

Abstract
Strategies to enhance response to poly(adenosine diphosphate–ribose) polymerase inhibitor (PARPi) in primary and acquired homologous recombination (HR)–proficient tumors would be a major advance in cancer care. We used a drug synergy screen that combined a PARPi, olaparib, with 20 well-characterized epigenetic drugs and identified bromodomain and extraterminal domain inhibitors (BETis; JQ1, I-BET762, and OTX015) as drugs that acted synergistically with olaparib in HR-proficient cancer cells. Functional assays demonstrated that repressed BET activity reduces HR and thus enhances PARPi-induced DNA damage in cancer cells. We also found that inhibition or depletion of BET proteins impairs transcription of BRCA1 and RAD51, two genes essential for HR. Moreover, BETi treatment sensitized tumors to PARP inhibition in preclinical animal models of HR-proficient breast and ovarian cancers. Finally, we showed that the BRD4 gene was focally amplified across 20 types of common cancers. Combination with BETi could greatly expand the utility of PARP inhibition to patients with HR-proficient cancer.
Introduction
Poly[adenosine diphosphate (ADP)–ribose] polymerase inhibitors (PARPis) have been approved by the U.S. Food and Drug Administration (FDA) as monotherapy for women with germline BRCA1/2-mutated advanced ovarian cancer (1–10). They have also shown promising clinical activity in breast (8, 10–12), prostate (8, 10, 13), and other solid cancers (8, 10), especially for tumors with defective DNA repair via homologous recombination (HR) (6, 7). PARP is an enzyme family that posttranslationally modifies its target proteins by conjugating polymeric chains of ADP-ribose [poly(ADP)-ribosylation (PARylation)] during a number of cellular processes including DNA repair, transcription, translation, cell signaling, and cell death (1–5, 14). The molecular basis of PARPi effectiveness in cancer treatment appears to mainly depend on the role of PARP, especially PARP1, in DNA damage repair and transcriptional regulation (1–5). Multiple mechanisms of PARPi have been proposed, including suppression of base excision repair (1–5), trapping of PARP1 on damaged DNA (15), defective DNA repair protein recruitment (16), activation of nonhomologous end joining (NHEJ) (17) and alternative end joining (18, 19), and regulation of transcription (20, 21). HR-deficient cancer cells, such as cells with BRCA mutations (6, 7) or PTEN loss (22), are extremely sensitive to PARPi. Despite promising clinical results (8–11), overcoming de novo and acquired HR proficiency is one of the major problems for PARPi (1–5). A large fraction of tumors are HR-proficient, suggesting that even in high-grade serous ovarian cancer, as many as 50% of patients may not benefit from this drug (1–5). Tumors that are initially HR-deficient commonly acquire HR proficiency after PARPi treatment by multiple mechanisms, including secondary mutations that restore BRCA1/2 functions (23, 24), and loss of expression of PARP1 (25), 53BP1 (26, 27), REV7 (28), or PTIP (29). Thus, the development of strategies to selectively impair HR in cancer cells and subsequently sensitize HR-proficient cancers to PARP inhibition may provide new clinical applications (1–5). In this regard, drug combination approaches have been designed and evaluated in preclinical and early clinical trials (30–39).
Potent and selective small-molecule modulators influencing chromatin-modifying proteins have been developed as first-in-class targeted therapies for patients with cancer (40, 41). For example, bromodomain (BRD) and extraterminal domain inhibitors (BETis) reversibly bind the BRDs of BET proteins and prevent their interaction with acetylated histones and transcription factors (40, 41). Because well-ordered, deep, hydrophobic pockets in BRDs provide a highly favorable locus for small-molecule compounds, a number of BETis have been developed and evaluated in multiple preclinical cancer models (42–54). BETis have advanced into clinical trials for cancer treatment, for example, in nuclear protein of the testis midline carcinoma (NMC) (55), acute leukemia (56), and multiple myeloma (57). BETis appear to preferentially affect the transcription of a relatively small subset of genes with super-enhancers (also called stretch-enhancers), such as MYC and BCL2 (42–49), suggesting that BET inhibition may specifically modulate the expression of a fraction of genes.
Results
Drug combination screen identifies BETi as acting synergistically with PARPi
To explore whether modulation of epigenetic regulators can sensitize cancer cells to PARPi, we performed a drug combination screen in a triple-negative breast cancer cell line, MDA-MB-231, which expresses wild-type BRCA1/2 and modestly responds to PARPi. An FDA-approved PARPi (olaparib/AZD2281) and 20 well-characterized epigenetic drugs targeting seven classes of epigenetic regulators (>2 compounds for each class; table S1 and Fig. 1A) were chosen for the initial screen. The average combination index (CI) values of olaparib and each epigenetic drug are summarized in Fig. 1B. Notably, all three BETis (JQ1, I-BET762, and OTX015) showed strong synergistic effects with olaparib in MDA-MB-231 cells. To confirm our observation, we validated the above results in cell lines originating from three cancer types, namely breast (MDA-MB-231), ovarian (OVCAR10), and prostate (VCaP) cancers, for which PARPi treatment has been proposed in clinical trials. Consistent with our initial screen, cell viability assays showed strong synergistic effects of JQ1 with olaparib in all three cancer cell lines (Fig. 1C, all CI < 0.5). Then, we took a genetic approach by simultaneously targeting BRD2, BRD3, and BRD4 using pooled small interfering RNAs (siRNAs) and found that the pooled siRNA treatment sensitized cells to PARP inhibition compared to the control siRNA (fig. S1). Furthermore, the synergistic effect was also confirmed by independent in vitro assays including anchorage-dependent colony formation assay (Fig. 1D) and anchorage-independent soft agar assay (Fig. 1E). Furthermore, we expanded our drug combination to several well-characterized BETis (JQ1, I-BET762, and OTX015) and PARPis (olaparib and veliparib/ABT888) in MDA-MB-231 cells. A strong synergistic effect was observed in all combinations tested in our study (Fig. 1F). Additionally, a synergistic effect of BETi and PARPi combination was observed in four HR-deficient cancer cell lines: UWB1.289, HCC1937, PEO1, and CAPAN-1 (fig. S2). We also found that JQ1 showed a synergistic effect in combination with the DNA-damaging drug cisplatin, but not with paclitaxel, which acts by stabilizing microtubules in MDA-MB-231 cells (fig. S3). Finally, although BETi represses the cell cycle (40, 41) and reduces the cancer stem cell population (58), the noncycling cancer cells and aldehyde dehydrogenase (ALDH)– positive cancer stem cells appeared insensitive to combination treatment with JQ1 and olaparib (figs. S4 and S5).
Fig. 1. Drug combination screen identifies BETi as acting synergistically with PARPi.
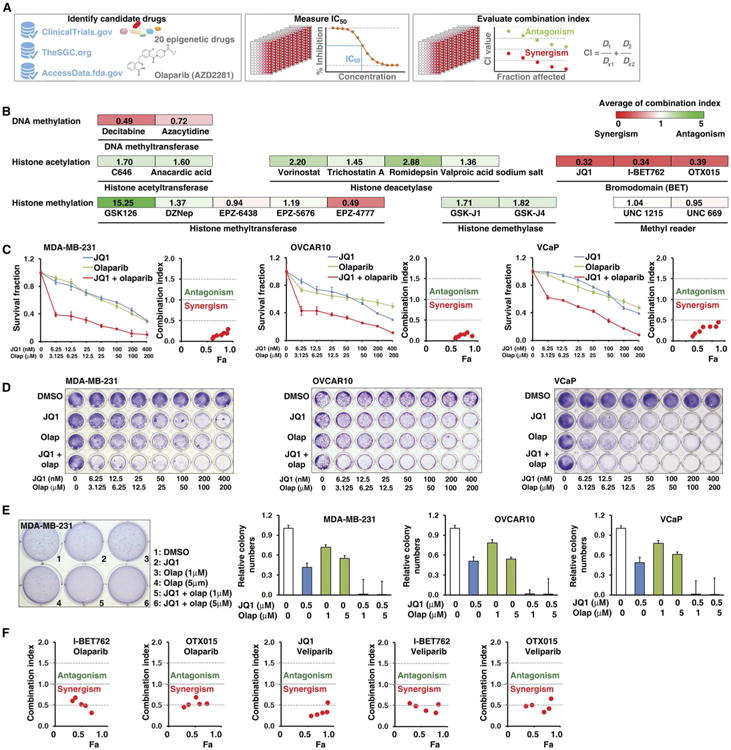
(A) The workflow of drug combination screen. IC50, median inhibitory concentration. (B) Twenty compounds targeting seven classes of epigenetic modulators were examined in a combination screen with the PARPi, olaparib, in MDA-MB-231 cells. The CI quantitatively depicts synergism (CI < 1), additive effect (CI = 1), and antagonism (CI > 1). The color intensity shows the average of CI (red, synergism; green, antagonism). DZNep, 3-deazaneplanocin A. (C) Sensitivity of MDA-MB-231, OVCAR10, and VCaP to olaparib (Olap) alone, JQ1 alone, or olaparib combined with JQ1. Survival fraction (left) and the CI (right) are shown for each of these three cell lines. Fa, fraction affected. Error bars represent means ± SD. (D) Crystal violet staining of anchorage-dependent colony formation assay indicates the sensitivity of cells to dimethyl sulfoxide (DMSO), JQ1, olaparib, or olaparib combined with JQ1. Effect of treatments is shown for MDA-MB-231, OVCAR10, and VCaP cells. (E) Anchorage-independent soft agar assay results for MDA-MB-231, OVCAR10, and VCaP cells treated with DMSO, JQ1, olaparib, or olaparib combined with JQ1. Crystal violet staining of MDA-MB-231 and colony quantification results of all three cell lines are shown. Error bars represent means ± SD. (F) Drug combination results (CI) for two PARPis (olaparib and veliparib) and three BETis (I-BET762, OTX015, and JQ1) examined in MDA-MB-231.
