

Abstract
A DNA vaccine encoding prM and E protein has been shown to induce protection against Zika virus (ZIKV) infection in mice and monkeys. However, its effectiveness in humans remains undefined. Moreover, identification of which immune cell types are specifically infected in humans is unclear. We show that human myeloid cells and B cells are primary targets of ZIKV in humanized mice. We also show that a DNA vaccine encoding full length prM and E protein protects humanized mice from ZIKV infection. Following administration of the DNA vaccine, humanized DRAG mice developed antibodies targeting ZIKV as measured by ELISA and neutralization assays. Moreover, following ZIKV challenge, vaccinated animals presented virtually no detectable virus in human cells and in serum, whereas unvaccinated animals displayed robust infection, as measured by qRT-PCR. Our results utilizing humanized mice show potential efficacy for a targeted DNA vaccine against ZIKV in humans.
Keywords: Zika virus (ZIKV), DNA vaccine, Humanized DRAG mice, Neutralizing antibody, B cell infection
Highlights
-
•
Zika DNA vaccine elicits protective antibody response in humanized DRAG mice.
-
•
Human myeloid cells and B cells are targets of Zika virus.
A Zika virus vaccine requires testing in human immune cells to determine its effectiveness. In this communication, we report that a DNA vaccine encoding Zika prM and E protein is able to elicit protective neutralizing antibodies against Zika virus in humanized DRAG mice, and that human myeloid cells and B cells are primary targets of Zika virus.
1. Introduction
Zika virus (ZIKV), a member of the flavivirus family was first isolated from a rhesus monkey in Uganda in 1947 (Dick et al., 1952). Although the first human case was described in 1952 in Nigeria (Macnamara, 1954), the virus is not known to have caused major epidemics until relatively recently. In 2007, ZIKV re-emerged to cause an epidemic in Micronesia (Duffy et al., 2009) and more recently an outbreak occurred in French Polynesia in 2013. ZIKV infection reached an almost a pandemic level in 2016 following its introduction to Brazil in 2015 (Zanluca et al., 2015). On February 1, 2016, the Zika virus was officially declared as public health emergency of international concern by the World Health Organization (WHO).
The primary mode of ZIKV transmission is through the bite of Aedes mosquitos, although recent studies suggest that sexual transmission is also a possibility (Foy et al., 2011, Musso et al., 2015). In fact, recent studies suggest that the virus can persist for long periods in the testes of infected mice leading to destruction of tissue resulting in sterility (Govero et al., 2016, Ma et al., 2016). What made the recent ZIKV epidemic alarming is its association with neurological illness such as Guillain-Barré syndrome in the French Polynesia and microcephaly in fetus and newborns (reviewed in (Retallack et al., 2016)) in the epidemic in Brazil and USA.
The implication of birth defects in infected pregnant mothers, neurological illness in adults as well as the possibility of sexual transmission has intensified efforts to develop a vaccine for ZIKV, as currently there is no approved vaccine for the virus. Great strides have already been made and several recent reports have demonstrated the efficacy of both inactivated ZIKV vaccine as well as of prM/E proteins expressed via DNA or adenoviral vectors in mice and monkey models, and most recently, a single dose of nanoparticle-encapsulated mRNA vaccine encoding prM-E protein also induced strong and durable protection in mice and monkeys (Abbink et al., 2016, Dowd et al., 2016, Larocca et al., 2016, Pardi et al., 2017). Moreover, human neutralizing antibodies have been shown to protect mice against ZIKV infection (Sapparapu et al., 2016). Although these results are encouraging, efficacy has so far not been demonstrated against human subjects or human cells in vivo.
Zika virus can infect multiple cell lines of different species in vitro. Primary human dermal fibroblasts, epidermal keratinocytes, and immature dendritic cells have been shown to be susceptible to ZIKV infection in vitro (Hamel et al., 2015). DC-SIGN (Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin) and members of TIM and TAM family of receptors, particularly AXL and Tyro3 have been also shown to facilitate infection and blocking antibodies to these receptors prevent infection, suggesting that they might serve as entry receptors (Hamel et al., 2015). Primary human neuronal stem cells and astrocytes, oligodendrocyte precursor cells, and microglia cells have also been shown to be susceptible to infection in vitro (Retallack et al., 2016). Other in vitro studies have shown that primary human placental cells and explants-cytotrophoblasts, endothelial cells, fibroblasts, Hofbauer cells in chorionic villi, amniotic epithelial cells and trophoblast progenitors can also be infected (Quicke et al., 2016, Tabata et al., 2016). However, which human cells are infected in human patients is not known.
We show that human myeloid cells and B cells are the major targets of ZIKV and that a DNA vaccine encoding the viral prM and E proteins elicits robust protection against ZIKV infection in humanized DRAG mice.
