

Abstract
Elevated amino acid catabolism is common to many cancers. Here, we show that glioblastoma are excreting large amounts of branched‐chain ketoacids (BCKAs), metabolites of branched‐chain amino acid (BCAA) catabolism. We show that efflux of BCKAs, as well as pyruvate, is mediated by the monocarboxylate transporter 1 (MCT1) in glioblastoma. MCT1 locates in close proximity to BCKA‐generating branched‐chain amino acid transaminase 1, suggesting possible functional interaction of the proteins. Using in vitro models, we demonstrate that tumor‐excreted BCKAs can be taken up and re‐aminated to BCAAs by tumor‐associated macrophages. Furthermore, exposure to BCKAs reduced the phagocytic activity of macrophages. This study provides further evidence for the eminent role of BCAA catabolism in glioblastoma by demonstrating that tumor‐excreted BCKAs might have a direct role in tumor immune suppression. Our data further suggest that the anti‐proliferative effects of MCT1 knockdown observed by others might be related to the blocked excretion of BCKAs.
Keywords: BCAT1, branched‐chain ketoacid, glioblastoma, MCT1, phagocytosis
Subject Categories: Cancer, Membrane & Intracellular Transport, Metabolism
Introduction
Rapidly proliferating types of cancers have been shown to exhibit characteristic alterations of metabolism including a shift away from oxidative phosphorylation and toward aerobic glycolysis, which is known as the “Warburg effect”, and an increased dependence on amino acid metabolism 1, 2. As a consequence, cancer cells typically display elevated uptake of amino acids, in particular glutamine and the branched‐chain amino acids (BCAAs) valine, leucine, and isoleucine 3. BCAAs participate directly and indirectly in a variety of crucial biochemical functions in the brain and other tissues. These include protein synthesis, the production of energy, the compartmentalization of glutamate, the transfer of nitrogen between tissues and synthesis of the amine neurotransmitters serotonin, and the catecholamines, dopamine and norepinephrine 4, 5. The first step of BCAA catabolism involves the transfer of the primary amino group to α‐ketoglutarate to generate glutamate and branched‐chain ketoacids (BCKAs). These transamination reactions are catalyzed by the branched‐chain aminotransferases BCAT1 and BCAT2 in the cytoplasm and the mitochondria, respectively. Genetic mutations that abolish BCAA catabolism result in the accumulation of toxic levels of BCAAs and BCKAs in tissues that has been proposed to block cell energy metabolism 6, 7, 8. We recently reported that BCAT1 is overexpressed in glioblastoma and required to support the sustained growth of these aggressive brain tumors. Interestingly, a large proportion of the glutamate produced by BCAT1 was excreted from the glioblastoma cells 9. Further, it is known that BCKAs also can be excreted from cells, for example, from cultured astrocytes 10, 11 and during overnight fasting when BCAAs are transaminated to BCKAs in muscle cells, excreted and transported to the liver for further oxidation 6.
Monocarboxylate transporters (MCTs) mediate the transport of monocarboxylates through plasma membranes of various cells. The family of MCTs comprises 14 members of which MCT1‐4 are known to mediate proton‐linked bidirectional transport 12, 13. Suppression of MCT1 and MCT4 has been shown to block cell proliferation of glioblastoma as well as other cancers 14, 15, 16, 17, 18, 19. In the cancer field, studies so far have mostly focused on the role of MCTs in the transport of lactate and pyruvate, but others have reported that MCTs also can mediate the influx of other hydroxy and ketoacids into Xenopus oocytes 20, 21, 22 and that an MCT protein is required for the transport of α‐ketoisocaproate (KIC) in neurons 23.
Recent studies suggested that different aspects of tumor metabolism, that is lactate production and consumption, are compartmentalized between tumor cells and cancer‐associated fibroblasts (CAFs) in mammary carcinoma 24. In this model, lactate is produced and excreted via MCT4 by stroma cells and taken up by tumor cells via MCT1 for ATP production. An analogous exchange of metabolites between different types of cells, coupling glutamate/glutamine and leucine/KIC cycles, was shown to regulate the maintenance of nitrogen balance in normal brain 10, 11. In glioblastoma, infiltrating macrophages and microglia constitute a substantial portion of the tumor mass that can be as high as one in every three cells 25. Whether and how these tumor‐associated macrophages are supporting tumor growth is still not entirely clear; however, it has been described that lactate can induce the polarization of macrophages into an M1‐ or M2‐like phenotype 26, suggesting that tumor‐derived metabolites can act as messengers between the tumor and its microenvironment.
Here, we are addressing the question whether glioblastoma cells with high BCAT1 expression are excreting BCKAs and whether MCTs can transport them across the cell membrane. Further, we are using isotope tracing and phenotypic analyses to evaluate potential effects that tumor‐derived BCKAs might exert on macrophages.
Results
Glioblastoma cells excrete BCKAs
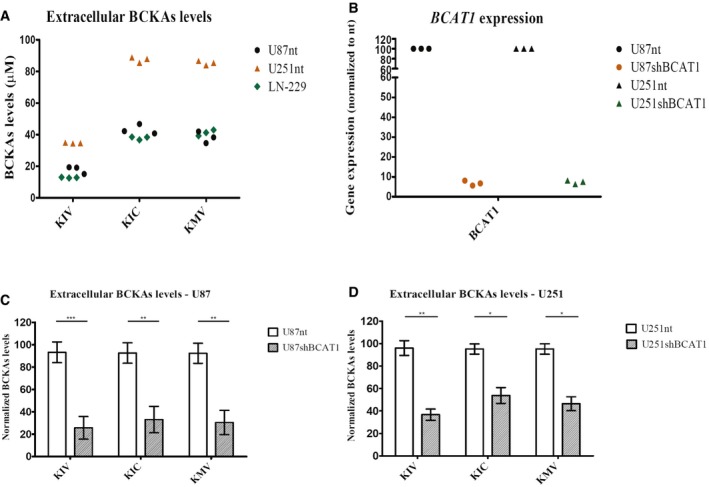
To be able to directly quantify the branched‐chain ketoacids (BCKAs) α‐ketoisocaproate (KIC), α‐ketoisovalerate (KIV), and α‐keto‐methylvalerate (KMV) in biological extracts, we established an ultra performance liquid chromatography (UPLC) protocol in which ketoacids were derivatized with either o‐phenylenediamine (OPD) or 1,2‐diamino‐4,5‐methylenedioxybenzene (DMB). While the very sensitive DMB method allows for the analysis of the usually low intracellular BCKA concentrations, the less sensitive OPD method was used to quantify the levels of BCKAs and pyruvate in cell culture supernatants (Fig EV1A and B, Appendix Table S3). This protocol then was applied to the analysis of cell culture media from three glioblastoma cell lines, U87‐MG, U251‐MG, and LN‐229, revealing accumulations of extracellular BCKAs to concentrations of up to 85 μM over a period of 24 h (Fig 1A). Supplementation of the culture media with BCKAs showed that even concentrations of up to 300 μM showed did not affect glioblastoma cell proliferation or viability (Fig EV2). Following shRNA‐mediated knockdown of BCAT1, the enzyme that generates BCKAs by transamination of BCAAs in the cytoplasm, BCKAs' excretion was reduced to about 70 and 50% in U87‐MG and U251‐MG, respectively (Fig 1C and D). These data indicate that glioblastoma is capable of producing and excreting large amounts of BCKAs over relatively short periods of time. It is not known, however, which transporters mediate BCKA efflux from glioblastoma cells.
Figure EV1. UPLC derivatization methods and heterologous expression in Xenopus oocytes.
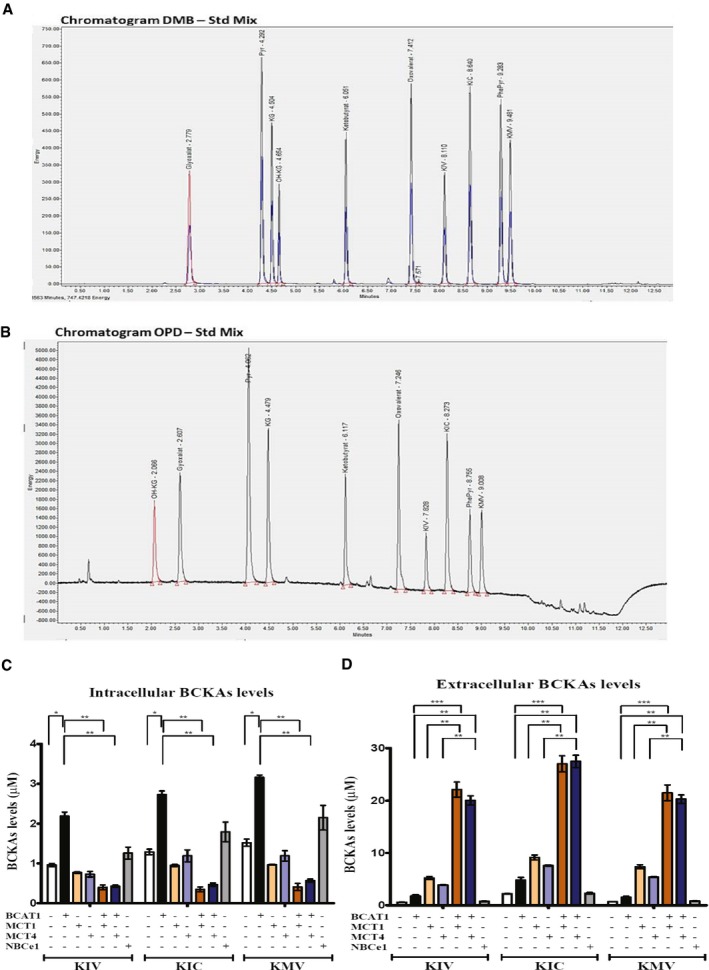
-
A, BBCKAs (KIV, KIC, KMV) are detected by ultra performance liquid chromatography (UPLC) coupled to fluorescence detection in cell extracts using the derivatization with DMB (A) or in cell culture supernatants using the OPD derivatization reagent (B).
