

Abstract
Kinesin spindle protein inhibition is known to be an effective therapeutic approach in several malignancies. Filanesib (ARRY-520), an inhibitor of this protein, has demonstrated activity in heavily pre-treated multiple myeloma patients. The aim of the work herein was to investigate the activity of filanesib in combination with pomalidomide plus dexamethasone backbone, and the mechanisms underlying the potential synergistic effect. The ability of filanesib to enhance the activity of pomalidomide plus dexamethasone was studied in several in vitro and in vivo models. Mechanisms of this synergistic combination were dissected by gene expression profiling, immunostaining, cell cycle and short interfering ribonucleic acid studies. Filanesib showed in vitro, ex vivo, and in vivo synergy with pomalidomide plus dexamethasone treatment. Importantly, the in vivo synergy observed in this combination was more evident in large, highly proliferative tumors, and was shown to be mediated by the impairment of mitosis transcriptional control, an increase in monopolar spindles, cell cycle arrest and the induction of apoptosis in cells in proliferative phases. In addition, the triple combination increased the activation of the proapoptotic protein BAX, which has previously been associated with sensitivity to filanesib, and could potentially be used as a predictive biomarker of response to this combination. Our results provide preclinical evidence for the potential benefit of the combination of filanesib with pomalidomide and dexamethasone, and supported the initiation of a recently activated trial being conducted by the Spanish Myeloma group which is investigating this combination in relapsed myeloma patients.
Introduction
The use of novel agents has resulted in a clear improvement in the survival of multiple myeloma (MM) patients. However, most patients eventually relapse,1 denoting the need for new drugs targeting key pathogenic mechanisms of the tumor plasma cell.2 CYCLIN D deregulation is a common oncogenic event found in 98% of MM patients,3 and considerable effort has been expended in trying to identify agents targeting the cell cycle of myeloma cells. Examples of such molecules are seliciclib (an inhibitor of CDK4/CDK6)4 and AURORA KINASE inhibitors.5 Unfortunately, thus far these agents have not proved to be sufficiently effective or were stopped due to toxicity6 in myeloma patients, who subsequently continued MM disease development.
Filanesib (ARRY-520), a first-in-class7 kinesin spindle protein (KSP) inhibitor, is a novel agent targeting this same pathogenic area.8 KSP (EG5/KIF11) is a member of the mitotic kinesin family that is only expressed in dividing cells9 and is essential for establishing the mitotic bipolar spindle and ensuring centrosome separation.10 Inhibition of KSP activity arrests cells in metaphase by forming aberrant monopolar spindles and impairing the separation of centrosomes.11 The activity of filanesib is determined by two main factors: first, the integrity of components of the spindle checkpoint, which arrests cells when an alteration in mitosis is found, and second, the loss of anti-apoptotic signals12 during mitotic blockade, particularly the decrease in the MCL-1 protein.13 This latter protein is essential for the survival of MM cells,13,14 therefore, myeloma cells might be particularly susceptible to filanesib treatment.12 This agent has already been explored in MM in a phase II clinical trial, in which it gave a 16% response rate (≥partial response (PR)) in heavily treated patients who had received all available agents and a median of six previous lines of therapy.7 This initial activity prompted the search for potential combinations that could enhance the activity of the current backbones of therapy in relapsed MM.
In this context, pomalidomide in combination with dexamethasone induces a 30% overall response rate and prolongs overall survival by up to one year in patients already exposed to immunomodulatory drugs (IMiDs) and proteasome inhibitors and refractory to the last line of therapy.15,16 However, novel partners for combination with this doublet are currently being sought, with the aim of improving these results.
In the present study we evaluated the preclinical anti-myeloma activity of the triple combination of pomalidomide+dexamethasone+filanesib (PDF). Preliminary data reported synergy of filanesib with pomalidomide in a xenograft mouse model.17
Herein, we demonstrate that filanesib is a good partner for combination with all IMiDs plus dexamethasone, the combination with pomalidomide being particularly potent in the dexamethasone sensitive MM.1S cell line, and very effective in a large panel of other MM cell lines. This synergistic effect is partly mediated by an increase in monopolar spindle formation and the simultaneous upregulated expression and activation of the proapoptotic protein BAX in actively proliferating myeloma cells. These findings supported the ongoing clinical trial (clinicaltrials.gov Identifier: 02384083) conducted by the Spanish MM group to evaluate the safety and efficacy of this triple combination in relapsed/refractory MM patients.
Methods
(For more specific information see the Online Supplementary File)
Reagents and drugs
Filanesib was provided by Array BioPharma Inc. (Boulder, CO, USA). Thalidomide, lenalidomide and pomalidomide were purchased from Selleckchem (Houston, TX, USA), dexamethasone from Sigma-Aldrich (St Louis, MO, USA) and bortezomib from LC Laboratories (Woburn, MA, USA).
MM cell lines, patient samples and cultures
Origin, authentication and in vitro growth conditions of MM cell lines have already been characterized.18,19 The study of the activity in the presence of interleukin (IL)-6, insulin-like growth factor (IGF)-1 or co-culture with stroma was performed as described.20,21 Bone marrow (BM) samples from MM patients were obtained following institutional approval and written informed consent.
Cell viability, cell cycle and apoptosis assays
Cell viability of MM cells was evaluated by the MTT assay.22 The cell cycle profile, apoptosis induction and mitochondrial membrane potential was analyzed by flow as described.21 In vitro synergism was quantitated using CalcuSyn software23 (Biosoft, Ferguson, MO, USA) via a constant ratio drug combination design. Ex vivo analysis of apoptosis in freshly isolated patient cells. Cytometry analyses of apoptosis in BM tumor plasma cells and lymphocytes were performed as described.24
Immunofluorescence study
MM.1S cells treated for 24h were fixed, blocked and incubated overnight with a primary anti-α-tubulin antibody (Sigma) and counterstained with Alexa Fluor 488 conjugated anti-mouse immunoglobulin G (IgG) secondary antibody (Life Technologies, Waltham, MA, USA) and a 4′,6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI) solution (Sigma).