2. Materials and Methods
2.1. Antibodies, Cell Lines and Viruses
Mouse anti-flavivirus antibody (clone D1-4G2-4-15 (4G2), RRID: Ab00230-2.0) and anti-ZIKV E protein DIII (LR) ZV-67 (RRID: Ab00812-2.0) was purchased from absolute antibody Inc., UK. FITC-conjugated goat anti-mouse polyclonal antibody was obtained from Sigma Inc. (MO, USA). Fluorescence-labeled anti-human CD45, CD4, CD19, CD14 and CD11c antibodies were purchased from BD Biosciences (CA, USA). Vero cells (strain Vero 76, clone E6, RRID: CVCL_0603) were obtained from ATCC and grown in Dulbecco's Modified Eagle medium (DMEM) containing Glutamax and supplemented with 10% fetal bovine serum (FBS) and 100 U/mL penicillin-streptomycin (PS) (Invitrogen) and maintained at 37 °C in the presence of 5% CO2. ZIKV (strain PRVABC59) was obtained from ATCC. The viral stocks were produced by infecting Vero cells in the laboratory using an MOI (Multiplicity of infection) of 0.01. The supernatants were collected at day 4–5, then clarified, filtered, aliquoted and stored at − 80 °C. These viral stocks were used for FRNT (focus reduction neutralization test) neutralization assays and mice challenge.
2.2. DNA Vector Constructs
ZIKV strain PRVABC59 (Genbank Accession number: KU501215.1) was used to design the DNA vaccine plasmid. Full-length pre-membrane and envelope gene (PrM-E, defined as amino acids 126–794 of the ZIKV polyprotein) or short version of PrM-E (216–794), as described in Larocca et al., 2016 were mammalian codon optimized, synthesized (Genscript, NJ) and cloned into mammalian expression vector pTTVH8G driven by the CMV immediate early promoter for mammalian expression. A Kozak sequence and a Japanese encephalitis (JE) signaling sequence were also included. The plasmid constructs were confirmed by DNA sequencing and amplified in E.coli DH5α, and the plasmid was prepared with Qiagen endo-free Maxi-prep kit.
2.3. Humanized DRAG Mice, Immunization and ZIKV Challenge of the Mice
Humanized DRAG mice (NSG mice transgenic for human DR4, RRID: IMSR_JAX:017914) were obtained from Jackson Laboratories and bred in TTUHSC animal facility. To increase scientific rigor where ever possible, experiments have been done in a blinded manner and to exclude sex bias, each experiment contains equal number of male and female mice. To generate humanized DRAG mice, animals were irradiated with a dose of 350 cGy and injected intravenously with 0.2 million of DR4 positive HSCs isolate from the fetal liver (Advanced Bioscience Inc.). After 10–12 weeks, the reconstitution will be confirmed by flow cytometry by staining the PBMCs with human CD45, CD3, CD4 and CD8. After confirming successful reconstitution, the DRAG mice were immunized intramuscularly (IM) by injection of 100 μg of plasmid DNA twice at week 0 and week 3. Sera collected at weeks 0, 3 and 6 were used for ELISA and Microneutralization assay. For ZIKV challenge, both unimmunized and immunized male and female DRAG mice were intraperitoneally administrated with 1 × 104 PFU of ZIKV strain PRVABC59. Three days after challenge, the animals were sacrificed, and the blood as well as spleen cells were collected for immunostaining assay. Also, the blood cells and sera were used to extract RNA for quantitative RT-PCR to determine the viral loads.
2.4. Quantitative RT-PCR
ZIKV viral loads were determined by quantitative RT-PCR assay using RNA extracted from cells or serum. Briefly, cellular RNA was extracted with RNAeasy Mini kit (Qiagen, Germany) and the serum RNA was extracted using QIAamp viral RNA mini kit (Qiagen, Germany) according to the manufacturer's instructions and the RNA concentration were determined by Nanodrop. The Zika virus RT-PCR detection kit was purchased from Primerdesign Ltd., UK and quantitative RT-PCR assays were performed according the manufacturer's instructions. The viral loads were calculated as copy numbers per ml according to the standard curve.
2.5. Negative Strand RT-PCR
Human B cells were isolated from human PBMCs using CD19 positive selection kit, then were activated and expanded with B cell expansion kit for 4 days. The activated B cells (1 × 106 cells) were infected 1 × 104 pfu of ZIKV for 4 h, following which the cells were extensively washed to remove free virus and cultured with fresh medium. The supernatants and aliquots of B cells were collected at days 0, 1, 3, 5 and 8. A two-step RT-PCR was used to detect the negative strand RNA. Cellular RNA was extracted as described earlier, and reverse transcribed using the forward primer complementary to the viral negative strand. The RT primer sequence is as follows: ggaggattccggattgtcaat. Reverse transcription was performed according the protocol of SuperScript III first-strand synthesis kit (Thermo Fisher Scientific, CA). The RT reactions were then treated at 95 °C for 10 mins to inactivate the enzyme. The PCR was then performed using the following primer pair: forward primer “ggaggattccggattgtcaat” and reverse primer “tgccgtgaatctcaaaaaggc”, which target a 150-bp-long fragment at the conserved region of capsid gene negative strand.
