

Abstract
Alterations in nuclear morphology are common in cancer progression. However, the degree to which gross morphological abnormalities translate into compromised higher-order chromatin organization is poorly understood. To explore the functional links between gene expression and chromatin structure in breast cancer, we performed RNA-seq gene expression analysis on the basal breast cancer progression model based on human MCF10A cells. Positional gene enrichment identified the major histone gene cluster at chromosome 6p22 as one of the most significantly upregulated (and not amplified) clusters of genes from the normal-like MCF10A to premalignant MCF10AT1 and metastatic MCF10CA1a cells. This cluster is subdivided into three sub-clusters of histone genes that are organized into hierarchical topologically associating domains (TADs). Interestingly, the sub-clusters of histone genes are located at TAD boundaries and interact more frequently with each other than the regions in-between them, suggesting that the histone sub-clusters form an active chromatin hub. The anchor sites of loops within this hub are occupied by CTCF, a known chromatin organizer. These histone genes are transcribed and processed at a specific sub-nuclear microenvironment termed the major histone locus body (HLB). While the overall chromatin structure of the major HLB is maintained across breast cancer progression, we detected alterations in its structure that may relate to gene expression. Importantly, breast tumor specimens also exhibit a coordinate pattern of upregulation across the major histone gene cluster. Our results provide a novel insight into the connection between the higher-order chromatin organization of the major HLB and its regulation during breast cancer progression.
Keywords: higher-order chromatin organization, histone locus body, MCF10 breast cancer progression series, topologically associating domain, CTCF
Introduction
The spatial arrangement of the eukaryotic genome plays central roles in cellular processes inherent to DNA. In particular, higher-order chromatin structures are critical to regulated gene expression by facilitating long range interactions between distal gene regulatory elements and target gene promoters (Fritz et al., 2016; Kadauke and Blobel, 2009; Krivega and Dean, 2012; Whalen et al., 2016). Coregulated genes aggregate upon their activation (Brown et al., 2008; Osborne et al., 2007) or silencing (Clowney et al., 2012; Guelen et al., 2008) via chromatin hubs and many studies demonstrate that chromosomes fold into megabase units called topologically associating domains (TADs) (Barutcu et al., 2016a; Dixon et al., 2016; Dixon et al., 2012). Even larger units termed compartments that represent euchromatic or heterochromatic states have been also been identified (Lieberman-Aiden et al., 2009; McCord et al., 2013; Seitan et al., 2013). While the specific interactions within these structures provides the basis for regulatory control in physiological states, little is known about how these structures are altered in disease.
Alterations in the morphology of the nucleus have long been used as diagnostic tools for identifying cancer (Dey, 2010; Zink et al., 2004). In addition, genetic and epigenetic alterations associated with aberrant gene expression are pronounced with cancer progression (Cancer Genome Atlas, 2012; Kandoth et al., 2013; Stephens et al., 2012; Suva et al., 2013) and may very well be linked with changes in genomic organization. For example, translocations are more frequent between regions of the genome that are in closer proximity in the nucleus (Rocha et al., 2012; Roix et al., 2003; Zhang et al., 2012). Moreover, the connection between spatial organization and gene expression may be a critical determinant for the progression of cancer. For instance, aberrant promoter-enhancer interactions may drive the progression of cancer (Dekker and Mirny, 2016). Since interactions between enhancers and promoters located in different TADs are less frequent (Dixon et al., 2012), alterations in TAD boundaries could be a mechanism driving cancer progression (Lupianez et al., 2016). One key chromatin organizing factor enriched at TAD boundaries is known as CCCTC-Binding Factor (CTCF) (Barutcu et al., 2017). Deletion of CTCF binding sites at TAD boundaries has been shown to result in altered TAD formation and aberrant promoter-enhancer interactions, expression, and spreading of active chromatin (Chen et al., 2015; Lupianez et al., 2015; Narendra et al., 2015; Nora et al., 2012). Distinct alterations in chromatin structure have clinical consequences. For example, Deng et al, were able to reactivate the expression of beta-globin by artificially forcing the looping of its locus of control region back into contact with its promoter (Deng et al., 2014). In this case, activation of globin genes may provide therapeutic strategies for overcoming sickle cell anemia. Given the importance of higher-order chromatin architecture and gene expression, similar strategies may also eventually prove useful for the understanding and prevention of cancer.
To investigate alterations in gene expression and higher-order chromatin organization in breast cancer cells, we adopted the MCF10 breast cancer progression model consisting of the normal-like, epithelial MCF10A (10A), premalignant MCF10AT1 (AT1), and metastatic MCF10CA1a (CA1a). This model provides a powerful tool for studying the stages of basal triple negative breast cancer (Dawson et al., 1996; Pauley et al., 1993; Santner et al., 2001). These cells share the same origin and have similar near-diploid karyotypes and relatively few chromosomal abnormalities. We identified many pathways involved in tumorigenesis, migration, invasion, and proliferation which are altered across the MCF10 series. Using this progression model and differential gene expression analysis, we identified transcriptional responses associated with cancer progression. Surprisingly, positional gene enrichment (De Preter et al., 2008) identified the major histone gene cluster on chr6p22 as one of the top upregulated clusters of genes across the MCF10 series. Using Hi-C analysis, we find that the chromatin landscape of the major histone gene cluster is contained within hierarchical structural domains. The major histone gene cluster is subdivided into three sub-clusters each of which is located at sub-TAD boundaries. Interestingly, these sub-clusters contact each other more frequently than the regions located between them. We further identified another downstream region which loops back to interact with these sub-clusters. There appear to be modest alterations which occur in the higher order chromatin organization of the major histone gene cluster. Histone gene clusters on 6p and 1q are known to be organized in sub-nucear domains termed histone locus bodies (HLBs) (Bongiorno-Borbone et al., 2008; Ghule et al., 2008; Miele et al., 2005; Zhao et al., 1998). Consistent with these results, CTCF, a major chromatin organizer, was associated in situ with the major HLB, which forms around the 6p22 histone gene cluster, at the nuclear matrix and was bound at the anchor sites of loops within this domain. Importantly, the pattern of upregulated histone expression found in the MCF10 series was similar to that found in breast tumor patient samples. Therefore, the mechanisms that orchestrate the chromatin organization and regulation of the major histone gene cluster body in the context of deregulated proliferation during breast cancer progression are of marked clinical relevance.
Results
Global gene expression analysis reveals the major histone gene cluster as highly-changed in the MCF10 breast cancer progression series
The human MCF10 series of breast cancer cell lines (i.e., parental MCF10A, premalignant MCF10-AT1, and metastatic MCF10-CA1a) are derived from a common origin and exhibit near normal karyotypes (Dawson et al., 1996; Pauley et al., 1993; Santner et al., 2001), thus providing a unique and informative model system to study altered gene expression across breast cancer progression. To identify pathways that contribute to breast cancer progression, we performed whole transcriptome RNAseq using actively proliferating MCF10 cell lines. Sequenced reads were mapped via TopHat2 (Kim et al., 2013), counted by htseq-count (Anders et al., 2015), and analyzed for differential expression by DESeq2 (Love et al., 2014) using a p-value cutoff of 0.05 and a 2-fold expression change. The RNAseq results were validated by analyzing the expression of several key genes by qPCR that are important in breast cancer progression (Fig S1). A pair-wise comparison between MCF10A (10A) cells and the premalignant MCF10AT1 (AT1) or the metastatic MCF10CA1a (CA1a) cells revealed 1282 downregulated and 869 upregulated genes that are shared across the series. 1383 genes are uniquely downregulated in AT1 or CA1a, and 1283 genes are uniquely upregulated (Fig 1A). This large degree of differentially expressed (DE) genes across the MCF10 series suggests a complex pattern of altered gene expression is coincident with the different stages of breast cancer progression.
Figure 1. Genome-wide expression analysis reveals pathways involved in tumorigenesis, migration, invasion and proliferation across the MCF10 breast cancer progression series.
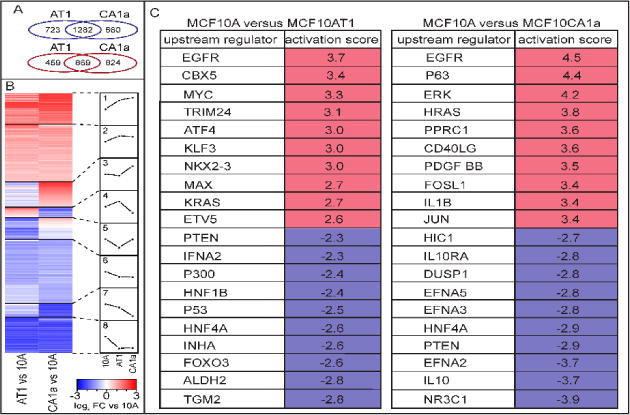
(A) Differential expression analysis of RNAseq from the normal-like mammary epithelial MCF10A (10A) cells versus the premalignant MCF10AT1 (AT1) cells or the metastatic MCF10CA1a (CA1a) cells identified 1282 downregulated and 869 upregulated genes that are shared in both AT1 and CA1a. In contrast, there are 723 downregulated genes from 10A to AT1 that are unique for that pairwise comparison; 660 are uniquely down in 10A versus CA1a; 459 are uniquely up in 10A vs AT1; 824 are uniquely up in 10A vs CA1a. (B) The pattern of differential expression is displayed in eight subsets of genes (1. highly upregulated, 2. moderately upregulated, 3. up specifically in CA1a, 4. up specifically in AT1, 5. down in AT1 and up in CA1a, 6. somewhat down, 7. moderately down, 8. highly downregulated). (C) The top ten activated and inhibited upstream regulators are displayed for both 10A vs AT1 and 10A vs CA1a.
