

Abstract
Type III secretion (T3S), a protein export pathway common to Gram‐negative pathogens, comprises a trans‐envelope syringe, the injectisome, with a cytoplasm‐facing translocase channel. Exported substrates are chaperone‐delivered to the translocase, EscV in enteropathogenic Escherichia coli, and cross it in strict hierarchical manner, for example, first “translocators”, then “effectors”. We dissected T3S substrate targeting and hierarchical switching by reconstituting them in vitro using inverted inner membrane vesicles. EscV recruits and conformationally activates the tightly membrane‐associated pseudo‐effector SepL and its chaperone SepD. This renders SepL a high‐affinity receptor for translocator/chaperone pairs, recognizing specific chaperone signals. In a second, SepD‐coupled step, translocators docked on SepL become secreted. During translocator secretion, SepL/SepD suppress effector/chaperone binding to EscV and prevent premature effector secretion. Disengagement of the SepL/SepD switch directs EscV to dedicated effector export. These findings advance molecular understanding of T3S and reveal a novel mechanism for hierarchical trafficking regulation in protein secretion channels.
Keywords: chaperone, EPEC, in vitro reconstitution, substrate switching, type III secretion
Subject Categories: Membrane & Intracellular Transport; Microbiology, Virology & Host Pathogen Interaction
Introduction
Several Gram‐negative pathogens use the type III protein secretion system (T3SS) to inject virulence proteins directly into eukaryotic host cytoplasms (Portaliou et al, 2016; Deng et al, 2017). T3SS comprises the “injectisome”, a complex, bacterial envelope‐associated, syringe‐like organelle, with four parts: (i) a membrane‐embedded translocase (or export apparatus) that associates with peripheral inner membrane structures including an ATPase. (ii) the basal body, comprising stacked rings spanning both bacterial membranes, connected by an inner rod, which transverses the periplasm. (iii) a protruding needle and (iv) the “translocon”; located at the tip of the needle to form a pore in the host plasma membrane through which bacterial toxins are injected.
T3SS‐exported proteins contain non‐homologous N‐terminal signal sequences (Deng et al, 2015) and are maintained in secretion‐competent states by cytoplasmic chaperones (Parsot, 2003; Wilharm et al, 2007). Chaperones are small, frequently dimeric, and fall in five classes; for example, class V binds early export substrates like the rod and needle subunits; class III and class IV preferentially bind middle substrates like translocators; I, interacts with late effectors.
A unique characteristic of T3S is that secretion through the translocase occurs in consecutive steps of defined, strict hierarchy, secured by poorly understood switching mechanisms that include gatekeeper proteins like SepL of EPEC (Portaliou et al, 2016; Deng et al, 2017). Gatekeepers carry their own secretion signals and class I heterodimeric chaperone suggesting they might act like pseudo‐effectors (Schubot et al, 2005; Botteaux et al, 2009; Burkinshaw et al, 2015).
To dissect the molecular mechanism of substrate targeting and switching during T3S, we used enteropathogenic Escherichia coli (EPEC) that attach to host cells, disrupt microvilli, cause actin rearrangements and infantile diarrhea (Dean & Kenny, 2009). Our model translocator exported protein EspA forms an outer needle sheath (Knutton et al, 1998) and is stabilized for export by CesAB, a 3‐helix, dimeric chaperone (Fig 1A; Creasey et al, 2003; Yip et al, 2005; Chen et al, 2011). Two helices from both EspA‐termini bind through coiled‐coil interactions with CesAB and monomerize it (Yip et al, 2005). EspA, although structurally distinct, shares sequence homology and presumably functions with needle tip proteins from other T3SS (Sato & Frank, 2011; Portaliou et al, 2016; Deng et al, 2017). Our model effector protein is Tir (translocated intimin receptor), chaperoned by CesT (Abe et al, 1999; Thomas et al, 2005).
Figure 1. CesAB comprises two structural and functional distinct domains.
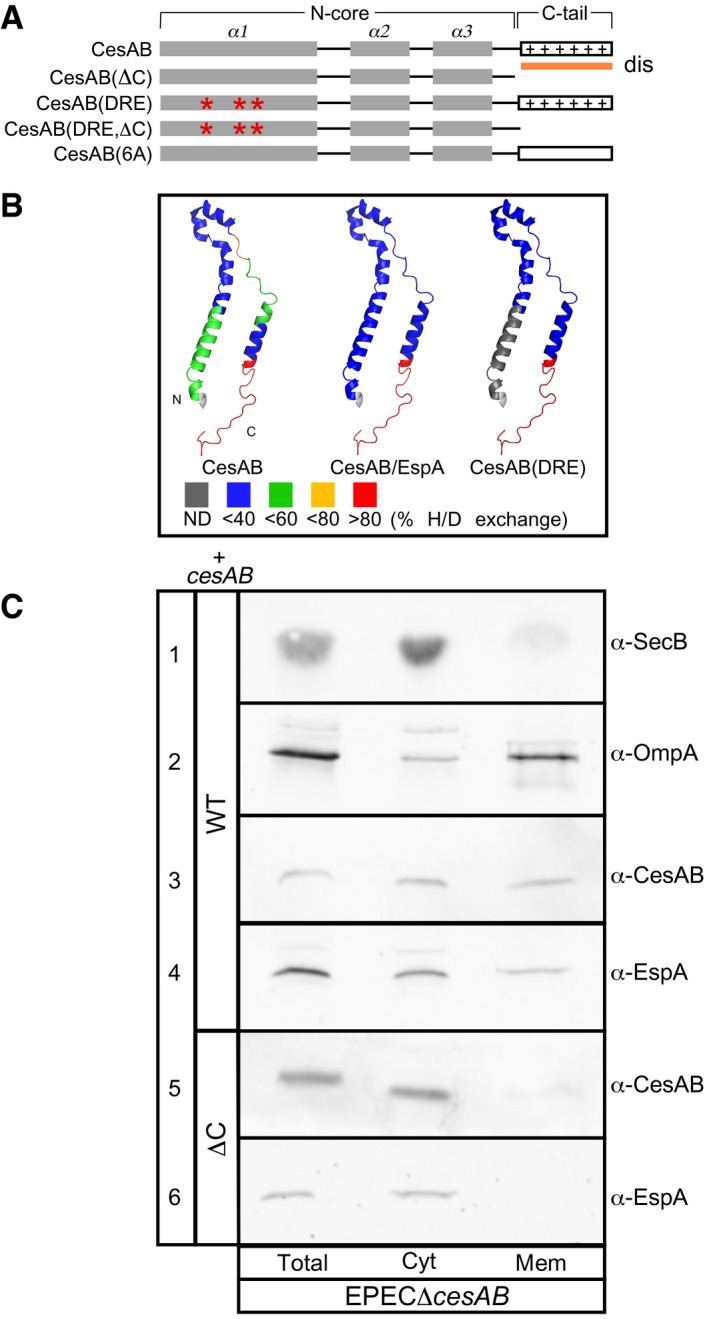
- Schematic representation of CesAB. The N‐core (α1–3 helices), C‐tail (aa: 86–107 that remain unresolved), and mutated derivatives are indicated. Dis, disorder prediction; *, mutations; +, positive charges R89, K91, R99, R101, R104, and K105.
- Local HDX‐MS analysis of CesAB, CesAB(DRE), and CesAB/EspA, mapped on a CesAB protomer. Cold colors: rigid regions (e.g., N‐core); hot colors: unstructured, solvent‐accessible, non‐hydrogen‐bonded regions (e.g., the C‐tail). ND: not determined. See Appendix Table S1 for peptides.
- Sub‐cellular localization of CesAB and EspA in EPECΔcesAB cells expressing plasmid‐borne cesAB or cesAB(ΔC) (see also Fig EV2A). Following fractionation, equal fraction volume (Total, cell proteins; Cyt, cytoplasmic; Mem, membrane) was analyzed on 15% SDS–PAGE and immunostained. Lane 1: cytoplasmic protein SecB; lane 2: membrane protein OmpA. n = 3.
Source data are available online for this figure.
We have now reconstituted T3SS targeting in vitro, using inverted inner membrane vesicles (IMVs) from EPEC, and revealed a novel mechanism that, on the one hand, governs translocator targeting and secretion and on the other the switching to effector export. While the “N‐core” of CesAB (aa 1–86) contains the EspA chaperoning function, its unstructured “C‐tail” is a previously unknown translocase targeting signal. High‐affinity CesAB docking occurs on a bipartite receptor that comprises the gatekeeper SepL (O'Connell et al, 2004), which associates tightly with the membrane and the C‐terminal domain of EscV, a membrane‐embedded subunit of the translocase (Gauthier et al, 2003; Abrusci et al, 2013). Binding of SepL on EscV reduces the affinity of chaperone/effector complexes, such as CesT/Tir, for EscV. SepL removal allows enhanced Tir affinity and secretion. Thus, SepL acts as a substrate affinity switch. SepD, one of the SepL chaperones (Deng et al, 2005), modulates the SepL‐EscV interaction, presumably by altering the conformation of SepL. SepD together with the CesAB C‐tail couple the receptor function of SepL to translocator secretion. Our findings provide quantitative molecular understanding of the T3S targeting process, a novel mechanism for regulating protein trafficking channels and set the stage for the complete mechanistic and biophysical dissection of the T3S machine.
Results
CesAB comprises two structurally and functionally distinct domains
To define chaperone and targeting elements in CesAB, we first analyzed its domain organization and flexibility (Fig 1A) by using hydrogen deuterium exchange mass spectrometry [HDX‐MS (Tsirigotaki et al, 2017b) (Fig 1B; Appendix Table S1)]. The N‐core is stabilized by either EspA binding (Fig 1B, middle) or mimicking mutations (right; i.e., CesAB(D14L/R18D/E20L), hereafter DRE (Chen et al, 2013). In contrast, the C‐tail remains unstructured in all cases (Fig 1B; Appendix Table S1). Limited trypsinolysis corroborated these results (Fig EV1A).
Figure EV1. The CesAB N‐core is essential for EspA stability (related to Fig 1).
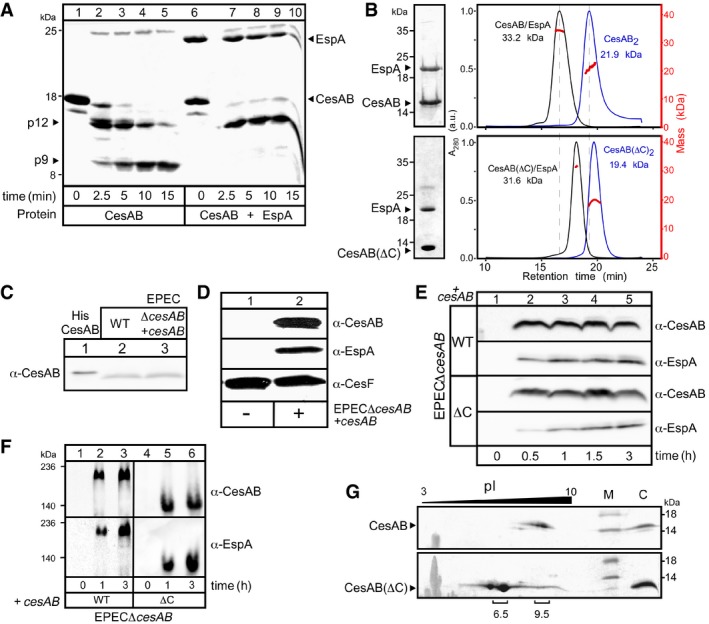
- Limited trypsinolysis of His‐CesAB, alone (lanes 1–5) or EspA‐bound (lanes 6–10). Peptides were analyzed on 5.5 M urea‐15% SDS–PAGE and Coomassie Blue‐stained. Tryptic peptides p12 and p9 were N‐terminally sequenced. CesAB alone was rapidly cleaved, and p12 was formed. Further cleavage of p12 generated p9. This last cleavage did not occur when CesAB was bound to EspA. A representative experiment is shown. n = 3.
- Co‐purified His‐CesAB/EspA (top) or His‐CesAB(ΔC)/EspA (bottom) complexes, analyzed by 15% SDS–PAGE (left) and GPC‐MALS (right; 50 μM; Buffer A). UV traces and masses are shown. Like wt CesAB, CesAB(ΔC) can form stable homodimers as well as heterodimers with EspA (compare blue and black lines on top and bottom panels). A representative experiment is shown. n = 5.
- Plasmid‐borne cesAB was expressed to similar levels with those of a chromosomal copy. The in vivo expression of cesAB from an EPECΔcesAB strain complemented in trans with a pASK‐IBA7plus vector carrying cesAB (lane 3), following addition of AHT (OD600 = 0.3; 37°C; 2.5 ng/ml AHT; 3 h), was compared to that of a wild‐type EPEC strain (lane 2). Equal amount of cells were analyzed on 15% SDS–PAGE and immunostained using α‐CesAB. Purified His‐CesAB (2 ng; lane 1) served as a molecular weight/protein amount marker. A representative experiment is shown. n = 3.
- CesAB is essential for the intracellular stabilization of EspA. EPECΔcesAB was complemented with either an empty pASK‐IBA7plus vector (left) or one carrying cesAB (right) (OD600 = 0.3; 37°C; 2.5 ng/ml AHT; 3 h). Equal amount of cells were analyzed on 15% SDS–PAGE and immunostained with the indicated polyclonal antisera. A representative experiment is shown. When CesAB was not present, EspA was not detected; once CesAB synthesis was restored, both proteins were detected (top and middle panels). CesF, a T3S chaperone whose stability was not affected by CesAB (Elliott et al, 2002), served as an internal control (bottom panel). n = 3.
