

Abstract
Formaldehyde-cross-linking underpins many of the most commonly used experimental approaches in the chromatin field, especially in capturing site-specific protein–DNA interactions. Extending such assays to assess the stability and binding kinetics of protein–DNA interactions is more challenging, requiring absolute measurements with a relatively high degree of physical precision. We previously described an experimental framework called the cross-linking kinetics (CLK) assay, which uses time-dependent formaldehyde–cross-linking data to extract kinetic parameters of chromatin binding. Many aspects of formaldehyde behavior in cells are unknown or undocumented, however, and could potentially affect CLK data analyses. Here, we report biochemical results that better define the properties of formaldehyde–cross-linking in budding yeast cells. These results have the potential to inform interpretations of “standard” chromatin assays, including chromatin immunoprecipitation. Moreover, the chemical complexity we uncovered resulted in the development of an improved method for measuring binding kinetics with the CLK approach. Optimum conditions included an increased formaldehyde concentration and more robust glycine-quench conditions. Notably, we observed that formaldehyde–cross-linking rates can vary dramatically for different protein–DNA interactions in vivo. Some interactions were cross-linked much faster than the in vivo macromolecular interactions, making them suitable for kinetic analysis. For other interactions, we found the cross-linking reaction occurred on the same time scale or slower than binding dynamics; for these interactions, it was sometimes possible to compute the in vivo equilibrium-binding constant but not binding on- and off-rates. This improved method yields more accurate in vivo binding kinetics estimates on the minute time scale.
Keywords: chromatin immunoprecipitation (ChiP), chromatin structure, nucleic acid chemistry, protein cross-linking, protein dynamic, transcription factor, formaldehyde chemistry
Introduction
Gene regulation is a complicated and highly regulated process involving the coordinated assembly of dozens of proteins on promoter DNA within the context of chromatin (1–4). In vitro studies have provided a structurally detailed paradigm for how the transcription preinitiation complex (PIC)4 is assembled and regulated (5–13), but less is known about the dynamic assembly of PICs in vivo or how transcription factors (TFs) contribute kinetically to PIC assembly or to the rate of the initiation of synthesis of individual RNAs. To develop molecular models for how these processes occur in vivo, estimates of on- and off-rates for TF binding to specific loci in vivo are required. In instances in which kinetic measurements cannot be made, biophysically rigorous estimates of site-specific in vivo affinity (as opposed to estimates of relative affinity) and fractional occupancy would be valuable.
Chromatin immunoprecipitation (ChIP) is quite possibly the most widely used assay for characterizing the interactions between TFs and specific sites on chromatin. ChIP typically uses formaldehyde to cross-link TFs to their chromatin sites (14), and although it is an undeniably powerful approach for determining transcription factor-binding locations with high precision (3), standard ChIP assays are static measurements that do not provide unambiguous insight into the in vivo kinetics of these dynamic interactions. Several assays have expanded ChIP to attempt to capture these relationships. We previously developed a ChIP-based method, the cross-linking kinetics (CLK) assay, which exploits the time dependence of formaldehyde–cross-linking to model chromatin-TF–binding dynamics on a broad time scale and at individual loci (15). In this approach, cells are incubated with formaldehyde for various periods of time; unreacted formaldehyde is then quenched, and the extent of DNA site cross-linking of a TF of interest at each time point is quantified by ChIP. The time-dependent increase in ChIP signal results from a combination of time-dependent formaldehyde reactivity and time-dependent binding of free TF molecules to unoccupied DNA sites in the cell population. To distinguish kinetic effects of cross-linking chemistry from kinetic effects of TF binding, measurements are made using congenic cells differing only in the concentration of TF, and the data are fit using both sets of data simultaneously (15, 16).
A challenge with the development of locus-specific kinetic assays such as CLK is that aspects of the effects of formaldehyde on cells largely remain a black box (17), and validation of the extracted dynamic parameters is difficult because complementary approaches are still being developed, and there are few “gold standard” interactions with convergent kinetic measurements obtained by different approaches. Support for the CLK approach was obtained by measurement of binding dynamics for two TFs with very different dynamic properties that had been assessed by live-cell imaging (15, 18, 19). However, live-cell imaging has its own technical challenges (20), and in most cases, it is not possible to identify particular single copy chromatin sites of interaction by live-cell imaging (8, 21, 22). An alternative approach is competition ChIP, an assay that measures the rate of turnover between an endogenous and inducible copy of a TF. Our recent work demonstrates that quantitative estimates of locus-specific binding kinetics can be obtained by modeling competition ChIP data, including the estimation of residence times much shorter than the time for full induction of the competitor TF (23). Importantly, comparison of CLK and competition ChIP data for TATA-binding protein (TBP) to a few specific loci shows that the time scales for chromatin interaction are similar as judged by the two methods, with residence times for promoter binding being in general on the order of several minutes (23).
Nonetheless, locus-specific TF-chromatin dynamics are just beginning to be explored, with only a small number of TFs and chromatin sites for which CLK, competition ChIP, and/or live-cell imaging kinetic data are available. A key aspect of the CLK assay involves the trapping of bound species using formaldehyde. Here, we report biochemical results that better define the chemical behavior of formaldehyde in yeast cells. An increased formaldehyde concentration led to more rapid cross-linking, which improved the time resolution and analytical ability of the assay to extract locus-specific binding kinetic information for some TFs. For other TFs, an increased formaldehyde concentration resulted in their depletion from the soluble pool, and in some cases a rapid depletion was observed. These observations emphasize the importance of optimizing the CLK approach for analysis of the dynamic behavior of a particular TF. We report the development of a general and improved CLK method framework with both more rapid cross-linking and more efficient quenching in yeast cells. We also report improved computational methods for data analysis and describe improved approaches for distinguishing contributions of cross-linking rate and binding kinetics to the time-dependent increases in ChIP signal.
Results
The CLK method relies on time-resolved formaldehyde–cross-linking ChIP data to assess the kinetics and thermodynamics of TF-chromatin binding. The original CLK method (15, 16) employed 1% formaldehyde (360 mm), and reactions were quenched with 250 mm glycine (24, 25). Under these conditions, the concentration of glycine is sub-stoichiometric to the formaldehyde concentration as added, but cross-linking was performed by adding formaldehyde to cells in YPD medium, which is made from an amino acid-rich extract of yeast cells. Therefore, the concentration of unreacted formaldehyde that reaches cells under these conditions is unknown and is most likely well below the initial concentration. Order-of-addition experiments showed that 250 mm glycine could block cross-linking of the Gal4–promoter interaction (15), but we noted in subsequent work that quenching may be variably efficient under these conditions (26). Indeed, we have noticed that for unknown reasons the quench efficiency can be variable from experiment to experiment for certain TFs (supplemental Fig. S1) (27). To better define time-dependent cross-linking behavior and the impact of different quenching conditions on the resulting ChIP signals, data were obtained using 1% (360 mm) formaldehyde and either 250 mm or 2.93 m glycine using the interaction between yeast TBP-myc and the URA1 promoter as a model interaction. The high concentration of 2.93 m glycine used in this and subsequent experiments was the maximum achievable based on the solubility of glycine in aqueous solution (∼3 m) and subsequent dilution resulting from addition of a relatively small volume of concentrated yeast cell culture to the quenching solution (see “Experimental procedures”). For this reason, we refer to this as the “max glycine” quench condition hereafter. As shown in Fig. 1A, the max glycine-quench conditions resulted in lower ChIP signals at each time point compared with 250 mm glycine. These results demonstrate that the concentration of glycine used in the quench can have a significant effect on the magnitude of the ChIP signal, suggesting that more robust quenching of formaldehyde can be achieved with a higher concentration of glycine.
Figure 1.

Effect of different formaldehyde and quench conditions on TBP-myc ChIP signal at the URA1 locus. A, TBP-myc strain was cross-linked for varying amounts of time with 1% (360 mm) formaldehyde followed by quenching with either low (0.25 m, red line) or high (2.9 m, blue line) glycine. ChIP was performed followed by analysis with real-time PCR at the URA1 locus. Normalized ChIP signal is the IP signal minus mock signal divided by an input signal; values were determined from a standard curve. B, similar to A, but TBP-myc–expressing cells were cross-linked for varying amounts of time with 1% (blue line) or 4.7% (red line) formaldehyde and quenched with high (2.9 m) glycine. C, similar to B, but 1% (blue line) or 4.4% (red line), formaldehyde was used for cross-linking and 600 mm Tris, pH 8, for quenching. For each plot, two replicates were collected, and the error bars represent the standard deviation.
In addition to lower signals at each time point obtained using max glycine conditions, some time-dependent datasets showed initial shallow slopes, which continuously increase until the curve reaches apparent linear behavior at longer times (Fig. 1). We refer to this as “positive curvature.” This type of behavior has several possible explanations (discussed below), but none are accounted for in the original CLK model. To better understand how glycine concentration affected the time course of formaldehyde–cross-linking, experiments were performed to test both the dependence of the reaction on formaldehyde concentration and how ChIP data were affected using Tris, rather than glycine, to quench the reaction. Tris has been reported to be a robust quencher of formaldehyde reactivity (27). As shown in Fig. 1B, using max glycine-quenching conditions, the ChIP signal depended on the formaldehyde concentration, as reaction with 4.7% formaldehyde increased the ChIP signal at each time point compared with reactions that employed 1% formaldehyde. A dependence on formaldehyde concentration was also seen in reactions using Tris as the quenching agent (Fig. 1C). However, in reactions that were quenched with Tris, the ChIP signals obtained for a given concentration of formaldehyde were reduced compared with the values obtained using glycine, and the resulting reaction progress curves showed positive curvature similar to reactions quenched with max glycine discussed above.
Although Tris is apparently a more efficient quencher than glycine, it also has the potential to reverse cross-links (28, 29). Cross-link reversal would be problematic for the CLK assay as it could lead to underestimates of ChIP signal, with potentially large percentage-wise effects on the modest levels of cross-linked material obtained after short cross-linking times. To test the potential for reversal with both Tris and glycine, samples were cross-linked, quenched, and incubated at room temperature for different periods of time in the quenching solution. As shown in Fig. 2A, incubation of cells in Tris-containing solution led to a loss of TBP ChIP signal over time. The greatest effect was observed with 750 mm Tris, but the ChIP signal diminished when cells were incubated in 50 mm Tris as well. In contrast, there was no detectable decrease in TBP ChIP signal over time when cross-linked cells were incubated in max glycine solution (Fig. 2B). Thus, although Tris is a robust quenching agent, we ruled out its use in the assay because it decreased the recovery of cross-linked complexes.
