

Abstract
An automated dried blood spot (DBS) elution coupled with solid phase extraction and tandem mass spectrometric analysis for multiple fentanyl analogs was developed and assessed. This method confirms human exposures to fentanyl, sufentanil, carfentanil, alfentanil, lofentanil, α-methyl fentanyl, and 3-methyl fentanyl in blood with minimal sample volume and reduced shipping and storage costs. Seven fentanyl analogs were detected and quantitated from DBS made from venous blood. The calibration curve in matrix was linear in the concentration range of 1.0 ng/mL to 100 ng/mL with a correlation coefficient greater than 0.98 for all compounds. The limit of detection varied from 0.15 ng/mL to 0.66 ng/mL depending on target analyte. Analysis of the entire DBS minimized the effects of hematocrit on quantitation. All quality control materials evaluated resulted in <15% error; analytes with isotopically labeled internal standards had <15% RSD, while analytes without matching standards had 15–24% RSD. This method provides an automated means to detect seven fentanyl analogs, and quantitate four fentanyl analogs with the benefits of DBS at levels anticipated from an overdose of these potent opioids.
Background
Dried blood spots (DBS) have many advantages over traditional clinical sample formats including increased analyte stability, small sample volume (<20 μL), decreased infection hazard for those handling samples, and reduced cost for shipping and storing. Used extensively for neonatal screening since the 1960s,1 improvements in analytical instrumentation sensitivity has made DBS a convenient biological matrix for pharmaceutical studies and exposure analyses.2–4
Traditional preparation of DBS often requires a manually intensive process of punching, extracting each punch individually, concentrating, and reconstituting prior to analysis of each spot. Automation of extraction processes minimizes the number of manual steps, reducing potential errors. Recent technologies for automating the extraction and/or mass spectrometric detection of DBS include paper spray,5 thin-layer chromatography,6,7 liquid-surface analysis,8 and on-line DBS sample extraction with and without additional clean-up.9–18 Flow-through extraction of DBS coupled to on-line SPE has also been developed;19 this system performs an automated desorption of DBS, complete with the addition of internal standard, followed by online SPE with subsequent analysis by a mass spectrometer. Incorporation of a large size (6-mm) clamp accommodates desorption of an entire small volume (~5 μL) DBS, minimizing quantitation challenges resulting from spot spread caused by hematocrit variations among blood samples.16,20,21 Additionally, the option for heated desorption solvent, has shown improvements to variations in extraction due to hematocrit.20,22,23 The automated desorption of DBS has been applied to multiple analytes with mass spectrometric analysis.15,17,20,24,25 More specifically, fentanyl has been quantitated from DBS using automated desorption but required separate SPE and LC separation.25 Fentanyl and two analogs have also been detected from DBS using manual extraction with HPLC-MS/MS.26,27
Fentanyl, initially synthesized by Janssen Pharmaceuticals in the 1960s, was the first in a class of potent opioid analgesics developed for chronic pain treatment, palliative care, and use as an anesthetic. Multiple analogs of fentanyl have been developed with varying potencies for use in the medical and veterinary fields.28 Analogs with no approved medical use have also been synthesized and are restricted as Schedule I compounds by the US Drug Enforcement Agency (DEA).29 As a strong opioid, fentanyl and its derivatives are frequently misused, resulting in overdoses and deaths.28 The drug poisoning rate involving opioid analgesics has approximately quadrupled from 1999 to 2013 with over 16,000 opioid-related deaths occurring in 2013.31 Fentanyl analogs have reportedly been misused in the US and Europe,30, 32–34 and as a result these compounds are an increasing public health concern.
Fentanyl exposures are confirmed through the analysis of clinical samples.35–41 Following overdose, the unaltered drug has been detected at concentrations ranging from 1.4–383 ng/mL in whole blood.35,36,37,38,40 Although metabolites may also be present, they are detected at lower levels in blood than the unaltered drug; the use of other pharmaceuticals may further reduce the measured amount of metabolites.39
This paper describes the development and validation of an automated method for the quantitation of seven fentanyl analogs in DBS using flow through desorption coupled to on-line solid phase extraction tandem mass spectrometry. The reported method incorporates isotopically labeled internal standards for four of the seven analogs. To our knowledge, no method has been developed for the analysis of multiple fentanyl analogs using automated DBS extraction. This work is critical for addressing the public health concern created by the misuse of these compounds.
Experimental
Appropriate safety control measures (including engineering, administrative, and personal protective equipment) were used for all procedures based on a site-specific risk assessment that identified physical, health, and procedural hazards.