BET inhibition enhances PARPi-induced DNA damage
To test whether JQ1 enhances the DNA damage induced by PARPi treatment, we measured the amount of DNA damage induced by olaparib alone and in combination with JQ1 in HR-proficient cancer cells using a comet assay. DNA damage was increased by olaparib alone in a dose-dependent manner, but the extent of DNA damage was greatly increased when cells were treated with a combination of JQ1 and olaparib (Fig. 2, A and B). Furthermore, we used immuno fluorescence staining to measure the number of phosphorylated his-tone H2AX (γH2AX)–positive foci formed in the cells in response to various treatments. Consistent with previous results, the numbers of γH2AX-positive foci were increased in cells subjected to combination treatment compared to cells treated with olaparib alone (Fig. 2, C and D). Finally, Western blot analysis also showed that the amounts of gH2AX were increased in cells subjected to combination drug treatment compared to cells treated with olaparib alone (Fig. 2E).
Fig. 2. BET inhibition enhances PARPi-induced DNA damage.
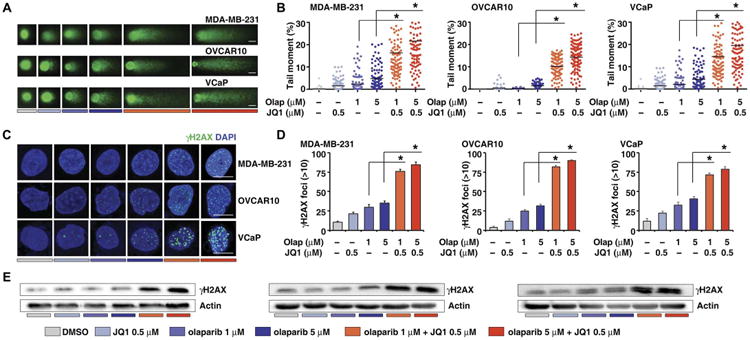
(A) DNA damage in MDA-MB-231, OVCAR10, and VCaP cells treated with DMSO, JQ1, olaparib, or JQ1 combined with olaparib, measured by the comet assay. Scale bars, 10 μm. (B) Extent of DNA damage, quantified by the tail moment in the comet assay. Statistical analysis by Student's t test, *P < 0.05; n = 3. Bars represent mean values of tail moment. (C) Representative images of γH2AX foci in MDA-MB-231, OVCAR10, and VCaP cells treated with DMSO, JQ1, olaparib, or JQ1 combined with olaparib. Scale bars, 10μm. DAPI, 4′,6-diamidino-2-phenylindole. (D) Quantification of the number of γH2AX-positive foci in MDA-MB-231, OVCAR10, and VCaP cells treated with DMSO, JQ1, olaparib, or JQ1 combined with olaparib. Statistical analysisbyStudent's t test, *P < 0.05;n = 3. Error bars represent means ± SD. (E) Western blot analysis of gH2AX in MDA-MB-231, OVCAR10, and VCaP cells treated with DMSO, JQ1, olaparib, or JQ1 combined with olaparib.
BET inhibition reduces HR
To test whether BET inhibition reduces HR and subsequently enhances PARPi response, we used an HR reporter assay (59) to monitor HR activity in response to ionizing radiation (IR)–induced DNA damage (Fig. 3A) and found that JQ1 decreased HR in cancer cells (Fig. 3B). This result was further confirmed by the pooled siRNAs targeting BRD2, BRD3, and BRD4 (fig. S6). Then, we used a single-stranded DNA (ssDNA) staining assay (Fig. 3C) to detect the formation of IR-induced ssDNA, which is promoted by BRCA1 and acts as a substrate for HR repair (60). Consistently, ssDNA staining assays indicated that JQ1 treatment reduces ssDNA formation in response to IR (Fig. 3D). Furthermore, immunofluorescence analyses indicated that JQ1 impaired HR by decreasing the foci formation of BRCA1 and RAD51 during IR-induced DNA damage (Fig. 3, E and F). Furthermore, we observed that BET inhibition did not affect PARP1 trapping (fig. S7) and cellular PARylation (fig. S8). We also found that treatment with JQ1 increased NHEJ repair efficiency (fig. S9), suggesting that JQ1 treatment may also increase the toxicity of PARPi by promoting error-prone NHEJ.
Fig. 3. BET inhibition reduces homologous recombination.
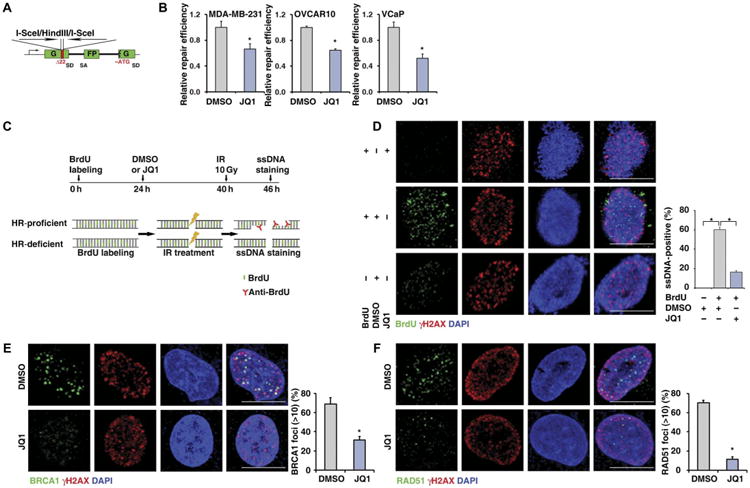
(A) Schematic illustration of HR reporter assay. SD, splice donor; SA, splice acceptor; G, green; FP, fluorescent protein; ATG, the triplet code for the amino acid methionine. (B) HR-mediated DNA repair activity, measured by HR reporter assay, in MDA-MB-231 (left), OVCAR10 (middle), and VCaP (right). Statistical analysis by Student's t test, *P < 0.05; n = 3. Error bars represent means ± SD. (C) Schematic illustration of ssDNA staining assay. BrdU, 5-bromo-2′-deoxyuridine. (D) Representative images (left) and quantitative results (right) of IR-induced ssDNA foci formation in DMSO- or JQ1-treated MDA-MB-231 cells. Cells without BrdU incorporation were used as negative control. Scale bars, 10 μm. (E and F) Representative images (left) and quantitative results (right) of IR-induced BRCA1 (E) and RAD51 (F) foci formation in DMSO- or JQ1-treated MDA-MB-231 cells. Scale bars, 10 μm. Ten gray (Gy) of IR was used for all three cell lines. Statistical analysis by Student's t test, *P < 0.05; n = 3 (D to F). Error bars represent means ± SD.
BET inhibition sensitizes HR-proficient tumors to PARPi treatment in vivo
Next, we tested the effect of combined BETi and PARPi treatment in xenograft tumors. In an orthotopic MDA-MB-231 breast cancer model, mice were randomly assigned to one of four groups to receive vehicle, JQ1, olaparib, or combinationof olaparib and JQ1 (Fig. 4A). The combination of JQ1 and olaparib resulted in a significant inhibition of tumor growth (P < 0.0001; Fig. 4, B to D). Next, in an OVCAR10 ovarian cancer orthotopic model (Fig. 4E), as compared with the vehicle group, all treatment groups displayed slower growth of peritoneal tumors and relief from the accumulation of ascites. Compared with single-agent treatment, combination of JQ1 and olaparib showed significantly decreased peritoneal ascites and tumor formation (P < 0.01; Fig. 4, F to H). All treatment protocols were well tolerated, and mean body weights were not significantly different among the four groups (fig. S10).
