

Abstract
Following exposure to biological milieus (e.g. after systemic administration), nanoparticles (NPs) get covered by an outer biomolecular corona (BC) that defines many of their biological outcomes, such as the elicited immune response, biodistribution, and targeting capabilities. In spite of this, the BC role in regulating the cellular uptake and the subcellular trafficking properties of NPs has remained elusive. Here, we tackle this issue by employing multicomponent (MC) lipid NPs, human plasma (HP) and HeLa cells as models for nanoformulation, biological fluid, and target cell (respectively). By confocal fluorescence microscopy experiments and image correlation analyses, we quantitatively demonstrate that the BC promotes a neat switch of cell entry mechanism and subsequent intracellular trafficking, from macropinocytosis to clathrin-dependent endocytosis. Nano liquid chromatography tandem mass spectrometry identifies Apolipoproteins as the most abundant components of the BC tested here. Interestingly, this class of proteins target the LDL receptors, which are overexpressed in clathrin-enriched membrane domains. Our results highlight the crucial role of BC as an intrinsic trigger of specific NP-cell interactions and biological responses and set the basis for a rational exploitation of the BC for targeted delivery.
Graphical abstract
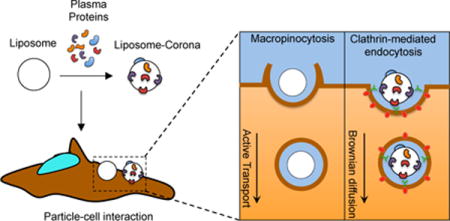
Biomolecular corona promotes a switch of both the cell entry mechanism and the intracellular dynamics of liposomes
Introduction
In the last decades, nanoparticle (NP)-based delivery systems have been extensively studied and employed for therapeutic purposes with many of the proposed solutions providing safe and efficient delivery of pharmaceutical nanoformulations to targeted cells and/or tissues.1–7. However, despite recent progresses and great expectations, few nanoformulations have reached clinical practice6. Such a wide gap between NP-based systems and advanced medicinal products was demonstrated to depend mainly on what occurs to NPs upon contact with a physiological environment (e.g. blood, interstitial fluids, saliva etc.).8 Under in vivo conditions, NPs get covered by an outer biomolecular corona (BC), which changes their “synthetic identity” including size, surface charge, and aggregation state9. It is precisely this new identity, usually referred to as “biological identity”, which controls the biological fate of NPs, including their circulation time, immune system activation, accumulation in liver and spleen, removal from the bloodstream via the mononuclear phagocytic system (MPS), biodistribution, and cellular interactions7. Concerning cellular interactions in particular, since the function of nanomaterials is expected to be carried out in a specific cellular location,10, 11 accurate characterization of BC12 and understanding NP-BC-cell interactions is a critical step to design safer and more efficient NPs as well as to predict possible toxicity effects13–16. After cellular association, most nanomaterials get internalized via endocytic pathways17–19. Some of them, such as clathrin-mediated endocytosis, are regulated by activation of transmembrane receptors, which activate intracellular signaling cascades that, in turn, control cellular processes such as cellular differentiation, proliferation, and survival10. Some other, such as micropinocytosis, entail a series of events initiated by extensive plasma membrane reorganization or ruffling to form an external macropinocytic structure that is then enclosed and internalized20. It thus appears crucial to understand if BC plays a role in mediating the interaction of NPs with cell machinery.
To fulfill this gap, in this work, we explored cellular uptake, endocytic pathways, and intracellular dynamics of NPs in HeLa cells, both in absence and presence of BC from human plasma (HP). We used multicomponent (MC) liposomes, in light of their proved high performances and potential applicability in a number of diseases17, 21–23. We focused on clathrin-mediated endocytosis (CME), caveolin-mediated endocytosis (CAV), and macropinocytosis (MCR) and detected significant differences between bare systems and NP-BC complexes. To date, the most efficient, reliable, and affordable tools for testing endocytosis of nanoparticles are two: i) chemical inhibition of internalization24; and ii) simultaneous labeling of NPs and endocytic vesicles25. In pharmacological blocking studies cells are pretreated with chemical drugs that specifically affect distinct uptake routes and intracellular pathways under well-established conditions. However, some drugs may induce unpredictable side effects (e.g. alteration of the cellular environment) and efficiency of inhibition is not always 100%. On the other hand, fluorescence confocal microscopy allows visualizing the intracellular trafficking of fluorescently labeled NPs (e.g. red labeled NPs) in living cells in the presence of various endocytic markers (e.g. green dyes). Controversial issues have been raised, however, that labeling is not completely specific. In addition, defects in uptake of endocytic dyes can give rise to significant background fluorescence. Thus, to rule out any possible side effect of these treatments, we previously explored the cellular uptake mechanisms of MC cationic liposomes by combining pharmacological and imaging tools26. Under optimized experimental conditions (e.g. concentration of chemical drugs and endocytic markers etc.) pharmacological and imaging results were in complete agreement. Thus, to evaluate the internalization mechanism of MC lipid NPs, we carried out fluorescence colocalization studies on two-channel images, then measured overlap and correlation of the signals corresponding to NPs and endocytic vesicles. Furthermore, we categorized the cytoplasmatic dynamics of the vesicles, by analyzing fluorescence image time-series. More precisely, we employed the iMSD method27, i.e., a fluorescence-based spatiotemporal fluctuation analysis that makes possible to detect the mode of motion of vesicles from imaging, in the form of a mean square displacement (MSD) vs. time-delay plot. The usage of fluorescence correlation spectroscopy (FCS) methods is emerging as a quantitative measure for following nano-sized objects being transported inside single living cells.28
By coupling results from colocalization studies and intracellular dynamics experiments, here we claim that the BC is responsible for a switch in the internalization processes of lipid NPs and affects their intracellular trafficking mechanisms. Notably, tandem nano liquid chromatography (nanoLC) tandem mass spectrometry (MS/MS) identified Apolipoproteins as the most abundant components of the BC tested here. Interestingly, Apolipoproteins target the LDL receptors, which are the most abundant receptors in clathrin-enriched membrane domains. Clearly, results of the present investigation cannot fully account for the complexity of the in vivo NP-BC-cellular interactions. To achieve this, comprehensive studies entailing libraries of NPs, BCs, and cells will be needed. However, we believe that basic studies like this will contribute to the design of specific targeted nanoformulations to exploit specific cellular pathways of interest. In this way, many off-target effects could be minimized or even fully avoided.