2.6. ELISA
The antibody titers in the DRAG mouse sera were determined by ELISA. To determine the ZIKV-specific IgG from human cells, the Human ZIKV Envelope ELISA kits were used, while mouse ZIKV Envelope ELISA kits were used for test the mouse ZIKV-specific IgG. Both ELISA kits were purchased from Alpha Diagnostic International, TX, USA. 96-well plates coated with ZIKV Env protein were first equilibrated at room temperature with 300 μL of kit working wash buffer for 5 min, then 1:20 dilution of DRAG mouse sera were added to the wells and incubated at room temperature for 1 h. After washing 4 times with wash buffer, 100 μL of HRP-conjugated anti-human IgG (or anti-mouse IgG if testing the mouse antibody) was then added to each well and the plates incubated for 1 h at room temperature. Plates were then washed 6 times, developed with 100 μL of TMB substrate for 15 min at room temperature then stopped by adding 100 μL of stop solution. OD450 measurements were then recorded on a FLUOstar Omega microplate reader. Antibody titers were expressed as thresholds of OD of 1 U/mL calibrator (> 1 threshold was considered as positive) and calculated according to the Manufacturer's instructions, and all data are reported as the average of three measurements.
To determine ZIKV protein E specificity of the vaccine constructs, 2 μg of full or short versions of DNA constructs were transfected into 293 T cells cultured in 12-well plates. After 3 days, the culture supernatants were collected and 100 μL of each (triplicate) were coated into 96-well immune plate overnight. After blocking with 5% non-fat milk (in PBS) for 2 h, the primary antibody ZV-67 (ZIKV E protein DIII region-specific, purchased from absolute antibody Inc., UK) were added for 1 h, followed by HRP-labeled anti-mouse IgG antibody. The color was then developed with TMB substrate, and OD450 were recorded as described earlier.
2.7. Western Blot
To assess the PrM-E expression of the vaccine constructs and the E protein specificity, cell lysates were collected after 72 h from PEI-transfected 293 T cells. The lysates were treated with lysis buffer, heated for 10 min at 100 °C, and then run on a precast 4–12% SDS-PAGE gel (BioRad, CA, USA). The gel was then transferred onto a PVDF membrane and then blocked overnight with 5% skim milk in PBS. The membrane was incubated with the primary antibody ZV-67 (1:2000) for 2 h, followed by washing 4 times using PBS-T buffer. Then, the membrane was incubated with secondary antibody anti-mouse IgG (HRP-labeled, 1:5000) for 1 h. After washing away the unbound secondary antibody with PBS-T for 5 times, the color was developed by using Pierce ECL Western Blotting Substrate (ThermoFisher, CA, USA).
2.8. Microneutralization Assay
Neutralizing activity of mouse sera was determined using a ZIKV microneutralization (MN) assay as described previously (Abbink et al., 2016, Larocca et al., 2016) with small modifications. In brief, serial dilutions of mouse sera were incubated with 100 μL of ZIKV PRVABC59 (containing 100 PFU of viruses) at 35 °C for 2 h. Next, the antibody-virus mixtures were added to overnight-cultured Vero cell monolayers in 96-well plates and incubated for 4–5 days until the CPE developed. The cells were fixed with ethanol/methanol (1:1), then stained with mouse-anti flavivirus antibody, followed by HRP-conjugated goat anti-mouse polyclonal antibody, and then developed by adding TMB substrate. The absorbance at 450 nm was recorded. The neutralizing titers were calculated as the average of mouse sera (triplicates for each serum) and presented as the reciprocal of serum dilution yielding 50% inhibition of the OD450 (compared with preimmune mouse serum values) after plotting the data with Sigmaplot software using a polynomial regression (quadratic) curve-fitting model (Hioe et al., 2010). Seropositivity was defined as a titer of ≥ 20.
2.9. Isolation and Staining of Immune Cells
The spleens harvested from DRAG mice were mashed and filtered through a 40 μm strainer twice. Then the human cells (CD45 + cells) were isolated using mouse/human chimera isolation kit (StemCell Technologies). Next, the human cells were used to isolate B cells using CD19 positive selection kit (StemCell Technologies), the rest of cells were used to further isolate CD3 + T cells with CD3 positive selection kit, and the final subpool of the cells were considered as myeloid cells. After isolation, the cells were checked for purity by staining with CD19, CD3 and CD14 antibodies respectively (The purities of the three cell types range from 95 to 99%, data not shown).
The above-purified immune cells were treated with fixation/permeabilization kit (BD Biosciences) for 30 min, then intracellularly stained with mouse anti-Flavivirus mAb for 1 h, followed by FITC-conjugated goat anti-mouse secondary antibody for 30 min. Then, the cells were washed once with FACS buffer (PBS + 2% FBS), then respectively stained with fluorescence-labeled human CD19, CD3 and CD14 antibody, and the cells were counted by flow cytometry.
2.10. In Vitro Infection of Human B Cells
Human B cells were isolated from human PBMCs purchased from Astarte Biologics (Bothell, WA) using CD19 positive selection kit (Stemcell Technologies, Canada). The purities of the isolated CD19 + cells are 95–100%. Human B cells were then activated using the B cell expansion kit (R&D System, Minneapolis, MN) according the manufacturers' instructions, immediately after isolation. For ZIKV in vitro infection, 1 × 106 of B cells were infected with a dose of 1 × 104 pfu of ZIKV for 4 h, then the cells were extensively washed (3–4 times), and cultured in medium supplemented with B cells expansion buffer. The day 0 samples were taken from this time.
2.11. Statistical Analysis
Statistical analyses were performed with GraphPad Prism version 6. Data between groups were compared using Unpaired Student t-tests with Welch's correction, or Mann-Whitney test. P < 0.05 was considered significant and P < 0.01 was considered extremely significant. Power analysis was used to determine animal size according to previous publications to assure significances.