Figure 2.
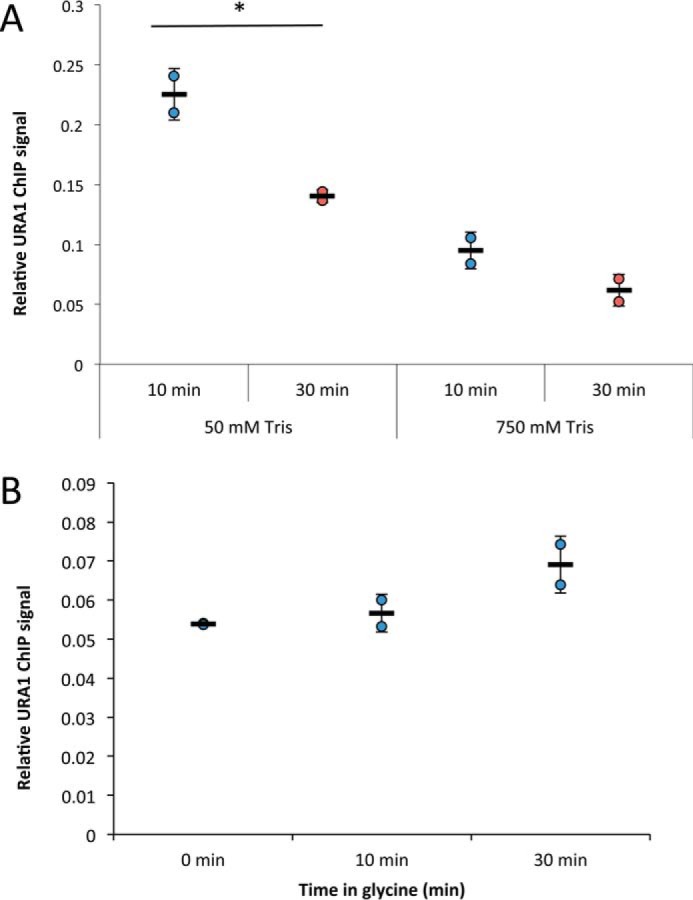
Tris, but not glycine, quenching reverses ChIP signal over time. A, average TBP ChIP signal at the URA1 locus in cells cross-linked with 1% formaldehyde, quenched with 250 mm glycine, and resuspended in either 50 or 750 mm Tris, pH 8. Samples were incubated at room temperature for 10 (blue circles) or 30 (red circles) min before processing. A Student's t test was performed to determine statistical significance between conditions. B, average ChIP signal of cells cross-linked with 5% formaldehyde and quenched with 2.93 m glycine, pH 5. Samples were incubated in glycine-quench solution for 0, 10, or 30 min before processing. All experiments were performed with two biological replicate samples, and error bars represent the standard deviation. *, p < 0.05.
The results thus far led to implementation of two significant changes in the CLK methodology. First, to obtain the most accurate time-resolved ChIP data, we employed the more robust quenching afforded by max glycine conditions, which lack the negative attributes of Tris as a quencher. Second, as the cross-linking rate is dependent on formaldehyde concentration, we employed 5% formaldehyde rather than 1% as used in previous work (Ref. 15 and most ChIP experiments published to date). Although 5% formaldehyde optimized the assay for analysis of several interactions in this study, it will be important to determine the optimal formaldehyde concentration for analysis of other types of interactions and in other cell types. We sought the highest feasible formaldehyde concentration for two reasons. First, experimentally, we wanted the ChIP signal to be minimally affected by noise. Second, because the overall cross-linking rate depends on the formaldehyde concentration, faster cross-linking would yield better time resolution between the cross-linking and binding dynamics time scales. To achieve the desired concentrations of reagents in the reactions and to obtain sufficient cellular material for analysis, cell cultures were concentrated by centrifugation; formaldehyde was added to the concentrated cell suspension, and then aliquots of cells were quenched by dilution in a much larger volume of glycine at high concentration. This approach also has the advantage that formaldehyde reactivity is reduced by dilution to 0.1% after glycine addition. Prior work showed that little cross-linking was detectable using 0.1% formaldehyde, so dilution alone was expected to have a substantial impact on formaldehyde reactivity (16). In addition, the glycine-quenching solution was adjusted to pH 5, which further improves the ability of glycine and formaldehyde to react (27). We refer to the experimental approach employing all of these modifications as CLKv2 (Fig. 3A) to distinguish it from the original CLK method.
Figure 3.

CLKv2 quenching conditions and overview of the updated method. A, flow chart of CLKv2 method focusing on sample collection. B, order of addition experiments to verify new excess glycine conditions are shown in the schematic. Three experiments were set up: 1) glycine alone added to samples; 2) glycine addition to samples then formaldehyde–cross-linking; and 3) formaldehyde–cross-linking followed by glycine-quenching. For all samples, 5% formaldehyde and 2.93 m glycine, pH 5, were used. C, real-time PCR read-out from experiments done in B; data were obtained from two biological replicates, and the error is the standard deviation. *, p < 0.05.
As shown in Fig. 3, B and C, order-of-addition experiments established that glycine was a very efficient quencher of formaldehyde reactivity when used in this way; the TBP-myc ChIP signal obtained in reactions in which formaldehyde was added first was ∼28-fold higher than in reactions with no formaldehyde. In contrast, the ChIP signal obtained when glycine was added before formaldehyde was not statistically different from the background ChIP signal obtained with no formaldehyde at all (p = 0.20). Next, the use of 5% formaldehyde prompted us to evaluate how this higher level of formaldehyde might generally impact cellular constituents. As shown in Fig. 4A, protein yields were reduced in whole-cell extracts prepared from cells treated with 5% formaldehyde for increasing periods of time. In contrast, there was no change in the yield of chromatin protein associated with extracts prepared as normally done for ChIP. In addition, there was little change in the pattern of protein bands or their relative intensities over a time course of formaldehyde incubation, indicating that the majority of proteins present in these chromatin extracts were not notably depleted or modified (Fig. 4B). This suggests that the reduced yield of protein in whole-cell extracts was due to cross-linked cells being refractory to lysis by rapid agitation with glass beads, whereas soluble protein contents were more efficiently released in the chromatin extract preparation procedure, which utilizes a combination of glass bead agitation plus sonication. Protein samples are typically heated to facilitate their denaturation prior to electrophoresis, but formaldehyde cross-links are also reversible by heat, so we analyzed protein extracts on gels with and without heating. There was relatively little difference in the overall protein-banding pattern when chromatin extract proteins were analyzed following brief heating to facilitate protein denaturation versus unheated samples (Fig. 4B). Heating did reduce an indistinct smear of protein toward the top of the lanes of unheated samples, consistent with heat improving denaturation of the samples. Brief heating had a dramatic effect on the ability to detect TBP in extracts by Western blotting (Fig. 4C). The formaldehyde cross-link reversal time is much longer than this brief heating period (30), suggesting that heating in this experiment facilitated disruption of TF-protein complexes and protein unfolding rather than cross-link reversal. In the case of TBP, it is likely that its association with the TATA-binding protein-associated factors and potentially other regulatory factors in extracts (31) make detection of monomeric TBP difficult or impossible without heating. MNase titration experiments were performed to examine the overall chromatin state resulting from treatment with cross-linker and quencher. Compared with untreated cells, we observed no difference in the overall chromatin digestion pattern when cells were treated with 2.93 m glycine, pH 5, with or without prior formaldehyde incubation (Fig. 4D). Thus, formaldehyde and glycine do not cause any detectable bulk structural changes in chromatin or affect chromatin accessibility.
Figure 4.

Effect of formaldehyde–cross-linking on proteins. A, relative concentration of protein in either whole-cell extract (blue circles) or chromatin (red circles) samples cross-linked with 5% formaldehyde for varying amounts of time. Bradford assays were used to determine the concentration. Two biological replicates were used for each time point, and samples were normalized to their respective zero time point. Error bars represent the standard deviation. B, Coomassie-stained SDS-polyacrylamide gel of AY146 whole-cell extract samples from cells cross-linked for varying amounts of time with 5% formaldehyde. Fifteen-microgram samples were heated (H) for 5 min at 95 °C or not heated (NH) before loading. C, samples from the AY146 strain were cross-linked for 0, 5, 10, or 15 min with 5% formaldehyde and either heated for 5 min at 95 °C or unheated before loading into an SDS-polyacrylamide gel. The Western blot was probed with a TBP antibody and visualized with chemiluminescence. C (control) is recombinant TBP protein. D, MNase digestion of chromatin from wild-type cells visualized on a 2% agarose gel. Cells were treated with no formaldehyde or glycine, 2.93 m glycine, pH 5, or 5% formaldehyde for 30 s followed by 2.93 m glycine, pH 5. The indicated amounts of MNase (units (U)) were added to aliquots of cells, and the resulting chromatin was resolved by gel electrophoresis. The base pair lengths of molecular weight standards are denoted to the left of the image.
A key requirement for the CLK method is that the unbound pool of the TF being investigated is not depleted significantly by formaldehyde incubation (15). This ensures that there are sufficient molecules available for interaction with unbound DNA sites and that the overall on-rate, which depends on the concentration of the free TF, does not change over the course of the reaction. To determine the effect of 5% formaldehyde on the soluble pools of particular TFs, Western blottings were performed using extracts obtained from cells treated with formaldehyde for various periods of time. Based on the results in Fig. 4, B and C, a brief heating step was used prior to loading samples on the gels to accurately estimate the relative amount of soluble TF without reversing any cross-links that had formed. Western blotting showed that 5% formaldehyde treatment resulted in depletion of some TFs and not others, and the rates of depletion among those that were depleted varied significantly (Fig. 5, A–F). TBP, Gal4, and Ace1 were not significantly depleted in these experiments, whereas Reb1, Cat8, Abf1, TFIIB, and Tfa1 were stable for ∼10 min and then were depleted. In contrast, the largest subunit of RNA polymerase II, Rpb1, and the TFIIF subunit Tfg1 were rapidly depleted. This indicates that some factors such as TBP and Gal4 are readily amenable to analysis by CLKv2. As shown below, others such as Tfa1 can be investigated as long as the cross-linking time course is confined to the period in which the levels of the factor are not depleted. Other factors such as Rpb1 and Tfg1 cannot be investigated at present using these conditions. (However, it should be noted that in principle one could incorporate the TF depletion rate into the dynamic model.)
