

Abstract
“Smart” hydrogels are an emerging class of biomaterials that respond to external stimuli and have been investigated for a range of biomedical applications, including therapeutic delivery and regenerative engineering. Stimuli-responsive nanogels constructed of thermoresponsive polymers such as poly(N-isopropylacrylamide-co-acrylamide) (poly(NIPAM-co-AM)) and magnetic nanoparticles (MNPs), have been developed as “smart carriers” for on-demand delivery of therapeutic biomolecules via magneto-thermal activation. However, due to their small size and systemic introduction, these poly(NIPAM-co-AM)/MNPs nanogels result in limited control over long-term, localized therapeutic delivery. Here, we developed an injectable nanoengineered hydrogel loaded with poly(NIPAM-co-AM)/MNPs for localized, on-demand delivery of therapeutics (doxorubicin (DOX)). We have engineered shear-thinning and self-recoverable hydrogels by modulating the crosslinking density of a gelatin methacrylate (GelMA) network. poly(NIPAM-co-AM)/MNP nanogels loaded with DOX were entrapped within a GelMA pre-polymer solution prior to crosslinking. The temperature and magnetic field dependent release of loaded DOX was observed from the nanoengineered hydrogels (GelMA/(poly(NIPAM-co-AM)/MNPs)). Finally, the in vitro efficacy of DOX released from injectable nanoengineered hydrogels was investigated using preosteoblast and osteosarcoma cells. Overall, these results demonstrated that the injectable nanoengineered hydrogels could be used for on-demand and localized therapeutic delivery for biomedical applications.
Graphical abstract
INTRODUCTION
Injectable hydrogels are investigated for a range of biomedical applications due to their ability to locally deliver therapeutics via minimally invasive approaches.1–6 A range of synthetic and natural polymers such as alginate, gelatin, chitosan, collagen, poly(ethylene glycol) are explored to mimic extracellular matrices.5, 6 For example, gelatin-based injectable hydrogels have been developed for gene delivery7, wound healing8, hemostatic agents8, stem cell delivery9, and tissue engineering10. Despite interesting biological characteristics, these injectable hydrogels lack response to external stimuli to control and release entrapped therapeutics.
Alternatively, stimuli-responsive hydrogels are aqueous-swollen polymer networks capable of undergoing a volume phase transition due to external stimuli such as temperature, pH, and sound.11–15 For example, hydrogels from pluronic F-12716, poly(trimethylene carbonate-b-poly(L-glutamic acid)17, oligo(poly(ethylene glycol) fumarate)18 and α-amino acid residues19 have ability to swell/deswell upon exposure to external stimuli. These polymeric hydrogels are extensively investigated for sustained or triggered delivery. However, many of these stimuli are not able to penetrate deep into tissue. Instead, alternating magnetic fields (AMF), commonly used for magnetic resonance imaging (MRI), can penetrate deep tissue and can potentially be used to design magnetically-responsive hydrogels.20–22
Magnetic nanoparticles (MNPs) exhibit superparamagnetic (SPM) behavior and can produce heat due to Brownian and Néel relaxation under AMF.23 MNPs are often combined with biocompatible, thermoresponsive polymers such as poly(N-isopropyl acrylamide) (PNIPAM) with relatively low critical solution temperature (LCST) of (~32 °C).24 To raise the LCST of PNIPAM above physiological temperature, copolymerizing it with another hydrophilic polymer such as acrylamide (AM) is well reported.25 By combining MNPs and PNIPAM, a range of magnetically-responsive hydrogels and nanogels are developed for controlled therapeutic delivery.26–30 PNIPAM can be used as a thermo-responsive matrix; however, due to biostability of PNIPAM, it is not readily cleared from the body under physiological conditions.24 To overcome this issue, a range of hydrogels are synthesized by combining PNIPAM and degradable functional groups31 or polymers32–34.
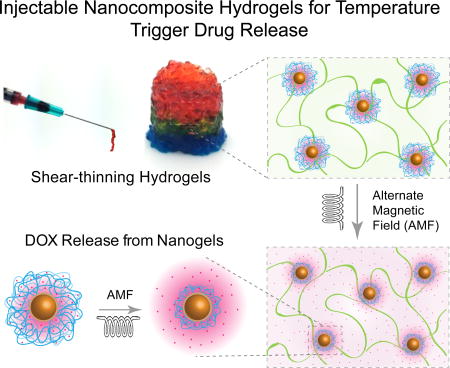
Here, we report injectable hydrogels loaded with stimuli-responsive nanogels for on-demand and localized delivery of therapeutics (Figure 1). For stimuli-responsive nanogels, we entrapped MNPs within a thermoresponsive poly(N-isopropylacrylamide-co-acrylamide) (poly(NIPAM-co-AM)) shell. The resulted poly(NIPAM-co-AM)/MNP nanogels can act as a drug reservoir. As these nanogels cannot be localized in the injected site for prolong duration, we incorporated these nanogels within a covalently crosslinked gelatin network. The gelatin based hydrogels allowed the construct to be maintained locally once injected35 and slowly degrade overtime to allow for tissue regeneration. Moreover, compared to synthetic polymers, gelatin have cell binding sites and able to integrate with the host tissue. Gelatin also have degradation site and surrounding cells can remodel the crosslinked network, which is not present in other synthetic gels. As the gelatin backbone can be modified easily, a close control over physical and chemical properties can be obtained. Based on these key characteristics, we selected gelatin-based hydrogels as injectable matrix. It is expected that the formulation of such injectable hydrogel systems can be used to deliver drugs locally and in a controlled manner.
Figure 1.

Schematic representation of fabrication of injectable nanocomposite hydrogels for stimuli-response release of therapeutics.