Western blot
All procedures were performed as described.24 All antibodies were purchased from Cell Signaling Technology (Boston, MA, USA) except for anti-MCL-1 (Santa Cruz Biotechnology. Santa Cruz, CA, USA), anti-CYTOCHROME C (Calbiochem, Billerica, MA, USA) and Horseradish peroxidase-conjugated secondary antibodies (GE Healthcare. Little Chalfont, UK).
Gene silencing with siRNA
MM.1S cells were transiently transfected with either 100 nM non-targeting control short interfering RNA (NT-siRNA) or 100 nM ON TARGET plus SMART pool siRNA targeting human BAX (BAX-siRNA; Dharmacon, Lafayette, CO, USA) using the Nucleofector II system (Lonza, Allendale, NJ, USA).
Animal models
The human subcutaneous plasmacytoma model in CB17-severe combined immunodeficiency (SCID) mice (The Jackson Laboratory, Bar Harbor, ME, USA) was used as described.21,25 Animal experiments were conducted with permission from the local Ethical Committee for Animal Experimentation.
Histological and immunohistochemistry (IHC) analyses
This technique has been previously described25 using an anti-BAX antibody (Cell Signaling, Boston, MA, USA) and the EnVision anti-mouse/rabbit peroxidase complexes (Dako, Glostrup, Denmark). Peroxidase activity was identified using the 3,3′-diaminobenzidine MAPO system (Ventana Medical Systems, Roche, Tucson, AZ, USA). Sections were counterstained with hematoxylin and analyzed by standard light microscopy.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) was performed using the In Situ Cell Death Detection Kit (Roche, Mannheim, Germany). Sections were counterstained with DAPI and visualized by confocal laser microscopy (Leica TCS SP2).
RNA isolation, cDNA synthesis and microarray hybridization and analysis
Tumors and MM.1S cells (n=3 for each treatment) were hybridized in Human Gene 2.0 ST arrays (Affymetrix) following manufacturer’s instructions. The raw intensity data were preprocessed using the RMA algorithm26 implemented in the Affymetrix Expression Console, version 1.4.1.46. Differentially expressed genes were identified using the significance analysis of microarrays (SAM) algorithm,27 version 4.01, selecting genes with q value <0.05. Microarray data are available in the Gene Expression Omnibus (GEO) repository database (Accession number GSE94341).
Statistical analyses
Statistical analyses were performed with IBM SPSS Statistics v.21.0 (IBM Corp., Armonk, NY, USA).
Results
Filanesib potentiates the anti-myeloma activity of pomalidomide in combination with dexamethasone
Firstly, the ability of filanesib (F) to enhance the activity of the different IMiDs in combination with dexamethasone (D) was evaluated. MM.1S cells were treated for 48h with different doses of thalidomide + dexamethasone (TD), lenalidomide + dexamethasone (LD) or pomalidomide + dexamethasone (PD), with or without different concentrations of filanesib, and viability was analyzed by MTT assay (Figure 1A). All IMiDs + dexamethasone had a synergistic effect with filanesib, with combination indexes (CIs) in the synergistic range (most synergic CIs of 0.112, 0.103, and 0.063 for TDF, LDF and PDF, respectively; Online Supplementary Figure S1). However, the most effective combination was that of F with PD (Figure 1A).
Figure 1.

IMiDs, especially pomalidomide and dexamethasone, strongly synergized with filanesib. A) Cell viability of MM.1S cells treated with F plus TD, LD, PD, or the triple combination for 48h. Results are shown as the percentage of control. Data are summarized as the mean ± SD (n=3). Combination indexes (CI) for the combination of filanesib and each of the IMiDs together with dexamethasone are shown in Online Supplementary Figure S1B & S1D. Tumor growth curves of SCID mice randomized and treated when their tumors became palpable (B; small plasmocytomas; mean volume around 125 mm3; range 30 – 237; n=6 per group) or reached a substantial volume (D; large plasmacytomas; mean volume around 2500 mm3; range 1617 – 3134; n=3 per group) at the moment of treatment initiation. Experimental groups included: C (vehicle control; intraperitoneal injection (i.p.) 5 days per week), F (10 mg/kg i.p., 2 days per week), D (0.5 mg/kg i.p., 2 days per week), T (50 mg/kg i.p., 5 days per week), L (25 mg/kg i.p., 5 days per week), and P (8 mg/kg i.p., 5 days per week) in monotherapy or in double or triple combination. Data are summarized as the mean ± SEM. C & E) Analysis of survival by Kaplan-Meier estimator from mice with (C) small or (E) large tumors. F) BM cells from 9 MM patients incubated with P 500 nM, D 10 nM and F 1 nM alone and in the different combinations for 48h. Apoptosis induction was analyzed by Annexin-V staining by flow cytometry assay, both in plasma cells and lymphocytes that were identified based on the surface expression of CD38 and CD45, respectively. A statistically significant difference (P<0.05) was found between plasma cells treated with PDF and control. Error bars indicate the last “non-outlier” value at each side. F: filanesib; D: dexamethasone; T: thalidomide; L: lenalidomide; P: pomalidomide; n.s.: not significant.