- CesAB C‐tail is not essential for the stabilization of EspA in vivo. EPECΔcesAB was complemented with a pASK‐IBA7plus vector carrying either cesAB (top panel) or cesAB(ΔC) (bottom) (OD600 = 0.3; 37°C; 2.5 ng/ml AHT). At the indicated time post‐induction, equal amount of cells were analyzed by 15% SDS–PAGE and immunostained with the indicated polyclonal antisera. A representative experiment is shown. n = 3.
- CesAB/EspA (left) and CesAB(ΔC)/EspA (right) complexes formed in vivo. At the indicated time post‐induction, equal amount of cytosolic proteins from EPECΔcesAB cells carrying either cesAB or cesAB(ΔC) plasmids were analyzed on 7% Native‐PAGE and immunostained with the indicated antisera. The difference in migration of the two complexes is probably due to the difference in the pI of CesAB and CesAB(ΔC) (see panel G). A representative experiment is shown. n = 3.
- The C‐tail of CesAB is positively charged. Thus, it shifts the pI of the protein to high values. CesAB (top) and CesAB(ΔC) (bottom) were separated (100 μg in rehydration buffer) according to their native pI in the 1st dimension (IPGphor strips; GE; 13 cm; pH range 3–10; 50 μA; 18,000 Vh) and their denatured mass in the 2nd dimension (15% SDS–PAGE). Proteins were visualized by Coomassie Blue staining. A representative gel is shown. Molecular weight markers (M) and purified proteins (C), run in the 2nd dimension, are indicated. n = 3.
Source data are available online for this figure.
Like CesAB, CesAB(ΔC) (Fig 1A), a C‐tail truncate, is dimeric and forms 1:1 stoichiometric complexes with EspA in vitro (Fig EV1B) and in vivo (Fig EV1C–F). Six basic C‐tail residues are responsible for CesAB's pI of 9.5 (Fig EV1G).
In summary, CesAB comprises the self‐dimerizing, EspA binding, N‐core (Yip et al, 2005; Chen et al, 2011) and an unstructured, positively charged C‐tail.
C‐tail‐dependent CesAB/EspA targeting to the membrane
To determine whether CesAB/EspA are targeted to the membrane, EPECΔcesAB cells expressing cesAB in trans were lysed, cytoplasmic and membrane fractions were separated by centrifugation, and polypeptides analyzed on SDS–PAGE and immunostained (Fig 1C). Fractionation efficiency was monitored by following a cytoplasmic (row 1; SecB) and an outer membrane (row 2; OmpA) protein. CesAB (row 3) and EspA (row 4) were detected in both fractions. In contrast, both proteins were found exclusively in the cytosol of EPECΔcesAB carrying a cesAB(ΔC) plasmid (Fig 1C, rows 5 and 6). Membrane‐bound CesAB associated peripherally since high ionic strength or urea removed it, unlike EspA that remained membrane‐associated (Fig EV2A).
Figure EV2. CesAB/EspA membrane localization in vivo and IMVs characterization (related to Figs 1 and 2).
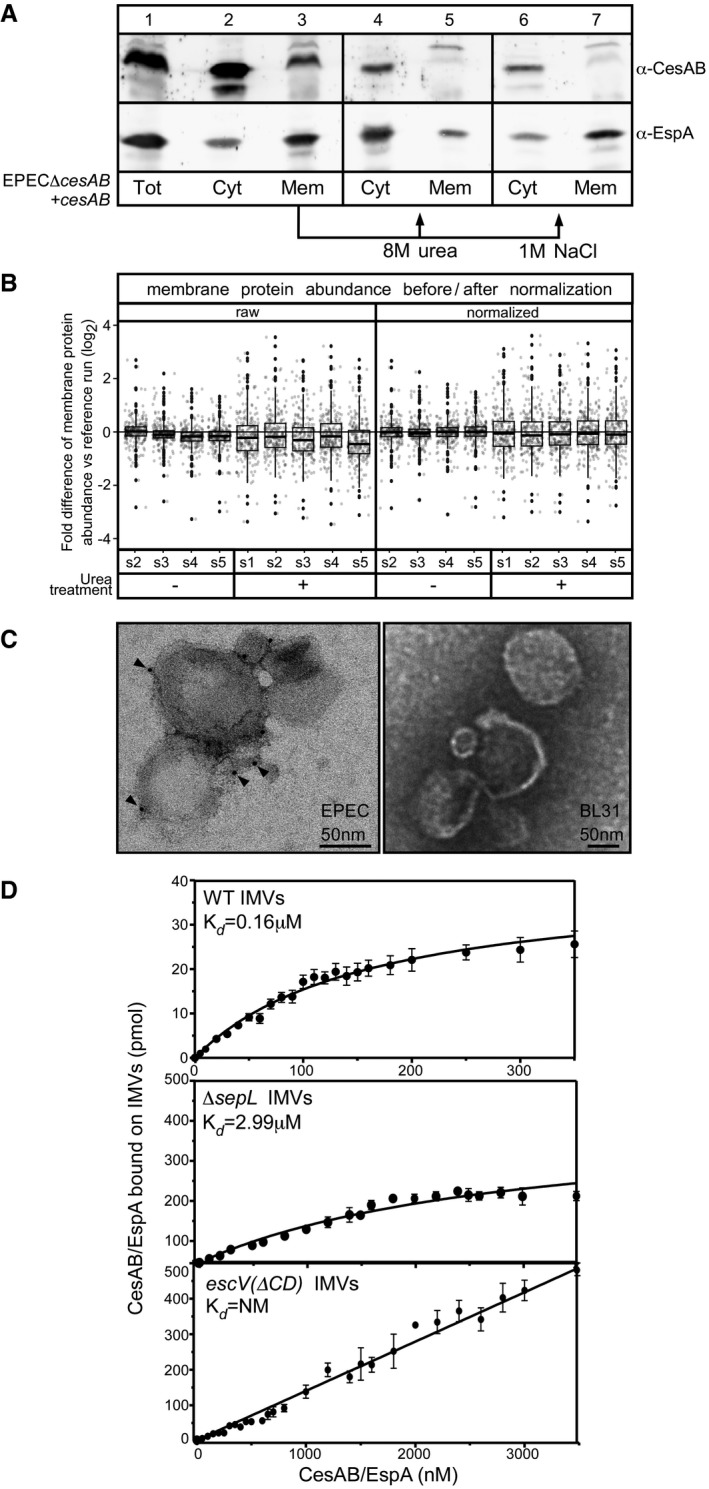
- Sub‐cellular localization of CesAB and EspA. EPECΔcesAB cells complemented with a plasmid carrying cesAB (OD600 = 0.3; 37°C; 2.5 ng/ml AHT; 3 h) were lysed and ultracentrifuged (300,000 g, 30 min, 4°C). A representative experiment is shown. CesAB and EspA were detected in both the cytosolic (Cyt) and membrane fraction (Mem). The majority of the membrane‐localized CesAB could be extracted by washing the membrane fraction with 8 M urea or high salt (as indicated), implying electrostatic interactions with either the membrane lipids or a protein component. On the contrary, the majority of membrane‐localized EspA could not be extracted by salt treatment, implying tight membrane association. n = 3.
- Quality control for the normalization of the different MS runs of IMVs used in this study. The normalization of the different runs was based on the peptide intensity of the integral membrane proteins, since these were not affected by the urea treatment, as described (Callister et al, 2006). A box plot, representing a single IMV run versus the reference run, with or without urea treatment (as indicated), is plotted before (raw) and after normalization (normalized). Clearly, following normalization the difference of mean protein abundance (i.e., the middle of the box plot) of integral membrane proteins was corrected (i.e., log of fold difference between runs is now close to zero). s1–s5: repeats of IMVs preparation from EPEC. Dots represent outliers (see Appendix); square box represents the value‐range between 1st and 3rd quartile; lines above and below boxes represent range from 1st quartile to the maximum value and from the 3rd quartile to the minimum value, respectively.
- Localization of EscV on IMVs, using electron microscopy. IMVs, derived either from EPEC (left) or BL31 (right) Escherichia coli strain, were probed with polyclonal antibodies against the cytoplasmic domain of the membrane‐embedded EscV protein, using goat anti‐rabbit secondary antibodies attached to 5‐nm nanogold particles and were negatively stained. A representative picture is shown. Gold particles, detected on the periphery of EPEC IMVs (left; arrowheads), indicate the presence of T3S injectisomes in the membrane vesicles, containing EscV with its C‐domain exposed and accessible. Gold particles were not detected on BL31‐derived IMVs (right). n = 3.
- Determination of equilibrium dissociation constants (K ds) of CesAB/EspA for wt (top panel), ΔsepL (middle panel), and escV(ΔCD) (bottom panel) EPEC IMVs. Data, analyzed by nonlinear regression, represent average values (for 20–23 concentration points); error bars: standard mean error (SEM), as in Fig 2E. escV(ΔCD) IMVs exhibited linear, non‐saturable binding, that did not converge to a K d. n = 12–14.
Source data are available online for this figure.
We concluded that (i) CesAB is targeted to the membrane, (ii) targeting requires the C‐tail, (iii) EspA is co‐targeted, and (iv) once there, EspA remains membrane‐associated independently of CesAB.
In vitro reconstitution of CesAB/EspA targeting
To determine its specificity for the T3S translocase and further dissect it, we reconstituted CesAB/EspA targeting in vitro. IMVs prepared from EPEC cells were urea‐treated, to strip away most of the peripheral and non‐specifically associated proteins (Papanastasiou et al, 2016; Tsolis & Economou, 2017a). The external side of the IMVs is the one that faced the cytoplasm in the cell; their lumen represents the periplasmic face. We demonstrated that EPEC‐derived IMVs were functionally similar to those prepared from the non‐EPEC strain BL31, using two functional assays derived from the ubiquitous Sec system (Gouridis et al, 2010): the proPhoA‐stimulated Sec translocase ATPase and proPhoA translocation in the lumen of the IMVs (Fig 2A and B).
Figure 2. In vitro reconstitution of type III chaperone/exported protein targeting.
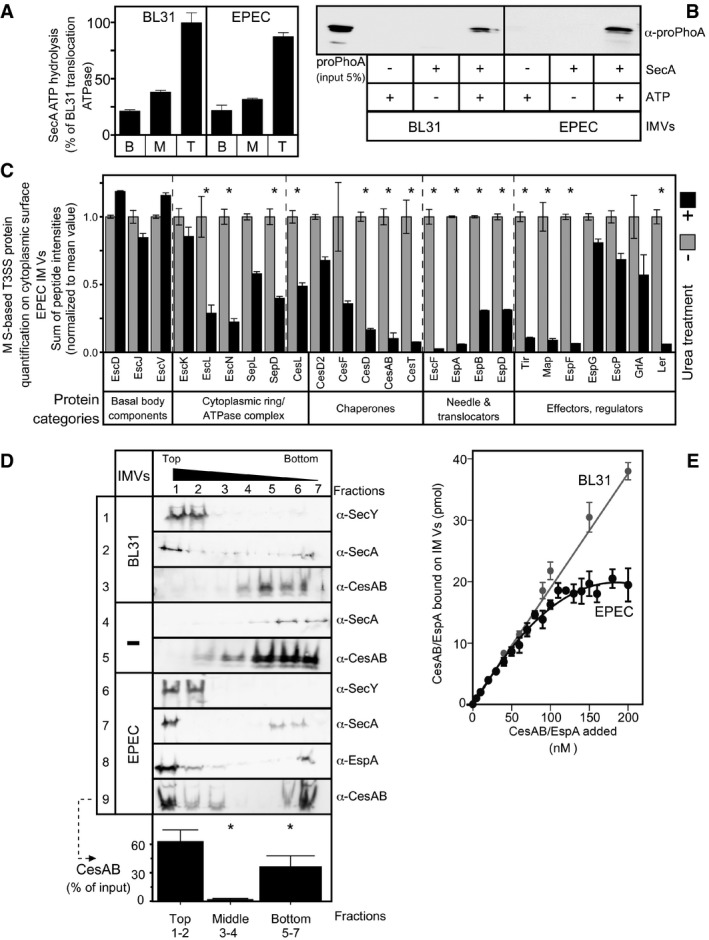
-
A, BUrea‐treated IMVs prepared from BL31 (left) and wild‐type EPEC (right) strain retained similar enzymatic properties, that is, stimulation of SecA ATP hydrolysis (B, basal; M, membrane; T, translocation ATPase) (A) and proPhoA translocation into the lumen of IMVs, in vitro (B). n = 5; bar graphs represent mean values; error bars standard deviation (SD).
-
CBottom‐up, liquid chromatography‐mass spectrometry‐based analysis of T3SS components on IMVs prior (light gray) or post (dark gray)‐urea treatment. The relative abundance of components (sum of peptide intensities) for the multiple repeats tested, from various T3SS proteins, grouped by function, is shown in bar graphs with error bars (standard error of the mean); n = 5. Differentially abundant proteins with P‐value < 0.05, as determined by t‐test and adjusted by BH and fold change > 2 (see Materials and Methods) are indicated with an asterisk (*). See Appendix Table S2 for details.