EXPERIMENTAL
Materials
Ammonium hydroxide solution (NH4 OH, ACS reagent, 28–30% NH3 basis), ammonium persulfate (APS, ACS reagent, 98+%), citric acid (99%), ferric chloride hexahydrate (FeCl3.6H2 O, ACS reagent, 97%), ferric chloride tetrahydrate (FeCl2 .4H2 O, ReagentPlus®, 98%), gelatin from porcine skin (gel strength 300, Type A), N,N’-Methylenebis(acrylamide) (BIS, 99%), N-isopropylacrylamide (NIPAM, 97%), and sodium metabisulfite (SBS, 99+%) were purchased from Sigma-Aldrich (USA). Doxorubicin hydrochloride (DOX, 98%) was purchased from Cayman Chemical (USA). Acrylamide (AM, 98+%) was purchased from Alfa Aesar (USA). Sodium dodecyl sulfate (SDS, Biotechnology grade) was purchased from Amresco (USA). Lithium acylphosphinate (LAP) photoinitiator was kindly provided by Dr. Daniel Alge’s laboratory at Texas A&M University (College Station, TX). All chemicals were used as received without further purification or processing. Ultra-pure water (17.8 MΩ.cm) was used for all experiments.
Synthesis of MNPs and Nanogels
Citric acid coated magnetic nanoparticles (MNPs) were synthesized via co-precipitation method as described previously.36 In short, FeCl3.6H2 O (4.44 grams) and FeCl2.4H2 O (1.732 grams) were mixed in 80 ml water a round bottom flask and then purged with N2 for 30 minutes. An oil bath was heated to 70 °C for 30 minutes and then a NH4 OH solution was added dropwise and a black precipitate was formed immediately. After 30 minutes, a solution of citric acid was added and the temperature was increased to 90 °C, left for 1 hour, cooled to room temperature (RT) and magnetically separated from the solution for washing 5 times with deionized water. The concentration of MNPs was then determined by measuring iron content in the resultant product using ion coupled plasma atomic emission spectroscopy (ICP-AES). The magnetization ability of MNPs was measured via superconducting quantum interference device (SQUID).
MNPs were then entrapped within poly(N-isopropylacrylamide-co-acrylamide) (poly(NIPAM-co-AM)) via free radical polymerization method described earlier to obtain poly(NIPAM-co-AM)/MNPs (nanogels).25, 28 The ratio of NIPAM to AM was specifically chosen to be 80:20 which ensures the LCST of the system to be above physiological temperature.25, 28 Briefly, solutions of NIPAM (1g), AM (0.15g) and MNPs (5ml) were added to 100 ml water in a flask and stirred and N2 purged for 30 minutes. BIS (0.027g) and SDS (0.2g) solutions were each added dropwise to the flask and purged for another 30 minutes. Then the oil bath was heated to 70 °C and APS (9.12 mg, 8 mL water) and SBS (2 mg, 8 mL of water) solutions were added dropwise. The reaction continued for 5 hours and was cooled to room temperature. To isolate nanogels, magnetic separation was performed overnight. The solution was then freeze-dried for storage.
Characterization of MNPs and Nanogels
Hydrodynamic particle size, Volume phase transition temperature (VPTT) determination, and zeta potential were performed with a Zetasizer Nano ZS (Malvern Instruments, UK). Attenuated Total Reflection - Fourier Transform Infrared (ATR-FTIR) measurement was performed with an FTIR spectrometer (ALPHA, Bruker, USA) on air-dried MNPs, NIPAM, AM, lyophilized poly(NIPAM-co-AM), and lyophilized nanogels. To determine shape and confirm the presence of an MNP core in the nanogels, transmission electron microscopy (TEM) was performed (JEM-2010, JEOL, JP). Nanogels were also imaged with scanning electron microscopy (SEM) (JSM-7500F, JEOL, JP). Thermal gravimetric analysis (TGA) measurement was performed with a TGA Q50 (TA Instruments, DE) to confirm copolymerization ratio and weight percentage of MNPs in nanogels.
Synthesis of Gelatin Methacrylate
Gelatin methacrylate (80% methacrylation) was synthesized as described previously.37 The final solution of gelatin methacrylate (GelMA) was dialyzed for 7 days, filtered with quantitative filter paper and then freeze-dried for storage purposes. Lyophilized GelMA was rehydrated in a 2.2 mM LAP solution. LAP was used as the photoinitiator due to noted problems within literature of photobleaching of DOX when more common photoinitiators such as Irgacure® 2959 are used.38 A UV light source (Omnicure S2000, Lumen Dynamics, Canada) set to an intensity of 10 mW/cm2 was used for gel crosslinking. Lyophilized crosslinked gels were imaged via SEM (JSM-7500F, JEOL, JP).
Preparation of Nanocomposite Hydrogels
Nanocomposites were prepared by suspending 5 mg/mL of nanogels were suspended in 1 mL of GelMA (5wt%) and subjecting to UV (10 mW/cm2) for 30 seconds. To perform stress, frequency, and shear stress sweeps, a 1 mm thick sheet was crosslinked in a sandwich mold and 7 mm diameter punches were made. In addition to this, extruded gels of 1 mL of gel from a syringe with an 18G needle were tested for comparison with stress and frequency sweeps. To study release, 5 mg of drug-loaded nanogels were well-dispersed and entrapped within a UV-crosslinked gel. To disperse nanogels GelMA, the pellet of washed DOX-loaded nanogels was re-suspended and vortexed within a 100 µL solution of GelMA+LAP, after which the nanocomposite precursor was exposed to UV light. Lyophilized crosslinked nanocomposites were imaged via SEM (JSM-7500F, JEOL, JP).
Rheological Studies
Shear stress and frequency sweeps were completed on an Anton Paar Physica MCR 301 rheometer (Anton-Paar GmbH, Austria). All experiments were completed at a physically relevant temperature (37 °C) in a humid atmosphere with solvent trap. Shear rate sweeps were performed from 0.1–100 1/s to determine the shear-thinning characteristics. The linear viscoelastic region (LVR) of the samples were determined via a frequency sweep executed at 1 Pa between 0.1–100 Hz. Shear stress sweep was performed at 1 Hz between 0.0–100 Pa. Frequency and shear stress sweeps were performed on as-prepared and extruded samples.
