzed to nucleosides with nuclease P1, snake venom phosphodiesterase and shrimp alkaline phosphatase. After the addition of commercially available nucleosides as standards (all Sigma-Aldrich, Missouri, US, except dm5C, Berry & Associates, Dexter, USA), the samples were concentrated and spotted on a 10 cm × 10 cm cellulose TLC plate (Merck ref#1.05577.000, Darmstadt, Germany) and subjected to 2-dimensional thin-layer chromatography. For the first dimension the solvent was isobutyric acid : concentrated ammonia : H2O (50 : 1.1 : 28.9 [v : v : v]) and for the second dimension isopropanol : concentrated HCl : H2O (68 : 18 : 14 [v : v : v]). After each run the TLC plate was dried for several hours in a fume hood. The nucleoside spots were visualized with a UV-lamp (256 nm), marked with a pencil, and the cellulose corresponding to a spot was scraped off with a scalpel and transferred to a reaction tube. The nucleosides were extracted with water and subjected to liquid scintillation counting.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgement
We are grateful to Frau Roth for excellent technical assistance with protein purification.
Funding
This work was supported by the DFG (HE 3397/6 to M.H, JE 252/8 to A.J) in the framework of the FOR1082 consortium “Biochemistry and biological function of Dnmt2 methyltransferases”
References
- 1. Rubio MA, Pastar I, Gaston KW, Ragone FL, Janzen CJ, Cross GA, Papavasiliou FN, Alfonzo JD. An adenosine-to-inosine tRNA-editing enzyme that can perform C-to-U deamination of DNA. Proc Natl Acad Sci U S A 2007; 104:7821–6; PMID:17483465; http://dx.doi.org/10.1073/pnas.0702394104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Harris RS, Petersen-Mahrt SK, Neuberger MS. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol Cell 2002; 10:1247–53; PMID:12453430; http://dx.doi.org/10.1016/S1097-2765(02)00742-6 [DOI] [PubMed] [Google Scholar]
- 3. Goll MG, Kirpekar F, Maggert KA, Yoder JA, Hsieh CL, Zhang X, Golic KG, Jacobsen SE, Bestor TH. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 2006; 311:395–8; PMID:16424344; http://dx.doi.org/10.1126/science.1120976 [DOI] [PubMed] [Google Scholar]
- 4. Jeltsch A, Ehrenhofer-Murray A, Jurkowski T, Lyko F, Reuter G, Ankri S, Nellen W, Schaefer M, Helm M. Mechanism and biological role of Dnmt2 in nucleic acid methylation. RNA Biol 2016:0; PMID:27232191; http://dx.doi.org/ 10.1080/15476286.2016.1191737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schaefer M, Pollex T, Hanna K, Tuorto F, Meusburger M, Helm M, Lyko F. RNA methylation by Dnmt2 protects transfer RNAs against stress-induced cleavage. Genes Dev 2010; 24:1590–5; PMID:20679393; http://dx.doi.org/10.1101/gad.586710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tuorto F, Liebers R, Musch T, Schaefer M, Hofmann S, Kellner S, Frye M, Helm M, Stoecklin G, Lyko F. RNA cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and protein synthesis. Nat Struct Mol Biol 2012; 19:900–5; PMID:22885326; http://dx.doi.org/10.1038/nsmb.2357 [DOI] [PubMed] [Google Scholar]
- 7. Shanmugam R, Fierer J, Kaiser S, Helm M, Jurkowski TP, Jeltsch A. Cytosine methylation of tRNA-Asp by DNMT2 has a role in translation of proteins containing poly-Asp sequences. Cell Discovery 2015; 1:15010; PMID:27462411; http://dx.doi.org/10.1038/celldisc.2015.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tuorto F, Herbst F, Alerasool N, Bender S, Popp O, Federico G, Reitter S, Liebers R, Stoecklin G, Gröne HJ, et al. The tRNA methyltransferase Dnmt2 is required for accurate polypeptide synthesis during haematopoiesis. EMBO J 2015; 34:2350–62; PMID:26271101; http://dx.doi.org/10.15252/embj.201591382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hermann A, Schmitt S, Jeltsch A. The human Dnmt2 has residual DNA-(cytosine-C5) methyltransferase activity. J Biol Chem 2003; 278:31717–21; PMID:12794065; http://dx.doi.org/10.1074/jbc.M305448200 [DOI] [PubMed] [Google Scholar]
