

Staphylococcus saprophyticus is an important cause of urinary tract infections (UTI) in women; such UTI are common, can be severe, and are associated with significant impacts to public health. In addition to being a cause of human UTI, S. saprophyticus can be found in the environment, in food, and associated with animals. After discovering that UTI strains of S. saprophyticus are for the most part closely related to each other, we sought to determine whether these strains are specially adapted to cause disease in humans. We found evidence suggesting that a mutation in the gene aas is advantageous in the context of human infection. We hypothesize that the mutation allows S. saprophyticus to survive better in the human urinary tract. These results show how bacteria found in the environment can evolve to cause disease.
KEYWORDS: Staphylococcus saprophyticus, adhesins, evolution, positive selection, urinary tract infection
ABSTRACT
Human-pathogenic bacteria are found in a variety of niches, including free-living, zoonotic, and microbiome environments. Identifying bacterial adaptations that enable invasive disease is an important means of gaining insight into the molecular basis of pathogenesis and understanding pathogen emergence. Staphylococcus saprophyticus, a leading cause of urinary tract infections, can be found in the environment, food, animals, and the human microbiome. We identified a selective sweep in the gene encoding the Aas adhesin, a key virulence factor that binds host fibronectin. We hypothesize that the mutation under selection (aas_2206A>C) facilitates colonization of the urinary tract, an environment where bacteria are subject to strong shearing forces. The mutation appears to have enabled emergence and expansion of a human-pathogenic lineage of S. saprophyticus. These results demonstrate the power of evolutionary genomic approaches in discovering the genetic basis of virulence and emphasize the pleiotropy and adaptability of bacteria occupying diverse niches.
IMPORTANCE Staphylococcus saprophyticus is an important cause of urinary tract infections (UTI) in women; such UTI are common, can be severe, and are associated with significant impacts to public health. In addition to being a cause of human UTI, S. saprophyticus can be found in the environment, in food, and associated with animals. After discovering that UTI strains of S. saprophyticus are for the most part closely related to each other, we sought to determine whether these strains are specially adapted to cause disease in humans. We found evidence suggesting that a mutation in the gene aas is advantageous in the context of human infection. We hypothesize that the mutation allows S. saprophyticus to survive better in the human urinary tract. These results show how bacteria found in the environment can evolve to cause disease.
INTRODUCTION
Urinary tract infections (UTI) are a global health problem of major significance, with an estimated annual incidence of 150 to 250 million and a lifetime risk of 50% among women (1–3). The associated costs for individuals and health care systems are substantial, with recent estimates from the United States numbering in the billions of dollars per year (4). UTI are also associated with severe complications such as pyelonephritis, sepsis, and premature labor (4). Staphylococcus saprophyticus is second only to Escherichia coli as a cause of UTI in reproduction-aged women (5, 6).
S. saprophyticus can be found in diverse niches, including the environment, foods, and livestock, and as a pathogen and commensal of humans. Several features of the epidemiology of S. saprophyticus suggest that infections leading to UTI are acquired from the environment rather than as a result of person-to-person transmission (7). This implies that adoption of the pathogenic niche by S. saprophyticus has not entailed a tradeoff in its ability to live freely in the environment. A recent PCR-based survey of virulence factors in clinical and animal-associated isolates showed that dsdA, a gene encoding d-serine deaminase that is important for survival in urine (8), and uafA and aas, genes encoding adhesins that mediate binding to uroepithelium (9, 10), were present in all isolates surveyed (11), suggesting an underlying pleiotropy, with these virulence factors playing important roles in the diverse environments occupied by S. saprophyticus.
The human urinary tract could represent an evolutionary dead end for S. saprophyticus, with “virulence factors” such as DsdA, UafA, and Aas serving an essential function in the primary environmental niche and enabling invasion of the urinary tract as an accidental by-product of this unknown primary function. In such a case, we would expect urinary isolates to be interspersed throughout a phylogeny of isolates from the primary niche(s). However, our previous research (7) indicated that human urinary tract infections are associated with a specific lineage of S. saprophyticus. Invasion of the human urinary tract enables S. saprophyticus to grow to high numbers in urine, isolated from competing bacterial species, before being redeposited in the environment. This is analogous to Vibrio cholerae, which cycles through human and environmental niches and grows to high abundance in the human gut before being deposited in the environment via stool (12, 13). Based on our previous observations and the example provided by other human pathogens that cycle through the environment, we hypothesized that the human urinary tract is an ecologically important niche for S. saprophyticus and sought to identify genetic signatures of adaptation to this niche.
The increased availability of sequencing data has enabled comparative genomic approaches that have led to identification of changes in gene content in association with pathogen emergence and shifts in host association. Several notable human pathogens, including Mycobacterium tuberculosis, Yersinia pestis, and Francisella tularensis, are the product of a single emergence characterized by gene loss and horizontal acquisition of virulence factors (14–16). Similarly, genomic analysis of Enterococcus faecium revealed gene gains and losses affecting metabolism and antibiotic resistance in the emergence of a hypermutable hospital-adapted clade that coincided with the profound shift in hospital ecology caused by the development of antibiotics (17). Gene gains via recombination have also allowed Staphylococcus aureus ST71 to emerge into a bovine-associated niche (18).
Using contemporary and ancient genomic data from strains of S. saprophyticus, we found previously that UTI-associated lineages of S. saprophyticus were not associated with specific gene gains or losses; the evolutionary genetic processes underlying the adoption by S. saprophyticus of the human-pathogenic niche are likely more subtle than those previously described for canonical pathogens (7). Here we have identified one of the mechanisms underlying the adaptation of S. saprophyticus to the uropathogenic niche: a selective sweep in the Aas adhesin, which is associated with an apparently large-scale expansion into the human-pathogenic niche. This is, to our knowledge, the first identification of a single nucleotide sweep in a bacterium.
RESULTS
We reconstructed the phylogeny of S. saprophyticus isolates (see Table S1 in the supplemental material) from a whole-genome alignment using maximum likelihood inference implemented in RAxML (Fig. 1). The bacterial isolates are separated into two clades, which we previously named clades P and E (7). In both clades, human-associated lineages are nested among isolates from diverse sources, including food (cheese rind, ice cream, meat), indoor and outdoor environments, and animals. Interestingly, cheese rinds harbor diverse strains of S. saprophyticus, which cluster with both human- and animal-pathogenic strains.
FIG 1 .

Maximum likelihood phylogeny of S. saprophyticus. Maximum likelihood phylogenetic analysis was performed in RAxML (92) using a whole-genome alignment with repetitive regions masked. The phylogeny is midpoint rooted, and nodes with bootstrap values of less than 90 are labeled. Branch lengths are scaled by the number of substitutions per site. Tips are colored based on the isolation source (pink, human; blue, animal; green, food; orange, environment). Tips are labeled with the isolate name and detailed source information. S. saprophyticus contains two major clades (clade P and clade E). Within clade P, there is a lineage enriched in human-pathogenic isolates (lineage U [branch labeled “U”]).
Accession numbers for S. saprophyticus isolates. Download TABLE S1, TXT file, 0.001 MB (1.9KB, txt) .
Copyright © 2017 Mortimer et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Thirty-three of 37 modern, human-pathogenic isolates are found within a single lineage (that we term lineage U [for "UTI associated"]) with respect to which bovine-pathogenic (mastitis), food-associated isolates, and an ancient genome are basal. Given the association between this lineage and illness in humans, we were curious about its potential adaptation to the human-pathogenic niche. The placement of the 800-year-old strain between bovine-associated and human-associated lineages suggests that it could represent a generalist intermediate between human-adapted and bovid-adapted strains.
Core genome analysis of the 58 isolates of S. saprophyticus in our sample showed substantial variability in gene content; the core genome is composed of 1,798 genes, and there are an additional 7,110 genes in the pan-genome. We found previously that uropathogenic isolates of S. saprophyticus were not associated with any unique gene content (7). Given the variability in accessory gene content among the members of this larger sample of isolates, we decided to test for relative differences in accessory gene content between human clinical isolates and other isolates using Scoary (19), which performs a genome-wide association study (GWAS) using gene presence and absence. We did not identify any genes that were significantly associated with the human-pathogenic niche after correction for multiple-hypothesis testing using the Bonferroni method.
In addition to the observed gene content variability, analyses of the core genome also indicated relatively frequent recombination among S. saprophyticus isolates (Fig. 2). We identified recombinant regions with Gubbins (20), which identifies regions with high densities of substitutions. These results indicated that 70% of sites in the S. saprophyticus alignment had been affected by recombination. Recombination can affect bacterial evolution both by introducing novel polymorphisms from outside the population and by reshuffling alleles without increasing overall diversity. Considering sites that are reshuffled within the S. saprophyticus sample to be recombinant sites, we estimated a ratio of recombinant to nonrecombinant single nucleotide polymorphisms (SNPs) of 3.4. Considering only the SNPs that introduce novel diversity to be recombinant SNPs, our estimate of the ratio of recombinant SNPs to nonrecombinant SNPs is 0.51. The mean recombination-per-mutation (r/m) value for branches in the phylogeny is 0.82 (range, 0 to 7.6) as estimated by Gubbins. Removal of recombinant SNPs did not affect the topology of the maximum likelihood phylogeny. We observed regional patterns in the amount of recombination inferred, and, as expected, recombination appears to be frequent at mobile elements such as the staphylococcal cassette chromosomes (SCC15305RM and SCC15305cap) and vSs15305 (10).