-
C, DCo‐expression of BCAT1 and either MCT1 or MCT4 facilitates the excretion of BCKAs from Xenopus oocytes. BCKAs (KIV, KIC, KMV) levels detected by ultra performance liquid chromatography (UPLC) coupled to fluorescence detection in native oocytes or oocytes expressing BCAT1, MCT1, MCT4, NBCe1, or co‐expressing MCT1 or MCT4 and BCAT1 stimulated with BCAAs (1 nmol L‐valine, 1 nmol L‐leucine, 1 nmol L‐isoleucine) and α‐ketoglutarate (3 nmol) for 2 h at RT (C) and in the oocytes culture medium (D). Heterologous expression of the human or rat proteins in the oocytes was performed by injection of the respective cRNA into the oocyte. The cRNA concentrations used are 5 ng rat MCT1, 5 ng rat MCT4, 12 ng human BCAT1, 7 ng human NBCe1 or 5 ng MCT1 or MCT4, and 12 ng BCAT1. Batches of 10 oocytes are used per condition. Values are mean ± SD of three independent biological replicates. KIV: α‐ketoisovalerate. KIC: α‐ketoisocaproate. KMV: α‐keto‐β‐methylvalerate. NBCe1: sodium/bicarbonate cotransporter. Unpaired Student's t‐test *P < 0.05; **P < 0.01; ***P < 0.001.
Figure 1. Glioblastoma cells excrete BCKAs.
-
ABCKAs (KIV, KIC, KMV) are detected by ultra performance liquid chromatography (UPLC) coupled to fluorescence detection in cell culture supernatants from U87‐MG and U251‐MG cells expressing normal BCAT1 levels, U87nt and U251nt, respectively, and from LN‐229 cells. n = 3 technical replicates.
-
BBCAT1 mRNA expression in U87‐MG and U251‐MG cells after BCAT1 knockdown. Expression levels are normalized to those from cells expressing normal BCAT1 levels (nt). n = 3 technical replicates.
-
C, DBCKAs levels after BCAT1 knockdown in supernatants from U87 (C) and U251 (D) cells. Values are mean ± SD of three independent biological replicates. BCKAs levels are normalized to the detected levels in U87nt or U251nt cells, respectively.
Figure EV2. BCKAs do not impact on glioblastoma cell phenotype.
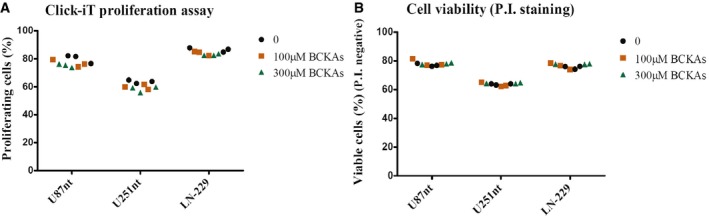
- Effect of 100 or 300 μM BCKAs on cell proliferation of U87nt, U251nt, and LN‐229 cells determined using the Click‐iT proliferation assay. n = 3 technical replicates.
- Effect of 100 or 300 μM BCKAs on cell proliferation of U87nt, U251nt, and LN‐229 cells on cell viability determined by propidium iodide (P.I) staining, and analysis of P.I. negative (viable) cells was done using flow cytometry (FACS Canto II, BD Biosciences). n = 3 technical replicates.
Previously, it has been shown that the monocarboxylate transporters MCT1 and MCT4 can mediate the influx of the BCKAs KIV and KIC into Xenopus oocytes, as indicated by the drop of intracellular pH due to the influx of protons that are co‐transported with KIC and KIV 21, 22. To determine which of the monocarboxylate transporters could be responsible for BCKA excretion from glioblastoma cells, we analyzed their RNA expression in published data of primary untreated glioblastoma (n = 480; TCGA dataset). SLC16A1 (MCT1) and SLC16A3 (MCT4) both were significantly overexpressed in glioblastoma compared to normal brain tissue (Fig 2A). SLC16A7 (MCT2) expression and SLC16A8 (MCT3) expression were variable but on average lower in tumor than in normal tissue (Fig 2A). Comparing glioblastoma subtypes, MCT4 expression was significantly higher in mesenchymal tumors than in others (Fig 2B). We also could confirm expression of MCT1 and MCT4 on RNA (Fig 2C) and protein (Fig 2D) levels in three glioblastoma cell lines, U87‐MG, U251‐MG, and LN‐229, which commonly are used as in vitro models. To confirm that MCT1 and MCT4 can mediate BCKA efflux from cells, we expressed combinations of BCAT1, MCT1, and MCT4 in Xenopus oocytes and analyzed media supernatant and cellular extracts. BCKAs accumulated in the oocytes upon BCAT1 expression and were released into the supernatant only after co‐expression of either MCT1 or MCT4 (Fig EV1C and D). BCKA concentrations in the supernatants reached about 25 μM within 2 h suggesting that MCT1 and MCT4 could mediate the observed efflux of BCKAs from glioblastoma cells.
Figure 2. MCT1 and MCT4 are upregulated in glioblastoma.
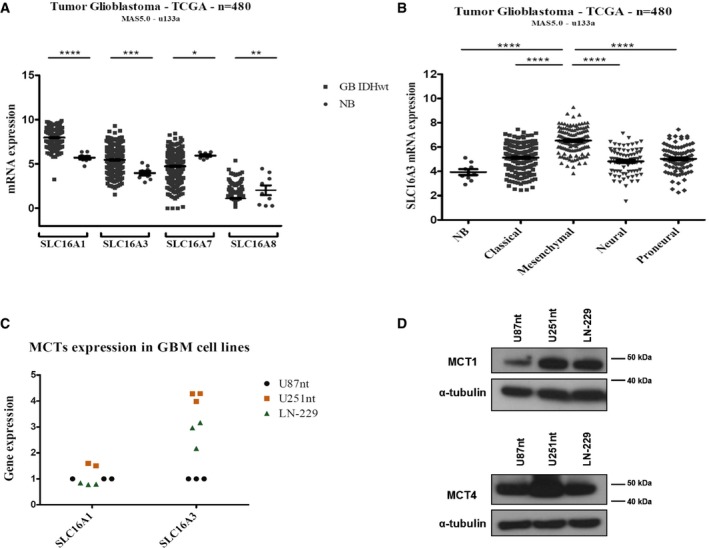
-
AmRNA expression of SLC16A1 (MCT1), SLC16A3 (MCT4), SLC16A7 (MCT2), SLC16A8 (MCT3) in glioblastoma (GB IDHwt) (n = 480) compared to normal brain (NB) tissue (n = 9) in samples from the TCGA dataset.
-
BmRNA expression of SLC16A3 in the four different subtypes of glioblastoma (n = 480) defined by gene expression patterns compared to normal brain (NB) tissue (n = 9) in samples from the TCGA dataset. Values are mean ± SEM.
-
C, DExpression of MCT1 and MCT4 in the glioblastoma cell lines U87nt, U251nt, and LN‐229, at mRNA (C) and protein level (D). (C) Values are normalized to the levels detected for U87nt. n = 3 technical replicates. (D) α‐tubulin was used as loading control.
Inhibition of MCT1 but not MCT4 reduces BCKA excretion from glioblastoma cells
To investigate the role of MCT1 and MCT4 in the transport of BCKAs in glioblastoma cells, we manipulated MCT function in the U87‐MG, U251‐MG, and LN‐229 cell lines by pharmacologic inhibition of MCT1 with the MCT1/MCT2‐specific inhibitor AR‐C155858 at concentrations that did not affect cell proliferation (Appendix Fig S1A and B). Application of AR‐C155858 reduced the excretion of individual BCKAs 20% in U87‐MG and 80% in LN‐229 (Fig 3A and B) while intracellular BCKA concentrations showed concurrent increases (Fig 3D and E). For unknown reasons, the effects of inhibition were less clear in U251‐MG cells, where in most cases, we could only observe trends for decreases of excreted and increases of intracellular BCKAs, respectively (Fig 3C and F). These findings were independently confirmed by siRNA‐mediated knockdown of MCT1 (Fig EV3).
Figure 3. Inhibition of MCT1 transporter decreases BCKAs excretion in glioblastoma cells.
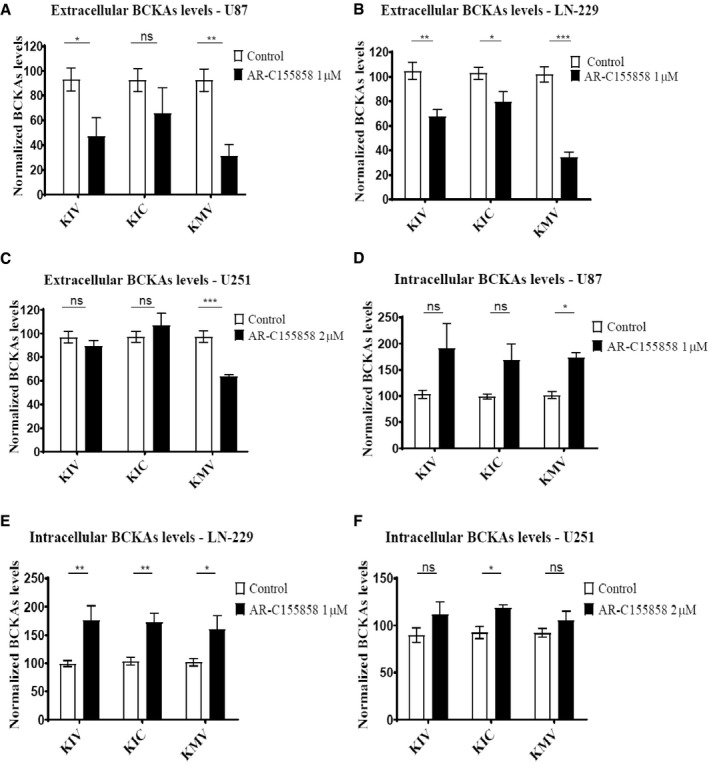
-
A–FBCKAs (KIV, KIC, KMV) levels are determined by ultra performance liquid chromatography (UPLC) coupled to fluorescence detection in supernatants from U87nt (A), LN‐229 (B), and U251nt (C) cells as well as in cell extracts (D–F) treated with AR‐C155858 for 24 h at 37°C 10% CO2. BCKAs levels are normalized to total protein content and to the detected levels in the respective cells treated with DMSO (Control). Values are mean ± SD of three independent experiments, run in triplicates. KIV: α‐ketoisovalerate. KIC: α‐ketoisocaproate. KMV: α‐keto‐β‐methylvalerate. Unpaired Student's t‐test; ns: not significant *P < 0.05; **P < 0.01; ***P < 0.001.
Figure EV3. MCT1 knockdown decreases BCKA excretion.