- 10. Jeltsch A, Nellen W, Lyko F. Two substrates are better than one: dual specificities for Dnmt2 methyltransferases. Trends Biochem Sci 2006; 31:306–8; PMID:16679017; http://dx.doi.org/10.1016/j.tibs.2006.04.005 [DOI] [PubMed] [Google Scholar]
- 11. Phalke S, Nickel O, Walluscheck D, Hortig F, Onorati MC, Reuter G. Retrotransposon silencing and telomere integrity in somatic cells of Drosophila depends on the cytosine-5 methyltransferase DNMT2. Nat Genet 2009; 41:696–702; PMID:19412177; http://dx.doi.org/10.1038/ng.360 [DOI] [PubMed] [Google Scholar]
- 12. Krauss V, Reuter G. DNA methylation in Drosophila–a critical evaluation. Prog Mol Biol Transl Sci 2011; 101:177–91; PMID:21507351; http://dx.doi.org/10.1016/B978-0-12-387685-0.00003-2 [DOI] [PubMed] [Google Scholar]
- 13. Schaefer M, Lyko F. Lack of evidence for DNA methylation of Invader4 retroelements in Drosophila and implications for Dnmt2-mediated epigenetic regulation. Nat Genet 2010; 42:920–1; author reply 1; PMID:20980983; http://dx.doi.org/10.1038/ng1110-920 [DOI] [PubMed] [Google Scholar]
- 14. Becker M, Muller S, Nellen W, Jurkowski TP, Jeltsch A, Ehrenhofer-Murray AE. Pmt1, a Dnmt2 homolog in Schizosaccharomyces pombe, mediates tRNA methylation in response to nutrient signaling. Nucleic Acids Res 2012; 40:11648–58; PMID:23074192; http://dx.doi.org/10.1093/nar/gks956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Muller S, Windhof IM, Maximov V, Jurkowski T, Jeltsch A, Forstner KU, Sharma CM, Gräf R, Nellen W. Target recognition, RNA methylation activity and transcriptional regulation of the Dictyostelium discoideum Dnmt2-homologue (DnmA). Nucleic Acids Res 2013; 41:8615–27; PMID:23877245; http://dx.doi.org/10.1093/nar/gkt634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schaefer M, Pollex T, Hanna K, Lyko F. RNA cytosine methylation analysis by bisulfite sequencing. Nucleic Acids Res 2009; 37:e12; PMID:19059995; http://dx.doi.org/10.1093/nar/gkn954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Burgess AL, David R, Searle IR. Conservation of tRNA and rRNA 5-methylcytosine in the kingdom Plantae. BMC Plant Biol 2015; 15:199; PMID:26268215; http://dx.doi.org/10.1186/s12870-015-0580-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shanmugam R, Aklujkar M, Schafer M, Reinhardt R, Nickel O, Reuter G, Lovley DR, Ehrenhofer-Murray A, Nellen W, Ankri S, et al. The Dnmt2 RNA methyltransferase homolog of Geobacter sulfurreducens specifically methylates tRNA-Glu. Nucleic Acids Res 2014; 42:6487–96; PMID:24711368; http://dx.doi.org/10.1093/nar/gku256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Motorin Y, Lyko F, Helm M. 5-methylcytosine in RNA: detection, enzymatic formation and biological functions. Nucleic Acids Res 2010; 38:1415–30; PMID:20007150; http://dx.doi.org/10.1093/nar/gkp1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Squires JE, Patel HR, Nousch M, Sibbritt T, Humphreys DT, Parker BJ, Suter CM, Preiss T. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res 2012; 40:5023–33; PMID:22344696; http://dx.doi.org/10.1093/nar/gks144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Edelheit S, Schwartz S, Mumbach MR, Wurtzel O, Sorek R. Transcriptome-wide mapping of 5-methylcytidine RNA modifications in bacteria, archaea, and yeast reveals m5C within archaeal mRNAs. PLoS Genet 2013; 9:e1003602; PMID:23825970; http://dx.doi.org/ 10.1371/journal.pgen.1003602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hussain S, Aleksic J, Blanco S, Dietmann S, Frye M. Characterizing 5-methylcytosine in the mammalian epitranscriptome. Genome Biol 2013; 14:215; PMID:24286375; http://dx.doi.org/10.1186/gb4143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Amort T, Souliere MF, Wille A, Jia XY, Fiegl H, Worle H, Micura R, Lusser A. Long non-coding RNAs as targets for cytosine methylation. RNA Biol 2013; 10:1003–8; PMID:23595112; http://dx.doi.org/10.4161/rna.24454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Militello KT, Chen LM, Ackerman SE, Mandarano AH, Valentine EL. A map of 5-methylcytosine residues in Trypanosoma brucei tRNA revealed by sodium bisulfite sequencing. Mol Biochem Parasitol 2014; 193:122–6; PMID:24389163; http://dx.doi.org/10.1016/j.molbiopara.2013.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Khoddami V, Cairns BR. Identification of direct targets and modified bases of RNA cytosine methyltransferases. Nat Biotechnol 2013; 31:458–64; PMID:23604283; http://dx.doi.org/10.1038/nbt.2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jurkowski TP, Jeltsch A. On the evolutionary origin of eukaryotic DNA methyltransferases and Dnmt2. PloS one 2011; 6:e28104; PMID:22140515; http://dx.doi.org/10.1371/journal.pone.0028104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tovy A, Hofmann B, Helm M, Ankri S. In vitro tRNA methylation assay with the Entamoeba histolytica DNA and tRNA methyltransferase Dnmt2 (Ehmeth) enzyme. J Vis Exp 2010; PMID:21048666; http://dx.doi.org/ 10.3791/2390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jurkowski TP, Meusburger M, Phalke S, Helm M, Nellen W, Reuter G, Jeltsch A. Human DNMT2 methylates tRNA(Asp) molecules using a DNA methyltransferase-like catalytic mechanism. RNA 2008; 14:1663–70; PMID:18567810; http://dx.doi.org/10.1261/rna.970408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dong A, Yoder JA, Zhang X, Zhou L, Bestor TH, Cheng X. Structure of human DNMT2, an enigmatic DNA methyltransferase homolog that displays denaturant-?esistant binding to DNA. Nucleic Acids Res 2001; 29:439–48; PMID:11139614; http://dx.doi.org/10.1093/nar/29.2.439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schmid K, Thuring K, Keller P, Ochel A, Kellner S, Helm M. Variable presence of 5-methylcytosine in commercial RNA and DNA. RNA Biol 2015; 12:1152–8; PMID:26274337; http://dx.doi.org/ 10.1080/15476286.2015.1076612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Motorin Y, Grosjean H. Multisite-specific tRNA:m5C-methyltransferase (Trm4) in yeast Saccharomyces cerevisiae: identification of the gene and substrate specificity of the enzyme. RNA 1999; 5:1105–18; PMID:10445884; http://dx.doi.org/10.1017/S1355838299982201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Aphasizhev R, Theobald-Dietrich A, Kostyuk D, Kochetkov SN, Kisselev L, Giege R, Fasiolo F. Structure and aminoacylation capacities of tRNA transcripts containing deoxyribonucleotides. Rna 1997; 3:893–904; PMID:9257648 [PMC free article] [PubMed] [Google Scholar]
- 33. Cheng X, Roberts RJ. AdoMet-dependent methylation, DNA methyltransferases and base flipping. Nucleic Acids Res 2001; 29:3784–95; PMID:11557810; http://dx.doi.org/10.1093/nar/29.18.3784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Grosjean H, Droogmans L, Roovers M, Keith G. Detection of enzymatic activity of transfer RNA modification enzymes using radiolabeled tRNA substrates. Methods Enzymol 2007; 425:55–101; PMID:17673079; http://dx.doi.org/10.1016/S0076-6879(07)25003-7 [DOI] [PubMed] [Google Scholar]