Encapsulation of DOX in Nanogels and Release
P(NIPAM-AM) nanogel was used to encapsulate and evaluate release of Doxorubicin (Dox). The system, in general, swells in aqueous media when it is lowered below its LCST and allows the encapsulation of aqueous soluble drug into its pores network. When the temperature is raised beyond its LCST, de-swelling of nanogel result in release of entrapped drugs.23 DOX absorbance and fluorescence was measured with an Infinite® 200 PRO microplate reader (Tecan, Switzerland). Various weights of lyophilized nanogels (2.5, 5, 10, and 15 mg) were re-suspended in 1 mL solutions of 100 µg DOX to obtain optimal loading. In addition to this, 5 mg of lyophilized nanogels were also rehydrated in 1 mL solutions of varying concentrations of DOX (25, 50, 100, 150 µg/mL). After overnight swelling at 4°C, the nanogels were centrifuged and washed 4x with chilled DI H2O. The DOX concentration loaded within the nanogels was found via encapsulation efficiency (EE) calculations (Eq. 1).
| (Eq. 1) |
Where, weight of feed cargo (Wf) and weight of the unloaded cargo (Wu). Wu were determined by measuring and summating fluorescence of the supernatant after each round of centrifugation.
As DOX is not stable in aqueous media for long time, so cumulative release of DOX from nanogel was monitor by replacing entire supernatant with PBS. This procedure ensured that released DOX does not stay in solution for long time. To measure release of DOX from nanogels, 5 mg of 100 µg/mL DOX loaded and washed nanogels were suspended in 1 mL of 1X PBS inside of a floating dialysis tube as a sink-reservoir system using dialysis membrane (Molecular weight cutoff ~ 10,000 Da). 1 mL was taken at predetermined time intervals, and then replaced with 1 mL of 1X PBS. These experiments were performed at 37 °C and 50 °C. Fluorescence was then measured using the microplate reader. Cumulative release was then calculated and plotted versus time.
An Ambrell® EasyHeat 2.4 kW induction heating system (Ambrell, UK) was utilized for generation of an AMF to be exposed to the nanocomposite system at RT. A 0.25 inch (0.635 cm) thick copper coil of 0.5 inch (1.27 cm) inner radius and 6 turns was utilized with a current of 400 A alternating at 170 kHz to produce a magnetic field of 2.99×104 A/m (0.0375 T). The magnetic strength required to produce a release from our system was calculated using the Biot-Savart Law (Eq. 2):
| (Eq. 2) |
Where H is magnetic field strength in A/m, N is number of turns, i is the current in amperes, R is the inner coil radius in meters, L is the coil length in meters.39 A DOX-loaded nanocomposite 100 µL droplet was submerged in 1 mL 1X PBS within an Eppendorf tube. A control tube was maintained at RT, while the other was exposed to the AMF. After 1 hour of exposure, sample and control tubes were centrifuged and the PBS solution was extracted and fluorescence measured in order to obtain the amount of DOX released.
2D Cell Exposure
Mouse preosteoblasts (MC3T3-E1 Subclone 4, ATCC) were cultured in normal growth media composed of α-minimal essential media (α-MEM, Hyclone), 16.5% fetal bovine serum (Atlanta Biologicals, USA), and 1% penicillin/streptomycin (100 U/100 µg/mL, Gibco). Similarly, mouse osteosarcoma cells (RFP-MOS-J) were cultured in Dulbecco’s modified eagle media (DMEM, Hyclone), 10% fetal bovine serum (Atlanta Biologicals, USA), and 1% penicillin/streptomycin (100 U/100 µg/mL, Gibco). RFP-MOS-J cell line was kindly provided by Dr. Roland Kaunas’ laboratory at Texas A&M University. Media was changed every 3–4 days and cells were passaged at ~70–80% confluency.
To test the cytotoxicity of doxorubicin, an MTT assay (MTT assay Kit, ATCC) was performed with the RFP-MOS-J cell line. Cells were seeded at a density of 10,000 cells per well in a 96 well plate and allowed to proliferate for 24 hours. Cells were then treated with varying concentrations of DOX for 6 hours. After 6 hour treatment, cells were washed with sterile 1X PBS (PBS, corning) and incubated in normal growth media for 48 hours. Next, 100 µL of fresh media was added to the samples along with 10 µL of MTT reagent. Samples were incubated at 37 °C for 3 hours. After incubation, media and MTT reagent were removed and the resulting crystal was dissolved with a 100 µL of a dimethyl sulfoxide (DMSO), isopropyl alcohol (IPA) solution (DMSO:IPA). Samples were well mixed and the absorbance was read at 540 nm. The IC50 curve for DOX exposed MOS-J cells was found through normalization to a control of untreated cells. Similarly, suspensions of 5 mg nanogels and DOX-loaded nanogels were exposed to the RFP MOS-J and MC3T3 cells for 6 hours and the cytotoxicity was determined using the same treatment procedure and MTT assay.
In addition, a Live/Dead assay was performed on both cell lines treated with solutions of DOX, the 5 mg nanogels, and DOX-loaded nanogels. The Live/Dead reagent was prepared using ethidium homodimer and Calcein AM (Santa Cruz Biotechnology Inc, USA). After cells had been subjected to similar treatment conditions discussed above, they were washed with 1X PBS and incubated at 37 °C with the Live/Dead reagent for 30 minutes. The samples were then washed with 1X PBS and imaged with an epifluorescence microscope (TE2000-S, Nikon, USA).
Finally, the cytotoxicity of the nanocomposite was tested using the RFP MOS-J cells. Cells were seeded into a 24 well plate and allowed to adhere for 24 hours. To demonstrate in vitro release behavior, nanocomposites were placed in transwell inserts to expose the cells to the GelMA matrix for 6 hours. After 6 hours, the inserts were removed and cells were incubated for 48 hours similar to the previous viability studies. Live/Dead and MTT assays were performed on the samples.
Statistical Analysis
The data is presented as the means ± standard deviations of the experiments (n=3–5). Statistical analysis was performed using with ANOVA with post-hoc Tukey method. The statistical significance was defined as *p<0.05, **p<0.01, ***p<0.001.