3. Results
While routinely used humanized mouse models, including BLT mice are good models to test for efficacy of drugs and gene therapy, they do not permit testing of vaccination approaches due to poor antibody response including class-switching. However, in recently described DRAG mice, which lack murine class II, but transgenically express human HLA-DR4, antibody response including class switching is robust (Allam et al., 2015). Therefore we tested the efficacy of the DNA vaccine in DRAG mice.
To generate humanized mice, DRAG mice were irradiated (350 gy) and intravenously injected with human DR4 + CD34 + hematopoietic stem cells (HSCs) isolated from fetal liver (obtained from Advanced Biosciences Inc.). Twelve weeks later, PBMCs (peripheral blood mononuclear cell) obtained from retro orbital bleeding were tested for human immune cell reconstitution by flow cytometry using human-specific antibodies. Human CD45 + cells in the spleen constituted ~ 50% and included CD4 and CD8 T cells, B cells, monocytes and dendritic cells (Fig. 1A). To characterize if ZIKV could be efficiently replicated in DRAG mice, a group of 4 mice were challenged with ZIKV (1 × 104 PFU (Plaque forming units) per mouse), disease progression was monitored daily and sera collected at days 0, 1, 3, 5 and 8 for viral load detection. ZIKV infection did not cause any significant symptoms or weight changes in infected mice when compared with uninfected mice (n = 2), however, infected mice did present with detectable viremia during an 8-day-follow-up period with peak viral load averaging 8 × 104 viral copies/ml at day 3, indicating effective viral replication in these mice (Fig. 1B).
Fig. 1.

Characterization of humanized DRAG mice and ZIKV infection. (A) Irradiated DRAG mice were intravenously injected with DR4-matched human CD34 + hematopoietic stem cells (HSCs). Twelve week later, PBMCs were stained with indicated human antibodies to verify reconstitution of T (human CD3 and CD4/or CD8 positive) and B cells (human CD19 positive), monocytes (human CD14 positive) and dendritic cells (human CD11c positive). (B) DRAG mice were intramuscularly administrated with 1 × 104 pfu of ZIKV (strain PRVABC59, n = 4), and the uninfected mice (n = 2) were used as negative control. The sera were collected at days 0, 1, 3, 5 and 8 to extract viral RNA and ZIKV RT-PCR was performed to determine the viral load.
Both partial prM-E (AAs 216–794) (Larocca et al., 2016) and full-length prM-E (AAs 126–794) coding sequence were used for DNA vaccines. We therefore tested both of these forms for E protein expression. We synthesized the prM-E of ZIKV (strain PRVABC59) and cloned it into a mammalian expression vector pTTVH8G. These constructs were transfected into 293 T cells and after 48 h, cells were stained with ZIKV-E protein specific antibody and examined by flow cytometry. As shown in Fig. 2A, both partial prM-E and full-length prM-E containing plasmid-transfected cells expressed E protein, although full-length prM-E expression was somewhat higher. We also tested the supernatants from transfected cell culture for antigenicity by ELISA (enzyme-linked immunosorbent assay). We found no significant difference between the partial and full-length constructs in terms of protein E specificity (Supplementary Fig. 1.). We further confirmed PrM-E protein expression by Western Blot (Supplementary Fig. 2). Therefore, we used the full-length plasmid for further studies.
Fig. 2.

ZIKV DNA vaccine induces antibody response in humanized DRAG mice.
(A) DNA vaccines encoding either a full-length or a short form of PrM-E gene were transfected into 293 T cells and the cells stained with ZIKV E antibody after 48 h. Representative flow cytometric data (left) and cumulative data on MFI from 3 independent experiments (right) are shown. Histograms from left to right represent vector control, partial PrM-E, and full length PrM-E respectively. Red histograms represent isotype control and blue histograms represent staining with E protein antibody. (B) DRAG mice were intraperitoneally administrated with 100 μg of full- length PrM-E DNA vaccine and boosted at week 3. Sera obtained at week 0 (preimmune), 3 and 6, were tested in triplicate for E-specific antibody response by ELISA. Each symbol represents mean of triplicate from individual mouse. (C) Pre-immune and vaccine boosted sera were tested for ZIKV neutralizing antibodies by microneutralization assay. The neutralizing titers were presented as the reciprocal of serum dilution yielding 50% inhibition of the OD450 (compared with preimmune mouse serum values).
DRAG mice were then immunized with a control plasmid (pTTVH8G vector only) or ZIKV DNA vaccine (100 μg per mouse) intramuscularly. The vaccination was repeated 3 weeks later. Sera collected 3 weeks after the first and second immunization was tested for ZIKV E protein-specific human IgG antibodies by ELISA using Alpha Diagnostic human ZIKV Envelope ELISA kit (with IgG-specific secondary antibody). As shown in Fig. 2B, human E protein-specific antibodies were detected following the second immunization. The sera were then tested for ZIKV neutralizing antibodies using previously described microneutralization assay (Abbink et al., 2016, Larocca et al., 2016). Significant titers of neutralizing antibodies were also detected after the second immunization (Fig. 2C). We further determined specific antibody isotypes development in DRAG mice after ZIKV vaccine immunization using Iso-gold rapid human antibody isotyping kit (Bioassay Works LLC, Ijamsville, MD) in pooled sera from twice immunized animals. The results demonstrated that all isotypes, including IgG (subclasses IgG 1, 2, 3), Ig A and IgM were present in the sera (Supplementary Fig. 3).
