

Abstract
Gayal (Bos frontalis), also known as mithan or mithun, is a large endangered semi-domesticated bovine that has a limited geographical distribution in the hill-forests of China, Northeast India, Bangladesh, Myanmar, and Bhutan. Many questions about the gayal such as its origin, population history, and genetic basis of local adaptation remain largely unresolved. De novo sequencing and assembly of the whole gayal genome provides an opportunity to address these issues. We report a high-depth sequencing, de novo assembly, and annotation of a female Chinese gayal genome. Based on the Illumina genomic sequencing platform, we have generated 350.38 Gb of raw data from 16 different insert-size libraries. A total of 276.86 Gb of clean data is retained after quality control. The assembled genome is about 2.85 Gb with scaffold and contig N50 sizes of 2.74 Mb and 14.41 kb, respectively. Repetitive elements account for 48.13% of the genome. Gene annotation has yielded 26 667 protein-coding genes, of which 97.18% have been functionally annotated. BUSCO assessment shows that our assembly captures 93% (3183 of 4104) of the core eukaryotic genes and 83.1% of vertebrate universal single-copy orthologs. We provide the first comprehensive de novo genome of the gayal. This genetic resource is integral for investigating the origin of the gayal and performing comparative genomic studies to improve understanding of the speciation and divergence of bovine species. The assembled genome could be used as reference in future population genetic studies of gayal.
Keywords: Bos frontalis, genome assembly, annotation, phylogeny
Data Description
Background
The gayal is a large-sized endangered semi-domesticated bovine species belonging to the family Bovidae, tribe Bovini, group Bovina, genus Bos, and species Bos frontalis (NCBI Taxon ID: 30 520). It is also called the mithan or mithun. Its distribution spans eastern Bhutan through the Arunachal Pradesh in India to the Naga and Chin hills in the Arakan Yomarange region that defines the borders between India, Bangladesh, Myanmar, and China [1, 2]. The gayal has unique characters and appearances compared to gaur, cattle, and other bovine species [3]. These features include a bony dorsal ridge on the shoulder and white stockings on all 4 legs (Figure 1). It has been previously held that gayal was domesticated from gaur and/or from a hybrid descendant from crossing domestic cattle (B. indicus or B. taurus) and wild gaur [2, 4, 5]. Karyotype analysis indicates that the Indian gayal has a 2n = 58 karyotype, same as the local gaur (2n = 58) [6, 7], but different from Chinese and Malaysian gaurs (B. gaurus, 2n = 56) as well as domesticated cattle (B. indicus and B. taurus, 2n = 60) [2, 6–10]. Phylogenetic analyses in multiple studies based on mtDNA or Y-chromosomal DNA place gayal in conflicting clustering positions with respect to cattle, zebu, and wild gaur. For example, Chinese gayal, or Dulong cattle, are known to harbor zebu or taurine mtDNA footprints, suggesting hybrid origin [5, 11], and more studies have shown a high mtDNA and Y-chromosomal DNA sequences similarity between gayal and guar [12–15]. One study has even placed the gayal as a distinct and separate species/subspecies [16]. In contrast, phylogenetic analyses based on single nucleotide polymorphisms (SNPs) from 20 randomly selected single-copy gene orthologs of B. taurus, B. mutus (wild yak), and Bubalus bubalis placed Chinese gayal off the B. mutus and B. taurus clade, indicating that gayal is distinct from the modern domestic cattle, B. taurus [5]. These authors further demonstrated from mtDNA analysis that the gayal is the most proximal to domesticated cattle (B. taurus and B. indicus), suggesting that the gayal could be a hybrid emanating from crossing of male wild gaur and female domestic cattle [5]. These differences illustrate the existence of unresolved uncertainties regarding the origin of gayal.
Figure 1:

A picture showing a female gayal (Bos frontalis, provided by Kai-Xing Qu).
