

ABSTRACT
Development of a vaccine to protect against cryptococcosis is a priority given the enormous global burden of disease in at-risk individuals. Using glucan particles (GPs) as a delivery system, we previously demonstrated that mice vaccinated with crude Cryptococcus-derived alkaline extracts were protected against lethal challenge with Cryptococcus neoformans and Cryptococcus gattii. The goal of the present study was to identify protective protein antigens that could be used in a subunit vaccine. Using biased and unbiased approaches, six candidate antigens (Cda1, Cda2, Cda3, Fpd1, MP88, and Sod1) were selected, recombinantly expressed in Escherichia coli, purified, and loaded into GPs. Three mouse strains (C57BL/6, BALB/c, and DR4) were then vaccinated with the antigen-laden GPs, following which they received a pulmonary challenge with virulent C. neoformans and C. gattii strains. Four candidate vaccines (GP-Cda1, GP-Cda2, GP-Cda3, and GP-Sod1) afforded a significant survival advantage in at least one mouse model; some vaccine combinations provided added protection over that seen with either antigen alone. Vaccine-mediated protection against C. neoformans did not necessarily predict protection against C. gattii. Vaccinated mice developed pulmonary inflammatory responses that effectively contained the infection; many surviving mice developed sterilizing immunity. Predicted T helper cell epitopes differed between mouse strains and in the degree to which they matched epitopes predicted in humans. Thus, we have discovered cryptococcal proteins that make promising candidate vaccine antigens. Protection varied depending on the mouse strain and cryptococcal species, suggesting that a successful human subunit vaccine will need to contain multiple antigens, including ones that are species specific.
KEYWORDS: Cryptococcus, bioinformatics, glucans, immunization, major histocompatibility complex
IMPORTANCE
The encapsulated fungi Cryptococcus neoformans and Cryptococcus gattii are responsible for nearly 200,000 deaths annually, mostly in immunocompromised individuals. An effective vaccine could substantially reduce the burden of cryptococcosis. However, a major gap in cryptococcal vaccine development has been the discovery of protective antigens to use in vaccines. Here, six cryptococcal proteins with potential as vaccine antigens were expressed recombinantly and purified. Mice were then vaccinated with glucan particle preparations containing each antigen. Of the six candidate vaccines, four protected mice from a lethal cryptococcal challenge. However, the degree of protection varied as a function of mouse strain and cryptococcal species. These preclinical studies identify cryptococcal proteins that could serve as candidate vaccine antigens and provide a proof of principle regarding the feasibility of protein antigen-based vaccines to protect against cryptococcosis.
INTRODUCTION
The encapsulated yeasts Cryptococcus neoformans and Cryptococcus gattii are major causes of morbidity and mortality in persons with qualitative and quantitative impairment of CD4+ T cells (1, 2). Worldwide, it has been estimated that close to 200,000 people with AIDS die from cryptococcal meningitis annually (3). The lifetime incidence of cryptococcosis in recipients of solid organ transplants is approximately 3% (4). Other high-risk groups have been identified, including persons with lymphoma, sarcoidosis, and idiopathic CD4 lymphocytopenia. C. gattii is more of a primary pathogen than C. neoformans, with many cases occurring in individuals with relatively intact immune function (1, 2). Of particular concern is the emergence and spread of hypervirulent C. gattii strains in parts of Canada and the United States (2, 5, 6).
Exposure to Cryptococcus is thought to most commonly occur following inhalation of fungi into the lungs (1, 7). If innate defenses fail, then an adaptive antigen-specific, T cell-mediated immune response becomes critical for preventing local spread and dissemination. Antibody responses, particularly those directed against glucuronoxylomannan (GXM) and other polysaccharide components in the capsule, appear to contribute to the adaptive response in at least some individuals (8).
The high global burden of cryptococcosis (3) has prompted research into the development of vaccines to protect individuals at high risk. Protection of mice using live and killed vaccines has been demonstrated using genetically altered whole yeast cells (9–12). Vaccines comprised of GXM and other cell wall carbohydrates conjugated to carrier proteins elicit antibody responses to GXM that protect in experimental models of cryptococcosis (13, 14). We and others have focused on the discovery of C. neoformans antigens that stimulate T cell responses and thus could serve as candidate antigens in cryptococcal vaccines (15–18). Moreover, because most vaccine adjuvants presently in clinical use stimulate predominantly protective antibody responses (19), we postulate that a successful vaccine will need to incorporate adjuvants and/or delivery systems that elicit strong T cell responses.
Our laboratories have been exploring glucan particles (GPs) as a combined delivery system and adjuvant for cryptococcal vaccines. GPs are hollow, porous yeast cell wall shells produced by treating Saccharomyces cerevisiae with a series of alkaline, acid, and solvent extractions (20–23). They are principally composed of β-1,3-glucan polymers with about 2% chitin and undetectable amounts of proteins, lipids, and mannans. Immune recognition of GPs is mediated by Dectin-1 and complement receptors (24). Immunization of mice with GPs containing trapped ovalbumin results in robust and durable antigen-specific antibody and Th1/Th17-biased CD4+ T cell responses (20, 21).
Recently, we performed whole-cell mild alkali extraction of acapsular mutant strains of C. neoformans and C. gattii. The soluble extracts were loaded into GPs and used to vaccinate mice. Mice so vaccinated exhibited CD4+ T cell recall responses in the lungs and were partially protected from pulmonary challenge with C. neoformans and C. gattii (15). In the present study, we sought to identify specific cryptococcal proteins responsible for protection. Candidate antigens were chosen using biased and unbiased approaches, recombinantly expressed in Escherichia coli, and loaded into GPs. Mouse strains were vaccinated with the antigen-laden GPs and then challenged with C. neoformans and C. gattii. We found that protection varied depending on the antigen, mouse strain, and cryptococcal species.
RESULTS
The six cryptococcal proteins selected for testing are described in Table 1. The immunogenic properties of three of the proteins (Cda2, Fpd1, and MP88) have previously been reported (16–18). Cda1 and Cda3 were chosen as they are chitin deacetylases (Cda) with protein sequence homology to Cda2 (25) and for the importance of Cda proteins for virulence (26). Sod1 was the most abundant protein in a mild alkaline extract that was immunoprotective (15) and contributes to virulence of C. gattii (27). With the exception of Sod1, the six proteins have little to no homology to human proteins. RNA encoding each of the proteins has been identified by transcriptome sequencing (RNA-seq) in C. neoformans obtained from human patients with cryptococcal meningitis (28). In addition, all six proteins have been identified as part of the secretome of the fungus (29, 30). The C. neoformans strain H99 proteins were expressed with a His tag in E. coli (Table 1) and purified over a nickel column. Five of the recombinant proteins were made from cDNA generated from isolated mRNA, while the cDNA for Cda3 expression was synthesized. Sequences for a predicted signal peptide (found in the full-length proteins, except for Sod1), as well as C-terminal serine/threonine-rich regions and a putative glycosylphosphatidylinositol (GPI) anchor motif (found in the proteins except Fpd1 and Sod1) were not included. Milligram quantities of each protein were purified, confirmed to be of the correct size by SDS-PAGE (Fig. 1), and loaded into GPs, typically at 10 μg per vaccine dose.
TABLE 1 .