FIG 2 .

Recombination in S. saprophyticus. Recombinant regions in the whole-genome alignment of S. saprophyticus were identified using Gubbins (20). Mobile genetic elements are highlighted in blue on the outer rim. The window with low Tajima’s D and π values is highlighted in pink. Few recombination events are inferred within this region.
Adaptation to a new environment may be facilitated by advantageous mutations that quickly rise in frequency, leaving a characteristic genomic imprint: reduced diversity at the target locus and nearby linked loci (i.e., selective sweep [21, 22]). In order for positive selection to be evident as a local reduction in diversity, there must be sufficient recombination for the target locus to be unlinked from the rest of the genome; for this reason, scans for sweeps have been used primarily for sexually reproducing organisms (23–26). As described above and in prior work (7), we found evidence of frequent recombination among S. saprophyticus isolates. We hypothesized that the transition of S. saprophyticus to the uropathogenic niche may have been driven by selection for one or more mutations that were advantageous in the new environment and that levels of recombination have been sufficient to preserve the signature of a selective sweep at loci under positive selection. We therefore used a sliding window analysis of diversity along the S. saprophyticus alignment as an initial screen for positive selection. We identified a marked regional decrease in nucleotide diversity (π) and in Tajima’s D (TD) that is specific to lineage U (Fig. 3); the TD values for this window were −0.38 and 0.94 for non-lineage U clade P isolates and clade E isolates, respectively. The region with a decreased π/TD ratio corresponded to bp 1760000 to 1820000 in S. saprophyticus ATCC 15305 and had the lowest values of π and TD in the entire alignment. We investigated the sensitivity of our sliding window analyses to sampling by randomly subsampling lineage U isolates to the same size as clade E (n = 10); we found the results to be robust with respect to changes in sampling scheme and size.
FIG 3 .

Sliding window analysis of diversity and neutrality statistics. Population genetic statistics were calculated for lineage U using EggLib (94). Windows were 50 kb in width with a step size of 10 kb. (A) Tajima’s D. (B) π (green) and θ (blue). The lowest values for Tajima’s D and π are found in the same window (1,760,000 to 1,820,000 bp; arrow).
To complement the sliding window analysis and pinpoint candidate variants under positive selection, we used an approach based on allele frequency differences between bacterial isolates from different niches. We calculated Weir and Cockerham’s FST (27) for single nucleotide polymorphisms (SNPs) in the S. saprophyticus genome using human association and nonhuman association to define populations. The region of low π/TD included three nonsynonymous variants in the top 0.05% of the FST values (Table 1). One of these variants was fixed among human-associated isolates in lineage U (position 1811777 in ATCC 15305; FST = 0.48) and distinct from the ancestral (anc) allele found in basal lineages of clade P, including the ancient strain of S. saprophyticus Troy. This suggests that the variant may have been important in adaptation to the human urinary tract. To assess the significance of the FST value for this variant, we performed permutation analyses by randomly assigning isolates as human associated, and we did not achieve FST values higher than 0.28 in 100 permutations.
TABLE 1 .
Single nucleotide polymorphisms with FST values in the top 0.05% between 1,760,000 and 1,820,000 bp in ATCC 15305
| Position | Frequency in human- associated isolates |
Frequency in non-human- associated isolates |
FST value | Type |
|---|---|---|---|---|
| 1772616 | 0.72 | 0.16 | 0.5 | Nonsynonymous |
| 1797190 | 0.9 | 0.37 | 0.48 | Synonymous |
| 1808274 | 0.8 | 0.21 | 0.52 | Synonymous |
| 1811585 | 0.72 | 0.16 | 0.5 | Synonymous |
| 1811777 | 0.9 | 0.37 | 0.48 | Nonsynonymous |
| 1813204 | 0.74 | 0.16 | 0.53 | Synonymous |
| 1816895 | 0.77 | 0.05 | 0.71 | Nonsynonymous |
| 1818150 | 0.77 | 0.16 | 0.56 | Intergenic |
| 1818151 | 0.77 | 0.16 | 0.56 | Intergenic |
| 1818156 | 0.77 | 0.16 | 0.56 | Intergenic |
Selective sweeps may be evident as a longer-than-expected haplotype block, since neutral variants linked to the adaptive mutation also sweep to high frequencies (28). Given the evidence suggesting that there was a selective sweep at this locus, we used haplotype-based statistics to test for such a signature in the S. saprophyticus alignment. Haplotype-based methods are hypothesized to not be applicable to bacteria due to differences between crossing-over and bacterial patterns of recombination (29), but the methods had not been tested in a scenario akin to a classical sweep, in which local changes in diversity and in the site frequency spectrum (SFS) have been observed. We found that the variant at position 181177 did show a signature of a sweep using the extended haplotype homozygosity (EHH) statistic (28) (Fig. 4). However, the variant did not have an extreme value of nSL, which compares haplotype homozygosity for ancestral and derived alleles (30), after normalization by the allele frequency.
FIG 4 .
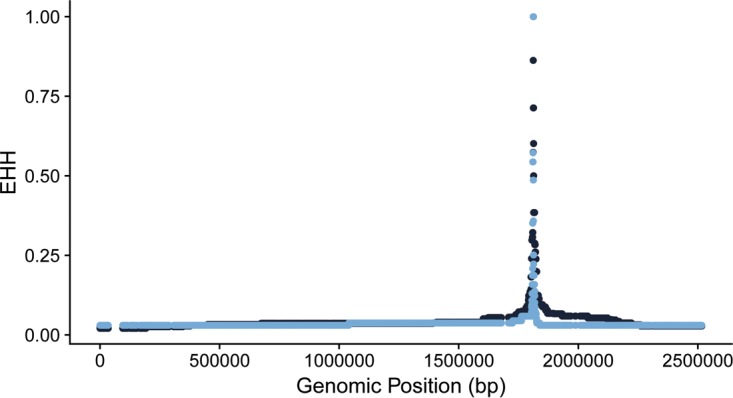
Extended haplotype homozygosity (EHH) of a single nucleotide polymorphism at position 1811777. EHH values for the ancestral allele are in light blue. EHH values for the derived allele are in dark blue.
The variant of interest (aas_2206A>C) causes a threonine-to-proline change in the amino acid sequence of Aas, a bifunctional autolysin with a fibronectin binding domain (Fig. 5) (31). There are 8 additional nonsynonymous polymorphisms in the fibronectin binding domain; however, none are as highly associated with human-pathogenic isolates. Adhesins such as Aas are important in the pathogenesis of S. saprophyticus urinary tract infections, and the gene encoding Aas has been previously implicated as a virulence factor (9, 31, 32).
FIG 5 .

Nonsynonymous variant in Aas fibronectin binding repeat. (Top) Domains of Aas protein (adapted from reference 31. R1ab is the peptide used in the fibronectin and thrombospondin binding experiments. (Bottom) Alignment of a portion of R1 showing amino acid sequences in Aas from selected S. saprophyticus strains. Amino acids are colored based on their propensity to form beta strands (light green, high propensity; light blue, low propensity). The alignment visualization was created in JalView.
The Aas variant is in a region known to bind fibronectin (Fig. 5) (31) and could be under selection because it affects adhesion to this host protein. We used enzyme-linked immunosorbent assays (ELISAs) to investigate potential effects of aas_2206A>C on binding to fibronectin and thrombospondin-1, which binds to this region of the homologous AtlE amidase from Staphylococcus epidermidis (33). Staphylococcal autolysins contains 3 C-terminal repeats (R1 to R3), which can each be divided into two subunits (a and b) based on structural information (34). We confirmed that Aas R1ab binds fibronectin and discovered that it also binds thrombospondin (Fig. 6); there was no detectable difference between the ancestral and derived R1ab alleles in binding to fibronectin or thrombospondin (human or bovine).
FIG 6 .