-
DCesAB/EspA binding to urea‐treated EPEC IMVs, using a flotation assay. Fractions were analyzed by 15% SDS–PAGE and immunostaining. SecY, membrane‐embedded protein; SecA, cytoplasmic protein that binds SecY, which are shown as controls. Soluble proteins remained at the bottom (fractions 5–7) or co‐migrated with IMVs once bound on them at the top of the gradient (fractions 1 and 2). The CesAB signal quantification (from row 9) is shown as % of the input, below the row. n = 6; bar graphs represent mean values; error bars standard deviation (SD). Significant difference with P‐values < 0.05, as determined by t‐test and adjusted by BH (see Materials and Methods) is indicated with an asterisk (*).
-
EDetermination of equilibrium dissociation constants (K d) of CesAB/EspA for EPEC (black) and BL31 (gray) IMVs. Data, analyzed by nonlinear regression, represent average values; error bars, standard mean error (SEM). BL31 IMVs exhibited linear, non‐saturable binding, which did not converge to a K d. n = 9–12.
Source data are available online for this figure.
Mass spectrometric (MS) analysis of surface‐accessible proteins from EPEC IMVs (Papanastasiou et al, 2013; Tsolis & Economou, 2017a) prior and post‐urea treatment confirmed the presence of the injectisome by identifying several integral membrane components (e.g., EscV, EscD, EscJ, Fig 2C, and EscU; Fig EV2B; Appendix Table S2). Immuno‐gold electron microscopy using an antibody against the cytoplasmic EscV C‐domain confirmed that it is surface‐exposed in EPEC IMVs. No non‐specific labeling was seen on BL31 IMVs (Fig EV2C). As expected, the chaotrope removed most of the T3SS proteins that are peripherally associated on IMVs, but not the integral membrane components (Fig 2C), except EscU, whose C‐terminal domain becomes cleaved and removable (Appendix Table S2).
We next used urea‐treated IMVs in a flotation assay (Karamanou et al, 2008). BL31 (Fig 2D, rows 1–3) or EPEC (rows 6–9) IMVs, loaded at the bottom of a sucrose gradient, floated up, to their corresponding density, during ultracentrifugation. Migration inside the gradient was monitored by immunostaining of an inner membrane protein (rows 1 and 6; SecY). The peripheral membrane protein SecA that binds to SecY (Tsirigotaki et al, 2017a) floated up in the presence of IMVs, co‐migrating with them (rows 2 and 7), but remained at the bottom in their absence (row 4). Under the same conditions, CesAB/EspA floated up to the top fractions in the presence of EPEC IMVs (rows 8 and 9; quantification of 9 is shown below the row) but remained at the bottom of the gradient in their absence (row 5) or in the presence of BL31 IMVs (row 3). These results demonstrated that CesAB/EspA bind specifically to a receptor on EPEC‐derived IMVs, presumably the injectisome, and do not bind non‐specifically to lipids or generic E. coli membrane components shared with BL31.
Next, we determined the equilibrium dissociation constants (K d) of CesAB/EspA for EPEC and BL31 IMVs, by concentration titrations of [35S]‐CesAB/EspA, using protocols routinely used for the Sec system (Gouridis et al, 2010, 2013). CesAB/EspA bind with high affinity (K d ~ 0.16 μM) to a saturable receptor on EPEC IMVs but only non‐specifically, non‐saturably to BL31 IMVs (Figs 2E and EV2D, top).
These data demonstrated successful reconstitution of targeting of T3S chaperone/exported proteins in vitro using EPEC‐derived IMVs.
CesAB/EspA targeting to the injectisome requires SepL and EscV
We hypothesized that the CesAB/EspA receptor comprises injectisome components that reside on the cytoplasmic face of the membrane and that would be surface‐exposed on the IMVs (Fig 3A, dark gray). To identify them, we generated whole and partial (i.e., encoding the large cytoplasmic domains of escV and escU) chromosomal gene deletions. None of the derivative strains secreted EspA in vivo (Fig 3B; Appendix Fig S1A), in agreement with previous results (Dziva et al, 2004; O'Connell et al, 2004; Nadler et al, 2006; Deng et al, 2015), nor formed actin pedestals on HeLa cells (Appendix Fig S1D–L), an assay reporting effector injection (e.g., Tir) via the EspA filaments and the EspB/D translocon pore (Creasey et al, 2003).
Figure 3. SepL and EscV form a bipartite CesAB/EspA receptor.
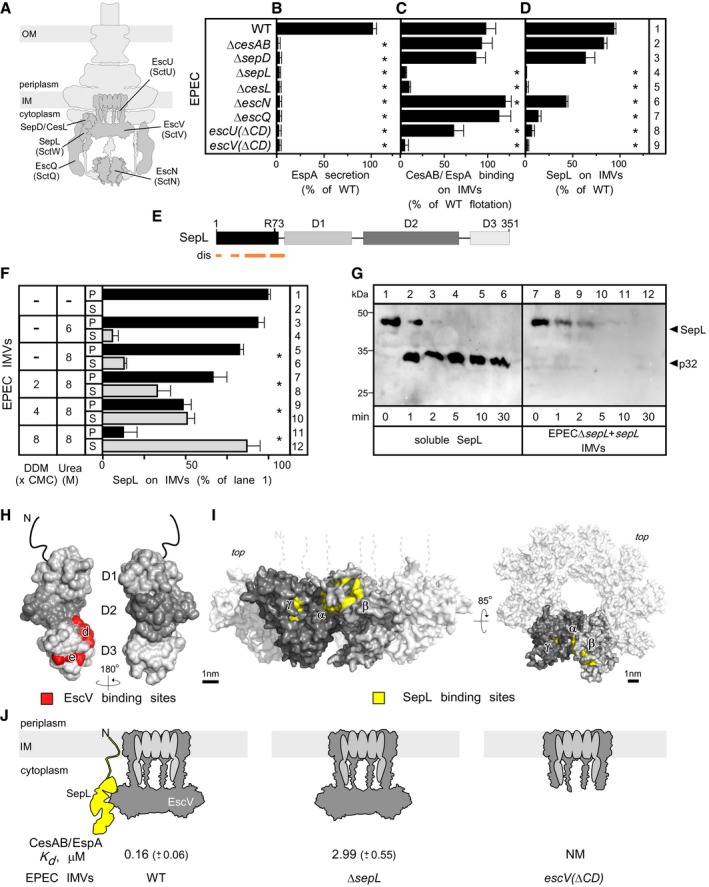
- The EspA secretion from various EPEC knockout strains (5 h post‐inoculation; Appendix Fig S1A) was quantified. WT secretion was considered 100%; all other values are expressed as % of this. n = 3; bar graphs represent mean values; error bars standard deviation (SD). Significant difference with P‐values < 0.05, as determined by t‐test and adjusted by BH (see Materials and Methods) is indicated with an asterisk (*).
- CesAB/EspA binding to urea‐treated IMVs, from various EPEC knockout strains, on flotation assays (Appendix Fig S1B) was quantified (as in lane 9, Fig 2D). The CesAB amount that migrated in the top gradient fraction upon incubation with wild‐type EPEC IMVs was considered 100%; all other values are expressed as % of this; n = 5; bar graphs represent mean values; error bars standard deviation (SD). Significant difference with P‐values < 0.05, as determined by t‐test and adjusted by BH (see Materials and Methods) is indicated with an asterisk (*).
- SepL content on IMVs. Following analysis of equal protein amount from various urea‐treated EPEC IMVs on 15% SDS–PAGE and immunostaining (Appendix Fig S1C), signals were quantified. SepL amount on wild‐type IMVs was considered 100%; all other values are expressed as % of this. n = 3; bar graphs represent mean values; error bars standard deviation (SD). Significant difference with P‐values < 0.05, as determined by t‐test and adjusted by BH (see Materials and Methods) is indicated with an asterisk (*).
- A linear map of SepL, indicating D1–3 domains, disordered predicted regions (dis) and the trypsin cleavage site in solution (R73).
- The association of SepL with the membrane is tight. EPEC IMVs (40 μg protein) were treated with the indicated urea or/and dodecyl maltoside (DDM) concentration (30 min; ice), ultracentrifuged (100,000 g, 30 min, 4°C; TLA100 rotor; Optima Max‐XP Beckman), analyzed on 15% SDS–PAGE, and immunostained (Fig EV3A). Signals were quantified. The SepL amount of untreated IMVs (lane 1) was considered 100%; all other values are expressed as % of this. S: supernatant; P: pellet; error bars represent the standard deviation. n = 4. Significant difference with P‐values < 0.05, as determined by t‐test and adjusted by BH (see Materials and Methods) is indicated with an asterisk (*).
- Limited trypsinolysis of SepL, in solution (lanes 1–6) or bound on IMVs (lanes 7–12), after immunostaining with α‐SepL (as in Fig EV1A); n = 6.
- On the surface structure of SepL (PDB accession number: 5C9E), the EscV C‐domain binding sites, as determined by peptide array analysis (Fig EV4A and B), are shown (red; Latin letters).
- CesAB/EspA targeting to the T3S translocase in wild‐type, ΔsepL and escV(ΔCD) IMVs; n = 6; mean ± SEM.
Source data are available online for this figure.
We next prepared urea‐treated IMVs from all the deletion strains and quantified CesAB/EspA targeting to them using flotation assays (Fig 3C; Appendix Fig S1B). Chromosomal deletions of cesAB, sepD, escN, escQ, and escU(ΔCD) did not or only moderately affect(ed) CesAB/EspA targeting to the injectisome (Fig 3C, rows 2, 3, 6, 7, and 8, respectively). On the contrary, CesAB/EspA did not bind to IMVs devoid of SepL, CesL, or the C‐domain of EscV (rows 4, 5, and 9). Since only some EPEC deletion strains were targeting‐deficient (compare Fig 3C to B), targeting can be biochemically dissected from downstream secretion steps.
Quantification of the SepL content of IMVs, by immunostaining (Fig 3D; Appendix Fig S1C), pointed out that three IMV preparations that were deficient in CesAB/EspA targeting (Fig 3C, rows 4, 5 and 9), also had low SepL levels (Fig 3D, rows 4, 5 and 9). CesL, a SepL chaperone (Younis et al, 2010), might stabilize SepL. EscU and EscQ appear also to contribute to SepL accumulation at the membrane and may have non‐essential regulatory roles in CesAB/EspA binding. The role of these proteins was not pursued further. Hereafter, we focused on SepL and EscV as potential CesAB/EspA receptor components.
SepL tightly associates with the membrane via its N‐terminus
Unlike the membrane‐embedded EscV, SepL has properties of a soluble protein, with a disordered N‐terminus (Fig 3E) and no extensive hydrophobic patches (Burkinshaw et al, 2015). Yet, a chaotrope (8 M urea) could only poorly extract SepL from the membrane; complete extraction required increasing concentration of non‐ionic detergent (Figs 3F and EV3A), suggesting a tight SepL–membrane association. While the N‐terminus of SepL is trypsin‐accessible in solution (Fig 3G, lanes 3–6), cleaved at R73 (Fig 3E), it becomes trypsin‐resistant upon membrane association (Fig 3G, lanes 7–9) suggesting SepL association with the membrane via its N‐terminus. Corroborating this, three C‐terminal truncated or mutated derivatives remained membrane‐bound (Fig EV3B). Membrane‐associated SepL is fully trypsinized after longer incubation (Fig 3G, lanes 10–12) and therefore fully exposed and peripheral.
Figure EV3. SepL receptor function determination (related to Figs 3, 4, 5).
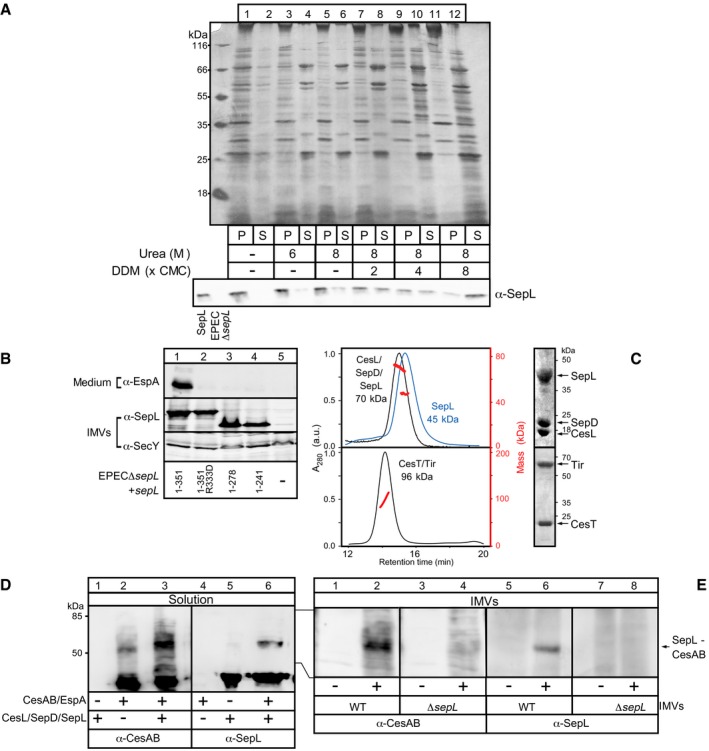
- The association of SepL with the membrane is tight. EPEC IMVs (40 μg of total membrane protein/sample) were incubated with the indicated urea or/and DDM concentration (30 min; on ice). Following ultracentrifugation (100,000 g, 30 min, 4°C; Optima Max‐XP, TLA100 rotor; Beckman Coulter), samples were analyzed on 15% SDS–PAGE and either silver‐ (top) or immunostained with α‐SepL (bottom). A representative experiment is shown. S = supernatant, P = pellet; n = 4.