Recombinant proteins of C. neoformans strain H99 expressed in E. coli and used for testing as vaccines
| CNAG no. | Name | Description | Region expressed |
Vector | Expressed protein mass (kDa) |
% of identity to C. gattii R265 homologa |
Human homologyb |
No. of RNA-seq readsc |
|---|---|---|---|---|---|---|---|---|
| CNAG_05799 | Cda1 | Chitin deacetylase | 20–374 | pET200 | 42.3 | 90 | None | 139,907 |
| CNAG_01230 | Cda2 | Chitin deacetylase | 20–378 | pET200 | 43.8 | 88 | None | 5,835 |
| CNAG_01239 | Cda3 | Chitin deacetylase | 19–336 | pET19b | 37.9 | 87 | 5.3 | 864 |
| CNAG_06291 | Fpd1 | Polysaccharide deacetylase | 20–249 | pET200 | 29.3 | 92 | None | 1,511 |
| CNAG_01019 | Sod1 | Superoxide dismutase | 2–154 | pET200 | 20.1 | 70 | 5−62 | 70,771 |
| CNAG_00776 | MP88 | Mannoprotein | 23–294 | pET200 | 32.9 | 94 | None | 24,244 |
C. gattii strain R265 protein sequences were retrieved from the Ensembl Fungi website (http://fungi.ensembl.org/index.html). Regions of C. gattii protein comparable to C. neoformans protein sequence expressed in E. coli were aligned to calculate the percentage of identity.
Homology to the human protein with the highest similarity (BLASTp analysis); the smaller the numerical value, the greater the similarity.
FIG 1 .
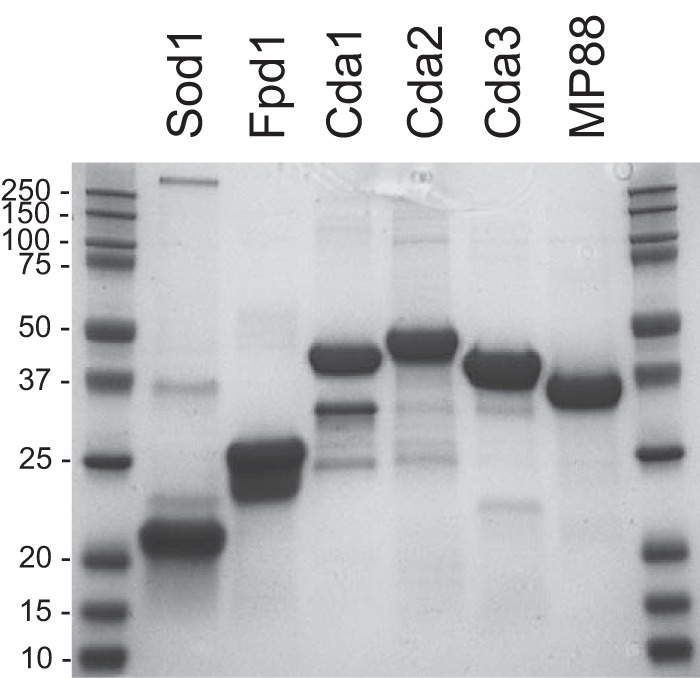
SDS-PAGE of purified recombinant proteins used to make GP vaccines. Each lane contains 10 μg of total protein. Proteins were stained with Coomassie. Molecular masses (in kilodaltons) of protein standards are indicated. The expected molecular masses of the proteins are listed in Table 1.
The vaccines were first tested in C57BL/6 mice. Each mouse received a prime and two boosts of GPs formulated with either one of the six recombinant proteins or with mouse serum albumin (MSA) as a control. Two weeks following the last boost, the mice were challenged with C. neoformans strain KN99 and monitored for 84 days. Significantly prolonged survival was observed following vaccination with Sod1, Cda1, and Cda2 (Fig. 2A). Some of the survivors had undetectable lung CFU when the study was terminated (Fig. 2B). Binary combinations of Sod1, Cda1, and Cda2 in GPs were then tested, with Cda1/Cda2 showing the most promising results (Fig. 2C and D). When the vaccine candidates were tested in BALB/c mice, complete and nearly complete protection was observed with Cda2 and Cda1, respectively (Fig. 3). Partial protection was again seen with Sod1. Cda3, which did not protect C57BL/6 mice, partially protected the BALB/c mice.
FIG 2 .

Vaccination studies with GP-recombinant protein vaccines in C57BL/6 mice. Mice were vaccinated 3 times with 10 µg/vaccine dose of the indicated recombinant protein in GPs and then challenged with 104 CFU of strain KN99. Survival curves and fungal burden in the lungs of mice that survived 84 days p.i. are shown for mice vaccinated with one recombinant protein (A and B) and with two recombinant proteins (C and D). Data were combined from two to three independent vaccination studies with five to six mice per vaccination group in each study. (A) P < 0.001 comparing MSA with Sod1, Cda1, or Cda2. (C) P < 0.001 comparing MSA with any other group. In panels B and D, the dotted line indicates the CFU of KN99 challenge; median CFU per lung for each group are indicated by a solid line.
FIG 3 .

Vaccination studies with GP-recombinant protein vaccines in BALB/c mice challenged with KN99. Studies were conducted as in Fig. 2A and B, except BALB/c mice were used, and the comparator was unvaccinated (UnVac) mice. Survival curves and fungal burden in the lungs of mice that survived 84 days p.i. are shown. Data were combined from two to three independent vaccination studies with four to five mice per vaccination group in each study. P < 0.001 comparing UnVac with Sod1, Cda1, Cda2, or Cda3. For panel B, the dotted line identifies CFU of KN99 challenge; median CFU per lung for each group are indicated by a solid line.
Next, we assessed the capacity of Cda1, Cda2, and Cda3 GP-based vaccines to protect following challenge with C. gattii strain R265. For C57BL/6 mice, the survival curves were nearly identical comparing lethal infections with C. neoformans to that with C. gattii; Cda1 and Cda2 were protective against both strains, whereas Cda3 was not (Fig. 4A). However, at the termination of the study, lung fungal loads and weights were significantly lower in the Cda1-vaccinated mice challenged with KN99 compared with R265 (Fig. 4B). In contrast to C57BL/6 mice, BALB/c mice challenged with R265 had only a minor prolongation of survival following vaccination with GPs containing Cda1, Cda2, or Cda3 (Fig. 4C). Moreover, BALB/c mice vaccinated with both GP-Cda1 and GP-Cda2 (Cda1/Cda2) and challenged with R265 all succumbed by day 52 postinfection (p.i.) (see Fig. S1 in the supplemental material).
FIG 4 .

Survival of vaccinated C57BL/6 and BALB/c mice following infection with C. gattii strain R265. C57BL/6 (A) and BALB/c (C) mice were vaccinated 3 times with 10 µg/vaccine dose of recombinant Cda1, Cda2, or Cda3 in GPs. Control mice were left unvaccinated (UnVac). Mice were then challenged with 104 CFU of KN99 (C. neoformans) or R265 (C. gattii). Panel B shows fungal burden in the lungs (left panel) and lung weights (right panel) of C57BL/6 mice vaccinated with GP-Cda1 that survived 84 days. Data were combined from two to four independent experiments, each with four to five mice per group. (A) P < 0.001 comparing UnVac with Cda1 or Cda2 for both KN99 and R265. (B) P < 0.01 and P < 0.001 comparing fungal load and lung weight, respectively, in KN99 with R265. (C) P < 0.05 comparing UnVac with any other group. In panel B, the dotted line indicates CFU of KN99 challenge; the median CFU per lung for each group are indicated by a solid line.
Survival of C57BL/6 and BALB/c mice vaccinated with Cda1 and Cda2 and challenged with C. neoformans strain KN99 or C. gattii strain R265. Mice were vaccinated with a mixture of Cda1 and Cda2 as in Fig. 2C, except the vaccination dose for each antigen was 6 µg. The challenge dose of KN99 and R265 was 104 CFU. (A) Survival curves of vaccinated and challenged mice. Data are from two independent experiments with 4 or 5 mice per group. P < 0.001 comparing C57BL/6 KN99 with C57BL/6 R265. P < 0.001 comparing BALB/c KN99 with BALB/c R265. Unvaccinated controls all died between 16 and 33 days postinfection (data not shown). (B) Lung fungal burdens of mice that survived for 12 weeks. For panel B, the dotted line identifies CFU of KN99 and R265 challenge; the median CFU per lung for each group are indicated by a solid line. Download FIG S1, PDF file, 0.4 MB (390.7KB, pdf) .