This potentiation was also maintained in vivo in a subcutaneous plasmacytoma model, as filanesib enhanced the effect of TD, LD and PD in terms of delay in tumor growth (Figure 1B). In this regard, the PDF combination was particularly potent, as the addition of low doses of filanesib significantly reduced the mean tumor volume from day 26 of treatment compared with the standard of care PD. Moreover, the triple combination of PDF completely controlled tumor growth for up to 50 days. PDF was also superior to the other two tested combinations, TDF and LDF (Student’s t-test, P<0.05, from days 8 and 10 of treatment, respectively). The tumor growth control observed with PDF translated into a statistically significant improvement in the survival of treated mice, with a median survival (CI 95%) of 74 (71–76) days for PDF compared with 56 (55–61) days for mice treated with the standard backbone PD (log-rank test, P=0.004; Figure 1C). The survival analysis also favored PDF over the other IMiD combinations (log-rank test, P<0.001 for PDF compared with TDF and LDF; Figure 1C). As filanesib exerts its activity on dividing cells, the PDF combination could be particularly active in highly proliferative cells. Therefore, we assessed the effect of PDF on mice bearing large plasmacytomas growing in the exponential phase. PDF was able to control these rapidly progressing tumors (Figure 1D), and there was also a statistically significant advantage with respect to the reduction of tumor volume compared with filanesib in monotherapy from day 15, and from day 5 when compared with the PD double combination (Student’s t-test, P<0.05). As shown in Figure 1E, there was a noticeable improvement in median survival (CI 95%), from 14 (11–17) and 18 (18–18) days for PD and F, respectively, to 59 (45–73) days for animals treated with PDF (log-rank test, P<0.05 for all comparisons). It is remarkable that PDF was able to improve the survival of these end-stage mice by almost two months. With regard to toxicity, the PDF triple combination was well tolerated by mice in the initial experiment, and the effect was not worse than that of PD alone. In mice with large plasmacytomas, treatment induced a moderate decrease in body weight (not reaching 20%), which was recovered on the days without treatment, and which was partly due to the poor clinical condition of the mice (Online Supplementary Figure S2).
Based on this data and the fact that the combination with pomalidomide and dexamethasone is particularly appealing in the clinical setting, we decided to expand our study of this combination in MM. First, its synergy was evaluated in some additional MM cell lines; MM144, JJN3, RPMI8226-LR5 and OPM-2; showing CIs in the synergistic range (Online Supplementary Figure S3). The potent anti-myeloma effect of the PDF triple combination was maintained in the presence of IGF-1 or IL-6, two important factors in the myeloma BM microenvironment. Additionally, the PDF combination was also effective when the MM.1S cells were co-cultured with either human mesenchymal stromal cells from myeloma patients (BMSCs) or the human mesenchymal stromal line (hMSC-TERT) (Online Supplementary Figure S4).
The effect of PDF was further evaluated ex vivo in BM aspirates of nine MM patients. Both filanesib alone as well as pomalidomide and dexamethasone were active in these patients. However, only the triple combination demonstrated a statistically significant difference when compared with the control (C) untreated cells (PDF vs. C, P=0.04; Figure 1F). Moreover, there was a clear therapeutic window, as the toxicity on the lymphocytes of these same patients was clearly lower.
It is of note that although the efficacy of the triple combination PDF was observed in vitro in several MM cell lines as well as in patients samples, its benefit over LDF or TDF was only explored in a single cell line.
The PDF combination deregulates genes necessary for mitosis
In order to investigate the mechanism of action of the triple combination, changes induced by the different treatments in the gene expression profiles of MM.1S cells in vitro or in tumors from treated mice were analyzed and compared with their respective untreated controls. In both studies, whereas filanesib alone induced a minimal genomic deregulation with respect to the control (6 and 4 genes significantly deregulated in the in vitro and in vivo experiments, respectively), treatment with PDF enhanced the effect of PD, particularly in the in vivo study (3460 vs. 3317 and 238 vs. 141 deregulated genes in the in vitro and in vivo studies, respectively; Figure 2A,B. See Online Supplementary Table S1 and Table S2 for differentially deregulated genes in the PDF combination in vitro and in vivo, respectively. A complete list of the 3460 genes deregulated in vitro is also provided as a supplement). Due to the variety of functions in which these genes were involved, a functional enrichment analysis was performed in order to identify the biological processes most significantly affected by the deregulated genes following exposure to PD, F and PDF treatments in both experiments (see Table 1 and Venn diagrams for biological processes in Figure 2C,D). Three biological processes were commonly deregulated by all treatments in the in vitro experiments (while no common process was discovered in the in vivo experiment), and all of them were related to mitosis and cell cycles (Table 1). Of interest, the top five biological processes exclusively deregulated in the PDF combination in vitro were involved in different stages of mitosis; in line with this, five out of the 17 biological processes uniquely altered in the PDF treatment in vivo were also associated with mitosis and nuclear division (see shaded processes in Table 1).
Figure 2.

Effect of PD, F and PDF treatments on gene expression profiles. A & B) Venn diagrams of significantly deregulated genes in MM.1S cells (A) or tumors from mice (B) after treatment with PD, F and PDF vs. control. C & D) Venn diagrams of the biological processes (Gene Ontology at level 5) deregulated by the PD, F and PDF treatments vs. control in in vitro (C) and in vivo (D) studies. A list of the most significant biological processes deregulated in the in vitro and in vivo studies is shown in Table 1; only processes from level 5 Gene Ontology categories (the most specific processes) were considered, in order to avoid redundancy. P: pomalidomide; D: dexamethasone; F: filanesib; C: control.
Table 1.
The most significant biological processes which were deregulated with the treatments.