Fig. 4. BET inhibition sensitizes HR-proficient tumors to PARPi treatment in vivo.
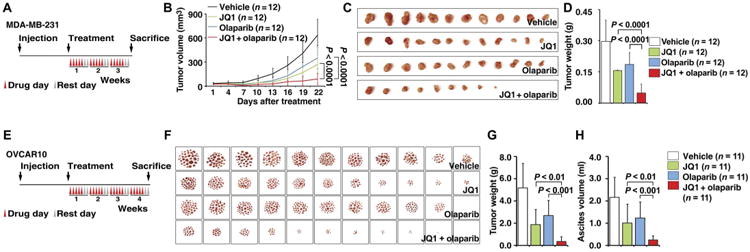
(A) Schematic illustrating the MDA-MB-231 mouse xenograft experimental design. MDA-MB-231 cells were implanted in the mammary fat pad and grown until tumors reached a size of about 30 mm3. Xenografted mice were randomized and then received vehicle, JQ1 (50 mg/kg), olaparib (50 mg/kg), or the combination of both agents as indicated (5 days a week for 3 weeks). Caliper measurements were taken every 3 days after the initiation of drug treatment. (B) Mean tumor volume is shown. Statistical analysis by Student's t test. Error bars represent means ± SD. (C) Images of tumors collected from animals receiving vehicle, JQ1, olaparib, or the combination of both agents. (D) Individual tumor weights from different treatment groups are shown. Statistical analysis by Student's t test. Error bars represent means ± SD. (E) Schematic illustrating the OVCAR10 mouse xenograft experimental design. OVCAR10 cells were implanted intraperitoneally in mice and grown for 2 weeks. Xenografted mice were randomized and then received vehicle, JQ1 (50 mg/kg), olaparib (50 mg/kg), or the combination of both agents as indicated (5 days a week for 4 weeks). (F) Images of the tumor nodes (diameter, >0.5 mm) collected from mice receiving different treatments. (G and H) Quantification of the weight of tumor nodules (G) and the volume of ascites (H) in mice receiving different treatments. Statistical analysis by Student's t test. Error bars represent means ± SD.
BET inhibition and depletion repress the expression of BRCA1 and RAD51
To investigate the molecular mechanism underlying the synergistic effect between BETi and PARPi, we used microarray profiles generated by Asangani et al. (50) to analyze transcriptome changes induced by JQ1 treatment of VCaP cells. Gene ontology analysis indicated that these JQ1 response genes were significantly enriched in cancer-associated pathways such as the DNA damage response (P = 0.00009; Fig. 5A). Notably, the expression of BRCA1 and RAD51, genes involved in the DNA damage repair via HR, was reduced about as much as that of MYC, a well-demonstrated JQ1 target gene (Fig. 5B). We further validated the above microarray results in two independent cell lines: MDA-MB-231 and OVCAR10. Real-time reverse transcription polymerase chain reaction (RT-PCR) analysis showed that JQ1 treatment reduced the mRNA expression of BRCA1 and RAD51 in a time- and dose-dependent manner (Fig. 5C). This result was confirmed at the protein level in MDA-MB-231 cells by Western blot (Fig. 5D). The tumor samples collected from the in vivo treatment experiment showed similar results (fig. S11). To test whether the repression of BRCA1 and RAD51 by JQ1 was mediated by BET proteins, we took a genetic approach by targeting each BET family member with two independent siRNAs. Knockdown specificity was confirmed by individual siRNA transfection (Fig. 5E). A reduction in both BRCA1 and RAD51 expression was observed when siRNAs targeting the three BETs were pooled together (Fig. 5E). This result was further confirmed at the protein level in MDA-MB-231, OVCAR10, and VCaP cells by Western blot (Fig. 5F). Notably, JQ1 treatment did not affect the expression of other core proteins in the DNA damage repair pathways (fig. S12).
Fig. 5. BET inhibition and depletion repress the expression of BRCA1 and RAD51.
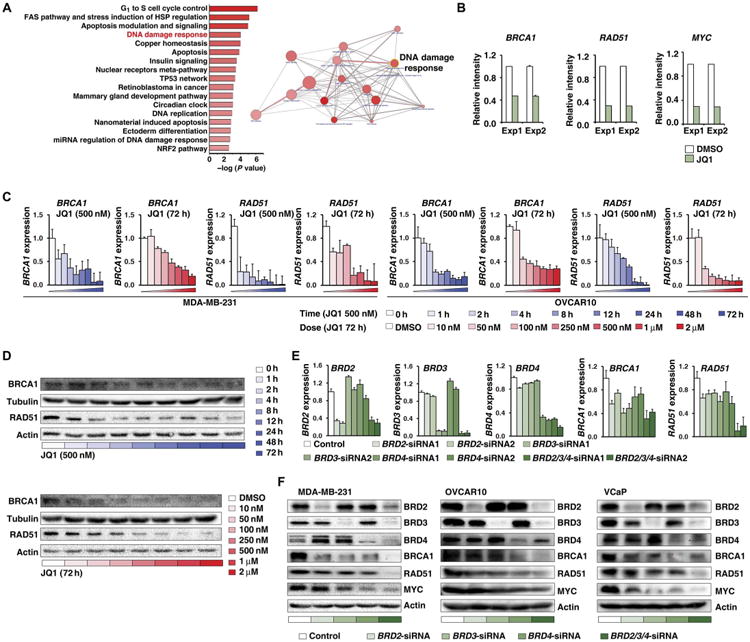
(A) Pathways overrepresented by JQ1 response genes (fold change, >2) in VCaP cells according to ConsensusPathDB analysis on the basis of gene ontology terms (left). Diagram depicts pathway interactions, grouped by pathways (right). Line thickness corresponds to the number of shared proteins between two pathways. Circle size corresponds to the number of JQ1 response genes in each pathway. The color intensity of circle indicates the P value for each pathway. miRNA, microRNA. (B) RNA expression of BRCA1, RAD51, and MYC in two independent microarray experiments in VCaP cells treated with DMSO or JQ1 (500 nM for 24 hours). (C) BRCA1 and RAD51 mRNA expression measured by real-time RT-PCR in MDA-MB-231 cells (left) and OVCAR10 cells (right) treated with 500 nM JQ1 for different time periods or with different concentrations of JQ1 for 72 hours. Error bars represent means ± SD. (D) BRCA1 and RAD51 expression assessed by Western blot in MDA-MB-231 cells treated with 500 nM JQ1 for different time periods (top) or with different concentrations of JQ1 for 72 hours (bottom). Tubulin and actin served as loading controls. (E) BRD2/3/4, BRCA1, and RAD51 mRNA expression in MDA-MB-231 cells treated with individual BRD2/3/4 siRNAs or with BRD2/3/4 siRNA pools. Error bars represent means ± SD. (F) BRD2/3/4, BRCA1, RAD51, and MYC expression measured by Western blot in cells treated with individual BRD2/3/4 siRNAs or with a BRD2/3/4 siRNA pool. Left: MDA-MB-231. Middle: OVCAR10. Right: VCaP. Actin served as a loading control.
BETi directly represses transcription of BRCA1 and RAD51 in cancer cells
To explore the mechanism by which JQ1 represses the expression of BRCA1 and RAD51, we analyzed chromatin immunoprecipitation (ChIP) with antibodies against BRD2/3/4 followed by ChIP sequencing (ChIP-seq) in VCaP cells treated with JQ1 (50). As expected, BRD2/3/4 immunoprecipitates shared overlapping enrichment for the promoter regions of BRCA1 and RAD51, and JQ1 treatment reduced the recruitment of all three proteins to these regions (Fig. 6, A and B). This observation was also validated by our ChIP–quantitative PCR (qPCR) analysis in MDA-MB-231 cells (Fig. 6, C and D). Next, we generated a luciferase reporter containing the DNA sequences of the BRCA1 or RAD51 promoter (Fig. 6, A and B). Consistent with ChIP experiments, the luciferase activity of BRCA1 and RAD51 promoters was repressed by JQ1 treatment but was unchanged byolaparib treatment. Compared to JQ1 treatment alone, there was no further reduction of the luciferase activity when the cells were treated with a combination of JQ1 and olaparib (Fig. 6, E and F). Furthermore, knocking down BRD2/3/4 by siRNAs genetically mimicked the effect of JQ1 in repressing the activity of BRCA1 and RAD51 promoters. Compared to the knockdown of individual BET family members, pooled siRNAs targeting all three BETs achieved greater repression in the activity of BRCA1 and RAD51 promoters (Fig. 6, G and H).