Research has revealed a high genomic divergence among bovine species [17, 18]. Consequently, mapping of resequencing data from 1 bovine species onto the reference genome of different species (for instance, gayal vs cattle) creates avenues for biases and/or errors in sequence alignment and SNP calling procedures. This challenge extends to species of great research interest like gayal, which so far have no de novo assembled reference genome. For instance, Mei et al. recently reported a whole-genome sequencing (resequencing) of Chinese gayal [5]. In their analysis, they retrieved variants based on mapping gayal sequencing reads (×13.06) to the cattle reference genome. Importantly, hydride gayals are hard to distinguish only through morphological characterization, yet Mei et al. did not examine the karyotype of the gayal they resequenced. In contrast to the gayal, de novo genome assembly has been accomplished for related species like cattle (Bos taurus) [19], yak (Bos grunniens) [17], wisent (Bison bonasus) [20], North American bison (Bison bison) [21], zebu (Bos indicus) [22], and water buffalo (Bubalus bubalis) [23]. This represents a critical resource toward mitigating the challenges inherent in resequencing approaches and provides great opportunities to refine the evolutionary history of bovine species. In this study, we for the first time report the draft genome assembly of the gayal with a high sequencing depth generated on the Illumina genome sequencing platform. This valuable resource is important to the research of the origin and evolution of this species, which has been classified as endangered by the International Union for Conservation of Nature (IUCN).
Sample collection and sequencing
The gayal (NCBI taxonomy ID: 30 520) used for genome sequencing came from a Dulong in Yunnan province, China (Figure 1). It was kept at Yunnan Academy of Grassland and Animal Science for breeding and research purposes. Karyotype examination showed that it has 2n = 58 chromosomes (Figure 2). We extracted total genomic DNA from skin fibroblast cell lines of the gayal using the Qiagen Blood and Tissue Kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions. The cells are maintained at the Cell Bank of Kunming Institute of Zoology (specimen ID: KCB201042). A total of 17 paired-end genomic sequence libraries were constructed with a gradient insert size ranging from 180 bp to 20 kb, and sequencing was carried out on the Illumina HiSeq 2000 platform according to the manufacturer's instructions. For short insert size libraries (180 bp, 250 bp, 450 bp, and 600 bp), sequencing was performed at the Central Laboratory of Kunming Institute of Zoology with read lengths of 100 bp. Sequencing of long insert size libraries (800 bp, 2, 5, 10 and 20 kb) was conducted at BGI-Shenzhen with read lengths of 49 bp, except for the 800-bp insert size library, which was sequenced with a read length of 85 bp. A total of 350.38 Gb of raw sequence data has been generated in our study (Additional file 1: Table S1). Before assembly, we performed strict quality control by removing poor-quality reads and/or bases using scripts from SOAPec (version 2.02) [24]. Reads were shortened by 2 bp at both the head and tail. We dropped any read plus its corresponding paired end if it contained more than 30 low-quality bases or more than 5% unknown base (usually denoted by N). Reads with duplications and adapters were also removed. We corrected for sequencing errors using the k-mer (13 used in this study) frequency method in SOAPec (version 2.02) [24]. After filtering and correction, we retained 276.86 Gb of high-quality sequences for genome assembly (Additional file 1: Table S2).
Figure 2:

Karyotype of the gayal used for genome sequencing (provided by Wen-Hui Nie).
De novo assembly of gayal genome
In order to have a basic knowledge about the genome size and attributes of the gayal genome, we performed a 17-mer analysis using clean and high-quality sequences from 180 and 450 bp insert size libraries. We extracted the 17-mer sequences using sliding windows with a size of 17 bp and calculated the frequency of each 17-mer. A clear peak at ×25 with 2 upward convex signals apart from it is evident, suggesting high heterozygosity. The genome size for gayal is estimated to be 3.15 Gb (Figure 3; Additional file 1: Table S3).
Figure 3:
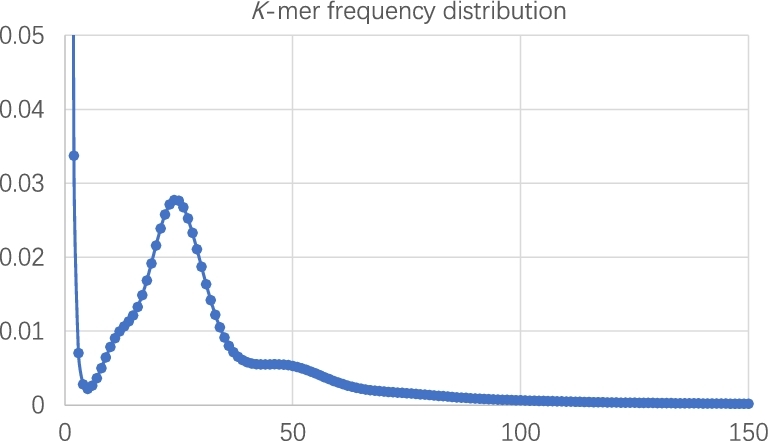
17-mer frequency distribution of sequencing reads.