Chemicals, standards, and reagents
Fentanyl and 2H5-fentanyl were purchased from Cerilliant (Round Rock, TX). Carfentanil, sufentanil, and their corresponding N-phenyl-2H5 labeled forms, as well as 13C6-alfentanil HCl were custom synthesized by Battelle (Columbus, OH). Lofentanil, alfentanil, 3-methyl fentanyl, and α-methyl fentanyl, were generous gifts from a variety of sources listed in the Acknowledgements. Structures of all target analytes are shown in Figure 1. High-performance liquid chromatography (HPLC)-grade methanol and acetonitrile were purchased from Tedia Company, Inc. (Fairfield, OH). Formic acid (99%) and ammonium hydroxide (28.6%) were purchased from Sigma Aldrich (Pittsburgh, PA). Deionized (DI) water (>18MΩ·cm) was prepared on-site using an installed water purification system (Aqua Solutions, Inc., Jasper, GA). Pooled whole blood and a convenience set of individual whole blood samples were purchased from Tennessee Blood Services (Memphis, TN). Use of deidentified spiked blood from a commercial source was not deemed to constitute human subject research.
Figure 1.
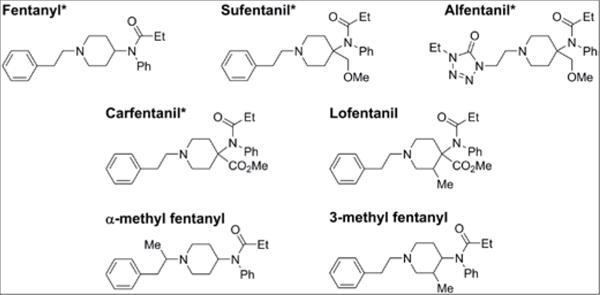
Structures of fentanyl analogs with * indicating isotopically labeled internal standard. Analogs with out matched internal standard used carfentanil as a suragate internal standard.
Stocks and working solutions
Individual stock solutions for each analyte were prepared at 10.0 μg/mL in DI water. From the stock solutions, a working solution containing all fentanyl analogs at 500 ng/mL was prepared volumetrically in DI water. This working solution was then diluted to create solution A at 50.0 ng/mL in DI water and solution B at 5.00 ng/mL. For internal standards, individual stock solutions were prepared at 100 μg/mL in DI water (2H5-fentanyl, 2H5-carfentanil, and 2H5-sufentanil) and at 20.0 μg/mL in DI water (13C6-alfentanil HCl). These stocks were diluted to prepare a mixture of all labeled compounds at 25.0 ng/mL in DI water. All solutions were stored at −20°C.
Materials preparation
Calibrators were prepared volumetrically in pooled whole human blood using the working solutions to achieve final concentrations of 1.00, 10.0, 25.0, 50.0, and 100 ng/mL. Two quality control standards (QCs) were prepared in the same manner as the calibrators at low (QL) and high (QH) concentrations of 7.50 and 75.0 ng/mL, respectively, also in pooled whole blood. All calibrators, QC samples, and individual blood samples were spotted at 5.00 μL onto FTA DMPK-C blood spot cards (GE LifeSciences, Pittsburgh, PA). The cards were allowed to dry at ambient temperature for a minimum of two hours before analysis or storage with desiccant at −20°C.
Instrumentation
Flow-through extraction of all dried blood spots was performed using a prototype device from Spark Holland (Emmen, Netherlands) consisting of an automated cartridge exchanger (ACE), a high pressure dispensing pump (HPD) for solvent delivery, two high performance liquid chromatography (HPLC) pumps, and a DBS card autosampler (DBSA) with 6-mm clamp head and 20 μL sample loop for internal standard addition. Analytes were detected using an AB Sciex 6500 Triple Quadrupole Mass Spectrometer (MS) (Foster City, CA). Individual whole blood samples were centrifuged in a capillary tube at 12000 rpm using a M24 Hematocrit Centrifuge (LW Scientific, Lawrenceville, GA) for measurement of hematocrit using a Micro-Capillary Reader (Damon/IEC Division, Needham Heights, MA).20
On-line DBS sample preparation, extraction procedure, and analysis
Analytes were desorbed using 1.2 mL of heated (100°C) 15:85 methanol: aqueous 1% formic acid. Internal standard was added using a 20 μL sample loop with the desorption solution prior to contact with the DBS. Analytes and internal standard were then trapped on a HySphereTM C18 HD SPE cartridge (7 μM, 10 × 2.0 mm ID) (Spark Holland, Emmen, Netherlands) which was preconditioned with 1 mL of methanol and equilibrated with 1 mL of aqueous 1% formic acid. The analytes were eluted off the SPE cartridge directly onto the mass spectrometer with a 3 minute gradient of 100% aqueous 1% formic acid to 100% acetonitrile at a flow rate of 0.5 mL/min. Each SPE cartridge was only used once.
Analytes were measured using TuboIonSpray® MS/MS in positive ion mode. Two transitions were monitored for each analyte and one transition was monitored for each internal standard, shown in supplemental table 1. Analyte-specific MS parameters were optimized for highest signal. Additional parameters used during analysis include the following values: curtain gas (CUR), 40 psi; nebulizer gas (GS1), 40 psi; turbo gas (GS2), 40 psi; turbo gas temperature (TEM), 550°C with the interface heater (IHE) on; collision gas (CAD), 7 producing a pressure reading of nitrogen @ 2.0 × 10–5 Torr; ionspray potential (IS), 4200 V; and entrance potential (EP), 10 V.