Fig. 6. JQ1 directly represses the promoter activities of BRCA1 and RAD51.
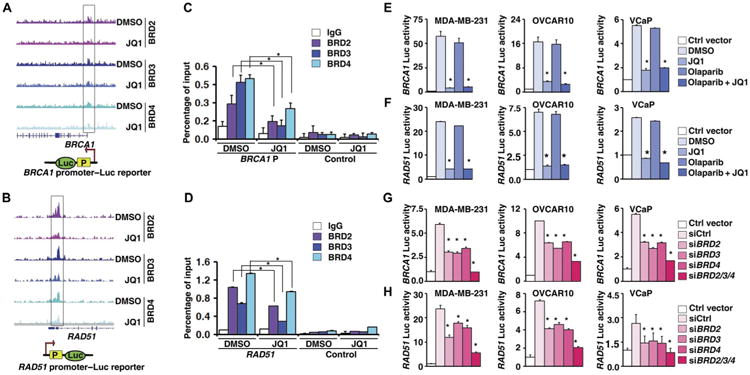
(A and B) BRD2/3/4 binding pattern in the BRCA1 (A) or RAD51 (B) promoter regions in VCaP cells treated with DMSO or JQ1. Bottom: Illustrations of BRCA1 promoter–luciferase (Luc) (A) and RAD51 promoter–Luc (B) reporters, in which BRCA1 or RAD51 promoter containing the BRD2/3/4 binding region was cloned into pGL3. (C and D) Quantification of the amount of BRCA1 (C) or RAD51 (D) promoter bound to BRD2/3/4 in MDA-MB-231 cells treated with DMSO or JQ1. Statistical analysis by Student's t test, *P < 0.05; n = 3. Error bars represent means ± SD. IgG, immunoglobulin G. (E and F) Luciferase reporter assay of the activities of the BRCA1 (E) and RAD51 (F) core promoter reporters in MDA-MB-231, OVCAR10, and VCaP cells treated with DMSO, JQ1, olaparib, or JQ1 combined with olaparib. Statistical analysis by Student's t test, *P < 0.05; n = 3. Error bars represent means ± SD. (G and H) Luciferase reporter assay of the activities of the BRCA1 (G) and RAD51 (H) core promoter reporters in MDA-MB-231, OVCAR10, and VCaP cells treated with individual siRNAs targeting BRD2/3/4 or pooled siRNAs. Statistical analysis by Student's t test, *P < 0.05; n = 3. Error bars represent means ± SD.
We also explored potential super-enhancer regions in the BRCA1 and RAD51 loci using ChIP-seq analysis. ChIP-seq data on H3K4me1, H3K4me3, and H3K27Ac indicated that a putative super-enhancer (a cluster of enhancers identified by high amounts of H3K27Ac/H3K4me1 and low amounts of H3K4me3) exists in the downstream region of the BRCA1 gene in MDA-MB-231 cells (Fig. 7A). A consistent H3K27Ac binding pattern was also observed in VCaP cells, in which BRD2/3/4 were enriched in the same regions (Fig. 7B). JQ1 treatment markedly decreased the binding ability of BRD2/3/4 in such enhancer regions in VCaP cells (Fig. 7B). Furthermore, an identical H3K4me1, H3K4me3, and H3K27Ac binding pattern was also found in nonmalignant cells such as GM12878 cells (Fig. 7C), in which the BRCA1 gene and this putative enhancer cluster are located in the same topologically associated domain, as defined by high-resolution Hi-C analysis (Fig. 7D). To confirm the physical interaction between the BRCA1 promoter and this potential enhancer cluster, we performed quantitative analysis of chromosome conformation capture assays (3C-qPCR) in three different cancer cell lines. Among the seven pairs of primers designed to detect the potential promoter-enhancer interaction (Fig. 7, A and E), the primers for the promoter and the enhancer locus 2 showed the greatest interaction strength and strongest response to JQ1 treatment in all three lines (Fig. 7E). The size of the corresponding PCR product was confirmed by electrophoresis (Fig. 7F). The above observations were also validated by ChIP-qPCR analysis in MDA-MB-231 cells (Fig. 7G). A strong transcriptional enhancing activity was observed in the cells transfected with the BRCA1 promoter-enhancer vector compared to the BRCA1 promoter vector, and the enhancer activity was inhibited by JQ1 alone or the combination of JQ1 and olaparib but not by olaparib alone (Fig. 7H). Finally, we confirmed the above result by siRNAs specifically targeting each BET family member (Fig. 7I).
Fig. 7. JQ1 represses the enhancer-promoter interaction of BRCA1.
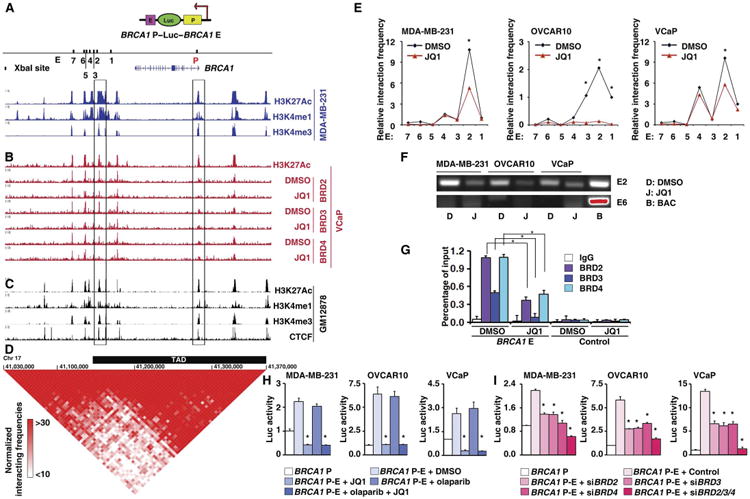
(A) H3K27Ac, H3K4me1, H3K4me3 profiles in MDA-MB-231. Right and left rectangle frame areas indicate the BRCA1 promoter and enhancer locus 2, respectively, which were used for the BRCA1 promoter-enhancer reporter construct. Upper schematic illustration indicates the construction of the BRCA1 promoter-enhancer reporter. Lower schematic illustrates the Xba I restriction sites in BRCA1 promoter and enhancer loci. P in red indicates promoter, and E in black indicates enhancer. Numbers 1 to 7 indicate seven Xba I restriction sites in the enhancer locus. The position of the BRCA1 gene is indicated below. (B) H3K27Ac in VCaP cells (top) and BRD2/3/4 profiles in VCaP treated with DMSO or JQ1 (bottom). (C) H3K27Ac, H3K4me1, H3K4me3, and CCCTC-binding factor (CTCF) profiles in GM12878. (D) A BRCA1 topologically associated domain (TAD) region (indicated by a thick black bar) was predicted on the basis of the Hi-C data. Normalized Hi-C interacting frequencies were shown as a triangle-like two-dimensional heat map, which was overlaid on ChIP-seq data (C) in GM12878 cells. Red indicates the intensity of the interacting frequencies of the Hi-C data. Chr 17, chromosome 17. (E) 3C-qPCR analysis of long-distance interactions between the BRCA1 promoter and seven enhancer loci [locus 1 to 7 indicated in (A)]. The relative amount of ligation product between the BRCA1 promoter and each of the seven different enhancer loci was plotted. Results from MDA-MB-231, OVCAR10, and VCaP cells treated with DMSO or JQ1 are shown. The data were normalized to a GAPDH loading control. (F) PCR products from the BRCA1 promoter and enhancer locus 2 (E2, top) and from the BRCA1 promoter and enhancer locus 6 (E6, bottom). Ligation sample from bacterial artificial chromosome (BAC) was used as a positive control. No specific PCR product was found in the ligation sample from the BRCA1 promoter and enhancer locus 6, even when the BAC control band was overexposed (shown in red). (G) Quantification of the amount of BRCA1 enhancer locus 2 bound to BRD2/3/4 in MDA-MB-231 cells treated with DMSO or JQ1. (H) Luciferase reporter assay of BRCA1 enhancer activity in MDA-MB-231, OVCAR10, and VCaP cells treated with DMSO, JQ1, olaparib, or JQ1 combined with olaparib. (I) Luciferase reporter assay of BRCA1 enhancer activity in MDA-MB-231, OVCAR10, and VCaP cells treated with BRD2/3/4 siRNA.