DRAG mice are devoid of murine T, B and NK cells (Danner et al., 2011), and thus presumably, non-humanized DRAG mice should not be able to elicit any antibody response. To confirm that mouse IgG did not contribute to the antibody response against Zika virus, we immunized non-humanized DRAG mice (without HSC (hematopoietic stem cells) injection) with full-length DNA vaccine, and 4 weeks later, tested the sera for murine antibody response by ELISA using non-immunized DRAG mouse sera as negative control. No ZIKV-specific antibody response was found in these mice (Supplemental Fig. 4). We also tested if any isotype of Ig is present in these mice using Iso-gold rapid mouse antibody isotyping kit (Bioassay Works LLC, Ijamsville, MD). We were unable to detect Ig of any isotype in these mice sera (Supplemental Fig. 5). Therefore, our data further confirms that ZIKV-specific antibody response is solely contributed by the engrafted human cells.
Fig. 4.

Susceptibility of different human immune cells to ZIKV infection. Control and DNA vaccine-immunized mice were challenged with ZIKV and after 3 days, human cells (CD45 +) were first isolated from the pooled spleens as described in the text (5 mice each treatment, 2 mice each were pooled into 2 groups, and 1 mice for third group). B cells (panel B), T cells (panel A) and myeloid cells (panel C) were further isolated and respectively stained with fluoro-labeled anti-E antibody (intracellular staining) together with fluoro-labeled anti-CD20, CD3 and CD14 (surface staining), and analyzed by flow cytometry. A representative flow cytometric data from each of 3 pools (left) and cumulative data from all mice tested (right) is shown. (D) The indicated isolated immune cell pools were tested in triplicate for ZIK viral load by qRT-PCR. Each symbol represents mean of triplicates from individual pools.
Fig. 5.

Confirmation of B cell infection in vivo and in vitro. DRAG mice were infected with 1 × 104 pfu of ZIKV and blood collected at days 0, 1, 3, 5 and 8. The PBMCs were stained with fluoro-labeled anti-humnan-CD45, CD20 and anti-ZIKV E antibodies. (A) A representative flow cytometric data of ZIKV infected B cells (CD20 and ZIKV E double positive cells) at indicated time points are shown. (B, C) Human B cells were isolated from human PBMCs using CD19 positive selection kit, then were activated and expanded with B cell expansion kit for 4 days. The activated B cells (1 × 106 cells) were infected 1 × 104 pfu of ZIKV for 4 h, following which the cells were extensively washed to remove free virus and cultured in fresh medium. The supernatants and aliquots of B cells were collected at days 0, 1, 3, 5 and 8. RNA extracted from the supernatants at indicated time points were tested for ZIKV by RT-PCR (B) and the cellular RNAs were tested for the presence of ZIKV negative strand RNA by two step RT-PCR to test for replicating virus (C). Lane 1: RNA from uninfected B cells; Lane 2–6: RNA from infected B cells sampled at days 0, 1, 3, 5 and 8; Lane 7: RNA from infected humanized DRAG mouse sera served as negative control; Lane 8: RNA from infected humanized DRAG mouse myeloid cells served as positive control.
Control and vaccinated animals were then challenged with ZIKV (1 × 104 PFU) and sacrificed 3 days after challenge. First, sera and blood cells were tested for the presence of ZIKV by RT PCR. While high levels of ZIKV were present in the control mice, virus was almost undetectable in the sera and blood cells in immunized mice (Fig. 3).
Fig. 3.
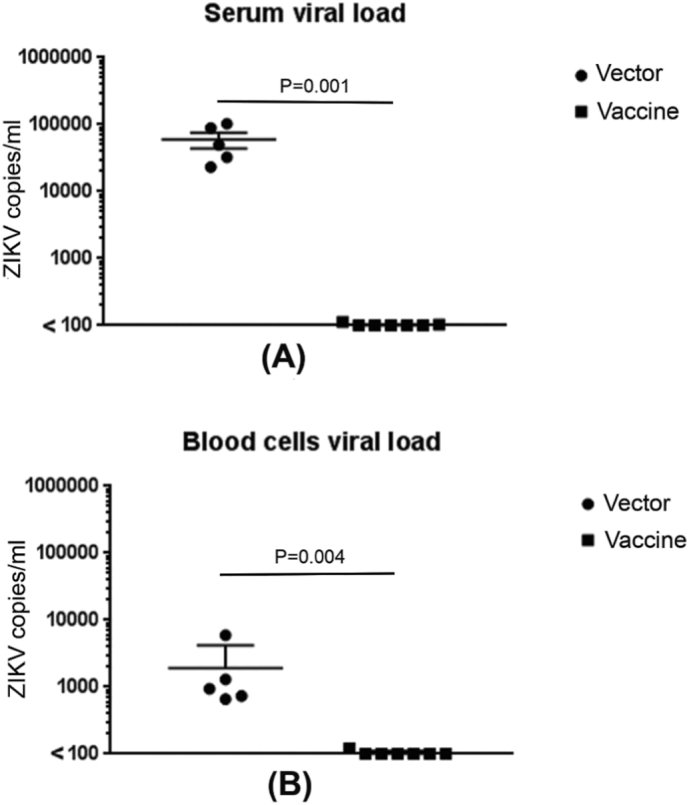
Immunization with DNA vaccine induces protection against ZIKV challenge. Vector control and DNA vaccine-immunized mice were challenged with 1 × 104 PFU of ZIKV 6 weeks later and after 3 days of challenge, viral load in the serum (A) and blood cells (B) tested by qRT-PCR. Each symbol represents an individual mouse.