Copyright © 2017 Specht et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
In the next set of experiments, we examined pulmonary fungal loads following challenge of GP-Cda1/Cda2-vaccinated C57BL/6 and BALB/c mice with 104 CFU of C. neoformans KN99. Control mice were immunized with GP-MSA. Mice were euthanized at selected time points. For both strains of mice immunized with GP-MSA, lung CFU rose inexorably (Fig. 5). Levels close to 109 CFU/lung were seen 21 days after challenge, a time point shortly before the mice typically succumb to infection. In contrast, for mice vaccinated with GP-Cda1/Cda2, lung CFU initially rose, but then eventually declined in the day 84 survivors. However, the mouse strains differed at the intermediate time points studied, with the C57BL/6 mice showing highly variable numbers of CFU, whereas the BALB/c mice had a steady, consistent decline 7 days after challenge.
FIG 5 .

Lung CFU of C57BL/6 and BALB/c mice vaccinated with GP-Cda1/Cda2 and challenged with C. neoformans strain KN99. C57BL/6 (A) and BALB/c (B) mice were vaccinated three times with 10 μg/vaccine dose of GP-Cda1 and GP-Cda2 (GP-Cda1/Cda2) and then challenged with 104 CFU of C. neoformans strain KN99. Control mice received GP-MSA. At the indicated time points, mice were euthanized and lung CFU determined. For panel A, GP-Cda1/Cda2 data were collected from three independent studies (does not include day 84 data from Fig. 2D), and for panel B, data were from two independent studies. Each datum point represents the CFU from one mouse.
Lung sections were prepared from BALB/c mice vaccinated with GP-Cda1/Cda2 or GP-MSA 7 and 14 days after challenge with KN99 and evaluated for fungal burden and extent of inflammation by a pathologist blind to experimental condition (Fig. 6A; see Fig. S2 in the supplemental material). Hematoxylin and eosin (H&E)-stained sections revealed mixed inflammatory infiltrates predominantly composed of histiocytes and small numbers of neutrophils and eosinophils within the airways, adjacent alveolar spaces, and walls in all the groups (Fig. 6A). Aggregates of lymphocytes consistent with the formation of inducible bronchus-associated lymphoid tissue (iBALT) were also noted in some of the airway walls in all groups, including the GP-MSA controls. The inflammation progressed to involve more distant airspaces and walls (in addition to airways and adjacent alveolar airspaces and walls) in mice sacrificed at day 14 compared to those sacrificed at day 7. In addition, we noted a slight increase in the proportion of neutrophils among the inflammatory infiltrates at day 14. When comparing the lung pathology at day 7 of mice vaccinated with GP-Cda1/Cda2 to that of controls that received GP-MSA, there was a trend toward greater inflammation in the vaccinated group, suggesting the vaccine promoted an earlier and more intense immune response to fungal microorganisms. No significant, morphological differences were noted in the extent of the inflammation in different mouse groups sacrificed at day 14 (Fig. S2). Mucicarmine-stained sections highlighted that yeast cells present within the airways and airspaces were surrounded by inflammatory cells (Fig. 6). Consistent with the quantitative CFU data, we noted significantly fewer microorganisms in mice vaccinated with GP-Cda1/Cda2 compared with controls that received GP-MSA on both days 7 and 14.
FIG 6 .

Histology of mouse lungs. Shown are representative H&E- and mucicarmine-stained sections of lungs from vaccinated mice. (A) Mice were vaccinated three times with 10 μg/vaccine dose of GP-Cda1 and GP-Cda2 (GP-Cda1/Cda2) and then challenged with 104 CFU of C. neoformans strain KN99. Control mice received GP-MSA. After 7 (A) and 84 (B) days, mice were euthanized and lung sections prepared. Scale bars on the photomicrographs are as follows: 20× = 1 mm, 100× = 100 µm, 200× = 50 µm, and 400× = 20 µm.
Pulmonary inflammation and fungal burden seen on pathology. BALB/c mice were vaccinated three times with 10 µg/vaccine dose each of GP-Cda1 and GP-Cda2 (Cda1/2) and then challenged with 104 CFU of C. neoformans strain KN99. Control mice received GP-MSA (MSA). At 7 and 14 days postchallenge, mice were euthanized, and stained slides prepared from sectioned lungs were scored by a pathologist blind as to group. n = 5 mice/group. For fungal burdens, P < 0.05 and P < 0.001, comparing MSA with Cda1/2 at days 7 (D7) and 14 (D14), respectively. Download FIG S2, PDF file, 0.1 MB (97KB, pdf) .
Copyright © 2017 Specht et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Lung sections from mice vaccinated with GP-Cda1/Cda2 and euthanized at day 84 post-cryptococcal challenge revealed scattered aggregates of lymphocytes in the wall of the airways (iBALT) (Fig. 6B). Rare collections of alveolar macrophages with foamy cytoplasm were also noted in lung sections. However, no fungal microorganisms were identified on H&E- and mucicarmine-stained sections. There were no surviving control GP-MSA mice at day 84 to study comparatively.
We next assessed whether the lead vaccine candidates could protect translationally relevant humanized DR4 mice against challenge with C. neoformans strain KN99. DR4 mice have a C57BL/6 background, but express human major histocompatibility complex class II (MHC-II) HLA-DR4 (DRB1*0401) while lacking endogenous murine MHC-II (31). GPs containing Cda1 or the combination of Cda1 and Cda2 induced protective responses in DR4 mice that were comparable to that seen following vaccination of C57BL/6 mice (Fig. 7). However, vaccines containing Cda2 and Sod1, which significantly protected C57BL/6 mice, afforded the DR4 mice no protection.
FIG 7 .

Protection of vaccinated DR4 mice from infection with C. neoformans strain KN99. DR4 and wild-type (WT) C57BL/6 mice were vaccinated three times with 10 μg/vaccine dose of GPs containing (A) Cda1, (B) Cda2, (C) Cda1 and Cda2 (Cda1/Cda2), and (D) Sod1. Control mice received GPs containing MSA or were left unvaccinated (UnVac). Mice were then challenged with 104 CFU of C. neoformans strain KN99 and monitored for survival. Data from vaccinated DR4 mice were combined from at least two independent experiments with a total of 10 to 20 mice per vaccination group. The number of control mice per experimental group ranged from 5 to 10. P < 0.001 comparing DR4 MSA with DR4 Cda1, P < 0.05 comparing DR4 MSA with DR4 Cda2, and P < 0.001 comparing DR4 UnVac with DR4 Cda1/2.
Finally, we performed individual epitope mapping of the six proteins of interest to try to understand the mechanisms responsible for the observed differences in vaccine responses among mouse strains and to assess the potential of the proteins as candidate antigens in human vaccines (see Fig. S3 in the supplemental material). Each protein contains several high-probability B cell linear epitopes and from 1 to 3 sequences comprising strong MHC-II binding for multiple human and murine alleles. In Fig. S3A to F, high-probability B cell epitopes and predicted MHC-II high-binding peptides are indicated with a score exceeding 1 standard deviation from the mean for that protein. Within these regions, multiple peptides are also predicted to be excised by cathepsin B, L, or S (data not shown) and so have the potential for presentation as T cell epitopes by antigen-presenting cells. When examined from the perspective of individual murine MHC-II allele binding, there are differences in the predicted binding between alleles, leading to differences in epitope dominance between mice. For instance, in Cda1, Fig. S3A shows similarity between binding by H2-I-Ab (C57BL/6) and H2-I-Ad (BALB/c) for the 15-mer with its index position at position 309, but similarity between H2-I-Ad and the human DRB1:0401 is carried by the DR4 mouse at positions 291 to 292. This may result in the observed differences in survival between C57BL/6, BALB/c, and DR4 mice. Thus, all of the recombinant proteins are characterized as good potential immunogens that are likely to elicit antibody responses and sustained cellular recall responses. The engineering in of the N-terminal His tag did not apparently bias the potential immunogenicity or dominance of the epitopes of the Cryptococcus protein.