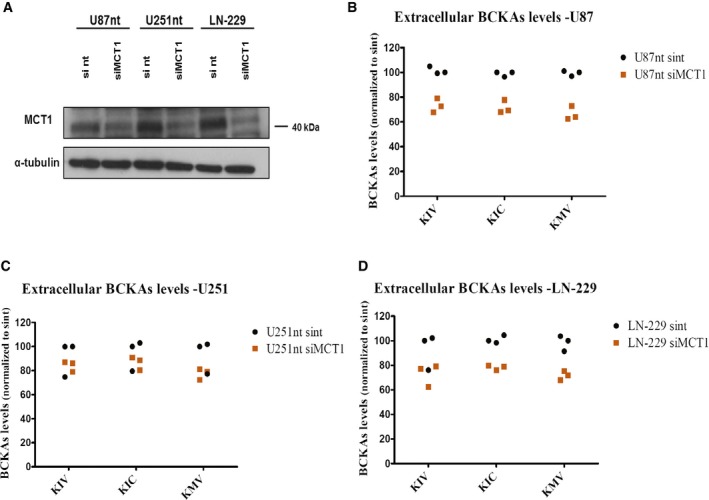
-
AWestern blot analysis of MCT1 levels in U87‐MG cells, U251‐MG expressing normal levels of BCAT1 (U87nt and U251nt, respectively) and LN‐229 cells 48 h post‐transfection using DharmaFECT 1 transfection reagent (Dharmacon) with 25 nmol of MCT1 siRNA smart pool (Dharmacon), or non‐target (nt) #2 siRNA pool (Dharmacon). Anti‐MCT1 antibody (AB3538P, Millipore) was used at 1:5,000 dilution. α‐tubulin was used as loading control.
-
B–DBCKAs (KIV, KIC, KMV) levels are determined by ultra performance liquid chromatography (UPLC) coupled to fluorescence detection in supernatants from U87nt (B), U251nt (C), and LN‐229 (D) cells 48 h after transfection. BCKAs levels are normalized to total protein content and to the levels detected in U87nt, U251nt, or LN‐229 cells transfected with non‐target #2 siRNA (U87nt sint, U251nt sint, or LN‐229 sint, respectively). n = 3 technical replicates. KIV: α‐ketoisovalerate. KIC: α‐ketoisocaproate. KMV: α‐keto‐β‐methylvalerate.
To examine the role of MCT4 in BCKA transport in glioblastoma cells, we used siRNA‐mediated knockdown since no specific inhibitors of MCT4 are commercially available. Despite good knockdown efficiencies in U87‐MG and U251‐MG cells using two different siRNA pools (Fig 4A), we did not observe any evident changes in the levels of BCKAs in the cell culture media (Fig 4B and C). To exclude the possibility that MCT4‐mediated transport activity lost due to the siRNA knockdown was compensated by increased activity of MCT1, we additionally performed MCT4 knockdown with and without concurrent MCT1 inhibition in U87‐MG cells. The combined inhibition of both MCTs did not result in any additive effect on BCKA excretion beyond that caused by MCT1 inhibition alone, except for KIC for which a small drop of secretion was observed (Appendix Fig S2). Neither pharmacological nor genetic perturbations of MCT1 and MCT4 lead to consistent downregulation of BCAT1 expression (Appendix Fig S3) suggesting that the observed reductions in BCKA excretions are not due to reduced BCKA production but indeed are reflecting reduced transmembrane transport. Interestingly, perturbation of MCT1 but not MCT4 also decreased the excretion of pyruvate in all three cell lines (Fig EV4). Together these data indicate that MCT1 is an important mediator of the excretion of BCKAs as well as pyruvate, whereas MCT4 seems to have only a very limited role in the excretion of these ketoacids from glioblastoma cells.
Figure 4. MCT4 knockdown does not impact on BCKAs excretion.
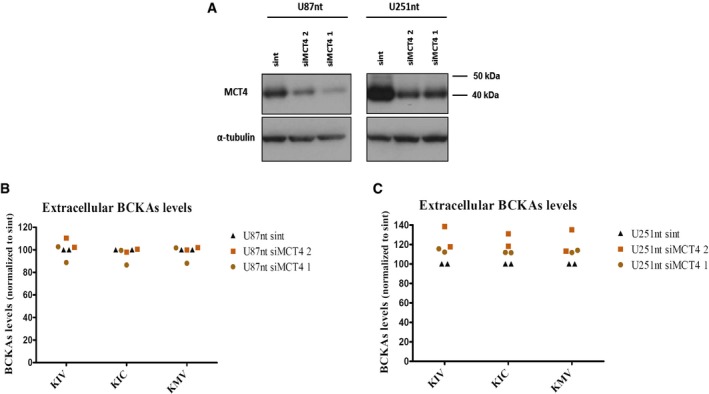
-
AWestern blot analysis of MCT4 levels in U87‐MG cells and U251‐MG expressing normal levels of BCAT1 (U87nt and U251nt) 48 h post‐transfection using DharmaFECT 1 transfection reagent (Dharmacon) with 25 nmol of MCT4 siRNA sc45892 (Santa Cruz) (siMCT41), MCT4 siRNA smart pool (Dharmacon) (siMCT4 2), or non‐target (nt) #2 siRNA pool (Dharmacon). Control cells were incubated with Opti‐MEM and DharmaFECT1 (no siRNA). Anti‐MCT4 sc50329 (Santa Cruz) antibody was used at 1:1,000 dilution. α‐tubulin was used as loading control.
-
B, CBCKAs (KIV, KIC, KMV) levels are determined by ultra performance liquid chromatography (UPLC) coupled to fluorescence detection in supernatants from U87nt (B) and U251nt (C) cells 48 h after transfection. BCKAs levels are normalized to total protein content and to the levels detected in U87nt or U251nt cells transfected with non‐target #2 siRNA (U87nt sint or U251nt sint, respectively). n = 2 independent biological replicates. KIV: α‐ketoisovalerate. KIC: α‐ketoisocaproate. KMV: α‐keto‐β‐methylvalerate.
Figure EV4. MCT1 inhibition or knockdown decreased pyruvate excretion in glioblastoma cells.
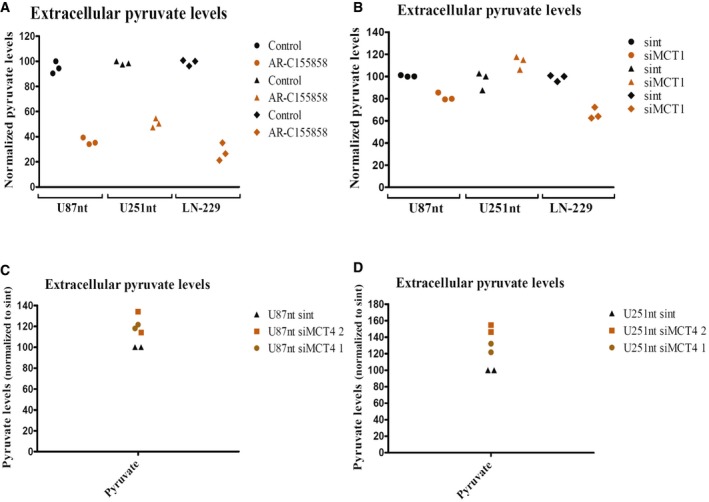
-
A, BPyruvate levels are determined by UPLC coupled to fluorescence detection in supernatants from U87nt, U251nt, and LN‐229 cells treated with AR‐C155858 for 24 h at 37°C 10% CO2 (A) and cells expressing normal MCT1 levels (sint) or low MCT1 levels (siMCT1) (B). n = 3 technical replicates.
-
C, DPyruvate levels are determined from U87nt (C) or U251nt (D) cells expressing normal MCT4 levels (sint) or low MCT4 levels (siMCT4). Pyruvate levels are normalized to total protein content and to the detected levels in the respective cells treated with DMSO (Control) or to the detected levels in cells expressing normal MCT1 or MCT4 levels (sint). n = 2 independent biological replicates.
BCAT1 and MCTs are in close proximity in glioblastoma cells
It has previously been shown that lactate‐transport activities of MCT1 and MCT4 are enhanced by their close association with carbonic anhydrases, which serve as proton donors or acceptors to help drive co‐transport of protons and lactate 27, 28. Here, we performed in situ proximity ligation assays (PLA) of MCT1, MCT4, and BCAT1 in U87‐MG and U251‐MG cells to test whether BCAT1, the enzyme producing the BCKAs in the cytoplasm, is located in close proximity to MCT1 and MCT4. We hypothesize that such an association might help increase local BCKA concentrations and enhance cross‐membrane transport. In both cell lines, we observed specific PLA signals indicating that BCAT1 and MCT1 indeed are located in close proximity to one another (Fig 5A and B). Close proximity of BCAT1 and MCT4 also was detected in U251‐MG cells (Fig 5D) but rarely in U87‐MG cells (Fig 5C). In both cell lines, BCAT1 significantly more often co‐localized with MCT1 than with MCT4 (Fig 5E and F). To control for non‐specific association of proteins, we performed PLA with MCT1, MCT4, and the cytoplasmic metabolic enzyme phosphoglycerate kinase 1 (PGK1), which is expressed at a similar level as BCAT1 in U251‐MG cells (NCI60 proteome resource: http://129.187.44.58:7070/NCI60/; Appendix Fig S4). Quantification of PLA signals confirmed specific interactions of BCAT1 with MCT1 in both cell lines and MCT4 in U251‐MG (Fig 5E and F).
Figure 5. BCAT1 and MCTs are in close proximity in glioblastoma cells.
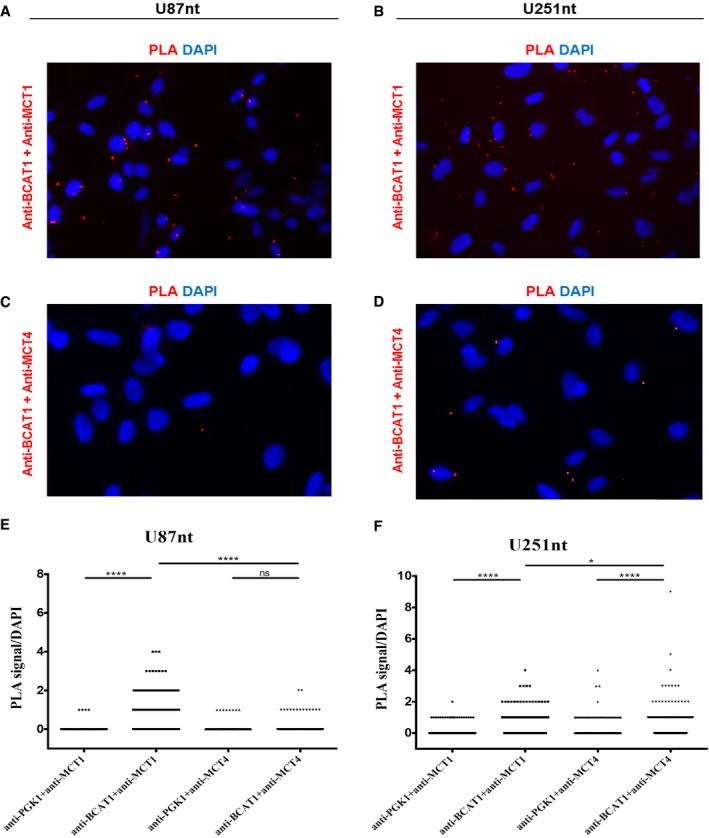
-
A–DRepresentative images of in situ proximity ligation assay (PLA) in the U87‐MG (A, C) and U251‐MG (B, D) cells expressing normal BCAT1 levels (U87nt, U251nt, respectively) are shown. Anti‐BCAT1 antibody (rabbit polyclonal antibody, Insight Biotechnology Limited) and anti‐MCT1 antibody (ab90582, Abcam) were used at 1:100 dilution. Anti‐MCT4 antibody (376140, Santa Cruz) and anti‐PGK1 antibody (GTX107614) were used at 1:200 dilution. Magnification: 200×. Red, PLA signal; blue, DAPI.