Figure 5.

Protein levels in cross-linked whole-cell extract or chromatin samples over time. A, Western blots of WCE samples for all factors except Gal4, which is a chromatin extract. Antibodies used are listed in supplemental Table S4, and molecular mass is denoted to the right in kDa. Samples were cross-linked with 5% formaldehyde for 0–15 min and quenched with excess glycine. B, quantification of WCE Western blot bands shown in A for TBP. Each sample was normalized to the zero time point as a percentage. Two replicates were averaged for the plot, and error bars represent standard deviation. C, same as B, except for Ace1. D, same as B, except for transcription factors Reb1, Cat8, Abf1, Sir2, and Sko1. E, same as B, except for preinitiation complex components TFIIB, Tfa1 (TFIIE), Tfg1 (TFIIE), and Rpb1 (RNA polymerase II). Independently performed Western blottings using chromatin rather than WCE samples showed the same trends. F, same as B, except for Gal4 chromatin extract. Gal4 was not abundant enough to be detected in WCEs.
To measure dynamics using the CLKv2 method, ChIP data for an interaction of interest are acquired in two different strains, each of which differ only in the concentration of the TF. One strain (“WT”) expresses the TF of interest at wild-type levels, and the other (“OE” for “overexpression”) typically harbors an additional copy of the TF gene, which increases the TF concentration ∼2–3-fold on average. The CLK model contains as variables the on-rate for TF-chromatin binding (ka), the off rate (kd), and the formaldehyde–cross-linking rate (kXL); the fractional occupancy (θb) and residence time (t½) are calculated from the variables and are not direct outputs of the fits. The saturation level of the ChIP signal (Ssat) is an additional parameter obtained from the fits, which we use to normalize the ChIP signal and, thereby, obtain an estimate of the fraction of cross-linked TF at a given site (θXL). The concentration of the TF in the nucleus (CTF) and the formaldehyde concentration (CFH) are experimentally measured quantities used in the fitting calculations. (For reference, all variables are defined in supplemental Table S5.) The CLK model makes no assumptions about the relative rates of chromatin binding or cross-linking, and indeed it provides a framework sufficiently flexible to model a wide range of chemical and dynamic behaviors (15, 16). Using the CLKv2 conditions, and as discussed in detail below, a wide range of behaviors was observed, including interactions with binding dynamics slower than cross-linking, comparable with cross-linking, or faster than cross-linking. In the binding dynamics-limited scenario (Fig. 6, A and D), cross-linking is much faster than the on- and off-rates for chromatin binding. The hallmarks of the binding-dynamics limited behavior (referred to as “TF-limited”) include two exponentials: a very steep exponential rise at short time scales (seconds), often manifesting as a non-zero y-intercept in the WT and OE data with a clear separation in the WT and OE y-intercepts, followed by a slower exponential rise. This clear separation in time scales makes it possible to extract binding dynamics, including the on- and off-rate (15). In contrast, if the rate of cross-linking is slower than the time scale of TF-binding dynamics, cross-linking–limited (referred to as “XL-limited”) data show a single exponential rise with a zero y-intercept for the WT and OE data (Fig. 6B). The simulation in Fig. 6B and schematic in Fig. 6E show that for XL-limited interactions, the cross-linking time scale is slower than for the TF-limited case, and under these conditions TF binding and unbinding can occur prior to cross-linking. If the cross-linking rate is so slow (Fig. 6F) that its associated time is longer than the latest cross-linking time (usually 1200 s for this study), the ChIP signal rises linearly (or nearly linearly) as shown in Fig. 6C. In the linear version of the XL-limited model, the theoretical curve shows a near-zero y-intercept, and no sign of saturation on the experimentally accessible time scale.
Figure 6.

Overview of CLKv2 possible model fits. A–C, simulations of CLKv2 fits. For each plot, blue represents the wild-type strain, and red is the overexpression strain. From left to right, the fits correspond to: binding dynamics (TF)-limited (A), cross-linking–limited (B), and linear cross-linking–limited behavior (C). D–F, schematic of binding dynamics for each of the three CLKv2 cases with formaldehyde–cross-linking over time: TF-limited (D), XL-limited (E), and linear XL-limited (F). In each square cell, the TF (blue circles) binds to its binding site (blue rectangles); red × symbols represent cross-linking by formaldehyde. Cross-linking time increases as the panels progress from top to bottom.
Once cross-linking time-dependent data have been acquired, determining which scenario describes the data and fitting to the model is described in the flow chart in Fig. 7. The fitting procedures themselves are described in detail under “Experimental procedures.” We note that different sets of parameters are gained from each type of fit as shown in the schematic as follows: TF-limited fits yield ka, kd, kXL, and Ssat from which the dissociation constant Kd, θb, and t½ can be derived. However, the XL-limited fit only gives Kd, kXL, and Ssat from which θb can be derived, and the linear model provides Kd and kXL·Ssat from which θb can be derived.
Figure 7.

Flow chart for fitting CLKv2 data. After visual inspection of the data, the fitting procedure in either Flow1 arm (blue) or Flow2 arm (red) was followed for a given locus. Fitting procedure for non-linear data were further broken down into arm 1-A or 1-B. Arm 1-A represents the TF full-model fit, whereas 1-B could be cross-linking–limited or full model fit. Flow2 is the linear cross-linking–limited fit.
Data were obtained for a number of TF–chromatin interactions using CLKv2. The interactions of TBP with the LOS1, ACT1, and URA1 promoters are shown in Fig. 8, A–C. Applying the flow chart shown in Fig. 7 revealed that these interactions were well-described by the TF-limited behavior (see “Computational modeling” under “Experimental procedures” for a definition of TF-limited behavior and the full model). At the URA1 promoter, TBP-myc displayed both a linear ChIP signal with cross-linking time and sensitivity to formaldehyde concentration consistent with XL-limited dynamics, suggesting that although Myc-tagged TBP complements growth, the Myc tag had a relatively strong effect on cross-linking and possibly TBP-binding dynamics. The data describing the interaction between LacI-GFP and an array of LacI sites is shown in Fig. 8D and was also well-described by TF-limited behavior. The fractional occupancies of the three TBP loci ranged from 0.04 to 0.07, while the residence times were about 60–90 s (Table 1). Consistent with prior work (15), this indicates that these promoters are unoccupied by TBP most of the time and that the TBP complexes that do form are not very long-lived. LacI fractional occupancy was lower still, but the complexes formed had half-lives of 1056 s. This long lifetime is consistent with both prior CLK and live-imaging data (15). TBP binding to NTS2 (the promoter for polymerase I transcription) and Ace1 binding to CUP1 were both best approximated by the linear model (Fig. 8, E and F). The linear behavior of Ace1 CLK data using the CLKv2 conditions is consistent with rapid binding dynamics (15, 32) being faster than the cross-linking rate. The high-fractional occupancy of Ace1 at CUP1 (0.83) is also consistent with prior observations (15, 32). The fractional occupancy of TBP at NTS2 (0.73) was much higher than TBP occupancies at the other promoters, consistent with the high-transcriptional activity of the rDNA in cells in log-phase growth in rich medium (33). Tables 1–3 provide all the measured kinetic parameters along with their associated errors. Notably, error analysis derived from multiple fits of simulated data (see “Experimental procedures”) showed that most parameters were associated with a single well-defined distribution (supplemental Figs. S2 and S3).
Figure 8.

CLKv2 fits of data for TBP, LacI, and Ace1. A–D, all fits shown are TF-limited full model fits. The blue line is the wild-type strain, and the red has the factor overexpressed; overexpression factors are listed in supplemental Table S3. TBP is shown at ACT1 (A), URA1 (B), and LOS1 (C); LacI is shown at a lac array (D). Occupancy (θb) and t½ are denoted on the plots. E and F, both fits are linear cross-link-limited. TBP is shown at NTS2 (E) and AceI is shown at CUP1 (F). Only occupancy (θb) is shown because residence time is not extracted with this fit. Each dataset resulted from two biological replicates, and the average is shown. Error bars represent the standard deviation.
Table 1.
Measurements for TBP-binding dynamics at select promoters with a full model fit
| TBP-full model | kxla | τxlb | ka·CTFc | kdd | Ssate | Kf | t½g | θbh |
|---|---|---|---|---|---|---|---|---|
| 1/mol s | s | 1/s | 1/s | mol | s | |||
| ACT1 | 0.14 (+31.2, −0.51) | 2.76 (+45.05, −0.73) | 3.21 (+1.3, −0.92) E-04 | 6.98 (+3.3, −2.2) E-03 | 1.35 (+0.35, −0.28) | 2.61 (+1.0, −0.74) E-04 | 99.3 (+99.32, −56.51) | 0.044 (+0.44, −0.016) |
| LOS1 | NAi | NA | 6.31 (+1.1, −0.93) E-04 | 8.58 (+2.3, −1.8) E-03 | 0.44 (+0.031, −0.029) | 1.63 (+0.32, −0.27) E-04 | 80.8 (+21.45, −16.94) | 0.069 (+0.012, 0.01) |
| URA1 | 0.30 (+1129.02, −4.74) | 1.29 (+19.22, −0.081) | 7.59 (+1.0, −0.91) E-04 | 1.0 (+0.29, −0.23) E-02 | 0.71 (+0.036, −0.035) | 1.62 (+0.36, −0.29) E-04 | 67.7 (+21.02, −16.2) | 0.069 (+0.015, −0.01) |
a Formaldehyde cross-linking rate is shown.
b Cross-linking time is shown.
c On-rate of transcription factor X nuclear concentration of factor is shown.
d Off-rate of transcription factor is shown.
e ChIP signal at saturation is shown.
f Dissociation constant, kd/ka is shown.
g Residence time of TF binding is shown.
h Occupancy is shown.
i NA means not applicable.
Table 2.