To evaluate human cell types that support infection, spleens of control and vaccinated mice were pooled into 3 groups (5 mice each treatment, 2 mice each were pooled into 2 groups, and 1 mice for third group). The CD45 + human cells were first isolated, followed by isolation of human T cells and B cells were further isolated using Miltenyi beads. The T and B cell-depleted cell population was considered to be mostly myeloid cells. The purities of the three cell types range from 95 to 99% (data not shown). All three populations were intracellularly stained for ZIKV E protein and examined by flow cytometry. The myeloid cells and surprisingly, B cell population from control mice were positive for E protein, whereas all cell types from the vaccinated mice were negative (Fig. 4 A, B, C). No infection was found for T cells in both vector and vaccine immunized mice. Similar results were also seen by qRT-PCR (Fig. 4D).
Human myeloid cells especially macrophages were reported to be infected with ZIKV (Quicke et al., 2016), but we find that B cells are also infected. To further confirm B cell infection, we infected another set of DRAG mice (n = 4) and collected blood at day 0, 1, 3, 5 and 8. The PBMCs were then stained with ZIKV E and B cell-specific antibody. As shown in Fig. 5A and Supplementary Fig. 6, the ZIKV E stained B cells were strongest at day 3, similar to the viral load curve in the sera as described in Fig. 1C. To investigate whether human B cells could be infected by ZIKV in vitro, CD19 + B cells were isolated from human peripheral blood mononuclear cells (PBMCs), and then activated using human B cell expansion kit. One million activated B cells were then infected with ZIKV at a dose of 1 × 104 pfu for 4 h, followed by extensive washing to remove free virus, at which time cells were cultured in fresh medium. Supernatants were collected at days 0, 1, 3, 5, 8 and were tested for viral load by RT-PCR. As shown in Fig. 5B, the viral RNA in the supernatants steadily increased with time, peaking at day 3–5, and then began to decline. To test for replicating virus, we examined B cells for ZIKV negative strand RNA by a two-step RT PCR. For this, 1 × 105 B cells were collected at different days after infection and cellular RNA was tested for ZIKV negative strand RNA. As shown in Fig. 5C, a 150 bp PCR product amplified from the ZIKV negative strand RNA could be seen at days 1, 3, 5 and 8 after infection but not in uninfected cells. It is worth noting that viral RNA was undetectable after 3 days of ZIKV infection in vitro in resting (non-activated) B cells (Supplementary Fig. 7), implicating that activation of B cells may promote expression of cellular receptors for ZIKV infection.
4. Discussion
Taken together, we have shown that a DNA vaccine encoding prM and E protein elicits protective antibody response in humanized DRAG mice, and thus, it is very likely to induce similar protection in humans. In addition, by using a humanized DRAG mouse model, we found that among the human immune cells, human myeloid cells and B cells are the major targets of Zika virus.
A number of Zika vaccine candidates have been tested in various animal models from mouse to monkey as has been previously described (Kublin and Whitney, 2017, Ming et al., 2016). Monkey models have experimental limitations such as high cost and difficulty in obtaining a large number of animals for a significant study, while the commonly used immunodeficient AG129 mice do not generate sufficient cellular and humoral immunity due to lack of IFN α/β/γ receptors. Moreover, they do not allow testing the human immune response. Humanized BLT mice have been used to test vaccine efficacy (Lavender et al., 2013), but in the commonly used BLT and similar models, the antibody response is weak and fails to switch class to IgG isotypes. Here we have shown that in the humanized DRAG mice, all kinds of immune cells including T cells, B cells, macrophages and DCs develop normally, and after vaccination, human ZIKV E-protein-specific antibodies of different Ig isotypes could be detected. Therefore, our results showed that DRAG is a potentially attractive animal model for testing human immune response to vaccine candidates.
We used PrM-E (AA 126–794) as an immunogen in our DNA vaccine construct, differing from the ones used by either Dowd et al., (2016) (AA 123–794) or Larocca et al. (2016) (AA 216–794). It has been reported that flaviviral PrM-E proteins can assemble together to form subviral particles, though such virus like particles are non-infectious, they are structurally similar to infectious virions (Chang et al., 2001, Ferlenghi et al., 2001), and are potentially important for eliciting a potent neutralizing antibody response. By careful examination of the PrM-E protein sequence, we found that there is a potential Golgi proteolytic cleavage site (Normally contains RR, KK or RK, as found in other flaviviruses (Stocks and Lobigs, 1998)) at R127 and R128, thus it is highly likely that the start amino acid of propeptide is from R128, suggesting that AA 123–127 may not be necessary for propeptide generation (although this remains untested). Nevertheless, our construct expressed ZIKV E protein — the major immunogenic component of the vaccine — as demonstrated by flow cytometry, ELISA, as well as Western blot after transfection of 293 T cells. Thus, we believe that our construct does not disturb correct expression and folding of the immunogen. It is worth noting that proteins of full length (which encoding AA 126–794) and shorter version (which encoding AA 216–794) constructs have the same size (around 60 kDa), indicating the successful cleavage at the furin cleavage site (AA 212–215, residues RSRR) after expression.