RESULTS AND DISCUSSION
Synthesis and Characterization of MNPs and poly(NIPAM-co-AM)/MNPs Nanogels
Citric acid-capped Fe3 O4 magnetic nanoparticles (MNPs) were synthesized using a co-precipitation method. TEM data revealed that MNPs of ~5–15nm were successfully synthesized (Figure 2a). Magnetic characterization of MNPs using SQUID indicate SPM behavior with a magnetization of ~65 emu/g. MNPs were later entrapped within poly(NIPAM-co-AM) to obtain poly(NIPAM-co-AM)/MNP nanogels. The size of resulting poly(NIPAM-co-AM)/MNP nanogels were characterized using TEM and SEM. TEM revealed that each dried nanogel was ~150nm, consisting of an MNP core and a poly(NIPAM-co-AM) shell (Figure 2b). Similarly, SEM images of core-shell poly(NIPAM-co-AM)/MNP nanogels corroborate TEM results.
Figure 2.

MNPs and Nanogel Characterizations. (a) TEM and SQUID characterization of MNPs revealing uniform size and superparamagnetic behavior respectively. (b) TEM and SEM of poly(NIPAM-co-AM)/MNP nanogels. (c) DLS comparing sized distribution of MNPs to poly(NIPAM-co-AM)/MNP nanogels. MNPs show a mean hydrodynamic diameter of 28 nm with a PDI of 0.40 while nanogels show a mean of 255 nm with a PDI of 0.21. (d) VPTT was determined to be 45°C from a temperature derivative of Dh.(e) TGA provides copolymer determination of approximately 80:20 (PNIPAM:AM) and MNP approx. 15 wt.%. Zeta potential of individual polymers compared to MNPs and nanocomposites demonstrates increased stability of nanocomposite.
Hydrodynamic diameter (Dh) of MNPs and poly(NIPAM-co-AM)/MNP nanogels were determined using DLS, Dh for MNPs~28 nm (PDI~0.4) and poly(NIPAM-co-AM)/MNP nanogels ~255 nm (PDI~0.21) (Figure 2c). To further characterize particle stability and subsequently electrophoretic mobility, the zeta potentials (ζ) of MNPs (−20.2±3.2 mV) and poly(NIPAM-co-AM)/MNPs nanogels (−8.9±2.3 mV) were measured (Figure 2e). DLS and electrophoretic mobility data confirm that the core consists of multiple MNP nanoparticles aggregated together in poly(NIPAM-co-AM) shell. These observed nanogel sizes are consistent with previously reported studies.28, 40, 41
In addition, hydrodynamic diameter of poly(NIPAM-co-AM)/MNP nanogels suspended in water was monitored using DLS over a range of temperatures to determine the thermoresponsive characteristics of nanogels (Figure 2d). At lower temperatures (25 to 40°C), poly(NIPAM-co-AM)/MNP nanogels had Dh ~255 nm and were swollen with the surrounding water solution. While at higher temperature (55°C), the nanogel sizes reduced to ~90nm and water was expelled. The VPTT, determined by obtaining the derivative of the DLS temperature sweep, was ~45°C for poly(NIPAM-co-AM)/MNP nanogels. The VPTT was chosen over the more commonly used LCST to describe the temperature response because the poly(NIPAM-co-AM) shell was a fully crosslinked network.42 Our results corroborate with previously reported studies that report a VPTT ~45°C for poly(NIPAM-co-AM) with similar copolymer ratio.25
The use of stimuli-responsive poly(NIPAM-co-AM) shell over MNPs core permits control over the release of entrapped drug via non-invasive approach. Thermo-responsive gel of poly(NIPAM-co-AM) polymer segments swells in water below LCST (<45°C) and collapse at higher temperatures (>45°C), defined as VPTT (Fig. 2d). The loaded drug can be released via application of alternating magnetic field which causes nanoparticles to heat-up due to Néel and Brownian relaxations and facilitates de-swelling of thermoresponsive shell. This release mechanism is therefore a combinatory approach of utilizing magnetic behavior to thermally activate our poly(NIPAM-co-AM) nanogels to accelerate drug diffusion into the targeted tissue site.
Finally, thermogravimetric analysis (TGA) of dried poly(NIPAM-co-AM)/MNPs nanogels was performed to determine the weight fraction of MNPs and the ratio between PNIPAM and AM (Figure 2e). Taking the derivative of the weight loss curve allowed for characterization of peaks at 275°C and 375°C corresponding to degradation of AM and NIPAM, respectively. In addition, the copolymerization ratio of ~80:20 (NIPAM:AM) was also ascertained from the weight loss data. The weight of MNPs within the nanogel sample was ~15%.
Shear-thinning and Injectable Nanocomposite Hydrogels
Injectable nanocomposite hydrogels were designed by loading P(NIPAM-co-AM)/MNP nanogels (5 mg/mL) within 1mL of GelMA pre-polymer (5 wt%) solution (Figure 3a). After subjecting GelMA to UV light, covalently crosslinked nanocomposite networks were obtained as determined from UV rheology (Figure 3b). Upon UV exposure, an increase in storage modulus (G’) was observed indicating formation of a covalently crosslinked network via acrylate bond formation. Covalent crosslinking of GelMA provides mechanical stability and high stiffness. Samples were subject to UV light and crosslinked within syringe to prepare for delivery using an 18-gauge needle. The mechanical integrity of as-prepared and extruded GelMA hydrogels were monitored via oscillatory stress sweeps (Figure 3c). Both as-prepared and extruded hydrogels had similar storage modulus at lower stress (<10Pa), but extruded gels exhibited a lower cross-over point, indicating a weaker network due to bond breakage after undergoing shear deformation. Covalent bond breakage due to large shear deformation (tearing/ripping) compromises the mechanical properties of the hydrogel. Previous studies have utilized nanoparticles or have processed gelatin below its solution temperature rather than crosslinking for injection purposes. 8, 35, 43 However for storage applications and ease of use in the clinic, pre-crosslinked materials are ideal. In anticipated, low stress applications injected crosslinked samples have similar modulus as freshly crosslinked samples. We speculate larger pieces of the hydrogel together are held together via unbroken covalent bonds and through interfacial interactions between solvent and hydrogel. Consequently, since the material has been previously extruded, lower stresses are necessary for flow/network breakage to occur. Holistically, we anticipate the once injected, the sample will have limited network break down across appropriate stresses. In addition, the concentration of GelMA (or crosslinking density) will strongly influences shear-thinning characteristics. For example, it is expected that higher GelMA concentrations (10 or 15 wt%) will results in stronger hydrogel network which will be difficult to inject though the syringe. At lower GelMA concentration (~3 wt%), crosslinked gel is injectable but will have low mechanical stability. In addition, no significant difference in microstructure was observed due to P(NIPAM-co-AM)/MNP nanogel addition (Figure 3d), which may result from the low concentration of nanogels within the crosslinked GelMA.