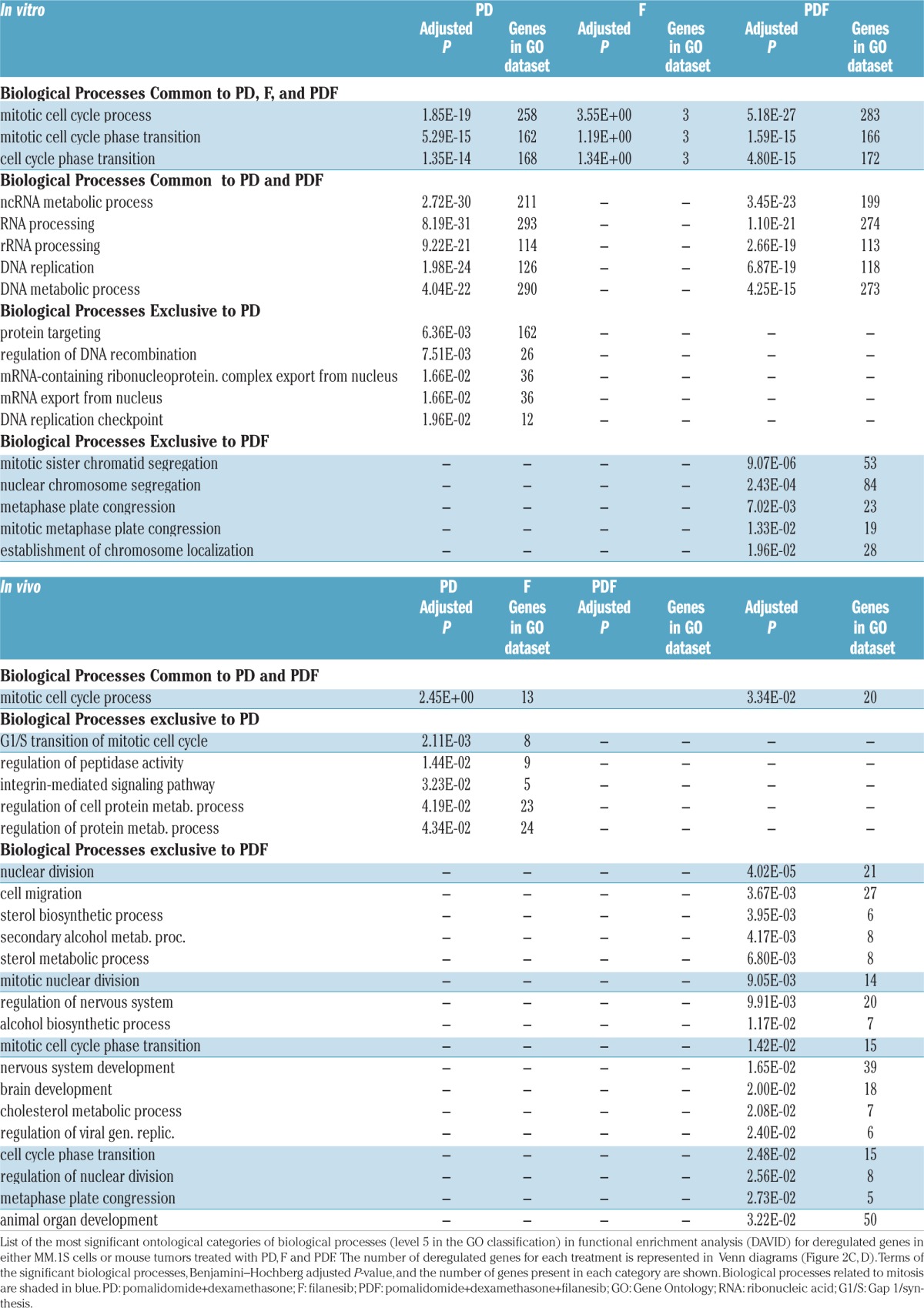
Next, we selected those genes deregulated in the triple combination in vitro which were involved in mitotic and cell cycle processes (Online Supplementary Table S3). Of note, CCNB1 and CCNB2, encoding for B-type cyclins and being major regulators of the G2/M transition of cell cycle, are upregulated in the PDF treatment. The upregulated expression of PSMD3, a 26S proteasome subunit, and NEK2, involved in the anaphase promoting complex and centrosomal separation, may also be mediating the increased mitotic and cell cycle progression processes in this combination. Similarly, CDC25C and CDC25B, required for entry into mitosis, show an increased expression in the PDF combination.
The PDF combination enhances the formation of aberrant monopolar spindles
One mechanism underlying the synergy of the triple combination could be the enhancement of the ability of filanesib to block mitosis and to induce monopolar spindles, the characteristic sequelae of KSP inhibition. To test this hypothesis, the formation of aberrant monopolar spindles was evaluated after a 24h treatment of MM.1S cells with vehicle, F, PD or PDF. Whereas the majority of untreated cells and those exposed to PD displayed normal mitoses with typical bipolar spindles, almost all F- and PDF-treated cells that were in mitosis showed a monopolar spindle phenotype (Figure 3A). Interestingly, and differentiating these latter two conditions, treatment with the triple combination increased the percentage of cells in mitosis, which resulted in a significant increase in the absolute number of monopolar spindles (0, 0, 8 and 19 aberrant spindles per 100 cells, for the control, PD, F and PDF, respectively (P=0.01 for the F vs. PDF comparison) (Figure 3A). These results were confirmed in vivo by immunohistochemistry, since no aberrant monopolar spindles were found in the control and PD-treated tumors, while F and PDF displayed an average of 6 and 11 monopolar spindles per field (P=0.008 for the F vs. PDF comparison; Figure 3B).
Figure 3.

PDF boosted the formation of monopolar spindles A) Quantitative determination of monopolar spindles in MM.1S cells treated with the vehicle (control), PD, F or PDF for 24h. Representative micrographs showing cells immunostained with anti-tubulin antibody (shown in green) to visualize microtubules and DAPI (blue) to evidence nuclei using confocal microcopy. Bar = 15 μm in lower magnification micrograph; bar = 5 μm in higher magnification insert). Data are presented as the mean ± SD of two independent experiments. Statistical significance was evaluated with Student’s t-test. B) Evaluation of monopolar spindles in paraffin sections of tumors from mice treated with vehicle (control=C), PD, F or PDF for two days and stained with hematoxylin and eosin (H&E). Thirty high-power fields (600×) were evaluated for each experimental condition (3 mice per condition). Monopolar spindles were characterized by chromosomes stained with hematoxylin (in purple) orientated in a ring. Bar = 10 μm). Data are presented as the mean ± SD. Statistical significance was evaluated with Student’s t-test. P: pomalidomide; D: dexamethasone; F: filanesib.