- EspA secretion of (top), and SepL localization on IMVs prepared from (middle), EPECΔsepL cells that were complemented with the indicated plasmid‐borne sepL derivatives (lanes 1–4) or an empty vector (lane 5) (as in Appendix Fig S1A and C). 1–351 is the full‐length, wild‐type SepL. Immunostaining SecY (bottom), an inner membrane protein unrelated to T3SS, served as loading control. Wild‐type (lane 1) and R333D (lane 2) SepL and its C‐terminal truncation mutants (lanes 3 and 4) were localized on the IMVs; gel migration differences are due to mass differences. However, SepL mutants did not restore EspA secretion. A representative experiment is shown. n = 3.
- The SepL alone or the co‐purified His‐CesL/SepD/SepL (top, blue, and black line, respectively) and His‐CesT/Tir (bottom) complexes were analyzed by 15% SDS–PAGE (right) or GPC‐MALS (left), as in Fig EV1B. The mass determined for His‐CesL/SepD/SepL indicated a stoichiometric ratio of 1:1:1 (top) while that for His‐CesT/Tir indicated a ratio of 2:1 (bottom), in agreement with previous results (Thomas et al, 2005; Younis et al, 2010).
- CesAB/EspA interacted with SepL in solution. His‐CesAB/EspA and His‐CesL/SepD/SepL protein complexes (10 μM each) were mixed (100‐μl reactions) and cross‐linked using a bifunctional chemical cross‐linker (SDA‐NHS‐diazirine; see Appendix Supplementary Materials and Methods). Protein samples were analyzed on 15% SDS–PAGE and immunostained using α‐CesAB and α‐SepL antisera. When the two complexes were mixed, a new protein species of ˜60 kDa appeared after cross‐linking, containing both CesAB and SepL (lanes 3 and 6), indicating a cross‐linked product between CesAB/SepL (theoretical mass 64 kDa). This band was not present when CesAB/EspA or CesL/SepD/SepL were mixed alone with the cross‐linker (lanes 1–2, and 4–5). A representative experiment is shown. n = 4.
- CesAB/EspA interacted with membrane‐bound SepL on IMVs. His‐CesAB/EspA (10 μM) was mixed (100 μl reactions) with IMVs (1 μg/μl total membrane protein) prepared from either wt or ΔsepL EPEC strains and cross‐linked using a bifunctional chemical cross‐linker (SDA‐NHS‐diazirine; see Appendix Supplementary Materials and Methods). Protein samples were analyzed on 15% SDS–PAGE and immunostained using α‐CesAB and α‐SepL. When wt IMVs were mixed with CesAB/EspA, a protein band of ˜60 kDa appeared after cross‐linking which contained both CesAB and SepL (lanes 2 and 6), indicating a cross‐linked product between CesAB/SepL (theoretical mass 64 kDa). This band was not present when wt (lanes 1 and 5) or ΔsepL (lanes 3 and 7) IMVs alone were mixed with the cross‐linker, or when CesAB/EspA was mixed and cross‐linked with ΔsepL IMVs (lanes 4 and 8). A representative experiment is shown. n = 4.
Source data are available online for this figure.
SepL physically interacts with the C‐domain of EscV
The absence of SepL from EPECescV(ΔCD) IMVs (Fig 3D, row 9) suggested that the EscV C‐domain might be a binding scaffold for SepL. To determine whether the two interact, we probed arrays of immobilized SepL peptides (13‐mers; 10 residue overlaps) with the EscV C‐domain (Fig EV4A, B, and E). Interacting residues localized primarily on two sites (d and e) on one face of SepL, in domains 2 and 3 (Fig 3H, red). In the reciprocal experiment, EscV C‐domain peptide arrays were probed with SepL (Fig EV4C, D, and F). When the linear peptide signals were mapped on the EscV monomer structure model derived from the structure of the Shigella homologue (Abrusci et al, 2013), it was revealed that SepL bound to three external surfaces. When mapped on the nonamer ring model, they could form a groove between two neighboring protomers containing two adjacent binding sites (α and β) on one protomer and site γ on its neighbor (Fig 3I, yellow; Fig EV4F). We concluded that SepL physically interacts with the C‐domain of EscV.
Figure EV4. SepL and EscV C‐domain peptide array analysis (related to Figs 3 and 4).
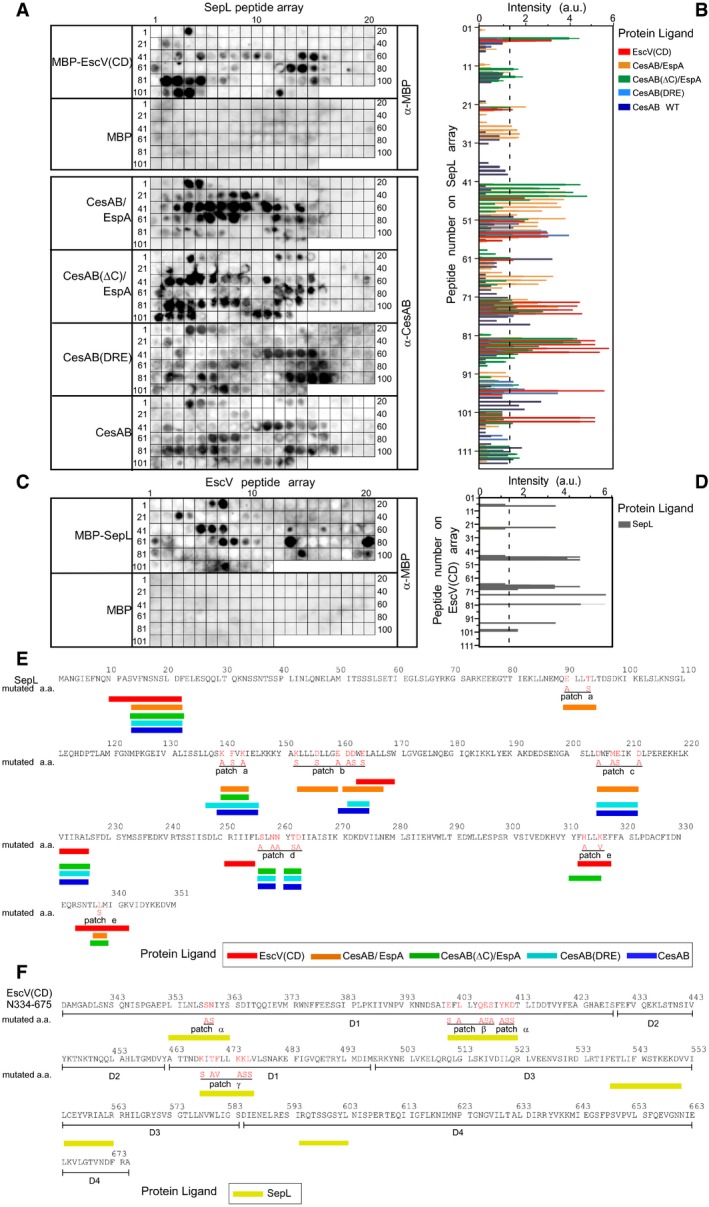
- SepL immobilized peptide arrays (13‐mers, 10 residues overlap) were probed with the indicated purified protein ligands (200 nM; 25 ml Buffer C; 25°C; 1 h): His‐MBP‐EscV C‐domain (CD), His‐MBP, His‐CesAB/EspA, His‐CesAB(ΔC)/EspA, His‐CesAB(DRE), and His‐CesAB, respectively, as described (Karamanou et al, 2008). Following washes, proteins that were bound to the array were electro‐transferred onto nitrocellulose membranes and immunostained with α‐CesAB or α‐MBP antibodies. Representative experiments are shown; n = 4–6.
- The intensity of signals from experiments like those presented in (A) was quantified using Image Quant software (GE), as described (Karamanou et al, 2008). Mean values were plotted using GraphPad Prism; signals below 1.2 (dashed line) were considered as background; n = 4; bar graphs represent mean values; error bars standard deviation (SD).
- EscV C‐domain immobilized peptide arrays (13‐mers; 10 residues overlap) were probed with the purified protein ligands (200 nM; 25 ml Buffer C; 25°C; 1 h): His‐MBP‐SepL and His‐MBP. Experiments were performed as in (A) and immunostained with α‐MBP antibodies. Representative experiments are shown; n = 4.
- The intensity of signals from experiments like those presented in (C) was quantified as described in (B); n = 4; bar graphs represent mean values; error bars standard deviation (SD).
- SepL sequence (1–351), CesAB, and EscV binding sites (a–e) identified by the peptide arrays are indicated (color code unique for each used ligand; as indicated). Residues mutated in this study (red) and their substitutions (red, below) are indicated.
Source data are available online for this figure.
SepL and EscV form a bipartite CesAB/EspA receptor
To quantify the contributions of SepL and EscV as CesAB/EspA receptor components, we compared the equilibrium dissociation constants (K ds) of CesAB/EspA for IMVs prepared from wild‐type EPEC, EPECΔsepL, and EPECescv(ΔCD) cells (Figs 3J and EV2D). Only when both SepL and EscV were present (Fig 3J, left), CesAB/EspA was targeted to the injectisome with high affinity (0.16 μM). When SepL was absent but EscV present (middle), CesAB/EspA exhibited only low affinity for the injectisome (~3 μM). When the EscV C‐domain was deleted (right), SepL did not associate with the membrane and the affinity of CesAB/EspA for the injectisome was non‐measurable.
We concluded that SepL and EscV form a bipartite receptor. SepL, apparently operates like an accessory injectisome subunit, associates with the membrane and with the C‐domain of EscV (Fig 3J, left), thus securing high‐affinity CesAB/EspA targeting to the injectisome.
CesAB/EspA binding sites on SepL are essential for its receptor function
Chemical cross‐linking of CesAB to soluble (Fig EV3D, lanes 3 and 6) or IMV‐associated SepL (Fig EV3E, lanes 2 and 6) suggested a direct interaction of CesAB/EspA with SepL. To identify the interaction sites, the SepL peptide array was incubated with CesAB/EspA, or CesAB, or their derivatives (Fig EV4A, B, and E). Three CesAB N‐core‐specific binding sites were identified on one face of SepL, in domains 1 and 3 (Fig 4A, left, green and B). Two of these sites (“a” and “e”) are composite, comprising residues that are distant in the primary but come together in the 3D structure. Some residues overlapped with the EscV C‐domain binding sites of SepL (Figs 3H and EV4A and E). The positively charged CesAB C‐tail seemed to recognize two negatively charged sites (b; c) on the other face of SepL (Fig 4A, right, green and B).
Figure 4. Requirements for CesAB/EspA targeting to SepL.
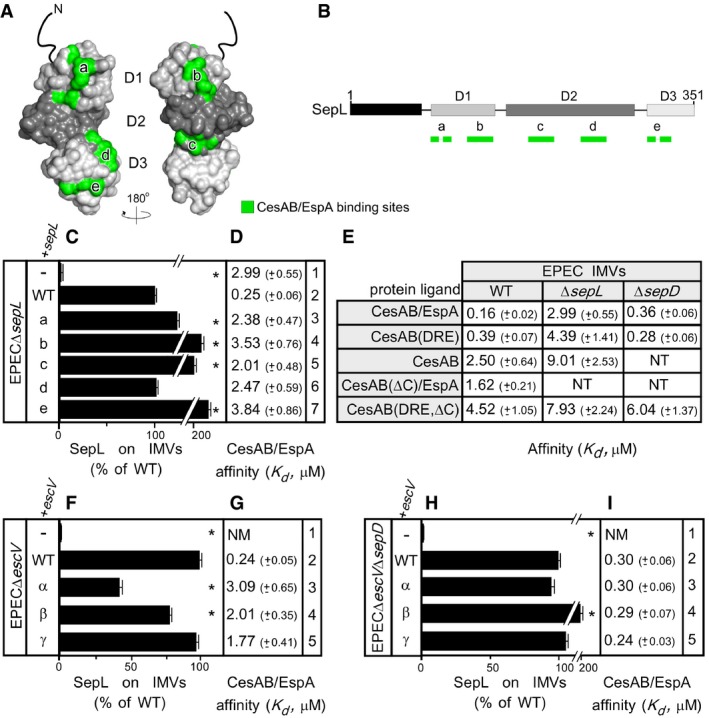
-
A, BThe CesAB/EspA binding sites (green), as determined by peptide arrays (Fig EV4A), are indicated on the surface structure (A) or the linear map (B) of SepL.
-
C–IThe SepL content (C, F, and H) for the indicated urea‐treated IMVs (Appendix Fig S2A, J and P, respectively) was quantified (as in Fig 3D); n = 3; bar graphs represent mean values; error bars standard deviation (SD). K ds of protein ligands (D, E, G, and I) for urea‐treated EPEC IMVs (as indicated; as in Figs 2E and EV2D); n = 6–9; mean ± SEM. Mutated sites on SepL (a–e) and EscV (α–γ) are detailed in Appendix Supplementary Materials and Methods. Significant difference with P‐values < 0.05, as determined by t‐test and adjusted by BH (see Materials and Methods) is indicated with an asterisk (*).
To test the importance of these associations, the implicated surface‐exposed SepL residues (Fig 4A) were alanyl or seryl substituted on plasmid‐borne sepL (Fig EV4E; detailed in Appendix Supplementary Materials and Methods). We transformed these plasmids in EPECΔsepL cells, prepared IMVs, and determined their SepL content and affinity for CesAB/EspA. All mutants retained the membrane association of SepL (Fig 4C; Appendix Fig S2A) but had lost high‐affinity CesAB/EspA binding (Fig 4D, compare rows 3–7 to 2).