Results and Discussion
The use of NPs offers effective solutions to a wide range of biomedical issues29. Their entry into target cells is a fundamental step towards boosting therapeutic efficacy. Upon contact with biological fluids, the surface of NPs is modified by the adsorption of biomolecules leading to formation of the so-called BC6. Key interactions are controlled by biomolecules residing at the interface between the NP surface and the biological target. Understanding how this BC could affect the NP-interactions is more important than studying each entity (i.e. NPs and cells) individually7. Detailed knowledge of the parameters regulating these interactions could help to manipulate the physiochemical properties of NPs in order to dictate selective accumulation in certain locations (e.g. tissues and organs) or to promote tunable cellular interactions. Even tough recent research suggests a strong relationship between the nature of BC and the NP-cell association13, 14, 30, 31, its precise role in regulating cellular uptake and subcellular processes has not been identified yet. The central aim of this investigation was therefore to explore the role played by the BC on the cellular uptake and intracellular trafficking of NPs.
MC liposomes exhibited significantly greater delivery efficiency in many cell lines as compared to commercially available formulations17 and were therefore used as a reference. Usually, a preliminary characterization of lipid NPs both before and after exposure to plasma proteins provides information about the impact of the BC on the chemical physical properties of the employed systems. In this regard, we characterized MC liposomes and MC liposome-BC complexes in terms of size and Zeta potential. We found that bare liposomes have mean hydrodynamic diameter of 144 ± 4 nm and positive Zeta potential (34.5 ± 1.1 mV). Following 1-hour exposure to HP, we detected an increase in particle size (178 ± 6 nm) and the inversion of surface charge (Zeta potential = − 18 ± 2 mV)1.
Size enlargement is in agreement with the results of recent investigations showing that the liposome–BC is roughly 10–40 nm thick depending on the lipid composition32, while ‘normalization’ of zeta potential has been frequently observed, given that most plasma proteins have a net negative charge at physiological pH1. Then, we administered liposomes and liposome-BC complexes to HeLa cells to evaluate cytotoxicity and cell uptake by MTT assay and flow cytometry, respectively. Results are listed in Table 1. Following administration, the percentage of living cells was extremely high for both liposomes and liposome-BC complexes. This finding ensured good cytocompatibility of the employed systems.
Table 1.
Preliminary characterization of the liposomes both in the absence (i.e. bare Liposomes) and in the presence of the biomolecular corona (BC) (i.e. Liposome-BC), in terms of average hydrodynamic diameter, Zeta potential, cell viability and uptake on HeLa cells.
| Liposome | Liposome-BC | |
|---|---|---|
| Particle size (nm) | 144±4 | 178±6 |
| Zeta potential (mV) | 34.5±1.1 | −18±2 |
| Cell viability (%) | 99±1 | 89±5 |
| Cell uptake (%) | 98.9±0.3 | 92.8±2.3 |
A similar trend was found in the outcomes of flow cytometry analysis, which quantifies the cell uptake as percentage of fluorescence cells. Measured values read 99% for bare liposomes and 93% for liposome-BC complexes. To investigate the role of BC on the cellular uptake and intracellular trafficking of MC liposomes, we applied fluorescence microscopy that provides useful tools to quantify spatial distributions of labeled particles within living cells and their intracellular motion. By fluorescence colocalization experiments, we measured overlap and correlation of the fluorescence signals arising from red-labeled particles and green-labeled endocytic vesicles, aiming at elucidating the possible impact of the BC on the internalization mechanisms of the employed NPs. Figure 1 shows some representative dual-color images depicting the colocalization of liposomes and liposome-BC complexes with endocytic vesicles, corresponding to clathrin-mediated endocytosis (CME), caveolae-mediated endocytosis (CAV), and macropinocytosis (MCR). Data processing is based on the evaluation of Manders and Pearson’s coefficients (i.e. M1, M2, and P, respectively) whose measured values are reported in a comprehensive 3D scatter plot (Figure 1 panels D, E, F).
Figure 1.
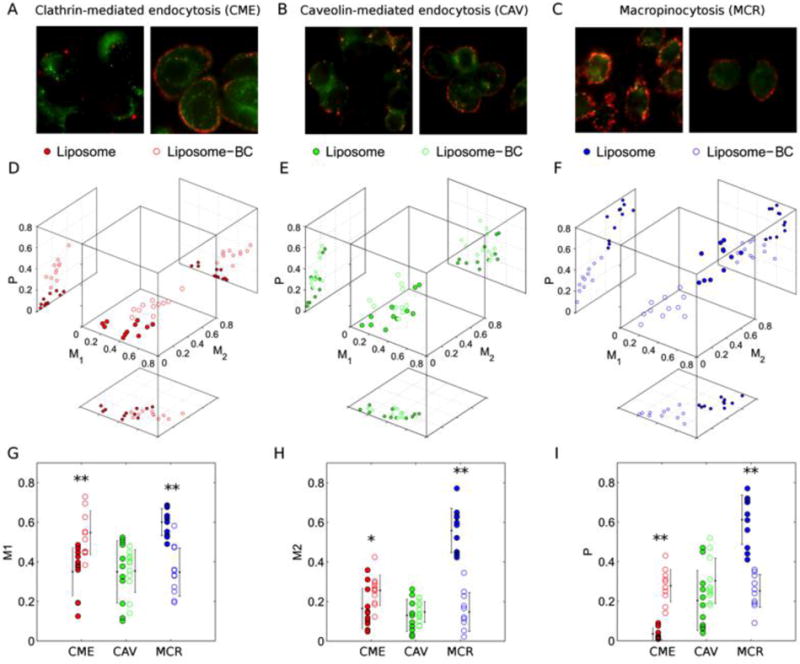
Dual color fluorescence images of red-labeled liposomes, liposome-BC complexes and green-labeled endocytic vesicles corresponding to (A) Clathrin-mediated endocytosis (CME), (B) Caveolin-mediated endocytosis (CAV) CAV and (C) Macropinocytosis (MCR). (D, E, F) Measured values of Manders’ (M1, M2) and Pearson’s (P) coefficients as multivariate distributions and (G, H, I) corresponding 1D projections. Asterisks refer to Student’s t-test significance, in terms of p-values below (*) 0.01 and (**) 0.005