Recurrent copy number amplification of the BRD4 gene is observed across common cancers
To characterize the genomic alterations of BET genes in cancer, we retrieved RNA sequencing (RNA-seq; n = 8298), single-nucleotide polymorphism (SNP) microarray (n = 9445), and exon sequencing (exon-seq; n = 8899) profiles across 20 common cancer types (table S2) from The Cancer Genome Atlas (TCGA). First, we found that gene fusion is a rare genomic alteration for BET genes across all cancer types. Only 10 fusion events (8 from BRD4 and 2 from BRD3) were identified across 8298 RNA-seq profiles of TCGA tumor specimens (Fig. 8A and table S3). No recurrent fusion events were identified. Second, we found that there was no recurrent mutation observed in BET genes, except for a low-frequency recurrent BRD2 mutation in colon cancer (3.01%) and BRDT in pancreatic (0.55%) and endometrial (8.12%) cancers, across 20 cancer types (n = 8899) based on exon-seq analysis (table S4). Last, we analyzed the somatic copy number alterations (SCNAs) of BET genes in cancer via SNP microarray analysis of 9445 tumors in 20 cancer types from TCGA. In a pan-cancer analysis, recurrent amplification of BRD4 was observed (Fig. 8, B and C). Analyses for each individual cancer type showed that BRD4 was recurrently amplified in breast, liver, ovarian, and endometrial cancers (Fig. 8, B and D). By contrast, BRD3 was recurrently deleted in breast and ovarian cancers at a low frequency (Fig. 8B). Although the BRDT gene (coding a testis-specific BET protein) was recurrently deleted and mutated in six and two cancer types, respectively, the RNA-seq analysis indicated that BRDT mRNA was not detectable in any cancer type (Fig. 8D). This suggests that deletion and mutation of BRDT may be a passenger event. In contrast, BRD2, BRD3, and BRD4 were widely expressed in all cancer types examined in our study (Fig. 8E). We found a positive correlation between BRD4 copy number and gene expression across all cancer types and in individual cancers (Fig. 8F). Cancer types that had higher expression of BRD4 SCNAs (such as ovarian and breast cancers) demonstrated stronger RNA-SCNA correlations than the cancer types with fewer SCNAs (such as prostate cancer) (Fig. 8F). We also found that the BRD4-amplified cancer cell lines have decreased sensitivity to PARPi (fig. S13). The recurrent focal amplification of the BRD4 gene provides a strong rationale for BRD4-targeted therapy for cancer patients.
Fig. 8. Recurrent copy number amplification of BRD4 gene was observed across common cancers.
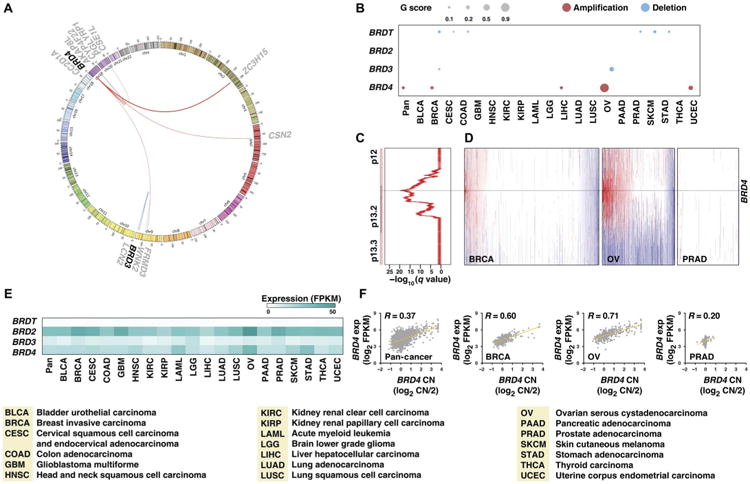
(A) Fusions of BET genes in TCGA samples are shown by a Circos plot. The fused genes are illustrated as a line that connects two parental genes. Line thickness corresponds to FFPM (fusion fragments per million total RNA-seq reads) value. (B) Plot of the recurrent copy number alterations in BET genes across 20 cancer types. Red and blue indicate amplification and deletion, respectively. Dot size indicates the G score, which represents frequency and amplitude of copy number alteration. (C) Significance of recurrent BRD4 amplification across 20 types of cancers. GISTIC q values (x axis) for amplifications are plotted across the genomic locus harboring the BRD4 gene (y axis). (D) Copy number profiles of the BRD4 locus from breast, ovarian, and prostate tumor specimens. Each sample is represented with a vertical line, and the positions of BRD4 are noted with black horizontal lines. Red, gain; blue, loss. (E) Heat map of the 50th percentile of BET gene mRNA expression across cancers. Each cancer type is represented by a column, and each BET gene is represented by a row. The intensity of blue indicates mRNA expression. FPKM, fragments per kilobase of exon per million. (F) A positive correlation between BRD4 gene copy number (CN) and RNA expression was observed in cancer specimens.
Discussion
Strategies that selectively disrupt HR in cancer cells may sensitize HR-proficient tumors to PARP inhibition. Furthermore, given that acquired HR proficiency is one of the most common resistance mechanisms to PARP inhibition, such approaches may also overcome resistance to PARPi treatment, which can develop in tumors initially sensitive to PARPi. In this regard, several approaches have been evaluated in preclinical and early clinical trials (30–39), such as combinations of PARPi with a CDK1 inhibitor that represses phosphorylation of BRCA1 (30) and combination of PARPi with PI3K (phosphatidylinositol 3-kinase)/AKT inhibitors that down-regulates BRCA1/2 expression (31) and suppresses RAD51 foci formation (32). Using a drug synergy screen, we identified BETi as acting synergistically with PARPi in HR-proficient cells. Our results indicate that BETi may synergize with PARPito treat patients with de novo HR-proficient cancer that is primarily resistant to PARPi because repressed BET activity impairs BRCA1 and RAD51 expression, subsequently converting HR-proficient tumors to HR-deficient tumors (fig. S14A). Furthermore, our results suggest that BETi may resensitize tumors with acquired HR proficiency to PARPi treatment, thus overcoming PARPi resistance. BETi may act by repressing the expression of secondary BRCA1 mutations that restores BRCA1 function (fig. S14B) or blocking the expression of BRCA1 and RAD51 in the context of other resistance mechanisms (such as 53BP/REV7 loss and BRCA2 reversion mutation) to overcome PARPi resistance. Because BET inhibition simultaneously represses the expression of BRCA1 and RAD51, BETi may also enhance the response to PARP inhibition in HR-deficient tumors such as BRCA1- or BRCA2-mutated cancers by further blocking RAD51 expression (fig. S14C). Finally, given that deficiency of HR increases the sensitivity of cancer cells to treatment with DNA damage agents, BETi may also act synergistically with classical chemotherapy drugs. Our study provides a strong rationale for clinical application of BETi in combination with PARPi or other DNA-damaging anticancer agents.
The molecular mechanism ofBET inhibitionin cancer treatment is a fundamental question. Considering the critical role of BET proteins in gene transcriptional elongation, the mechanism underlying the antitumor activity of BETi may be mainly mediated by transcriptional repression (40, 41). The observation that BET inhibition only represses the transcription of a selected subset of genes was unexpected, indicating that BETi may specifically impair the expression of a fraction of cancer-associated genes such as MYC (42–49). We showed that expression of BRCA1 and RAD51 was repressed by BETi to a comparable extent as MYC repression in multiple cancer models. A potential super-enhancer of BRCA1 was identified in epithelial cancer cells, and it is conserved in nontransformed normal cells. We demonstrated that repression of BET proteins impairs expression of BRCA1 and RAD51, subsequently disrupting HR in tumor cells. Our study thus provides a mechanistic insight into the development of BET-targeted therapy in cancer.
Our study has been limited to focusingon the BETi targeting genes, which are directly involved in DNA damage repair. Although we believe that direct transcriptional repression of HR genes is the dominant mechanism, we cannot exclude other indirect mechanisms that may also cooperate or contribute to this synergistic effect. For example, BETi represses expression of MYC (42–49), a transcriptional amplifier, and thus, MYC reduced by BETi might also indirectly contribute to reduction of HR gene expression and enhancement of NHEJ activity, especially in the MYC-hyperactivated tumors. Additionally, BETi may also directly influence the DNA damage response by interacting with chromatin remodeling complexes. A recent study identified a short isoform of BRD4, which serves as an insulator of chromatin, modulating the signaling response to DNA damage (61). Therefore, it is still necessary to further characterize how other mechanisms cooperate or contribute to the BETi/PARPi synergistic effect in some cellular contexts. Despite these limitations, our findings provide a strong rationale for clinical application of PARPi in the setting of combination with BETi to treat cancers with de novo or acquired resistance to PARP inhibition.