Measurements for TBP-binding dynamics at select promoters with a linear model fit
| TBP-linear | kxl·Ssata | τxlb | ka·CTFc | kdd | Ssate | Kf | t½g | θbh |
|---|---|---|---|---|---|---|---|---|
| 1/mol s | s2 | 1/s | 1/s | mol | s | |||
| NTS2 | 1.1 (+0.084, −0.078) E-03 | NAi | NA | NA | NA | 4.51 (+2.21, −1.44) E-06 | NA | 0.73 (+0.077, −0.07) |
| SNR6 | 1.7 (+0.12, −0.11) E-03 | NA | NA | NA | NA | 4.46 (+2.0, −1.37) E-06 | NA | 0.73 (+0.074, −0.067) |
| HSC82 | 3.4 (+0.54, −0.47) E-04 | NA | NA | NA | NA | 8.96 (+5,73, −3.51) E-06 | NA | 0.57 (+0.13, −0.1) |
a Formaldehyde cross-linking rate × ChIP signal at saturation is shown.
b Cross-linking time is shown.
c On-rate of transcription factor X nuclear concentration of factor is shown.
d Off-rate of transcription factor is shown.
e ChIP signal at saturation is shown.
f Dissociation constant, kd/ka is shown.
g Residence time of TF binding is shown.
h Occupancy is shown.
i NA means not applicable.
Table 3.
Measurements for Ace1- and LacI-binding dynamics
| kxla | τxlb | ka·CTFc | kdd | Ssate | Kf | t½g | θbh | |
|---|---|---|---|---|---|---|---|---|
| 1/mol s | s | 1/s | 1/s | mol | s | |||
| Ace1 (linear) at CUP1 | 2.28 (+0.1, −0.098) E-03 | NA | NA | NA | NA | 2.07 (+1.14, −0.77) E-07 | NA | 0.83 (+0.055, −0.052) |
| LacI (full model) at LacO | 3323 (+5.26E09, −767979) | 1.2E-04 (+0.0034, −5.0 E-07) | 5.33 (+5.09, −2.37) E-07 | 6.57 (+1.51, −1.22) E-04 | 906.9 (+893.7, −473.1) | 1.23 (+1.75, −0.79) E-03 | 1055.5 (+254.6, −206.1) | 8.10 (+8.3, −3.8) E-04 |
a Formaldehyde cross-linking rate is shown.
b Cross-linking time is shown.
c On-rate of transcription factor X nuclear concentration of factor is shown.
d Off-rate of transcription factor is shown.
e ChIP signal at saturation is shown.
f Dissociation constant, kd/ka is shown.
g Residence time of TF binding is shown.
h Occupancy is shown.
i NA means not applicable.
Datasets obtained for TBP binding to the HSC82 and SNR6 promoters were not obviously linear or non-linear; these ambiguous cases required a more rigorous selection process for the best fit (see flowchart, Fig. 9A, and under “Experimental procedures” for detailed explanation). These datasets were fit with both the TF-limited and linear models, and the sum of squared residuals (SSR) derived from the fits were compared for the appropriate fit (Fig. 9, B and C). Both loci had a better fit with the linear model; the SSR for the TF-limited/linear models for HSC82 and SNR6 were 0.11/0.042 and 3.35/0.43, respectively. The occupancy of TBP at HSC82 and SNR6 was 0.57 and 0.73, respectively.
Figure 9.

Resolution of ambiguous TBP fits. A, flow chart to determine best fit for ambiguous data. Data were fit with both linear and full models, and F tests or SSR was then used to differentiate the best fit. B and C, TBP fits at HSC82 (B) and SNR6 (C) were fit with both full (top) and linear (bottom) models. SSR derived from the fits was used to find that both datasets were best represented with the linear fit; SSR (labeled res) is shown on all four plots and occupancy (θb) for the linear fit. Each dataset resulted from two biological replicates, and the average is shown. Error bars represent the standard deviation.
As mentioned earlier, it is possible to model kinetic behavior of TFs that are depleted by formaldehyde by focusing measurements on the formaldehyde incubation time period when the levels remained stable. TFIIE was significantly depleted by about 10 min (Fig. 5, A and E), but the protein levels were not detectably changed through 7 min of formaldehyde incubation (Fig. 10A). This allowed us to measure TFIIE interaction with the ACT1, LOS1, and URA1 promoters (Fig. 10, B–D, and Table 4). TFIIE binding to URA1 and LOS1 was best described by a cross-linking–limited model, whereas a full model fit described binding to ACT1. Fractional occupancies were well below saturation for all three sites, and at ACT1 we compute a residence time of about 6 min, which is within the bounds of an estimate of its 95% confidence interval (supplemental Table S8) and on par with the time scale for TBP interaction at this site.
Figure 10.

CLKv2 for TFIIE on a shorter experimental time scale. A, Western blot of Tfa1-TAP chromatin using an anti-TAP antibody. Samples were cross-linked for 30 s to 7 min. Wild-type and overexpression strains were both tested for depletion. Quantification of the signal was plotted below; two replicates were averaged, and the standard deviation is shown as error bars. The wild-type strain was normalized to its 30-s time point; the overexpression strain was normalized to its 30-s time point and multiplied by the overexpression factor (supplemental Table S3). The overexpression factor was determined by running four 5% formaldehyde–cross-linked time points for the wild-type and overexpression strains on the same gel and blotting for TAP tag (data not shown). Bands were quantified with ImageJ and compared with determined overexpression. B, TFIIE at ACT1 resulted in a full model fit; occupancy and residence time are denoted. C and D, TFIIE at both URA1 and LOS1 gave cross-link–limited fits; only the occupancy is shown. Each dataset resulted from two biological replicates, and the average is shown. Error bars represent the standard deviation.
Table 4.
Measurements for TFIIE-binding dynamics at select promoters
| TFIIE | kxla | τxlb | ka·CTFc | kdd | Ssate | Kf | t½ g | θbh |
|---|---|---|---|---|---|---|---|---|
| 1/mol s | s | 1/s | 1/s | mol | ||||
| Full model fit | ||||||||
| ACT1 | 4.80E-03 (+1.3, −0.22) | 80.2 (+992.3, −16.85) | 3.5 (+9.4, −2.7) E-04 | 2.0 (+7.2, −2.2) E-03 | 3.11 (+3.21, −2.94) | 7.80 (+14, −6.2) E-05 | 346.6 (+523.9, −156.8) | 0.15 (+0.48, −0.01) |
| XL-limited fit | ||||||||
| URA1 | 1.96 (+1.0, −0.69) E-03 | 206.5 (+76.52, −52.94) | NAi | NA | 2.71 (+2.71, −0.48) | 7.17 (+18.0, −5.5) E-06 | NA | 0.66 (+0.15, −0.12) |
| LOS1 | 4.3 (+5.4, −2.5) E-03 | 90.1 (+95.4, −44.2) | NA | NA | 0.29 (+0.08, −0.05) | 4.14 (+2.4, −1.0) E-05 | NA | 0.31 (+0.14, −0.1) |
a Formaldehyde cross-linking rate is shown.
b Cross-linking time is shown.
c On-rate of transcription factor X nuclear concentration of factor is shown.
d Off-rate of transcription factor is shown.
e ChIP signal at saturation is shown.
f Dissociation constant, kd/ka is shown.
g Residence time of TF binding is shown.
h Occupancy is shown.
i NA means not applicable.
Discussion
The CLK assay was conceived to provide biophysically rigorous on- and off-rates for TF binding to single copy loci in vivo (15). We sought to develop an approach that would also be generally applicable and potentially scalable to genome-wide analysis. The biggest obstacle to implementation of this assay has been to develop general experimental conditions and a companion model that accurately account for the many effects occurring in cells that undergo formaldehyde–cross-linking and to distinguish them from the contributions of binding kinetics to the time-dependent change in ChIP signal. Here, we extend our understanding of the effects of formaldehyde on yeast cells and use our observations to both improve the CLK assay conditions and to improve the approach to data analysis. Formaldehyde–cross-linking is ubiquitous in the chromatin field, so the results that we report here may contribute to the understanding and interpretation of ChIP and related types of experimental results in general as well.
Our results demonstrate improvement in formaldehyde quenching using a higher concentration of glycine than was used previously. The residual unquenched formaldehyde that remains following addition of 250 mm glycine as commonly used and in the original CLK procedure likely inflated the ChIP signal values at short cross-linking times as the unquenched formaldehyde continued to capture complexes during the centrifugation step that follows quenching. However, despite this, the relative differences in ChIP signal change with time apparent in the original CLK data do capture the relative differences in binding dynamics validated by other methods. For example, the rapid rise in Ace1 ChIP signal with short cross-linking times observed originally is consistent with the known highly dynamic behavior of Ace1 binding to its sites in the CUP1 promoter (18), whereas the shallow slope and gradual approach to saturation seen with LacI time-dependent ChIP signals are consistent with its long residence time (19), which we confirmed by live-cell imaging (15). Remarkably, the residence times for TBP binding to particular promoters reported here are also broadly consistent with the residence times obtained with the original version of the CLK assay (15). The results argue that TBP has residence times at these promoters on the order of one to a several minute time scale. Thus, although the original CLK data were modeled assuming infinitely fast quenching, we nonetheless captured the relative time scale of dynamic behavior as validated by both live-cell imaging and in this study using CLKv2.
Based on the results presented here, although Tris is highly effective in quenching unreacted formaldehyde, it is unsuited for use in this type of kinetic analysis due to its ability to reverse cross-links. The cross-link reversal that we observed is consistent with a prior report (28) and is exacerbated by the relatively high concentration of Tris required to completely react with a relatively high concentration of added formaldehyde. We also show that time-dependent increases in ChIP signal can be affected by the concentration of formaldehyde. The use of a formaldehyde concentration that is as high as possible boosts the cross-linking rate, thereby extending the useful range of the assay. Although we employed 5% formaldehyde here, this may not be advisable or appropriate for analysis of other TFs or in other types of cells. The best formaldehyde concentration ought to be determined empirically by choosing the concentration that yields the best separation between the cross-linking and binding dynamics time scales, and which does not impact overall recovery of soluble components or deplete the unbound TF in the soluble pool over the kinetic time course. Those factors that are stable constituents of multisubunit complexes such as Rpb1 may be impossible to assess using this approach; what is observed by Western blotting as their rapid depletion from extracts may be due to rapid cross-linking to other biologically relevant polypeptides with which they stoichiometrically co-associate.