Comparison of B cell linear epitopes and human and murine MHC-II binding patterns. For each protein tested as a candidate vaccine antigen, the probability of participation in a B cell linear epitope and predicted MHC-II binding affinities of sequential 15-mer peptides are shown in the five panels. Panels should be read from top to bottom. Panel 1 shows the probability of linear B cell epitopes for both mice and humans. Panel 2 shows the permuted human MHC-II binding, smoothed across 16 DRB alleles. Panel 3 shows MHC-II binding for allele H-2-IAb (C57BL/6 mice). Panel 4 shows MHC-II binding for allele H-2-IAd (BALB/c mice). Panel 5 shows MHC-II binding for human allele DRB1-0401 found in transgenic mouse strain DR4. y-axis units are the inverted standard deviation of linear B cell epitope probability, and for MHC binding panels, they are standard deviation units below the mean of the natural log binding affinity (ln of the 50% inhibitory concentration [lnIC50]) for that protein. The vertical black line in each set of panels indicates the boundary between vector sequence that includes the His tag and each Cryptococcus protein sequence. The epitope predictions illustrated in panels A to F were generated using the ioGenetics’ EigenBio platform. The EigenBio algorithms are written on JMP (SAS Institute). The rationale underlying the predictions is further described in several publications by R. D. Bremel and E. J. Homan as cited in Text S1 in the supplemental material (Immunome Res 6:8, 2010, https://doi.org/10.1186/1745-7580-6-8; Immunome Res 6:7, 2010, https://doi.org/10.1186/1745-7580-6-7; PLoS One, 8:e70115, 2013, https://doi.org/10.1371/journal.pone.0070115; Front Immunol 5:541, 2014, https://doi.org/10.3389/fimmu.2014.00541). Download FIG S3, PDF file, 0.4 MB (452.7KB, pdf) .
Copyright © 2017 Specht et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Methods specific to Fig. S3. Download TEXT S1, DOCX file, 0.1 MB (17.5KB, docx) .
Copyright © 2017 Specht et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DISCUSSION
Compared with vaccines featuring whole organisms or crude antigen preparations, subunit vaccines have the advantage of being well defined and less likely to elicit adverse reactions, including autoimmune responses. Here, we identify several antigens that, when used as part of a GP-based vaccine formulation, protect mice against lethal cryptococcal challenge. Cda2 (also known as MP98) was originally identified as the antigen that stimulated a T cell hybridoma generated from immunized mice (16). Subsequent studies using a peptide–MHC-II tetramer containing a 13-amino-acid peptide from Cda2 revealed that 6.5% of Th cells stained with the tetramer 14 days postinfection with C. neoformans strain KN99 (32). In addition to being identified on our biased screen of immunostimulatory proteins, Cda2 was the fifth most common protein found in the protective alkaline extracts (15). The two other definitive Cdas, Cda1 and Cda3, were also tested in our vaccine model. Each of the three recombinant CDAs, when delivered in a GP-based vaccine, protected at least one mouse strain against C. neoformans challenge, with rCda1 and rCda2 being the most promising of the antigens. Interestingly, it was recently shown that vaccination with a strain of C. neoformans genetically deficient in CDAs protected mice against a subsequent lethal C. neoformans challenge (9). Taken together, these data highlight that while the CDAs are promising vaccine candidates, other protective antigens exist.
One such protective antigen, identified as one of the top two hits in our unbiased screen (15), is Sod1. These results validate the unbiased approach, and we are presently testing the other abundant proteins identified in the alkaline extracts. While the CDAs have little to no homology with human proteins, Sod1 may not be an ideal candidate vaccine antigen due to its significant homology with human superoxide dismutase (SOD). However, the immunoinformatic analysis supports the prospect that epitope mapping will identify a protective region within the antigen that has minimal homology with human antigens. Indeed, future studies will seek to find such regions within the various protective antigens and then test the efficacy of chimeric antigens containing the protective epitopes.
The recombinant proteins were made in E. coli and thus are likely lacking in glycosylation. Cryptococcal N-linked and O-linked glycans generally are terminally mannosylated. Upon infection, the mannose groups are recognized by C-type lectin receptors with affinity for mannose (22). Notably, this interaction of mannosylated proteins with host mannose receptors augments immunity. Diminution of T cell responses was observed following chemical deglycosylation of cryptococcal mannoproteins or by blocking mannose receptors with mannan (33, 34). In addition, Cda2-specific cytokine production was greater when cells were stimulated with recombinant Cda2 (rCda2) produced in the yeast Pichia pastoris, compared with E. coli-derived protein (35). Nevertheless, in the present study, the mice were protected following vaccination with proteins made in E. coli. We posit this is because the antigens are encased in GPs that are phagocytosed by Dectin-1 and complement receptors (24), obviating the participation of mannose receptors. Nevertheless, it would be interesting to compare responses of native and unglycosylated cryptococcal antigens encased in GPs.
A challenge in the development of vaccines designed to stimulate T cell-mediated protection is the diversity of MHC alleles within the human population (19). We tested our GP-based vaccines using three different mouse strains: C57BL/6, BALB/c, and DR4. We found that for a given antigen, the degree of observed protection varied as a function of mouse strain. The importance of MHC-II is evident comparing C57BL/6 and DR4 mice; GP-Cda2 and GP-Sod1 were partially protective vaccines in C57BL/6, but not DR4 mice. These mice share genetic backgrounds, with the exception that in transgenic DR4 mice, native MHC class II molecules have been replaced by human HLA-DR4 (36). GP-Cda1 and GP-Cda2 vaccines afforded complete or nearly complete protection in BALB/c mice, with many of the mice having undetectable CFU in the lungs at the time of euthanasia. Moreover, the GP-Cda3 vaccine, which was not protective in C57BL/6 and DR4 mice, was partially protective in BALB/c mice. As shown in Fig. S3, the predicted MHC-II binding affinity of individual sequential peptides within each protein varies considerably between the MHC-II alleles of BALB/c and C57BL/6 and DR4. Similar differences may be expected between individual human MHC-II alleles; the permuted average of the predicted binding of each peptide across multiple human MHC-II alleles provides an indication (Fig. S3) of which mouse epitopes may be most likely to elicit a comparable result in the human population. However, taken together, these results suggest that a successful cryptococcal vaccine that stimulates T cell protective responses in humans will require a combination of antigens or epitopes.
Pathology and CFU data demonstrate that vaccinated mice are able to mount an inflammatory response during the first week of infection and that this inflammatory response is correlated with a reduction in the fungal burden compared with challenged mice that received control vaccine. At day 7, vaccinated mice had 3- to 8-fold fewer CFU in the lung than control mice. By 14 days postinfection, the inflammatory responses in control and vaccinated mice are similar, yet CFU continue to rise in control mice while plateauing or falling in the vaccinated animals. This suggests there are differences in the qualitative nature of the inflammatory response not detected on H&E staining. We have shown that GP-based vaccines promote robust antigen-specific antibody, Th1, and Th17 responses (15, 20, 21). Importantly, Th1 and Th17 recall responses to the lungs occur following pulmonary challenge of vaccinated mice (15). Ongoing work is directed at further defining the mechanisms responsible for the protection observed with the GP-based recombinant protein vaccines.
It is estimated that C. neoformans and C. gattii diverged between 16 and 160 million years ago (37). Both species share major virulence determinants, although a comparison of their genomes reveals only 86% identity (37–39). Differences in immunopathology have been noted in experimental murine models when comparing infections with the two species (38, 40, 41). Thus, it was of interest to see whether vaccines that protected against C. neoformans would provide cross-protection in C. gattii models of infection. Remarkably, we found vaccination with GP-Cda1 and GP-Cda2 provided almost identical levels of protection against both cryptococcal species in C57BL/6 mice. However, in BALB/c mice, the same vaccines provided robust protection against challenge with C. neoformans but no protection against C. gattii. Comparative epitope mapping of C. neoformans and C. gattii is under way to determine if differences in epitopes engendered by the 35- to 42-amino-acid differences in each protein assist in identifying the basis for protection. The cryptococcal strains we used, KN99 and R265, are hypervirulent and thus provide a stringent test for any vaccine; future studies also will examine epitope differences in additional cryptococcal strains that commonly cause disease in humans.