For clarity, the superiority of Manders vs. Pearson’s coefficients is under debate33. As establishing a ranking between Manders and Pearson’s coefficients is beyond our scopes, we decided to calculate and report all of them. In this representation, each data point corresponds to a dual-color image and points belonging to the same class define a specific “cluster”, i.e. a multivariate distribution that is uniquely characterized by average and covariance matrix. Interestingly, for CME and MCR, the distributions of liposome-BC complexes are clearly distinguishable with respect to those of bare liposomes (Figure 1 panels D, F). Colocalization of liposome-BC complexes with CME-vesicles yields higher values of all the coefficients, with respect to their bare counterparts (i.e. pristine MC liposomes). Projections along the coordinate axes are reported to compare all the measured values of M1 (Figure 1 panel G), M2 (Figure 1 panel H), and P (Figure 1 panel I). Of note, this trend is inverted for colocalization experiments with MCR-vesicles. By contrast, colocalization with CAV-vesicles yields almost superimposed distributions (Figure 1 panel E). Indeed, no significant difference is found for the colocalization parameters of liposome and liposome-BC complexes with CAV-vesicles. In summary, confocal imaging inspection followed by quantitative colocalization assays clarified that the cell entry of MC liposomes occurs mainly through MCR, while both clathrin- and caveolae-mediated endocytosis play a minor role in the process. This result is in very good agreement with previous findings by some of us17. MCR is a pinocytosis process resulting in the formation of large vesicles of irregular size and shape, generated by actin-driven invagination of the plasma membrane10. This suggests that the cellular uptake of bare MC liposomes does not involve the utilization of lipid rafts or pit-forming proteins. Of note, BC clearly induces a switch in the mechanism of internalization of MC liposomes, from MCR to CME. This is noteworthy, as these clathrin-enriched domains cover a minor fraction of the cell surface (typically between 0.5–2%)10. Recently, it became clear that the mechanism of intracellular trafficking of NPs is crucial to sort components of the endocytic pathway to a number of subcellular destinations.
Motion characterization at intracellular level requires acquisition and post-processing of time series exploring the behavior of the investigated particles at high spatial and temporal resolution. In this regard, confocal microscopy and spatiotemporal image correlation analysis allowed us to categorize the intracellular dynamics of liposomes and liposome-BC complexes.
In detail, we acquired fluorescence image-stacks (technical details are reported in the Methods section) and processed them through the iMSD method27 that allowed us to categorize the particle mode of motion (Brownian diffusion vs. active transportation) and determine the main system’s dynamic parameters, i.e. diffusion coefficient (D) and speed (v) of particles. The measured values of D and v for bare Figure 2 panel A, where each data point corresponds to an image time-series and ellipses describe the 2D distributions liposomes and liposome-BC complexes are reported in in terms of average values and covariance matrices. Distributions are located and oriented in distinct regions of the (D, v)-space, revealing significant differences between the intracellular dynamics of the investigated systems. Particularly, iMSD shows that bare liposomes are actively transported in the cytosol, while liposome-BC complexes mainly undergo Brownian diffusion (see supplementary Figure S1 for details). Similarly, we carried out iMSD analyses on CME, CAV and MCR vesicles. Measured values of the dynamic parameters are reported in Figure 2 panel B, which depicts the experimental data as 2D multivariate distributions. Dynamics of MCR vesicles (mainly active transport) is remarkably different as compared to that of, CME (mainly Brownian diffusion). Of note, a global comparison of these outcomes suggests that the intracellular dynamics of bare liposomes is similar to that of MCR vesicles as well as the motion of liposome-BC complexes resembles that of CME-vesicles, in keeping with colocalization results. These trends can be also recognized in the 1D distributions of diffusion coefficient (Figure 2 panel C) and particle speed (Figure 2 panel D).
Figure 2.
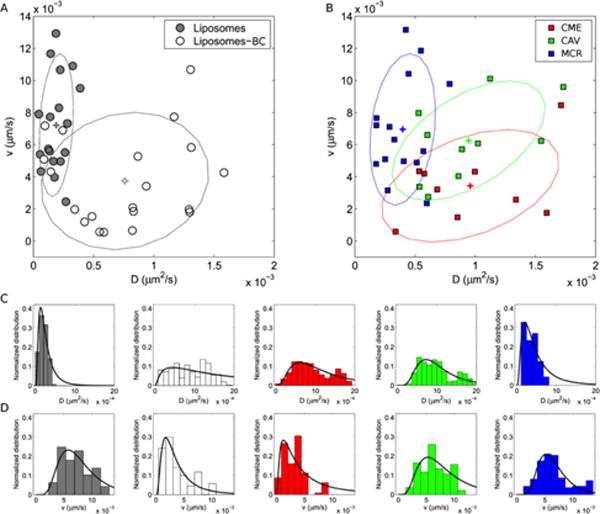
Distributions of the measured dynamic parameters for liposomes and liposome-BC complexes (A) and (B) endocytic vesicles: clathrin-mediated endocytic vesicles (CME), caveolae (CAV) and micropinosomes (MCR). Corresponding weighted distributions of diffusion coefficient, D, and particle speed, v. Crosses indicate the distributions’ centroids.
To provide quantitative estimation of this behavior, we compared multivariate distributions of NPs and endocytic vesicles in terms of Mahalanobis distance34, 35. The Mahalanobis distance, here referred to as λM, is a statistical parameter representing a non-Euclidean distance between a data point and a distribution. In the employed representation, λM-values anisotropically increase from the average position of the 2D distribution, where λM =0. Points on the ellipse have λM =1 and λM >1 outside the ellipse. Notably, the statistical significance of the difference between λM = 1 and λM = 2 is the same as that of a data point located at σ or 2σ far from the mean value of a normal distribution of variance σ2.
We report in Figure 3 panel A the measured λM between data points corresponding to liposomes and the distributions of endocytic vesicles. Results confirm that the dynamic parameters of liposomes are located “far” from the distribution of CME-vesicles (maximum <λM>=1.95) and “near” that of MCR-vesicles (minimum <λM>=1.01). This trend is inverted for liposome-BC complexes (Figure 3 panel B).
Figure 3.