-
E, FQuantification of PLA signals per cell in (E) U87nt (n > 340 cells) and (F) U251nt (n > 270 cells). Mann–Whitney test; ns: not significant; *P < 0.05; ****P < 0.0001.
BCKAs are taken up and aminated by macrophages
After characterizing the transport of BCKAs from glioblastoma cells, we wanted to know whether the predominant cells of the glioblastoma stroma, that is tumor‐associated macrophages, can take up and metabolize tumor‐secreted BCKAs, as recently had been shown for tumor‐derived lactate in mammary carcinoma cells 24. For this purpose, we performed non‐radioactive tracing of BCKA carbon in human monocyte‐derived, M‐CSF‐differentiated macrophages incubated with 100 and 300 μM pools of BCKAs containing 1,2‐13C2‐αKIC and 13C5‐αKIV. These BCKA concentrations are well within the range observed in tissues with aberrant BCAA catabolism (see Discussion) and did not reduce cell viability in our in vitro model (Fig EV5A). We detected labeling of 10–30% of macrophage leucine and valine (Fig 6A and B), but not of succinate (Fig 6C) or other TCA intermediates (Appendix Table S2). These data indicate that macrophages can take up tumor‐derived BCKAs and aminate them to BCAAs. Consistent with this interpretation of the data, exposure of macrophages to BCKAs was associated with reduced BCAA uptake (Fig EV5B). Uptake of pyruvate and hexoses also was reduced (Fig EV5C). Uptake of BCKAs potentially could involve the MCT1 and MCT4 transporters, which also are expressed on macrophages (Appendix Fig S5).
Figure EV5. Effects of BCKAs on macrophage viability and nutrient consumption.
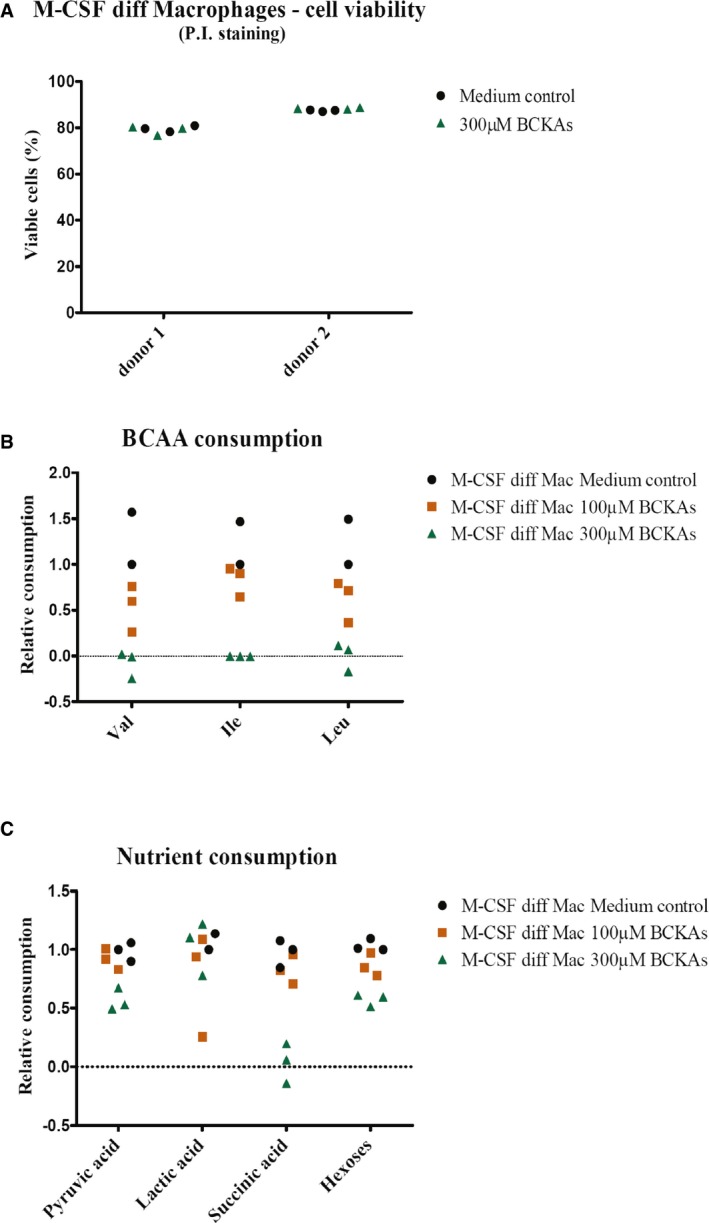
-
AThe effect of BCKAs on macrophage viability was assessed using propidium iodide (P.I.) staining, and analysis of P.I. negative (viable) was done using flow cytometry (FACS Canto II, BD Biosciences). M‐CSF differentiated macrophages were incubated in the absence (medium control) or presence of 300 μM BCKAs (KIV, KIC, KMV) at 37°C for 24 h. Cells were isolated from two independent donors, run in triplicates.
-
B, CConsumption of BCAAs (B), pyruvic acid, lactic acid, succinic acid, and hexoses (C) by monocyte‐derived macrophages incubated in the absence (medium control) or presence of 100 or 300 μM BCKAs (KIV, KIC, KMV) at 37°C for 24 h was determined by GC‐MS in the supernatants collected from macrophage cultures. Nutrient levels are normalized to protein content and to the levels detected in the supernatant of macrophages incubated in the absence of BCKAs (Medium control: DMEM 6046 10% FCS 1%P/S). Val: valine. Ile: isoleucine. Leu: leucine.
Figure 6. BCKAs are taken up by human monocyte‐derived macrophages and reduce phagocytosis.
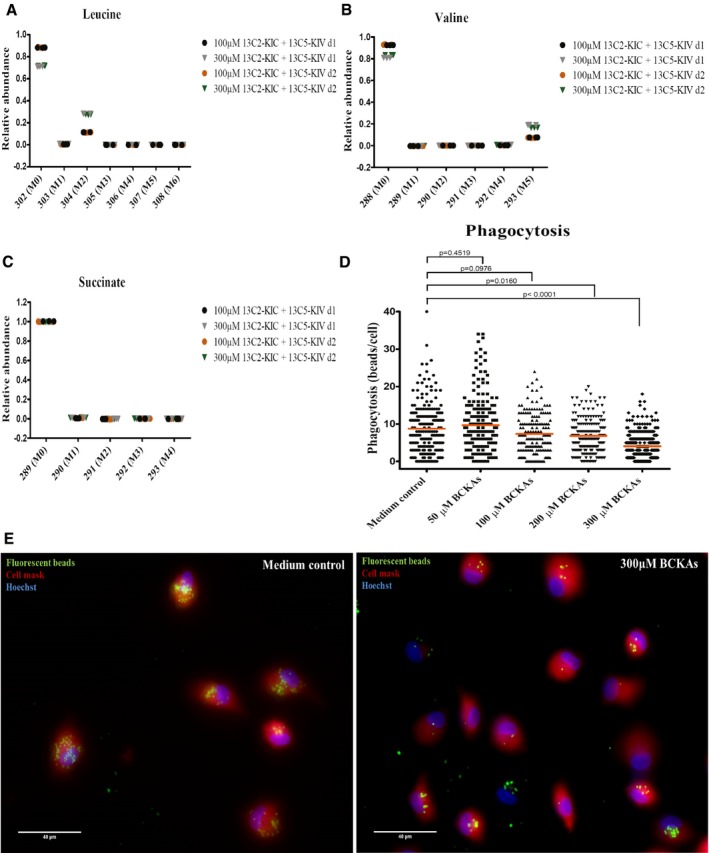
-
A–CLeucine (A), valine (B), and succinate (C) labeling patterns were determined by GC‐MS in cell extracts from M‐CSF differentiated macrophages cultured with 100 or 300 μM of 13C‐αKIC and 13C‐αKIV. n = 3 technical replicates. Monocytes were isolated from two independent donors (d1, d2). M0, M1 to Mn: M is the base mass of an ion fragment, and the following number from 0 to n (active carbon number) indicates the mass shift from M.
-
D, EMonocyte‐derived macrophages previously incubated in the absence (medium control) or presence of 50, 100, 200, or 300 μM of BCKAs (KIV, KIC, and KMV) at 37°C for 24 h were incubated with 1‐μm‐diameter fluorescent beads at 37°C for 2 h, and phagocytosis of beads was examined by fluorescence microscopy. (D) The number of beads engulfed by each cell is quantified using ImageJ software. Line represents mean. Cells were isolated from two independent donors, and eight fields were examined per condition (n > 165 cells). Mann–Whitney test. (E) Fluorescent beads (green). Plasma membrane (red) is stained with Cell Mask stain. The Hoechst stains nuclei (blue). Scale bar: 40 μm.
BCKAs mitigate macrophage phagocytosis
Glioblastoma exert strong immuno‐suppressive effects on their microenvironment including the suppression of macrophage phagocytic activities 29. It is unclear what role tumor‐derived metabolites might have in this process. Here, we considered whether BCKAs could directly affect the phagocytic properties of macrophages. For this purpose, we assessed the engulfment of fluorescent beads by monocyte‐derived macrophages in the presence and absence of BCKAs (Fig 6D and E). Once the beads were biotin‐labeled, we first discriminated between engulfed and non‐engulfed beads using streptavidin PE (Appendix Fig S6). Considering only engulfed beads, we observed a decrease in the number of beads in macrophages treated with 100 μM of BCKAs (P = 0.0976), that is even more pronounced with increasing concentrations of BCKAs [i.e., 200 μM (P = 0.0160) or 300 μM (P < 0.0001)] compared with the cultures not exposed to BCKAs (Fig 6D and E). These data suggest that tumor cell‐derived BCKAs can affect glioblastoma microenvironment by suppressing phagocytosis by tumor‐associated macrophages.