Using the model antigen ovalbumin encapsulated in GPs, we previously demonstrated robust CD4+ T cell and antibody responses occur even after immunization with submicrogram doses of antigen (21). Our new studies extend these findings to demonstrate the utility of using the GP-based delivery system as an effective tool for vaccine antigen discovery. Four of the six antigens tested, when administered as a GP-based vaccine, provided significant protection in at least one mouse model. We are currently using this approach to identify other protective antigens based on our biased and unbiased screens. Eventually, the antigens must be tested in human systems. Moreover, as the greatest risk factor for cryptococcosis is T cell dysfunction (7), we hypothesize that a successful cryptococcal vaccine for use in humans will need to elicit not only T cell responses to protein antigens but also antibody responses. Regarding the latter, antibody to capsular and cell wall carbohydrates has been shown to be protective in some models of cryptococcosis (13, 14, 42). Using the GP delivery system, we are currently testing whether the addition of GXM conjugated to a protein carrier will boost protection in mouse models.
MATERIALS AND METHODS
Chemicals and culture media.
Chemical reagents were from Thermo Fisher Scientific (Pittsburgh, PA), unless stated otherwise. The media used for culturing of Cryptococcus were YPD (Difco yeast extract, Bacto peptone, dextrose, with and without 2% agar) and Sabouraud dextrose agar Emmons (Remel). E. coli strains TOP10 and BL21 were cultured at 37°C in LB broth or agar. Transformed strains of BL21 were used for protein expression by being cultured for 18 h at 30°C with shaking in Overnight Express Instant TB medium (EMD Millipore, Billerica, MA). Antibiotic selection was with ampicillin (100 μg/ml) or kanamycin (100 μg/ml).
Strains of Cryptococcus.
C. neoformans var. grubii strains KN99 (43) and cap59 (44) and C. gattii strain R265 (45) were maintained as glycerol stocks at −80°C and initially cultured on YPD agar. To prepare Cryptococcus for in vivo challenge studies, KN99 and R265 were cultured in liquid YPD for 15 to 22 h at 30°C with shaking. Yeast cells were then harvested by centrifugation, washed once with phosphate-buffered saline (PBS), counted using a hemocytometer, and finally suspended in PBS at 2 × 105 cells/ml. CFU were determined on Sabouraud dextrose agar.
Expression of cryptococcal proteins in E. coli.
Six C. neoformans strain H99 proteins were expressed in E. coli (Table 1) for subsequent testing as vaccine candidates. When present, cDNA was deleted of regions coding for a signal peptide and C terminus rich in serine and threonine and the GPI anchoring motif. Five proteins were expressed following cloning of PCR-amplified cDNA in pET200/D-TOPO (Invitrogen), and for the sixth protein its cDNA was synthesized and cloned in pET19b by GenScript (Piscataway, NJ). Both vectors are designed to fuse vector-encoded sequence that contains a His tag to the N terminus of the cloned cDNA.
Briefly, strain cap59 was cultured in YPD at 30°C with shaking to the mid-log phase. Approximately 108 cells were collected by centrifugation in a 2.0-ml microcentrifuge tube, suspended in 0.7 ml RLT Plus buffer containing β-mercaptoethanol, and homogenized using 0.6 ml of 0.7-mm zirconium beads (BioSpec Products, Bartlesville, OK). Purification of RNA from the homogenate was performed according to the manufacturer’s instructions (RNeasy Plus minikit; Qiagen, Hilden, Germany). Purified RNA was used to make cDNA using the iScript cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA). The cDNA was then used as the template for amplification by PCR with Platinum Pfx DNA polymerase (Invitrogen) with gene-specific primers designed for directional cloning. Each PCR product was purified using a QIAquick PCR purification kit (Qiagen) and then ligated to pET200/D-TOPO according to the manufacturer’s instructions that accompanied the Champion pET 200 Directional TOPO expression kit (Invitrogen). The ligated DNA was transformed into E. coli TOP10 cells and plated for single-colony isolation. Several colonies were screened by PCR using PCR SuperMix (Invitrogen) and gene-specific primers. PCR products of the predicted size following electrophoresis in agarose were confirmed, and plasmid DNA was purified from select clones grown in 4 ml LB-kanamycin medium using a QIAprep spin miniprep kit (Qiagen). DNA then was sequenced (GeneWiz, South Plainfield, NJ) using vector forward and reverse primers. Plasmids confirmed to have the expected cDNA sequence ligated correctly were transformed into E. coli BL21 cells for protein expression. Glycerol stocks of E. coli were made for storage at −80°C.
Purification of proteins.
Each recombinant BL21 strain was grown in 50 ml of Overnight Express medium with antibiotic in 250-ml baffled culture flasks (Bellco Glass, Vineland, NJ) at 30°C for 18 h. Cultures were inoculated with approximately 25 μl of −80°C glycerol stock. The buffers used in purification of protein on His·Bind resin were recommended by the manufacturer (EMD Millipore). Cells were harvested by centrifugation at 6,000 × g for 10 min, suspended in 5 ml binding buffer (BB: 0.5 M NaCl, 20 mM Tris-HCl, 5 mM imidazole [pH 7.9]), sonicated for 30 s, and collected by centrifugation. The cell pellets were suspended in 5 ml BB containing 6 M urea and sonicated again. Following centrifugation, the supernatant was collected and centrifuged at 20,000 × g for 5 min to remove residual insoluble material. Clarified supernatants from two 50-ml cultures were combined (ca. 9 ml) and applied to a 3-ml bed volume of His·bind resin that had been equilibrated with BB plus 6 M urea. The column was washed with 15 ml BB–6 M urea and 10 ml wash buffer (0.5 M NaCl, 20 mM Tris-HCl, 60 mM imidazole, 6 M urea [pH 7.9]), and protein was eluted with a mixture of 0.5 M NaCl, 20 mM Tris-HCl, 1 M imidazole, and 6 M urea (pH 7.9). The eluate was collected in 1-ml fractions, which were each analyzed by SDS-PAGE to determine which fractions to pool—typically 3 to 4 fractions. The purified protein was dialyzed against 6 M urea–20 mM Tris-HCl and concentrated to 10 mg/ml using Amicon Ultra-15 centrifugal filters (10-kDA cutoff; Merck Millipore, Cork, Ireland). All procedures following the growth of cells were at 22°C. Protein determinations were by the bicinchoninic acid assay. Samples of concentrated protein were stored at −80°C. For SDS-PAGE, proteins were separated on 4 to 20% gradient gels (BioRad) in Tris-glycine-SDS running buffer and stained with Coomassie (Expedeon, Ltd., Cambridgeshire, United Kingdom); protein standards were from BioRad.
GP vaccines.
Saccharomyces cerevisiae cells were purified into GPs following a series of hot alkali, organic, and aqueous extraction steps, as described previously (20, 21, 46, 47). The final GP vaccine consisted of recombinant protein, mouse serum albumin (MSA; Equitech-Bio, Kerrville, TX) and yeast RNA (yRNA; Sigma-Aldrich, St. Louis, MO) complexed within the glucan shells. Each recombinant protein at 10 mg/ml in 6 M urea–20 mM Tris-HCl was loaded into GPs and complexed with MSA and yRNA as described previously (20, 21, 46). Vaccines were diluted in sterile 0.9% saline for injection to deliver 10 μg of antigen and 25 μg of MSA complexed with yRNA in 200 μg GPs (approximately 108 particles) per 0.1-ml dose. Control vaccine was prepared identically with GPs loaded with MSA and yRNA, but without recombinant protein. Vaccines were stored in 0.6-ml aliquots at −80°C and briefly vortexed prior to use. Vaccine formulations were quality controlled by manual counting of GPs per milliliter using a hemocytometer, microscopically assessing for intact GPs with a phase-distinct protein-yRNA complex. Vaccines were also extracted and analyzed by 10% SDS-PAGE to verify loading of antigens and MSA (15).