Figure 3.

Nanocomposite Characterizations. (a) Schematic of poly(NIPAM-co-AM)/MNP nanogels suspended within GelMA. (b) UV gelatin kinetics of GelMA. (c) comparison of GelMA as-prepared and extruded gel demonstrating no significant difference in network stability. (d) Microstructures of 5 wt.% GelMA and 5 wt.% GelMA with 5 mg nanogels added (Nanocomposites) observed via SEM. (e,f) Frequency and stress sweeps of GelMA and Nanocomposite demonstrating stability of networks overtime. G’ is approximately 200 Pa and there is no significant difference between GelMA and Nanocomposites. (f) A shear rate sweep demonstrating shear-thinning behavior for both GelMA and Nanocomposites. (g) Injected Nanocomposite exhibited recoverability of original strain after 3 cycles demonstrating thixotropic behavior.
To further investigate the effect of P(NIPAM-co-AM)/MNP nanogel addition on the mechanical properties of nanocomposite hydrogels, stress and frequency sweeps were performed to monitor the storage (G′) and loss modulus (G″). Injection of the hydrogel destroys covalent bonds thereby shifting oscillatory stress data left (lower yield point). Stress sweeps performed on pristine samples (Figure 3e) are contrary to injected samples and data points are shifted to the right within the linear viscoelastic region as determined via frequency sweep. At low stress (<10 Pa), both GelMA and nanocomposite hydrogels exhibited linear viscoelastic regions (plateau of G′). At higher stress, a decrease in storage modulus and an increase in loss modulus were observed, indicating network breakage. The crossover point, where G″ was higher than G′, was determined for GelMA and nanocomposite hydrogels to evaluate the effect of nanogel addition on network stability. Interestingly, the addition of nanogels to GelMA showed to increase network stability. Nanogels are more highly cross-linked compared to the surrounding hydrogel which imparts both a slightly higher modulus and an increase in the yield stress.44 Serendipitously, we have chosen a concentration that does not appear to be frequency dependent and viscous dissipation appears low across all frequencies.
Shear-thinning characteristics of GelMA and nanocomposite hydrogels were determined by monitoring the change in viscosity over different shear rates (0.1–100 1/s). Both GelMA and nanocomposite hydrogels exhibited shear-thinning properties as evident by a decrease in viscosity with an increase in shear rates (Figure 3f). This property is critical for developing a minimally invasive hydrogel delivery system as shear-thinning has shown to allow for injection.8, 45, 46 The addition of nanogels to GelMA did not significantly affect the shear-thinning characteristics. This might be due to the low concentration of nanogels. The stability of the injected nanocomposite hydrogel was determined by subjecting it to low (1%) and high (100%) strain (Figure 3g). Over multiple cycles, the nanocomposite hydrogel recovered to its original modulus, indicating self-recovery characteristics. This thixotropic characteristic is critical as solidification of hydrogels after injection is necessary to remain localized.8 It is also important to note that the shear- recovery was performed at physiological temperatures (37°C), that may contribute to the elastomeric property of nanocomposites after injection.
Release of Therapeutics from Nanogels and Nanocomposite Hydrogels
To test the system as a delivery vehicle, DOX was loaded in the poly(NIPAM-co-AM)/MNP nanogels and release kinetics studies with and without presence of stimuli, thermal and magnetic, were performed (Figure 4a). Prior to quantifying release, calibration was performed with varying DOX concentrations within the expected range. The weight of nanogels and DOX used were based on encapsulation efficiency (EE) studies (Figure 4b, c). 5 mg nanogels rehydrated with 100 µg/mL DOX exhibited the best loading efficiency of 42.1±8.1% (or 42.1±8.1 µg loading), and corresponded with previous studies16, 47 so subsequent release studies were performed with these nanogel and DOX concentrations. These nanogel and DOX concentrations provided sufficient surface area to load the highest amount of DOX. In addition, this ratio exhibited the maximal encapsulation efficiency compared to 50 and 150 µg/mL DOX. This decrease in EE with concentrations below 100 µg/mL may result from supersaturation of the DOX solution. When EE of 100 µg/mL was tested with various nanogel concentrations, a significant increase was observed at 5 mg as compared to 2.5 and 15 mg nanogel weights (Figure 4c). Above 5 mg, the decrease in EE could be attributed to packing of the lyophilized nanogels, which would limit surface area available for uptake. Below 5 mg, the pellet of DOX-loaded nanogels was observed to be too small to utilize this method of encapsulation. In addition, the concentrations tested were saturated for the lower amounts of nanogels to absorb a considerable amount of DOX. This method of loading DOX within the nanogels also leads to low EE due to rate limiting step (diffusion).17, 18 Another limiting factor in EE is aggregation of DOX molecules that further slower down diffusion of DOX in nanogels.48
Figure 4.

DOX loading and release in nanogels and nanocomposites. (a) Schematic representation of loading DOX in nanogels. (b) Encapsulation efficiency of DOX in 5mg nanogels. 100 µg/mL proved to be the most efficient concentration. (c) Encapsulation efficiency of 100 µg/mL DOX in various concentrations of nanogels. 5 and 10 mg nanogels provided a significant increase in loading (***p<0.001). (d) Release kinetics from nanogels were observed to be significantly greater (**p<0.01) when exposed to temperatures above the VPTT. (e) When DOX loaded nanogels were encapsulated in GelMA matrix, release was also significantly greater (*p<0.05) when exposed to temperatures above VPTT. (f) Nanocomposites were exposed to an AMF at RT for 1 hour and cumulative release was observed to be significantly greater (***p<0.001) than nanocomposites exposed to thermal stimulus.