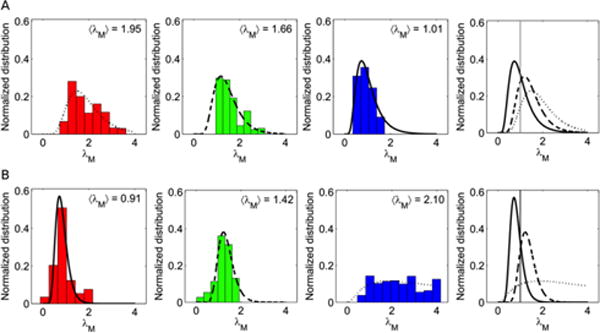
Histograms representing the Mahalanobis distances of data points corresponding to (A) liposomes and (B) liposome-BC complexes, from the distributions of clathrin-mediated endocytic vesicles (red), caveolae (green) and micropinosomes (blue).
In summary, our body of evidence shows that: i) pristine liposomes are mainly internalized by MCR and, once inside HeLa cells, their mode of motion resembles that of macropinosomes (active transportation); ii) The preferential uptake pathway of liposome-BC complexes is CME and their intracellular trafficking is similar to that of CME-vesicles. Collectively, our data show that BC triggers favorable interactions of MC liposomes with clathrin-coated pits. CME, among others, has the function to regulate the mechanism by which cells achieve nutrients and plasma membrane components, such as cholesterol, via low-density lipoproteins (LDL) and vesicles containing proteins with receptor binding sites10 While clarifying the exact molecular mechanisms regulating the internalization of liposome-BC complexes within HeLa cells is beyond the scope of this investigation, our findings are likely to suggest that some BC components may favorably interact with receptor molecules in turn internalized by CME. To verify this speculation, we characterized the BC of MC liposomes by liquid chromatography (LC) tandem mass spectrometry (MS/MS).
Table 2 shows the 25 most abundant proteins identified in the BC (the full list of identified proteins is given in Table S1): interestingly, MC liposomes are mainly covered by apolipoproteins (Apo A-I, Apo C−II, Apo E, Apo C−III, Apo A−IV are the most enriched). Remarkably, it is well known that Apolipoproteins can be targets of the LDL receptor, which in turn is preferentially localized within clathrin-enriched domains36.
Table 2.
List of the twenty five most abundant plasma proteins identified in the biomolecular corona of MC liposomes. Relative protein abundance (RPA) is the average of nine measurements ±elative protein abundance (RPA)
| # | Identified Proteins | Accession Number | Molecular Weight (kDa) | RPA(%) | St. Dev. (%) |
|---|---|---|---|---|---|
|
| |||||
| 1 | Apolipoprotein A–I | APOA1 | 31 | 2.69 | 0.07 |
| 2 | Apolipoprotein C–II | APOC2 | 11 | 2.66 | 0.07 |
| 3 | Apolipoprotein E | APOE | 36 | 2.44 | 0.06 |
| 4 | Apolipoprotein C–III | APOC3 | 11 | 2.37 | 0.05 |
| 5 | Apolipoprotein A–IV | APOA4 | 45 | 2.22 | 0.05 |
| 6 | Apolipoprotein C–I | APOC1 | 9 | 1.74 | 0.04 |
| 7 | Apolipoprotein A–II | APOA2 | 11 | 1.67 | 0.04 |
| 8 | Complement C3 | CO3 | 187 | 1.66 | 0.04 |
| 9 | Keratin, type II cytoskeletal 1 | K2C1 | 66 | 1.50 | 0.04 |
| 10 | Prothrombin | THRB | 70 | 1.50 | 0.04 |
| 11 | Complement C4-B | CO4B | 193 | 1.47 | 0.04 |
| 12 | Fibrinogen beta chain | FIBB | 56 | 1.46 | 0.04 |
| 13 | Tropomyosin alpha-4 chain | TPM4 | 29 | 1.44 | 0.04 |
| 14 | Ig kappa chain C region | IGKC | 12 | 1.38 | 0.03 |
| 15 | Serum albumin | ALBU | 69 | 1.35 | 0.04 |
| 16 | Vitronectin | VTNC | 54 | 1.29 | 0.03 |
| 17 | Inter-alpha-trypsin inhibitor heavy chain H2 | ITIH2 | 106 | 1.29 | 0.03 |
| 18 | Transthyretin | TTHY | 16 | 1.27 | 0.03 |
| 19 | Hemoglobin subunit beta | HBB | 16 | 1.26 | 0.04 |
| 20 | Ig kappa chain V–III region HAH | KV312 | 14 | 1.24 | 0.03 |
| 21 | Ig lambda-2 chain C regions | LAC2 | 11 | 1.23 | 0.03 |
| 22 | Fibrinogen gamma chain | FIBG | 52 | 1.21 | 0.03 |
| 23 | Keratin, type I cytoskeletal 9 | K1C9 | 62 | 1.19 | 0.03 |
| 24 | Clusterin | CLUS | 52 | 1.18 | 0.03 |
| 25 | Fibrinogen alpha chain | FIBA | 95 | 1.17 | 0.03 |
ApoA-I and apoE4 are two relevant members of the family of soluble Apolipoproteins. While apoA-I is the principal component of high-density lipoprotein (HDL), apoE4 preferentially binds to very low-density lipoprotein (VLDL). Of note, both ApoA-1 and ApoE4 show high affinity for the LDL receptor. Recently, absorption of Apo A-I and ApoE4 to negatively charged FePt NPs has been investigated37. Of note, the affinity of ApoE4 for the particle surface was found to be almost four orders of magnitude higher than that of Apo A-I. On the other hand, results reported in Table 2 show that both proteins seem to be present on the liposome surface to the same amount. This finding confirms that many factors shaping BC exist leading to different BCs. Our suggestion is supported by recent literature showing that LDL-enriched NPs present functional motifs that allow recognition by LDL receptor38. Our future biological understanding of how NPs interact with cells will confirm these conclusions more in depth by benefiting from a detailed knowledge of the arrangements of functional motifs of BC components at the nanoscale39–41.
Figure 4 summarizes our present understanding of the role played by BC on the MC liposome association with HeLa cells. BC controls the mechanism of NP-cell association and directs the cellular uptake via specific internalization pathways, CME in our case. This is crucial point, since each internalization route has its own intracellular dynamics (i.e. active transport vs. Brownian diffusion) and final fate. Thus, when one designs NPs for biomedical applications42 the BC should be carefully considered, since it greatly influences the mechanism of cellular uptake. In addition partial conservation of the original BC on the nanoparticle surface may play a key role in determining subsequent cellular dynamics43. Basic studies like this shall contribute to the design of specific nanodelivery systems to exploit specific endocytic pathways of interest. This aspect may have a dramatic application in the emerging field of “personalized nanomedicine”44.