Although recent studies have determined differential expression of transcripts and microRNAs in this progression model (Bhardwaj et al., 2017), no systematic pathway analyses was performed. To address which pathways are activated and inhibited across the series, the DE genes were clustered into eight subsets (e.g., highly upregulated, moderately upregulated, highly downregulated; Fig 1B, Table S1). Using Ingenuity pathway analysis (IPA), we identified many signaling cascades that are involved in proliferation and cell growth (Table S2). Consistent with the observation of tumorigenic phenotypes associated with cancer progression, the top ten activated upstream regulators of genes that change across the series include the oncogenes EGFR, MYC, and RAS, while putative inhibited upstream regulators include tumor suppressor PTEN, as well as P300. Furthermore, the AP-1 transcription factor complex proteins JUN and FOSL1 are among activated upstream regulators that are uniquely within the top ten for the 10A-CA1a pairwise comparison suggesting a role for these factors in metastasis. Interestingly, P53 is among the inhibited upstream regulators from 10A to AT1, while its family member P63 is activated in CA1a. This suggests a switch from downregulating genes regulated by P53 in AT1 to upregulation of genes regulated by P63 in CA1a (Fig 1C, Table S3). Taken together, the differential expression across the MCF10 series reflects higher tumorigenesis, invasion, migration and proliferation indicative of their progressive cancer phenotypes (as determined by IPA diseases and functions; Table S4).
To interrogate potential coordinate control of gene expression that is altered across the MCF10 series, we applied Positional Gene Enrichment analysis (De Preter et al., 2008) to identify clusters of genes along chromosomes. Among the top clusters that were coordinately upregulated was the major histone gene cluster on chr6p22 (Fig 2A). The region is comprised of 3 histone sub-clusters (termed A, B and C) that have non-histone genes interspersed between them. Notably, although the histone genes were upregulated, the non-histone genes between these three sub-clusters were more frequently downregulated (Fig 2B–C). Importantly, this region is not amplified across MCF10 (Fig S2), suggesting the upregulation is not simply due to increased numbers of histone locus bodies, but increased activity within the histone locus bodies across the population. Importantly, since histone mRNA are not polyadenylated, it is necessary to note that our RNAseq protocol was performed using random hexamer cDNA synthesis. Furthermore, since the histone genes share considerable sequence similarity we used only uniquely mapping reads to quantify these genes in our RNA-seq analysis. Together, these data indicate increased expression of histone genes across MCF10 progression series is required to accommodate increased proliferation of MCF10 cell lines.
Figure 2. Positional gene clustering identified the major histone locus as a highly-upregulated cluster across MCF10 breast cancer.
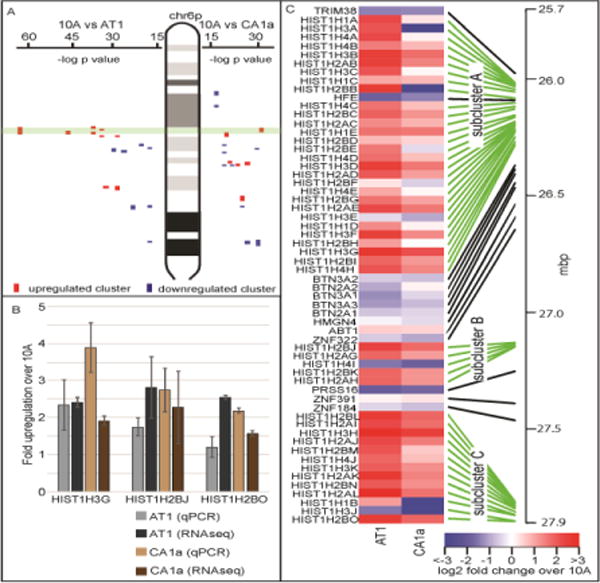
(A) Positional gene enrichment (De Preter et al., 2008) was applied to the differential expression from RNAseq from 10A to AT1 or 10A to CA1a to identify coordinate regulation of gene expression across the chromosomes. The major histone gene locus on chr6p22 was among the most significantly upregulated clusters across the MCF10 series (upregulated clusters are in red, downregulated clusters are in blue, the major histone gene cluster is outlined in green). (B) The major histone gene cluster is subdivided into three sub-clusters denoted A, B and C. One histone gene from each sub-cluster was randomly chosen for qPCR validation. qPCR was consistent in determining upregulation for all three histone genes chosen. (C) A heatmap of this region based upon RNA-seq demonstrates that the histone sub-clusters are upregulated while the non-histone genes located between the histone sub-clusters are more frequently downregulated.
The sub-clusters of the major histone gene locus are occupied by the chromatin organizer CTCF and configured in a hierarchy of topologically associating domains
The global increase in histone mRNA expression within the major histone gene cluster across the MCF10 series raises compelling questions regarding the nuclear microenvironment within and around this region during breast cancer progression. To examine the higher order chromatin organization within this region, we determined pairwise DNA-DNA interactions using the Hi-C approach (see methods). Interestingly, the major histone gene cluster is organized into hierarchical topologically associating domains (TADs) wherein sub-cluster A is localized within a sub-TAD adjacent to another sub-TAD encompassing sub-clusters B and C (Fig 3A). These two sub-TADs are present within a TAD that in turn resides within a mega-TAD including downstream genes. All three histone sub-clusters are located at sub-TAD boundaries. Interestingly, there are two histone genes located upstream of sub-cluster A which are located outside the major histone gene cluster mega-TAD (Figure 3A) and their expression was not detected in our RNA-seq analysis. Since intra-chromosomal DNA interactions are generally higher the closer two sequences are on the chromosome, the degree that the observed interactions occur above this expectation was determined (Fig 3B). The enriched interactions were subdivided into blocks representing the sub-clusters, the regions containing non-histone genes, and another region which demonstrated strong contact with the major histone gene cluster sub-clusters. This analysis revealed more frequent contacts between the histone sub-clusters when compared with the interspersed non-histone genes (Fig 3C, Fig S3). The downstream region (termed region D) that is included within the major histone gene cluster mega-TAD contacted sub-clusters A and B more frequently than sub-cluster C. While TAD boundaries are largely unchanged across MCF10 breast cancer progression, there are subtle changes occurring within these TAD boundaries (Fig 3D). For example, although the interaction frequency between sub-clusters B and C is relatively constant, interactions between A and the downstream regions B and D increase across the series. Local interactions are constant within sub-cluster A, but increase in sub-clusters B and C. Together, this data suggests a higher-order chromatin structure within the major histone gene cluster coincident with the high expression of the histone genes relative to the interspersed non-histone regions. The sub-clusters come together in an active chromatin hub. We also identified a fourth region which loops back into the major histone gene cluster (as represented in a schematic illustration in Fig 3E).
Figure 3. The higher order chromatin organization of the major histone locus.
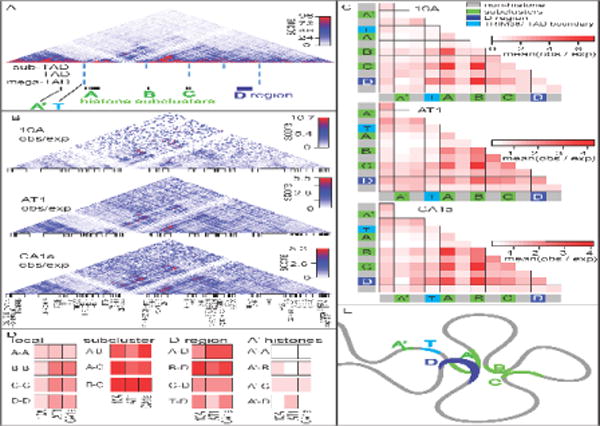
(A) Pairwise DNA-DNA interactions were analyzed via Hi-C within the major histone gene cluster across the MCF10 series. The structure of the region is organized into hierarchical topologically associating domains. The histone sub-cluster A is contained within a sub-TAD adjacent to another sub-TAD containing sub-clusters B and C. Interestingly the histone sub-clusters are located at the TAD boundaries. A downstream region termed region D loops back to interact with the histone sub-clusters. (B) Interactions that are enriched above expected linear sequence-interactions are shown. (C) The major histone gene cluster was subdivided into sub-regions of interest including two histone genes which are upstream of sub-cluster A and located outside the major histone gene cluster-megaTAD, the TRIM38 gene which is located at the megaTAD boundary, histone sub-clusters, non-histone regions which are located between, and the D region which interacts with the major histone gene sub-clusters. Interestingly, the histone sub-clusters interact with each other more frequently than the non-histone regions. (D) For ease of comparison across the MCF10 breast cancer progression series, specific sub-region interactions are displayed side-by-side. Importantly, the overall pattern is dissimilar for different subregion-subregion interactions. Local interactions: 1) within each cluster, 2) inter-subcluster interactions, 3) interactons to the D region, and 4) to the A’ histone genes outside the major histone gene cluster TAD are shown (E) A schematic diagram of the higher-order chromatin organization of the major histone gene locus is shown.