Mice.
Six-week-old female C57BL/6 and BALB/c mice were obtained from either Charles River Laboratories, Inc. (Kingston, NY), or The Jackson Laboratory (Bar Harbor, ME). The Abb knockout/transgenic HLA-DR4 (DR4) mice were from Taconic Biosciences (Hudson, NY). Mice were bred and housed in a specific-pathogen-free environment in the animal facilities at the University of Massachusetts Medical School. Vaccinations commenced when the mice were 6 to 10 weeks of age; both male and female mice were used. All animal procedures were carried out under a protocol approved by the University of Massachusetts Medical School Institutional Use and Care of Animals Committee.
Vaccination and challenge studies.
The GP-based vaccines (0.1-ml dose) were administered three times at 2-week intervals as a subcutaneous injection at the midline of the abdomen, as described previously (20, 21). Two weeks following the third vaccination, the mice were challenged with 104 CFU of C. neoformans strain KN99 or C. gattii strain R265 in 50 µl of PBS. Mice were anesthetized with 2% isoflurane (Piramal Health Care, Andrah Pradesh, India) in a VetEquip laboratory animal anesthesia system (VetEquip, Livermore, CA) and inoculated orotracheally. For survival studies, mice were monitored twice daily and euthanized if they developed advanced signs of disease, including ataxia, listlessness, and failure to groom. At the termination of the study, the surviving mice were euthanized, and their lungs and brains were removed. Lungs were weighed, and the organs were homogenized in PBS containing 100 U/ml penicillin and 100 µg/ml streptomycin (lungs in 4 ml and brains in 2 ml) using an Omni TH tissue homogenizer and plastic homogenizing probes (Omni, Kennesaw, GA). Undiluted and diluted homogenates were plated on Sabouraud medium and incubated at 30°C for 2 to 3 days, at which time fungal CFU were enumerated. In other studies, mice were euthanized at various times after fungal challenge to enumerate CFU. The lower limit of detection was 20 CFU. Lungs with no CFU were arbitrarily assigned a value of 20 CFU.
Histology.
Lungs from euthanized mice were inflated with 0.5 ml 4% paraformaldehyde in PBS and then excised. Following embedding in paraffin, 5-μm sections obtained from each of five lobes were mounted on glass slides and stained with H&E or mucicarmine (Sigma-Aldrich). A pathologist, blind as to experimental group, then scored the slides on a scale of 0 to 3 for pulmonary inflammation and fungal burden as shown in Fig. S2.
Immunoinformatics.
Each of the six proteins was analyzed as previously described to evaluate the distribution of B cell linear epitopes, MHC binding for human and murine alleles (48, 49), and the presence or absence of possible suppressive T cell epitope motifs (50, 51). In addition, both wild-type and recombinant sequences of each candidate protein were evaluated to determine the content of high-frequency T-cell-exposed motifs relative to all other proteins in the C. neoformans proteome (51). More details are presented following the legend to Fig. S2.
Statistics.
Data were analyzed using GraphPad Prism, version 6.0 (GraphPad Software, Inc., La Jolla, CA). Kaplan-Meier survival curves were compared using the Mantel-Cox log rank test. The unpaired two-tailed t test was used for comparisons of two groups with the Bonferroni correction applied for multiple comparisons. The one-way analysis of variance (ANOVA) with the Tukey multiple-correction test was used to compare more than two groups.
ACKNOWLEDGMENTS
We acknowledge NIH grants AI025780 to Stuart M. Levitz, AI102618 to Stuart M. Levitz, AI125045 to Jennifer K. Lodge, Stuart M. Levitz, and Charles A. Specht, HL112671 to Stuart M. Levitz, and AI072195 to Jennifer K. Lodge and Charles A. Specht.
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Citation Specht CA, Lee CK, Huang H, Hester MM, Liu J, Luckie BA, Torres Santana MA, Mirza Z, Khoshkenar P, Abraham A, Shen ZT, Lodge JK, Akalin A, Homan J, Ostroff GR, Levitz SM. 2017. Vaccination with recombinant Cryptococcus proteins in glucan particles protects mice against cryptococcosis in a manner dependent upon mouse strain and cryptococcal species. mBio 8:e01872-17. https://doi.org/10.1128/mBio.01872-17.
REFERENCES
- 1.Heitman J, Kozel TR, Kwon-Chung KJ, Perfect JR, Casadevall A (ed). 2011, Cryptococcus: from human pathogen to model yeast. ASM Press, Washington, DC. [Google Scholar]
- 2.Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, White TC. 2012. Hidden killers: human fungal infections. Sci Transl Med 4:165rv13. doi: 10.1126/scitranslmed.3004404. [DOI] [PubMed] [Google Scholar]
- 3.Rajasingham R, Smith RM, Park BJ, Jarvis JN, Govender NP, Chiller TM, Denning DW, Loyse A, Boulware DR. 2017. Global burden of disease of HIV-associated cryptococcal meningitis: an updated analysis. Lancet Infect Dis 17:873–881. doi: 10.1016/S1473-3099(17)30243-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh N, Dromer F, Perfect JR, Lortholary O. 2008. Cryptococcosis in solid organ transplant recipients: current state of the science. Clin Infect Dis 47:1321–1327. doi: 10.1086/592690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Byrnes EJ III, Li W, Lewit Y, Ma H, Voelz K, Ren P, Carter DA, Chaturvedi V, Bildfell RJ, May RC, Heitman J. 2010. Emergence and pathogenicity of highly virulent Cryptococcus gattii genotypes in the northwest United States. PLoS Pathog 6:e1000850. doi: 10.1371/journal.ppat.1000850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byrnes EJ, Li W, Ren P, Lewit Y, Voelz K, Fraser JA, Dietrich FS, May RC, Chaturvedi S, Chaturvedi V, Heitman J. 2011. A diverse population of Cryptococcus gattii molecular type VGIII in southern Californian HIV/AIDS patients. PLoS Pathog 7:e1002205. doi: 10.1371/journal.ppat.1002205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Williamson PR, Jarvis JN, Panackal AA, Fisher MC, Molloy SF, Loyse A, Harrison TS. 2017. Cryptococcal meningitis: epidemiology, immunology, diagnosis and therapy. Nat Rev Neurol 13:13–24. doi: 10.1038/nrneurol.2016.167. [DOI] [PubMed] [Google Scholar]
- 8.Rohatgi S, Pirofski LA. 2015. Host immunity to Cryptococcus neoformans. Future Microbiol 10:565–581. doi: 10.2217/fmb.14.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Upadhya R, Lam WC, Maybruck B, Specht CA, Levitz SM, Lodge JK. 2016. Induction of protective immunity to cryptococcal infection in mice by a heat-killed, chitosan-deficient strain of Cryptococcus neoformans. mBio 7:e00547-16. doi: 10.1128/mBio.00547-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fromtling RA, Blackstock R, Hall NK, Bulmer GS. 1979. Immunization of mice with an avirulent pseudohyphal form of Cryptococcus neoformans. Mycopathologia 68:179–181. doi: 10.1007/BF00578527. [DOI] [PubMed] [Google Scholar]
- 11.Wormley FL Jr, Heinrich G, Miller JL, Perfect JR, Cox GM. 2005. Identification and characterization of an SKN7 homologue in Cryptococcus neoformans. Infect Immun 73:5022–5030. doi: 10.1128/IAI.73.8.5022-5030.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhai B, Wozniak KL, Masso-Silva J, Upadhyay S, Hole C, Rivera A, Wormley FL Jr, Lin X. 2015. Development of protective inflammation and cell-mediated immunity against Cryptococcus neoformans after exposure to hyphal mutants. mBio 6:e01433-15. doi: 10.1128/mBio.01433-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Devi SJ. 1996. Preclinical efficacy of a glucuronoxylomannan-tetanus toxoid conjugate vaccine of Cryptococcus neoformans in a murine model. Vaccine 14:841–844. doi: 10.1016/0264-410X(95)00256-Z. [DOI] [PubMed] [Google Scholar]