Using the CLKv2 method, we find that cross-linking rates are highly variable and depend on the particular TF-DNA site of interaction (Tables 1–3). Prior to our measurement of formaldehyde–cross-linking rates in vivo, cross-linking of ChIP complexes was generally thought to be rapid (14, 34–36), and this was supported qualitatively by the differences in ChIP signals that were observed at closely spaced time points (26) and that highly transient interactions (residence times on the ∼ second scale) could nonetheless be captured by formaldehyde–cross-linking in ChIP experiments (8, 15, 16, 37). In addition, there is a global correlation between steady-state ChIP signals and in vitro binding affinity (38, 39) consistent with the overall ChIP signal level not being merely proportional to the rate of capture by cross-linking. In vitro, the rate of formaldehyde reaction with DNA bases is relatively slow (40), but reactivity could be greatly accelerated when DNA and amino acids were present together (41). Interestingly, the rates of TBP cross-linking to the URA1 and ACT1 promoters calculated by CLKv2 (kXL, Tables 1–3) are in the same range as in vitro cross-linking rates obtained in reactions containing DNA and amino acids (41). Experiments measuring formaldehyde reactivity with amino acids and proteins have shown that formaldehyde adducts tend to be mainly formed with cysteine, lysine, and tryptophan side chains as well as the N-terminal group of polypeptide chains (42, 43). In reactions containing both nucleic acids and protein/amino acid substrates, the most efficient cross-linking was found to occur between lysine and deoxyguanosine (34, 42, 43). We suggest that the wide range in cross-linking rates reported here reflects the variation in reactive chemical groups on the TF surface and their proximity and orientation to reactive groups on DNA bases at or near binding sites.
Although some factors of interest were eventually depleted from extracts following formaldehyde treatment, our results with the TFIIE subunit Tfa1 show that it is still possible to investigate them kinetically if the cross-linking time course is confined to a temporal window in which their overall levels are not affected by formaldehyde. A possible limitation in this approach is that a shorter time course may make it more difficult to determine the saturation level of the ChIP signal, an estimate that is required for confident fitting of the data and accurate estimates of the parameters. An alternative approach for future work is to extend the current model to include the depletion of the TF of interest in the fitting. Conceptually, by quantifying the rate of TF depletion from Western blottings such as those shown in Fig. 5, the decrease in the overall level of the TF with cross-linking time could be modeled and the level of the TF at different times included explicitly as a parameter during the analysis of the data.
In instances in which the cross-linking rate is much slower than TF-DNA binding, CLKv2 yields the fractional occupancy as well as the equilibrium-binding constant. Although the residence time cannot be estimated from the data in these situations, the fractional occupancy and binding constant are useful parameters as they provide insight into the variation in site occupancy across the cell population, which could have implications for understanding the molecular basis of transcriptional noise (44, 45), as well as energetic barriers in the intracellular environment that reduce binding from in vitro values obtained using purified components. If the cross-linking rate can be determined, this can be used to set an upper limit for binding dynamics. For many biological systems, knowing whether binding is occurring faster or equal to the second, minute, or tens of minutes time scale would be valuable for developing dynamic models for the order of events underlying transcriptional responses.
Experimental procedures
Yeast strains and growth conditions
Many of the Saccharomyces cerevisiae strains used in this study were described previously (15). Other strains were newly developed for the work presented here, and all are listed in supplemental Table S1. Plasmids used for strain construction are listed in supplemental Table S2. TBP ChIP was performed in two ways: 1) using a monoclonal antibody that recognizes untagged TBP, and 2) using an antibody that recognizes the epitope tag on TBP-myc. Chromatin-associated Myc-tagged TBP was measured using the epitope-tagged strain YAD154. TBP ChIP using the monoclonal TBP antibody was performed in various strains as described below. YAD154 cells used for the TBP-myc ChIP experiments were grown in YPD overnight at 30 °C and harvested at A600 ∼1. For other TBP ChIP experiments comparing strains with two different levels of TBP, AY146 (wild-type TBP levels) and YSC018 (harboring a 2-micron TBP overexpression plasmid) were obtained from the TBP shuffling strain YAD165 as described previously (15). Cells were grown in synthetic medium without leucine plus 2% glucose overnight at 30 °C. Culture volumes for each type of experiment are noted below and range from 100 to 450 ml depending on the experiment. When an A600 of ∼0.8 was reached, cells were pelleted and resuspended in an equivalent volume of YEP plus 2% glucose medium. They were grown at 30 °C for approximately 1 h until an A600 of 1.0 was reached, and cells were then formaldehyde cross-linked as described below. This regimen allowed cells to be initially grown under plasmid selection but then transferred to YPD to standardize ChIP results, which could otherwise be potentially influenced by effects of growth medium. In addition, growth in YPD prior to cross-linking permitted direct comparison with previously published work (2, 3).
For TFIIE ChIP, strains were used with WT or elevated levels of the two TFIIE subunits Tfa1 and Tfa2 in which the Tfa1 subunit was TAP-tagged. YRV006 (TFA1-TAP, Dharmacon) (15) was transformed with an empty pRS315 vector (46) (AY151, WT TFIIE levels) or a pRS315-based plasmid carrying copies of TFA1-TAP and TFA2 under control of their endogenous promoters (AY152, overexpressed levels of TFIIE). Cells were grown at 30 °C overnight in synthetic medium without leucine and with 2% glucose. When an A600 of ∼0.8 was reached, cells were pelleted, resuspended in an equivalent volume of YPD, and grown at 30 °C to an A600 of 1.0 as described above. Strain construction for AceI (YTK539 and YSC002) and LacI (YTK260 and YSC001) as well as collection for ChIP was described previously (15).
For Western blotting, strains YGR186W, YBR049C, YRV018, and ML307-1 were grown overnight in YPD at 30 °C to A600 of 1; YRV005 was grown in YEP + 2% raffinose at 30 °C overnight to A600 of 0.8, and then 2% galactose was added, and cells were incubated to A600 of ∼1.0. YTK539 cells were grown under conditions of copper induction as described previously (15).
Quenching and cross-linking conditions
Different cross-linking and quenching conditions were tested with the TBP-myc strain (YAD154) to explore the relationship between cross-linking rate and formaldehyde concentration, as well as quenching efficiency. In all experiments, cells were first grown in YPD at 30 °C overnight to an A600 of 1.0. To test the effect of 250 mm glycine, 100-ml cell cultures were incubated with 2.7 ml of 37% formaldehyde (1% final, Fisher) followed by addition of 10 ml of 2.5 m glycine, pH 6.3, at various times. To test the effect of 2.93 m glycine, 450-ml cultures were grown in YPD overnight at 30 °C to A600 of 1. Cells were then concentrated 5-fold by centrifugation and resuspended in 90 ml of YPD. The concentrated cultures were then incubated with 2.7 ml of 37% formaldehyde (1% final concentration) by addition of formaldehyde to the culture while rapidly mixed using a stir bar. At various times thereafter, 10-ml aliquots were removed and added to 440 ml of glycine, pH 5, contained in 450-ml Sorvall centrifuge bottles. Bottles were capped by hand as quickly as possible and vigorously shaken. Samples were washed and worked up as detailed below.
To test different formaldehyde concentrations and other quenching conditions, TBP-myc cells were grown as described above. To test formaldehyde concentrations at 1% or lower, in most cases the appropriate volume of 37% formaldehyde was added to a rapidly stirring 100-ml culture, and the reaction was then quenched after specific incubation times by addition of 3 m glycine or 3 m Tris-HCl, pH 8, to achieve the indicated final quencher concentration. For reactions in which formaldehyde was added to a final concentration greater than 1%, cells were concentrated 5-fold in YPD as described above; 37% formaldehyde was added to achieve the indicated final concentration, and after particular incubation times, 10-ml aliquots were removed to centrifuge bottles or tubes containing 3 m glycine or Tris yielding the final concentration of the quencher indicated in the figure legends. Cell samples quenched in Tris were worked up and analyzed as described above except that the first TBS wash contained 120 mm Tris-HCl, pH 8, rather than glycine.
Quenching reversal experiments
To determine the stability of cross-linked material in the presence of quencher, cross-linked cells were incubated in solution containing glycine or Tris for different periods of time prior to ChIP work-up. For Tris-quenched samples, 100-ml cultures of AY146 cells were grown overnight in synthetic media lacking leucine and containing 2% glucose at 30 °C. Cells were then transferred to YPD at an A600 of 0.8 and grown until reaching an A600 of 1. Each sample was cross-linked by adding formaldehyde to 1% for 5 min and then quenched by adding 10 ml of 2.5 m glycine to each 100 ml of culture. Cells were pelleted and resuspended in either 750 mm Tris-HCl, pH 8, or TBS buffer (which contains 50 mm Tris-HCl, pH 8, as described above) and incubated at room temperature for 10 or 30 min. Subsequent steps were carried out as described below.
To test cross-link stability in the presence of glycine, 250-ml replicate cultures of AY146 cells in synthetic media plus 2% glucose and without leucine were incubated overnight at 30 °C, then resuspended in YPD, and grown to an A600 of 1 as described above. Three aliquots of 50 ml were taken from each culture and pelleted at room temperature. Each pellet was then resuspended in 10 ml of YPD and transferred to a flask on a stir plate. Formaldehyde was then added to a 5% final concentration to each sample and mixed at room temperature for 5 min. 10 ml from each sample were quenched in 440 ml of 3 m glycine, pH 5, at room temperature for 0, 10, or 30 min. The 0-min sample was pelleted at 4 °C immediately after quenching; the other time point samples were pelleted the same way after glycine incubation of 10 or 30 min. Following incubation of the cross-linked cells in glycine solution for the indicated times, the cells were processed for ChIP as described below.
Order-of-addition experiments
Order-of-addition experiments were performed to test quenching efficiency using the TBP-myc strain, YAD154. Replicate cultures of YAD154 cells (300 ml) were grown overnight at 30 °C in YPD to an A600 of 1.0 and then concentrated by resuspension in 60 ml of YPD. In each experiment, three 10-ml aliquots were collected in duplicate as follows: 1) no formaldehyde control samples in which 3 m glycine, pH 5, was added to 2.93 m final concentration; 2) samples in which 3 m glycine was added to 2.93 m final concentration before 5% formaldehyde addition for 8 min; and 3) 5% formaldehyde incubation for 8 min followed by addition of 3 m glycine, pH 5, to 2.93 m final concentration. Following these treatments, cell samples were washed in 50 ml of TBS plus 300 mm glycine, pH 5, followed by washing in 50 ml of TBS, both washes at 4 °C. Subsequent work-up for ChIP and real-time PCR for TBP binding to the URA1 locus were performed as described above.