The histone genes contained within the major histone gene cluster localize within the nucleus at histone locus bodies (HLBs). HLBs contain the regulatory factors (e.g., NPAT, HINFP, FLASH) involved in the transcriptional and post-transcriptional regulation of the histone genes (Bongiorno-Borbone et al., 2008; Ghule et al., 2008; Zhao et al., 1998) that are required for physiological control of cell proliferation (Bongiorno-Borbone et al., 2010; Dominski and Marzluff, 2007; Ghule et al., 2014; Salzler et al., 2013; Yang et al., 2014; Ye et al., 2003; Zhao et al., 2000). These sub-nuclear bodies are essential for cell cycle-specific expression of histone genes, however the mechanisms and potential factors that contribute to the higher order chromatin organization of the major histone gene cluster is unclear. Given its known role as a chromatin organizer, we investigated whether CTCF has a role in organizing the major HLB that forms around the major histone gene cluster on chr6p22. We first performed immunofluorescence microscopy to assess whether CTCF, a nuclear protein that associates with the nuclear matrix, localizes to Histone locus bodies in the MCF10 series. The histone gene co-regulator NPAT exclusively localizes to histone gene clusters on chromosomes 1 and 6. Since NPAT foci are greater in intensity around the major histone gene cluster, it was used as a surrogate to identify the major HLB (Fig 4A). Immunofluorescence before and after nuclear matrix preparation revealed that CTCF is present within the major HLB and associates with NPAT at the nuclear matrix. This observation suggested that CTCF plays an important role in organizing the major histone gene cluster body (Fig 4B). To address whether CTCF is integral to the organization of histone gene cluster at the molecular level, chromatin immunoprecipitation was performed using a CTCF specific antibody, followed by deep sequencing (ChIP-seq). As expected, CTCF occupied many sites across the genome, including in and around the major histone gene cluster in the MCF10 breast cancer series (Fig 4C). This genomic occupancy was further investigated in relation to the higher order chromatin organization around this locus by analyzing pairwise DNA interactions as measured by Hi-C around this region. This analysis identified that CTCF is coincident with looping of chromatin at the major histone gene cluster (Fig 4C). These findings provide evidence that CTCF may play a key role in organizing the higher order chromatin within and around nuclear microenvironment of the major HLB.
Figure 4. CTCF is a structural component of the major histone locus body.
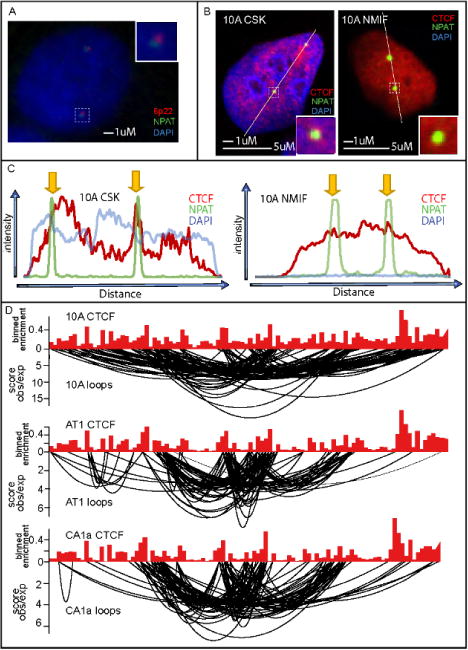
(A) ImmunoFISH was performed to 6p22 (red) and NPAT (green), a coregulatory protein involved in histone expression to demonstrate that NPAT can be used a surrogate for the major histone locus body (major HLB) within the nucleus. (B) Immunofluorescence before (CSK) and after (NIMF) nuclear matrix extraction shows that CTCF associates with the major HLB at the nuclear matrix. (C) ChIPseq for CTCF compared with looping interactions from the Hi-C demonstrates that CTCF is present at the anchor sites of looping interactions within the major HLB.
Breast tumor patient specimens exhibit a coordinate pattern of elevated major histone gene cluster expression
To determine the clinical relevance of increased histone gene expression and whether these sub-clusters are upregulated independently of the intervening non-histone genes, we utilized publicly available patient breast tumor TCGA data (Fig 5). Microarrays from 527 tumor samples (based on random hexamer generated cDNA) were analyzed for expression across the major histone gene cluster. As a validation, we clustered the expression of the histone genes to identify any that are co-expressed to an unusually high degree and therefore may be artifacts. Normal tissue, taken adjacent to tumor samples, clustered distinctly within the lower expression of the major histone gene cluster (Fig S4). Given this clustering we next determined the fold-change of tumor samples versus their normal matched tissue. Interestingly, the pattern of upregulation across this locus was comparable to that across the MCF10 breast cancer progression series (Fig 5). Similar to the MCF10 series, this chromosome band containing this region is not significantly amplified or deleted in TCGA breast tumor samples (Ciriello et al., 2015), only 1.09% of tumors had an amplification in this region. Next, we implemented survivorship curves for each individual gene across the region. Almost all the histone genes had similar trends in that the higher expression of histone genes had higher survivorship (Fig S5). The non-histone genes were more mixed in their patterns (Fig S6). Taken as a cumulative group, the histone genes in this region significantly indicate better prognosis with higher expression (Fig S7) while the non-histone genes did not show a significant relationship to survivorship (Fig S8).
Figure 5. TCGA datasets for breast tumors confirms upregulation across the major histone locus in breast cancer patient samples.
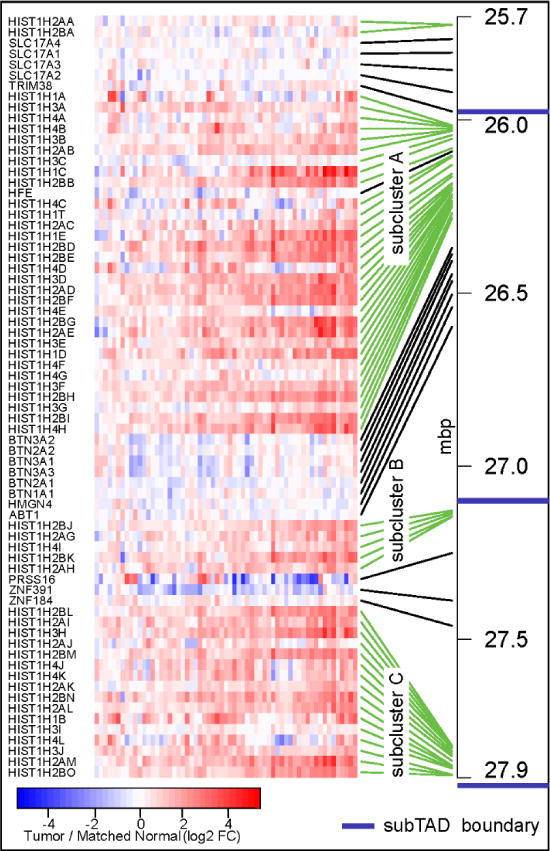
Comparison of normal tissue adjacent to tumors demonstrates upregulation of the histone genes within the major histone gene locus in breast cancer tumors. The pattern is reminiscent of the MCF10 breast cancer progression series. The blue lines indicate the location of sub-TAD boundaries in the MCF10 series.
Discussion
In this study, we used a comprehensive and unbiased approach to analyze altered gene expression across the MCF10 breast cancer progression series consisting of normal-like MCF10A, premalignant MCF10AT1, and metastatic MCF10CA1a. Genome-wide expression analysis revealed the major histone gene cluster on chr6 as one of the most significantly upregulated clusters across the MCF10 breast cancer progression series and in breast cancer tumor specimens. The chromatin of this region is organized into hierarchical domains with the histone gene sub-clusters putatively configured in an active chromatin anchored by CTCF. This work provides a novel insight into potential mechanisms by which the higher order chromatin organization within the major histone gene cluster may contribute to its regulation and function in breast cancer progression.
The higher order chromatin organization of the genome is functionally linked to the regulation of gene expression in order to establish and maintain physiological states. Physical interactions between promoters and enhancers, that may be located very distal in their linear sequence, have been demonstrated to be critical for the expression of many genes (Fritz et al., 2016; Kadauke and Blobel, 2009; Krivega and Dean, 2012; Whalen et al., 2016). Furthermore, multiple genes that are co-expressed congregate into active chromatin hubs upon their activation (Brown et al., 2008; Osborne et al., 2007) or silenced domains upon their inactivation (Clowney et al., 2012; Guelen et al., 2008). These interactions have since been determined to be primarily constrained within so-called topologically associating domains (TADs) which are regions of local higher frequency interactions approximately one to ten megabases in size (Barutcu et al., 2016a; Dixon et al., 2016; Dixon et al., 2012). At the largest level, chromosomes are organized into discrete domains within the nucleus called chromosome territories (CTs) (Fritz et al., 2016). Active TADs are more frequently located at periphery of CTs (Nagano et al., 2013). Interchromosomal interactions are different in cell and tissue lineages (Fritz et al., 2014a; Mayer et al., 2005; Parada et al., 2004). These profiles, in turn, suggest that genomic organization could explain the high frequency of particular translocations prevalent in different cancer types. Interestingly, altered interchromosomal interactions have been determined across breast cancer progression (Fritz et al., 2014b) and specifically between small chromosomes (Barutcu et al., 2015). Since active TADs are present at the CT boundaries (Nagano et al., 2013) altered interchromosomal interactions may be a result of different TADs interacting between CTs at their borders. Furthermore, Depletion of specific transcription factors such as RUNX1 (Barutcu et al., 2016b), chromatin modifiers such as SMARCA4 (Barutcu et al., 2016c), and Histone H1 (Geeven et al., 2015) have demonstrated genome-wide alterations in higher order chromatin organization. Despite this progress, much more work is required to fully understand the interplay between gene expression and higher-order chromatin organization and the alterations which occur as a result of dysregulated gene expression in breast cancer.