PDF causes cell cycle arrest in G2/M phases and specifically induces apoptosis in cells arrested in proliferative phases
The increase in mitotic cells observed with the triple combination was intriguing, and therefore its specific effect on the cell cycle profile of treated tumor cells was studied (Figure 4A). To check whether there was a particular susceptibility to apoptosis in cells at certain phases of the cell cycle, simultaneous staining with Draq5 and Annexin-V was performed (Figure 4B). The treatment with filanesib in monotherapy arrested cells in the proliferative phases of the cell cycle, with 49% of cells in synthesis (S) and gap 2 mitosis (G2-M) phases compared with 36% in control cells (two-tailed Student’s t-test from three independent experiments P=0.03). The PDF triple combination did not show much difference in this regard compared with filanesib alone, with 44% of cells in G2-M arrest (Figure 4A). However, treatment with filanesib specifically induced apoptosis of cells in G2-M phases with 58% of them being apoptotic compared with only 33% in non-proliferating phases (Figure 4B,C). This fact was significantly enhanced in the triple combination as most apoptotic cells (88%) were seen to be in proliferative phases (G2 or M) (Figure 4B,C). In order to confirm these findings, various cell cycle markers were biochemically analyzed. MM.1S cells treated with filanesib and the triple combination showed an accumulation of CYCLIN B1 levels at 12h and 24h as shown by western blot, this being indicative of the activation of the spindle assembly checkpoint (SAC),28 which correlated with the percentage of cells arrested in G2-M phases. In contrast to treatment with filanesib, CYCLIN B1 levels rapidly decreased after 48h in the PDF combination, possibly indicating an attempt of the cells to exit from mitosis.29,30 However, they proved unable to do so, as after 48h cells treated with PDF died in mitosis via apoptosis, as shown by an increase in phosphorylated histone H3 protein levels, a marker of mitotic cells,31 and PARP cleavage and degradation32 (Figure 4D).
Figure 4.

PDF triggered cell cycle arrest and apoptosis of MM cells in pro-liferative phases. (A) Analysis of cell cycle by flow cytometry in MM.1S cells treated with the vehicle (control=C), PD, F or PDF for 48h after Draq5 staining (Draq5 binds to DNA from both living and non-living cells). B) Simultaneous analysis of cell cycle profile and apoptosis induction in MM.1S cells treated with C, PD, F or PDF for 48h by flow cytometry after Draq5/Annexin-V staining. C) Percentage of apoptotic cells in each phase of the cell cycle was also calculated. Data shown is for a representative experiment that was repeated at least twice. D) Western blot analysis of cell cycle proteins CYCLIN B1, phosphorylated HH3 and PARP in MM.1S cells treated with indicated conditions for 12h to 48h. E) Apoptosis induction in MM.1S cells treated with C, PD, F or PDF for 48h and analyzed by flow cytometry after staining with Annexin-V. F) Micrographs of tumor sections from mice treated for two days with C, PD, F or PDF and histochemically stained with TUNEL assay. Quantitative determination of TUNEL-positive cells (30 630× fields per experimental condition, 3 mice per condition). Data are expressed as the mean ± SD (n=3); **P<0.01 (Student’s t-test). P: pomalidomide; D: dexamethasone; F: filanesib.
The increase in apoptotic induction was confirmed by Annexin-V/IP staining, as treatment of MM.1S with both PD and filanesib for 48h induced an increase of Annexin-V-positive cells (23% and 56%, respectively) compared with the control (5%), and this effect was much greater with the triple combination (70% apoptosis; Figure 4E). In the same manner, the TUNEL assay of treated tumors revealed a significant increase in apoptotic cells in the triple combination compared with filanesib or PD alone (Figure 4F).
Anti-myeloma efficacy of filanesib in monotherapy depends on the levels of both the anti-apoptotic protein MCL-1 and the proapoptotic protein BAX
To gain insight into the mechanism underlying the enhanced apoptotic response in dividing cells with PDF treatment, key molecular features associated with sensitivity to filanesib alone were investigated. A dose response to filanesib at 48h in 11 cell lines showed that, although all of them were sensitive to this agent, some cell lines were more sensitive to filanesib treatment, with almost no viable cells at 2.5 nM (Figure 5A). Other cells, however, had higher IC50 values and 30–50% of cells remained viable even at higher doses of the drug (Figure 5A). Given that the sensitivity to filanesib has previously been correlated with levels of MCL-1, the basal expression of six representative BCL-2 family members was evaluated in the same panel of MM cell lines showing different patterns of expression (Figure 5B). No correlation was observed between MCL-1 expression and filanesib sensitivity. However, the sensitivity to filanesib showed a direct correlation with basal expression levels of BAX, as shown in Figure 5C (P<0.05), whereby cell lines with higher basal BAX levels were particularly sensitive to this agent. In contrast, no correlation was observed between BAX expression and sensitivity to the proteasome inhibitor bortezomib, which we used here as a control (Online Supplementary Figure S5). To confirm the role of BAX in filanesib-induced cell death, MM.1S cells were transfected with BAX-specific siRNA. BAX knockdown induced resistance to filanesib treatment, reducing the number of apoptotic cells by half (Figure 5D). A similar effect related to BAX levels was observed in OPM-2 cells (data not shown).
Figure 5.