Our results demonstrated that CesAB/EspA physically associated with specific SepL sites that are essential for its high‐affinity binding to the translocase. Peptide arrays provide neither temporal order nor stoichiometries.
High‐affinity CesAB binding to SepL requires the CesAB C‐tail
Do both CesAB and EspA bind on SepL? Since EspA aggregates in the absence of its chaperone [Fig EV1D, E and F, compare lanes 2 and 3 to lane 1 (Creasey et al, 2003)], we could only probe this question using CesAB/EspA, or CesAB alone, or their derivatives.
CesAB alone had low affinity for the translocase (Fig 4E, 2.5 μM). Only when loaded with EspA in a 1:1 heterodimer, CesAB acquired its high‐affinity binding conformation (0.16 μM). CesAB(DRE), a mutant that mimics this conformation (Chen et al, 2013), bound with high affinity on wild‐type EPEC IMVs (0.39 μM) and with low affinity on EPECΔsepL IMVs (4.39 μM), arguing that CesAB is the component of the complex that is specifically recognized by SepL.
Deletion of the CesAB C‐tail reduced the affinity of either CesAB(ΔC)/EspA or CesAB(DRE,ΔC) for EPEC IMVs by 10‐fold (Fig 4E, 1.62 and 4.52 μM, respectively), pointing out that the C‐tail is a main element responsible for high‐affinity binding on SepL, in agreement with the fractionation results (Fig 1C). EspA had an additional but minor contribution, directly or indirectly, to CesAB/EspA binding (Fig 4E, compare CesAB/EspA to CesAB(DRE) in wt or ΔsepL IMVs).
EscV controls the SepL receptor function through their physical interaction
To test whether the EscV‐SepL interaction affects the receptor function of SepL, surface‐exposed residues of EscV located on its SepL‐binding patches (Fig 3I) were alanyl or seryl substituted on plasmid‐borne escV (Fig EV4F; detailed in Appendix Supplementary Materials and Methods) and transformed into EPECΔescV cells. IMVs were prepared, and their SepL content (Fig 4F; Appendix Fig S2J) and affinity for CesAB/EspA (Fig 4G) were determined.
Wild‐type, plasmid‐borne escV restored both the membrane association between SepL and CesAB/EspA affinity (Fig 4F and G, compare row 1 to 2). All escV mutants supported almost wild‐type levels of membrane‐associated SepL (Fig 4F, rows 3–5), presumably since SepL does not depend solely on its interaction with the C‐domain of EscV to remain membrane‐associated but also makes use of its N‐terminal regions. Nevertheless, all derivatives lost high‐affinity CesAB/EspA binding (Fig 4G, compare rows 3–5 to 2). Mutating the SepL‐binding interfaces of EscV significantly affected the receptor function of wild‐type SepL (Fig 4G, rows 3–5). Presumably, EscV alters the conformation of SepL, implying a dominant EscV‐SepL allosteric cross‐talk.
The SepL‐EscV conformational cross‐talk requires SepD
SepL is chaperoned by CesL and SepD [Fig EV3C, top (Younis et al, 2010)]. While the absence of CesL destabilized membrane‐bound SepL (Fig 3D, row 5 and data not shown), that of SepD affected neither membrane association nor receptor function of SepL (Figs 3C and D, row 3, and 4E).
To test whether SepD affected the SepL‐EscV interaction, we constructed the double deletion strain EPECΔescVΔsepD, transformed it with plasmids encoding escV mutants, prepared IMVs, and determined their SepL content and CesAB/EspA affinity. In all cases, SepL remained membrane‐associated (Fig 4H; Appendix Fig S2P). Notably, removal of SepD restored high‐affinity CesAB/EspA binding to the injectisomes with mutated EscVs (compare Fig 4I and G, rows 3–5) arguing that SepL and EscV were (Fig 4G), but no longer are (Fig 4I), in conformational cross‐talk.
Our experiments suggest that by binding to SepL (O'Connell et al, 2004), SepD establishes the SepL‐EscV conformational cross‐talk that allows EscV to control SepL receptor function (Fig 4G and I).
SepL and SepD establish a translocator/effector affinity switch on the injectisome
Given that the SepL‐EscV physical interaction determines CesAB/EspA targeting to SepL (Fig 3J), while SepD controls the SepL‐EscV cross‐talk, we wondered whether the same mechanism regulates switching from translocators to effectors (O'Connell et al, 2004; Deng et al, 2005). To test this, we co‐purified a chaperone/effector pair, CesT/Tir [translocated intimin receptor (Kenny et al, 1997)] (Fig EV3C, bottom), and determined its affinity (K d) for various IMVs.
Wild‐type EPEC IMVs bind CesAB/EspA with high and CesT/Tir with low affinity (Fig 5A, 0.16 and 2.23 μM, respectively). CesT alone exhibited twofold lower affinity (4.45 μM). Therefore, Tir binding enhanced the affinity of CesT for the translocase (Fig 5A), but not as drastically as EspA did for CesAB (Fig 4E). While EPECΔsepL IMVs lost their high affinity for CesAB/EspA, they “gained” high affinity for CesT/Tir (Fig 5A, 2.99 and 0.38 μM, respectively). SepL seems to be an affinity switch not only for translocator but also for effector complexes: positive for translocators and negative for effectors. EPECescV(ΔCD) IMVs did not exhibit any affinity for CesT/Tir (Fig 5A), arguing that EscV is the primary receptor for the chaperone/effector pair.
Figure 5. SepL acts as an affinity switch between translocators and effectors.
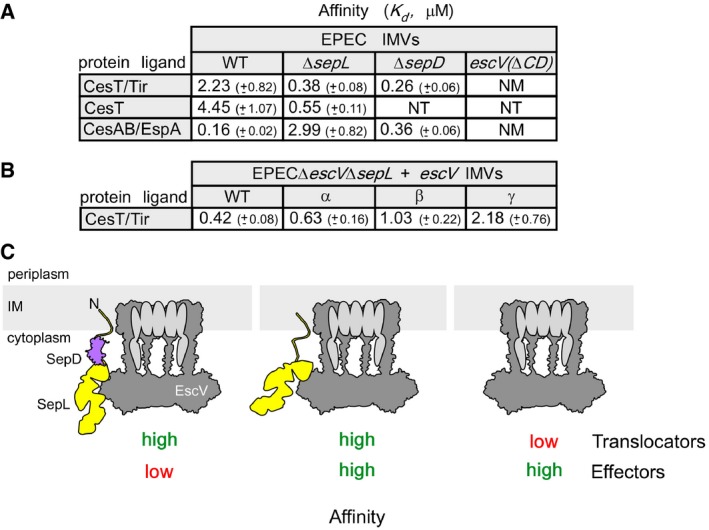
-
A, BK ds of protein ligands for urea‐treated EPEC IMVs (as indicated); n = 9; mean ± SEM.
-
CSchematic representation of the affinity switch between translocators/effectors.
EPECΔsepD IMVs exhibited as high affinity for CesT/Tir as for CesAB/EspA (Fig 5A, 0.26 and 0.36 μM, respectively), demonstrating that once the SepL‐EscV cross‐talk is disrupted, SepL cannot act anymore as a repressor of CesT/Tir binding, while it fully retains its function as a translocator receptor.
Are the SepL and the CesT/Tir interaction sites on EscV common? To address this, we generated an EPECΔsepLΔescV strain and transformed it with wild‐type escV or derivative plasmids carrying mutations on one of the three EscV binding sites for SepL (Fig 3I). IMVs were prepared and K ds for CesT/Tir determined. Binding of CesT/Tir (Fig 5B) was compromised, particularly for site γ, suggesting these may be universal receptor sites on the translocase.
Our results demonstrated that a single mechanism regulates targeting of both T3S translocators and effectors (Fig 5C). SepD binding to SepL establishes SepL‐EscV conformational cross‐talk that ensures translocator targeting, while repressing effector targeting (left). SepD release disrupts this cross‐talk, thus leading to non‐selective targeting: translocators and effectors bind to the injectisome equally well (middle). Release of the translocator receptor SepL leads to exclusive effector targeting (right).
Targeting is an essential, early step of secretion/infection
To test whether the targeting mechanism that was revealed by the in vitro experiments is directly related to the secretion and infection processes in vivo, we monitored secretion of EspA in the medium of bacterial cultures (Fig 6A, C, E, G, and I) and infection of HeLa cells by the same bacterial strains (Fig 6B, D, F, H, and J).
Figure 6. Affinity switching in T3S targeting underlies secretion switching.

-
A–JEspA and Tir secretion (A, C, E, G, and I) from various EPEC strains carrying, or not, plasmids (as indicated; Appendix Fig S2A and J; Fig EV5C, F, and H) was quantified (as in Fig 3B); n = 3; bar graphs represent mean values; error bars standard deviation (SD). +, chromosomal level expression; +++, overexpression. The HeLa cell infection by the indicated EPEC strains (B, D, F, H, and J) was quantified for: colonies formed/HeLa cell, bacteria found/colony and actin pedestal formation (for representative micrographs, see Appendix Fig S1F–H; Fig S2B–I and K–O; Fig EV5I–N; ˜100 HeLa cells/experiment were examined). Results are presented as % of wild type, with error bars. n = 3; bar graphs represent mean values; error bars standard deviation (SD). Significant difference with P‐values < 0.05, as determined by t‐test and adjusted by BH (see Materials and Methods) is indicated with an asterisk (*).
-
KSchematic representation of the secretion switch between translocators/effectors.
Bacteria devoid of CesAB, neither secreted EspA nor formed actin pedestals on HeLa cells (Fig 6A and B, compare row 2 to 1; Fig EV5I and J). Wild‐type cesAB expressed in trans restored both phenotypes (Fig 6A and B, compare row 3 to 1; Fig EV5F lanes 2–4, and K). On the contrary, cesAB derivatives with mutated C‐tails that exhibited targeting defects [i.e., cesAB(ΔC) and cesAB(6A), a mutant in which the six positive charges of the C‐tail were substituted by alanines; data not shown] complemented neither phenotype (Fig 6A and B, rows 4 and 5; Fig EV5F, L and M). These defects were not corrected even after CesAB(ΔC)/espA were over‐synthesized, several‐fold over their K d (Fig 6A and B, row 6; Fig EV5A–C), demonstrating a role of the C‐tail not only in the targeting but also in the downstream secretion step.
Figure EV5. The CesAB C‐tail acts as an activator for EspA secretion (related to Fig 6).
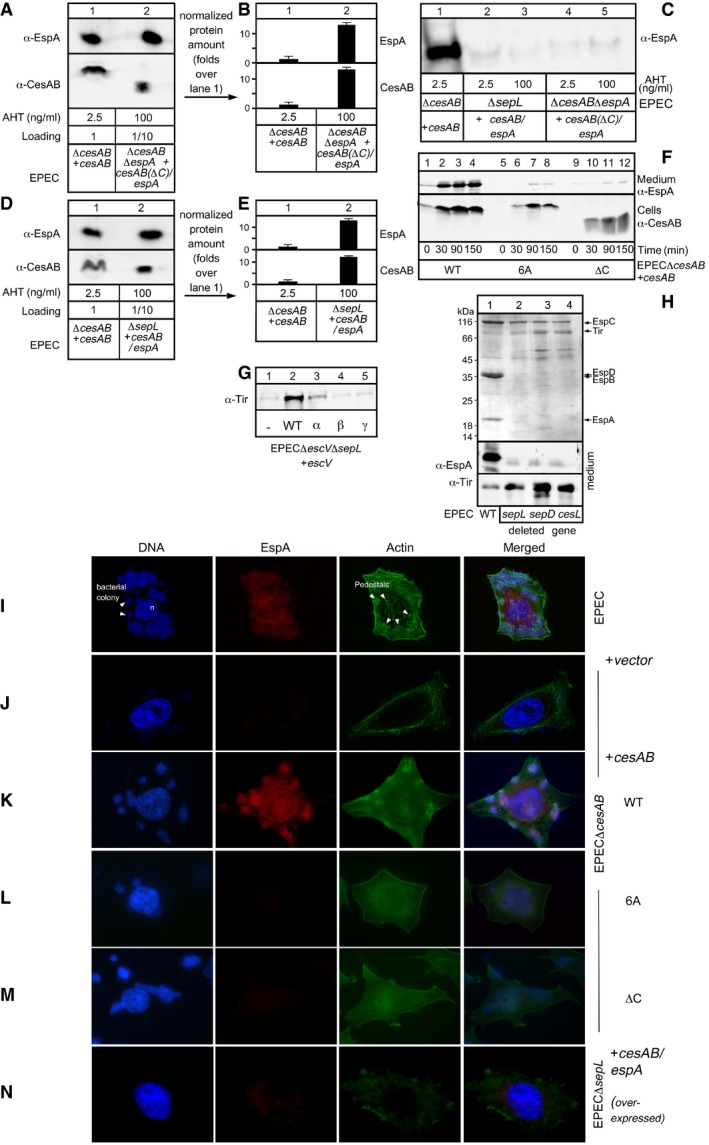
-
AEPECΔcesAB cells transformed with a pASK‐IBA7plus vector carrying cesAB and ΔcesABΔespA cells transformed with a pASK‐IBA7plus vector carrying cesAB(ΔC)/espA were grown (OD600 = 0.3; 37°C), and plasmid gene expression was induced (2.5 ng/ml or 100 ng/ml AHT as indicated; 3 h). Cells were separated from the spent growth medium by centrifugation (50 ml; 5,000 g, 20 min, 4°C) and resuspended in 50 mM Tris pH 8, to reach the same OD600. The indicated relative amount of cells was loaded on 15% SDS–PAGE and probed with α‐EspA and α‐CesAB. EPECΔcesABΔespA cells transformed with cesAB(ΔC)/espA plasmid (lane 2; 100 ng/ml AHT) synthesized ˜10 times more CesAB(ΔC) and EspA compared to EPECΔcesAB cells transformed with a cesAB plasmid (lane 1; 2.5 ng/ml AHT). A representative experiment is shown. n = 3.