Breast cancer is the second leading cause of cancer mortality in women. Despite significant progress in early detection and treatment, understanding the mechanisms of breast cancer progression and metastasis still require rigorous study. To study these important processes, model systems are required due to the advantages that they are relatively easy to grow, inexpensive, and can be used in genome editing/mechanistic studies as well as in high-throughput assays of therapeutic treatments (Goodspeed et al., 2016). While there are differences between patient tumor samples and cancer cell lines, the vast majority of the alterations that occur in breast cancer progression are shared in these model systems. For example, tumor patient samples and breast cancer cell lines, share 72% of the genes that are significantly altered in both gene expression and copy number (Neve et al., 2006). In this study, we adopted the MCF10 breast cancer progression model series of cells which consist of the normal-like MCF10A, premalignant MCF10AT1, and metastatic MCF10CA1a (Dawson et al., 1996; Pauley et al., 1993; Santner et al., 2001). The strength of this series lies in the fact that all three cell lines are derived from the same origin and have similar near-diploid karyotypes. Previous studies have identified altered gene expression resulting in an epithelial to mesenchymal transition across the MCF10 series (Hong et al., 2017) and miRNA (Bhardwaj et al., 2017) involved in the progression of this series. Despite this progress, no systematic studies have been done to identify all the pathways which are putatively altered across this series. We used a comprehensive and unbiased approach to identify altered gene expression across these stages of progression. Interestingly, we identified key upstream regulators which play a major role in this progression including the oncogenes EGFR, MYC, and RAS and the tumor suppressors P53 and PTEN, as well as other factors including P300 and the AP-1 transcription factor complex proteins JUN and FOSL. Importantly, this unbiased approach identified signatures indicative of increased proliferation, migration, invasion and tumorigenesis across the MCF10 series reminiscent of their progressive tumorigenic potential.
There is a direct connection between alterations that occur in gene expression across breast cancer and the physical location of genes within the genome. For example, certain regions of the genome are more frequently amplified or deleted in breast cancer (Ciriello et al., 2015) suggesting specific positional clusters of genes which may be relevant to breast cancer progression. We investigated whether there are clusters of genes which are altered in expression, but not in their copy number. We used a positional gene enrichment tool to identify altered gene expression across MCF10. We found that the major histone gene cluster on chr6p22 is one of the most significant highly-upregulated clusters which is not altered in its copy number across MCF10. This upregulation within the major histone cluster is likely due to increased proliferation resulting in an increased demand for histone mRNA expression in breast cancer.
Within the nucleus the major histone gene cluster is contained within a sub-nuclear domain termed the major histone locus body that contains the factors necessary for histone mRNA expression such as HINFP, NPAT, and FLASH and histone mRNA 3′ end processing factors such as SLBP and LSM10 (Bongiorno-Borbone et al., 2008; Ghule et al., 2008; Zhao et al., 1998). The precise regulation of these factors is required for physiological control of cell proliferation (Bansal et al., 2013; Bongiorno-Borbone et al., 2010; Dominski and Marzluff, 2007; Ghule et al., 2014; Krishnan et al., 2012; Salzler et al., 2013; Yang et al., 2014; Ye et al., 2003; Zhao et al., 2000). Interestingly, LSM10 and NPAT have been shown to be decoupled in several breast cancer cell lines indicating an altered microenvironment within HLBs (Ghule et al., 2009). This decoupling may suggest dysregulation and spatial separation between histone transcription and processing.
Understanding the higher order chromatin organization of HLB and any alterations that occur within these sub-nuclear bodies across breast cancer progression may provide mechanistic insight into its regulation and dysregulation in disease. The upregulation of the major histone gene cluster across breast cancer progression, together with the finding that there is an altered microenvironment within the sub-nuclear domain formed around this region raises compelling questions with regard to its higher order chromatin organization. The major histone gene cluster on chr6p22 is itself composed of three sub-clusters denoted A, B, C. We determined that the major histone gene cluster is hierarchically organized into domains. Two histone genes upstream of sub-cluster A are excluded from the mega-TAD encompassing the major HLB and are not detectably expressed. Other than these two histone genes, we determined that the histone sub-clusters interact with each other more frequently than non-histone genes located between them. This organization suggests that the major histone gene cluster forms an active chromatin hub excluding the non-histone genes. Interestingly, we determined a fourth region which loops back to interact with the major HLB (Fig 3). This region may contain an unknown regulatory function within the HLB. While the overall structure of the major HLB is maintained, we detected subtle changes in its structure. The largest changes were seen in the local contacts within the histone sub-clusters B or C. Since the histones genes are in an active state, only to be increased in breast cancer progression the overall structure of the major HLB might be important for the function of the histone sub-cluster active chromatin hub.
Given its known role as a chromatin organizer, we determined that CTCF may play an important role in organizing the major HLB within the nucleus. Both CTCF (Dunn et al., 2003) and NPAT (Dobson et al., 2017) have been shown to be associated with the nuclear matrix. We extended these findings and determined that the major HLB is associated with CTCF at the nuclear matrix. Consistent with its insulator role at TAD boundaries (Ali et al., 2016; Phillips-Cremins and Corces, 2013) and its enrichment at the anchor sites of chromatin loops (Rao et al., 2014), we find that this is the case within the major histone gene cluster. Since CTCF binding sites are frequently mutated in cancer (Katainen et al., 2015), we considered whether any changes in the structure of the major HLB are due to differences in CTCF binding. While the major TAD boundaries are unchanged across breast cancer progression, there may be some modest differences in sub-TAD boundaries contained within the largest TAD structure at this region. Since the binding of CTCF is unchanged within this region any changes in sub-TADs or looping are independent of CTCF occupancy. The fact that CTCF binding is maintained suggests that it may be critical for the active chromatin state within histone sub-cluster chromatin hub and may be involved in insulating it from the neighboring TADs.
This work provides insight into coordinate regulation and higher order chromatin organization of the major HLB during breast cancer progression. Despite these important observations, further work will be required to determine the mechanisms that drive the assembly and structure-function relationships of the major HLB to regulate expression of histone genes.
Materials and methods
Cell culture
MCF10A and MCF10AT1 cells were grown in DMEM: F12 (Hyclone-SH30271), 5% (v/v) horse serum (Gibco #16050) + 10ug/ml human insulin (Sigma I-1882)+ 20ng/ml recombinant hEGF (Peprotech AF-100-15) + 100ng/ml Cholera toxin (Sigma C-8052) + 0.5 ug/ml Hydrocortisone (Sigma H-0888) Pen/Strep (Life Technologies) and Glutamine (Life Technologies). MCF10CA1a cells were grown in DMEM: F12 (Hyclone-SH30271) + 5% (v/v) horse serum (Gibco #16050) +Pen/Strep (Life Technologies) and Glutamine (Life Technologies). Cells were validated by STR and karyotype analysis (Fig S9).
Nuclear matrix extraction and microscopy
ImmunoFISH for NPAT and chr6p22 (RP11-2p4) was performed as previously described (Ghule, 2008 PNAS). For immunofluorescene, cells were grown overnight on 0.5% gelatin coated coverslips. One set of coverslips were fixed in 3.7% formaldeyde, another was CSK (100 mM NaCl, 1 mM EGTA, 10 mM PIPES pH 6.8, 3 mM MgCl2, 300 mM sucrose, 0.5% v/v Triton X-100) treated for 5 minutes on ice, and a third were treated for isolation of the nuclear matrix. The nuclear matrix was isolated using DNaseI in a DNase buffer (50 mM NaCl, 1 mM EGTA, 10 mM PIPES pH 6.8, 3 mM MgCl2, 300 mM sucrose, 0.5% v/v Triton X-100) for 30 minutes at 30°C. The maintenance of the nuclear matrix was confirmed by DIC and presence of the UBF protein which has been shown to be maintained in the nuclear matrix (Fig S10). The images were obtained using Zeiss LSM 510 META confocal microscope using 63× oil immersion objective. Images were pseudo-colored. Image analyses were performed either using ImageJ.
Hi-C
Hi-C was performed as previously described (Belton et al., 2012). Briefly, cells were fixed with 1% methanol free formaldehyde for 10′ at room temperature, treated with glycine on ice for 15 minutes, PBS rinsed, and snap frozen in liquid nitrogen. HINDIII restriction digested products were blunt ligated after incorporation of biotinylated dCTP, The DNA was sonicated and crosslinks were reversed. Beads were used to pull down biotinylated di-tags. Sheered ends were repaired, A-tailed, and barcoded adapters were ligated. High ligation efficiency was determined by nhel restriction digestion (blunt end formation creates a nheI site). The belton et al., 2012 protocol was modified to increase the ligation step to 10 hours with a second spike-in of t4 ligase at hour 4. We found a dramatic increase in ligation efficiency using these conditions.