Data Processing
Data analysis was performed using Analyst software (Sciex, Version 1.5.1). Linear regression analysis of the calibrator concentration versus the ratio of the quantification ion area to the internal standard ion area with 1/x weighting was used for quantitation. Isotopically-labeled analytes were used as internal standards for corresponding unlabeled analogs. Isotopically-labeled carfentanil was also used as internal standard for lofentanil, α-methyl fentanyl, and 3-methyl fentanyl due to similar structure and retention time. All calibration curves meeting the correlation coefficient requirement of 0.980 or greater were accepted for use.
Method Characterization
Data from 20 replicate calibration curves and QC samples were evaluated to assess accuracy and precision over 8 weeks with two analysts running a maximum of two curves per day. Potential matrix interferences (selectivity) were evaluated by analyzing 20 unidentified patient samples from individuals with no anticipated exposure to fentanyls. Ten individual whole blood samples were also fortified at 6.0 and 30 ng/mL, five at each spiking level. Each individual blood sample was analyzed in triplicate.
Matrix Effects
Matrix effects were evaluated by adding the analyte mixtures (solution A and solution B) to the desorption solvent using the internal standard sample loop described above. Both solutions were passed through a blank card as well as DBS from two different blood pools. Area counts were used to calculate matrix effects for each compound using the following equation:
Matrix Effects (%) = ((Response with blank card matrix -Response with DBS matrix)/(Response with blank card matrix))×100%
Limit of Detection
The limit of detection (LOD) was determined from the 20 replicate results obtained for the four lowest calibrators. The deviation of the blank (S0) for each analyte in this method was extrapolated from the plot of the standard deviations of the lowest four calibrators versus their respective concentrations. S0, the y-intercept of the linear regression analysis of these data points, was multiplied by three to determine the LOD.41 These results were confirmed by the analysis of low level spikes at the estimated LOD values made in the same manner as the calibrators and QCs.
Results and Discussion
Automated DBS extraction, online-SPE and MS conditions
Automated DBS extraction was applied to the analysis of multiple fentanyl analogs to promote the use of DBS as a sampling mechanism. This novel instrument design introduced internal standard to the sample during desorption of the analytes, reducing manual steps required for analysis and possibly enabling field collection of DBS. Initial mass spectrometric parameters were determined by individually infusing each compound into the electrospray source and optimizing for highest response. Two mass spectrometric transitions were selected per compound to achieve maximum sensitivity and minimal matrix interferences.
Multiple online-SPE cartridges were investigated for compatibility with the automated DBS desorption. These sorbents included Oasis® HLB (Waters, Milford, MA), which was previously used for these compounds41 Resin SH, C2-SE, CN-SE, C8-SE, C18 HD, Resin GP, MM anion, and MM cation (HySphere™) (Spark, Emmen, Netherlands). The C18 HD cartridge, which has been used previously with this system for similar compounds25 gave the highest response with baseline resolution from matrix peaks for all analytes, and thus was selected for further development.
Desorption of fentanyl analogs from the DBS was optimized for several solvent conditions including composition, volume, temperature, and dispensing speed. To ensure retention of the analytes on reverse phase SPE, water was initially used for desorption. Formic acid was added to release any analogs potentially bound to proteins in the DBS;43 the analytes were found to be poorly desorbed with this solution. Given the high solubility of fentanyl in methanol, the addition of methanol to the desorption solvent was investigated to improve recovery. A mixture of 85% aqueous 1% formic acid and 15% methanol was selected to maximize desorption while maintaining sufficient water content to also maximize retention on the SPE cartridge for all analytes. Solvent extraction volume was evaluated from 0.50 to 1.5 mL; an increase in response for all analytes was seen until 1.2 mL, so this volume was selected. The desorption solvent temperature was tested at 50°C, 75°C, 100°C, and at ambient temperature (~25°C). A slight increase in response was seen as temperatures increased, so 100°C was chosen as the optimal temperature. This higher solvent temperature may overcome extraction challenges resulting from hematocrit variations.20 Dispensing speed of the solvent did not impact desorption; therefore, a nominal value of 2000 μL/min was used.
Elution from the online SPE cartridge was evaluated using both methanol and acetonitrile. Acetonitrile was selected as the strong elution solvent and resulted in improved peak shape and rapid elution of all compounds (Figure 2). Baseline separation was not achieved for all compounds, of particularly concern, the constitutional isomers α-methyl fentanyl and 3-methyl fentanyl were not separated under these conditions. To address this issue, unique quantitation transitions were selected for each compound to prevent signal contribution between all analytes, including the isomers as previously documented.42 Because baseline resolution from detectable matrix interferences was achieved with SPE alone, no additional separation was deemed necessary for this method.
Figure 2.