Materials and Methods
Study design
The overall goal of this study was to identify epigenetic modulators that can act synergistically with PARPis in preclinical models of human cancer. Further experiments were designed to characterize the molecular and cellular mechanisms of the combination. The selection of cancer cell lines was based on the clinical application of PARPi: We chose cell lines originating from ovarian (OVCAR10), breast (MDA-MB-231), and prostate (VCaP) cancers, for which PARPi treatment has been applied or proposed in clinic. Given that olaparib (AZD2281) was the first PARPi approved by FDA, we chose it in our initial screen. For all xenograft studies, sample sizes were decided using data from preliminary caliper measurements of tumor growth. We performed preliminary experiments using a few mice per group to decide the sample size, followed by larger studies to perform the in vivo drug combination experiments with statistical analysis. We also repeated experiments to ensure reproducibility. Xenografted mice were randomized before receiving different treatments. The resulting tumors were analyzed in a blinded manner. For all in vitro experiments, no randomization or blinding was used because they were deemed unfeasible because of the nature of the performed experiments.
Statistical analysis
Statistical analysis was performed using SPSS and R software. All results are expressed as means ± SD, and P < 0.05 indicates significance.
Supplementary Material
Fig. S1. BET depletion increases PARPi sensitivity.
Fig. S2. BETi synergizes with PARPi in HR-deficient cancer cell lines.
Fig. S3. BETi synergizes with cisplatin but not paclitaxel in MDA-MB-231 cells.
Fig. S4. Combination treatment with BETi and PARPi does not act synergistically in noncycling cancer cells.
Fig. S5. BET inhibition decreases the ALDH+ tumor-initiating cell population.
Fig. S6. BET depletion decreases HR.
Fig. S7. BET inhibition does not affect PARP trapping.
Fig. S8. BET inhibition does not affect total cellular PARylation and Ku80 PARylation.
Fig. S9. BET inhibition increases NHEJ repair efficiency.
Fig. S10. Combining JQ1 and olaparib does not increase toxicity to mice.
Fig. S11. BET inhibition represses the expression of BRCA1 and RAD51 in vivo.
Fig. S12. The effect of BET inhibition on the expression of other proteins was analyzed by Western blot.
Fig. S13. BRD4 amplification results in PARPi resistance.
Fig. S14. The study provides a strong rationale for clinical application of PARPi in combination with BETi.
Table S1. Epigenetic compounds used in drug combination screen (provided as an Excel file).
Table S2. TCGA specimens used in this study (provided as an Excel file).
Table S3. Fusion events of BET genes in TCGA (provided as an Excel file).
Table S4. Mutation frequency of BET genes in TCGA (provided as an Excel file).
Acknowledgments
We thank J. E. Bradner and J. Qi (Harvard Medical School) for providing JQ1.
Funding: This work was supported, in whole or in part, by the Basser Center for BRCA (to L.Z.), the Harry Fields Professorship (to L.Z.), the NIH (R01CA142776, R01CA190415, P50CA083638, and P50CA174523 to L.Z., R01CA148759, R21CA198558, and U01CA200495 to Q.H., and R01NS094533 to Y.F.), the Breast Cancer Alliance (to L.Z. and C.V.D.), the Ovarian Cancer Research Fund (to X.H. and L.Z.), the Foundation for Women's Cancer (to X.H. and L.Z.), the Kaleidoscope of Hope Ovarian Cancer Foundation (to L.Z.), and the Marsha Rivkin Center for Ovarian Cancer Research (to L.Z.). L.Y. and W.S. were supported by the China Scholarship Council.
Footnotes
Author contributions: L.Y. and Y.Z. performed the experiments, analyzed the data, and wrote the article. W.S., L.F., Z.T., C.L., J.P., Y.W., and X.H. performed the experiments and analyzed the data. Z.H. and J.Y. performed bioinformatics analyses. J.L.T. and K.M. reviewed the clinical and pathological data of TCGA. Q.H. and Y.F. provided experimental materials and cell-based models. C.V.D. and L.Z. designed the experiments and wrote the article.
Competing interests: The authors declare that they have no competing interests.
References and Notes
- 1.Lord CJ, Tutt ANJ, Ashworth A. Synthetic lethality and cancer therapy: Lessons learned from the development of PARP inhibitors. Annu Rev Med. 2015;66:455–470. doi: 10.1146/annurev-med-050913-022545. [DOI] [PubMed] [Google Scholar]
- 2.Scott CL, Swisher EM, Kaufmann SH. Poly (ADP-ribose) polymerase inhibitors: Recent advances and future development. J Clin Oncol. 2015;33:1397–1406. doi: 10.1200/JCO.2014.58.8848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D'Andrea AD. Homologous recombination deficiency: Exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov. 2015;5:1137–1154. doi: 10.1158/2159-8290.CD-15-0714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feng FY, de Bono JS, Rubin MA, Knudsen KE. Chromatin to clinic: The molecular rationale for PARP1 inhibitor function. Mol Cell. 2015;58:925–934. doi: 10.1016/j.molcel.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pommier Y, O'Connor MJ, de Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med. 2016;8:362ps17. doi: 10.1126/scitranslmed.aaf9246. [DOI] [PubMed] [Google Scholar]
- 6.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 7.Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NMB, Jackson SP, Smith GCM, Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 8.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, Ashworth A, Carmichael J, Kaye SB, Schellens JHM, de Bono JS. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 9.Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, Scott C, Weitzel JN, Oaknin A, Loman N, Lu K, Schmutzler RK, Matulonis U, Wickens M, Tutt A. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: A proof-of-concept trial. Lancet. 2010;376:245–251. doi: 10.1016/S0140-6736(10)60893-8. [DOI] [PubMed] [Google Scholar]
- 10.Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmaña J, Mitchell G, Fried G, Stemmer SM, Hubert A, Rosengarten O, Steiner M, Loman N, Bowen K, Fielding A, Domchek SM. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33:244–250. doi: 10.1200/JCO.2014.56.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, Wardley A, Mitchell G, Earl H, Wickens M, Carmichael J. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet. 2010;376:235–244. doi: 10.1016/S0140-6736(10)60892-6. [DOI] [PubMed] [Google Scholar]
- 12.Rugo HS, Olopade OI, DeMichele A, Yau C, van't Veer LJ, Buxton MB, Hogarth M, Hylton NM, Paoloni M, Perlmutter J, Symmans WF, Yee D, Chien AJ, Wallace AM, Kaplan HG, Boughey JC, Haddad TC, Albain KS, Liu MC, Isaacs C, Khan QJ, Lang JE, Viscusi RK, Pusztai L, Moulder SL, Chui SY, Kemmer KA, Elias AD, Edmiston KK, Euhus DM, Haley BB, Nanda R, Northfelt DW, Tripathy D, Wood WC, Ewing C, Schwab R, Lyandres J, Davis SE, Hirst GL, Sanil A, Berry DA, Esserman LJ. I-SPY 2 Investigators, Adaptive randomization of veliparib–carboplatin treatment in breast cancer. N Engl J Med. 2016;375:23–34. doi: 10.1056/NEJMoa1513749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N, Boysen G, Porta N, Flohr P, Gillman A, Figueiredo I, Paulding C, Seed G, Jain S, Ralph C, Protheroe A, Hussain S, Jones R, Elliott T, Mc Govern U, Bianchini D, Goodall J, Zafeiriou Z, Williamson CT, Ferraldeschi R, Riisnaes R, Ebbs B, Fowler G, Roda D, Yuan W, Wu YM, Cao X, Brough R, Pemberton H, A'Hern R, Swain A, Kunju LP, Eeles R, Attard G, Lord CJ, Ashworth A, Rubin MA, Knudsen KE, Feng FY, Chinnaiyan AM, Hall E, de Bono JS. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373:1697–1708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu Y, Petit SA, Ficarro SB, Toomire KJ, Xie A, Lim E, Cao SA, Park E, Eck MJ, Scully R, Brown M, Marto JA, Livingston DM. PARP1-driven poly-ADP-ribosylation regulates BRCA1 function in homologous recombination–mediated DNA repair. Cancer Discov. 