ChIP-seq
ChIP-seq was performed as previously described (O’Geen et al., 2010). We performed independent replicates for MCF10A, MCF10AT1 and MCF10CA1a using 20ul of antibody against CTCF (Cell Signaling Technologies, 3418S) and 150ug of chromatin for each sample.
Gene expression analysis
Total RNA was isolated from cells using Trizol (Life Technologies) and purified using the Direct-zol RNA kit (Zymo Research, Irvine, CA, USA: R2050). RNA quality and quantity were assessed using the RNA 6000 Nano Kit with the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). RNA quantity was further assessed using a Nanodrop2000 (Thermo Scientific, Lafayette, CO) and Qubit HS RNA assay (Thermo Fisher Scientific). Total RNA was depleted of ribosomal RNA, reverse transcribed using a random hexamer strategy, and strand-specific adapters were added following manufacturer’s protocol (TruSeq Stranded Total RNA Library Prep kit with Ribo-Zero Gold, Illumina, San Diego, CA, USA) with the exception that the final cDNA libraries were amplified using the Real-time Library Amplification Kit (Kapa Biosystems, Wilmington, MA, USA) to reduce over-amplification of libraries. Generated cDNA libraries were assayed for quality using the High Sensitivity DNA Kit on the Agilent 2100 Bioanalyzer (Agilent Technologies) then sequenced as single-end 100 bp reads (IlluminaHiSeq1000, UVM Advanced Genome Technologies Core).
Complementary DNA (cDNA) was synthesized from isolated RNA using the Superscript III First-Strand Synthesis System according to the manufacturer’s instructions (Life Technologies). Quantitative RT-PCR (qPCR) was performed using gene-specific primers and SYBR Green Master Mix (Bio Rad, Hercules, CA) on a Viia 7 system (Applied Biosystems, Foster City, CA, USA). After normalization to the reference genes glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and actin, relative expression levels of each target gene were calculated using the comparative CT (CT) method and normalized relative to expression in MCF10A. Oligonucleotide primer sequences used for qPCR are as follows: hGAPDH forward primer, 5′-ATGTTCGTCATGGGTGTGAA-3′; hGAPDH reverse primer, 5′-TGTGGTCATGAGTCCTTCCA-3′; hACTB forward primer, 5′-AGCACAGAGCCTCGCCTTT-3′; hACTB reverse primer, 5′-CGGCGATATCATCATCCAT-3′; HIST1H3G forward primer, 5′-CAGACTGCACGCAAGTCCA-3′; HIST1H3G reverse primer, 5′-CGGAACTCTGAAAGCGCAGAT-3′; HIST1H2BJ forward primer, 5′-CTGACACCGGCATTTCGTC-3′; HIST1H2BJ reverse primer, 5′-CGGCCTTAGTACCCTCGGA-3′; HIST1H2BO forward primer, 5′-GACCCGGCTAAATCTGCTCC-3′, HIST1H2BO reverse primer- 5′- GGCCTTGGTTACGGCTTTC-3′; CDH1 Forward: GGAAGTCAGTTCAGAGCATC, CDH1 Reverse: AGGCCTTTTGACTGTAATCACACC; CDH2 Forward: TGTTTGACTATGAAGGCAGTGG, CDH2 Reverse: TCAGTCATCACCTCCACCAT; Vimentin Forward: AGGAAATGGCTCGTCACCTTCGTGAATA, Vimentin Reverse: GGAGTGTCGGTTGTTAAGAACTAGAGCT; GAPDH Forward: TGTGGTCATGAGTCCTTCCA, GAPDH Reverse: ATGTTCGTCATGGGTGTGAA; Zeb2 Forward AAGCCAGGGACAGATCAGC; Zeb2 Reverse: CCACACTCTGTGCATTTGAACT; Twist1 Forward: TGAGCAAGATTCAGACCCTCA; Twist 1 Reverse: ATCCTCCAGACCGAGAAGG; Snail1 Forward: CCAATCGGAAGCCTAACTACAG; Snail 1 Reverse: GACAGAGTCCCAGATGAGCATT; Zeb1 Forward: GGGAGGAGCAGTGAAAG; Zeb1 Reverse: TTTCTTGCCCTTCCTTTCT.
Bioinformatics Analyses
Sequencing base calls were generated on the HiSeq 1500 instrument in the UVM Advanced Genome Technologies Core Massively Parallel Sequencing Facility. Fastq conversion and demultiplexing were done using bcl2fastq (Illumina, v1.8.4) and evaluated using Fastqc. MCF10A HiC data was downloaded from GEO series GSE66733.
CTCF ChIP-seq
Adapters were cut (cutadapt v1.11) and low quality reads trimmed (Galaxy FASTQ Quality Trimmer 1.0.0; window 10, step 1, minimum quality 20). Reads were mapped to the human genome (hg38 canonical) using STAR version 2.4 (Dobin et al., 2013) with splicing disabled (–alignIntronMax 1) (Dobin et al., 2013). Enriched regions (narrowPeak calls) for each replicate were generated using MACS2 (Feng et al., 2012) and replicates were then evaluated using deepTools (Ramírez et al., 2016) to correlate alignments and IDR (Li et al., 2011) to evaluate peak call reproducibility (Table S5). After pooling replicates, MACS2 (Zhang et al., 2008) was used on H3K4me3 to call narrowPeak at high stringency (P-value <10e-5), these peaks were further filtered according to IDR cutoffs. FE wiggle tracks were generated using MACS2’s bdgcmp and UCSC’s bedGraphToBigwig utility. CTCF ChIP-seq datasets have been deposited in the Gene Expressions Omnibus (GEO) under accession code GSE98551.
HiC data processing and analysis
Reads were aligned to canonical hg38 chromosomes using HiC-Pro 2.8.1 (Servant et al., 2015) calling bowtie2 2.1.0 (Langmead and Salzberg, 2012) with HiC-Pro’s default options. The full HiC-Pro pipeline was run for biological replicates and pooled samples. The merged valid pair files from HiC-Pro were used to create JuiceBox (Durand et al.) .hic files using the hicpro2juicebox.sh script (slightly modified to include the 40kb resolution for convenience with HiC-Pro output) distributed with HiC-Pro and juicer_tools 0.7.5. Observed vs. Expected matrices were exported from JuiceBox for plotting using custom R scripts (Sushi v1.12.0 package for rotated interaction matrix and looping line plots). Observed vs Expected heatmaps had values capped below the .999 quantile to control outliers; the Observed heatmap was similarly capped at 0.975. Looping line plots were limited to lines representing the 100 strongest bin interaction that were between bins more than 4 bins apart. Hi-C datasets have been deposited in the Gene Expressions Omnibus (GEO) under accession code GSE98552. Quality control metrics can be found in supplementary material (Table S6).
TAD Calling
TADtool 0.61 (Kruse et al., 2016) was used to determine appropriate TAD calling parameters and for final TAD calls. The region of chr6 from 23 Mb to 31 Mb was selected for TAD calling. ICE matrices of 40 kb bins from HiC-Pro were used as input after masking bins from 26,680,001–26,800,000 due to an apparent mappability issue that caused artificial TAD boundaries to be called. The directionality algorithm was applied at windows of 0.8 Mb, 2Mb, and 3 Mb with varying cutoffs as appropriate for each cell line.
RNA-seq Data Processing and DE analysis
Sequence files (fastq) were mapped to the most recent assemblies of the human genome (hg38) using TopHat2 (Kim et al., 2013). Expression counts were determined by HTSeq (Anders et al., 2015) with recent gene annotations (Gencode v23/v24) (Harrow et al., 2012). Differential expression was analyzed by DESeq2 (Love et al., 2014). Correlation between replicates and differential gene expression between samples was assessed by principal component analysis (PCA). RNA-Seq datasets have been deposited in the Gene Expressions Omnibus (GEO) under accession code GSE98393. DE call on protein coding genes only, minimally 2 fold-change and p-adjusted value of 0.05 (Tracy et al, 2017 et al, in press). K-means clustering was performed on the log2 FC vs MCF10A of the average normalized counts from DESEQ2 for DE genes only. 14 genes (8 HIST1), in the region analyzed did not have enough uniquely aligned reads (>= 5) to be considered (Table S7). TCGA data presented is level3 microarray data (downloaded March 23, 2016) log2 lowess normalized (cy5/cy3) collapsed by gene symbol for all 593 samples this data is available for. Data for 3 genes (1 HIST1) was not present in this dataset (Table S8).
Supplementary Material
Figure S1. QPCR validation of RNA-seq. We validated the expression of eight mRNA that are critical to breast cancer progression. qPCR and RNAseq demonstrate similar expression patterns across the MCF10 progression series.
Figure S10. Maintenance of the nuclear matrix and colocalization of NPAT and CTCF in breast cancer. (A) The pattern of UBF and overall nuclear morphology in DIC images were used as positive controls for proper nuclear matrix extraction. (B) CTCF and NPAT colocalize and are associated with the nuclear matrix across breast cancer progression. Similar results to those found in figure 4B are shown.
Figure S2. The major histone cluster is not amplified in MCF10. NPAT foci, which label the HLB, are not increased in number across the MCF10 breast cancer progression series. Background signal and/or inability to visualize the minor HLB are likely causes of 3 NPAT foci and greater than 4 foci.
Figure S3. Subdividing the major histone gene cluster and non-histone genes to determine intra and inter sub-cluster interactions. The matrix resulting from expected versus observed interactions was subdivided into the regions containing the sub-cluster and non-histone regions located between sub-clusters.