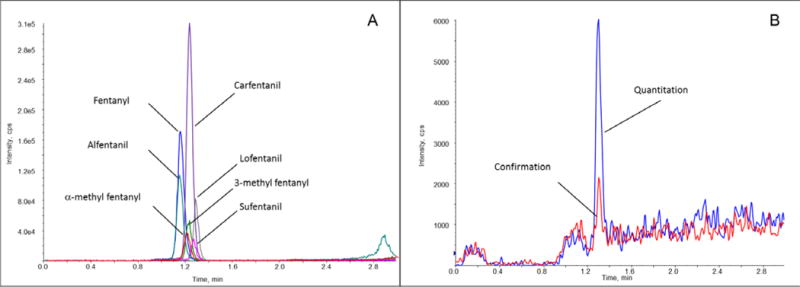
SPE-MS/MS chromatogram of fentanyl analogs eluted from 5 μL dried blood spots. Extracted ion chromatograms of (A) all analytes at a concentration of 7.5 ng/mL in pooled whole blood and of (B) sufentanil at a concentration of 1.0 ng/mL.
Matrix effects
Matrix effects were investigated in two blood pools at two concentrations, 5.0 ng/mL and 50 ng/mL and analysed in triplicate. Data shown in Table 1 is from one pool at a single concentration, as both blood pools gave comparable results. As shown in Table 1, the matrix effects were high (68–96%) for all analytes at the 50 ng/mL concentration; similar matrix effects were seen at the lower concentration ranging from 58–91%. These high matrix effects were not unexpected as the DBS was extracted with a significant aqueous content, solubilizing many components of the blood which can suppress signal. Comparable matrix effects have been noted in urine for reverse phase SPE followed by LC-MS/MS.42 Additionally, an in-house evaluation of plasma using similar SPE-LC-MS/MS parameters42 resulted in matrix effects ranging 19–93% for the same fentanyls and normetabolites. The combination of SPE and HPLC has not significantly decreased matrix effects for these analytes from clinical samples. For this study, further reduction of matrix effects was not expected with the addition of HPLC separation. The main concern with high matrix effects remains the loss of sensitivity; however, the limit of detection (Table 1) for each analyte was lower than values expected following overdose (1.4–383 ng/mL).35,36,37,38,40
Table 1.
Matrix effects in % and limit of detection (LOD). Matrix effects were calculated at a concentration of 50 ng/mL (N=3).
| Analyte | Matrix Effects (%) | Calculated LOD (ng/mL) |
|---|---|---|
|
| ||
| Fentanyl | 80 ± 4 | 0.16 |
| Sufentanil | 96 ± 1 | 0.24 |
| Carfentanil | 83 ± 4 | 0.25 |
| Alfentanil | 92 ± 1 | 0.15 |
| Lofentanil | 68 ± 3 | 0.35 |
| α-methyl fentanil | 83 ± 4 | 0.66 |
| 3-methyl fentanil | 88 ± 3 | 0.56 |
Method Characterization
This analytical method was characterized by evaluating 20 sets of calibration curves and QC samples in matrix at concentrations of 7.5 ng/mL and 75 ng/mL. Calibrators in dried blood spots were extracted and analyzed with the QC samples over the course of two months by two analysts. Sensitivity, accuracy, and precision were calculated based on the quantitation ion transition. The precision, defined by the %RSD, was <15% for analytes with matched isotopically labeled internal standards; for analytes without labeled internal standard, precision ranged from 15–24% (Table 2). The accuracies for the QC samples and the lowest calibrator were within ±10% of the expected value for all analytes. For the four analytes with matched internal standards, accuracy and precision were within the FDA guidelines for biomedical testing.44
Table 2.
Accuracy and precision for QC samples and lowest calibrator, N=20.
| Lowest Calibrator (1.0 ng/mL) | QC Low (7.5 ng/mL) | QC High (75 ng/mL) | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| % RSD | % Error | % RSD | % Error | % RSD | % Error | |
|
| ||||||
| Fentanyl* | 9.5 | −7.8 | 6.6 | −2.3 | 5.1 | −4.1 |
| Sufentanil* | 11 | −6.3 | 9.8 | −7.4 | 6.5 | −4.9 |
| Carfentanil* | 14 | −8.3 | 11 | −7.4 | 6.4 | −5.9 |
| Alfentanil* | 10 | −9.1 | 6.8 | −4.4 | 6.6 | −3.6 |
| Lofentanil | 16 | −4.3 | 15 | −5.6 | 15 | −4.2 |
| α-methyl fentanll | 25 | −5.0 | 24 | −4.6 | 19 | −7.2 |
| 3-methyl fentanil | 20 | −2.3 | 21 | −5.3 | 18 | −5.3 |
Represents analogs with isotopically labeled matched internal standards.