-
BCesAB and EspA signals from (A) were quantified using Image Quant (GE). Expression levels of espA (lane 1, top) and cesAB (bottom) using 2.5 ng/ml AHT were considered as 1; all other values (lane 2) were expressed as folds over this (y‐axis). n = 3; bar graphs represent mean values; error bars standard deviation (SD).
-
CSecreted polypeptides from the spent growth medium from (A) and (D) were treated and analyzed as described in Appendix Fig S1A. Proteins were immunostained with α‐EspA. EspA was not secreted in the absence of either SepL (lanes 2 and 3) or the CesAB C‐tail (lanes 4 and 5), even though in both cases CesAB and EspA were over‐expressed (see A + B and D + E, respectively). A representative experiment is shown. n = 3.
-
DEPECΔcesAB transformed with a pASK‐IBA7plus vector carrying cesAB and EPECΔsepL transformed with a pASK‐IBA7plus vector carrying cesAB/espA were grown (OD600 = 0.3; 37°C), and plasmid gene expression was induced (2.5 ng/ml or 100 ng/ml AHT as indicated; 3 h). Cells were separated from the spent growth medium by centrifugation (50 ml; 5,000 g, 20 min, 4°C) and resuspended in 50 mM Tris pH 8, to reach the same OD600. The indicated relative amounts of cells were loaded on 15% SDS–PAGE and probed with α‐EspA and α‐CesAB. CesAB and EspA were synthesized at least 10 times more in EPECΔsepL cells carrying cesAB/espA plasmid (lane 2; 100 ng/ml AHT) compared to EPECΔcesAB complemented with a cesAB plasmid (lane 1; 2.5 ng/ml AHT). A representative experiment is shown. n = 3.
-
ECesAB and EspA signals from (D) were quantified using Image Quant (GE). Expression levels of espA (lane 1, top) and cesAB (bottom) using 2.5 ng/ml AHT were considered as 1; all other values (lane 2) were expressed as folds over this (y‐axis). n = 3; bar graphs represent mean values; error bars standard deviation (SD).
-
FThe C‐tail of CesAB is essential for EspA secretion. EPECΔcesAB cells, transformed with a pASK‐IBA7plus vector carrying cesAB (lanes 1–4), or cesAB(ΔC) (lanes 5–8), or cesAB(6A) (lanes 9–12) were grown, as in Fig EV1D. At the indicated time post‐induction, 50 ml culture was centrifuged, to separate cells from the spent growth medium (5,000 g, 20 min, 4°C). EspA secretion was monitored in the spent growth medium, as in Appendix Fig S1A (top panel). CesAB expression was monitored in total cells, using α‐CesAB (bottom). A representative experiment is shown. n = 4.
-
GSecretion of Tir from EPECΔescVΔsepL strain transformed with a pASK‐IBA7plus vector empty or carrying the indicated escV derivatives, analyzed as in Appendix Fig S1A using α‐Tir; n = 3.
-
HSecretome analysis of the indicated EPEC strains (5 h post‐inoculation; as in Appendix Fig S1A), visualized by Coomassie Blue‐ (top) or immunostaining (as indicated); n = 3.
-
I–MInfection of HeLa cells by EPEC wt or EPECΔcesAB cells transformed with a pASK‐IBA7plus vector empty or carrying the indicated cesAB derivatives, as described in Appendix Fig S2B (without induction of plasmid gene expression). n = nucleus; white arrowheads indicate bacterial colonies (in blue) and actin‐pedestals (in green). n = 4.
-
NInfection of HeLa cells by EPECΔsepL cells transformed with a pASK‐IBA7plus vector carrying cesAB/espA, as described in Appendix Fig S2B. Plasmid gene expression was induced with 100 ng/ml AHT prior to the incubation with HeLa cells to ensure cesAB/espA overexpression (as in A). As anticipated, HeLa cells were not infected due to lack of EspA secretion. n = 4.
Source data are available online for this figure.
In agreement with these results, cells devoid of the CesAB/EspA‐targeting receptor SepL show no EspA secretion or HeLa infection even under conditions where CesAB/EspA was over‐expressed (Fig 6C and D; Fig EV5C–E and N). In trans expression of wild‐type sepL restored these defects (Fig 6E and F, row 1; Appendix Fig S2A, lane 2, top panel; Appendix Fig S2B and C). In contrast, under identical conditions, sepL derivatives with defective receptor sites for CesAB/EspA (Fig 4D) exhibited defects in either EspA secretion or HeLa cell infection (Fig 6E and F, rows 2–6; Appendix Fig S2A and D–I).
Moreover, mutations on escV that affected targeting of both CesAB/EspA (Fig 4G) and CesT/Tir (Fig 5B) practically abolished secretion of EspA as well as that of Tir (Fig 6G; Appendix Fig S2J; Fig EV5G) and infection of HeLa cells (Fig 6H; Appendix Fig S2K–O).
Clearly, targeting is an essential, early step of T3SS secretion/infection. In vitro targeting defects are directly correlated with compromised in vivo secretion and infection.
Affinity switching in T3S targeting underlies substrate secretion switching
SepD controls the SepL‐EscV conformational cross‐talk underlying the receptor function of SepL. To test whether this is coupled to secretion, we examined the secretion of EspA and Tir in the medium of bacterial cultures (Fig 6I, as indicated; Fig EV5H) of wild‐type (row 1), ΔsepL (row 2), ΔsepD (row 3), and ΔcesL (row 4) EPEC cells. None of these deletion strains grown in bacterial cultures secreted translocators (i.e., EspA, EspB, and EspD; Fig EV5H). All three had switched to effector secretion (e.g., Tir, NleA; Fig EV5H), as anticipated by the in vitro targeting results (Fig 5A and data not shown). However, in the absence of translocator‐built channels, none of them infected HeLa cells (Fig 6J; Appendix Fig S1F–H).
We concluded that SepD couples the translocator receptor function of SepL to the biochemically distinct, succeeding, secretion step (Fig 6K, left). This coupling mechanism most likely also involves chaperone elements, such as the CesAB C‐tail. Release of SepD alters the SepL‐EscV interaction, thus uncoupling the translocator receptor function of SepL from that of secretion (middle). This new state allows now effector binding to EscV and secretion. SepL release may consolidate switching of the injectisome to dedicated binding and secretion of late substrates (right).
Discussion
How exported proteins are targeted to the injectisome of T3SS‐harboring bacteria, how the translocase is activated for secretion, and how the hierarchy of the appropriate exported substrate is imposed have remained elusive. The absence of in vitro systems has hindered dissection of the molecular mechanism. Here, we present the first in vitro reconstitution of T3S targeting that allowed us to determine the order of events, quantify binding interactions, dissect receptor functions from contribution to secretion, and determine the molecular basis of translocator‐effector switching.
CesAB targets EspA to the membrane mainly via its dynamic and unstructured C‐tail (Fig 1B and C). In inactive CesAB dimers, the C‐tail is solvent‐accessible but presumably unavailable for high‐affinity targeting. EspA binding to CesAB relieves this auto‐inhibition and conformationally primes CesAB for two events: electrostatic docking of the C‐tail on the membrane‐associated SepL (Fig 4A, D, and E; Fig EV2A) and molecular recognition of the N‐core by the EscN ATPase (Chen et al, 2013). Since chaperone/substrate docking on SepL does not require EscN (Fig 3C, lane 6), CesAB/EspA association with SepL precedes that with EscN. This simple mechanism couples membrane targeting to the next, ATPase‐catalyzed step, generally believed to cause dissociation of the exported protein from the chaperone and initiate its translocation (Akeda & Galan, 2005; Chen et al, 2013).
That SepL acts as a CesAB receptor was both unexpected and remarkable. SepL and homologous gatekeepers (e.g., MxiC, CopN, YopN/TyeA) are considered as exported, effector‐like proteins with N‐terminal signal peptides (Fields & Hackstadt, 2000; Botteaux et al, 2009; Younis et al, 2010; Amer et al, 2016). However, if and how their own export relates to that of translocators or effectors is unclear (Roehrich et al, 2017). Gatekeepers might recognize specific sequences that lie downstream of the N‐terminal signal sequences of translocators, for example, in EspB (Deng et al, 2015; Roehrich et al, 2017). We anticipate that the novel receptor role revealed here and analyzed in detail for CesAB might be universal for translocator chaperones, even those structurally distinct from CesAB [e.g., the CesD and CesD2 class III chaperones of EspB and D (Wainwright & Kaper, 1998; Neves et al, 2003)]. This would be in agreement with, structures of gatekeepers co‐complexed with chaperone homologues (Archuleta & Spiller, 2014), pull‐down assays (Kubori & Galan, 2002; Silva‐Herzog et al, 2011), and the abolished secretion of all translocators in SepL, SepD, and CesL mutants (Fig EV5H; Wang et al, 2008; Younis et al, 2010; Deng et al, 2015). In this capacity, SepL may act indeed as an early pseudo‐effector (Younis et al, 2010), that occupies EscV, switches it to preferentially accepting translocators and blocks binding of all other effectors.
While EspA‐activated CesAB alone, specifically recognizes SepL with high affinity (Figs 4E and EV4A and B), EspA marginally enhances this binding (~2‐ to 3‐fold). This effect could be mediated indirectly, for example, by EspA optimizing the conformation of CesAB for SepL docking, or directly, for example, through recognition of EspA by EscV. Such interaction would also rationalize the low‐affinity binding component of CesAB/EspA for EscV (i.e., ΔsepL, Fig 3J). In the flagellum system, the C‐terminal region of substrate loaded‐chaperones targets the chaperone/secretory protein complex to the C‐domain of the EscV homologue, FlhA with higher affinity than the chaperone alone (Bange et al, 2010; Minamino et al, 2012; Kinoshita et al, 2013, 2016; Furukawa et al, 2016). Defining the structural basis of such interactions with EscV is a major challenge due to its multimeric and membrane‐embedded nature. Targeting modes of T3S chaperone/exported protein pairs may be variable, but EscV is unique, and hence, entrance and movement through its 5‐nm‐wide C‐domain ante‐chamber and the transmembrane EscRST channel around which the nonamer seems assembled (Abrusci et al, 2013; Hu et al, 2017) are expected to be universally conserved for all T3S exported proteins (early, middle, or late). Attesting to this, mutations on the SepL‐binding sites on EscV severely compromised secretion of Tir (Fig EV5G). Analogous promiscuity is seen in the universal Sec system where the single SecA receptor recognizes the 505 secreted proteins of E. coli K12 on two adjacent sites (Chatzi et al, 2017; Sardis et al, 2017).
SepL participates in multiple interactions (i.e., association with membranes, with EscV, with three different translocator chaperones, with its own two chaperones) and is flexible enough to become secreted through the translocase. These features hampered the use of internal SepL mutations to biochemically dissect its receptor function from its downstream role on EscV activation/EspA secretion. However, dissection was achieved by removing SepD (Figs 4E, H and I, and 5A). This provided important mechanistic insight: Targeting can be completely dissected from secretion reactions. Although limited trypsinolysis demonstrated that SepL harbors a structured 3‐domain core (Fig 3G, lanes 3–6), several observations suggest that SepL is conformationally flexible: the multiplicity of its ligands and topology, its protease susceptibility while membrane‐bound (Fig 3G, lanes 10–12) contrasts the 3‐domain core stability, while in the soluble state (lanes 2–6), its aggregation propensity in the absence of its chaperones (Deng et al, 2005; Younis et al, 2010; Burkinshaw et al, 2015), the multiple conformational states seen crystallographically (Burkinshaw et al, 2015) and the Yersinia homologue being assembled from two distinct polypeptides (Iriarte et al, 1998). We anticipate that SepD, the CesAB C‐tail, and EscV exploit these dynamics to couple SepL receptor function to EspA secretion through EscV (Figs 4E and 6). SepD might have additional, direct roles in the secretion process and, as with other late T3SS chaperones, may directly bind to EscV. These possibilities remain to be explored.
Flux of the hundreds of house‐keeping exported proteins to the bacterial cell envelope that commonly follow the Sec pathway, need not be inter‐dependent, may be largely under transcriptional control and may be fine‐tuned by internal conformational optimization of the secretory molecules (De Geyter et al, 2016; Sardis et al, 2017). In contrast, a hallmark of T3S is its strict secretion hierarchy. This has been imposed because a third of the injectisome components and regulators are themselves T3S substrates that can only be assembled in a defined order, as the machine is self‐built and because its released effectors need to attack host processes in an orchestrated manner (Buttner, 2012; Portaliou et al, 2016; Deng et al, 2017). SepL and SepD determine translocator to effector switching as they repress the affinity of EscV for chaperone‐effector complexes until translocator secretion is complete. This mechanism provides for the first‐time molecular understanding of how effector molecules are prevented from being exported, while they have been already synthesized and diffusing in the cytoplasm (Deng et al, 2005). The structural basis of this repression is not currently known but will undoubtedly be determined by the conformational states of the nonameric translocase component EscV. We consider two likely modes: In an “allosteric regulation” model, SepL/SepD acquire a particular conformation and bind externally to an EscV intraprotomeric groove. This causes long range allosteric changes that affect EscV conformation along all nine subunits and alter a CesT/Tir receptor site located elsewhere on the ring (Fig 7A, left). The finding that sub‐stoichiometric amounts of SepL are sufficient for CesAB/EspA binding (Fig 3D, lanes 7 and 8) is consistent with this model. In a “receptor occlusion” model (Fig 7A, right), SepL and CesT/Tir bind to overlapping surfaces on EscV. Whenever SepL is bound, access of CesT/Tir to the receptor cleft is sterically prevented. Future biochemical and structural studies will be required to resolve these possibilities.