Figure 4.
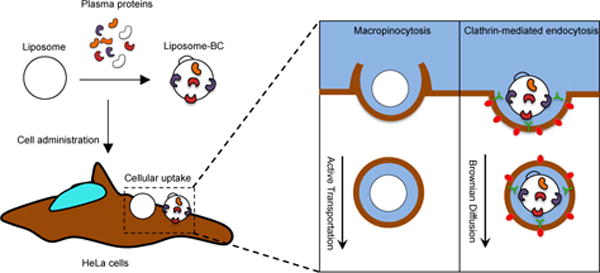
(A) Following exposure to plasma proteins, liposomes’ surface is decorated by a biomolecular corona (BC), which depends on several factors such liposome’ physiochemical properties (i.e. surface chemistry, size, charge etc.), protein source (e.g. human plasma vs. mouse plasma) and environmental factors (i.e. temperature, pH, etc.). To the sake of clarify, we underline that proteins cover liposome’ surface entirely, but, for simplicity of representation, we left the liposome surface only partly decorated by plasma proteins. (B) When particles are given to HeLa cells, they are efficiently internalized with cellular uptake being higher than 90% as quantified by flow cytometry. Despite similar levels of cellular uptake, BC promotes a neat switch of cell entry mechanism of liposomes, from macropinocytosis to clathrin-dependent endocytosis (C). BC has a major impact on the intracellular dynamics of liposomes. Our results highlight the crucial role of BC as an intrinsic trigger of specific NP-cell interactions and biological responses.
Some of us have shown that alterations in concentration and structure of plasma proteins as those produced by clinical manifestations of disease lead to formation of “personalized BC” (PBC)5, 45. Among other implications, this means that various cell types may employ different pathways to internalize NPs with their own PBC. To dictate selective accumulation of NPs at the desired target site, comprehensive studies involving libraries of NPs, PBCs, and cell lines will be needed. In this way, many off-target effects that typically occur in vivo could be minimized or fully prevented.
Materials and Methods
Human Plasma collection
Lyophilized HP was purchased by Sigma-Aldrich (Milan, Italy, P9523 SIGMA) and dissolved in 1 mL of ultrapure water. Plasma is the liquid part of the blood and lymphatic fluid, which makes up about half of its volume. It is prepared from whole blood that is collected with anticoagulants (9:1) and centrifuged to remove cells and cellular debris. P9523 SIGMA is prepared from pooled human blood and contains 4% trisodium citrate as an anticoagulant. It is tested for clotting, which implies that the clotting factors are active. However, it is not analyzed to control whether other enzymes are native or denatured. Before analysis, HP was thawed at 4°C and then warmed at room temperature.
Preparation of liposomes
Cationic lipids (3β-[N-(N′,N′-dimethyl-aminoethane)-carbamoyl])-cholesterol (DC-Chol) and 1,2-Dioleoyl-3-trimethyl-ammonium-propane (DOTAP), zwitterionic lipids dioleoyl-phosphocholine (DOPC) and dioleoyl-phosphatidyl-ethanol-amine (DOPE) were purchased from Avanti Polar Lipids (Alabaster, AL) and used without further refinement. For the preparation of MC liposomes, DC-Chol, DOTAP, DOPC and DOPE were dissolved in chloroform (molar ratios 1:1:1:1) and the solvent was evaporated under vacuum for 2 hours. Lipid films were then hydrated with ultrapure water to obtain a final concentration of 1 mg/ml. Finally, the obtained liposome solutions were extruded 20 times through a 0.1 μm polycarbonate filter with an Avanti Mini-Extruder (Avanti Polar Lipids, Alabaster, AL). For fluorescence microscopy experiment liposomal formulations were synthesized with the addition of Texas red-DOPE (Life Technologies, Carlsbad, CA) (Concentration 7 10−3 mg/mL; fluorescent lipid/total lipid molar ratio = 5/1000).
Preparation of liposome-BC complexes
MC liposomes-BC complexes were obtained by incubating MC liposomes with human plasma (HP) for one h at a fixed ratio of 1:1 v/v.
Characterization of complexes
A preliminary characterization of the complexes has been carried out in terms of hydrodynamic radius and zeta potential. All size and zeta potential measurements were made on a Zetasizer Nano ZS90 (Malvern, U.K.) equipped with a 5 mW HeNe laser (wavelength = 632.8 nm) and a digital logarithmic correlator. All the results are given as mean ± standard deviation of three independent replicates.
Cell culture
Human cervical cancer cell line (HeLa), derived from human cervix adenocarcinoma, was purchase from ATCC (Manassas, VA, USA). HeLa cells were maintained in Eagle’s Minimum Essential Medium (EMEM) supplemented with 2 mM L-glutamine, 100 IU/mL penicillin/streptomycin, 1 mM sodium pyruvate, 10 mM hepes, 1.5 mg/L sodium bicarbonate and 10% fetal bovine serum (FBS).
Flow cytometry
To investigate cellular uptake of nanoparticles in HeLa cells, liposomal formulations were synthesized with the addition of Texas red-DOPE (Life Technologies, Carlsbad, CA) (Concentration 7 10−3 mg/mL; fluorescent lipid/total lipid molar ratio = 5/1000). Bare liposomes and liposome-HP complexes were administered to cells cultured with serum-free medium. HeLa cells were plated 200,000 cells/mL in 12-well dishes. After 24 hours, cells were incubated for 3 hours with 10 μg/mL of Texas res-labeled liposomes in serum-free Optimem medium. After the treatment cells were detached with trypsin/ethylenediaminetetraacetic acid (EDTA), washed two times with cold PBS, and run on a BD LSFORTESSA (BD Biosciences, San Jose, CA, USA). Cells were gated using forward vs side scatter to exclude debris. Data were analyzed using FlowJo software (FlowJo LLC data analysis software, Ashland, OR, USA).