- 14.Casadevall A, Feldmesser M, Pirofski LA. 2002. Induced humoral immunity and vaccination against major human fungal pathogens. Curr Opin Microbiol 5:386–391. doi: 10.1016/S1369-5274(02)00337-5. [DOI] [PubMed] [Google Scholar]
- 15.Specht CA, Lee CK, Huang H, Tipper DJ, Shen ZT, Lodge JK, Leszyk J, Ostroff GR, Levitz SM. 2015. Protection against experimental cryptococcosis following vaccination with glucan particles containing Cryptococcus alkaline extracts. mBio 6:e01905-15. doi: 10.1128/mBio.01905-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levitz SM, Nong S, Mansour MK, Huang C, Specht CA. 2001. Molecular characterization of a mannoprotein with homology to chitin deacetylases that stimulates T cell responses to Cryptococcus neoformans. Proc Natl Acad Sci U S A 98:10422–10427. doi: 10.1073/pnas.181331398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang C, Nong SH, Mansour MK, Specht CA, Levitz SM. 2002. Purification and characterization of a second immunoreactive mannoprotein from Cryptococcus neoformans that stimulates T-cell responses. Infect Immun 70:5485–5493. doi: 10.1128/IAI.70.10.5485-5493.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Biondo C, Beninati C, Bombaci M, Messina L, Mancuso G, Midiri A, Galbo R, Teti G. 2003. Induction of T helper type 1 responses by a polysaccharide deacetylase from Cryptococcus neoformans. Infect Immun 71:5412–5417. doi: 10.1128/IAI.71.9.5412-5417.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levitz SM, Golenbock DT. 2012. Beyond empiricism: informing vaccine development through innate immunity research. Cell 148:1284–1292. doi: 10.1016/j.cell.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang H, Ostroff GR, Lee CK, Specht CA, Levitz SM. 2010. Robust stimulation of humoral and cellular immune responses following vaccination with antigen-loaded beta-glucan particles. mBio 1:e00164-10. doi: 10.1128/mBio.00164-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang H, Ostroff GR, Lee CK, Specht CA, Levitz SM. 2013. Characterization and optimization of the glucan particle-based vaccine platform. Clin Vaccine Immunol 20:1585–1591. doi: 10.1128/CVI.00463-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levitz SM, Huang H, Ostroff GR, Specht CA. 2015. Exploiting fungal cell wall components in vaccines. Semin Immunopathol 37:199–207. doi: 10.1007/s00281-014-0460-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soto ER, Caras AC, Kut LC, Castle MK, Ostroff GR. 2012. Glucan particles for macrophage targeted delivery of nanoparticles. J Drug Deliv 2012:143524. doi: 10.1155/2012/143524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang H, Ostroff GR, Lee CK, Agarwal S, Ram S, Rice PA, Specht CA, Levitz SM. 2012. Relative contributions of dectin-1 and complement to immune responses to particulate beta-glucans. J Immunol 189:312–317. doi: 10.4049/jimmunol.1200603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baker LG, Specht CA, Donlin MJ, Lodge JK. 2007. Chitosan, the deacetylated form of chitin, is necessary for cell wall integrity in Cryptococcus neoformans. Eukaryot Cell 6:855–867. doi: 10.1128/EC.00399-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baker LG, Specht CA, Lodge JK. 2011. Cell wall chitosan is necessary for virulence in the opportunistic pathogen Cryptococcus neoformans. Eukaryot Cell 10:1264–1268. doi: 10.1128/EC.05138-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Narasipura SD, Ault JG, Behr MJ, Chaturvedi V, Chaturvedi S. 2003. Characterization of Cu,Zn superoxide dismutase (SOD1) gene knock-out mutant of Cryptococcus neoformans var. gattii: role in biology and virulence. Mol Microbiol 47:1681–1694. doi: 10.1046/j.1365-2958.2003.03393.x. [DOI] [PubMed] [Google Scholar]
- 28.Chen Y, Toffaletti DL, Tenor JL, Litvintseva AP, Fang C, Mitchell TG, McDonald TR, Nielsen K, Boulware DR, Bicanic T, Perfect JR. 2014. The Cryptococcus neoformans transcriptome at the site of human meningitis. mBio 5:e01087-13. doi: 10.1128/mBio.01087-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eigenheer RA, Jin Lee Y, Blumwald E, Phinney BS, Gelli A. 2007. Extracellular glycosylphosphatidylinositol-anchored mannoproteins and proteases of Cryptococcus neoformans. FEMS Yeast Res 7:499–510. doi: 10.1111/j.1567-1364.2006.00198.x. [DOI] [PubMed] [Google Scholar]
- 30.Geddes JM, Croll D, Caza M, Stoynov N, Foster LJ, Kronstad JW. 2015. Secretome profiling of Cryptococcus neoformans reveals regulation of a subset of virulence-associated proteins and potential biomarkers by protein kinase A. BMC Microbiol 15:206. doi: 10.1186/s12866-015-0532-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ito K, Bian HJ, Molina M, Han J, Magram J, Saar E, Belunis C, Bolin DR, Arceo R, Campbell R, Falcioni F, Vidović D, Hammer J, Nagy ZA. 1996. HLA-DR4-IE chimeric class II transgenic, murine class II-deficient mice are susceptible to experimental allergic encephalomyelitis. J Exp Med 183:2635–2644. doi: 10.1084/jem.183.6.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wiesner DL, Specht CA, Lee CK, Smith KD, Mukaremera L, Lee ST, Lee CG, Elias JA, Nielsen JN, Boulware DR, Bohjanen PR, Jenkins MK, Levitz SM, Nielsen K. 2015. Chitin recognition via chitotriosidase promotes pathologic type-2 helper T cell responses to cryptococcal infection. PLoS Pathog 11:e1004701. doi: 10.1371/journal.ppat.1004701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mansour MK, Latz E, Levitz SM. 2006. Cryptococcus neoformans glycoantigens are captured by multiple lectin receptors and presented by dendritic cells. J Immunol 176:3053–3061. doi: 10.4049/jimmunol.176.5.3053. [DOI] [PubMed] [Google Scholar]
- 34.Mansour MK, Schlesinger LS, Levitz SM. 2002. Optimal T-cell responses to Cryptococcus neoformans mannoprotein are dependent on recognition of conjugated carbohydrates by mannose receptors. J Immunol 168:2872–2879. doi: 10.4049/jimmunol.168.6.2872. [DOI] [PubMed] [Google Scholar]
- 35.Specht CA, Nong S, Dan JM, Lee CK, Levitz SM. 2007. Contribution of glycosylation to T cell responses stimulated by recombinant Cryptococcus neoformans mannoprotein. J Infect Dis 196:796–800. doi: 10.1086/520536. [DOI] [PubMed] [Google Scholar]
- 36.Pan S, Trejo T, Hansen J, Smart M, David CS. 1998. HLA-DR4 (DRB1*0401) transgenic mice expressing an altered CD4-binding site: specificity and magnitude of DR4-restricted T cell response. J Immunol 161:2925–2929. [PubMed] [Google Scholar]