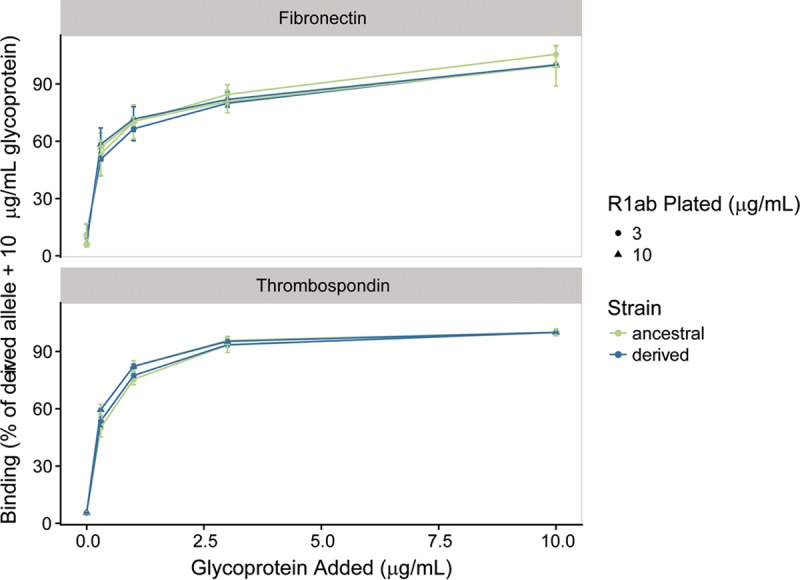
Fibronectin and thrombospondin binding to human-associated and ancestral strain Aas R1ab. ELISAs detecting the binding of soluble human fibronectin and thrombospondin to plates coated with Aas R1ab at 3 and 10 µg/ml were performed. Results have been normalized to the percentage of binding of 10 µg/ml glycoprotein to human-associated strain R1ab. Human and bovine fibronectin and human thrombospondin bound to the two constructs equally well.
Interestingly, we observed several instances of recombination of the aas variant. In each case, the recombination event reinforced the association of the derived allele with human infection. Two of the non-human-associated bacterial isolates in lineage U—an isolate from a pig and a second from cheese rind—had evidence of a recombination event at the aas locus resulting in acquisition of the ancestral allele. Conversely, one of the human UTI isolates in clade E (for which the ancestral allele is otherwise fixed) acquired the derived aas variant.
Several human pathogens appear to have undergone recent population expansion (35–38). We wondered whether the uropathogenic lineage of S. saprophyticus might also have undergone a recent change in its effective size. The value for the genome-wide estimate of TD for lineage U was negative (−0.58), which is consistent with population expansion. We used the methods implemented in ∂a∂i (39) to identify the demographic model that best fit the observed synonymous SFS of lineage U (Fig. 7).
FIG 7 .
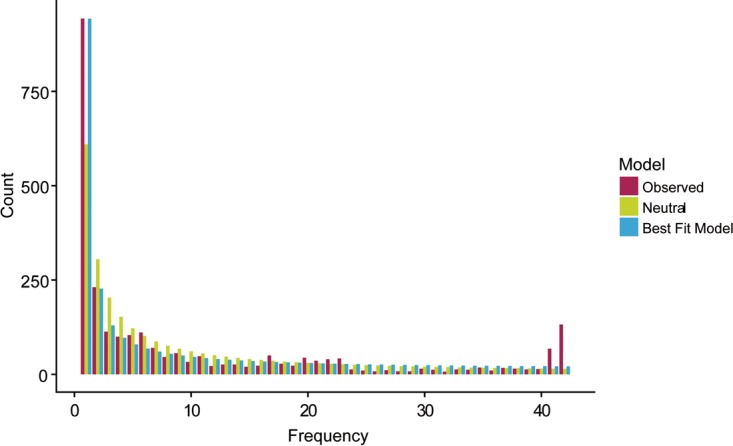
Site frequency spectrum of lineage U. The ancient genome (Troy) was used as the outgroup to determine the ancestral state. Synonymous, nonsynonymous, and intergenic sites were identified with SnpEff (98). The observed synonymous SFS contained an excess of singletons and high-frequency-derived variants. Both the observed SFS and the SFS predicted by the best-fitting model have an excess of singletons compared to the SFS predicted by the standard neutral model with no population size change.
The synonymous SFS showed an unexpected excess of high-frequency-derived alleles, which we hypothesized were the result of gene flow from populations with ancestral variants. Within-population recombination has been shown to have no effect on SFS-based methods of demographic inference in bacteria (40). However, external sources of recombination were not modeled in previous studies. We used SimBac (41) to simulate bacterial populations with a range of internal and external recombination rates. Similarly to previous studies, we found that within-population recombination had no effect on the value of Tajima’s D. However, we did find that recombinant tracts from external sources resulted in positive values of Tajima’s D (Fig. 8). Positive values of Tajima’s D are also associated with population bottlenecks and balancing selection.
FIG 8 .

Effects of internal and external recombination on Tajima’s D values. Bacterial populations with a range of recombination rates were simulated with SimBac. (A) Tajima's D values from simulations of internal recombination rates ranging from 0 to 0.03 in the absence of external recombination. (B) Tajima’s D values from simulations performed with an internal recombination rate of 0.003 (r/m = 1) and external recombination rates ranging from 0 to 0.003. Points are filled according to the upper limit of diversity in external recombinant fragments.
We used fastGEAR (42) to identify recombinant tracts that originated outside lineage U, and these sites were removed from the analysis prior to demographic inference. We compared five demographic models (constant size, instantaneous population size change, exponential population size change, instantaneous population size change followed by exponential population size change, and two instantaneous population size changes; Fig. 9) and used bootstrapping to estimate the uncertainty of the parameters and to adjust the composite likelihoods using the Godambe Information Matrix implemented in ∂a∂i (43). We found significant evidence of expansion in all models (Table 2). The best-fitting model was an instantaneous contraction followed by an instantaneous expansion, in which the population underwent a tight bottleneck followed by a 15-fold expansion without recovering to its ancestral (anc) size (ν, Ne/Nanc; τ, number of generations/Nanc; νA, 2.9 × 10−2; νB, 4.5 × 10−1; τA, 1.2 × 10−1; τB, 3.1 × 10−3).
FIG 9 .

Cartoon of fitted demographic models. The observed synonymous SFS was fitted to 5 demographic models, including constant size, instantaneous population size change, exponential population size change, instantaneous population size change followed by exponential population size change, and two instantaneous population size changes. The parameters for the instantaneous and exponential models are the magnitude of the population size change (ν = Ne/Nancestral) and the timing of the change (τ = number of generations/Nancestral). For the models with two population size changes, magnitudes are reported as νA = Nb/Nancestral and νb = Ne/Nancestral.
TABLE 2 .
Results of demographic inferencea
| Model | Optimized parameters (SD) | Log likelihood |
P value (comparison model) |
|---|---|---|---|
| Constant size | −562 | ||
| Instantaneous change | ν, 9.9 × 104 (6.0 × 104); τ, 4.4 × 10−2 (4.7 × 10−3) | −455 | 0.0 (constant size) |
| Exponential change | ν, 9.7 × 104 (9.8 × 104); τ, 4.1 × 10−2 (4.9 × 10−3) | −455 | 0.0 (constant size) |
| Instantaneous change followed by exponential change |
νA, 2.1 × 10−2 (2.9 × 10−2); νB, 1.1 (3.9 × 10−2); τ, 1.3 × 10−1 (1.6 × 10−1) |
−404 | 1.6 × 10−4 (exponential change) |
| Two instantaneous size changes | νA, 2.9 × 10−2 (1.2 × 10−2); νB, 4.5 × 10−1 (2.1 × 10−1); τA, 1.2 × 10−1 (3.4 × 10−2); τB, 3.1 × 10−3 (5.3 × 10−3) |
−393 | 4.6 × 10−7 (instantaneous change) |
ν = Ne/Nancestral; τ = number of generations/Nancestral.
Recombination and positive selection are known to confound the inference of bacterial demography (40), so we used simulations to investigate their effects on our demographic inference performed for uropathogenic S. saprophyticus. We used SFS_CODE (44) to simulate positive selection (with a range of recombination rates) and to evaluate its effects on the accuracy of demographic inference with ∂a∂i. The method implemented in ∂a∂i relies on inference from the synonymous SFS, but it is possible for synonymous variation to be affected by selection, particularly at low rates of recombination (40, 45). Neutral simulations with gene conversion did not affect demographic inference. We did find that positive selection can affect the synonymous SFS, resulting in inference of population size changes. In simulations of positive selection in a population of constant size, we found the spurious inference to be a bottleneck rather than an expansion. This suggests that the observed synonymous SFS of lineage U has been affected both by positive selection and by demographic expansion.
DISCUSSION
A central issue in the population biology of infectious diseases is how and why pathogenic traits emerge in microbes. Addressing this issue is important for understanding novel disease emergence and for identifying the genetic basis of virulence. Here we present evidence suggesting that a mutation in the S. saprophyticus aas gene, which binds host matrix proteins, is under positive selection and has enabled the emergence and spread of a human-pathogenic, UTI-associated lineage of this bacterium.
S. saprophyticus is familiar to medical microbiologists and clinicians as a common cause of UTI (46), which are associated with significant morbidity, economic costs, and severe complications (4). Despite its strong association with UTI in humans, S. saprophyticus can also be isolated from diverse environments, including livestock, food and food processing plants, and the environment (47, 48). Our previous research suggested that pathogenicity to humans is a derived trait in the species (7).