Figure S4. The histone genes within the major histone gene cluster are upregulated in breast tumor specimens. Clustering of the histone gene expression revealed that normal adjacent tissue is lower in expression for the histone genes. This clustering also reveals which histone genes are likely cross-reacting with each other in the microarray.
Figure S5. Survivorship curves for individual histone genes within the major histone gene cluster region. TCGA data was used to determine survivorship curves for individual histone genes along the major histone gene cluster.
Figure S6. Survivorship curves for individual non-histone genes within the major histone gene cluster region. TCGA data was used to determine survivorship curves for individual non-histone genes along the major histone gene cluster.
Figure S7. Survivorship curves for the histone genes taken as a whole within the major histone gene cluster region. TCGA data was used to determine survivorship curves for the overall expression of the histone genes within the major histone gene cluster.
Figure S8. Survivorship curves for the non-histone genes taken as a whole within the major histone gene cluster region. TCGA data was used to determine survivorship curves for the overall expression of the non-histone genes within the major histone gene cluster.
Figure S9. Maintenance of the nuclear matrix and colocalization of NPAT and CTCF in breast cancer. (A) STR analysis of the MCF10 breast cancer progression series validated that the cell lines are properly identified. (B) Karyotype analysis showed that the karyotype is similar and near-diploid across the MCF10 series.
Table S1. Gene subsets in MCF10 breast cancer progression. Genes that fall within each subcluster of expression patterns are reported. Each tab within the file represents a subset of genes that correspond to figure 1B.
Table S2. Pathway analysis (IPA) in MCF10 breast cancer progression. IPA was used to identify pathways that are present within the total differential expression or that are represented within each subset of genes found in table S1 and figure 1B.
Table S3. Upstream regulators in MCF10 breast cancer progression. IPA was used to identify upstream regulators that are present within the total differential expression or that are represented within each subset of genes found in table S1 and figure 1B.
Table S4. Diseases and functions altered across MCF10 breast cancer progression. IPA was used to identify diseases and functions that are represented within the total differential expression or that are represented within each subset of genes found in table S1 and figure 1B.
Table S5. CTCF ChIP-seq quality control metrics. Quality control metrics were analyzed on the CTCF ChIP-seq and are presented.
Table S6. Hi-C Quality control metrics. Quality control metrics were analyzed for the Hi-C and are presented.
Table S7. Histone genes below the RNA-seq detection threshold. We applied a threshold of 5 normalized counts to determine expression of genes within the major histone gene cluster.
Table S8. Genes not in the TCGA dataset. Genes in the RNAseq were not present in the TCGA dataset are displayed.
Acknowledgments
We would like to acknowledge the Microscopy Imaging Core, Advanced Genomics Technology Core, and Cytogenetics Facilities at UVM Larner College of Medicine. We would also like to acknowledge Dr Jeffrey A. Nickerson for providing the cells used in this study.
References
- Ali T, Renkawitz R, Bartkuhn M. Insulators and domains of gene expression. Curr Opin Genet Dev. 2016;37:17–26. doi: 10.1016/j.gde.2015.11.009. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal N, Zhang M, Bhaskar A, Itotia P, Lee E, Shlyakhtenko LS, Lam TT, Fritz A, Berezney R, Lyubchenko YL, Stafford WF, Thapar R. Assembly of the SLIP1-SLBP complex on histone mRNA requires heterodimerization and sequential binding of SLBP followed by SLIP1. Biochemistry. 2013;52(3):520–536. doi: 10.1021/bi301074r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barutcu AR, Fritz AJ, Zaidi SK, van Wijnen AJ, Lian JB, Stein JL, Nickerson JA, Imbalzano AN, Stein GS. C-ing the Genome: A Compendium of Chromosome Conformation Capture Methods to Study Higher-Order Chromatin Organization. J Cell Physiol. 2016a;231(1):31–35. doi: 10.1002/jcp.25062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barutcu AR, Hong D, Lajoie BR, McCord RP, van Wijnen AJ, Lian JB, Stein JL, Dekker J, Imbalzano AN, Stein GS. RUNX1 contributes to higher-order chromatin organization and gene regulation in breast cancer cells. Biochim Biophys Acta. 2016b;1859(11):1389–1397. doi: 10.1016/j.bbagrm.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barutcu AR, Lajoie BR, Fritz AJ, McCord RP, Nickerson JA, van Wijnen AJ, Lian JB, Stein JL, Dekker J, Stein GS, Imbalzano AN. SMARCA4 regulates gene expression and higher-order chromatin structure in proliferating mammary epithelial cells. Genome Res. 2016c;26(9):1188–1201. doi: 10.1101/gr.201624.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barutcu AR, Lajoie BR, McCord RP, Tye CE, Hong D, Messier TL, Browne G, van Wijnen AJ, Lian JB, Stein JL, Dekker J, Imbalzano AN, Stein GS. Chromatin interaction analysis reveals changes in small chromosome and telomere clustering between epithelial and breast cancer cells. Genome Biol. 2015;16:214. doi: 10.1186/s13059-015-0768-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barutcu AR, Lian JB, Stein JL, Stein GS, Imbalzano AN. The connection between BRG1, CTCF and topoisomerases at TAD boundaries. Nucleus. 2017;8(2):150–155. doi: 10.1080/19491034.2016.1276145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belton JM, McCord RP, Gibcus JH, Naumova N, Zhan Y, Dekker J. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods. 2012;58(3):268–276. doi: 10.1016/j.ymeth.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj A, Singh H, Rajapakshe K, Tachibana K, Ganesan N, Pan Y, Gunaratne PH, Coarfa C, Bedrosian I. Regulation of miRNA-29c and its downstream pathways in preneoplastic progression of triple-negative breast cancer. Oncotarget. 2017;8(12):19645–19660. doi: 10.18632/oncotarget.14902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bongiorno-Borbone L, De Cola A, Barcaroli D, Knight RA, Di Ilio C, Melino G, De Laurenzi V. FLASH degradation in response to UV-C results in histone locus bodies disruption and cell-cycle arrest. Oncogene. 2010;29(6):802–810. doi: 10.1038/onc.2009.388. [DOI] [PubMed] [Google Scholar]
- Bongiorno-Borbone L, De Cola A, Vernole P, Finos L, Barcaroli D, Knight RA, Melino G, De Laurenzi V. FLASH and NPAT positive but not Coilin positive Cajal Bodies correlate with cell ploidy. Cell Cycle. 2008;7(15):2357–2367. doi: 10.4161/cc.6344. [DOI] [PubMed] [Google Scholar]
- Brown JM, Green J, das Neves RP, Wallace HA, Smith AJ, Hughes J, Gray N, Taylor S, Wood WG, Higgs DR, Iborra FJ, Buckle VJ. Association between active genes occurs at nuclear speckles and is modulated by chromatin environment. The Journal of cell biology. 2008;182(6):1083–1097. doi: 10.1083/jcb.200803174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Yu H, Wang J, Zhang Z, Gao Z, Chen Z, Lu Y, Liu W, Jiang D, Zheng SL, Wei GH, Issacs WB, Feng J, Xu J. Systematic enrichment analysis of potentially functional regions for 103 prostate cancer risk-associated loci. Prostate. 2015;75(12):1264–1276. doi: 10.1002/pros.23008. [DOI] [PubMed] [Google Scholar]
- Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, Zhang H, McLellan M, Yau C, Kandoth C, Bowlby R, Shen H, Hayat S, Fieldhouse R, Lester SC, Tse GM, Factor RE, Collins LC, Allison KH, Chen YY, Jensen K, Johnson NB, Oesterreich S, Mills GB, Cherniack AD, Robertson G, Benz C, Sander C, Laird PW, Hoadley KA, King TA, Network TR. Perou CM. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell. 2015;163(2):506–519. doi: 10.1016/j.cell.2015.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clowney EJ, LeGros MA, Mosley CP, Clowney FG, Markenskoff-Papadimitriou EC, Myllys M, Barnea G, Larabell CA, Lomvardas S. Nuclear aggregation of olfactory receptor genes governs their monogenic expression. Cell. 2012;151(4):724–737. doi: 10.1016/j.cell.2012.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson PJ, Wolman SR, Tait L, Heppner GH, Miller FR. MCF10AT: a model for the evolution of cancer from proliferative breast disease. Am J Pathol. 1996;148(1):313–319. [PMC free article] [PubMed] [Google Scholar]
- De Preter K, Barriot R, Speleman F, Vandesompele J, Moreau Y. Positional gene enrichment analysis of gene sets for high-resolution identification of overrepresented chromosomal regions. Nucleic Acids Res. 2008;36(7):e43. doi: 10.1093/nar/gkn114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J, Mirny L. The 3D Genome as Moderator of Chromosomal Communication. Cell. 2016;164(6):1110–1121. doi: 10.1016/j.cell.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Rupon JW, Krivega I, Breda L, Motta I, Jahn KS, Reik A, Gregory PD, Rivella S, Dean A, Blobel GA. Reactivation of developmentally silenced globin genes by forced chromatin looping. Cell. 2014;158(4):849–860. doi: 10.1016/j.cell.2014.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey P. Cancer nucleus: morphology and beyond. Diagn Cytopathol. 2010;38(5):382–390. doi: 10.1002/dc.21234. [DOI] [PubMed] [Google Scholar]