Among cells of the immune system, we have shown that in addition to myeloid cells, B cells also potentially support ZIKV infection. Even though we showed evidences of B cell infection in cell culture and humanized mouse model, it must be cautioned, however, that there is always a possibility that studies in experimental models may not necessarily reflect the situation in humans. Particularly, we cannot exclude the possibility that in the scenarios of either cell culture or humanized mouse model, the susceptible human cell types for Zika infection is highly restricted, thus distinct from the clinical situation in ZIKV infection. Therefore, the relevance of B cell infection as it pertains to clinical ZIKV infection remains to be further dissected. In addition, if the B cell infection indeed occurs in humans, whether the virus remains in memory B cells and seeds other tissues such as testis remains to be tested in future experiments.
Ethics Statement
All work including animal studies were conducted following the guidelines for the Care and Use of Laboratory Animals of National Research Council, USA. The authors received approval from the IACUC of the Texas Tech University Health Sciences Center (protocol number 8007).
Funding Source
The work is supported by Texas Tech University Health Sciences Center at El Paso Seed Grant (183267) (to G.Y.).
Disclosure of Conflicts of Interest
The authors have declared that no competing interests exist. Funders played no role in study design, data collection, data analysis, interpretation or writing of this report.
Authorship Contributions
G.Y. and X.X. contributed equally to this work as well as performed most of the experiments. N.M., G.Y. and P.S. designed the experiments, S.A., H.G. generated the humanized mice, N.O. performed molecular cloning experiments. S·P performed the Western blot experiment and edited the manuscript. G.Y., N.M. and P.S. interpreted the data and wrote the manuscript.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ebiom.2017.10.006.
Contributor Information
Guohua Yi, Email: g.yi@ttuhsc.edu.
N. Manjunath, Email: pariswamy100@gmail.com.
Appendix A. Supplementary Data
Supplementary material
References
- Abbink P., Larocca R.A., De La Barrera R.A., Bricault C.A., Moseley E.T., Boyd M., Kirilova M., Li Z., Ng'ang'a D., Nanayakkara O. Protective efficacy of multiple vaccine platforms against Zika virus challenge in rhesus monkeys. Science. 2016;353(6304):1129–1132. doi: 10.1126/science.aah6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allam A., Majji S., Peachman K., Jagodzinski L., Kim J., Ratto-Kim S., Wijayalath W., Merbah M., Kim J.H., Michael N.L. TFH cells accumulate in mucosal tissues of humanized-DRAG mice and are highly permissive to HIV-1. Sci Rep. 2015;5 doi: 10.1038/srep10443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang G.J., Davis B.S., Hunt A.R., Holmes D.A., Kuno G. Flavivirus DNA vaccines: current status and potential. Ann. N. Y. Acad. Sci. 2001;951:272–285. [PubMed] [Google Scholar]
- Danner R., Chaudhari S.N., Rosenberger J., Surls J., Richie T.L., Brumeanu T.D., Casares S. Expression of HLA class II molecules in humanized NOD.Rag1KO.IL2RgcKO mice is critical for development and function of human T and B cells. PLoS One. 2011;6:e19826. doi: 10.1371/journal.pone.0019826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick G.W., Kitchen S.F., Haddow A.J. Zika virus. I. Isolations and serological specificity. Trans. R. Soc. Trop. Med. Hyg. 1952;46:509–520. doi: 10.1016/0035-9203(52)90042-4. [DOI] [PubMed] [Google Scholar]
- Dowd K.A., Ko S.Y., Morabito K.M., Yang E.S., Pelc R.S., DeMaso C.R., Castilho L.R., Abbink P., Boyd M., Nityanandam R. Rapid development of a DNA vaccine for Zika virus. Science. 2016;354:237–240. doi: 10.1126/science.aai9137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy M.R., Chen T.H., Hancock W.T., Powers A.M., Kool J.L., Lanciotti R.S., Pretrick M., Marfel M., Holzbauer S., Dubray C. Zika virus outbreak on Yap Island, Federated States of Micronesia. N. Engl. J. Med. 2009;360:2536–2543. doi: 10.1056/NEJMoa0805715. [DOI] [PubMed] [Google Scholar]
- Ferlenghi I., Clarke M., Ruttan T., Allison S.L., Schalich J., Heinz F.X., Harrison S.C., Rey F.A., Fuller S.D. Molecular organization of a recombinant subviral particle from tick-borne encephalitis virus. Mol. Cell. 2001;7:593–602. doi: 10.1016/s1097-2765(01)00206-4. [DOI] [PubMed] [Google Scholar]