That pattern was replicated here, where phylogenetic analyses linked human UTI with two lineages of S. saprophyticus that are nested among isolates from diverse, nonhuman niches (i.e., the free-living and food- and animal-associated niches). The aas mutation arose in lineage U, which contains most of the UTI isolates. Two lineages are basal to lineage U; one is bovine associated, and the other contains an ancient bacterial sequence from a pregnancy-related infection in Late Byzantine Troy. The Troy bacterium has the ancestral, bovine-associated aas allele, and we have previously hypothesized (7) that this lineage could be associated with human infections in regions where humans have close contacts with animals—e.g., sharing living quarters with livestock, as they did at Troy during that time.
A second cluster of UTI isolates appears in clade E. One isolate has acquired the derived aas allele, which parallels our finding that two nonhuman isolates in lineage U acquired the ancestral variant; all of the recombination events that we observed at this locus reinforced the idea of an association between aas_2206A>C and human infection.
Several UTI isolates in clade E do not have the derived aas allele, and the clustering of UTI isolates suggests there may be a distinct adaptive path to virulence in this clade. Larger and more-comprehensive samples will be needed to investigate this hypothesis and to identify the factors shaping the separation of clades P and E.
The aas mutation has characteristics associated with a classical selective sweep driven by positive selection, namely, a regional reduction in diversity (21) and in Tajima’s D (22, 49). With the exception of the interesting allelic replacements noted above, there was also relatively little recombination at this locus, consistent with it being functionally important. To our knowledge, this is the first description of a single nucleotide sweep in a bacterium.
Depending on the strength of selection and the recombination rate, positive selection in bacteria has been observed to affect the entire genome, resulting in clonal replacements, or to affect only specific regions of the genome (50). For example, multiple clonal replacements have occurred in Shigella sonnei populations in Vietnam due to acquisition of resistance to antimicrobials and environmental stress (51). Recurrent clonal replacements have also been observed within single hosts during chronic infection of cystic fibrosis patients by Pseudomonas aeruginosa (52). Environmental bacterial populations can also be subject to clonal replacements; a metagenomic time course study of Trout Bog found evidence of clonal replacement occurring in natural bacterial populations but not gene- or region-specific sweeps (53). However, large regions of low diversity were also observed, suggesting that gene-specific selective sweeps had occurred prior to the start of the study. Shapiro et al. identified genomic loci that differentiated Vibrio cyclitrophicus isolates that were associated with distinct niches but that had limited diversity within niches; they concluded that differentiation of these populations had been enabled by recombination events that reinforced the association of alleles with the niche in which they were advantageous (54).
The aas_2206A>C mutation is among the genetic variants that differentiate bacteria associated with human-pathogenic niches from those associated with other niches (i.e., it is an FST outlier). SNPs associated with specific clinical phenotypes in the pathogen Streptococcus pyogenes were described recently (55), which is consistent with our finding that clinical phenotypes can represent distinct niche spaces preferentially occupied by subpopulations of bacteria. There is also precedent for a single nucleotide polymorphism to affect host tropism of bacteria (56).
In sexually reproducing organisms, haplotype-based statistics are frequently used to identify selective sweeps because positively selected alleles also increase the frequency of nearby linked loci faster than recombination can disrupt linkage, producing longer haplotypes for selected alleles (28, 57). We found that aas_2206A>C had a longer haplotype than the ancestral variant, but this difference was not extreme relative to the results seen with other regions of the genome (assessed with the nSL statistic). Haplotype-based statistics have been found to perform poorly in analyses of purebred dogs, where linkage across the genome is high (58). Relatively low levels of recombination may also contribute to a lack of sensitivity when haplotype-based detection methods are applied to bacteria; linkage of sites is also likely to be disrupted in a less predictable way by bacterial gene conversion than by crossing over (29). Based on our findings, we conclude that screening for regional decreases in diversity and distortions of the SFS (i.e., sliding window analyses) and identification of genetic variants with extreme differences in frequency between niches can be useful in identifying candidate sites of positive selection in bacteria.
S. saprophyticus encodes a number of adhesins, including UafA, UafB, SdrI, and Aas. UafA and Aas are found in all isolates, suggesting that they play important roles in the diverse niches occupied by S. saprophyticus. Aas has autolytic, fibronectin binding, and hemagluttinating functions (9, 31, 32, 59). We identified a single, nonsynonymous polymorphism as a target of selection in the fibronectin binding repeats of Aas. This variant is predicted to affect the repeat’s structure, as proline has a more rigid structure than other amino acids. Adhesins are plausible candidates for adaptation to the uropathogenic niche, as they are known to be important virulence factors in pathogens causing urinary tract infections (60). Fibronectin binding proteins, including Aas, have been identified as virulence factors in S. saprophyticus and Enterococcus faecalis (32, 61, 62). Adhesion to the uroepithelium is essential for uropathogens to establish themselves in the bladder, where they are subject to strong shear stress (63): we hypothesize that S. saprophyticus strains with the derived aas variant are better able to colonize the human bladder.
Invasion of the human urinary tract may provide a fitness advantage by allowing relative enrichment of S. saprophyticus in a site with little competition from other bacterial species and by providing a mechanism of dispersal in the environment. In analyses of selection in E. coli, another bacterium occupying diverse niches, residues in the FimH adhesin were found to be subject to positive selection in uropathogenic strains (64–66). FimH binds mannose, providing protection from shear stress through a catch bond mechanism (67). Interestingly, the vascular adherence and resistance to shear stress of Borrelia burgdorferi were recently found to be enabled by interactions between a bacterial adhesin and host fibronectin that also use a catch bond mechanism (68). There are also precedents in Staphylococcus aureus for polymorphisms in bacterial fibronectin-binding adhesins to affect the strength of binding and for these polymorphisms to associate with specific clinical phenotypes (69).
Further experiments are needed to investigate the effects of variation in Aas on S. saprophyticus biology. In our preliminary investigations of binding using ELISAs of recombinant bacterial peptides, we did not detect differences between ancestral and derived alleles in binding of the R1a1b repeat with respect to fibronectin. The variant could still affect fibronectin binding by altering the conformation of the protein in a manner analogous to that seen with FimH in E. coli (66). It is also possible that variants in the peptide affect binding under specific conditions that we did not test. Another possibility is that the variant affects autolysis or other as-yet-undescribed functions of Aas. The roles of adhesins and other virulence factors in the colonization by S. saprophyticus of niches in livestock and the environment are also interesting topics for further study.
Our demographic analysis of the uropathogenic lineage of S. saprophyticus showed evidence of a population bottleneck and subsequent expansion. Bottlenecks and expansion of drug-resistant clones have previously been shown to affect the population structure of Streptococcus agalactiae (70), demonstrating the effects of positive selection on the demographic trajectories of bacterial subpopulations. However, previous work has also shown that selection—and recombination—can produce spurious results from demographic inference in bacteria (40, 71). We used an SFS-based method to reconstruct the demographic history of S. saprophyticus; the accuracy of demographic inference using these methods has been shown to be unaffected by within-population recombination (40), and this was confirmed in our analyses of simulated data. We found that recombination from external sources may result in an excess of intermediate frequency variants, which is also a signature of population bottlenecks, so we masked externally imported sites. However, the frequency of synonymous variants could still be affected by selection on linked nonsynonymous sites, including the selective sweep in aas that we have described. We performed simulations to address these potential confounders and to aid in the interpretation of our demographic inferences. Simulation of a single site under conditions of positive selection resulted in the inference of a bottleneck (Ne/Na = 0.01 to 0.42), indicating that, at the recombination rates that we simulated, diversity was lost from neutrally evolving sites due to their linkage to the site under selection. In inferences from our observed data, a bottleneck was followed by a 15-fold expansion, suggesting that lineage U has undergone both a selective sweep and demographic expansion.
Here we have described an adaptation of S. saprophyticus that may have enabled its expansion into a human-pathogenic niche. Mutation of a single nucleotide within the aas adhesin appears to have driven a selective sweep, and allele frequency differences at the locus are consistent with niche-specific adaptation. Lateral gene transfer events in aas reinforced the association of the positively selected allele with human infection. These results provide new insights into the emergence of virulence in bacteria and outline an approach for discovering the molecular basis of adaptation to the human-pathogenic niche.
MATERIALS AND METHODS
DNA extraction.
After overnight growth in tryptic soy broth (TSB) at 37°C in a shaking incubator, cultures were pelleted and resuspended in 140 μl of Tris-EDTA (TE) buffer. Cells were incubated overnight with 50 units of mutanolysin. We used a MasterPure Gram-positive DNA purification kit (EpiCentre) for DNA extraction. For DNA precipitation, we used 1 ml 70% ethanol and centrifugation at 4°C for 10 min. We additionally used a SpeedVac for 10 min to ensure that pellets were dry before resuspending the pellet in 50 µl of water.