- Dixon JR, Gorkin DU, Ren B. Chromatin Domains: The Unit of Chromosome Organization. Mol Cell. 2016;62(5):668–680. doi: 10.1016/j.molcel.2016.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485(7398):376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson JR, Hong D, Barutcu AR, Wu H, Imbalzano AN, Lian JB, Stein JL, van Wijnen AJ, Nickerson JA, Stein GS. Identifying Nuclear Matrix-Attached DNA Across the Genome. J Cell Physiol. 2017;232(6):1295–1305. doi: 10.1002/jcp.25596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominski Z, Marzluff WF. Formation of the 3′ end of histone mRNA: getting closer to the end. Gene. 2007;396(2):373–390. doi: 10.1016/j.gene.2007.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn KL, Zhao H, Davie JR. The insulator binding protein CTCF associates with the nuclear matrix. Exp Cell Res. 2003;288(1):218–223. doi: 10.1016/s0014-4827(03)00185-x. [DOI] [PubMed] [Google Scholar]
- Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, Aiden EL. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Systems. 3(1):99–101. doi: 10.1016/j.cels.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz AJ, Barutcu AR, Martin-Buley L, van Wijnen AJ, Zaidi SK, Imbalzano AN, Lian JB, Stein JL, Stein GS. Chromosomes at Work: Organization of Chromosome Territories in the Interphase Nucleus. J Cell Biochem. 2016;117(1):9–19. doi: 10.1002/jcb.25280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz AJ, Stojkovic B, Ding H, Xu J, Bhattacharya S, Berezney R. Cell type specific alterations in interchromosomal networks across the cell cycle. PLoS Comput Biol. 2014a;10(10):e1003857. doi: 10.1371/journal.pcbi.1003857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz AJ, Stojkovic B, Ding H, Xu J, Bhattacharya S, Gaile D, Berezney R. Wide-scale alterations in interchromosomal organization in breast cancer cells: defining a network of interacting chromosomes. Hum Mol Genet. 2014b;23(19):5133–5146. doi: 10.1093/hmg/ddu237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geeven G, Zhu Y, Kim BJ, Bartholdy BA, Yang SM, Macfarlan TS, Gifford WD, Pfaff SL, Verstegen MJ, Pinto H, Vermunt MW, Creyghton MP, Wijchers PJ, Stamatoyannopoulos JA, Skoultchi AI, de Laat W. Local compartment changes and regulatory landscape alterations in histone H1-depleted cells. Genome Biol. 2015;16:289. doi: 10.1186/s13059-015-0857-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghule PN, Dominski Z, Lian JB, Stein JL, van Wijnen AJ, Stein GS. The subnuclear organization of histone gene regulatory proteins and 3′ end processing factors of normal somatic and embryonic stem cells is compromised in selected human cancer cell types. J Cell Physiol. 2009;220(1):129–135. doi: 10.1002/jcp.21740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghule PN, Dominski Z, Yang XC, Marzluff WF, Becker KA, Harper JW, Lian JB, Stein JL, van Wijnen AJ, Stein GS. Staged assembly of histone gene expression machinery at subnuclear foci in the abbreviated cell cycle of human embryonic stem cells. Proc Natl Acad Sci U S A. 2008;105(44):16964–16969. doi: 10.1073/pnas.0809273105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghule PN, Xie RL, Medina R, Colby JL, Jones SN, Lian JB, Stein JL, van Wijnen AJ, Stein GS. Fidelity of histone gene regulation is obligatory for genome replication and stability. Mol Cell Biol. 2014;34(14):2650–2659. doi: 10.1128/MCB.01567-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodspeed A, Heiser LM, Gray JW, Costello JC. Tumor-Derived Cell Lines as Molecular Models of Cancer Pharmacogenomics. Mol Cancer Res. 2016;14(1):3–13. doi: 10.1158/1541-7786.MCR-15-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, van Steensel B. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453(7197):948–951. doi: 10.1038/nature06947. [DOI] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, Barnes I, Bignell A, Boychenko V, Hunt T, Kay M, Mukherjee G, Rajan J, Despacio-Reyes G, Saunders G, Steward C, Harte R, Lin M, Howald C, Tanzer A, Derrien T, Chrast J, Walters N, Balasubramanian S, Pei B, Tress M, Rodriguez JM, Ezkurdia I, van Baren J, Brent M, Haussler D, Kellis M, Valencia A, Reymond A, Gerstein M, Guigo R, Hubbard TJ. GENCODE: the reference human genome annotation for The ENCODE Project. Genome research. 2012;22(9):1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong D, Messier TL, Tye CE, Dobson JR, Fritz AJ, Sikora KR, Browne G, Stein JL, Lian JB, Stein GS. Runx1 stabilizes the mammary epithelial cell phenotype and prevents epithelial to mesenchymal transition. Oncotarget. 2017;8(11):17610–17627. doi: 10.18632/oncotarget.15381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadauke S, Blobel GA. Chromatin loops in gene regulation. Biochim Biophys Acta. 2009;1789(1):17–25. doi: 10.1016/j.bbagrm.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MD, Miller CA, Welch JS, Walter MJ, Wendl MC, Ley TJ, Wilson RK, Raphael BJ, Ding L. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katainen R, Dave K, Pitkanen E, Palin K, Kivioja T, Valimaki N, Gylfe AE, Ristolainen H, Hanninen UA, Cajuso T, Kondelin J, Tanskanen T, Mecklin JP, Jarvinen H, Renkonen-Sinisalo L, Lepisto A, Kaasinen E, Kilpivaara O, Tuupanen S, Enge M, Taipale J, Aaltonen LA. CTCF/cohesin-binding sites are frequently mutated in cancer. Nat Genet. 2015;47(7):818–821. doi: 10.1038/ng.3335. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome biology. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan N, Lam TT, Fritz A, Rempinski D, O’Loughlin K, Minderman H, Berezney R, Marzluff WF, Thapar R. The prolyl isomerase Pin1 targets stem-loop binding protein (SLBP) to dissociate the SLBP-histone mRNA complex linking histone mRNA decay with SLBP ubiquitination. Mol Cell Biol. 2012;32(21):4306–4322. doi: 10.1128/MCB.00382-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivega I, Dean A. Enhancer and promoter interactions-long distance calls. Curr Opin Genet Dev. 2012;22(2):79–85. doi: 10.1016/j.gde.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse K, Hug CB, Hernández-Rodríguez B, Vaquerizas JM. TADtool: visual parameter identification for TAD-calling algorithms. Bioinformatics. 2016;32(20):3190–3192. doi: 10.1093/bioinformatics/btw368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Meth. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Brown JB, Huang H, Bickel PJ. Measuring reproducibility of high-throughput experiments. 2011:1752–1779. [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326(5950):289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupianez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, Santos-Simarro F, Gilbert-Dussardier B, Wittler L, Borschiwer M, Haas SA, Osterwalder M, Franke M, Timmermann B, Hecht J, Spielmann M, Visel A, Mundlos S. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015;161(5):1012–1025. doi: 10.1016/j.cell.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupianez DG, Spielmann M, Mundlos S. Breaking TADs: How Alterations of Chromatin Domains Result in Disease. Trends Genet. 2016;32(4):225–237. doi: 10.1016/j.tig.2016.01.003. [DOI] [PubMed] [Google Scholar]
- Mayer R, Brero A, von Hase J, Schroeder T, Cremer T, Dietzel S. Common themes and cell type specific variations of higher order chromatin arrangements in the mouse. BMC Cell Biol. 2005;6:44. doi: 10.1186/1471-2121-6-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCord RP, Nazario-Toole A, Zhang H, Chines PS, Zhan Y, Erdos MR, Collins FS, Dekker J, Cao K. Correlated alterations in genome organization, histone methylation, and DNA-lamin A/C interactions in Hutchinson-Gilford progeria syndrome. Genome Res. 2013;23(2):260–269. doi: 10.1101/gr.138032.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miele A, Braastad CD, Holmes WF, Mitra P, Medina R, Xie R, Zaidi SK, Ye X, Wei Y, Harper JW, van Wijnen AJ, Stein JL, Stein GS. HiNF-P directly links the cyclin E/CDK2/p220NPAT pathway to histone H4 gene regulation at the G1/S phase cell cycle transition. Mol Cell Biol. 2005;25(14):6140–6153. doi: 10.1128/MCB.25.14.6140-6153.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, Laue ED, Tanay A, Fraser P. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 2013;502(7469):59–64. doi: 10.1038/nature12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra V, Rocha PP, An D, Raviram R, Skok JA, Mazzoni EO, Reinberg D. CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science. 2015;347(6225):1017–1021. doi: 10.1126/science.1262088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, Speed T, Spellman PT, DeVries S, Lapuk A, Wang NJ, Kuo WL, Stilwell JL, Pinkel D, Albertson DG, Waldman FM, McCormick F, Dickson RB, Johnson MD, Lippman M, Ethier S, Gazdar A, Gray JW. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10(6):515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, Gribnau J, Barillot E, Bluthgen N, Dekker J, Heard E. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485(7398):381–385. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Geen H, Frietze S, Farnham PJ. Using ChIP-seq technology to identify targets of zinc finger transcription factors. Methods Mol Biol. 2010;649:437–455. doi: 10.1007/978-1-60761-753-2_27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne CS, Chakalova L, Mitchell JA, Horton A, Wood AL, Bolland DJ, Corcoran AE, Fraser P. Myc dynamically and preferentially relocates to a transcription factory occupied by Igh. PLoS Biol. 2007;5(8):e192. doi: 10.1371/journal.pbio.0050192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parada LA, McQueen PG, Misteli T. Tissue-specific spatial organization of genomes. Genome Biol. 2004;5(7):R44. doi: 10.1186/gb-2004-5-7-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauley RJ, Soule HD, Tait L, Miller FR, Wolman SR, Dawson PJ, Heppner GH. The MCF10 family of spontaneously immortalized human breast epithelial cell lines: models of neoplastic progression. Eur J Cancer Prev. 1993;2(Suppl 3):67–76. [PubMed] [Google Scholar]