2014;4:1430–1447. doi: 10.1158/2159-8290.CD-13-0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murai J, Huang SyN, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72:5588–5599. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li M, Yu X. Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell. 2013;23:693–704. doi: 10.1016/j.ccr.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patel AG, Sarkaria JN, Kaufmann SH. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Natl Acad Sci USA. 2011;108:3406–3411. doi: 10.1073/pnas.1013715108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mateos-Gomez PA, Gong F, Nair N, Miller KM, Lazzerini-Denchi E, Sfeir A. Mammalian polymerase q promotes alternative NHEJ and suppresses recombination. Nature. 2015;518:254–257. doi: 10.1038/nature14157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MIR, O'Connor KW, Konstantinopoulos PA, Elledge SJ, Boulton SJ, Yusufzai T, D'Andrea AD. Homologous-recombination-deficient tumours are dependent on Polq-mediated repair. Nature. 2015;518:258–262. doi: 10.1038/nature14184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brenner JC, Ateeq B, Li Y, Yocum AK, Cao Q, Asangani IA, Patel S, Wang X, Liang H, Yu J, Palanisamy N, Siddiqui J, Yan W, Cao X, Mehra R, Sabolch A, Basrur V, Lonigro RJ, Yang J, Tomlins SA, Maher CA, Elenitoba-Johnson KSJ, Hussain M, Navone NM, Pienta KJ, Varambally S, Feng FY, Chinnaiyan AM. Mechanistic rationale for inhibition of poly(ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer. Cancer Cell. 2011;19:664–678. doi: 10.1016/j.ccr.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schiewer MJ, Goodwin JF, Han S, Brenner JC, Augello MA, Dean JL, Liu F, Planck JL, Ravindranathan P, Chinnaiyan AM, McCue P, Gomella LG, Raj GV, Dicker AP, Brody JR, Pascal JM, Centenera MM, Butler LM, Tilley WD, Feng FY, Knudsen KE. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012;2:1134–1149. doi: 10.1158/2159-8290.CD-12-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dedes KJ, Wetterskog D, Mendes-Pereira AM, Natrajan R, Lambros MB, Geyer FC, Vatcheva R, Savage K, Mackay A, Lord CJ, Ashworth A, Reis-Filho JS. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci Transl Med. 2010;2:53ra75. doi: 10.1126/scitranslmed.3001538. [DOI] [PubMed] [Google Scholar]
- 23.Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, Boyd J, Reis-Filho JS, Ashworth A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–1118. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- 24.Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ, Couch FJ, Urban N, Taniguchi T. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116–1120. doi: 10.1038/nature06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pettitt SJ, Rehman FL, Bajrami I, Brough R, Wallberg F, Kozarewa I, Fenwick K, Assiotis I, Chen L, Campbell J, Lord CJ, Ashworth A. A genetic screen using the PiggyBac transposon in haploid cells identifies Parp1 as a mediator of olaparib toxicity. PLOS ONE. 2013;8:e61520. doi: 10.1371/journal.pone.0061520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, Xu X, Deng CX, Finkel T, Nussenzweig M, Stark JM, Nussenzweig A. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA, Drost R, Wientjens E, Ji J, Aly A, Doroshow JH, Cranston A, Martin NMB, Lau A, O'Connor MJ, Ganesan S, Borst P, Jonkers J, Rottenberg S. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013;3:68–81. doi: 10.1158/2159-8290.CD-12-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P, Bartkova J, Gogola E, Warmerdam D, Barazas M, Jaspers JE, Watanabe K, Pieterse M, Kersbergen A, Sol W, Celie PHN, Schouten PC, van den Broek B, Salman A, Nieuwland M, de Rink I, de Ronde J, Jalink K, Boulton SJ, Chen J, van Gent DC, Bartek J, Jonkers J, Borst P, Rottenberg S. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature. 2015;521:541–544. doi: 10.1038/nature14328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, John S, Day A, Crespo AV, Shen B, Starnes LM, de Ruiter JR, Daniel JA, Konstantinopoulos PA, Cortez D, Cantor SB, Fernandez-Capetillo O, Ge K, Jonkers J, Rottenberg S, Sharan SK, Nussenzweig A. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature. 2016;535:382–387. doi: 10.1038/nature18325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson N, Li YC, Walton ZE, Cheng KA, Li D, Rodig SJ, Moreau LA, Unitt C, Bronson RT, Thomas HD, Newell DR, D'Andrea AD, Curtin NJ, Wong KK, Shapiro GI. Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nat Med. 2011;17:875–882. doi: 10.1038/nm.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ibrahim YH, García-García C, Serra V, He L, Torres-Lockhart K, Prat A, Anton P, Cozar P, Guzmán M, Grueso J, Rodríguez O, Calvo MT, Aura C, Díez O, Rubio IT, Pérez J, Rodón J, Cortés J, Ellisen LW, Scaltriti M, Baselga J. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012;2:1036–1047. doi: 10.1158/2159-8290.CD-11-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Juvekar A, Burga LN, Hu H, Lunsford EP, Ibrahim YH, Balmaña J, Rajendran A, Papa A, Spencer K, Lyssiotis CA, Nardella C, Pandolfi PP, Baselga J, Scully R, Asara JM, Cantley LC, Wulf GM. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2012;2:1048–1063. doi: 10.1158/2159-8290.CD-11-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bajrami I, Kigozi A, Van Weverwijk A, Brough R, Frankum J, Lord CJ, Ashworth A. Synthetic lethality of PARP and NAMPT inhibition in triple-negative breast cancer cells. EMBO Mol Med. 2012;4:1087–1096. doi: 10.1002/emmm.201201250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu JF, Barry WT, Birrer M, Lee JM, Buckanovich RJ, Fleming GF, Rimel BJ, Buss MK, Nattam S, Hurteau J, Luo W, Quy P, Whalen C, Obermayer L, Lee H, Winer EP, Kohn EC, Ivy SP, Matulonis UA. Combination cediranib and olaparibversus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: A randomised phase 2 study. Lancet Oncol. 2014;15:1207–1214. doi: 10.1016/S1470-2045(14)70391-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ha K, Fiskus W, Choi DS, Bhaskara S, Cerchietti L, Devaraj SGT, Shah B, Sharma S, Chang JC, Melnick AM, Hiebert S, Bhalla KN. Histone deacetylase inhibitor treatment induces ‘BRCAness’ and synergistic lethality with PARP inhibitor and cisplatin against human triple negative breast cancer cells. Oncotarget. 2014;5:5637–5650. doi: 10.18632/oncotarget.2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Du Y, Yamaguchi H, Wei Y, Hsu JL, Wang HL, Hsu YH, Lin WC, Yu WH, Leonard PG, Lee GR, IV, Chen MK, Nakai K, Hsu MC, Chen CT, Sun Y, Wu Y, Chang WC, Huang WC, Liu CL, Chang YC, Chen CH, Park M, Jones P, Hortobagyi GN, Hung MC. Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat Med. 2016;22:194–201. doi: 10.1038/nm.4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jdey W, Thierry S, Russo C, Devun F, Al Abo M, Noguiez-Hellin P, Sun JS, Barillot E, Zinovyev A, Kuperstein I, Pommier Y, Dutreix M. Drug-driven synthetic lethality: Bypassing tumor cell genetics with a combination of AsiDNA and PARP inhibitors. Clin Cancer Res. 2017;23:1001–1011. doi: 10.1158/1078-0432.CCR-16-1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson SF, Cruz C, Greifenberg AK, Dust S, Stover DG, Chi D, Primack B, Cao S, Bernhardy AJ, Coulson R, Lazaro JB, Kochupurakkal B, Sun H, Unitt C, Moreau LA, Sarosiek KA, Scaltriti M, Juric D, Baselga J, Richardson AL, Rodig SJ, D'Andrea AD, Balmaña J, Johnson N, Geyer M, Serra V, Lim E, Shapiro GI. CDK12 inhibition reverses de novo and acquired PARP inhibitor resistance in BRCA wild-type and mutated models of triple-negative breast cancer. Cell Rep. 2016;17:2367–2381. doi: 10.1016/j.celrep.2016.10.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun C, Fang Y, Yin J, Chen J, Ju Z, Zhang D, Chen X, Vellano CP, Jeong KJ, Ng PKS, Eterovic AKB, Bhola NH, Lu Y, Westin SN, Grandis JR, Lin SY, Scott KL, Peng G, Brugge J, Mills GB. Rational combination therapy with PARP and MEK inhibitors capitalizes on therapeutic liabilities in RAS mutant cancers. Sci Transl Med. 2017;9:eaal5148. doi: 10.1126/scitranslmed.aal5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shu S, Polyak K. BET bromodomain proteins as cancer therapeutic targets. Cold Spring Harb Symp Quant Biol. 2017;81:1–9. doi: 10.1101/sqb.2016.81.030908. [DOI] [PubMed] [Google Scholar]