- Foy B.D., Kobylinski K.C., Chilson Foy J.L., Blitvich B.J., Travassos da Rosa A., Haddow A.D., Lanciotti R.S., Tesh R.B. Probable non-vector-borne transmission of Zika virus, Colorado, USA. Emerg. Infect. Dis. 2011;17:880–882. doi: 10.3201/eid1705.101939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govero J., Esakky P., Scheaffer S.M., Fernandez E., Drury A., Platt D.J., Gorman M.J., Richner J.M., Caine E.A., Salazar V. Zika virus infection damages the testes in mice. Nature. 2016;540:438–442. doi: 10.1038/nature20556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel R., Dejarnac O., Wichit S., Ekchariyawat P., Neyret A., Luplertlop N., Perera-Lecoin M., Surasombatpattana P., Talignani L., Thomas F. Biology of Zika virus infection in human skin cells. J. Virol. 2015;89:8880–8896. doi: 10.1128/JVI.00354-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hioe C.E., Wrin T., Seaman M.S., Yu X., Wood B., Self S., Williams C., Gorny M.K., Zolla-Pazner S. Anti-V3 monoclonal antibodies display broad neutralizing activities against multiple HIV-1 subtypes. PLoS One. 2010;5 doi: 10.1371/journal.pone.0010254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kublin J.L., Whitney J.B. Zika virus research models. Virus Res. 2017 doi: 10.1016/j.virusres.2017.07.025. pii: S0168-1702(17)30622-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larocca R.A., Abbink P., Peron J.P., Zanotto P.M., Iampietro M.J., Badamchi-Zadeh A., Boyd M., Ng'ang'a D., Kirilova M., Nityanandam R. Vaccine protection against Zika virus from Brazil. Nature. 2016;536(7617):474–478. doi: 10.1038/nature18952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavender K.J., Pang W.W., Messer R.J., Duley A.K., Race B., Phillips K., Scott D., Peterson K.E., Chan C.K., Dittmer U. BLT-humanized C57BL/6 Rag2 −/− gammac −/− CD47 −/− mice are resistant to GVHD and develop B- and T-cell immunity to HIV infection. Blood. 2013;122:4013–4020. doi: 10.1182/blood-2013-06-506949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W., Li S., Ma S., Jia L., Zhang F., Zhang Y., Zhang J., Wong G., Zhang S., Lu X. Zika virus causes testis damage and leads to male infertility in mice. Cell. 2016;167:1511–1524. doi: 10.1016/j.cell.2016.11.016. (e1510) [DOI] [PubMed] [Google Scholar]
- Macnamara F.N. Zika virus: a report on three cases of human infection during an epidemic of jaundice in Nigeria. Trans. R. Soc. Trop. Med. Hyg. 1954;48:139–145. doi: 10.1016/0035-9203(54)90006-1. [DOI] [PubMed] [Google Scholar]
- Ming G.L., Tang H., Song H. Advances in Zika virus research: stem cell models, challenges, and opportunities. Cell Stem Cell. 2016;19:690–702. doi: 10.1016/j.stem.2016.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musso D., Roche C., Robin E., Nhan T., Teissier A., Cao-Lormeau V.M. Potential sexual transmission of Zika virus. Emerg. Infect. Dis. 2015;21:359–361. doi: 10.3201/eid2102.141363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardi N., Hogan M.J., Pelc R.S., Muramatsu H., Andersen H., DeMaso C.R., Dowd K.A., Sutherland L.L., Scearce R.M., Parks R. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature. 2017;543(7644):248–251. doi: 10.1038/nature21428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quicke K.M., Bowen J.R., Johnson E.L., McDonald C.E., Ma H., O'Neal J.T., Rajakumar A., Wrammert J., Rimawi B.H., Pulendran B. Zika virus infects human placental macrophages. Cell Host Microbe. 2016;20:83–90. doi: 10.1016/j.chom.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retallack H., Di Lullo E., Arias C., Knopp K.A., Laurie M.T., Sandoval-Espinosa C., Mancia Leon W.R., Krencik R., Ullian E.M., Spatazza J. Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc. Natl. Acad. Sci. U. S. A. 2016;113:14408–14413. doi: 10.1073/pnas.1618029113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapparapu G., Fernandez E., Kose N., Bin C., Fox J.M., Bombardi R.G., Zhao H., Nelson C.A., Bryan A.L., Barnes T. Neutralizing human antibodies prevent Zika virus replication and fetal disease in mice. Nature. 2016;540:443–447. doi: 10.1038/nature20564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocks C.E., Lobigs M. Signal peptidase cleavage at the flavivirus C-prM junction: dependence on the viral NS2B-3 protease for efficient processing requires determinants in C, the signal peptide, and prM. J. Virol. 1998;72:2141–2149. doi: 10.1128/jvi.72.3.2141-2149.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata T., Petitt M., Puerta-Guardo H., Michlmayr D., Wang C., Fang-Hoover J., Harris E., Pereira L. Zika virus targets different primary human placental cells, suggesting two routes for vertical transmission. Cell Host Microbe. 2016;20:155–166. doi: 10.1016/j.chom.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanluca C., Melo V.C., Mosimann A.L., Santos G.I., Santos C.N., Luz K. First report of autochthonous transmission of Zika virus in Brazil. Mem. Inst. Oswaldo Cruz. 2015;110(4):569–572. doi: 10.1590/0074-02760150192. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material