Utilizing the optimal concentrations of DOX and nanogels, the system was exposed to temperature above the VPTT (50°C) and after six hours, 74.6±6.3% of encapsulated DOX was released (Figure 4d). Similarly, DOX loaded nanogels were tested at 37°C; however, release was limited to less than 25%. These release profiles are comparable to previous studies.47 This limited release may result from surface adhered DOX not fully encapsulated within the nanogel, or from a slight decrease in hydrodynamic diameter at 37°C, which was previously observed in the DLS temperature sweep at the beginning of the transition temperature range (Figure 2c). The significantly enhanced release at a higher temperature, particularly above VPTT, confirmed the temperature-triggered behavior of the poly(NIPAM-co-AM) shell. Release from nanocomposites was similarly tested and an average of 75.1% of the loaded DOX was released after 24 hours of thermal exposure above the VPTT (Figure 4e). A significantly lower release was observed in the samples exposed to 37°C compared to the release profiles of the nanogels; the encapsulation of the nanogels within the GelMA matrix slowed the initial release. This delay in release is observed in previous reports that utilize macroscopic gel matrices for drug release studies.49
Doxorubicin can potentially interact with the citric acid-capped MNPs50 and thus result in ~75% cumulative release (Figure 4d&e). In addition, it is expected that the steric hindrance due to polymeric chain movements against the MNP core prevents full deswelling of the nanogel system and retains some of the loaded therapeutic drug (DOX). It should also be noted that the plateau observed in Figures 4d&e is in alignment with previously reported hydrogel release studies.51 In addition, earlier studies also reported ~80% release of DOX from negatively charged oligo(poly(ethylene glycol) fumarate) hydrogels.18 They attributed strong electrostatic interaction between DOX and polymer for sustained release due to an ion-exchange mechanism. Other studies also support that dimerization and aggregation from DOX-DOX interactions potentially reduced the DOX removed from the nanogels.48
An average release of 23.4±1.4% was observed after 1 hour of exposure to the AMF, and in the same environmental conditions (37°C) or higher temperature (50°C) with no AMF-exposure, a lower burst release was observed (Figure 4f). Compared to previous results, the release after AMF exposure, as expected, was lower in the present study;28 which can be attributed to the highly crosslinked hydrogel network. In addition, the induction heating system utilized had a lower frequency of 170 kHz compared to previous experiments (230 kHz).28 It is also worth noting that the field strength utilized for this experiment (0.0375 T) was lower than the AMF strength found in commercial MRIs (0.05 to 3 T), so exposure to a larger field would improve the release rate. This increased release after 1 h magnetic exposure, suggests the potential of this magnetically-responsive nanocomposite to be used as a sustained delivery vehicle. Similar observations have been made by others in which a more pronounced burst release was observed in magnetically-triggered nanogels as compared to their thermally triggered nanogels.52 These results provide evidence that this nanocomposite delivery system could be utilized for controlled drug delivery applications.
In vitro Evaluation of Nanogels and Nanocomposite Hydrogels
To evaluate the effectiveness of the nanocomposite delivery system, in vitro studies utilizing osteosarcomas (MOS-J) and preostoblasts (MC3T3) were conducted. First, an MTT assay was performed to determine the IC50 concentration (Figure 5a). At concentrations ~5µg/mL, more than 50% of the cells were no longer viable; therefore, for further experiments, 30 µg/mL of DOX was chosen as the “Free DOX” concentration and acted as a positive control. With this concentration, cell viability decreased to approximately 17.7±2.5% of normalized mitochondrial activity. DOX acts as an anti-cancer drug by entering the nucleus and intercalating with DNA to inhibit DNA replication, ultimately leading to cytostasis.53, 54 Therefore, free DOX exposure on fast-replicating cell lines such as MOS-J cause significant cell death.
Figure 5.

In vitro DOX delivery and analysis. (a) MOS-J osteosarcoma normalized mitochondrial activity or viability after exposure to various concentrations of DOX. IC50 of DOX was observed to be at concentrations above 10 µg/mL. (b) Live/Dead staining of MOS-Js revealed a greater number of dead cells when exposed to free DOX as compared to DOX-loaded nanogels and nanocomposites (Scale bar 100µm). (c) MTT assay of MOS-Js exposed to various experimental groups. DOX-loaded nanogels significantly increased (***p<0.001) cell viability in comparison to free DOX. Additionally, incorporation of DOX-loaded nanogels in the GelMA matrix further increased cell viability. (d) MC3T3s were also exposed to free DOX and DOX-loaded nanogels and a similar response in cell viability was observed (Scale bar 100µm). (e) comparison of MC3T3 viability of various experimental groups normalized to untreated control. A significant increase (***p<0.001) in cell viability was observed in the DOX-loaded nanogels.
To evaluate the effect of the nanoengineered system on cell viability, MOS-J cells were treated with free DOX, poly(NIPAM-co-AM)/MNP nanogels, DOX-loaded nanogels, GelMA, and DOX-loaded nanocomposites for 6 hours. After 48 hours, live/dead staining qualitatively revealed that free DOX caused significant cell death (100%). Exposure of cells to nanogels, GelMA and DOX-loaded nanocomposites maintained cell viability (Figure 5b, c). This indicated that hydrogels could shield the entrapped DOX from release. Similarly, MC3T3s were exposed to the various treatment groups for 6 hours. After 48 hours, cell viability was assessed with live/dead staining (Figure 5d). Similar to the MOS-Js, free DOX exposure resulted in significant cell death; however, exposure to the DOX-loaded nanogels did not affect MC3T3 viability to the extent to which they affected MOS-J viability. An MTT assay further confirmed a significant increase in metabolic activity or viability in the nanogels and DOX-loaded nanogels (Figure 5e). Comparing the two cell types, viability was observed to be lower on average for MOS-Js exposed to DOX-loaded nanogels than for MC3T3s. A previous study observed a similar response, in which cell viability was compared in two cell lines grown in different media but both uptake the same amount and type of nanoparticles.55 Another possibility is that some DOX adhered to the surface of the nanogels instead of being loaded within. However, by suspending and fixing the nanogels within the GelMA matrix, cell viability was improved while still maintaining sustained release. It is expected that upon AMF exposure, entrapped DOX will release and thus can be used for stimuli-responsive release.