Linearity of the calibrator curves was confirmed through the back calculation of the calibrators, which resulted in mean accuracies within 10% for all analytes. Corresponding RSDs from the 20 analytical runs was less than 16% for all analytes, with the exception of α- and 3-methyl fentanyl with variances up to 25%. All curves achieved a correlation coefficient of 0.980 or higher. To confirm the selection of regression weighting, the sum of residuals were compared between no weighting, 1/×, and 1/×2; 1/× weighting resulted in a lower sum of residuals than no weighting. Although 1/×2 weighting had a lower sum of residuals overall, the correlation coefficient did not meet the criteria; therefore, 1/× weighting was selected.
The linear range for this method was established to confirm overdose exposures to these fentanyls, with calibrators from 1.0–100 ng/mL. The calculated LODs ranged from 0.15–0.66 ng/mL (Table 1); however, to ensure the confirmation ion was detected for all analytes, the lowest calibrator was set at 1.0 ng/mL (Figure 2 and Supplementary Figure 1). It should also be noted that sensitivity was greatly impacted by the small sample volume used in this method. While past urinary methods had a lower limit of quantitation of 0.050 ng/mL, these methods required a 100-fold increase in sample volume (500 μL) plus a concentration step.42 For a 1.0 ng/mL sample the DBS method evaluated here injected 0.005 ng on cartridge, whereas 0.1 ng were injected on column for the urinary SPE-LC-MS/MS method. This significant difference confirms that sample volume plays a critical role in sensitivity and is the most limiting factor for this DBS method.
Carry-over between samples was also investigated, as this was identified as a concern in a previous study with this instrument.25 A blank DBS sample was evaluated following the highest calibrator on every run. There were no detectable responses in this blank sample, confirming that carry-over using a 5 μL spot was not an issue for this method.
A convenience set of twenty individual blood samples with no known exposure to these compounds were analyzed to measure any endogenous interferences. No interferences were observed at the anticipated retention time for either the quantitation or confirmation transition for any of the fentanyl analytes. These results indicated that this method is selective for fentanyl analogs and should not result in false positives.
Individual blood samples fortified at 6.0 ng/mL and 30 ng/mL were evaluated for accuracy and precision (N=3). The hematocrit of these samples was measured and ranged from 34.8% to 48.0% which is in the normal range for healthy adults. The 30 ng/mL spike level demonstrated high precision and accuracy for fentanyl, sufentanil, carfentanil, lofentanil, and alfentanil; however, α-methyl fentanyl and 3-methyl fentanyl showed more variability (Figure 3). Similar results were observed for all analytes for the 6.0 ng/mL spike level with the exception of alfentanil, which was biased low at this concentration. Although hematocrit has been documented to negatively impact quantitation from DBS,21–23,45,46 no trends associated between concentration and hematocrit level was observed, therefore, no correlation was identified.
Figure 3.
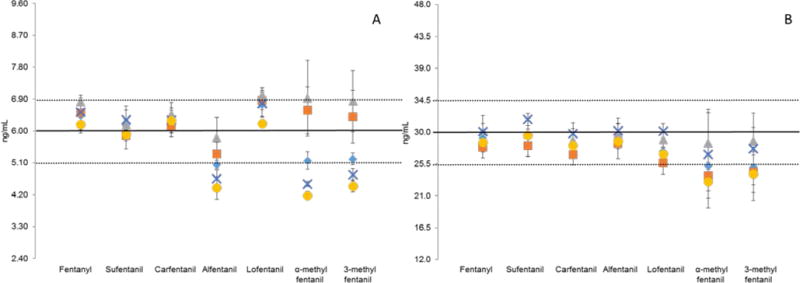
Analysis of individual blood samples (A) five fortified at 6.0 ng/mL and (B) five fortified at 30 ng/mL, N=3. The dotted lines represent 15% precision from spiked value.
Conclusions
An automated method for desorption, extraction, and detection of seven fentanyl analogs from DBS has been developed and validated. Accuracy values for all analytes and precision for analytes with matching isotope enriched internal standards, except for alfentanil at 6.00 ng/mL, were within the FDA specifications for bioanalytical methods.43 Thus this method can be used to quantitate four fentanyl analogs. With a reportable range of 1.0–100 ng/mL, concentrations measured in blood products from overdose cases can be detected.33–36,38 Although, additional evaluation of clinical samples could necessitate a lower limit of detection as the potency for these analogs can vary greatly. Analysis of simulated clinical samples confirmed the ability of this method to positively identify fentanyl analogs in individual samples. Use of automated preparation with online SPE amenable to DBS samples provided a streamlined laboratory workflow and minimized analyst exposure to potentially hazardous substances while maximizing sample throughput. Future work should include analysis of clinical samples to determine the clinical relevant range for all analytes and investigation of samples with hematocrit outside the normal range.