Figure 7. Working model of T3S targeting and translocator‐effector switching.
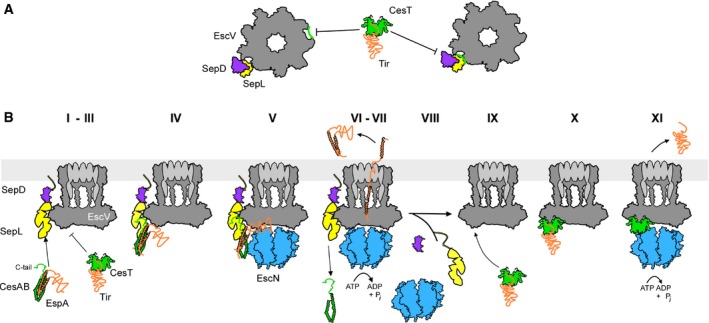
- Allosteric (left) and occlusion (right) models for SepL repression of CesT/Tir targeting. The EscV nonameric ring (dark gray) is in top down view.
- Step‐wise dissection of translocator/effector secretion through the T3SS (see text for details). Green: CesAB, CesT; orange: EspA, Tir; dark gray: EscV; yellow: SepL; magenta: SepD; blue: EscN. CesL (not shown) probably forms heterodimers with SepD. The EscV nonameric ring (dark gray) is in side view.
Whichever the SepL‐mediated suppression mechanism is, at some point, repression ceases and effector secretion proceeds. Given the physical SepL/EscV contact, SepL would have to detach from EscV, at least partially, to allow for effector secretion. Three possible ways to gradually empty EscV sites will be probed in future studies: (i) SepD dissociation; (ii) SepL secretion (Botteaux et al, 2009; Younis et al, 2010; Roehrich et al, 2017); and (iii) SepL detachment from the membrane.
Synthesis of our data and those of others allowed us to propose a working model for targeting and secretion through the T3S translocase: (i) EspA binding to the N‐core of CesAB monomerizes (Chen et al, 2011) and activates it for membrane targeting. (ii) SepL bound to SepD/CesL diffuses to the membrane where it binds to a location adjacent to EscV and thereby (iii) represses CesT/Tir binding to EscV. (iv) CesAB/EspA bind to EscV‐associated SepL mainly via the CesAB C‐tail and N‐core contacts, while EspA might also weakly associate with EscV. (v) The ATPase binds to the activated CesAB N‐core (Chen et al, 2013) and (vi) cycles of ATP hydrolysis dissociate EspA from CesAB (Akeda & Galan, 2005) and release CesAB to the cytoplasm. (vii). Multiple cycles of ATP hydrolysis and the PMF (Andrade et al, 2007; Lee & Rietsch, 2015) drive secretion of EspA. (viii) The SepL/D affinity switch becomes disengaged from EscV and no longer represses CesT/Tir binding. (ix) The affinity of EscV for CesT/Tir increases, (x) CesT/Tir associate with high affinity to EscV and (xi) become secreted after re‐docking of the ATPase and a repeat of the cycle described for CesAB/EspA.
The availability of an in vitro system for CesAB/EspA and CesT/Tir targeting paves the way for a complete functional in vitro reconstitution of the T3S machinery that will allow systematic dissection of all the reaction steps and testing of mechanistic predictions. Together with structural and biophysical data, this promises to provide mechanistic insight in this complex, tightly regulated protein trafficking machine.
Materials and Methods
For the complete list of strains, plasmids, mutants, primers, buffers, antibodies, immobilized peptides on peptide arrays, peptide and protein list identified by MS and HDX‐MS analysis, see Appendix and Source Data.
Cell growth, induction of gene expression, in vivo secretion
EPEC strains were grown (DMEM; 37°C; 5 h); plasmid gene expression was induced (OD600 = 0.3; AHT; 2.5 ng/ml; 3 h or as indicated). Cells were harvested (5,000 g; 20 min; 4°C); the spent medium was TCA‐precipitated (20% w/v) and resuspended in volumes adjusted according to OD600 (1.5 M Tris–HCl pH 8.8). An equal amount of cells or supernatant derived from equal amount of cells were analyzed on SDS–PAGE, and immuno‐ or Coomassie Blue‐stained.
Intracellular complexes and subcellular protein localization
Bacteria were sonicated (3 h post‐induction; Buffer A; ethanol‐dry‐ice bath; 30 s × 15 cycles). Unbroken cells were removed (5,000 g; 20 min; 4°C) before ultracentrifugation (300,000 g; 30 min; 4°C). Equal amount of cytosolic or membrane proteins was analyzed (15% SDS–PAGE; 7% Native‐PAGE; 30 mA; 4°C; 15 h) and immunostained.
Limited trypsinolysis and N‐terminal sequencing
Purified soluble protein (1 μg) mixed with 29 μg BSA or 1 μg of the same IMV‐bound protein/30 μg of total membrane protein were incubated with trypsin (Roche; 0.5 μg/sample) before Pefabloc addition (0.01 M; Roche). Following 5.5 M urea‐15% SDS–PAGE, peptides were electro‐transferred onto ProBlott (Applied Biosystems; 10 mM CAPS pH 11; 10% methanol), Coomassie Blue‐stained and N‐terminally sequenced (Alta Bioscience; Birmingham, UK).
Purification of inverted Inner membrane vesicles
As described (Chatzi et al, 2017; Tsolis & Economou, 2017a); detailed in Appendix Supplementary Materials and Methods.
Flotation assays
As described (Papanikou et al, 2005; Karamanou et al, 2008) with a gradient modification (2 M loading; 30 μl 1.8 M; 75 μl 1.5 M; 20 μl 1 M sucrose in Buffer C); 7 × 25 μl fractions were collected.
Equilibrium dissociation constants (K d)
As described (Gouridis et al, 2009, 2010, 2013; Karamanou et al, 2008); detailed in Appendix Supplementary Materials and Methods. Briefly, urea‐treated IMVs (20 μg/sample) were incubated with a wide range of protein ligand concentration (0–5,000 nM) mixed with a tracer amount of [35S]‐labeled protein ligand (Buffer C; 4°C).
Peptide array experiments
As described (Karamanou et al, 2008) with three sequential blotting transfers to PVDF (30 min; 1.5 h; 1.5 h). Following immunostaining, Image Quant (GE Healthcare) was used to quantify the intensity of ligand binding‐signals on peptide arrays.
Quantification of signals
Expressed or secreted, soluble or IMV‐bound proteins, wherever indicated, were quantified using Western blot analysis, against a standard curve of purified protein and Image Quant software (GE). Protein amount is relative to wt control or input, present on the same gel, which represented 100%. For significance test, a two‐side t‐test was used without assuming equal variance. Calculated P‐values were adjusted for multiple hypothesis testing error using the “Benjamini–Hochberg” (Benjamini & Hochberg, 1995) method, as previously described (Tsolis et al, 2016; Tsolis & Economou, 2017b).
Miscellaneous
Ni2+‐affinity chromatography was as per the manufacturer's instructions (Qiagen). SDS–PAGE, protein transfer to nitrocellulose, Western blot analysis was as per manufacturer's instructions (Bio‐Rad instruction manual; see Appendix). GPC‐MALS analysis, SecA ATPase assays, in vitro proPhoA translocation assays (Gouridis et al, 2010; Karamanou et al, 1999), and HeLa cell infection (Creasey et al, 2003) were as previously described.
Data availability
MS data are available via ProteomeXchange with identifier PXD007087.
Author contributions
AGP performed in vivo and in vitro assays and molecular cloning; KCT performed proteomics analysis, molecular cloning, genetic knockouts, statistical analysis, and antibody preparation; MSL and VB contributed in molecular cloning, antibody preparation, genetic knockouts, secretion and infection assays; JR performed cross‐linking experiments; AT performed HDX‐MS experiments; VFC, CGK, and GF contributed in training, strains, and molecular cloning; SK trained and supervised biochemical and biophysical experiments; SK and AE wrote the paper with contributions from AGP and KCT and editing from GF and CGK; AE and SK conceived and supervised the project.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Expanded View
Acknowledgements
We thank L. Persoons and K. Lepaige for HeLa cells, P. Baatsen for EM microscopy, V. Zorzini for modeling the EscV C‐Domain nonamer, S. Munck and N. Corthout for access to fluorescence microscopes and guidance, S. Carpentier for help with Mass spectrometry, M. Koukaki for plasmids, O. Tomoaki for strains. This work was supported by grants (to AE): KUL‐Spa (Onderzoekstoelagen 2013; Bijzonder Onderzoeksfonds; KU Leuven); RiMembR (Vlaanderen Onderzoeksprojecten; #G0C6814N; FWO); T3RecS (#G002516N; FWO); and DIP‐BiD (#AKUL/15/40–G0H2116N; Hercules/FWO) and (to SK): #G0B4915N; FWO and (to JR): R&D Pilot project‐2015 [Instruct, part of the European Strategy Forum on Research Infrastructures (ESFRI) and supported by national member subscriptions], (to GF): Wellcome Trust and (to C.G.K.): NIH Grant AI094623.
The EMBO Journal (2017) 36: 3517–3531
Contributor Information
Spyridoula Karamanou, Email: lily.karamanou@kuleuven.be.
Anastassios Economou, Email: tassos.economou@kuleuven.be.