- Phillips-Cremins JE, Corces VG. Chromatin insulators: linking genome organization to cellular function. Mol Cell. 2013;50(4):461–474. doi: 10.1016/j.molcel.2013.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Research. 2016;44:W160–W165. doi: 10.1093/nar/gkw257. Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159(7):1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha PP, Micsinai M, Kim JR, Hewitt SL, Souza PP, Trimarchi T, Strino F, Parisi F, Kluger Y, Skok JA. Close proximity to Igh is a contributing factor to AID-mediated translocations. Mol Cell. 2012;47(6):873–885. doi: 10.1016/j.molcel.2012.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roix JJ, McQueen PG, Munson PJ, Parada LA, Misteli T. Spatial proximity of translocation-prone gene loci in human lymphomas. Nat Genet. 2003;34(3):287–291. doi: 10.1038/ng1177. [DOI] [PubMed] [Google Scholar]
- Salzler HR, Tatomer DC, Malek PY, McDaniel SL, Orlando AN, Marzluff WF, Duronio RJ. A sequence in the Drosophila H3-H4 Promoter triggers histone locus body assembly and biosynthesis of replication-coupled histone mRNAs. Dev Cell. 2013;24(6):623–634. doi: 10.1016/j.devcel.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santner SJ, Dawson PJ, Tait L, Soule HD, Eliason J, Mohamed AN, Wolman SR, Heppner GH, Miller FR. Malignant MCF10CA1 cell lines derived from premalignant human breast epithelial MCF10AT cells. Breast Cancer Res Treat. 2001;65(2):101–110. doi: 10.1023/a:1006461422273. [DOI] [PubMed] [Google Scholar]
- Seitan VC, Faure AJ, Zhan Y, McCord RP, Lajoie BR, Ing-Simmons E, Lenhard B, Giorgetti L, Heard E, Fisher AG, Flicek P, Dekker J, Merkenschlager M. Cohesin-based chromatin interactions enable regulated gene expression within preexisting architectural compartments. Genome Res. 2013;23(12):2066–2077. doi: 10.1101/gr.161620.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant N, Varoquaux N, Lajoie BR, Viara E, Chen C-J, Vert J-P, Heard E, Dekker J, Barillot E. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biology. 2015;16(1):259. doi: 10.1186/s13059-015-0831-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, Yates LR, Papaemmanuil E, Beare D, Butler A, Cheverton A, Gamble J, Hinton J, Jia M, Jayakumar A, Jones D, Latimer C, Lau KW, McLaren S, McBride DJ, Menzies A, Mudie L, Raine K, Rad R, Chapman MS, Teague J, Easton D, Langerod A, Oslo Breast Cancer C. Lee MT, Shen CY, Tee BT, Huimin BW, Broeks A, Vargas AC, Turashvili G, Martens J, Fatima A, Miron P, Chin SF, Thomas G, Boyault S, Mariani O, Lakhani SR, van de Vijver M, van’t Veer L, Foekens J, Desmedt C, Sotiriou C, Tutt A, Caldas C, Reis-Filho JS, Aparicio SA, Salomon AV, Borresen-Dale AL, Richardson AL, Campbell PJ, Futreal PA, Stratton MR. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486(7403):400–404. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suva ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. 2013;339(6127):1567–1570. doi: 10.1126/science.1230184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen S, Truty RM, Pollard KS. Enhancer-promoter interactions are encoded by complex genomic signatures on looping chromatin. Nat Genet. 2016;48(5):488–496. doi: 10.1038/ng.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XC, Sabath I, Kunduru L, van Wijnen AJ, Marzluff WF, Dominski Z. A conserved interaction that is essential for the biogenesis of histone locus bodies. J Biol Chem. 2014;289(49):33767–33782. doi: 10.1074/jbc.M114.616466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Franco AA, Santos H, Nelson DM, Kaufman PD, Adams PD. Defective S phase chromatin assembly causes DNA damage, activation of the S phase checkpoint, and S phase arrest. Mol Cell. 2003;11(2):341–351. doi: 10.1016/s1097-2765(03)00037-6. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based Analysis of ChIP-Seq (MACS) Genome Biology. 2008;9(9):R137–R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, McCord RP, Ho YJ, Lajoie BR, Hildebrand DG, Simon AC, Becker MS, Alt FW, Dekker J. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 2012;148(5):908–921. doi: 10.1016/j.cell.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Dynlacht B, Imai T, Hori T, Harlow E. Expression of NPAT, a novel substrate of cyclin E-CDK2, promotes S-phase entry. Genes Dev. 1998;12(4):456–461. doi: 10.1101/gad.12.4.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Kennedy BK, Lawrence BD, Barbie DA, Matera AG, Fletcher JA, Harlow E. NPAT links cyclin E-Cdk2 to the regulation of replication-dependent histone gene transcription. Genes Dev. 2000;14(18):2283–2297. [PMC free article] [PubMed] [Google Scholar]
- Zink D, Fischer AH, Nickerson JA. Nuclear structure in cancer cells. Nat Rev Cancer. 2004;4(9):677–687. doi: 10.1038/nrc1430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. QPCR validation of RNA-seq. We validated the expression of eight mRNA that are critical to breast cancer progression. qPCR and RNAseq demonstrate similar expression patterns across the MCF10 progression series.
Figure S10. Maintenance of the nuclear matrix and colocalization of NPAT and CTCF in breast cancer. (A) The pattern of UBF and overall nuclear morphology in DIC images were used as positive controls for proper nuclear matrix extraction. (B) CTCF and NPAT colocalize and are associated with the nuclear matrix across breast cancer progression. Similar results to those found in figure 4B are shown.
Figure S2. The major histone cluster is not amplified in MCF10. NPAT foci, which label the HLB, are not increased in number across the MCF10 breast cancer progression series. Background signal and/or inability to visualize the minor HLB are likely causes of 3 NPAT foci and greater than 4 foci.
Figure S3. Subdividing the major histone gene cluster and non-histone genes to determine intra and inter sub-cluster interactions. The matrix resulting from expected versus observed interactions was subdivided into the regions containing the sub-cluster and non-histone regions located between sub-clusters.
Figure S4. The histone genes within the major histone gene cluster are upregulated in breast tumor specimens. Clustering of the histone gene expression revealed that normal adjacent tissue is lower in expression for the histone genes. This clustering also reveals which histone genes are likely cross-reacting with each other in the microarray.
Figure S5. Survivorship curves for individual histone genes within the major histone gene cluster region. TCGA data was used to determine survivorship curves for individual histone genes along the major histone gene cluster.
Figure S6. Survivorship curves for individual non-histone genes within the major histone gene cluster region. TCGA data was used to determine survivorship curves for individual non-histone genes along the major histone gene cluster.
Figure S7. Survivorship curves for the histone genes taken as a whole within the major histone gene cluster region. TCGA data was used to determine survivorship curves for the overall expression of the histone genes within the major histone gene cluster.
Figure S8. Survivorship curves for the non-histone genes taken as a whole within the major histone gene cluster region. TCGA data was used to determine survivorship curves for the overall expression of the non-histone genes within the major histone gene cluster.
Figure S9. Maintenance of the nuclear matrix and colocalization of NPAT and CTCF in breast cancer. (A) STR analysis of the MCF10 breast cancer progression series validated that the cell lines are properly identified. (B) Karyotype analysis showed that the karyotype is similar and near-diploid across the MCF10 series.
Table S1. Gene subsets in MCF10 breast cancer progression. Genes that fall within each subcluster of expression patterns are reported. Each tab within the file represents a subset of genes that correspond to figure 1B.
Table S2. Pathway analysis (IPA) in MCF10 breast cancer progression. IPA was used to identify pathways that are present within the total differential expression or that are represented within each subset of genes found in table S1 and figure 1B.
Table S3. Upstream regulators in MCF10 breast cancer progression. IPA was used to identify upstream regulators that are present within the total differential expression or that are represented within each subset of genes found in table S1 and figure 1B.
Table S4. Diseases and functions altered across MCF10 breast cancer progression. IPA was used to identify diseases and functions that are represented within the total differential expression or that are represented within each subset of genes found in table S1 and figure 1B.
Table S5. CTCF ChIP-seq quality control metrics. Quality control metrics were analyzed on the CTCF ChIP-seq and are presented.
Table S6. Hi-C Quality control metrics. Quality control metrics were analyzed for the Hi-C and are presented.
Table S7. Histone genes below the RNA-seq detection threshold. We applied a threshold of 5 normalized counts to determine expression of genes within the major histone gene cluster.
Table S8. Genes not in the TCGA dataset. Genes in the RNAseq were not present in the TCGA dataset are displayed.