Library preparation and sequencing.
For SSC01, SSC02, and SSC03, library preparation was performed using a modified Nextera protocol as described by Baym et al. (72) with a reconditioning PCR with fresh primers and polymerase for an additional 5 PCR cycles to minimize chimeras and two-step bead-based size selection with a target fragment size of 650 bp and sequencing on an Illumina HiSeq 2500 sequencer (paired end; 150 bp). For 43, SSC04, SCC05, and SSMast, DNA was submitted to the University of Wisconsin—Madison Biotechnology Center for library preparation and was prepared using a TruSeq Nano DNA LT Library Prep kit (Illumina Inc., San Diego, CA) with minor modifications. A maximum of 200 ng of each sample was sheared using a Covaris M220 Ultrasonicator (Covaris Inc., Woburn, MA). Sheared samples were size selected for an average insertion size of 550 bp using Spri bead-based size exclusion. The quality and quantity of the finished libraries were assessed using an Agilent DNA High Sensitivity chip (Agilent Technologies, Santa Clara, CA) and an Qubit dsDNA HS assay kit, respectively. Libraries were standardized to 2 μM. Paired-end, 150-bp sequencing was performed using v2 SBS chemistry on an Illumina MiSeq sequencer. Images were analyzed using the Illumina Pipeline, version 1.8.2.
Reference guided mapping.
We mapped reads to ATCC 15305 via the use of a pipeline (available at https://github.com/pepperell-lab/RGAPepPipe). Briefly, read quality was assessed and reads were trimmed with TrimGalore! v 0.4.0 (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore), which runs both FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) and cutadapt. Reads were mapped using BWA-MEM v 0.7.12 (73) and sorted using Samtools v 1.2 (74). We used Picard v 1.138 (http://broadinstitute.github.io/picard/) to add read group information and removed duplicates. Reads were locally realigned using GATK v 2.8.1 (75). We identified variants using Pilon v 1.16 (76) (minimum read depth, 10; minimum mapping quality, 40; minimum base quality, 20).
Assembly.
We used the iMetAMOS pipeline for de novo assembly (77). We chose to compare assemblies from SPAdes (78), MaSurCA (79), and Velvet (80). KmerGenie (81) was used to select kmer sizes for assembly. iMetAMOS uses FastQC, QUAST (82), REAPR (83), LAP (84), ALE (85), FreeBayes (86), and CGAL (87) to evaluate the quality of reads and assemblies. We also used Kraken (88) to detect potential contamination in sequence data. For all newly assembled isolates (43, SSC01-05, SSMast), the SPAdes assembly was the highest quality. Assembly statistics are reported in Table S2 in the supplemental material.
Assembly statistics for S. saprophyticus genomes. Download TABLE S2, TXT file, 0.01 MB (12.6KB, txt) .
Copyright © 2017 Mortimer et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Annotation and gene content analyses.
We annotated the de novo assemblies using Prokka v 1.11 (89) and used Roary (90) to identify orthologous genes in the core and accessory genomes. To look for associations between accessory gene content and human association, we used Scoary (19). For the analysis, we used human association as the trait of interest, and we adjusted the P value for multiple comparisons using the Bonferroni method.
Alignment.
When short-read data for reference guided mapping were unavailable, whole-genome alignment of genomes to ATCC 15305 was performed using Mugsy v 2.3 (91). Repetitive regions in the reference genome that were greater than 100 bp in size were identified using nucmer, and these regions were masked in the alignment used in downstream analyses.
Maximum likelihood phylogenetic analysis.
Maximum likelihood phylogenetic trees were inferred using RAxML 8.0.6 (92). We used the GTRGAMMA substitution model and performed bootstrapping using the autoMR convergence criteria. Tree visualizations were created in ggtree (93).
Population genetics statistics.
To calculate π and Tajima’s D, we used EggLib v 2.1.10 (94), a Python package for population genetic analyses. A script to perform the sliding window analysis is available at https://github.com/tatumdmortimer/popgen-stats/blob/master/slidingWindowStats.py. We used vcflib (https://github.com/vcflib/vcflib) to calculate FST and EHH and selscan v 1.1.0b (95) to calculate nSL.
Recombination analyses.
To identify recombinant regions in the S. saprophyticus alignment, we used Gubbins v 2.1.0 (20). fastGEAR (42) was used with the recommended input specifications to identify recombination events between major lineages of S. saprophyticus. We used Circos (96) for visualization of recombinant tracts.
Site frequency spectrum.
We used SNP-sites v 2.0.3 (97) to convert the alignment of S. saprophyticus isolates to a multisample variant call format (VCF). SnpEff v 4.1j (98) was used to annotate variants in this VCF as synonymous, nonsynonymous, or intergenic. Using the Troy genome as an outgroup, we calculated an unfolded site frequency spectrum (SFS) for lineage U for each category of sites. To reduce the impact of lateral gene transfer on the SFS, we removed sites where the origin was outside lineage U based on results of fastGEAR analysis.
Ancestral reconstruction of aas_2206A>C.
We used TreeTime (99) to reconstruct the evolutionary history of the variant in aas using the maximum likelihood phylogeny inferred using RAxML.
Demography.
We performed demographic inference with the synonymous SFS using ∂a∂i (39). The models tested were the standard neutral model and the expansion and exponential-growth models. The parameters ν (Ne/Na) and τ (time scaled by 2) were optimized for both the expansion and exponential-growth models. The statistical significance of results of comparisons of data from the expansion and exponential-growth models to data from the standard neutral model was evaluated using a likelihood ratio test. The scripts used to perform this analysis are available at https://github.com/tatumdmortimer/popgen-stats.
Simulations.
Simulations were performed in SimBac (41) to evaluate the effect of external recombination on the SFS. Populations were simulated with sample size and θ equivalent to those seen with our sample of lineage U (n = 44; θ = 0.003). The length of internal recombinant tracts was 6,500 bp (median of Gubbins output), and the length of external recombination events was 3,000 bp (median of fastGEAR output). Internal recombination was simulated at rates ranging from 0 to 0.03 (r/m = 10). External recombination was simulated at rates ranging from 0 to 0.003. The lower bound of difference for external recombination was 0, and the upper bound was simulated at values ranging from 0.25 to 1.0. Simulations were performed in SFS_CODE (release date, 10 September 2015) (44) to evaluate the power of ∂a∂i to accurately estimate demographic parameters in the presence of gene conversion and selection. We simulated a locus of length of 100 kb with theta of 0.003, a gene conversion tract length of 1,345 bp, and a range of recombination/mutation ratios (0.0002 to 2.0). In addition to neutral simulations with gene conversion, we also performed simulations with a single site under conditions of selection (γ = 10 to 1,000) with the same parameters as those used for the neutral simulations.
Expression of R1ab.
Human-associated and ancestral strain R1ab bacteria were cloned into expression vector pET-ELMER (102), transformed into BL21(DE3) cells (EMD, Gibbstown, NJ) for expression, and induced with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside). Bacteria were lysed in lysis buffer consisting of 100 mM NaH2PO4, 10 mM Tris, 8 M urea, 1 mM β-mercaptoethanol, and 5 mM imidazole (pH 8.0). The cleared lysate was incubated overnight with nickel-nitrilotriacetic acid agarose (Qiagen), washed, and eluted in lysis buffer (pH 7.0)–300 mM imidazole.
ELISA.
Antigen was diluted to 10 µg/ml in Tris-buffered saline (TBS; 10 mM Tris, 150 mM NaCl, pH 7.4) and used to coat 96-well microtiter plates (Costar 3590 high binding; Corning Inc., Corning, NY) with 50 µl per well for 16 h at 4°C. The plates were blocked with 1% bovine serum albumin (BSA)–TBS–0.05% Tween 20 (TBST) for 1 h. After washing with TBST was performed three times, purified plasma fibronectin (100) or platelet-derived thrombospondin-1 (101) diluted to 10, 3, 1, or 0.3 µg/ml in TBST–0.1% BSA was added to the plates and the reaction mixture was incubated for 2 h. Plates were washed four times with TBST. Rabbit anti-fibronectin and rabbit anti-thrombospondin antibodies diluted in TBST–0.1% BSA were added to the appropriate wells, and the reaction mixtures were incubated for 1 h. Plates were washed four times with TBST. Peroxidase-conjugated secondary antibody was incubated with the plates for 1 h. Plates were washed four times with TBST, and 50 µl of SureBlue TMB peroxidase substrate (KLP) per well was added to each well. Color development was monitored for 10 to 30 min, and 50 µl of TMB stop solution (KLP) was added, followed by measurement of absorbance at 450 nm.
Accession number(s).
The new accession numbers for the data from the S. saprophyticus isolates are provided in Table S1 in the supplemental material.
ACKNOWLEDGMENTS
We thank Andrew Kitchen (University of Iowa), Jeniel Nett (University of Wisconsin—Madison [UW-Madison]), J. D. Sauer (UW-Madison), and Rod Welch (UW-Madison) for their helpful input on this study. We also thank the University of Wisconsin Biotechnology Center DNA Sequencing Facility for providing library preparation and sequencing facilities and services.