Conclusions
Overall, we have developed an injectable stimuli-responsive hydrogel for on-demand delivery of therapeutic biomolecules via magneto-thermal activation. The shear-thinning and self-recoverable hydrogels were obtained by modulating the crosslinking density of the GelMA network. poly(NIPAM-co-AM)/MNP nanogels were incorporated within the hydrogel network for stimuli-responsive, controlled release of therapeutics. The temperature and magnetic field dependent release of loaded DOX was observed from the nanoengineered hydrogels (GelMA/(poly(NIPAM-co-AM)/MNPs)). The in vitro efficacy of DOX released from injectable nanoengineered hydrogels was investigated using preosteoblast and osteosarcoma cells. Overall, these results demonstrated that the injectable nanoengineered hydrogels could be used for on-demand and localized therapeutic delivery for biomedical applications.
Acknowledgments
We would like to thank Madyson Muscarello, Roshni Nambiar, and Pournima Prabhakaran for assistance in data collection. Ceylan Hayrettin and Dr. Ibrahim Karaman (Department of Materials Science and Engineering, Texas A&M University) for assistance in using their induction heating system for AMF experiments and finally Dr. Anup Bandyopadhyay for assistance in collecting SQUID data. A.K.G would like to acknowledge funding support from National Institute of Health (R03 EB023454), Texas Engineering Experiment Station, Texas A&M University Seed Grant.
Footnotes
Conflicts of interest
There are no conflicts of interest to declare.
References
- 1.Langer R, Tirrell DA. Nature. 2004;428:487–492. doi: 10.1038/nature02388. [DOI] [PubMed] [Google Scholar]
- 2.Kretlow JD, Klouda L, Mikos AG. Advanced drug delivery reviews. 2007;59:263–273. doi: 10.1016/j.addr.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 3.Gaharwar AK, Peppas NA, Khademhosseini A. Biotechnology and bioengineering. 2014;111:441–453. doi: 10.1002/bit.25160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buwalda SJ, Vermonden T, Hennink WE. Biomacromolecules. 2017;18:316–330. doi: 10.1021/acs.biomac.6b01604. [DOI] [PubMed] [Google Scholar]
- 5.Peppas NA, Hilt JZ, Khademhosseini A, Langer R. Advanced materials. 2006;18:1345–1360. [Google Scholar]
- 6.Park KM, Lewis D, Gerecht S. Annual Review of Biomedical Engineering. 2017;19 doi: 10.1146/annurev-bioeng-071516-044619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paul A, Hasan A, Kindi HA, Gaharwar AK, Rao VT, Nikkhah M, Shin SR, Krafft D, Dokmeci MR, Shum-Tim D. ACS nano. 2014;8:8050–8062. doi: 10.1021/nn5020787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaharwar AK, Avery RK, Assmann A, Paul A, McKinley GH, Khademhosseini A, Olsen BD. ACS Nano. 2014;8:9833–9842. doi: 10.1021/nn503719n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park H, Temenoff JS, Tabata Y, Caplan AI, Mikos AG. Biomaterials. 2007;28:3217–3227. doi: 10.1016/j.biomaterials.2007.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xavier JR, Thakur T, Desai P, Jaiswal MK, Sears N, Cosgriff-Hernandez E, Kaunas R, Gaharwar AK. ACS Nano. 2015;9:3109–3118. doi: 10.1021/nn507488s. [DOI] [PubMed] [Google Scholar]
- 11.Bawa P, Pillay V, Choonara YE, du Toit LC. Biomed. Mater. 2009;4:15. doi: 10.1088/1748-6041/4/2/022001. [DOI] [PubMed] [Google Scholar]
- 12.De SK, Aluru NR, Johnson B, Crone WC, Beebe DJ, Moore J. J. Microelectromech. Syst. 2002;11:544–555. [Google Scholar]
- 13.Klouda L, Mikos AG. European Journal of Pharmaceutics and Biopharmaceutics. 2008;68:34–45. doi: 10.1016/j.ejpb.2007.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kwok CS, Mourad PD, Crum LA, Ratner BD. J. Biomed. Mater. Res. 2001;57:151–164. doi: 10.1002/1097-4636(200111)57:2<151::aid-jbm1154>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 15.Zhao X, Ding X, Deng Z, Zheng Z, Peng Y, Long X. Macromolecular Rapid Communications. 2005;26:1784–1787. [Google Scholar]
- 16.Missirlis D, Kawamura R, Tirelli N, Hubbell JA. European Journal of Pharmaceutical Sciences. 2006;29:120–129. doi: 10.1016/j.ejps.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 17.Sanson C, Schatz C, Le Meins J-F, Soum A, Thévenot J, Garanger E, Lecommandoux S. Journal of Controlled Release. 2010;147:428–435. doi: 10.1016/j.jconrel.2010.07.123. [DOI] [PubMed] [Google Scholar]
- 18.Dadsetan M, Liu Z, Pumberger M, Giraldo CV, Ruesink T, Lu L, Yaszemski MJ. Biomaterials. 2010;31:8051–8062. doi: 10.1016/j.biomaterials.2010.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Casolaro M, Casolaro I, Bottari S, Del Bello B, Maellaro E, Demadis KD. European Journal of Pharmaceutics and Biopharmaceutics. 2014;88:424–433. doi: 10.1016/j.ejpb.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 20.Mura S, Nicolas J, Couvreur P. Nature materials. 2013;12:991–1003. doi: 10.1038/nmat3776. [DOI] [PubMed] [Google Scholar]
- 21.Pankhurst QA, Connolly J, Jones SK, Dobson J. Journal of physics D: Applied physics. 2003;36:R167. [Google Scholar]