- 37.Chen SC, Meyer W, Sorrell TC. 2014. Cryptococcus gattii infections. Clin Microbiol Rev 27:980–1024. doi: 10.1128/CMR.00126-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng PY, Sham A, Kronstad JW. 2009. Cryptococcus gattii isolates from the British Columbia cryptococcosis outbreak induce less protective inflammation in a murine model of infection than Cryptococcus neoformans. Infect Immun 77:4284–4294. doi: 10.1128/IAI.00628-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.D’Souza CA, Kronstad JW, Taylor G, Warren R, Yuen M, Hu G, Jung WH, Sham A, Kidd SE, Tangen K, Lee N, Zeilmaker T, Sawkins J, McVicker G, Shah S, Gnerre S, Griggs A, Zeng Q, Bartlett K, Li W, Wang X, Heitman J, Stajich JE, Fraser JA, Meyer W, Carter D, Schein J, Krzywinski M, Kwon-Chung KJ, Varma A, Wang J, Brunham R, Fyfe M, Ouellette BFF, Siddiqui A, Marra M, Jones S, Holt R, Birren BW, Galagan JE, Cuomo CA. 2011. Genome variation in Cryptococcus gattii, an emerging pathogen of immunocompetent hosts. mBio 2:e00342-10. doi: 10.1128/mBio.00342-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Angkasekwinai P, Sringkarin N, Supasorn O, Fungkrajai M, Wang YH, Chayakulkeeree M, Ngamskulrungroj P, Angkasekwinai N, Pattanapanyasat K. 2014. Cryptococcus gattii infection dampens Th1 and Th17 responses by attenuating dendritic cell function and pulmonary chemokine expression in the immunocompetent hosts. Infect Immun 82:3880–3890. doi: 10.1128/IAI.01773-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ngamskulrungroj P, Chang Y, Sionov E, Kwon-Chung KJ. 2012. The primary target organ of Cryptococcus gattii is different from that of Cryptococcus neoformans in a murine model. mBio 3:e00103-12. doi: 10.1128/mBio.00103-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rachini A, Pietrella D, Lupo P, Torosantucci A, Chiani P, Bromuro C, Proietti C, Bistoni F, Cassone A, Vecchiarelli A. 2007. An anti-beta-glucan monoclonal antibody inhibits growth and capsule formation of Cryptococcus neoformans in vitro and exerts therapeutic, anticryptococcal activity in vivo. Infect Immun 75:5085–5094. doi: 10.1128/IAI.00278-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nielsen K, Cox GM, Wang P, Toffaletti DL, Perfect JR, Heitman J. 2003. Sexual cycle of Cryptococcus neoformans var. grubii and virulence of congenic a and alpha isolates. Infect Immun 71:4831–4841. doi: 10.1128/IAI.71.9.4831-4841.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nelson RT, Hua J, Pryor B, Lodge JK. 2001. Identification of virulence mutants of the fungal pathogen Cryptococcus neoformans using signature-tagged mutagenesis. Genetics 157:935–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fraser JA, Giles SS, Wenink EC, Geunes-Boyer SG, Wright JR, Diezmann S, Allen A, Stajich JE, Dietrich FS, Perfect JR, Heitman J. 2005. Same-sex mating and the origin of the Vancouver Island Cryptococcus gattii outbreak. Nature 437:1360–1364. doi: 10.1038/nature04220. [DOI] [PubMed] [Google Scholar]
- 46.Huang H, Ostroff GR, Lee CK, Wang JP, Specht CA, Levitz SM. 2009. Distinct patterns of dendritic cell cytokine release stimulated by fungal beta-glucans and Toll-like receptor agonists. Infect Immun 77:1774–1781. doi: 10.1128/IAI.00086-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Soto ER, Ostroff GR. 2008. Characterization of multilayered nanoparticles encapsulated in yeast cell wall particles for DNA delivery. Bioconjug Chem 19:840–848. doi: 10.1021/bc700329p. [DOI] [PubMed] [Google Scholar]
- 48.Bremel RD, Homan EJ. 2010. An integrated approach to epitope analysis. II. A system for proteomic-scale prediction of immunological characteristics. Immunome Res 6:8. doi: 10.1186/1745-7580-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bremel RD, Homan EJ. 2013. Recognition of higher order patterns in proteins: immunologic kernels. PLoS One 8:e70115. doi: 10.1371/journal.pone.0070115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bremel RD, Homan EJ. 2014. Frequency patterns of T-cell exposed amino acid motifs in immunoglobulin heavy chain peptides presented by MHCs. Front Immunol 5:541. doi: 10.3389/fimmu.2014.00541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bremel RD, Homan EJ. 2015. Extensive T-cell epitope repertoire sharing among human proteome, gastrointestinal microbiome, and pathogenic bacteria: implications for the definition of self. Front Immunol 6:538. doi: 10.3389/fimmu.2015.00538. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Survival of C57BL/6 and BALB/c mice vaccinated with Cda1 and Cda2 and challenged with C. neoformans strain KN99 or C. gattii strain R265. Mice were vaccinated with a mixture of Cda1 and Cda2 as in Fig. 2C, except the vaccination dose for each antigen was 6 µg. The challenge dose of KN99 and R265 was 104 CFU. (A) Survival curves of vaccinated and challenged mice. Data are from two independent experiments with 4 or 5 mice per group. P < 0.001 comparing C57BL/6 KN99 with C57BL/6 R265. P < 0.001 comparing BALB/c KN99 with BALB/c R265. Unvaccinated controls all died between 16 and 33 days postinfection (data not shown). (B) Lung fungal burdens of mice that survived for 12 weeks. For panel B, the dotted line identifies CFU of KN99 and R265 challenge; the median CFU per lung for each group are indicated by a solid line. Download FIG S1, PDF file, 0.4 MB (390.7KB, pdf) .
Copyright © 2017 Specht et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Pulmonary inflammation and fungal burden seen on pathology. BALB/c mice were vaccinated three times with 10 µg/vaccine dose each of GP-Cda1 and GP-Cda2 (Cda1/2) and then challenged with 104 CFU of C. neoformans strain KN99. Control mice received GP-MSA (MSA). At 7 and 14 days postchallenge, mice were euthanized, and stained slides prepared from sectioned lungs were scored by a pathologist blind as to group. n = 5 mice/group. For fungal burdens, P < 0.05 and P < 0.001, comparing MSA with Cda1/2 at days 7 (D7) and 14 (D14), respectively. Download FIG S2, PDF file, 0.1 MB (97KB, pdf) .
Copyright © 2017 Specht et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Comparison of B cell linear epitopes and human and murine MHC-II binding patterns. For each protein tested as a candidate vaccine antigen, the probability of participation in a B cell linear epitope and predicted MHC-II binding affinities of sequential 15-mer peptides are shown in the five panels. Panels should be read from top to bottom. Panel 1 shows the probability of linear B cell epitopes for both mice and humans. Panel 2 shows the permuted human MHC-II binding, smoothed across 16 DRB alleles. Panel 3 shows MHC-II binding for allele H-2-IAb (C57BL/6 mice). Panel 4 shows MHC-II binding for allele H-2-IAd (BALB/c mice). Panel 5 shows MHC-II binding for human allele DRB1-0401 found in transgenic mouse strain DR4. y-axis units are the inverted standard deviation of linear B cell epitope probability, and for MHC binding panels, they are standard deviation units below the mean of the natural log binding affinity (ln of the 50% inhibitory concentration [lnIC50]) for that protein. The vertical black line in each set of panels indicates the boundary between vector sequence that includes the His tag and each Cryptococcus protein sequence. The epitope predictions illustrated in panels A to F were generated using the ioGenetics’ EigenBio platform. The EigenBio algorithms are written on JMP (SAS Institute). The rationale underlying the predictions is further described in several publications by R. D. Bremel and E. J. Homan as cited in Text S1 in the supplemental material (Immunome Res 6:8, 2010, https://doi.org/10.1186/1745-7580-6-8; Immunome Res 6:7, 2010, https://doi.org/10.1186/1745-7580-6-7; PLoS One, 8:e70115, 2013, https://doi.org/10.1371/journal.pone.0070115; Front Immunol 5:541, 2014, https://doi.org/10.3389/fimmu.2014.00541). Download FIG S3, PDF file, 0.4 MB (452.7KB, pdf) .
Copyright © 2017 Specht et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Methods specific to Fig. S3. Download TEXT S1, DOCX file, 0.1 MB (17.5KB, docx) .
Copyright © 2017 Specht et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.