Order-of-addition experiments for Gal4 with the previously published CLK conditions (15) were done in the same way as order-of-addition experiments described above, except different glycine and formaldehyde concentrations were used. For each sample set, three 100-ml YPH499 cultures were grown overnight at 30 °C in YEP + 2% raffinose. When an A600 of 0.8 was reached, each culture was induced with 2% galactose. At an A600 of 1.0, samples were collected in duplicate. The following experimental parameters were used: 1) 2.5 m glycine, pH 6.3, was added to 250 mm final concentration; 2) 2.5 m glycine, pH 6.3, was added to 250 mm final concentration for 5 min before addition of 1% formaldehyde for 8 min; and 3) 1% formaldehyde incubation for 8 min before addition of 2.5 m glycine, pH 6.3, to 250 mm final concentration for a 5-min incubation. The subsequent steps were the same as above, except analysis was performed for interaction at the GAL3 locus.
MNase digestion
The protocol was adapted from a published MNase ChIP-sequencing method (47). Five-ml YPD primary cultures were grown overnight at 30 °C. These were added to 495 ml of YPD media the next day, and cells were grown until the A600 was ∼0.8. Each culture was then split into three 150-ml aliquots. Cells in 1 aliquot were collected by centrifugation for 5 min at 4000 rpm in an Eppendorf 5810R benchtop centrifuge. Cells in the 2nd aliquot were cross-linked with 5% formaldehyde for 30 s and then quenched with 2.93 m glycine, pH 5, for 1 min, and then the cells were pelleted by centrifugation. The 3rd aliquot was mixed with 2.93 m glycine, pH 5, for 1 min, and cells were collected by centrifugation. Each cell pellet was washed with 50 ml of ice-cold TBS, and cells were collected by centrifugation as above. Samples were processed and digested as described (47) with the following exceptions. Aliquots were taken for the undigested (input) samples, and digestions were performed by adding 500, 1000, or 2000 units of MNase (Worthington) as indicated in the figure. Samples were then resolved on 2% agarose gels, stained with ethidium bromide, and imaged.
Whole-cell extract preparation and Western blotting
Strains were grown in 300 ml of YPD overnight to an A600 = 1.0 in YPD and then concentrated 5-fold as described above. Following removal of a 0-min (no formaldehyde) control, formaldehyde was added to 5%, and cells were incubated for various times at room temperature as indicated in the figures, and then 10-ml aliquots were quenched in 440 ml of 3 m glycine, pH 5. Samples were spun down and then prepared as either chromatin or whole-cell extracts (WCE); the Benoit's buffer (200 mm Tris-HCl, pH 8.0, 400 mm (NH4)2SO4, 10 mm MgCl2, 1 mm EDTA, 10% glycerol, 7 mm β-mercaptoethanol) lysis extraction protocol was employed for whole-cell extracts (2, 3). Chromatin extracts for Western blotting were prepared in the same way as chromatin was prepared for ChIP. The only difference for chromatin samples was the use of 300 mm glycine, pH 5, in the first TBS wash instead of 250 mm glycine, pH ∼6.3 (2, 3). Both chromatin and WCE protein levels were quantified with Bradford protein dye (Bio-Rad) using bovine serum albumin as the standard. 8 or 10% denaturing protein gels were used to resolve 15 μg of protein for each sample. Unless otherwise noted, before loading the gel, samples were incubated at 95 °C for 5 min. This heating step was left out for unheated samples. Coomassie staining or membrane transfer was performed following electrophoresis. For staining, the gel was incubated with Coomassie dye (Research Organics Inc) for 1 h at room temperature with gentle shaking, followed by overnight destaining (40% methanol, 10% acetic acid) at room temperature. The gel was imaged with the FluorChemQ system (protein simple). For gel transfer, proteins were transferred to Immobilon P, and Western detection of particular protein species was performed using the antibodies listed in supplemental Table S4 and detection with Amersham Biosciences ECL Prime (GE Healthcare). Quantification of bands on the blots was done using ImageJ software (National Institutes of Health).
Collection of cross-linking time points and preparation of chromatin samples
We found that collection of eight cross-linking time points in a single experiment was manageable. A single eight-time point experiment performed with optimized glycine-quenching required nearly 4 liters of 3 m glycine, which was made by adding 900.84 g of glycine (Bio-Rad) to a total volume of 4 liters of water. The solution was gently heated on a hot plate to help the glycine dissolve. The pH of the resulting solution was then adjusted to 5 using a few milliliters of concentrated HCl (Fisher). The glycine was then aliquoted into eight 500-ml bottles, each of which contained 440 ml of the solution. The flask containing 90 ml of cell culture was rapidly mixed with a stir bar, and 14 ml of 37% formaldehyde (Fisher) was added to the culture (resulting in 5% final formaldehyde concentration) at time 0. 10-ml aliquots of culture were then removed from the flask using a Pipet-Aid (Drummond) and immediately added to the aliquoted glycine solution. For each sample, bottles were immediately capped and vigorously shaken for a few seconds to ensure good mixing. All subsequent steps were performed at 4 °C by keeping the samples on ice and using buffers and centrifuges chilled to 4 °C. Quenched cell samples were pelleted by centrifugation for 7 min at 5000 rpm in an SLA-3000 rotor and Sorvall RC 5B centrifuge. Cell pellets were resuspended in 50 ml of TBS plus 300 mm glycine and transferred to 50-ml conical tubes. The tubes were centrifuged for 5 min at 4000 rpm in an Eppendorf 5810R benchtop centrifuge. Cell pellets were then washed with 50 ml of TBS (40 mm Tris-HCl, pH 7.5, 300 mm NaCl) and spun as before. Each pellet was transferred to a FastPrep tube, and cell pellets were stored at −80 °C for later work-up or resuspended in 600 μl of 140 mm ChIP lysis buffer (50 mm Hepes, pH 7.5, 140 mm NaCl, 1% Triton X-100, 0.1% sodium deoxycholate) with protease inhibitors (Complete Protease inhibitor mixture tablet (Roche Applied Science) or 1.0 mm phenylmethylsulfonyl fluoride, 2.0 mm benzamidine, 2.0 μm pepstatin, 0.6 μm leupeptin, and 2.0 μg of chymostatin per ml of buffer) for bead beading.
Once the pellets were resuspended in ChIP lysis buffer, acid-washed beads (Sigma) were added to just above the liquid line, and samples were processed for seven cycles of 45 s on and 1 min off in a FastPrep machine (MP Biomedicals). Tube bottoms were punctured with an 18-gauge needle (BD Precision Glide) and placed in 13 × 100-mm glass tubes, and the liquid was recovered by centrifugation for 5 min at 3000 rpm in an Eppendorf 5810R benchtop centrifuge. Each sample was briefly vortexed and transferred to a 1.5-ml Eppendorf tube on ice. Samples were then sonicated with a Branson Sonifier 250 with microtip probe for seven cycles of five pulses each with 30% output and 90% duty cycle. This was followed with a 5-min spin at 14,000 rpm and 4 °C in an Eppendorf 5415C benchtop centrifuge. The supernatant was transferred to a new Eppendorf tube. Following a second spin for 20 min at 14,000 rpm, supernatants were collected, and the protein was quantified by Bradford protein assay as described above.
ChIP and real-time PCR
Chromatin immunoprecipitation was performed with 1 mg of total protein for each sample. For each time point IP, mock, and total (input) samples were assayed. IP and mock sample volumes were adjusted to 500 μl with 140 mm ChIP lysis buffer with protease inhibitors added. For TBP ChIP, 2.5 μl of anti-TBP antibody (catalog no. ab61411, Abcam) was used in the IP. For TBP-myc, 2.5 μl of anti-Myc antibody (catalog no. ab32, Abcam) was used. For LacI and AceI, 5 μl of anti-GFP antibody (catalog no. A11122 Life Technologies, Inc.) was added to the samples. The IP and mock samples were inverted overnight at 4 °C. Following overnight incubation, the IP and mock samples were then incubated with 40 μl of Sepharose A Fast Flow 4 beads (GE Healthcare) for 2 h at 4 °C. Samples were washed twice with 1 ml of 140 mm ChIP lysis buffer, 500 mm ChIP lysis buffer (same as 140 mm ChIP lysis buffer but containing 500 mm NaCl), LiCl wash buffer (10 mm Tris, pH 8.0, 250 mm LiCl, 0.5% Nonidet P-40, 0.5% sodium deoxycholate, 1 mm EDTA), and 1× TE (10 mm Tris-Cl, pH 8.0, 1 mm EDTA). Two elutions of the bound material were performed by adding 75 μl of elution buffer (50 mm Tris-HCl, pH 8.0, 1% SDS, 10 mm EDTA) to each sample for 10 min at 65 °C. The two eluates were combined and incubated overnight at 65 °C along with the total samples, which consisted of 0.1 mg of input chromatin protein combined with 150 μl of elution buffer. The following day, samples were cleaned up using the QiaQuick PCR cleanup kit (Qiagen) following the manufacturer's instructions, and DNA was eluted with 50 μl of diethyl pyrocarbonate water pre-warmed at 55 °C.
ChIP for TFA1-TAP was performed as described above, except 40 μl of a 50% slurry of IgG-Sepharose 6 Fast Flow beads (GE Healthcare) was added to the IP sample, and 40 μl of a 50% slurry of Sepharose 6 Fast Flow beads (GE Healthcare) was used for the mock samples. An overnight IP was carried out at 4 °C followed by washing the bead pellet the next day as described above.
To quantify the ChIP DNA, real-time PCR was performed using appropriate primer sets, iQ SYBR Green Supermix, and a MyiQ instrument (Bio-Rad). The standard curve inputs were run in duplicate, and all unknowns (IP, mock, and total samples) were run in triplicate. The relative ChIP signal for each time point was calculated by subtracting the mock signal from the IP signal and then dividing by the total signal. The kinetic data reported here represent the average from at least two independent experiments for each strain and condition.
Statistical analysis
Student's t tests (non-paired, two-tail, and equal variance) were used to compare data sets represented on scatter plots and marked with an asterisk for significance (p < 0.05) (see Figs. 2 and 3 and supplemental Fig. S1).