Cell viability
To evaluate the potential toxicity arising from the administration of NPs, cell viability of HeLa cells was assessed by 3-(4,5-dymethyl thiazol 2-y1)-2,5-diphenyl tetrazolium bromide (MTT, mitochondrial respiration analysis; Sigma-Aldrich), according to Mosmann protocol. Briefly, HeLa cells were seeded on 96-wells plate (10,000 cells/well). The day after, cells were treated with 10 g/mL of each formulation in Optimem medium (Life Technologies, Carlsbad, CA) for 24 hours. Then, MTT was added to each well at the final concentration of 0.5 mg/mL and after 4 hours of incubation at 37 °C, the formazan salt was dissolved with 100 μL isopropylic alcohol. The absorbance of each well was measured with Glomax Discover System (Promega, Madison, WI, USA), a ready-to-use high performance multimode detection instrument.
Colocalization analysis
Fluorescence colocalization experiments were performed with an Olympus FV1000 (Olympus, Tokyo, Japan) confocal microscope (objective: 60×, 1.40 NA, oil-immersion), equipped with an X-light spinning disk (pinhole aperture: 70 μm) and Lumencor Spectra X-LED illumination. Hela cells were seeded onto 12 mm round glass coverlips and incubated for 1h in Optimem medium. MC liposomes and MC liposome-BC complexes were labeled with Texas red-DOPE (excitation/emission wavelengths: 595/615 nm). Lysosomes, clathrin, caveolae and macropinosomes were labeled with lysotracker, transferrin, cholera-toxin and dextran fluorescent dyes, respectively (excitation/emission wavelengths: 495/519 nm). Measurements of colocalization are based on the evaluation of Pearson’s and Manders coefficients46, 47 of dual-color fluorescence images, i.e. red (R) channel for complexes and green (G) channel for endocytic vesicles. The Pearson’s coefficient measures statistic correlation between the detected intensities of the two channels and is defined as
| (1) |
where Rk and Gk refer to the intensity values of the k-th pixel from red and green channel respectively and the brackets indicate averaging operations over the entire image48. P values range from 1 (i.e. perfect linear correlation) to -1 (anticorrelation). Values near zero reflect distributions that are not correlated with one another. Manders coefficients M1 and M2 evaluate the overlap of the detected signals from the two channels. We carried out a pre-processing procedure according to the Costes method48 to determine the threshold intensities, below which a pixel can be regarded as dark (i.e. with zero intensity). Thus, thresholds have been iteratively calculated and uniquely defined by the correlation of the raw channels. The Costes method is considered a robust and reproducible method that eliminates user bias and can be easily automated 48. Finally, Manders coefficients have been obtained as46, 48:
| (2) |
where Rk′=Rk if Gk>0 and Rk′=0 if Gk=0 and
| (3) |
where Gk′=Gk if Rk>0 and Gk′=0 if Rk=0.
These split parameters quantify the percentage of not-dark pixels in both channels with respect to the total number of not-dark pixels in Channel R and Channel G, separately. Colocalization analyses were performed using Coloc2 plugin on ImageJ software (https://imagej.nih.gov/ij/).
Measurements of intracellular dynamics
To study the intracellular dynamics of complexes, fluorescence microscopy experiments were performed with an Olympus FV1000 (Olympus, Tokyo, Japan) confocal microscope (objective: 100×, 1.40 NA, oil-immersion), provided with an X-light spinning disk (pinhole aperture: 70 μm) and Lumencor Spectra X-LED illumination. To study the intracellular dynamics of endocytic vesicles, fluorescence microscopy experiments were performed with an Olympus FluoView FV1000 confocal microscope with a 60× NA 1.20 water immersion objective. All experiments were carried out at 37 °C and 5% CO2 using an incubation chamber enclosing the microscope stage and body. The diameter of the detection pinhole was set to the size of 1 Airy. Sequential image series at 16 bits were collected at a fixed pixel size of 69 nm selecting a region of interest of 256×256 pixels and by varying the pixel dwell time from 0.5 to 2 or 4 μs per pixel depending on the characteristic diffusivity of the structure under study. The overall acquisition time varied from 30 to 60 seconds. Image-stacks of at least 300 frames were collected at 1 Hz and the dynamics of about 7000 detected particles was determined by custom scripts working on MATLAB (MathWorks Inc., Natick, MA).
Protein Identification and quantification
MC liposomes (100 μL, concentration =1 mg/mL) were incubated with 100 μL of HP at 37 °C. After 1-h incubation, liposome−BC complexes were centrifuged (14,000 rpm for 15 min) to remove loosely bound proteins. Subsequently, pellets were washed three times with 100 μL of the dissolving buffer (Tris-HCl pH 7.4, 10 mmol L−1; NaCl, 150 mmol L−1; EDTA, 1 mmol L−1). Protein denaturation, digestion, and desalting were carried out by a robust methodology that is commonly used to separate liposome−BC complexes from unbound proteins51. Afterwards, lyophilization was performed by a Speed-Vac apparatus (mod. SC 250 Express; Thermo Savant, Holbrook, NY, USA). Then samples were reconstituted with 0.1% HCOOH solution (final concentration 0.32 mg/mL) and stored at − 80 °C until LC MS/MS was done. Tryptic peptides were examined by a nano-LC system (Dionex Ultimate 3000, Sunnyvale, CA, USA) connected to a hybrid mass spectrometer (Thermo Fisher Scientific Bremen, Germany), equipped with a nanoelectrospray ion source. Experimental details have been already reported.51 Xcalibur (v.2.07, ThermoFisher Scientific) raw data files were submitted to Proteome Discover (1.2 version, Thermo Scientific) for database search using Mascot (version 2.3.2 Matrix Science). Data were searched against SwissProt database (v 57.15, 20 266 sequences) using the decoy search option of Mascot. Finally, protein quantification was made by Scaffold software. For each identified protein, the mean value of (normalized spectral countings, NSCs) was normalized to the protein molecular weight (MWNSC) to obtain the relative protein abundance (RPA). Statistical significance of data was guaranteed by replicating the procedure for nine samples (three technical replicates for three independent biological samples). Data of relative protein abundance were provided as mean ± standard deviation.