Role of the increase and activation of the proapoptotic protein BAX on the anti-myeloma efficacy of F and PDF treatments. (A) Cell viability of a panel of MM cell lines incubated with increasing concentrations of F for 48h and analyzed by MTT assay. Data are expressed as a percentage of control values. B) Western blot analysis of basal levels of six BCL-2 family members (BCL-2, BCL-XL, MCL-1, BAX, BAK, BAD) studied in the same MM cell lines. C) Expression levels for BAX were determined by densitometry analysis of bands (using ImageJ software) and relative to those of β-ACTIN. Relation between the basal levels of BAX protein and drug sensitivity to F was calculated with respect to IC50 for 48h. Statistical analysis was performed with the Mann-Whitney U test. D) Immunoblot of MM.1S cells 24h and 48h after transfection with BAX-specific siRNA. Nontransfected cells (NT) were also analyzed for comparison. 24h after siRNA transfection, cells were treated with either filanesib (F) or bortezomib (B) for an additional 24h, and apoptosis was assessed by Annexin-V+ cells and evaluated by flow cytometry. E) MM.1S cells were treated for 12–48h with 10 nM F, and expression levels of MCL-1 and BAX in the mitochondrial and cytosolic fractions were analyzed by western blot. COX IV and GAPDH expression were used as loading controls for mitochondrial and cytosolic, respectively. F & G) Immunoblot of total (F), mitochondrial and cytosolic (G) extracts from MM.1S cells treated for 12–48h with the vehicle (control=C), PD, F or PDF for 48h. The expression of COX IV and GAPDH proteins were used as mitochondrial and cytosolic loading controls, respectively. H) Analysis of mitochondrial membrane potential in MM.1S cells treated with different conditions as assessed by flow cytometry after TMRE staining. I) Immunohistochemical staining of BAX in tumors from mice treated for two days under the indicated conditions. Bar = 20 μm. In Figure E, “BAX over” indicates overexposure of the film to better visualize mild changes in the levels of this protein; “p18 BAX” indicates the proapoptotic 18 kDa BAX fragment. P: pomalidomide; D: dexamethasone.
Moreover, subcellular fractionation studies indicated that treatment with filanesib triggered the translocation of BAX from the cytoplasm to the mitochondria, where it was cleaved into the very potent proapoptotic 18 kDa fragment33 (Figure 5E). Simultaneously, the levels of the prosurvival protein MCL-1 also decreased in both subcellular compartments (Figure 5E). These findings suggest the potential importance of the modulation of both BAX and MCL-1 protein levels in filanesib-induced apoptosis.
The PDF triple combination induces augmented expression and activation of BAX protein
Interestingly, and unlike filanesib alone, MCL-1 levels were not significantly affected after treatment with PDF. In contrast, two events were clearly potentiated in the triple combination compared with filanesib alone: first, PDF induced the expression of total BAX levels in vitro (Figure 5F), and second, PDF enhanced the presence of this protein in the mitochondria, where it exerts its apoptotic activity (Figure 5G). This event led to the permeabilization of the external mitochondrial membrane, resulting in the release of the apoptogenic factor CYTOCHROME C into the cytosol (Figure 5G), and an associated decrease in mitochondrial membrane potential, as observed by flow cytometry with tetramethylrhodamine, ethyl ester (TMRE; Figure 5H). Finally, in vivo experiments confirmed the PDF-induced BAX upregulation (Figure 5I).
Discussion
MM remains an incurable disease in most patients34 and there is an urgent need for novel drugs to improve this situation. In the work herein, we have preclinically evaluated the combination of various IMiDs together with dexamethasone and filanesib, a novel KSP inhibitor that particularly affects dividing cells and depends on the survival protein MCL-1, which is essential for MM cell endurance.13,14 This novel therapeutic agent has already demonstrated an anti-myeloma effect with dexamethasone,7 and is currently being explored in combination with protea-some inhibitors.35 Preliminary data have demonstrated synergy of filanesib with pomalidomide in a mouse xenograft model,17 and our in vitro, ex vivo and, notably, in vivo results clearly confirm this effect and provide evidence of the benefit of the addition of dexamethasone. This strong synergistic effect was confirmed in several MM cell lines, even in the presence of stromal cells. The potency of the combination with pomalidomide was shown to be better than those including thalidomide or lenalidomide, however, one limitation of the present study is that the advantages bestowed by PDF over TDF or LDF was only demonstrated in one cell line, that of MM.1S, particularly in the in vivo model. Nevertheless, taking into account that pomalidomide and dexamethasone is currently considered a backbone for combinations we decided to elaborate on this particular combination.
Since filanesib is an agent designed to interfere with the mechanisms of cell division, it is reasonable to think that it might be particularly effective in actively dividing cells. In line with this, the in vivo effect of the triple combination was particularly potent in mice bearing large tumors in the exponential phase of growth. Interestingly, this combination resulted in an extended survival of almost two months, on average, in these end-stage mice. Analysis of the gene expression profile of both MM cells and PDF-substantial proportion of upregulated genes to be involved in the centrosome separation (NEK2), control of spindle assembly checkpoint (CCNB1, CCNB2) and entry into mitosis (CDC25 C and CDC25 B), implying that the triple combination significantly alters mitotic processes. Recently, the overexpression of CCNB2 was associated with an acceleration in centrosome separation.36 Based on these data, the antimyeloma effect of the triplet could be due to an increased mitosis and an increased monopolar spindle formation. In order to investigate this hypothesis, the phenotype of cells in metaphase was evaluated by immunohistochemistry. The triple combination induced an increase in the frequency of cells with an aberrant monopolar spindle phenotype compared with filanesib monotherapy. Accordingly, we demonstrated that the PDF combination generated the same degree of G2-M arrest as did filanesib alone, but the combination provoked a substantially more specific apoptosis of these cells when arrested in proliferative phases. A delicate balance between pro-apoptotic and anti-apoptotic members of the BCL-2 family and their subcellular localization generally determines the fate of proliferative cells treated with anti-mitotic agents.37–40 In particular, it has been reported that apoptosis is enhanced by the activation of BAX in cancer cells using other KSP inhibitors.41,42 Accordingly, silencing BAX prompted cell survival under filanesib treatment in monotherapy. In line with this, PDF treatment induced increased apoptosis in proliferative phases that was associated with a significantly higher expression of the proapoptotic BAX protein, promoting its activation. In parallel with these results, the tumors of mice treated with PDF also showed higher immunoreactivity for BAX. Moreover, we detected a release of the apoptogenic factor CYTOCHROME C into the cytosol, possibly due to permeabilization of the mitochondrial membrane induced by BAX. These experiments suggest that the expression levels and activation of BAX are crucial in determining the fate of MM cells treated with PDF.