Discussion
Glioblastoma are characterized by striking overexpression of the metabolic enzyme BCAT1 and associated excretion of glutamate, features that are well conserved in cell line models 9. In glioma patients, glutamate excretion was shown to cause tumor‐associated epilepsy 30. Here, we for the first time show that BCKAs, which are generated by BCAT1 alongside glutamate, are excreted by glioblastoma cells. For this purpose, we established a UPLC protocol to directly determine BCKA concentrations in biological extracts and validated it by determining intra‐ and extracellular BCKA concentrations in cultures of Xenopus oocytes expressing mammalian BCAT1, MCT1, and/or MCT4. This heterologous expression system previously had been used to identify MCT1 and MCT4 substrates, including the BCKAs KIC and KIV, based on changes of intracellular pH caused by proton co‐transport 10, 11; our experimental approach differs from this originally used one in that we directly determined the concentrations of all three BCKAs to analyze BCKA efflux rather than influx. Our analysis showed that glioblastoma cells are excreting BCKAs, resulting in their accumulation to concentrations of up to 85 μM in the culture media within 24 h. Extracellular BCKA levels in tumors are unknown but BCKA concentrations of 0.4–4.6 mM have been observed in patients with maple syrup urine disease (MSUD), a heritable defect of BCAA catabolism which is associated with increased cellular BCKA excretion 31.
To analyze whether both MCT1 and MCT4 are involved in BCKA excretion from glioblastoma cells, we used pharmacologic and genetic perturbation of MCT1 and MCT4 function, respectively. Suppression of MCT1 by the MCT1/2‐specific inhibitor AR‐C155858 32 in glioblastoma cell lines indicated that BCKAs are excreted from the tumor cells via MCT1. Inhibition of MCT2‐mediated transport by AR‐C155858 appears unlikely given that the inhibition of MCT2 by this inhibitor was only observed when MCT2 is associated with the ancillary protein CD147 (which is not the usual partner for MCT2) but not when it is associated with embigin (the usual partner of MCT2) 32. Furthermore, MCT2 is downregulated in tumor compared to normal brain. Inhibition of MCT1 also reduced the efflux of pyruvate in all three glioblastomas cell lines consistent with similar observations reported for mammary carcinoma cell lines 33. Knockdown of MCT4 did not appear to affect the efflux of BCKAs or pyruvate, and we did not observe any MCT1‐mediated compensatory effects as previously had been reported for pancreatic cancer 34. These observations indicate that while both MCT1 and MCT4 are in principle capable of transporting BCKAs, MCT4‐mediated transport appears to be attenuated in glioblastoma cells. In part, this might be explained by differences in substrate‐dependent transport kinetics. Estimates of Michaelis–Menten constants for MCT1‐ and MCT4‐mediated transport in Xenopus oocytes (summarized in 35) are suggesting preferred transport of KIC, KIV, and pyruvate by MCT1 (KIC, KIV, and pyruvate ~1 mM versus lactate 3.5 mM) and of lactate by MCT4 (lactate 28 mM versus KIC, KIV, and pyruvate ~100–150 mM). This is consistent with the idea that in cells that express both MCT1 and MCT4, BCKAs and pyruvate, which are present in the cell at lower concentrations, will preferentially be transported by the high‐affinity carrier MCT1, while lactate, which is produced in vast amounts in cancer cells, will be excreted via the high‐capacity transporter MCT4.
Metabolite‐specific transport might be further regulated through spatial association of proteins resulting in locally increased metabolite concentrations. This was shown for carbonic anhydrase II (CAII) which supports proton/lactate co‐transport by MCT1 and MCT4 by increasing the concentration of protons in the vicinity of MCTs 36, 37, 38. Statistical analysis of our PLA data indeed showed that BCAT1 appears to more frequently associate with MCT1 than MCT4 in U87‐MG and U251‐MG cells. To investigate a potential preferential binding of BCAT1 with MCT1 and LDHA with MCT4, we performed PLA of lactate dehydrogenase and the MCTs. We showed that LDHA was significantly more often associated with MCT1 than with MCT4 in U87‐MG cells, suggesting no preferential co‐localization of LDHA and MCT4. In U251‐MG cells, which do not express LDHA, we did not detect any preferential association of LDHB with either of the MCTs (Appendix Fig S7).
A similar pattern of metabolite transport as observed in this study was recently reported for mammary carcinoma 33. In triple‐negative breast cancer (TNBC) cell lines, Hong and colleagues found a segregation of metabolite excretion, with pyruvate and lactate export being exclusively mediated by MCT1 and MCT4, respectively. Further, suppression of MCT1‐mediated transport switched tumor metabolism to a more oxidative state and blocked mammary carcinoma growth in vitro and in a xenograft model 33. A query of published microarray data (GSE76675) of the three breast cancer cell lines (HS578T, SUM149, and SUM159) used in this study revealed all as having high levels of BCAT1 expression. Consistent with this observation, we found intracellular BCKA contents and levels of BCKA excretion of SUM149 and SUM159 cells to be similar to those of glioblastoma cells (Appendix Fig S8). This raises the interesting possibility that intracellular accumulation of BCKAs in addition to pyruvate might be causing the growth inhibition that was observed following MCT1 suppression in several studies 19, 33. It will be difficult to address this question since transport of pyruvate and BCKAs via MCT1 might co‐occur in many tumors. However, accumulation of BCKAs in MSUD has been shown to affect cell energy metabolism and cell survival 7, 8, 39 suggesting that blocked BCKA export might be responsible for at least part of the observed phenotype.
Recent studies suggested that different aspects of tumor metabolism, that is, lactate production and consumption are compartmentalized between tumor cells and CAFs in mammary carcinoma 24. In this model, lactate is produced and excreted via MCT4 by stroma cells and taken up by oxygenated tumor cells via MCT1 for ATP production. An analogous exchange of metabolites, in this case the amino acids glutamate/glutamine and a BCAT cycle, has been proposed to occur in the normal brain for nitrogen balance maintenance 10, 11, 40, 41.
Here, we found that BCKA carbon did not contribute to macrophage TCA metabolites; however, 13C‐tracing analysis revealed that macrophages did take up BCKAs that were supplied in the cell culture media and aminated them to BCAAs. This generation of BCAAs was accompanied by a drop of uptake of BCAAs, pyruvate, and hexoses from the media, but no change in macrophage viability. These observations suggest that a similar exchange and effect on macrophage metabolism could occur in glioblastoma tumors. The purpose of KIC uptake in tumor–stroma interaction is still unclear but could be related to buffering of intracellular glutamate 42. Furthermore, we could show BCKAs can mitigate phagocytosis in vitro. These data suggest that in glioblastoma, tumor cell‐secreted BCKAs might be able to modulate the tumor‐associated macrophages/microglia, contributing to their role in tumor immune suppression and supporting glioblastoma tumor growth and progression as described by others 43.
These results expand upon a growing literature on MCT1 involvement in the transport of tumor‐derived metabolites and provide further evidence for the eminent role of BCAA catabolism in glioblastomas. Our data indicate that tumor cell‐derived BCKAs are taken up and metabolized by tumor‐associated macrophages. BCKAs attenuate macrophages' phagocytic activity and thereby potentially can contribute to tumor immune suppression. Furthermore, our data suggest that the anti‐proliferative effects of MCT1 knockdown observed by others 19, 33 could in part be due to intracellular accumulation of BCKAs in addition to pyruvate, adding additional support for the concept of using MCT1 inhibitors as anti‐cancer therapeutics.
Materials and Methods
Cell culture
Inducible BCAT1 knockdown cells were established as described previously 9 by infecting U87‐MG and U251‐MG cells with pLKO‐Tet‐On non‐targeting (nt) shRNA and pLKO‐Tet‐On BCAT1 shRNA2 lentiviral particles and selecting with puromycin (1 μg/ml) for 7 days. Induction was achieved by adding 0.1 μg/ml doxycycline to the medium for at least 3 days. Cells were cultured in DMEM containing 1 g/l glucose (D5921, Sigma) supplemented with 10% tetracycline‐free fetal bovine serum (Clontech), 1% penicillin and streptomycin (P/S) mix, and glutamine (0.5 mM). LN‐229 cells were cultured in DMEM containing 4.5 g/l glucose (D5796, Sigma) supplemented with 10% fetal calf serum (FCS) and 1% P/S.
All cell lines were cultured under 10% CO2 at 37°C. U87‐MG cell line was used as a glioblastoma model considering the recent studies identifying it as a bona fide human glioblastoma cell line of unknown origin 44.
RNA extraction, cDNA preparation, and qRT–PCR
Total RNA was isolated using RNeasy Microkit, Qiagen, according to the manufacturer's instructions. Total RNA (250 ng) was reverse transcribed using random primers and Superscript II Reverse Transcriptase (Invitrogen) according to manufacturer's instructions. Each cDNA sample was analyzed in triplicate with the Applied Biosystems Prism 7900HT Fast Real‐Time PCR System using Absolute SYBR Green ROX Mix (ABgene). The relative amount of specific mRNA was normalized to ARF1 and TBP mRNA. Expression levels were calculated according to ΔCt method. Primer sequences are given in the Appendix Table S1.
Protein quantification
Protein content was determined using the bicinchoninic acid (BCA) method with bicinchoninic acid (Sigma) and copper sulfate (Sigma) solution mixed in a ratio of 50:1. Absorption was measured at 562 nm. A bovine serum albumin (BSA)‐based standard curve was used for regression analysis to determine protein concentration.
Western blot analysis
Total protein of cell lines was extracted using RIPA buffer (Sigma) with phosphatase inhibitor cocktail. 15 μg of protein was separated by 4–12% SDS–PAGE and transferred to a polyvinylidene difluoride membrane (Millipore). The membrane was blocked in blocking solution (5% milk in TBS and 0.1% Tween 20) and incubated with primary antibodies overnight at 4°C. Horseradish peroxidase (HRP)‐conjugated secondary antibodies were incubated for 1 h at RT before chemiluminescent detection of protein (ECL kit, GE Healthcare). The following antibodies were used: monoclonal mouse antibody to α‐tubulin (1:3,000 dilution) (clone DM1A, #T9026, Sigma‐Aldrich), anti‐MCT4 (sc50329, Santa Cruz) antibody (1:1,000 dilution), anti‐MCT1 (AB3538P, Millipore) (1:500 dilution), HRP‐conjugated antibody to mouse IgG (1:5,000, #7076, Cell Signaling Technology), and HRP‐conjugated antibody to rabbit IgG (1:2,000, #7074, Cell Signaling Technology).