Conclusions
We probe the role of BC in directing liposomes interaction with living cells. The switch from the synthetic identity (MC liposomes) to the biological one (MC liposomes with absorbed BC) is proved to be responsible for a switch in the uptake mechanism and intracellular trafficking properties of NPs, from macropinocytosis to clathrin-mediated endocytosis. Since Apolipoproteins are the main components of the BC, we suggest that this protein class may be active player in triggering receptor-mediated uptake of liposome-BC into HeLa cells. This finding sheds new light on the basic molecular determinants of NP interaction with living matter and, potentially, paves the way to a new era of studies aimed at creating targeted nanoformulations to exploit specific cellular pathways of interest. The knowledge obtained from this study will contribute to understand basic principles of the uptake mechanism. This will allow better prediction and control of the biodistribution of liposomes, which could lead to engineer such nanomaterials for specific targeting of tissues and/or organs of interest.
Supplementary Material
Acknowledgments
MD and EG where supported in part by the following grants NIH P41-GM103540 and NIH P50-GM076516
Footnotes
Electronic Supplementary Information (ESI) available: Figure S1. (A) Sequential image series collected by a confocal microscope. (B) The spatiotemporal correlation function g of the fluorescence fluctuations δi. (C) The image-derived Mean Square Displacement (iMSD). Table S1. The full list of the most abundant corona proteins associated with MC liposomes as identified by NanoLC-MS/MS.
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Caracciolo G. Nanomedicine: Nanotechnology, Biology and Medicine. 2015;11:543–557. doi: 10.1016/j.nano.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 2.Docter D, Westmeier D, Markiewicz M, Stolte S, Knauer S, Stauber R. Chemical Society reviews. 2015;44:6094–6121. doi: 10.1039/c5cs00217f. [DOI] [PubMed] [Google Scholar]
- 3.Mahmoudi M, Sheibani S, Milani AS, Rezaee F, Gauberti M, Dinarvand R, Vali H. Nanomedicine. 2015;10:215–226. doi: 10.2217/nnm.14.69. [DOI] [PubMed] [Google Scholar]
- 4.Maiolo D, Del Pino P, Metrangolo P, Parak WJ, Baldelli Bombelli F. Nanomedicine. 2015;10:3231–3247. doi: 10.2217/nnm.15.163. [DOI] [PubMed] [Google Scholar]
- 5.Corbo C, Molinaro R, Parodi A, Toledano Furman NE, Salvatore F, Tasciotti E. Nanomedicine. 2016;11:81–100. doi: 10.2217/nnm.15.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caracciolo G, Farokhzad OC, Mahmoudi M. Trends in Biotechnology. 2017;35:257–264. doi: 10.1016/j.tibtech.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 7.Polo E, Collado M, Pelaz B, del Pino P. ACS nano. 2017;11:2397–2402. doi: 10.1021/acsnano.7b01197. [DOI] [PubMed] [Google Scholar]
- 8.Salvati A, Pitek AS, Monopoli MP, Prapainop K, Bombelli FB, Hristov DR, Kelly PM, Åberg C, Mahon E, Dawson KA. Nature nanotechnology. 2013;8:137–143. doi: 10.1038/nnano.2012.237. [DOI] [PubMed] [Google Scholar]
- 9.Walkey CD, Chan WCW. Chemical Society Reviews. 2012;41:2780–2799. doi: 10.1039/c1cs15233e. [DOI] [PubMed] [Google Scholar]
- 10.Behzadi S, Serpooshan V, Tao W, Hamaly MA, Alkawareek MY, Dreaden EC, Brown D, Alkilany AM, Farokhzad OC, Mahmoudi M. Chemical Society Reviews. 2017 doi: 10.1039/c6cs00636a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Silvio D, Maccarini M, Parker R, Mackie A, Fragneto G, Bombelli FB. Journal of Colloid and Interface Science. 2017 doi: 10.1016/j.jcis.2017.05.086. [DOI] [PubMed] [Google Scholar]
- 12.Carrillo-Carrion C, Carril M, Parak WJ. Current Opinion in Biotechnology. 2017;46:106–113. doi: 10.1016/j.copbio.2017.02.009. [DOI] [PubMed] [Google Scholar]
- 13.Walkey CD, Olsen JB, Song F, Liu R, Guo H, Olsen DWH, Cohen Y, Emili A, Chan WC. ACS nano. 2014;8:2439–2455. doi: 10.1021/nn406018q. [DOI] [PubMed] [Google Scholar]
- 14.Liu R, Jiang W, Walkey CD, Chan WC, Cohen Y. Nanoscale. 2015;7:9664–9675. doi: 10.1039/c5nr01537e. [DOI] [PubMed] [Google Scholar]
- 15.Sahneh FD, Scoglio CM, Monteiro-Riviere NA, Riviere JE. Nanomedicine. 2015;10:25–33. doi: 10.2217/nnm.14.60. [DOI] [PubMed] [Google Scholar]
- 16.Treuel L, Docter D, Maskos M, Stauber RH. Beilstein journal of nanotechnology. 2015;6:857–873. doi: 10.3762/bjnano.6.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pozzi D, Marchini C, Cardarelli F, Rossetta A, Colapicchioni V, Amici A, Montani M, Motta S, Brocca P, Cantù L. Molecular pharmaceutics. 2013;10:4654–4665. doi: 10.1021/mp400470p. [DOI] [PubMed] [Google Scholar]
- 18.Ojeda E, Puras G, Agirre M, Zarate J, Grijalvo S, Eritja R, Digiacomo L, Caracciolo G, Pedraz JL. International Journal of Pharmaceutics. 2016;503:115–126. doi: 10.1016/j.ijpharm.2016.02.043. [DOI] [PubMed] [Google Scholar]
- 19.Rejman J, Bragonzi A, Conese M. Molecular Therapy. 2005;12:468–474. doi: 10.1016/j.ymthe.2005.03.038. [DOI] [PubMed] [Google Scholar]
- 20.Swanson JA, Watts C. Trends in cell biology. 1995;5:424–428. doi: 10.1016/s0962-8924(00)89101-1. [DOI] [PubMed] [Google Scholar]
- 21.Caracciolo G, Pozzi D, Amenitsch H, Caminiti R. Langmuir. 2005;21:11582–11587. doi: 10.1021/la052077c. [DOI] [PubMed] [Google Scholar]