This material is based on work supported by the National Science Foundation Graduate Research Fellowship Program under grant no. DGE-1256259 to T.D.M. and M.B.O. Any opinions, findings, and conclusions or recommendations expressed in this material are ours and do not necessarily reflect the views of the National Science Foundation. T.D.M. is also supported by a National Institutes of Health National Research Service award (T32 GM07215). C.S.P. is supported by the National Institutes of Health (R01AI113287).
REFERENCES
- 1.Stamm WE, Norrby SR. 2001. Urinary tract infections: disease panorama and challenges. J Infect Dis 183:S1–S4. doi: 10.1086/318850. [DOI] [PubMed] [Google Scholar]
- 2.Ronald AR, Nicolle LE, Stamm E, Krieger J, Warren J, Schaeffer A, Naber KG, Hooton TM, Johnson J, Chambers S, Andriole V. 2001. Urinary tract infection in adults: research priorities and strategies. Int J Antimicrob Agents 17:343–348. doi: 10.1016/S0924-8579(01)00303-X. [DOI] [PubMed] [Google Scholar]
- 3.Barber AE, Norton JP, Spivak AM, Mulvey MA. 2013. Urinary tract infections: current and emerging management strategies. Clin Infect Dis 57:719–724. doi: 10.1093/cid/cit284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Foxman B. 2014. Urinary tract infection syndromes: occurrence, recurrence, bacteriology, risk factors, and disease burden. Infect Dis Clin North Am 28:1–13. doi: 10.1016/j.idc.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 5.Wallmark G, Arremark I, Telander B. 1978. Staphylococcus saprophyticus: a frequent cause of acute urinary tract infection among female outpatients. J Infect Dis 138:791–797. doi: 10.1093/infdis/138.6.791. [DOI] [PubMed] [Google Scholar]
- 6.Kahlmeter G; ECO.SENS . 2003. An international survey of the antimicrobial susceptibility of pathogens from uncomplicated urinary tract infections: the ECO.SENS project. J Antimicrob Chemother 51:69–76. doi: 10.1093/jac/dkg028. [DOI] [PubMed] [Google Scholar]
- 7.Devault AM, Mortimer TD, Kitchen A, Kiesewetter H, Enk JM, Golding GB, Southon J, Kuch M, Duggan AT, Aylward W, Gardner SN, Allen JE, King AM, Wright G, Kuroda M, Kato K, Briggs DE, Fornaciari G, Holmes EC, Poinar HN, Pepperell CS. 2017. A molecular portrait of maternal sepsis from Byzantine Troy. eLife 6:e20983. doi: 10.7554/eLife.20983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gatermann S, John J, Marre R. 1989. Staphylococcus saprophyticus urease: characterization and contribution to uropathogenicity in unobstructed urinary tract infection of rats. Infect Immun 57:110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meyer HG, Wengler-Becker U, Gatermann SG. 1996. The hemagglutinin of Staphylococcus saprophyticus is a major adhesin for uroepithelial cells. Infect Immun 64:3893–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuroda M, Yamashita A, Hirakawa H, Kumano M, Morikawa K, Higashide M, Maruyama A, Inose Y, Matoba K, Toh H, Kuhara S, Hattori M, Ohta T. 2005. Whole genome sequence of Staphylococcus saprophyticus reveals the pathogenesis of uncomplicated urinary tract infection. Proc Natl Acad Sci U S A 102:13272–13277. doi: 10.1073/pnas.0502950102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kleine B, Gatermann S, Sakinc T. 2010. Genotypic and phenotypic variation among Staphylococcus saprophyticus from human and animal isolates. BMC Res Notes 3:163. doi: 10.1186/1756-0500-3-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nelson EJ, Harris JB, Morris JG, Calderwood SB, Camilli A. 2009. Cholera transmission: the host, pathogen and bacteriophage dynamic. Nat Rev Microbiol 7:693–702. doi: 10.1038/nrmicro2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boucher Y, Orata FD, Alam M. 2015. The out-of-the-delta hypothesis: dense human populations in low-lying river deltas served as agents for the evolution of a deadly pathogen. Front Microbiol 6:1120. doi: 10.3389/fmicb.2015.01120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Veyrier FJ, Dufort A, Behr MA. 2011. The rise and fall of the Mycobacterium tuberculosis genome. Trends Microbiol 19:156–161. doi: 10.1016/j.tim.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 15.McNally A, Thomson NR, Reuter S, Wren BW. 2016. ‘Add, stir and reduce’: Yersinia spp. as model bacteria for pathogen evolution. Nat Rev Microbiol 14:177–190. doi: 10.1038/nrmicro.2015.29. [DOI] [PubMed] [Google Scholar]
- 16.Larsson P, Elfsmark D, Svensson K, Wikström P, Forsman M, Brettin T, Keim P, Johansson A. 2009. Molecular evolutionary consequences of niche restriction in Francisella tularensis, a facultative intracellular pathogen. PLoS Pathog 5:e1000472. doi: 10.1371/journal.ppat.1000472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lebreton F, van Schaik W, McGuire AM, Godfrey P, Griggs A, Mazumdar V, Corander J, Cheng L, Saif S, Young S, Zeng Q, Wortman J, Birren B, Willems RJL, Earl AM, Gilmore MS. 2013. Emergence of epidemic multidrug-resistant Enterococcus faecium from animal and commensal strains. mBio 4:e00534-13. doi: 10.1128/mBio.00534-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spoor LE, Richardson E, Richards AC, Wilson GJ, Mendonca C, Gupta RK, McAdam PR, Nutbeam-Tuffs S, Black NS, O’Gara JP, Lee CY, Corander J, Fitzgerald JR. 2015. Recombination-mediated remodelling of host-pathogen interactions during Staphylococcus aureus niche adaptation. Microb Genom 1:e000036. doi: 10.1099/mgen.0.000036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brynildsrud O, Bohlin J, Scheffer L, Eldholm V. 2016. Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary. Genome Biol 17:238. doi: 10.1186/s13059-016-1108-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Croucher NJ, Page AJ, Connor TR, Delaney AJ, Keane JA, Bentley SD, Parkhill J, Harris SR. 2015. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res 43:e15. doi: 10.1093/nar/gku1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith JM, Haigh J. 1974. The hitch-hiking effect of a favourable gene. Genet Res 23:23–35. doi: 10.1017/S0016672300014634. [DOI] [PubMed] [Google Scholar]
- 22.Braverman JM, Hudson RR, Kaplan NL, Langley CH, Stephan W. 1995. The hitchhiking effect on the site frequency spectrum of DNA polymorphisms. Genetics 140:783–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harr B, Kauer M, Schlötterer C. 2002. Hitchhiking mapping: a population-based fine-mapping strategy for adaptive mutations in Drosophila melanogaster. Proc Natl Acad Sci U S A 99:12949–12954. doi: 10.1073/pnas.202336899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nair S, Williams JT, Brockman A, Paiphun L, Mayxay M, Newton PN, Guthmann JP, Smithuis FM, Hien TT, White NJ, Nosten F, Anderson TJC. 2003. A selective sweep driven by pyrimethamine treatment in Southeast Asian malaria parasites. Mol Biol Evol 20:1526–1536. doi: 10.1093/molbev/msg162. [DOI] [PubMed] [Google Scholar]
- 25.Wright SI, Gaut BS. 2005. Molecular population genetics and the search for adaptive evolution in plants. Mol Biol Evol 22:506–519. doi: 10.1093/molbev/msi035. [DOI] [PubMed] [Google Scholar]
- 26.de Simoni Gouveia JJ, da Silva MVGB, Paiva SR, de Oliveira SMP. 2014. Identification of selection signatures in livestock species. Genet Mol Biol 37:330–342. doi: 10.1590/S1415-47572014000300004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weir BS, Cockerham CC. 1984. Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 28.Sabeti PC, Reich DE, Higgins JM, Levine HZP, Richter DJ, Schaffner SF, Gabriel SB, Platko JV, Patterson NJ, McDonald GJ, Ackerman HC, Campbell SJ, Altshuler D, Cooper R, Kwiatkowski D, Ward R, Lander ES. 2002. Detecting recent positive selection in the human genome from haplotype structure. Nature 419:832–837. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
- 29.Shapiro BJ. 2014. Signatures of natural selection and ecological differentiation in microbial genomes, p 339–359. In Landry CR, Aubin-Horth N (ed), Ecological genomics. Springer, Dordrecht, Netherlands. [DOI] [PubMed] [Google Scholar]
- 30.Ferrer-Admetlla A, Liang M, Korneliussen T, Nielsen R. 2014. On detecting incomplete soft or hard selective sweeps using haplotype structure. Mol Biol Evol 31:1275–1291. doi: 10.1093/molbev/msu077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hell W, Meyer HG, Gatermann SG. 1998. Cloning of aas, a gene encoding a Staphylococcus saprophyticus surface protein with adhesive and autolytic properties. Mol Microbiol 29:871–881. doi: 10.1046/j.1365-2958.1998.00983.x. [DOI] [PubMed] [Google Scholar]