Cell viability assay
Cell viability was assessed through the use of dye exclusion using propidium iodide (P.I.) membrane impermeant dye. Cells were harvested into FACS tubes washed with FACS buffer (containing bovine serum albumin (BSA) and sodium azide) and centrifuged at 300 g for 5 min, and then, cells were resuspended in 200 μl of FACS buffer, 1 μl of P.I. solution (P4864, Sigma) (1.0 mg/ml) was added, and P.I. fluorescence was determined using FACS Canto II. Unstained cells were used to adjust acquisition parameters.
MCT4 and MCT1 siRNA‐mediated knockdown
A total of 2.5 × 105 U87nt, U251nt or LN‐229 cells were seeded onto 6‐well plates in medium without P/S and incubated at 37°C 10% CO2 overnight. Cells were transfected using DharmaFECT 1 transfection reagent (Dharmacon) with 25 nmol of MCT4 siRNA (sc45892, Santa Cruz) (siMCT4 1), MCT4 siRNA smart pool (M‐005126‐04‐0005, Dharmacon) (siMCT4 2), 25 nmol of MCT1 siRNA (M‐007402‐02‐0005, Dharmacon), or non‐target #2 siRNA pool (D‐001206‐14‐05, Dharmacon). Control cells were incubated with Opti‐MEM medium (Gibco) and DharmaFECT1 (no siRNA added). Cells were incubated at 37°C 10% CO2 for 48 h with medium change at 24 h post‐transfection. At 48 h post‐transfection, cells were harvested for RNA isolation (RNeasy Microkit, Qiagen) and protein extraction (using RIPA buffer) and the knockdown efficiency was evaluated by Western blot. Supernatants from U87nt or U251nt cells 48 h post‐transfection using non‐target siRNA or MCT4 siRNAs were centrifuged at 913 g for 5 min, filtered using 0.22‐μm filters, and analyzed by UPLC for BCKAs determination.
MCT1 inhibition and cell proliferation assay
A total of 1 × 105 U87nt or U87shBCAT1, U251nt or U251shBCAT1, and 0.4 × 105 LN‐229 cells were seeded onto 12‐well plates, medium was replaced after 16 h, fresh medium with AR‐C155858 (Tocris) or DMSO was added, and cells were incubated at 37°C 10% CO2 for 24 h. U87 and LN‐229 cells were treated with 1 μM AR‐C155858, whereas for U251 cells, 2 μM AR‐C155858 was used.
To assess the effect on proliferation of glioblastoma cells after treatment with MCT1 inhibitor (AR‐C155858, Tocris), the Click‐iT EdU cell proliferation assay (Invitrogen) was used following the manufacturer's instructions. The cells were incubated with 10 μM of the nucleoside analog EdU for 8–10 h. Quantification of cells that incorporated EdU was performed using a FACS Canto II (BD Biosciences).
BCKA treatment, cell proliferation, and viability assay
To assess the effect on proliferation of glioblastoma cells after treatment with 100 μM or 300 μM of KIV (198994‐5G, Sigma‐Aldrich), KIC (K0629‐16, Sigma‐Aldrich), and KMV (sc‐214140, Santa Cruz) for 24 h, the Click‐iT EdU cell proliferation assay (Invitrogen) was used following the manufacturer's instructions. Glioblastoma cell viability after treatment with KIV, KIC, and KMV (100 or 300 μM) for 24 h was assessed using P.I. staining.
Cell viability of M‐CSF differentiated monocyte‐derived macrophages treated with DMEM 6046 10% FCS 1% P/S (medium control) or DMEM 6046 10% FCS 1% P/S supplemented with 300 μM of KIV, KIC, and KMV for 24 h was assessed through the use of P.I. staining.
Heterologous protein expression in Xenopus oocytes
Plasmid DNA of MCT1, MCT4, human BCAT1 version 6, or NBCe1 cloned into the oocyte expression vector pGEM‐He‐Juel, which contains the 5′ and the 3′ untranscribed regions of the Xenopus β‐globin flanking the multiple cloning site, was used. DNA was linearized with SalI and transcribed in vitro with T7 RNA‐polymerase (mMessage mMachine, Ambion Inc., USA) as described earlier 45. Xenopus laevis females were purchased from Xenopus Express. Segments of ovarian lobules were removed surgically under sterile conditions from frogs anesthetized with 1 g/l of 3‐amino‐benzoic acid ethyl ester and rendered hypothermic. The procedure was approved by the Landesuntersuchungsamt Rheinland‐Pfalz, Koblenz, Germany (23 177–07/A07‐2–003 §6). Oocytes at stages V were injected with 5 ng of cRNA coding for rat MCT1 or rat MCT4, 12 ng of cRNA coding for human BCAT1, 7 ng cRNA coding for human NBCe1, 5 ng of cRNA for rat MCT1 or MCT4, and 12 ng of cRNA for human BCAT1 5 days before the experiments.
Native oocytes or oocytes expressing the specified proteins were stimulated with BCAAs (1 nmol L‐valine, 1 nmol L‐leucine, 1 nmol L‐isoleucine) and α‐ketoglutarate (3 nmol) for 2 h at RT. After the incubation time, culture medium was removed and stored for posterior quantification of BCKAs by ultra performance liquid chromatography (UPLC). Cells were lysed by pipetting, and the lysate was centrifuged for 10 min at 10,000 g at 4°C and stored at −80°C before quantification of intracellular BCKAs by UPLC.
Batches of 10 oocytes were used per condition, and the experiment was performed three times.
BCKAs and pyruvate quantification
Supernatants from glioblastoma cells treated with MCT1 inhibitor (AR‐C155858) or DMSO were centrifuged at 913 g for 5 min and filtered using 0.22‐μm filters and analyzed by UPLC. Cells were harvested by trypsinization, centrifuged at 228 g for 5 min, and washed once with ice‐cold 0.9% NaCl, and the cells pellets were frozen at −80°C before analysis by UPLC. Supernatants from U87nt, U251nt, or LN‐229 cells 48 h post‐transfection using non‐target (nt) siRNA or MCT4 or MCT1 siRNAs were centrifuged at 2,000 rpm for 5 min and filtered using 0.22‐μm filters and analyzed by UPLC. Intracellular levels were normalized to total protein content.
In situ proximity ligation assay (PLA)
In situ PLA was performed in the U87nt and U251nt cells using the Duolink in situ proximity ligation assay (Olink Bioscience, Uppsala, Sweden) according to manufacturer's protocol.
In short, cells were seeded onto eight‐well chamber slides (Lab‐TekII, Thermo Scientific Nunc) and incubated overnight at 37°C 10% CO2. Cells were fixed with 4% paraformaldehyde solution for 15 min at RT, permeabilized with PBS 0.1% Triton X‐100 for 15 min at RT and dehydrated using ethanol series (70, 85, 100%) for 2 min at RT. Before the blocking step, cells were rehydrated with 1× PBS for 15 min at RT. Blocking solution (Olink Bioscience) was added, and slides were incubated for 30 min at 37°C. The primary antibodies used were anti‐BCAT1 (rabbit polyclonal antibody, Insight Biotechnology Limited (Wembley, UK)) (provided by Myra Conway), anti‐MCT1 (ab90582, Abcam), anti‐MCT4 (376140, Santa Cruz) at 1:100 dilution in antibody diluent (Olink Bioscience), anti‐PGK1 antibody (GTX107614, GeneTex) at 1:200 dilution, anti‐LDHA (SAB1100050, Sigma) at 1:2,400 dilution, and anti‐LDHB (PA527505, Invitrogen) at 1:200 dilution. Cells incubated without primary antibodies or with only one of each of the antibodies tested in combination were used as procedure controls. The results were visualized using Axioplan 2 imaging (Zeiss) microscope and Hokawo software. The images were processed, and PLA signals/cells were quantified using ImageJ software.
Generation of human monocyte‐derived macrophages in vitro
Human PBMCs were isolated from buffy coats from normal donors obtained from the Institut für Klinische Transfusionsmedizin und Zelltherapie (IKTZ) (Heidelberg, Germany) by density gradient using Biocoll (Biochrom) separation solution according to standard procedures.
Monocytes were enriched from PBMCs by plastic adhesion for 2 h at 37°C 5% CO2 and differentiated in the presence of 20 ng/ml human M‐CSF (Miltenyi Biotec) for 7–10 days.
Metabolic tracing using 13C stable isotopes
After differentiation, medium was replaced by DMEM 5921 1% HS 1% P/S and glutamine (0.5 mM) with 100 or 300 μM 13C α‐KIV (13C5, 98%, Cambridge Isotope Laboratories) and 100 or 300 μM 13C α‐KIC (1,2‐13C2, 99%, Cambridge Isotope Laboratories) and cells were incubated for 48 h at 37°C 10% CO2. After 48 h, cells were collected and intracellular metabolites were extracted for gas chromatography–mass spectrometry (GC–MS) analysis. Monocytes were isolated from two different donors.
Nutrient depletion analysis
After differentiation, medium was replaced by DMEM 6046 10% FCS 1% P/S (medium control) or DMEM 6046 10% FCS 1% P/S supplemented with 100 or 300 μM KIV (198994‐5G, Sigma‐Aldrich), KIC (K0629‐16, Sigma‐Aldrich), and KMV (sc‐214140, Santa Cruz), and cells were incubated for 24 h at 37°C 10% CO2. After 24 h, cell culture supernatants were collected for gas chromatography–mass spectrometry (GC–MS) analysis.
Ultra performance liquid chromatography (UPLC) analysis
For derivatization with OPD (o‐phenylenediamine) reagent, 100 μl of cell culture supernatant was mixed with 200 μl ice‐cold 1 M perchloric acid for protein precipitation. After centrifugation at 4°C and 16,400 g for 10 min, 150 μl of the supernatant was mixed with an equal volume of OPD derivatization solution (25 mM OPD in 2 M HCl) and derivatized by incubation at 50°C for 30 min. After centrifugation for 10 min, the derivatized ketoacids were separated by reversed phase chromatography on an Acquity HSS T3 column (100 mm × 2.1 mm, 1.7 μm, Waters) connected to an Acquity H‐Class UPLC system. Prior separation, the column was heated to 40°C and equilibrated with five column volumes of solvent A (0.1% formic acid in 10% acetonitrile) at a flow rate of 0.55 ml/min. Separation of ketoacid derivates was achieved by increasing the concentration of solvent B (acetonitrile) in solvent A as follows: 2 min 2% B, 5 min 18% B, 5.2 min 22% B, 9 min 40% B, 9.1 min 80% B and hold for 2 min, and return to 2% B in 2 min.