In conclusion, we report the strong synergy of the PDF triple combination in vitro and especially in vivo in preclinical models of MM. The activity of PDF relies on the induction of monopolar spindles and arrest in mitosis, characterized by primary activation of SAC; increased apoptosis in these mitosis-arrested cells is characterized by the increased expression and activation of BAX, which could be a useful predictive biomarker of response to PDF in MM. These results support the phase II clinical trial POMDEFIL, which is currently being conducted by the Spanish MM group to evaluate this combination in refractory MM patients.
Supplementary Material
Acknowledgments
The authors thank Phil Mason for his help in reviewing the English language of our manuscript.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/12/2113
Funding
This work was funded in part by Array BioPharma, by the Spanish ISCIII-FIS (PI 15/0067 and PI 15/2156) and FEDER, the Spanish RTICC (RD12/0036/0058), Spanish Association Against Cancer (AECC, GCB120981SAN) and the Regional Council of Castilla y León (GRS 1029/A/14, GRS 1175/A/15 and FIC335U14).
References
- 1.Kumar SK, Lee JH, Lahuerta JJ, et al. Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: a multi-center international myeloma working group study. Leukemia. 2012;26(1):149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ocio EM, Richardson PG, Rajkumar SV, et al. New drugs and novel mechanisms of action in multiple myeloma in 2013: a report from the International Myeloma Working Group (IMWG). Leukemia. 2014; 28(3):525–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bergsagel PL, Kuehl WM, Zhan F, Sawyer J, Barlogie B, Shaughnessy J., Jr Cyclin D dysregulation: an early and unifying pathogenic event in multiple myeloma. Blood. 2005; 106(1):296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Niesvizky R, Lentzsch S, Badros AZ, et al. A phase I study of PD 0332991: complete CDK4/6 inhibition and tumor response in sequential combination with bortezomib and dexamethasone for relapsed and refractory multiple myeloma. ASH Annual Meeting Abstracts. 2010; 116(21):860. [Google Scholar]
- 5.Rosenthal A, Kumar S, Hofmeister C, et al. A phase Ib study of the combination of the aurora kinase inhibitor alisertib (MLN8237) and bortezomib in relapsed multiple myeloma. Br J Haematol. 2016; 174(2):323–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kollareddy M, Zheleva D, Dzubak P, Brahmkshatriya PS, Lepsik M, Hajduch M. Aurora kinase inhibitors: progress towards the clinic. Invest New Drugs. 2012; 30(6):2411–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lonial S, Shah JJ, Zonder J, Bensinger WI, Cohen AD, Kaufman JL, et al. Prolonged survival and improved response rates with ARRY-520 in relapsed/refractory multiple myeloma (RRMM) patients with low α-1 acid glycoprotein (AAG) levels: results from a phase 2 study. Blood. 2013; 122(21):285. [Google Scholar]
- 8.Ocio EM, Mitsiades CS, Orlowski RZ, Anderson KC. Future agents and treatment directions in multiple myeloma. Expert Rev Hematol. 2014;7(1):127–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jackson JR, Patrick DR, Dar MM, Huang PS. Targeted anti-mitotic therapies: can we improve on tubulin agents¿ Nat Rev Cancer. 2007;7(2):107–117. [DOI] [PubMed] [Google Scholar]
- 10.Blangy A, Lane HA, d’Herin P, Harper M, Kress M, Nigg EA. Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell. 1995;83(7):1159–1169. [DOI] [PubMed] [Google Scholar]
- 11.Stern BM, Murray AW. Lack of tension at kinetochores activates the spindle checkpoint in budding yeast. Curr Biol. 2001; 11(18):1462–1467. [DOI] [PubMed] [Google Scholar]
- 12.Tunquist BJ, Woessner RD, Walker DH. Mcl-1 stability determines mitotic cell fate of human multiple myeloma tumor cells treated with the kinesin spindle protein inhibitor ARRY-520. Mol Cancer Ther. 2010;9(7):2046–2056. [DOI] [PubMed] [Google Scholar]
- 13.Peperzak V, Vikstrom I, Walker J, et al. Mcl-1 is essential for the survival of plasma cells. Nat Immunol. 2013;14(3):290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang B, Gojo I, Fenton RG. Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood. 2002;99(6):1885–93. [DOI] [PubMed] [Google Scholar]
- 15.San Miguel J, Weisel K, Moreau P, et al. Pomalidomide plus low-dose dex-amethasone versus high-dose dexamethasone alone for patients with relapsed and refractory multiple myeloma (MM-003): a randomised, open-label, phase 3 trial. Lancet Oncol. 2013;14(11):1055–1066. [DOI] [PubMed] [Google Scholar]
- 16.Dimopoulos MA, Palumbo A, Corradini P, et al. An updated analysis of the STRATUS trial (MM-010): safety and efficacy of pomalidomide plus low-dose dexamethasone (POM + LoDEX) in patients (Pts) with relapsed/refractory multiple myeloma (RRMM). Blood. 2015;126(23):4225. [Google Scholar]
- 17.Humphries MJ, Anderson D, Williams L, Rieger R, Tunquist B, Walker D. ARRY-520 combined with pomalidomide displays enhanced anti-tumor activity in preclinical models of multiple myeloma. Blood. 2013;122(21):3167. [Google Scholar]