- 35. Ogawa T, Tomita K, Ueda T, Watanabe K, Uozumi T, Masaki H. A cytotoxic ribonuclease targeting specific transfer RNA anticodons. Science 1999; 283:2097–100; PMID:10092236; http://dx.doi.org/10.1126/science.283.5410.2097 [DOI] [PubMed] [Google Scholar]
- 36. Durdevic Z, Mobin MB, Hanna K, Lyko F, Schaefer M. The RNA methyltransferase Dnmt2 is required for efficient Dicer-2-dependent siRNA pathway activity in Drosophila. Cell Rep 2013; 4:931–7; PMID:24012760; http://dx.doi.org/10.1016/j.celrep.2013.07.046 [DOI] [PubMed] [Google Scholar]
- 37. Klimasauskas S, Kumar S, Roberts RJ, Cheng X. HhaI methyltransferase flips its target base out of the DNA helix. Cell 1994; 76:357–69; PMID:8293469; http://dx.doi.org/10.1016/0092-8674(94)90342-5 [DOI] [PubMed] [Google Scholar]
- 38. Ni J, Tien AL, Fournier MJ. Small nucleolar RNAs direct site-specific synthesis of pseudouridine in ribosomal RNA. Cell 1997; 89:565–73; PMID:9160748; http://dx.doi.org/10.1016/S0092-8674(00)80238-X [DOI] [PubMed] [Google Scholar]
- 39. Ganot P, Bortolin ML, Kiss T. Site-specific pseudouridine formation in preribosomal RNA is guided by small nucleolar RNAs. Cell 1997; 89:799–809; PMID:9182768; http://dx.doi.org/10.1016/S0092-8674(00)80263-9 [DOI] [PubMed] [Google Scholar]
- 40. Kiss-Laszlo Z, Henry Y, Bachellerie JP, Caizergues-Ferrer M, Kiss T. Site-specific ribose methylation of preribosomal RNA: a novel function for small nucleolar RNAs. Cell 1996; 85:1077–88; PMID:8674114; http://dx.doi.org/10.1016/S0092-8674(00)81308-2 [DOI] [PubMed] [Google Scholar]
- 41. Tycowski KT, You ZH, Graham PJ, Steitz JA. Modification of U6 spliceosomal RNA is guided by other small RNAs. Mol Cell 1998; 2:629–38; PMID:9844635; http://dx.doi.org/10.1016/S1097-2765(00)80161-6 [DOI] [PubMed] [Google Scholar]
- 42. Blum B, Bakalara N, Simpson L. A model for RNA editing in kinetoplastid mitochondria: "guide" RNA molecules transcribed from maxicircle DNA provide the edited information. Cell 1990; 60:189–98; PMID:1688737; http://dx.doi.org/10.1016/0092-8674(90)90735-W [DOI] [PubMed] [Google Scholar]
- 43. Benne R. RNA editing in trypanosomes. The us(e) of guide RNAs. Mol Biol Rep 1992; 16:217–27; PMID:1454054; http://dx.doi.org/10.1007/BF00419661 [DOI] [PubMed] [Google Scholar]
- 44. Graber D, Moroder H, Steger J, Trappl K, Polacek N, Micura R. Reliable semi-synthesis of hydrolysis-resistant 3'-peptidyl-tRNA conjugates containing genuine tRNA modifications. Nucleic Acids Res 2010; 38:6796–802; PMID:20525967; http://dx.doi.org/10.1093/nar/gkq508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jurkowski TP, Shanmugam R, Helm M, Jeltsch A. Mapping the tRNA binding site on the surface of human DNMT2 methyltransferase. Biochemistry 2012; 51:4438–44; PMID:22591353; http://dx.doi.org/10.1021/bi3002659 [DOI] [PubMed] [Google Scholar]
- 46. Wang J, Dong H, Chionh YH, McBee ME, Sirirungruang S, Cunningham RP, Shi PY, Dedon PC. The role of sequence context, nucleotide pool balance and stress in 2'-deoxynucleotide misincorporation in viral, bacterial and mammalian RNA. Nucleic Acids Res 2016; PMID:27365049; http://dx.doi.org/ 10.1093/nar/gkw572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hengesbach M, Meusburger M, Lyko F, Helm M. Use of DNAzymes for site-specific analysis of ribonucleotide modifications. RNA 2008; 14:180–7; PMID:17998290; http://dx.doi.org/10.1261/rna.742708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kellner S, Seidu-Larry S, Burhenne J, Motorin Y, Helm M. A multifunctional bioconjugate module for versatile photoaffinity labeling and click chemistry of RNA. Nucleic Acids Res 2011; 39:7348–60; PMID:21646334; http://dx.doi.org/10.1093/nar/gkr449 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.