Supplementary Material
Acknowledgments
This study was possible due to the support of a number of people and organizations who provided instrumentation and analytical standards. For instrumentation, we would like to thank Spark Holland and iChrom Solutions for the generous loan of the dried blood spot autosampler as well as for providing instrumental support for the set up and running of the autosampler. For the valuable contribution of analytical standards, we would like to thank Dr. Rita McManamon, Atlanta Fulton County Zoo (Atlanta, GA); the DEA Western Laboratory (San Francisco, CA); Dr. C. Randall Clark, School of Pharmacy, Auburn University (Auburn, AL); Dr. Thomas Tobin, Department of Veterinary Science, University of Kentucky (Lexington, KY); Dr. France Varin, Faculty of Pharmacy, University of Montreal (Quebec, Canada); Dr. Jim Baron, Battelle Columbus Laboratories (Columbus, OH); and the NIDA Drug Supply Program.
Footnotes
Disclaimer
The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention. Use of trade names is for identification only and does not imply endorsement by the Centers for Disease Control and Prevention, the Public Health Service, or the US Department of Health and Human Services.
Notes and references
- 1.Gunthrie R, Susi A. A Simple Phenylalanine Method for Detecting Phenylketonuria in Large Populations of Newborn Infants. Pediatrics. 1963;32:338–343. [PubMed] [Google Scholar]
- 2.Demirev PA. Dried Blood Spots: Analysis and Applications. Anal Chem. 2013;85:779–789. doi: 10.1021/ac303205m. [DOI] [PubMed] [Google Scholar]
- 3.Meesters RJ, Hooff GP. State-of-the-art dried blood spot analysis: an overview of recent advances and future trends. Bioanalysis. 2016;5(17):2187–2208. doi: 10.4155/bio.13.175. [DOI] [PubMed] [Google Scholar]
- 4.Zimmer JS, Christianson CD, Johnson CJ, Needham SR. Recent advances in the bioanalytical applications of dried matrix spotting for the analysis of drugs and their metabolites. Bioanalysis. 2013;5(20):2581–2588. doi: 10.4155/bio.13.214. [DOI] [PubMed] [Google Scholar]
- 5.Yang Q, Manicke NE, Wang H, Petucci C, Cooks RG, Ouyang Z. Direct and quantitative analysis of underivatized acylcarnitines in serum and whole blood using paper spray mass spectrometry. Anal Bioanal Chem. 2012;404(5):1389–1397. doi: 10.1007/s00216-012-6211-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abu-Rabie P, Spooner N. Direct quantitative bioanalysis of drugs in dried blood spot samples using a thin-layer chromatography mass spectrometer interface. Anal Chem. 2009;81(24):10275–10284. doi: 10.1021/ac901985e. [DOI] [PubMed] [Google Scholar]
- 7.Abu-Rabie P, Spooner N. Dried matrix spot direct analysis: evaluating the robustness of a direct extraction technique for use in quantiative bioanalysis. Bioanalysis. 2011;3(24):2769–2781. doi: 10.4155/bio.11.270. [DOI] [PubMed] [Google Scholar]
- 8.Kertesz V, Van Berkel GJ. Liquid microjunction surface sampling coupled with high-pressure liquid chromatography-electrospray ionization-mass spectrometry for analysis of drugs and metabolites in whole-body thin tissue sections. Anal Chem. 2010;82(14):5917–5921. doi: 10.1021/ac100954p. [DOI] [PubMed] [Google Scholar]
- 9.Deglon J, Thomas A, Cataldo A, Mangin P, Staub C. On-line desorption of dried blood spot: a novel approach for the direct LC/MS analysis of micro-whole blood samples. J Pharm Biomed Anal. 2009;49(4):1034–1039. doi: 10.1016/j.jpba.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 10.Deglon J, Thomas A, Mangin P, Staub C. Direct analysis of dried blood spots coupled with mass spectrometry: concepts and biomedical applications. Anal Bioanal Chem. 2012;402(8):2485–2498. doi: 10.1007/s00216-011-5161-6. [DOI] [PubMed] [Google Scholar]
- 11.Miller JH, 4th, Poston PA, Karnes HT. Direct analysis of dried blood spots by in-line desorption combined with high-resolution chromatography and mass spectrometry for quantification of maple syrup urine disease biomarkers leucine and isoleucine. Anal Bioanal Chem. 2011;400(1):237–244. doi: 10.1007/s00216-011-4740-x. [DOI] [PubMed] [Google Scholar]