- 22.Caracciolo G, Pozzi D, Caminiti R, Amenitsch H. Applied Physics Letters. 2005;87:1–3. [Google Scholar]
- 23.Palchetti S, Pozzi D, Marchini C, Amici A, Andreani C, Bartolacci C, Digiacomo L, Gambini V, Cardarelli F, Di Rienzo C, Peruzzi G, Amenitsch H, Palermo R, Screpanti I, Caracciolo G. Nanomedicine: Nanotechnology, Biology, and Medicine. 2017;13:681–691. doi: 10.1016/j.nano.2016.08.019. [DOI] [PubMed] [Google Scholar]
- 24.Kastl L, Sasse D, Wulf V, Hartmann R, Mircheski J, Ranke C, Carregal-Romero S, Martínez-López JA, Fernández-Chacón R, Parak WJ, Elsasser H-P, Rivera_Gil P. ACS Nano. 2013;7:6605–6618. doi: 10.1021/nn306032k. [DOI] [PubMed] [Google Scholar]
- 25.Vercauteren D, Deschout H, Remaut K, Engbersen JFJ, Jones AT, Demeester J, De Smedt SC, Braeckmans K. ACS Nano. 2011;5:7874–7884. doi: 10.1021/nn2020858. [DOI] [PubMed] [Google Scholar]
- 26.Cardarelli F, Pozzi D, Bifone A, Marchini C, Caracciolo G. Molecular Pharmaceutics. 2012;9:334–340. doi: 10.1021/mp200374e. [DOI] [PubMed] [Google Scholar]
- 27.Digiacomo L, Digman MA, Gratton E, Caracciolo G. Acta Biomaterialia. 2016;42:189–198. doi: 10.1016/j.actbio.2016.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Åberg C, Varela JA, Fitzpatrick LW, Dawson KA. Scientific reports. 2016;6 doi: 10.1038/srep34457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heath JR. Proceedings of the National Academy of Sciences. 2015;112:14436–14443. doi: 10.1073/pnas.1515202112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bigdeli A, Palchetti S, Pozzi D, Hormozi-Nezhad MR, Bombelli FBaldelli, Caracciolo G, Mahmoudi M. ACS nano. 2016;10:3723–3737. doi: 10.1021/acsnano.6b00261. [DOI] [PubMed] [Google Scholar]
- 31.Palchetti S, Digiacomo L, Pozzi D, Peruzzi G, Micarelli E, Mahmoudi M, Caracciolo G. Nanoscale. 2016;8:12755–12763. doi: 10.1039/c6nr03898k. [DOI] [PubMed] [Google Scholar]
- 32.Caracciolo G, Pozzi D, Capriotti AL, Cavaliere C, Piovesana S, La Barbera G, Amici A, Laganà A. Journal of Materials Chemistry B. 2014;2:7419–7428. doi: 10.1039/c4tb01316f. [DOI] [PubMed] [Google Scholar]
- 33.Adler J, Parmryd I. Cytometry Part A. 2010;77:733–742. doi: 10.1002/cyto.a.20896. [DOI] [PubMed] [Google Scholar]
- 34.De Maesschalck R, Jouan-Rimbaud D, Massart DL. Chemometrics and intelligent laboratory systems. 2000;50:1–18. [Google Scholar]
- 35.Xiang S, Nie F, Zhang C. Pattern Recognition. 2008;41:3600–3612. [Google Scholar]
- 36.Goldstein JL, Brown MS, Anderson RG, Russell DW, Schneider WJ. Annual review of cell biology. 1985;1:1–39. doi: 10.1146/annurev.cb.01.110185.000245. [DOI] [PubMed] [Google Scholar]
- 37.Maffre P, Nienhaus K, Amin F, Parak WJ, Nienhaus GU. Beilstein Journal of Nanotechnology. 2011;2:374–383. doi: 10.3762/bjnano.2.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lara S, Alnasser F, Polo E, Garry D, Lo Giudice MC, Hristov DR, Rocks L, Salvati A, Yan Y, Dawson KA. ACS nano. 2017;11:1884–1893. doi: 10.1021/acsnano.6b07933. [DOI] [PubMed] [Google Scholar]
- 39.Kelly PM, Åberg C, Polo E, O’Connell A, Cookman J, Fallon J, Krpetić Ž, Dawson KA. Nature nanotechnology. 2015;10:472–479. doi: 10.1038/nnano.2015.47. [DOI] [PubMed] [Google Scholar]
- 40.O’Connell D, Bombelli FB, Pitek A, Monopoli M, Cahill D, Dawson K. Nanoscale. 2015;7:15268–15276. doi: 10.1039/c5nr01970b. [DOI] [PubMed] [Google Scholar]
- 41.Giudice MCL, Herda LM, Polo E, Dawson KA. Nature Communications. 2016;7:13475. doi: 10.1038/ncomms13475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hristov DR, Rocks L, Kelly PM, Thomas SS, Pitek AS, Verderio P, Mahon E, Dawson KA. Scientific reports. 2015;5:17040. doi: 10.1038/srep17040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bertoli F, Garry D, Monopoli MP, Salvati A, Dawson KA. ACS nano. 2016;10:10471–10479. doi: 10.1021/acsnano.6b06411. [DOI] [PubMed] [Google Scholar]
- 44.Zhang XQ, Xu X, Bertrand N, Pridgen E, Swami A, Farokhzad OC. Advanced drug delivery reviews. 2012;64:1363–1384. doi: 10.1016/j.addr.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mo ZC, Ren K, Liu X, Tang ZL, Yi GH. Advanced drug delivery reviews. 2016;106:132–147. doi: 10.1016/j.addr.2016.04.030. [DOI] [PubMed] [Google Scholar]
- 46.Manders E, Verbeek F, Aten J. Journal of microscopy. 1993;169:375–382. doi: 10.1111/j.1365-2818.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]
- 47.Bolte S, Cordelieres F. Journal of microscopy. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 48.Dunn KW, Kamocka MM, McDonald JH. American Journal of Physiology-Cell Physiology. 2011;300:C723–C742. doi: 10.1152/ajpcell.00462.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Di Rienzo C, Gratton E, Beltram F, Cardarelli F. Proceedings of the National Academy of Sciences. 2013;110:12307–12312. doi: 10.1073/pnas.1222097110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hebert B, Costantino S, Wiseman PW. Biophysical journal. 2005;88:3601–3614. doi: 10.1529/biophysj.104.054874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barrán-Berdón AL, Pozzi D, Caracciolo G, Capriotti AL, Caruso G, Cavaliere C, Riccioli A, Palchetti S, Laganaì A. Langmuir. 2013;29:6485–6494. doi: 10.1021/la401192x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.