- 32.Gatermann S, Meyer HG. 1994. Staphylococcus saprophyticus hemagglutinin binds fibronectin. Infect Immun 62:4556–4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kohler TP, Gisch N, Binsker U, Schlag M, Darm K, Völker U, Zähringer U, Hammerschmidt S. 2014. Repeating structures of the major staphylococcal autolysin are essential for the interaction with human thrombospondin 1 and vitronectin. J Biol Chem 289:4070–4082. doi: 10.1074/jbc.M113.521229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zoll S, Schlag M, Shkumatov AV, Rautenberg M, Svergun DI, Götz F, Stehle T. 2012. Ligand-binding properties and conformational dynamics of autolysin repeat domains in staphylococcal cell wall recognition. J Bacteriol 194:3789–3802. doi: 10.1128/JB.00331-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He M, Sebaihia M, Lawley TD, Stabler RA, Dawson LF, Martin MJ, Holt KE, Seth-Smith HMB, Quail MA, Rance R, Brooks K, Churcher C, Harris D, Bentley SD, Burrows C, Clark L, Corton C, Murray V, Rose G, Thurston S, van Tonder A, Walker D, Wren BW, Dougan G, Parkhill J. 2010. Evolutionary dynamics of Clostridium difficile over short and long time scales. Proc Natl Acad Sci U S A 107:7527–7532. doi: 10.1073/pnas.0914322107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nübel U, Dordel J, Kurt K, Strommenger B, Westh H, Shukla SK, Žemličková H, Leblois R, Wirth T, Jombart T, Balloux F, Witte W. 2010. A timescale for evolution, population expansion, and spatial spread of an emerging clone of methicillin-resistant Staphylococcus aureus. PLoS Pathog 6:e1000855. doi: 10.1371/journal.ppat.1000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pepperell CS, Casto AM, Kitchen A, Granka JM, Cornejo OE, Holmes EC, Holmes EC, Birren B, Galagan J, Feldman MW. 2013. The role of selection in shaping diversity of natural M. tuberculosis populations. PLoS Pathog 9:e1003543. doi: 10.1371/journal.ppat.1003543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davies MR, Holden MT, Coupland P, Chen JHK, Venturini C, Barnett TC, Zakour NLB, Tse H, Dougan G, Yuen KY, Walker MJ. 2015. Emergence of scarlet fever Streptococcus pyogenes emm12 clones in Hong Kong is associated with toxin acquisition and multidrug resistance. Nat Genet 47:84–87. doi: 10.1038/ng.3147. [DOI] [PubMed] [Google Scholar]
- 39.Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD. 2009. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet 5:e1000695. doi: 10.1371/journal.pgen.1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lapierre M, Blin C, Lambert A, Achaz G, Rocha EPC. 2016. The impact of selection, gene conversion, and biased sampling on the assessment of microbial demography. Mol Biol Evol 33:1711–1725. doi: 10.1093/molbev/msw048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brown T, Didelot X, Wilson DJ, De Maio N. 2016. SimBac: simulation of whole bacterial genomes with homologous recombination. Microb Genom 2:e000044. doi: 10.1099/mgen.0.000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mostowy R, Croucher NJ, Andam CP, Corander J, Hanage WP, Marttinen P. 2016. Analysis of recent and ancestral recombination reveals high-resolution population structure in Streptococcus pneumoniae. bioRxiv https://doi.org/10.1101/059642. [Google Scholar]
- 43.Coffman AJ, Hsieh PH, Gravel S, Gutenkunst RN. 2016. Computationally efficient composite likelihood statistics for demographic inference. Mol Biol Evol 33:591–593. doi: 10.1093/molbev/msv255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hernandez RD. 2008. A flexible forward simulator for populations subject to selection and demography. Bioinformatics 24:2786–2787. doi: 10.1093/bioinformatics/btn522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neher RA, Hallatschek O. 2013. Genealogies of rapidly adapting populations. Proc Natl Acad Sci U S A 110:437–442. doi: 10.1073/pnas.1213113110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Widerström M, Wiström J, Ferry S, Karlsson C, Monsen T. 2007. Molecular epidemiology of Staphylococcus saprophyticus isolated from women with uncomplicated community-acquired urinary tract infection. J Clin Microbiol 45:1561–1564. doi: 10.1128/JCM.02071-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hedman P, Ringertz O, Lindström M, Olsson K. 1993. The origin of Staphylococcus saprophyticus from cattle and pigs. Scand J Infect Dis 25:57–60. [PubMed] [Google Scholar]
- 48.Kastman EK, Kamelamela N, Norville JW, Cosetta CM, Dutton RJ, Wolfe BE. 2016. Biotic interactions shape the ecological distributions of Staphylococcus species. mBio 7:e01157-16. doi: 10.1128/mBio.01157-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tajima F. 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shapiro BJ, David LA, Friedman J, Alm EJ. 2009. Looking for Darwin’s footprints in the microbial world. Trends Microbiol 17:196–204. doi: 10.1016/j.tim.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 51.Holt KE, Thieu Nga TV, Thanh DP, Vinh H, Kim DW, Vu Tra MP, Campbell JI, Hoang NV, Vinh NT, Minh PV, Thuy CT, Nga TT, Thompson C, Dung TT, Nhu NT, Vinh PV, Tuyet PT, Phuc HL, Lien NT, Phu BD, Ai NT, Tien NM, Dong N, Parry CM, Hien TT, Farrar JJ, Parkhill J, Dougan G, Thomson NR, Baker S. 2013. Tracking the establishment of local endemic populations of an emergent enteric pathogen. Proc Natl Acad Sci U S A 110:17522–17527. doi: 10.1073/pnas.1308632110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Diaz Caballero JD, Clark ST, Coburn B, Zhang Y, Wang PW, Donaldson SL, Tullis DE, Yau YCW, Waters VJ, Hwang DM, Guttman DS. 2015. Selective sweeps and parallel pathoadaptation drive Pseudomonas aeruginosa evolution in the cystic fibrosis lung. mBio 6:e00981-15. doi: 10.1128/mBio.00981-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bendall ML, Stevens SL, Chan LK, Malfatti S, Schwientek P, Tremblay J, Schackwitz W, Martin J, Pati A, Bushnell B, Froula J, Kang D, Tringe SG, Bertilsson S, Moran MA, Shade A, Newton RJ, McMahon KD, Malmstrom RR. 2016. Genome-wide selective sweeps and gene-specific sweeps in natural bacterial populations. ISME J 10:1589–1601. doi: 10.1038/ismej.2015.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shapiro BJ, Friedman J, Cordero OX, Preheim SP, Timberlake SC, Szabó G, Polz MF, Alm EJ. 2012. Population genomics of early events in the ecological differentiation of bacteria. Science 336:48–51. doi: 10.1126/science.1218198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bao YJ, Shapiro BJ, Lee SW, Ploplis VA, Castellino FJ. 2016. Phenotypic differentiation of Streptococcus pyogenes populations is induced by recombination-driven gene-specific sweeps. Sci Rep 6:36644. doi: 10.1038/srep36644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Viana D, Comos M, McAdam PR, Ward MJ, Selva L, Guinane CM, González-Muñoz BM, Tristan A, Foster SJ, Fitzgerald JR, Penadés JR. 2015. A single natural nucleotide mutation alters bacterial pathogen host tropism. Nat Genet 47:361–366. doi: 10.1038/ng.3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vitti JJ, Grossman SR, Sabeti PC. 2013. Detecting natural selection in genomic data. Annu Rev Genet 47:97–120. doi: 10.1146/annurev-genet-111212-133526. [DOI] [PubMed] [Google Scholar]
- 58.Schlamp F, van der Made J, Stambler R, Chesebrough L, Boyko AR, Messer PW. 2016. Evaluating the performance of selection scans to detect selective sweeps in domestic dogs. Mol Ecol 25:342–356. doi: 10.1111/mec.13485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meyer HG, Müthing J, Gatermann SG. 1997. The hemagglutinin of Staphylococcus saprophyticus binds to a protein receptor on sheep erythrocytes. Med Microbiol Immunol 186:37–43. doi: 10.1007/s004300050044. [DOI] [PubMed] [Google Scholar]
- 60.Flores-Mireles AL, Walker JN, Caparon M, Hultgren SJ. 2015. Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat Rev Microbiol 13:269–284. doi: 10.1038/nrmicro3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.King NP, Beatson SA, Totsika M, Ulett GC, Alm RA, Manning PA, Schembri MA. 2011. UafB is a serine-rich repeat adhesin of Staphylococcus saprophyticus that mediates binding to fibronectin, fibrinogen and human uroepithelial cells. Microbiology 157:1161–1175. doi: 10.1099/mic.0.047639-0. [DOI] [PubMed] [Google Scholar]