- 12.Ganz N, Singrasa M, Nicolas L, Gutierrez M, Dingemanse J, Dobelin W, Glinski M. Development and validation of a fully automated online human dried blood spot analysis of bosentan and its metabolites using the Sample Card And Prep DBS System. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;885:50–60. doi: 10.1016/j.jchromb.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 13.Oliveira RV, Henion J, Wickremsinhe E. Fully-automated approach for online dried blood spot extraction and bioanalysis by two-dimentional-liquid chromatography coupled with high-resolution quadrupole time-of-flight mass spectrometry. Anal Chem. 2014;86(2):1246–1253. doi: 10.1021/ac403672u. [DOI] [PubMed] [Google Scholar]
- 14.Crawford E, Gordon J, Wu JT, Musselman B, Liu R, Yu S. Direct analysis in real time coupled with dried blood spot sampling for bioanalysis in a drug-discovery setting. Bioanalysis. 2011;3(11):1217–1226. doi: 10.4155/bio.11.99. [DOI] [PubMed] [Google Scholar]
- 15.Oliveira RV, Henion J, Wickremsinhe ER. Automated direct extraction and analysis of dired blood spots employing on-line SPE high-resolution accurate mass bioanalysis. Bioanalysis. 2014;6(15):2027–2041. doi: 10.4155/bio.14.162. [DOI] [PubMed] [Google Scholar]
- 16.Oliveira RV, Henion J, Wickremsinhe ER. Automated high-capacity on-line extraction and bioanalysis of dried blood spot samples using liquid chromatography/high-resolution accurate mass spectrometry. Rapid Commun Mass Spectrom. 2014;28(22):2415–2426. doi: 10.1002/rcm.7033. [DOI] [PubMed] [Google Scholar]
- 17.Ooms JA, Knegt L, Koster EH. Exploration of new concept for automated dried blood spot analysis using flow-through extraction and online SPE-MS/MS. Bioanalysis. 2011;3(20):2311–2320. doi: 10.4155/bio.11.214. [DOI] [PubMed] [Google Scholar]
- 18.Verplaetse R, Tytgat J. Development and validation of a sensitive UPLC-MS/MS method for the analysis of narcotic analgesics in urine and whole blood in forensic context. Forensic Sci Int. 2012;215(1–3):136–145. doi: 10.1016/j.forsciint.2011.01.047. [DOI] [PubMed] [Google Scholar]
- 19.Spark Holland. http://www.sparkholland.com (accessed Oct 26, 2016)
- 20.Hempen CM, Maarten Koster EH, Ooms JA. Hematocrit-idependent recovery of immunosuppressants from DBS using heated flow-through desorption. Bioanalysis. 2015;7(16):2019–2029. doi: 10.4155/bio.15.97. [DOI] [PubMed] [Google Scholar]
- 21.Denniff P, Spooner N. The effect of hematocrit on assay bias when using DBS samples for the quantitative bioanalysis of drugs. Bioanalysis. 2010;2(8):1385–1395. doi: 10.4155/bio.10.103. [DOI] [PubMed] [Google Scholar]
- 22.De Kesel PMM, Sadones N, Capiau S, Lambert WE, Stove CP. Hemato-critical issues in quantitative analysis of dried blood spots: challenges and solutions. Bioanalysis. 2013;5(16):2023–2041. doi: 10.4155/bio.13.156. [DOI] [PubMed] [Google Scholar]
- 23.Abu-Rabie P, Denniff P, Spooner N, Chowdhry BZ, Pullen FS. Investigation of Different Approaches to Incorporating Internal Standard in DBS Quantitative Bioanalytical Workflows and Their Effect on Nullifying Hematocrit-Based Assay Bias. Anal Chem. 2015;87:4496–5003. doi: 10.1021/acs.analchem.5b00908. [DOI] [PubMed] [Google Scholar]
- 24.Thompson JW, Zhang H, Smith P, Hillman S, Moseley MA, Millington DS. Extraction and analysis of carnitine and acylcarnitines by electrospray ionization tandem mass spectrometry directly from dried blood and plasma spots using a novel autosampler. Rapid Commun Mass Spectrom. 2012;26(21):2548–2554. doi: 10.1002/rcm.6370. [DOI] [PubMed] [Google Scholar]
- 25.Verplaetse R, Henion J. Quantitative determindation of opioids in whole blood using fully automated dried blood spot desorption coupled to on-line SPE-LC-MS/MS. Drug Test Anal. 2016;8:30–38. doi: 10.1002/dta.1927. [DOI] [PubMed] [Google Scholar]
- 26.Odoardi S, Anzillotti L, Strano-Rossi S. Simplifying sample pretreatment: application of dried blood spot (DBS) method to blood samples, including postmortem, for UHPLC-MS/MS analysis of drugs of abuse. Forensic Sci Int. 2014;243:61–67. doi: 10.1016/j.forsciint.2014.04.015. [DOI] [PubMed] [Google Scholar]
- 27.Clavijo CF, Thomas JJ, Cromie M, Schniedewind B, Hoffman KL, Christians U, Galinkin JL. A low blood volume LC-MS/MS assay for the quantification of fentanyl and its major metabolites norfentanyl and despropionyl fentanyl in children. J Sep Sci. 2011;34(24):3568–3577. doi: 10.1002/jssc.201100422. [DOI] [PubMed] [Google Scholar]