- 41.Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell. 2014;54:728–736. doi: 10.1016/j.molcel.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung Cw, Hopf C, Savitski MM, Huthmacher C, Gudgin E, Lugo D, Beinke S, Chapman TD, Roberts EJ, Soden PE, Auger KR, Mirguet O, Doehner K, Delwel R, Burnett AK, Jeffrey P, Drewes G, Lee K, Huntly BJP, Kouzarides T. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi JW, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, Chesi M, Schinzel AC, McKeown MR, Heffernan TP, Vakoc CR, Bergsagel PL, Ghobrial IM, Richardson PG, Young RA, Hahn WC, Anderson KC, Kung AL, Bradner JE, Mitsiades CS. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L, Sims RJ., III Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci USA. 2011;108:16669–16674. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, Taylor MJ, Johns C, Chicas A, Mulloy JC, Kogan SC, Brown P, Valent P, Bradner JE, Lowe SW, Vakoc CR. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lockwood WW, Zejnullahu K, Bradner JE, Varmus H. Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proc Natl Acad Sci USA. 2012;109:19408–19413. doi: 10.1073/pnas.1216363109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, Nekritz EA, Zeid R, Gustafson WC, Greninger P, Garnett MJ, McDermott U, Benes CH, Kung AL, Weiss WA, Bradner JE, Stegmaier K. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 2013;3:308–323. doi: 10.1158/2159-8290.CD-12-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MGM, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, Reichert E, Kung AL, Rodig SJ, Young RA, Shipp MA, Bradner JE. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell. 2013;24:777–790. doi: 10.1016/j.ccr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, Escara-Wilke J, Wilder-Romans K, Dhanireddy S, Engelke C, Iyer MK, Jing X, Wu YM, Cao X, Qin ZS, Wang S, Feng FY, Chinnaiyan AM. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014;510:278–282. doi: 10.1038/nature13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baratta MG, Schinzel AC, Zwang Y, Bandopadhayay P, Bowman-Colin C, Kutt J, Curtis J, Piao H, Wong LC, Kung AL, Beroukhim R, Bradner JE, Drapkin R, Hahn WC, Liu JF, Livingston DM. An in-tumor genetic screen reveals that the BET bromodomain protein, BRD4, is a potential therapeutic target in ovarian carcinoma. Proc Natl Acad Sci USA. 2015;112:232–237. doi: 10.1073/pnas.1422165112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM, Janiszewska M, Huh SJ, Liang Y, Ryan J, Doherty E, Mohammed H, Guo H, Stover DG, Ekram MB, Peluffo G, Brown J, D'Santos C, Krop IE, Dillon D, McKeown M, Ott C, Qi J, Ni M, Rao PK, Duarte M, Wu SY, Chiang CM, Anders L, Young RA, Winer EP, Letai A, Barry WT, Carroll JS, Long HW, Brown M, Liu XS, Meyer CA, Bradner JE, Polyak K. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016;529:413–414. doi: 10.1038/nature16508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mazur PK, Herner A, Mello SS, Wirth M, Hausmann S, Sánchez-Rivera FJ, Lofgren SM, Kuschma T, Hahn SA, Vangala D, Trajkovic-Arsic M, Gupta A, Heid I, Noël PB, Braren R, Erkan M, Kleeff J, Sipos B, Sayles LC, Heikenwalder M, Heβmann E, Ellenrieder V, Esposito I, Jacks T, Bradner JE, Khatri P, Sweet-Cordero EA, Attardi LD, Schmid RM, Schneider G, Sage J, Siveke JT. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat Med. 2015;21:1163–1171. doi: 10.1038/nm.3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tasdemir N, Banito A, Roe JS, Alonso-Curbelo D, Camiolo M, Tschaharganeh DF, Huang CH, Aksoy O, Bolden JE, Chen CC, Fennell M, Thapar V, Chicas A, Vakoc CR, Lowe SW. BRD4 connects enhancer remodeling to senescence immune surveillance. Cancer Discov. 2016;6:612–629. doi: 10.1158/2159-8290.CD-16-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stathis A, Zucca E, Bekradda M, Gomez-Roca C, Delord JP, de La Motte Rouge T, Uro-Coste E, de Braud F, Pelosi G, French CA. Clinical response of carcinomas harboring the BRD4–NUT oncoprotein to the targeted bromodomain inhibitor OTX015/MK-8628. Cancer Discov. 2016;6:492–500. doi: 10.1158/2159-8290.CD-15-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Berthon C, Raffoux E, Thomas X, Vey N, Gomez-Roca C, Yee K, Taussig DC, Rezai K, Roumier C, Herait P, Kahatt C, Quesnel B, Michallet M, Recher C, Lokiec F, Preudhomme C, Dombret H. Bromodomain inhibitor OTX015 in patients with acute leukaemia: A dose-escalation, phase 1 study. Lancet Haematol. 2016;3:e186–e195. doi: 10.1016/S2352-3026(15)00247-1. [DOI] [PubMed] [Google Scholar]
- 57.Amorim S, Stathis A, Gleeson M, Iyengar S, Magarotto V, Leleu X, Morschhauser F, Karlin L, Broussais F, Rezai K, Herait P, Kahatt C, Lokiec F, Salles G, Facon T, Palumbo A, Cunningham D, Zucca E, Thieblemont C. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: A dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016;3:e196–e204. doi: 10.1016/S2352-3026(16)00021-1. [DOI] [PubMed] [Google Scholar]
- 58.Yokoyama Y, Zhu H, Lee JH, Kossenkov AV, Wu SY, Wickramasinghe JM, Yin X, Palozola KC, Gardini A, Showe LC, Zaret KS, Liu Q, Speicher D, Conejo-Garcia JR, Bradner JE, Zhang Z, Sood AK, Ordog T, Bitler BG, Zhang R. BET inhibitors suppress ALDH activity by targeting ALDH1A1 super-enhancer in ovarian cancer. Cancer Res. 2016;76:6320–6330. doi: 10.1158/0008-5472.CAN-16-0854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mao Z, Seluanov A, Jiang Y, Gorbunova V. TRF2 is required for repair of nontelomeric DNA double-strand breaks by homologous recombination. Proc Natl Acad Sci USA. 2007;104:13068–13073. doi: 10.1073/pnas.0702410104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schlegel BP, Jodelka FM, Nunez R. BRCA1 promotes induction of ssDNA by ionizing radiation. Cancer Res. 2006;66:5181–5189. doi: 10.1158/0008-5472.CAN-05-3209. [DOI] [PubMed] [Google Scholar]
- 61.Floyd SR, Pacold ME, Huang Q, Clarke SM, Lam FC, Cannell IG, Bryson BD, Rameseder J, Lee MJ, Blake EJ, Fydrych A, Ho R, Greenberger BA, Chen GC, Maffa A, Del Rosario AM, Root DE, Carpenter AE, Hahn WC, Sabatini DM, Chen CC, White FM, Bradner JE, Yaffe MB. The bromodomain protein Brd4 insulates chromatin from DNA damage signalling. Nature. 2013;498:246–250. doi: 10.1038/nature12147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. BET depletion increases PARPi sensitivity.
Fig. S2. BETi synergizes with PARPi in HR-deficient cancer cell lines.
Fig. S3. BETi synergizes with cisplatin but not paclitaxel in MDA-MB-231 cells.
Fig. S4. Combination treatment with BETi and PARPi does not act synergistically in noncycling cancer cells.
Fig. S5. BET inhibition decreases the ALDH+ tumor-initiating cell population.
Fig. S6. BET depletion decreases HR.
Fig. S7. BET inhibition does not affect PARP trapping.
Fig. S8. BET inhibition does not affect total cellular PARylation and Ku80 PARylation.
Fig. S9. BET inhibition increases NHEJ repair efficiency.
Fig. S10. Combining JQ1 and olaparib does not increase toxicity to mice.
Fig. S11. BET inhibition represses the expression of BRCA1 and RAD51 in vivo.
Fig. S12. The effect of BET inhibition on the expression of other proteins was analyzed by Western blot.
Fig. S13. BRD4 amplification results in PARPi resistance.
Fig. S14. The study provides a strong rationale for clinical application of PARPi in combination with BETi.
Table S1. Epigenetic compounds used in drug combination screen (provided as an Excel file).
Table S2. TCGA specimens used in this study (provided as an Excel file).
Table S3. Fusion events of BET genes in TCGA (provided as an Excel file).
Table S4. Mutation frequency of BET genes in TCGA (provided as an Excel file).