Computational modeling
The CLK model is described in equations 7, 11, and 16, Section 2.2, supplemental material of Poorey et al. (15). The model is characterized by the transcription factor association rate (ka) and disassociation rate (kd) of binding to chromatin, the formaldehyde-transcription factor cross-linking rate (kXL), the saturation level of the ChIP signal (Ssat), the transcription factor concentration in vivo (CTF), and the formaldehyde concentration (CFH). The ChIP signal, S(t), is related to the in vivo fraction of a given binding site cross-linked by the TF across cells (θXL) by the relationship θXL(t) = S(t)/Ssat, where Ssat is the saturation value of the ChIP signal. This scaling of the ChIP signal ensures that θXL(t) approaches 1 as cross-linking time goes to infinity, as required by the CLK model. Two physically interpretable parameter regimes of the model are the TF-limited regime, where TF dynamics are much slower than cross-linking dynamics (i.e. ka·CTF ≪ kXL·CFH and kd ≪ kXL·CFH), and the XL-limited regime where cross-linking dynamics are much slower than TF dynamics (i.e. kXL·CFH ≪ ka·CTF and kXL·CFH ≪ kd), as detailed in Section 2.3, supplemental material of Poorey et al. (15). Finally, for extremely slow cross-linking dynamics that occur on the time scale of the full range of cross-linking times or longer (i.e. kXL·CFH·θb·tl ≪ 1, where tl is the last cross-linking time point, which is usually 1200 s), the CLK model predicts that θXL(t) will be a linear function of cross-linking time, θXL ∼ kXL·CFH·θb·t. Notably, we observe TF-limited, XL-limited, and linear in cross-linking time CLK curves depending on the TF and locus examined.
The simulations presented in Fig. 6 show the expected CLK curves in the TF-limited, the XL-limited, and the linear regimes, whereas the schematic diagram shows the physical interpretation of the in vivo dynamics in these regimes. The hallmarks of the TF-limited model are a relatively fast exponential rise at time scales of less than ∼100 s but often less than 5 s (first cross-linking time point in the experiment) followed by a slower exponential rise (see Fig. 6A). Notably, when the first relatively fast exponential rise is less than 5 s, we observe a non-zero y-intercept in the WT and OE data with a clear separation between the WT and OE y-intercepts. When the rise in the first relatively fast exponential is ∼100 s, we find a zero y-intercept, an initial fast exponential rise in the data followed by a slower exponential rise, hence forming what looks like a “knee” in the data around the transition from the fast to the slow exponential for both the WT and OE data. Interestingly, the y-intercept for very fast cross-linking or knee for modestly fast cross-linking in the WT data yields an excellent approximation of the in vivo occupancy, θb. The XL-limited model shows a single exponential rise with a zero y-intercept for the WT and OE data (see Fig. 6B). The linear model shows a near-zero y-intercept at t = 5 s, and no sign of saturation on the experimental time scale of 700–1200 s (see Fig. 6C). Importantly, the two cross-linking dynamics-limited models, XL-limited and linear, display relatively high sensitivity to formaldehyde concentration (as shown in Fig. 1, B and C), although the TF-limited (which we also refer to as the “full model” (see Tables 1–4) for reasons described below) does not. Although the full mathematical model presented in equations 11 and 16 in the supplemental material of Poorey et al. (15) can be used to fit and represent all of these parameter regimes, we use and refer to a “full model” fit for data that clearly show the double exponential behavior (i.e. relatively fast cross-linking rise followed by a second TF-dynamics limited rise with a relatively clear kink or knee between the two). Moreover, in the case of XL-limited behavior, we use the single exponential XL-limited model shown in equation 21 of the supplemental material of Poorey et al. (15), which is a highly accurate approximation of the “full” model (equations 11 and 16 in the supplemental material of Poorey et al. (15)) in the XL-limited parameter regime. Finally, for linear in cross-linking time data, we use the linear model shown in equation 22 of the supplemental material of Poorey et al. (15), which is a highly accurate approximation of the “full” model in the very slow cross-linking dynamics parameter regime.
For data that showed negative curvature (i.e. TF-limited or XL-limited), we started by visually estimating Ssat to be close to the late time point overexpression ChIP signal. Hence, our initial guess was normally Ssat between 1 and 5, except for LacI, where we started with Ssat ∼10. In the case of data that visually showed TF-limited behavior (e.g. TBP at ACT1, LOS1, and URA1), we estimated the initial value for kXL by looking at the time (τXL) around which the data showed a knee. Setting ln(2)/kXL ∼τXL gives an estimate for kXL. The y-intercept of a linear extrapolation of the late-time S(t) data points (i.e. linear extrapolation of the S(t) data points that are approximated by the second exponential) divided by Ssat gives an initial estimate for θb. The in vivo occupancy, θb, is expressed in terms of ka and kd as θb = ka·CTF/(ka·CTF + kd). For a given θb, we can sweep over a wide range of ka and Ssat values to see where the theoretical curves match with the WT and OE experimental data. Importantly, the overall on-rate, ka·CTF, dominates the rate at which the second exponential rises. With these starting estimates for the kinetic parameters, we run the non-linear ModelFit routine in Mathematica (48) to fit the full model to the data using least squares. The fit reliably gives us ka, kd, and Ssat (equivalently, Ssat, θb = ka·CTF/(ka·CTF + kd), and t½ = ln(2)/kd).
For data that did not show TF-limited behavior (but still showed negative curvature, as opposed to a purely linear response, for example, TFIIE at ACT1 and URA1), there were two possibilities as follows: either the data were XL-limited (showing a single exponential), or the knee was not markedly visible by inspection because of the experimental time scales. We started by fitting a straight line to the short cross-linking time data to estimate kXL·Ssat and θb. With these estimates, we swept over a wide range of ka, kXL, and Ssat values to match the theoretical full model with the data. With these tuned estimates, we fit both the XL-limited model and the full model to the data, and we determined which model yielded a better fit of the data by looking at the validity of parameters obtained, the sum of squared residuals (SSR), or by conducting an F test.
For data that fit the linear XL-limited model best (e.g. TBP at NTS2 and ACE1), we subtracted the y-intercept (extrapolated ChIP signal at t = 0 s) from the data as background, and we fit a line to each of the WT and OE data using least squares. The overexpression factor is known, so we could extract kXL·Ssat and θb from the two slopes.
For some loci it was not obvious whether the data would fit the full model/TF-limited model or the linear XL-limited model (e.g. TBP at HSC82 and U6). It was important to answer the question of the better fit because the two models have a different number of effective parameters as follows: the full-model fit has four free parameters (Ssat, ka, kd, and kXL), whereas the linear regime has only one, Ssat·kXL·θb. The SSR with the linear fit (with fewer degrees of freedom) was lower than the SSR with the full model fit (with more degrees of freedom); hence, the linear fit was chosen without the need to conduct an F test comparing the two models. The full model fit gave worse SSR values because we were explicitly starting with estimates close to the TF-limited regime when fitting the full model, which lead the minimization of the difference between the model and data to a suboptimal, local minimum. Note that the SSR was calculated without normalizing the data using Ssat because the SSR scales with Ssat and Ssat is unknown in the linear fit case.
For TFIIE at ACT1, the final parameters from the XL-limited fit were unphysical (θb ∼0 and Kd = kd/ka ∼107 mol); hence, the full model fit was chosen. An F test was performed to choose the XL-limited fit for TFIIE at LOS1 over the full model fit. For TFIIE at URA1, the parameter estimates from a full model fit satisfied XL-limited binding dynamic conditions. Therefore, the TFIIE data at LOS1 and URA1 were fit with the XL-limited model.
To estimate the errors associated with our output parameters, we ran our fitting procedure on simulated data for each locus. Specifically, we simulated the data at each locus with the mean value at each time point given by the theoretical fit and the variance given by the mean of the squared residuals. We simulated and fit the data at each locus for 1000 successful fitting iterations. The standard deviation in the simulated fit parameters was calculated on the log scale and was transformed back from the log scale to determine the lower and upper bounds on the error bars quoted in Tables 1–3. Error bars for kXL could not be estimated in the case of TBP at LOS1 because the fit parameters were TF-limited and fitting the full-model to the simulated data gave spurious values for kXL in addition to failing often. Hence, the error bars for ka*CTF, kd, and Ssat for TBP at LOS1 were calculated by fitting the TF-limited model to the simulated LOS1 data.
In addition to the parameter error analysis, we assessed the significance of each of the fits as well as the parameters derived from the fits by calculating their associated adjusted R2 and p values, respectively, as shown in supplemental Tables S6–8. Importantly, the estimation of p values relies on an approximation that is equivalent to the model being linear in the parameters, which it is not. Nevertheless, we estimated parameter p values as guides to their relative significance. We found all the fits to significantly explain the variance in the data, accounting for the number of fitting parameters, with the adjusted R2 ranging from 0.93 to 0.99 for unambiguous fits (i.e. excluding full model fits of TBP at SNR6 and HSC82). We also found all kinetic parameters were significantly different from 0 except the on-rate for LacI at the lac array and TFIIE fits, suggesting that these were the least significant parameters. In the case of TFIIE, we also estimated the 95% confidence intervals of the parameters, which take into account significance information via the linearized model assumptions. For a number of the kinetic parameters, the linearized model assumption produced a lower limit of the confidence interval that yielded an unphysical, negative value, which we set to 0. For linear fits, we performed linear regression analysis separately for the wild-type and overexpressed TF data from which we derived linear coefficients (i.e. β = θb·kXL·Ssat·CFH) shown in supplemental Tables S6 and S7 along with each fit's adjusted R2 value. We calculate the occupancy and association constants shown in Tables 1–3 using the wild-type and overexpressed β values shown in supplemental Tables S6 and S7.
Author contributions
D. T. A. designed, performed, and analyzed the data for Fig 1. S. J. S. performed and analyzed the data for Figs. 2–5 and collected data for Figs. 8 and 9. E. A. H. performed and analyzed data for Figs. 5 and 10 and supplemental Fig. S1. H. Z. and S. B. developed the model and analyzed the data in Figs. 8–10 and supplemental Figs. S2 and S3. H. Z. and E. A. H. made the figures. D. T. A., E. A. H., H. Z., and S. B. wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We are grateful to Nicole Francis for discussions and for input on development of the method and to Patrick Grant for consultation on MNase digestion and chromatin accessibility.
This work was supported by National Institutes of Health Grant R01 GM55763 (to D. T. A.), Grant R21 GM110380 (to S. B. and D. T. A.), and NCI Cancer Training Grant T32 CA009109-38 and a Wagner Fellowship (to E. A. H.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Tables S1–S8 and Figs. S1–S3.
- PIC
- preinitiation complex
- TF
- transcription factors
- CLK
- cross-linking kinetics assay
- TBP
- TATA-binding protein
- OE
- overexpression
- CTF
- concentration of the TF in the nucleus
- CFH
- formaldehyde concentration
- SSR
- sum of squared residuals
- WCE
- whole-cell extract
- IP
- immunoprecipitation
- MNase
- micrococcal nuclease.