- 28.Romagnoli A. Duration of Action of Fentanyl. Anesthesiology. 1973;39(5):568–569. doi: 10.1097/00000542-197311000-00034. [DOI] [PubMed] [Google Scholar]
- 29.Controlled Substance By DEA Drug Code Number. https://www.deadiversion.usdoj.gov/schedules/orangebook/d_cs_drugcode.pdf (Accessed Oct 26, 2016)
- 30.Centers for Disease Control and Prevention. Notes from the Field: Increase in Fentanyl-Related Overdose Deaths - Rhode Island, November 2013-March 2014. MMWR Morb Mortal Wkly Rep. 2014;63(24):531. [PMC free article] [PubMed] [Google Scholar]
- 31.Centers for Disease Control and Prevention. Quickstats: Rate of deaths from drug poisoning and drug poisoning involving opiod analgesics - United States, 1999–2013. MMWR Morb Mortal Wkly Rep. 2015;64:32. [Google Scholar]
- 32.Centers for Disease Control and Prevention. Notes from the Field: Acetyl Fentanyl Overdose Fatalities - Rhode Island, March-May 2013. MMWR Morb Mortal Wkly Rep. 2016;62(34):703–704. [PMC free article] [PubMed] [Google Scholar]
- 33.Marinetti LJ, Ehlers BJ. A series of forensic toxicology and drug seizure cases involving illicit fentanyl alone and in combination with heroin, cocaine or heroin and cocaine. J Anal Toxicol. 2014;38(8):592–598. doi: 10.1093/jat/bku086. [DOI] [PubMed] [Google Scholar]
- 34.Mounteney J, Giraudon I, Denissov G, Griffiths P. Fentanyls: Are we missing the signs? Highly potent and on the rise in Europe. Int J Drug Policy. 2015;26(7):626–631. doi: 10.1016/j.drugpo.2015.04.003. [DOI] [PubMed] [Google Scholar]
- 35.McIntyre IM, Gary RD, Estrada J, Nelson CL. Antemortem and postmortem fentanyl concentrations: a case report. Int J Legal Med. 2014;128(1):65–67. doi: 10.1007/s00414-013-0897-5. [DOI] [PubMed] [Google Scholar]
- 36.Moore PW, Palmer RB, Donovan JW. Fatal fentanyl patch misuse in a hospitalized patient with a postmortem increase in fentanyl blood concentration. J Forensic Sci. 2015;60(1):243–246. doi: 10.1111/1556-4029.12559. [DOI] [PubMed] [Google Scholar]
- 37.Smialek JE, Levine B, Chin L, Wu SC, Jenkins AJ. A fentanyl epidemic in Maryland 1992. J Forensic Sci. 1994;39:159–164. [PubMed] [Google Scholar]
- 38.Martin TL, Woodall KL, McLellan BA. Fentanyl-related deaths in Ontario, Canada: toxicological findings and circumstances of death in 112 cases (2002–2004) J Anal Toxicol. 2006;30(8):603–610. doi: 10.1093/jat/30.8.603. [DOI] [PubMed] [Google Scholar]
- 39.Saari TI, Laine K, Neuvonen M, Neuvonen PJ, Olkkola KT. Effect of voriconazole and fluconazole on the pharacokinetics of intravenous fentanyl. Eur J Clin Pharmacol. 2008;64:25–30. doi: 10.1007/s00228-007-0398-x. [DOI] [PubMed] [Google Scholar]
- 40.Garriott JC, Rodriguez R, Di Maio VJ. A death from fentayl overdose. J Anal Toxicol. 1984;8(6):288–289. doi: 10.1093/jat/8.6.288. [DOI] [PubMed] [Google Scholar]
- 41.Taylor JK. Quality Assurance of Chemical Measurements. Lewis Publishers; Boca Raton, Florida: 1987. pp. 79–82. [Google Scholar]
- 42.Shaner RL, Kaplan P, Hamelin EI, Bragg WA, Johnson RC. Comparison of two automated solid phase extractions for the detection of ten fentanyl analogs and metabolites in human urine using liquid chromatography tandem mass spectrometry. J Chromatogr B: Anal Technol Biomed Life Sci. 2014;962:52–58. doi: 10.1016/j.jchromb.2014.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meuldermans WE, Hurkmans RM, Heykants JJ. Plasma Protein Binding and Distrubution of Fentanyl, Sufentanil, Alfentanil and Lofentanil in Blood. Arch Int Pharmacodyn Ther. 1982;257(1):4–19. [PubMed] [Google Scholar]
- 44.Food and Drug Administration. UCM368107. FDA; MD, USA: 2013. Guidance For Industry - Bioanalytical Method Validation; pp. 1–34. [Google Scholar]
- 45.Jopling J, Henry E, Wiedmeier SE, Christensen RD. Reference ranges for hematocrit and blood hemoglobin concentration during neonatal period: data from a multihospital health care system. Pediatrics. 2009;123(2):e333–337. doi: 10.1542/peds.2008-2654. [DOI] [PubMed] [Google Scholar]
- 46.Fan L, Lee JA. Managing the effect of hematocrit on DBS analysis in a regulated environment. Bioanalysis. 2012;4(4):345–347. doi: 10.4155/bio.11.337. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.