- 62.Torelli R, Serror P, Bugli F, Paroni Sterbini F, Florio AR, Stringaro A, Colone M, De Carolis E, Martini C, Giard JC, Sanguinetti M, Posteraro B. 2012. The PavA-like fibronectin-binding protein of Enterococcus faecalis, EfbA, is important for virulence in a mouse model of ascending urinary tract infection. J Infect Dis 206:952–960. doi: 10.1093/infdis/jis440. [DOI] [PubMed] [Google Scholar]
- 63.Wright KJ, Hultgren SJ. 2006. Sticky fibers and uropathogenesis: bacterial adhesins in the urinary tract. Future Microbiol 1:75–87. doi: 10.2217/17460913.1.1.75. [DOI] [PubMed] [Google Scholar]
- 64.Chen SL, Hung CS, Xu J, Reigstad CS, Magrini V, Sabo A, Blasiar D, Bieri T, Meyer RR, Ozersky P, Armstrong JR, Fulton RS, Latreille JP, Spieth J, Hooton TM, Mardis ER, Hultgren SJ, Gordon JI. 2006. Identification of genes subject to positive selection in uropathogenic strains of Escherichia coli: a comparative genomics approach. Proc Natl Acad Sci U S A 103:5977–5982. doi: 10.1073/pnas.0600938103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen SL, Hung CS, Pinkner JS, Walker JN, Cusumano CK, Li Z, Bouckaert J, Gordon JI, Hultgren SJ. 2009. Positive selection identifies an in vivo role for FimH during urinary tract infection in addition to mannose binding. Proc Natl Acad Sci U S A 106:22439–22444. doi: 10.1073/pnas.0902179106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schwartz DJ, Kalas V, Pinkner JS, Chen SL, Spaulding CN, Dodson KW, Hultgren SJ. 2013. Positively selected FimH residues enhance virulence during urinary tract infection by altering FimH conformation. Proc Natl Acad Sci U S A 110:15530–15537. doi: 10.1073/pnas.1315203110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yakovenko O, Sharma S, Forero M, Tchesnokova V, Aprikian P, Kidd B, Mach A, Vogel V, Sokurenko E, Thomas WE. 2008. FimH forms catch bonds that are enhanced by mechanical force due to allosteric regulation. J Biol Chem 283:11596–11605. doi: 10.1074/jbc.M707815200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Niddam AF, Ebady R, Bansal A, Koehler A, Hinz B, Moriarty TJ. 2017. Plasma fibronectin stabilizes Borrelia burgdorferi–endothelial interactions under vascular shear stress by a catch-bond mechanism. Proc Natl Acad Sci U S A 114:E3490–E3498. doi: 10.1073/pnas.1615007114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Messina JA, Thaden JT, Sharma-Kuinkel BK, Fowler VG Jr. 2016. Impact of bacterial and human genetic variation on Staphylococcus aureus infections. PLoS Pathog 12:e1005330. doi: 10.1371/journal.ppat.1005330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Da Cunha V, Davies MR, Douarre PE, Rosinski-Chupin I, Margarit I, Spinali S, Perkins T, Lechat P, Dmytruk N, Sauvage E, Ma L, Romi B, Tichit M, Lopez-Sanchez MJ, Descorps-Declere S, Souche E, Buchrieser C, Trieu-Cuot P, Moszer I, Clermont D, Maione D, Bouchier C, McMillan DJ, Parkhill J, Telford JL, Dougan G, Walker MJ; Devani Consortium, Holden MTG, Poyart C, Glaser P. 2014. Streptococcus agalactiae clones infecting humans were selected and fixed through the extensive use of tetracycline. Nat Commun 5:4544. doi: 10.1038/ncomms5544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hedge J, Wilson DJ. 2014. Bacterial phylogenetic reconstruction from whole genomes is robust to recombination but demographic inference is not. mBio 5:e02158-14. doi: 10.1128/mBio.02158-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Baym M, Kryazhimskiy S, Lieberman TD, Chung H, Desai MM, Kishony R. 2015. Inexpensive multiplexed library preparation for megabase-sized genomes. PLoS One 10:e0128036. doi: 10.1371/journal.pone.0128036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv https://arxiv.org/abs/1303.3997. [Google Scholar]
- 74.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup . 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, Earl AM. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Koren S, Treangen TJ, Hill CM, Pop M, Phillippy AM. 2014. Automated ensemble assembly and validation of microbial genomes. BMC Bioinformatics 15:126. doi: 10.1186/1471-2105-15-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zimin AV, Marçais G, Puiu D, Roberts M, Salzberg SL, Yorke JA. 2013. The MaSuRCA genome assembler. Bioinformatics 29:2669–2677. doi: 10.1093/bioinformatics/btt476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chikhi R, Medvedev P. 2014. Informed and automated k-mer size selection for genome assembly. Bioinformatics 30:31–37. doi: 10.1093/bioinformatics/btt310. [DOI] [PubMed] [Google Scholar]
- 82.Gurevich A, Saveliev V, Vyahhi N, Tesler G. 2013. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29:1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hunt M, Kikuchi T, Sanders M, Newbold C, Berriman M, Otto TD. 2013. REAPR: a universal tool for genome assembly evaluation. Genome Biol 14:R47. doi: 10.1186/gb-2013-14-5-r47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ghodsi M, Hill CM, Astrovskaya I, Lin H, Sommer DD, Koren S, Pop M. 2013. De novo likelihood-based measures for comparing genome assemblies. BMC Res Notes 6:334. doi: 10.1186/1756-0500-6-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Clark SC, Egan R, Frazier PI, Wang Z. 2013. ALE: a generic assembly likelihood evaluation framework for assessing the accuracy of genome and metagenome assemblies. Bioinformatics 29:435–443. doi: 10.1093/bioinformatics/bts723. [DOI] [PubMed] [Google Scholar]
- 86.Garrison E, Marth G. 2012. Haplotype-based variant detection from short-read sequencing. ArXiv https://arxiv.org/abs/1207.3907. [Google Scholar]
- 87.Rahman A, Pachter L. 2013. CGAL: computing genome assembly likelihoods. Genome Biol 14:R8. doi: 10.1186/gb-2013-14-1-r8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wood DE, Salzberg SL. 2014. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol 15:R46. doi: 10.1186/gb-2014-15-3-r46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 90.Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MTG, Fookes M, Falush D, Keane JA, Parkhill J. 2015. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31:3691–3693. doi: 10.1093/bioinformatics/btv421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Angiuoli SV, Salzberg SL. 2011. Mugsy: fast multiple alignment of closely related whole genomes. Bioinformatics 27:334–342. doi: 10.1093/bioinformatics/btq665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yu G, Smith DK, Zhu H, Guan Y, Lam TT-Y 22 September 2016. ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol doi: 10.1111/2041-210X.12628. [DOI] [Google Scholar]
- 94.De Mita SD, Siol M. 2012. EggLib: processing, analysis and simulation tools for population genetics and genomics. BMC Genet 13:27. doi: 10.1186/1471-2156-13-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Szpiech ZA, Hernandez RD. 2014. selscan: an efficient multithreaded program to perform EHH-based scans for positive selection. Mol Biol Evol 31:2824–2827. doi: 10.1093/molbev/msu211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res 19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Page AJ, Taylor B, Delaney AJ, Soares J, Seemann T, Keane JA, Harris SR. 2016. SNP-sites: rapid efficient extraction of SNPs from multi-FASTA alignments. Microb Genom 2:e000056. doi: 10.1099/mgen.0.000056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sagulenko P, Puller V, Neher R. 2017. TreeTime: maximum likelihood phylodynamic analysis. bioRxiv https://doi.org/10.1101/153494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Maurer LM, Tomasini-Johansson BR, Mosher DF. 2010. Emerging roles of fibronectin in thrombosis. Thromb Res 125:287–291. doi: 10.1016/j.thromres.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Annis DS, Murphy-Ullrich JE, Mosher DF. 2006. Function-blocking antithrombospondin-1 monoclonal antibodies. J Thromb Haemost 4:459–468. doi: 10.1111/j.1538-7836.2006.01723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Maurer LM, Tomasini-Johansson BR, Ma W, Annis DS, Eickstaedt NL, Ensenberger MG, Satyshur KA, Mosher DF. 2010. Extended binding site on fibronectin for the functional upstream domain of protein F1 of Streptococcus pyogenes. J Biol Chem 285:41087–41099. doi: 10.1074/jbc.M110.153692. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Accession numbers for S. saprophyticus isolates. Download TABLE S1, TXT file, 0.001 MB (1.9KB, txt) .
Copyright © 2017 Mortimer et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Assembly statistics for S. saprophyticus genomes. Download TABLE S2, TXT file, 0.01 MB (12.6KB, txt) .
Copyright © 2017 Mortimer et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.