For determination of intracellular BCKA content, cells were extracted with 200 μl cold 1 M perchloric acid. Insoluble material was removed by centrifugation for 10 min at 25,000 g. For derivatization with DMB (1,2‐diamino‐4,5‐methylenedioxybenzene), 30 μl extract was mixed with 30 μl DMB derivatization reagent (5 mM DMB, 20 mM sodium hydrosulfite, 1 M 2‐mercaptoethanol, 1.2 M HCl) and incubated at 100°C for 45 min. After 10‐min centrifugation, the reaction was diluted with 240 μl 10% acetonitrile. UPLC system, column, and solvent were used as described above. Baseline separation of DMB derivates was achieved by increasing the concentration of acetonitrile (B) in buffer A as follows: 2 min 2% B, 4.5 min 15% B, 10.5 min 38% B, 10.6 min 90% B, hold for 2 min, and return to 2% B in 3.5 min.
BCKAs levels in cell lysates and in supernatants from Xenopus oocytes were detected using the derivatization with DMB derivatization reagent as described above.
The separated derivates were detected by fluorescence (Acquity FLR detector, Waters, OPD: excitation: 350 nm, emission: 410 nm; DMB: excitation: 367 nm, emission: 446 nm) and quantified using ultrapure standards (Sigma). Data acquisition and processing was performed with the Empower3 software suite (Waters).
Extraction of intracellular metabolites for GC‐MS (isotope tracing analysis)
Extraction was according to Sapcariu et al 46. Briefly, cells were cultivated in 6‐well plates, medium was retained for quantification of extracellular metabolites, cells were washed with 2 ml of 0.9% NaCl, and then quenched with 0.2 ml of −20°C methanol, and after adding an equal volume of 4°C cold water, cells were collected with a cell scraper and transferred to tubes containing 0.2 ml −20°C chloroform. The extracts were shaken at 1,400 rpm for 20 min at 4°C (Thermomixer Eppendorf) and centrifuged at 16,000 g for 5 min at 4°C. 0.2 ml of the upper aqueous (polar) phase was collected and stored at −80°C. After collecting polar phase, the non‐polar phase was also collected. Interphase was washed with 0.3 ml −20°C methanol and centrifuged at 16,000 g for 10 min at 4°C. After removing the methanol, 50 μl of methanol was added and interphase was stored at −80°C. Polar phase was subsequently subjected to GC‐MS analysis.
Extraction of cell culture supernatants for GC‐MS (nutrient depletion analysis)
Macrophage cell culture supernatants (100 μl) were extracted in 180 μl of 100% MeOH for 15 min. at 70°C with vigorous shaking. As internal standard, 5 μl ribitol (0.2 mg/ml) was added to each sample. After the addition of 100 μl chloroform, samples were shaken at 37°C for 5 min. To separate polar and organic phases, 200 μl water was added and samples were centrifuged for 5 min at 12,000 g. For metabolite derivatization, 350 μl of the polar (upper) phase was transferred to a fresh tube and dried in a speed‐vac (Eppendorf vacuum concentrator) without heating.
Gas chromatography–mass spectrometry (GC–MS) analysis
Isotope tracing analysis
Derivatization was performed with a Gerstel autosampler directly before measurement on the GC‐MS. Dried metabolites were dissolved in 15 μl of 2% methoxyamine hydrochloride in pyridine at a temperature of 40°C for 60 min. Then, 15 μl of 2,2,2‐trifluoro‐N‐methyl‐N‐trimethylsilyl‐acetamide + 1% chloro‐trimethyl‐silane was added and incubated at 40°C for 30 min. The metabolite extracts were measured on an Agilent 7890 GC containing with a 30 m DB‐35MS capillary column. The GC was connected to an Agilent 5975C MS operating in electron ionization (EI) at 70 eV. 1 μl of derivatized sample was hot injected at 270°C in splitless mode. Helium was used as the carrier gas at a flow rate of 1 ml/min. The GC oven temperature was kept constant at 100°C for 2 min and then increased to 300°C at 10°C/min, where it was held for 4 min. The total GC‐MS run time of one sample was 26 min. For relative quantification of metabolite levels, an alkane mix was run with the experimental sequence in order to provide retention index calibration for the experimental samples. The MS source was kept at a constant temperature of 230°C and the quadrupole at 150°C. For relative quantification of metabolite levels, the detector was operated in scan mode with an m/z range of 70–800. For analysis of stable isotope labeling, the detector was operated in selected ion monitoring (SIM) mode. Using this approach, we analyzed the metabolites present in Appendix Table S2.
Nutrient depletion analysis
For derivatization, pellets were re‐dissolved in 20 μl methoximation reagent containing 20 mg/ml methoxyamine hydrochloride (Sigma 226904) in pyridine (Sigma 270970) and incubated for 30 min at 50°C with shaking. For silylation, 32.2 μl N‐methyl‐N‐(trimethylsilyl)trifluoroacetamide (MSTFA; Sigma M7891) and 2.8 μl alkane standard mixture (50 mg/l C10–C40; Fluka 68281) were added to each sample. After incubation for 30 min at 37°C, samples were transferred to glass vials for GC‐MS analysis.
A GC‐MS‐QP2010 Plus (Shimadzu®) fitted with a Zebron ZB 5MS column (Phenomenex®; 30 meter × 0.25 mm × 0.25 μm) was used for GC‐MS analysis. The GC was operated with an injection temperature of 230°C, and 1 μl sample was injected with split mode (1:30). The GC temperature program started with a 1 min hold at 40°C followed by a 6°C/min ramp to 210°C, a 20°C/min ramp to 330°C, and a bake‐out for 5 min at 330°C using helium as carrier gas with constant linear velocity. The MS was operated with ion source and interface temperatures of 250°C, a solvent cut time of 6.95 min and a scan range (m/z) of 40–700 with an event time of 0.2 s. The “GCMS solution” software (Shimadzu®) was used for data processing.
Phagocytosis assay
Monocyte‐derived macrophages differentiated in the presence of 20 ng/ml human M‐CSF (Miltenyi Biotec) previously incubated in the absence (medium control) or presence of 50, 100, 200 or 300 μM of KIV (198994‐5G, Sigma‐Aldrich), KIC (K0629‐16, Sigma‐Aldrich), and KMV (sc‐214140, Santa Cruz) at 37°C for 24 h were incubated with 1‐μm‐diameter fluorescent biotin‐labeled beads (F8768, Molecular probes, Life technologies) at a ratio of 50 beads/cell at 37°C for 2 h. After 2 h, medium was removed and cells were washed five times with 1× PBS and stained with Cell Mask stain (C10046, Molecular probes, Life Technologies) for 10 min at 37°C prior to fixation in 4% paraformaldehyde for 20 min at RT. In parallel, cells were stained with streptavidin PE (554061, BD Biosciences) at 1:200 dilution for 30 min at 4°C prior to fixation in 4% paraformaldehyde for 20 min at RT. Hoechst dye (Molecular probes, Invitrogen) was used to stain cell nuclei. Phagocytosis of fluorescent beads was examined by fluorescence microscopy (Cell Observer, Zeiss). The number of beads engulfed by each cell was quantified using ImageJ software. Cells were isolated from two independent donors, and eight fields per condition were examined.
Statistical analysis
Statistical analysis was performed using GraphPad Prism software. Differences in means as determined by Student's t‐test, Mann–Whitney test, or two‐way ANOVA were considered statistically significant at P < 0.05. Furthermore, the following convention was used: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Author contributions
LSS performed all cell culture experiments, collected, and analyzed experimental data, and prepared the manuscript. GP performed metabolite quantification using UPLC and prepared the manuscript. HMB performed heterologous expression experiments using Xenopus oocytes. YN and SS performed 13C‐BCKAs tracing experiments using GC‐MS. A‐CG assisted with monocyte‐derived macrophage culture experiments. MS assisted with siRNA‐mediated knockdown experiments and cell viability and proliferation experiments. YW assisted with monocyte‐derived macrophage culture experiments and provided valuable feedback. NK assisted with isotope tracing experiments. MS provided valuable feedback. RH and KH provided valuable feedback and prepared the manuscript. PL reviewed all experimental data and co‐prepared the manuscript. BR designed the study, reviewed all experimental data, and co‐prepared the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
This work was funded in part by grants of the Heidelberg University EcTop6 network within the framework of the German Excellence Initiative (#EC81‐) and the German Ministry for Education and Science (BMBF) eMed initiative (#031A425A) to B.R and P.L. We would like to thank the Metabolomics Core Technology Platform of the Excellence Cluster CellNetworks for support with UPLC‐based metabolite quantification. Support by the DKFZ Light Microscopy Facility is gratefully acknowledged. We thank Myra Conway (Faculty of Health and Life Sciences, Bristol, UK) for providing anti‐BCAT1 antibody. We thank Ola Söderberg for providing in situ PLA reagents. We thank Michaela Kirchgäßner and Irene Helbing for technical assistance and helpful discussions and the members from the Tumor Metabolism group (DKFZ) for helpful discussions.