References
- Abe A, de Grado M, Pfuetzner RA, Sanchez‐Sanmartin C, Devinney R, Puente JL, Strynadka NC, Finlay BB (1999) Enteropathogenic Escherichia coli translocated intimin receptor, Tir, requires a specific chaperone for stable secretion. Mol Microbiol 33: 1162–1175 [DOI] [PubMed] [Google Scholar]
- Abrusci P, Vergara‐Irigaray M, Johnson S, Beeby MD, Hendrixson DR, Roversi P, Friede ME, Deane JE, Jensen GJ, Tang CM, Lea SM (2013) Architecture of the major component of the type III secretion system export apparatus. Nat Struct Mol Biol 20: 99–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akeda Y, Galan JE (2005) Chaperone release and unfolding of substrates in type III secretion. Nature 437: 911–915 [DOI] [PubMed] [Google Scholar]
- Amer AA, Gurung JM, Costa TR, Ruuth K, Zavialov AV, Forsberg A, Francis MS (2016) YopN and TyeA hydrophobic contacts required for regulating Ysc‐Yop Type III secretion activity by yersinia pseudotuberculosis. Front Cell Infect Microbiol 6: 66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade A, Pardo JP, Espinosa N, Perez‐Hernandez G, Gonzalez‐Pedrajo B (2007) Enzymatic characterization of the enteropathogenic Escherichia coli type III secretion ATPase EscN. Arch Biochem Biophys 468: 121–127 [DOI] [PubMed] [Google Scholar]
- Archuleta TL, Spiller BW (2014) A gatekeeper chaperone complex directs translocator secretion during type three secretion. PLoS Pathog 10: e1004498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bange G, Kummerer N, Engel C, Bozkurt G, Wild K, Sinning I (2010) FlhA provides the adaptor for coordinated delivery of late flagella building blocks to the type III secretion system. Proc Natl Acad Sci USA 107: 11295–11300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57: 289–300 [Google Scholar]
- Botteaux A, Sory MP, Biskri L, Parsot C, Allaoui A (2009) MxiC is secreted by and controls the substrate specificity of the Shigella flexneri type III secretion apparatus. Mol Microbiol 71: 449–460 [DOI] [PubMed] [Google Scholar]
- Burkinshaw BJ, Souza SA, Strynadka NC (2015) Structural analysis of SepL, an enteropathogenic Escherichia coli type III secretion‐system gatekeeper protein. Acta Crystallogr F Struct Biol Commun 71: 1300–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttner D (2012) Protein export according to schedule: architecture, assembly, and regulation of type III secretion systems from plant‐ and animal‐pathogenic bacteria. Microbiol Mol Biol Rev 76: 262–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callister SJ, Barry RC, Adkins JN, Johnson ET, Qian WJ, Webb‐Robertson BJ, Smith RD, Lipton MS (2006) Normalization approaches for removing systematic biases associated with mass spectrometry and label‐free proteomics. J Proteome Res 5: 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzi KE, Sardis MF, Tsirigotaki A, Koukaki M, Sostaric N, Konijnenberg A, Sobott F, Kalodimos CG, Karamanou S, Economou A (2017) Preprotein mature domains contain translocase targeting signals that are essential for secretion. J Cell Biol 216: 1357–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Balabanidou V, Remeta DP, Minetti CA, Portaliou AG, Economou A, Kalodimos CG (2011) Structural instability tuning as a regulatory mechanism in protein‐protein interactions. Mol Cell 44: 734–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Ai X, Portaliou AG, Minetti CA, Remeta DP, Economou A, Kalodimos CG (2013) Substrate‐activated conformational switch on chaperones encodes a targeting signal in type III secretion. Cell Rep 3: 709–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creasey EA, Friedberg D, Shaw RK, Umanski T, Knutton S, Rosenshine I, Frankel G (2003) CesAB is an enteropathogenic Escherichia coli chaperone for the type‐III translocator proteins EspA and EspB. Microbiology 149: 3639–3647 [DOI] [PubMed] [Google Scholar]
- De Geyter J, Tsirigotaki A, Orfanoudaki G, Zorzini V, Economou A, Karamanou S (2016) Protein folding in the cell envelope of Escherichia coli . Nat Microbiol 1: 16107 [DOI] [PubMed] [Google Scholar]
- Dean P, Kenny B (2009) The effector repertoire of enteropathogenic E. coli: ganging up on the host cell. Curr Opin Microbiol 12: 101–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Li Y, Hardwidge PR, Frey EA, Pfuetzner RA, Lee S, Gruenheid S, Strynakda NC, Puente JL, Finlay BB (2005) Regulation of type III secretion hierarchy of translocators and effectors in attaching and effacing bacterial pathogens. Infect Immun 73: 2135–2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Yu HB, Li Y, Finlay BB (2015) SepD/SepL‐dependent secretion signals of the type III secretion system translocator proteins in enteropathogenic Escherichia coli . J Bacteriol 197: 1263–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Marshall NC, Rowland JL, McCoy JM, Worrall LJ, Santos AS, Strynadka NCJ, Finlay BB (2017) Assembly, structure, function and regulation of type III secretion systems. Nat Rev Microbiol 15: 323–337; Corrigendum: 15: 379 [DOI] [PubMed] [Google Scholar]
- Dziva F, van Diemen PM, Stevens MP, Smith AJ, Wallis TS (2004) Identification of Escherichia coli O157: H7 genes influencing colonization of the bovine gastrointestinal tract using signature‐tagged mutagenesis. Microbiology 150: 3631–3645 [DOI] [PubMed] [Google Scholar]
- Elliott SJ, O'Connell CB, Koutsouris A, Brinkley C, Donnenberg MS, Hecht G, Kaper JB (2002) A gene from the locus of enterocyte effacement that is required for enteropathogenic Escherichia coli to increase tight‐junction permeability encodes a chaperone for EspF. Infect Immun 70: 2271–2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields KA, Hackstadt T (2000) Evidence for the secretion of Chlamydia trachomatis CopN by a type III secretion mechanism. Mol Microbiol 38: 1048–1060 [DOI] [PubMed] [Google Scholar]
- Furukawa Y, Inoue Y, Sakaguchi A, Mori Y, Fukumura T, Miyata T, Namba K, Minamino T (2016) Structural stability of flagellin subunit affects the rate of flagellin export in the absence of FliS chaperone. Mol Microbiol 102: 405–416 [DOI] [PubMed] [Google Scholar]
- Gauthier A, Puente JL, Finlay BB (2003) Secretin of the enteropathogenic Escherichia coli type III secretion system requires components of the type III apparatus for assembly and localization. Infect Immun 71: 3310–3319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouridis G, Karamanou S, Gelis I, Kalodimos CG, Economou A (2009) Signal peptides are allosteric activators of the protein translocase. Nature 462: 363–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouridis G, Karamanou S, Koukaki M, Economou A (2010) In vitro assays to analyze translocation of the model secretory preprotein alkaline phosphatase. Methods Mol Biol 619: 157–172 [DOI] [PubMed] [Google Scholar]
- Gouridis G, Karamanou S, Sardis MF, Scharer MA, Capitani G, Economou A (2013) Quaternary dynamics of the SecA motor drive translocase catalysis. Mol Cell 52: 655–666 [DOI] [PubMed] [Google Scholar]
- Hu B, Lara‐Tejero M, Kong Q, Galan JE, Liu J (2017) In situ molecular architecture of the salmonella type III secretion machine. Cell 168: 1065–1074.e1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iriarte M, Sory MP, Boland A, Boyd AP, Mills SD, Lambermont I, Cornelis GR (1998) TyeA, a protein involved in control of Yop release and in translocation of Yersinia Yop effectors. EMBO J 17: 1907–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karamanou S, Vrontou E, Sianidis G, Baud C, Roos T, Kuhn A, Politou AS, Economou A (1999) A molecular switch in SecA protein couples ATP hydrolysis to protein translocation. Mol Microbiol 34: 1133–1145 [DOI] [PubMed] [Google Scholar]
- Karamanou S, Bariami V, Papanikou E, Kalodimos CG, Economou A (2008) Assembly of the translocase motor onto the preprotein‐conducting channel. Mol Microbiol 70: 311–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny B, DeVinney R, Stein M, Reinscheid DJ, Frey EA, Finlay BB (1997) Enteropathogenic E. coli (EPEC) transfers its receptor for intimate adherence into mammalian cells. Cell 91: 511–520 [DOI] [PubMed] [Google Scholar]
- Kinoshita M, Hara N, Imada K, Namba K, Minamino T (2013) Interactions of bacterial flagellar chaperone‐substrate complexes with FlhA contribute to co‐ordinating assembly of the flagellar filament. Mol Microbiol 90: 1249–1261 [DOI] [PubMed] [Google Scholar]
- Kinoshita M, Nakanishi Y, Furukawa Y, Namba K, Imada K, Minamino T (2016) Rearrangements of alpha‐helical structures of FlgN chaperone control the binding affinity for its cognate substrates during flagellar type III export. Mol Microbiol 101: 656–670 [DOI] [PubMed] [Google Scholar]
- Knutton S, Rosenshine I, Pallen MJ, Nisan I, Neves BC, Bain C, Wolff C, Dougan G, Frankel G (1998) A novel EspA‐associated surface organelle of enteropathogenic Escherichia coli involved in protein translocation into epithelial cells. EMBO J 17: 2166–2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubori T, Galan JE (2002) Salmonella type III secretion‐associated protein InvE controls translocation of effector proteins into host cells. J Bacteriol 184: 4699–4708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PC, Rietsch A (2015) Fueling type III secretion. Trends Microbiol 23: 296–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamino T, Kinoshita M, Hara N, Takeuchi S, Hida A, Koya S, Glenwright H, Imada K, Aldridge PD, Namba K (2012) Interaction of a bacterial flagellar chaperone FlgN with FlhA is required for efficient export of its cognate substrates. Mol Microbiol 83: 775–788 [DOI] [PubMed] [Google Scholar]
- Nadler C, Shifrin Y, Nov S, Kobi S, Rosenshine I (2006) Characterization of enteropathogenic Escherichia coli mutants that fail to disrupt host cell spreading and attachment to substratum. Infect Immun 74: 839–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves BC, Mundy R, Petrovska L, Dougan G, Knutton S, Frankel G (2003) CesD2 of enteropathogenic Escherichia coli is a second chaperone for the type III secretion translocator protein EspD. Infect Immun 71: 2130–2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell CB, Creasey EA, Knutton S, Elliott S, Crowther LJ, Luo W, Albert MJ, Kaper JB, Frankel G, Donnenberg MS (2004) SepL, a protein required for enteropathogenic Escherichia coli type III translocation, interacts with secretion component SepD. Mol Microbiol 52: 1613–1625 [DOI] [PubMed] [Google Scholar]
- Papanastasiou M, Orfanoudaki G, Koukaki M, Kountourakis N, Sardis MF, Aivaliotis M, Karamanou S, Economou A (2013) The Escherichia coli peripheral inner membrane proteome. Mol Cell Proteomics 12: 599–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papanastasiou M, Orfanoudaki G, Kountourakis N, Koukaki M, Sardis MF, Aivaliotis M, Tsolis KC, Karamanou S, Economou A (2016) Rapid label‐free quantitative analysis of the E. coli BL21(DE3) inner membrane proteome. Proteomics 16: 85–97 [DOI] [PubMed] [Google Scholar]
- Papanikou E, Karamanou S, Baud C, Frank M, Sianidis G, Keramisanou D, Kalodimos CG, Kuhn A, Economou A (2005) Identification of the preprotein binding domain of SecA. J Biol Chem 280: 43209–43217 [DOI] [PubMed] [Google Scholar]
- Parsot C (2003) The various and varying roles of specific chaperones in type III secretion systems. Curr Opin Microbiol 6: 7–14 [DOI] [PubMed] [Google Scholar]
- Portaliou AG, Tsolis KC, Loos MS, Zorzini V, Economou A (2016) Type III secretion: building and operating a remarkable nanomachine. Trends Biochem Sci 41: 175–189 [DOI] [PubMed] [Google Scholar]
- Roehrich AD, Bordignon E, Mode S, Shen DK, Liu X, Pain M, Murillo I, Martinez‐Argudo I, Sessions RB, Blocker AJ (2017) Steps for shigella gatekeeper protein MxiC function in hierarchical type III secretion regulation. J Biol Chem 292: 1705–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo‐Hamano Y, Imada K, Minamino T, Kihara M, Shimada M, Kitao A, Namba K (2010) Structure of the cytoplasmic domain of FlhA and implication for flagellar type III protein export. Mol Microbiol 76: 260–268 [DOI] [PubMed] [Google Scholar]
- Sardis MF, Tsirigotaki A, Chatzi KE, Portaliou AG, Gouridis G, Karamanou S, Economou A (2017) Preprotein conformational dynamics drive bivalent translocase docking and secretion. Structure 25: 1056–1067.e1056 [DOI] [PubMed] [Google Scholar]
- Sato H, Frank DW (2011) Multi‐functional characteristics of the Pseudomonas aeruginosa type III needle‐tip protein, PcrV; comparison to orthologs in other gram‐negative bacteria. Front Microbiol 2: 142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubot FD, Jackson MW, Penrose KJ, Cherry S, Tropea JE, Plano GV, Waugh DS (2005) Three‐dimensional structure of a macromolecular assembly that regulates type III secretion in Yersinia pestis . J Mol Biol 346: 1147–1161 [DOI] [PubMed] [Google Scholar]
- Silva‐Herzog E, Joseph SS, Avery AK, Coba JA, Wolf K, Fields KA, Plano GV (2011) Scc1 (CP0432) and Scc4 (CP0033) function as a type III secretion chaperone for CopN of Chlamydia pneumoniae . J Bacteriol 193: 3490–3496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas NA, Deng W, Puente JL, Frey EA, Yip CK, Strynadka NC, Finlay BB (2005) CesT is a multi‐effector chaperone and recruitment factor required for the efficient type III secretion of both LEE‐ and non‐LEE‐encoded effectors of enteropathogenic Escherichia coli . Mol Microbiol 57: 1762–1779 [DOI] [PubMed] [Google Scholar]
- Tsirigotaki A, De Geyter J, Sostaric N, Economou A, Karamanou S (2017a) Protein export through the bacterial Sec pathway. Nat Rev Microbiol 15: 21–36 [DOI] [PubMed] [Google Scholar]
- Tsirigotaki A, Papanastasiou M, Trelle MB, Jørgensen TJD, Economou A (2017b) Chapter four – Analysis of translocation‐competent secretory proteins by HDX‐MS In Methods in enzymology, Arun KS. (ed), Vol. 586, pp 57–83. Cambridge, MA: Academic Press; [DOI] [PubMed] [Google Scholar]
- Tsolis KC, Bagli E, Kanaki K, Zografou S, Carpentier S, Bei ES, Christoforidis S, Zervakis M, Murphy C, Fotsis T, Economou A (2016) Proteome changes during transition from human embryonic to vascular progenitor cells. J Proteome Res 15: 1995–2007 [DOI] [PubMed] [Google Scholar]
- Tsolis KC, Economou A (2017a) Chapter two – Quantitative proteomics of the E. coli membranome In Methods in enzymology, Arun KS. (ed), Vol. 586, pp 15–36. Cambridge, MA: Academic Press; [DOI] [PubMed] [Google Scholar]
- Tsolis KC, Economou A (2017b) Quantitative proteomics of the E. coli membranome. Methods Enzymol 586: 15–36 [DOI] [PubMed] [Google Scholar]
- Wainwright LA, Kaper JB (1998) EspB and EspD require a specific chaperone for proper secretion from enteropathogenic Escherichia coli . Mol Microbiol 27: 1247–1260 [DOI] [PubMed] [Google Scholar]
- Wang D, Roe AJ, McAteer S, Shipston MJ, Gally DL (2008) Hierarchal type III secretion of translocators and effectors from Escherichia coli O157:H7 requires the carboxy terminus of SepL that binds to Tir. Mol Microbiol 69: 1499–1512 [DOI] [PubMed] [Google Scholar]
- Wilharm G, Dittmann S, Schmid A, Heesemann J (2007) On the role of specific chaperones, the specific ATPase, and the proton motive force in type III secretion. Int J Med Microbiol 297: 27–36 [DOI] [PubMed] [Google Scholar]
- Yip CK, Finlay BB, Strynadka NC (2005) Structural characterization of a type III secretion system filament protein in complex with its chaperone. Nat Struct Mol Biol 12: 75–81 [DOI] [PubMed] [Google Scholar]
- Younis R, Bingle LE, Rollauer S, Munera D, Busby SJ, Johnson S, Deane JE, Lea SM, Frankel G, Pallen MJ (2010) SepL resembles an aberrant effector in binding to a class 1 type III secretion chaperone and carrying an N‐terminal secretion signal. J Bacteriol 192: 6093–6098 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Expanded View
Data Availability Statement
MS data are available via ProteomeXchange with identifier PXD007087.