References
- 1. Kim T. H., Barrera L. O., Zheng M., Qu C., Singer M. A., Richmond T. A., Wu Y., Green R. D., and Ren B. (2005) A high-resolution map of active promoters in the human genome. Nature 436, 876–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. ENCODE Project Consortium. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rhee H. S., and Pugh B. F. (2012) Genome-wide structure and organization of eukaryotic pre-initiation complexes. Nature 483, 295–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dowen J. M., Fan Z. P., Hnisz D., Ren G., Abraham B. J., Zhang L. N., Weintraub A. S., Schujiers J., Lee T. I., Zhao K., and Young R. A. (2014) Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 159, 374–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Horn A. E., Kugel J. F., and Goodrich J. A. (2016) Single molecule microscopy reveals mechanistic insight into RNA polymerase II preinitiation complex assembly and transcriptional activity. Nucleic Acids Res. 44, 7132–7143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Coulon A., Chow C. C., Singer R. H., and Larson D. R. (2013) Eukaryotic transcriptional dynamics: from single molecules to cell populations. Nat. Rev. Genet. 14, 572–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cramer P. (2014) A tale of chromatin and transcription in 100 structures. Cell 159, 985–994 [DOI] [PubMed] [Google Scholar]
- 8. Hager G. L., McNally J. G., and Misteli T. (2009) Transcription dynamics. Mol. Cell 35, 741–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. He Y., Fang J., Taatjes D. J., and Nogales E. (2013) Structural visualization of key steps in human transcription initiation. Nature 495, 481–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Luse D. S. (2014) The RNA polymerase II preinitiation complex. Transcription 5, e27050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zawel L., and Reinberg D. (1992) Advances in RNA polymerase II transcription. Curr. Opin. Cell Biol. 4, 488–495 [DOI] [PubMed] [Google Scholar]
- 12. Conaway R. C., and Conaway J. W. (1993) General initiation factors for RNA polymerase II. Annu. Rev. Biochem. 62, 161–190 [DOI] [PubMed] [Google Scholar]
- 13. Roeder R. G. (1996) The role of general initiation factors in transcription by RNA polymerase II. Trends Biochem. Sci. 21, 327–335 [PubMed] [Google Scholar]
- 14. Solomon M. J., and Varshavsky A. (1985) Formaldehyde-mediated DNA-protein cross-linking: a probe for in vivo chromatin structures. Proc. Natl. Acad. Sci. U.S.A. 82, 6470–6474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Poorey K., Viswanathan R., Carver M. N., Karpova T. S., Cirimotich S. M., McNally J. G., Bekiranov S., and Auble D. T. (2013) Measuring chromatin interaction dynamics on the second time scale at single-copy genes. Science 342, 369–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Viswanathan R., Hoffman E. A., Shetty S. J., Bekiranov S., and Auble D. T. (2014) Analysis of chromatin binding dynamics using the cross-linking kinetics (CLK) method. Methods 70, 97–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gavrilov A., Razin S. V., and Cavalli G. (2015) In vivo formaldehyde–cross-linking: it is time for black box analysis. Brief. Funct. Genomics 14, 163–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Karpova T. S., Kim M. J., Spriet C., Nalley K., Stasevich T. J., Kherrouche Z., Heliot L., and McNally J. G. (2008) Concurrent fast and slow cycling of a transcriptional activator at an endogenous promoter. Science 319, 466–469 [DOI] [PubMed] [Google Scholar]
- 19. Robinett C. C., Straight A., Li G., Willhelm C., Sudlow G., Murray A., and Belmont A. S. (1996) In vivo localization of DNA sequences and visualization of large-scale chromatin organization using lac operator/represor recognition. J. Cell Biol. 135, 1685–1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mueller F., Stasevich T. J., Mazza D., and McNally J. G. (2013) Quantifying transcription factor kinetics: at work or at play? Crit. Rev. Biochem. Mol. Biol. 48, 492–514 [DOI] [PubMed] [Google Scholar]
- 21. Larson D. R., Singer R. H., and Zenklusen D. (2009) A single molecule view of gene expression. Trends Cell Biol. 19, 630–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morisaki T., Müller W. G., Golob N., Mazza D., and McNally J. G. (2014) Single-molecule analysis of transcription factor binding at transcription sites in live cells. Nat. Commun. 5, 4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zaidi H. A., Auble D. T., and Bekiranov S. (2017) RNA synthesis is associated with multiple TBP-chromatin binding events. Sci. Rep. 7, 39631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kuo M. H., and Allis C. D. (1999) In vivo cross-linking and immunoprecipitation for studying dynamic protein:DNA associations in a chromatin environment. Methods 19, 425–433 [DOI] [PubMed] [Google Scholar]
- 25. Wu C. H., Chen S., Shortreed M. R., Kreitinger G. M., Yuan Y., Frey B. L., Zhang Y., Mirza S., Cirillo L. A., Olivier M., and Smith L. M. (2011) Sequence-specific capture of protein-DNA complexes for mass spectrometric protein identification. PLoS ONE 6, e26217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hoffman E. A., Frey B. L., Smith L. M., and Auble D. T. (2015) Formaldehyde–cross-linking: a tool for the study of chromatin complexes. J. Biol. Chem. 290, 26404–26411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sutherland B. W., Toews J., and Kast J. (2008) Utility of formaldehyde–cross-linking and mass spectrometry in the study of protein-protein interactions. J. Mass Spectrom. 43, 699–715 [DOI] [PubMed] [Google Scholar]
- 28. Kawashima Y., Kodera Y., Singh A., Matsumoto M., and Matsumoto H. (2014) Efficient extraction of proteins from formalin-fixed paraffin-embedded tissues requires higher concentration of tris(hydroxymethyl)aminomethane. Clin. Proteomics 11, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shi S.-R., Taylor C. R., Fowler C. B., and Mason J. T. (2013) Complete solubilization of formalin fixed, paraffin-embedded tissue may improve proteomic studies. Proteomics Clin. Appl. 7, 264–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Quievryn G., and Zhitkovich A. (2000) Loss of DNA-protein cross-links from formaldehyde-exposed cells occurs through spontaneous hydrolysis and an active repair process linked to proteosome function. Carcinogenesis 21, 1573–1580 [PubMed] [Google Scholar]
- 31. Thomas M. C., and Chiang C.-M. (2006) The general transcription machinery and general cofactors. Crit. Rev. Biochem. Mol. Biol. 41, 105–178 [DOI] [PubMed] [Google Scholar]
- 32. Karpova T. S., Chen T. Y., Sprague B. L., and McNally J. G. (2004) Dynamic interactions of a transcription factor with DNA are accelerated by a chromatin remodeller. EMBO Rep. 5, 1064–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dammann R., Lucchini R., Koller T., and Sogo J. M. (1993) Chromatin structures and transcription of rDNA in yeast Saccharomyces cerevisiae. Nucleic Acids Res. 21, 2331–2338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lu K., Ye W., Zhou L., Collins L. B., Chen X., Gold A., Ball L. M., and Swenberg J. A. (2010) Structural characterization of formaldehyde-induced cross-links between amino acids and deoxynuc leosides and their oligomers. J. Am. Chem. Soc. 132, 3388–3399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Solomon M. J., Larsen P. L., and Varshavsky A. (1988) Mapping protein–DNA interactions in vivo with formaldehyde: evidence that histone H4 is retained on a highly transcribed gene. Cell 53, 937–947 [DOI] [PubMed] [Google Scholar]
- 36. Aparicio O., Geisberg J. V., Sekinger E., Yang A., Moqtaderi Z., and Struhl K. (2005) Chromatin immunoprecipitation for determining the association of proteins with specific genomic sequences in vivo. Curr. Protoc. Mol. Biol. Chapter 21, Unit 21.3 [DOI] [PubMed] [Google Scholar]
- 37. Chen J., Zhang Z., Li L., Chen B. C., Revyakin A., Hajj B., Legant W., Dahan M., Lionnet T., Betzig E., Tjian R., and Liu Z. (2014) Single-molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell 156, 1274–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Toth J., and Biggin M. D. (2000) The specificity of protein–DNA cross-linking by formaldehyde: in vitro and in Drosophila embryos. Nucleic Acids Res. 28, e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kaplan T., Li X.-Y., Sabo P. J., Thomas S., Stamatoyannopoulos J. A., Biggin M. D., and Eisen M. B. (2011) Quantitative models of the mechanisms that control genome-wide patterns of transcription factor binding during early Drosophila development. PLoS Genet. 7, e1001290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McGhee J. D., and von Hippel P. H. (1975) Formaldehyde as a probe of DNA structure. I. Reaction with exocyclic amino groups of DNA Bases. Biochemistry 14, 1281–1296 [DOI] [PubMed] [Google Scholar]
- 41. Siomin Y. A., Simonov V. V., and Poverenny A. M. (1973) The reaction of formaldehyde with deoxynucleotides and DNA in the presence of amino acids and lysine-rich histone. Biochim. Biophys. Acta 331, 27–32 [DOI] [PubMed] [Google Scholar]
- 42. Metz B., Kersten G. F., Hoogerhout P., Brugghe H. F., Timmermans H. A., de Jong A., Meiring H., ten Hove J., Hennink W. E., Crommelin D. J., and Jiskoot W. (2004) Identification of formaldehyde-induced modifications in proteins: reactions with model peptides. J. Biol. Chem. 279, 6235–6243 [DOI] [PubMed] [Google Scholar]
- 43. Toews J., Rogalski J. C., Clark T. J., and Kast J. (2008) Mass spectrometric identification of formaldehyde-induced peptide modifications under in vivo protein cross-linking conditions. Anal. Chim. Acta 618, 168–183 [DOI] [PubMed] [Google Scholar]
- 44. Stewart-Ornstein J., Weissman J. S., and El-Samad H. (2012) Cellular noise regulons underlie fluctuations in Saccharomyces cerevisiae. Mol. Cell 45, 483–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ravarani C. N., Chalancon G., Breker M., de Groot N. S., and Babu M. M. (2016) Affinity and competition for TBP are molecular determinants of gene expression noise. Nat. Commun. 7, 10417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sikorski R. S., and Hieter P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wal M., and Pugh B. F. (2012) Genome-wide mapping of nucleosome positions in yeast using high-resolution MNase ChIP-Seq. Methods Enzymol. 513, 233–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wolfram Research, Inc. (2016) Mathematica, Version 10.6, Champaign, IL [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.