EMBO Reports (2017) 18: 2172–2185
References
- 1. Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646–674 [DOI] [PubMed] [Google Scholar]
- 2. Koppenol WH, Bounds PL, Dang CV (2011) Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer 11: 325–337 [DOI] [PubMed] [Google Scholar]
- 3. Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, Kafri R, Kirschner MW, Clish CB, Mootha VK (2012) Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 336: 1040–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Daikhin Y, Yudkoff M (2000) Compartmentation of brain glutamate metabolism in neurons and glia. J Nutr 130: 1026S–1031S [DOI] [PubMed] [Google Scholar]
- 5. Suryawan A, Hawes JW, Harris RA, Shimomura Y, Jenkins AE, Hutson SM (1998) A molecular model of human branched‐chain amino acid metabolism. Am J Clin Nutr 68: 72–81 [DOI] [PubMed] [Google Scholar]
- 6. Hutson SM, Sweatt AJ, Lanoue KF (2005) Branched‐chain [corrected] amino acid metabolism: implications for establishing safe intakes. J Nutr 135: 1557S–1564S [DOI] [PubMed] [Google Scholar]
- 7. Amaral AU, Leipnitz G, Fernandes CG, Seminotti B, Schuck PF, Wajner M (2010) Alpha‐ketoisocaproic acid and leucine provoke mitochondrial bioenergetic dysfunction in rat brain. Brain Res 1324: 75–84 [DOI] [PubMed] [Google Scholar]
- 8. Sgaravatti AM, Rosa RB, Schuck PF, Ribeiro CAJ, Wannmacher CMD, Wyse ATS, Dutra‐Filho CS, Wajner M (2003) Inhibition of brain energy metabolism by the α‐keto acids accumulating in maple syrup urine disease. Biochim Biophys Acta 1639: 232–238 [DOI] [PubMed] [Google Scholar]
- 9. Tönjes M, Barbus S, Park YJ, Wang W, Schlotter M, Lindroth AM, Pleier SV, Bai AH, Karra D, Piro RM et al (2013) BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild‐type IDH1. Nat Med 19: 901–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yudkoff M, Daikhin Y, Lin ZP, Nissim I, Stern J, Pleasure D, Nissim I (1994) Interrelationships of leucine and glutamate metabolism in cultured astrocytes. J Neurochem 62: 1192–1202 [DOI] [PubMed] [Google Scholar]
- 11. Yudkoff M, Daikhin Y, Nissim I, Pleasure D, Stern J, Nissim I (1994) Inhibition of astrocyte glutamine production by alpha‐ketoisocaproic acid. J Neurochem 63: 1508–1515 [DOI] [PubMed] [Google Scholar]
- 12. Halestrap AP (2013) The SLC16 gene family ‐ structure, role and regulation in health and disease. Mol Aspects Med 34: 337–349 [DOI] [PubMed] [Google Scholar]
- 13. Halestrap AP, Meredith D (2004) The SLC16 gene family‐from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflugers Arch 447: 619–628 [DOI] [PubMed] [Google Scholar]
- 14. Miranda‐Goncalves V, Honavar M, Pinheiro C, Martinho O, Pires MM, Pinheiro C, Cordeiro M, Bebiano G, Costa P, Palmeirim I et al (2013) Monocarboxylate transporters (MCTs) in gliomas: expression and exploitation as therapeutic targets. Neuro Oncol 15: 172–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baenke F, Dubuis S, Brault C, Weigelt B, Dankworth B, Griffiths B, Jiang M, Mackay A, Saunders B, Spencer‐Dene B et al (2015) Functional screening identifies MCT4 as a key regulator of breast cancer cell metabolism and survival. J Pathol 237: 152–165 [DOI] [PubMed] [Google Scholar]
- 16. Kim Y, Choi JW, Lee JH, Kim YS (2015) Expression of lactate/H(+) symporters MCT1 and MCT4 and their chaperone CD147 predicts tumor progression in clear cell renal cell carcinoma: immunohistochemical and The Cancer Genome Atlas data analyses. Hum Pathol 46: 104–112 [DOI] [PubMed] [Google Scholar]
- 17. Lee JY, Lee I, Chang WJ, Ahn SM, Lim SH, Kim HS, Yoo KH, Jung KS, Song HN, Cho JH et al (2016) MCT4 as a potential therapeutic target for metastatic gastric cancer with peritoneal carcinomatosis. Oncotarget 7: 43492–43503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lim KS, Lim KJ, Price AC, Orr BA, Eberhart CG, Bar EE (2014) Inhibition of monocarboxylate transporter‐4 depletes stem‐like glioblastoma cells and inhibits HIF transcriptional response in a lactate‐independent manner. Oncogene 33: 4433–4441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Takada T, Takata K, Ashihara E (2016) Inhibition of monocarboxylate transporter 1 suppresses the proliferation of glioblastoma stem cells. J Physiol Sci 66: 387–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Manning Fox JE, Meredith D, Halestrap AP (2000) Characterisation of human monocarboxylate transporter 4 substantiates its role in lactic acid efflux from skeletal muscle. J Physiol 529(Pt 2): 285–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Broer S, Schneider HP, Broer A, Rahman B, Hamprecht B, Deitmer JW (1998) Characterization of the monocarboxylate transporter 1 expressed in Xenopus laevis oocytes by changes in cytosolic pH. Biochem J 333(Pt 1): 167–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dimmer KS, Friedrich B, Lang F, Deitmer JW, Broer S (2000) The low‐affinity monocarboxylate transporter MCT4 is adapted to the export of lactate in highly glycolytic cells. Biochem J 350: 219–227 [PMC free article] [PubMed] [Google Scholar]
- 23. Mac M, Nehlig A, Nalecz MJ, Nalecz KA (2000) Transport of alpha‐ketoisocaproate in rat cerebral cortical neurons. Arch Biochem Biophys 376: 347–353 [DOI] [PubMed] [Google Scholar]
- 24. Nakajima EC, Van Houten B (2013) Metabolic symbiosis in cancer: refocusing the Warburg lens. Mol Carcinog 52: 329–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sarkar S, Doring A, Zemp FJ, Silva C, Lun X, Wang X, Kelly J, Hader W, Hamilton M, Mercier P et al (2014) Therapeutic activation of macrophages and microglia to suppress brain tumor‐initiating cells. Nat Neurosci 17: 46–55 [DOI] [PubMed] [Google Scholar]
- 26. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM et al (2014) Functional polarization of tumour‐associated macrophages by tumour‐derived lactic acid. Nature 513: 559–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Klier M, Andes FT, Deitmer JW, Becker HM (2014) Intracellular and extracellular carbonic anhydrases cooperate non‐enzymatically to enhance activity of monocarboxylate transporters. J Biol Chem 289: 2765–2775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jamali S, Klier M, Ames S, Barros LF, McKenna R, Deitmer JW, Becker HM (2015) Hypoxia‐induced carbonic anhydrase IX facilitates lactate flux in human breast cancer cells by non‐catalytic function. Sci Rep 5: 13605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hambardzumyan D, Gutmann DH, Kettenmann H (2016) The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci 19: 20–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Buckingham SC, Campbell SL, Haas BR, Montana V, Robel S, Ogunrinu T, Sontheimer H (2011) Glutamate release by primary brain tumors induces epileptic activity. Nat Med 17: 1269–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tanaka K, Rosenberg LE (1983) Disorders of branched‐chain amino acid and organic acid metabolism In The metabolic basis of inherited disease, Stanbury JB, Wyngaarden JB, Fredrickson D, Goldstein JL, Brown MS (eds), pp 440–473. New York: McGraw Hill; [Google Scholar]
- 32. Nancolas B, Sessions RB, Halestrap AP (2015) Identification of key binding site residues of MCT1 for AR‐C155858 reveals the molecular basis of its isoform selectivity. Biochem J 466: 177–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hong CS, Graham NA, Gu W, Espindola Camacho C, Mah V, Maresh EL, Alavi M, Bagryanova L, Krotee PA, Gardner BK et al (2016) MCT1 modulates cancer cell pyruvate export and growth of tumors that co‐express MCT1 and MCT4. Cell Rep 14: 1590–1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Baek G, Tse YF, Hu Z, Cox D, Buboltz N, McCue P, Yeo CJ, White MA, DeBerardinis RJ, Knudsen ES et al (2014) MCT4 defines a glycolytic subtype of pancreatic cancer with poor prognosis and unique metabolic dependencies. Cell Rep 9: 2233–2249 [DOI] [PubMed] [Google Scholar]
- 35. Halestrap AP (2012) The monocarboxylate transporter family–Structure and functional characterization. IUBMB Life 64: 1–9 [DOI] [PubMed] [Google Scholar]
- 36. Becker HM, Klier M, Schuler C, McKenna R, Deitmer JW (2011) Intramolecular proton shuttle supports not only catalytic but also noncatalytic function of carbonic anhydrase II. Proc Natl Acad Sci USA 108: 3071–3076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stridh MH, Alt MD, Wittmann S, Heidtmann H, Aggarwal M, Riederer B, Seidler U, Wennemuth G, McKenna R, Deitmer JW et al (2012) Lactate flux in astrocytes is enhanced by a non‐catalytic action of carbonic anhydrase II. J Physiol 590: 2333–2351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Noor SI, Dietz S, Heidtmann H, Boone CD, McKenna R, Deitmer JW, Becker HM (2015) Analysis of the binding moiety mediating the interaction between monocarboxylate transporters and carbonic anhydrase II. J Biol Chem 290: 4476–4486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jouvet P, Rustin P, Taylor DL, Pocock JM, Felderhoff‐Mueser U, Mazarakis ND, Sarraf C, Joashi U, Kozma M, Greenwood K et al (2000) Branched chain amino acids induce apoptosis in neural cells without mitochondrial membrane depolarization or cytochrome c release: implications for neurological impairment associated with maple syrup urine disease. Mol Biol Cell 11: 1919–1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bak LK, Waagepetersen HS, Sorensen M, Ott P, Vilstrup H, Keiding S, Schousboe A (2013) Role of branched chain amino acids in cerebral ammonia homeostasis related to hepatic encephalopathy. Metab Brain Dis 28: 209–215 [DOI] [PubMed] [Google Scholar]
- 41. Leke R, Bak LK, Anker M, Melo TM, Sorensen M, Keiding S, Vilstrup H, Ott P, Portela LV, Sonnewald U et al (2011) Detoxification of ammonia in mouse cortical GABAergic cell cultures increases neuronal oxidative metabolism and reveals an emerging role for release of glucose‐derived alanine. Neurotox Res 19: 496–510 [DOI] [PubMed] [Google Scholar]
- 42. Yudkoff M, Daikhin Y, Grunstein L, Nissim I, Stern J, Pleasure D, Nissim I (1996) Astrocyte leucine metabolism: significance of branched‐chain amino acid transamination. J Neurochem 66: 378–385 [DOI] [PubMed] [Google Scholar]
- 43. Zhou W, Bao S (2014) Reciprocal supportive interplay between glioblastoma and tumor‐associated macrophages. Cancers 6: 723–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Allen M, Bjerke M, Edlund H, Nelander S, Westermark B (2016) Origin of the U87MG glioma cell line: good news and bad news. Sci Transl Med 8: 354re3 [DOI] [PubMed] [Google Scholar]
- 45. Becker HM, Broer S, Deitmer JW (2004) Facilitated lactate transport by MCT1 when coexpressed with the sodium bicarbonate cotransporter (NBC) in Xenopus oocytes. Biophys J 86: 235–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sapcariu SC, Kanashova T, Weindl D, Ghelfi J, Dittmar G, Hiller K (2014) Simultaneous extraction of proteins and metabolites from cells in culture. MethodsX 1: 74–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File