- 22.Stuart MAC, Huck WT, Genzer J, Müller M, Ober C, Stamm M, Sukhorukov GB, Szleifer I, Tsukruk VV, Urban M. Nature materials. 2010;9:101–113. doi: 10.1038/nmat2614. [DOI] [PubMed] [Google Scholar]
- 23.Subhankar B, Wolfgang K. Journal of Physics D: Applied Physics. 2009;42:013001. [Google Scholar]
- 24.Schild HG. Progress in polymer science. 1992;17:163–249. [Google Scholar]
- 25.Zhang J, Chen H, Xu L, Gu Y. Journal of Controlled Release. 2008;131:34–40. doi: 10.1016/j.jconrel.2008.07.019. [DOI] [PubMed] [Google Scholar]
- 26.Meenach SA, Shapiro JM, Hilt JZ, Anderson KW. Journal of Biomaterials Science, Polymer Edition. 2013;24:1112–1126. doi: 10.1080/09205063.2012.741321. [DOI] [PubMed] [Google Scholar]
- 27.Wong JE, Gaharwar AK, Müller-Schulte D, Bahadur D, Richtering W. Journal of Magnetism and Magnetic Materials. 2007;311:219–223. [Google Scholar]
- 28.Jaiswal MK, De M, Chou SS, Vasavada S, Bleher R, Prasad PV, Bahadur D, Dravid VP. ACS applied materials & interfaces. 2014;6:6237–6247. doi: 10.1021/am501067j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jalili NA, Muscarello M, Gaharwar AK. Bioengineering & Translational Medicine. 2016 doi: 10.1002/btm2.10034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Behrens S. Nanoscale. 2011;3:877–892. doi: 10.1039/c0nr00634c. [DOI] [PubMed] [Google Scholar]
- 31.Patenaude M, Hoare T. ACS Macro Letters. 2012;1:409–413. doi: 10.1021/mz200121k. [DOI] [PubMed] [Google Scholar]
- 32.Wang M, Qiang J, Fang Y, Hu D, Cui Y, Fu X. Journal of Polymer Science Part A: Polymer Chemistry. 2000;38:474–481. [Google Scholar]
- 33.Ohya S, Nakayama Y, Matsuda T. Biomacromolecules. 2001;2:856–863. doi: 10.1021/bm010040a. [DOI] [PubMed] [Google Scholar]
- 34.Li C, Mu C, Lin W, Ngai T. ACS applied materials & interfaces. 2015;7:18732–18741. doi: 10.1021/acsami.5b05287. [DOI] [PubMed] [Google Scholar]
- 35.Liu W, Heinrich MA, Zhou Y, Akpek A, Hu N, Liu X, Guan X, Zhong Z, Jin X, Khademhosseini A, Zhang YS. Advanced Healthcare Materials. 2017 doi: 10.1002/adhm.201601451. 1601451-n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nigam S, Barick KC, Bahadur D. Journal of Magnetism and Magnetic Materials. 2011;323:237–243. doi: 10.1016/j.jmmm.2009.02.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nichol JW, Koshy ST, Bae H, Hwang CM, Yamanlar S, Khademhosseini A. Biomaterials. 2010;31:5536–5544. doi: 10.1016/j.biomaterials.2010.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fairbanks BD, Schwartz MP, Bowman CN, Anseth KS. Biomaterials. 2009;30:6702–6707. doi: 10.1016/j.biomaterials.2009.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rudnev V. Handbook of induction heating. Marcel Dekker; New York: 2003. [Google Scholar]
- 40.Legendre JY, Szoka FC. Pharm. Res. 1992;9:1235–1242. doi: 10.1023/a:1015836829670. [DOI] [PubMed] [Google Scholar]
- 41.Lee RJ, Low PS. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1995;1233:134–144. doi: 10.1016/0005-2736(94)00235-h. [DOI] [PubMed] [Google Scholar]
- 42.Constantin M, Cristea M, Ascenzi P, Fundueanu G. Express Polym Lett. 2011;5:839–848. [Google Scholar]
- 43.Avery RK, Albadawi H, Akbari M, Zhang YS, Duggan MJ, Sahani DV, Olsen BD, Khademhosseini A, Oklu R. Science Translational Medicine. 2016;8:365ra156–365ra156. doi: 10.1126/scitranslmed.aah5533. [DOI] [PubMed] [Google Scholar]
- 44.Maitland D, Campbell SB, Chen J, Hoare T. RSC Advances. 2016;6:15770–15781. [Google Scholar]
- 45.Thakur A, Jaiswal MK, Peak CW, Carrow JK, Gentry J, Dolatshahi-Pirouz A, Gaharwar AK. Nanoscale. 2016;8:12362–12372. doi: 10.1039/c6nr02299e. [DOI] [PubMed] [Google Scholar]
- 46.Marquardt LM, Heilshorn SC. Current Stem Cell Reports. 2016;2:207–220. doi: 10.1007/s40778-016-0058-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shah SA, Asdi MH, Hashmi MU, Umar MF, Awan S-U. Materials Chemistry and Physics. 2012;137:365–371. [Google Scholar]
- 48.Gaurav R. Master of Science. University of Toronto; 2012. [Google Scholar]
- 49.Dong Z, Wang Q, Du Y. Journal of Membrane Science. 2006;280:37–44. [Google Scholar]
- 50.Nawara K, Romiszewski J, Kijewska K, Szczytko J, Twardowski A, Mazur M, Krysinski P. Journal of Physical Chemistry C. 2012;116:5598–5609. [Google Scholar]
- 51.Singh M, Kundu S, Reddy MA, Sreekanth V, Motiani RK, Sengupta S, Srivastava A, Bajaj A. Nanoscale. 2014;6:12849–12855. doi: 10.1039/c4nr04064c. [DOI] [PubMed] [Google Scholar]
- 52.Liu TY, Hu SH, Liu KH, Shaiu RS, Liu DM, Chen SY. Langmuir. 2008;24:13306–13311. doi: 10.1021/la801451v. [DOI] [PubMed] [Google Scholar]
- 53.Tacar O, Sriamornsak P, Dass CR. Journal of Pharmacy and Pharmacology. 2013;65:157–170. doi: 10.1111/j.2042-7158.2012.01567.x. [DOI] [PubMed] [Google Scholar]
- 54.Mizutani H, Tada Oikawa S, Hiraku Y, Kojima M, Kawanishi S. Life sciences. 2005;76:1439–1453. doi: 10.1016/j.lfs.2004.05.040. [DOI] [PubMed] [Google Scholar]
- 55.Maiorano G, Sabella S, Sorce B, Brunetti V, Malvindi MA, Cingolani R, Pompa PP. ACS Nano. 2010;4:7481–7491. doi: 10.1021/nn101557e. [DOI] [PubMed] [Google Scholar]