- 18.Paino T, Garcia-Gomez A, Gonzalez-Mendez L, et al. The novel pan-PIM kinase inhibitor, PIM447, displays dual antimyeloma and bone-protective effects, and potently synergizes with current standards of care. Clin Cancer Res. 2017;23(1):225–238. [DOI] [PubMed] [Google Scholar]
- 19.Herrero AB, San Miguel J, Gutierrez NC. Deregulation of DNA double-strand break repair in multiple myeloma: implications for genome stability. PLoS One. 2015;10(3):e0121581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia-Gomez A, Quwaider D, Canavese M, et al. Preclinical activity of the oral proteasome inhibitor MLN9708 in Myeloma bone disease. Clin Cancer Res. 2014;20(6):1542–1554. [DOI] [PubMed] [Google Scholar]
- 21.Ocio EM, Maiso P, Chen X, et al. Zalypsis: a novel marine-derived compound with potent antimyeloma activity that reveals high sensitivity of malignant plasma cells to DNA double-strand breaks. Blood. 2009;113(16):3781–3791. [DOI] [PubMed] [Google Scholar]
- 22.Paino T, Sarasquete ME, Paiva B, et al. Phenotypic, genomic and functional characterization reveals no differences between CD138++ and CD138low sub-populations in multiple myeloma cell lines. PLoS One. 2014;9(3):e92378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010; 70(2):440–446. [DOI] [PubMed] [Google Scholar]
- 24.Maiso P, Carvajal-Vergara X, Ocio EM, et al. The histone deacetylase inhibitor LBH589 is a potent antimyeloma agent that overcomes drug resistance. Cancer Res. 2006;66(11):5781–5789. [DOI] [PubMed] [Google Scholar]
- 25.Ocio EM, Vilanova D, Atadja P, et al. In vitro and in vivo rationale for the triple combination of panobinostat (LBH589) and dexamethasone with either bortezomib or lenalidomide in multiple myeloma. Haematologica. 2010;95(5):794–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31(4):e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98(9):5116–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8(5):379–393. [DOI] [PubMed] [Google Scholar]
- 29.Pines J. Cubism and the cell cycle: the many faces of the APC/C. Nat Rev Mol Cell Biol. 2011;12(7):427–438. [DOI] [PubMed] [Google Scholar]
- 30.Murray A. Cyclin ubiquitination: the destructive end of mitosis. Cell. 1995;81(2):149–152. [DOI] [PubMed] [Google Scholar]
- 31.Schimming TT, Grabellus F, Roner M, et al. pHH3 immunostaining improves interobserver agreement of mitotic index in thin melanomas. Am J Dermatopathol. 2012;34(3):266–269. [DOI] [PubMed] [Google Scholar]
- 32.Kaufmann SH, Desnoyers S, Ottaviano Y, Davidson NE, Poirier GG. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res. 1993;53(17):3976–3985. [PubMed] [Google Scholar]
- 33.Cao X, Deng X, May W. Cleavage of Bax to p18 Bax accelerates stress-induced apoptosis, and a cathepsin-like protease may rapidly degrade p18 Bax. Blood. 2003;102(7):2605–2614. [DOI] [PubMed] [Google Scholar]
- 34.Barlogie B, Mitchell A, van Rhee F, Epstein J, Morgan GJ, Crowley J. Curing myeloma at last: defining criteria and providing the evidence. Blood. 2014;124(20):3043–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shah JJ, Thomas S, Weber DM, Wang M, Orlowski R. Novel kinesin spindle protein inhibitor ARRY–520 + carfilzomib(Car) in patients with relapsed and/or refractory multiple myeloma (RRMM). Haematologica. 2013;98(S1):Abstract-S579. [Google Scholar]
- 36.Nam HJ, van Deursen JM. Cyclin B2 and p53 control proper timing of cen-trosome separation. Nat Cell Biol. 2014;16(6):538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferlini C, Cicchillitti L, Raspaglio G, et al. Paclitaxel directly binds to Bcl-2 and functionally mimics activity of Nur77. Cancer Res. 2009;69(17):6906–6914. [DOI] [PubMed] [Google Scholar]
- 38.Vijapurkar U, Wang W, Herbst R. Potentiation of kinesin spindle protein inhibitor-induced cell death by modulation of mitochondrial and death receptor apoptotic pathways. Cancer Res. 2007;67(1):237–245. [DOI] [PubMed] [Google Scholar]
- 39.Haschka MD, Soratroi C, Kirschnek S, et al. The NOXA-MCL1-BIM axis de-fines lifespan on extended mitotic arrest. Nat Commun. 2015;6:6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang P, Lindsay J, Owens TW, et al. Phosphorylation of the proapoptotic BH3-only protein bid primes mitochondria for apoptosis during mitotic arrest. Cell Rep. 2014;7(3):661–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tao W, South VJ, Zhang Y, et al. Induction of apoptosis by an inhibitor of the mitotic kinesin KSP requires both activation of the spindle assembly check-point and mitotic slippage. Cancer Cell. 2005;8(1):49–59. [DOI] [PubMed] [Google Scholar]
- 42.Tao W, South VJ, Diehl RE, et al. An inhibitor of the kinesin spindle protein activates the intrinsic apoptotic pathway independently of p53 and de novo protein synthesis. Mol Cell Biol. 2007;27(2):689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.