We then performed de novo assembly of the gayal genome using Platanus (version 2.0; Platanus, RRID:SCR_015531) [25] in 3 steps: contig construction, scaffolding, and gap filling. To construct contigs based on short insert size libraries (180, 250, 450, 600, and 800 bp), we used Platanus (version 2.0) [25], which includes a series of procedures such as constructing de Bruijn graphs, clipping tips, merging bubbles, and removing low coverage links. In the scaffolding step, reads from both small and large insert libraries were mapped to contig sequences to construct scaffolds using distance information from read pairs. An additional local assembly of reads, with 1 end of a read pair uniquely aligned to a contig and the other end located within the gap, was performed using GapCloser (version 1.12; GapCloser, RRID:SCR_015026) [24]. These processes yielded a final draft gayal genome assembly with a total length of 2.85 Gb, contig N50 of 14.4 kb, and scaffold N50 of 2.74 Mb (Table 1). The assembled genome size is similar to that reported for cattle [26] and yak [17]. To assess the completeness of the assembled gayal genome, we performed BUSCO analysis (BUSCO, RRID:SCR_015008) [27] by searching against the arthropod universal benchmarking single-copy orthologs (BUSCOs, version 2.0). Overall, 85.2% and 7.8% of the 4104 expected vertebrate genes are identified in the assembled genome as complete and partial, respectively. Approximately 291 genes could be considered missing in our assembly. Of the expected complete vertebrate genes, 3434 and 60 are identified as single-copy and duplicated BUSCOs, respectively (Table 2). Our newly assembled gayal genome has a slightly lower completeness rate compared to genomes of yak [17], wisent [20], bison [21], zebu [22], and buffalo [23] (Table 2).
Table 1:
Statistics of the completeness of the hybrid de novo assembly of Bos frontalis genome
| Terms | Contig | Scaffold | ||
|---|---|---|---|---|
| Size | Number | Size | Number | |
| N90 | 2461 | 211 577 | 158 610 | 1357 |
| N80 | 5335 | 140 237 | 1 060 177 | 800 |
| N70 | 8109 | 99 930 | 1 668 147 | 587 |
| N60 | 11 044 | 71 764 | 2 170 469 | 437 |
| N50 | 14 405 | 50 585 | 2 737 757 | 320 |
| Max length | 208 099 | 13 764 521 | ||
| Total length | 2 669 378 334 | 2 848 570 279 | ||
| Total number | 583 373 | 460 059 | ||
| Average length | 4575 | 6191 | ||
| Number ≥ 500 bp | 394 757 | 116 481 | ||
| Number ≥ 1000 bp | 300 178 | 53 989 | ||
| Number ≥ 2000 bp | 229 796 | 19 915 | ||
| Number ≥ 5000 bp | 146 493 | 5387 | ||
Table 2:
Statistics of the completeness of the assembled genomes for Bos frontalis and close related species by BUSCO (version 2)
| Species | Terms | Complete (C) | Complete and single-copy (S) | Complete and duplicated (D) | Fragmented (F) | Missing (M) |
|---|---|---|---|---|---|---|
| Gayal | Number | 3494 | 3434 | 60 | 319 | 291 |
| Proportion, % | 85.14 | 83.67 | 1.46 | 7.77 | 7.09 | |
| Zebu | Number | 3698 | 3644 | 54 | 158 | 248 |
| Proportion, % | 90.11 | 88.79 | 1.32 | 3.85 | 6.04 | |
| Wisent | Number | 3794 | 3763 | 31 | 180 | 130 |
| Proportion, % | 92.45 | 91.69 | 0.76 | 4.39 | 3.17 | |
| Yak | Number | 3841 | 3809 | 32 | 138 | 125 |
| Proportion, % | 93.59 | 92.81 | 0.78 | 3.36 | 3.05 | |
| Buffalo | Number | 3817 | 3780 | 37 | 142 | 145 |
| Proportion, % | 93.01 | 92.11 | 0.90 | 3.46 | 3.53 | |
| Bison | Number | 3779 | 3735 | 44 | 165 | 160 |
| Proportion, % | 92.08 | 91.01 | 1.07 | 4.02 | 3.90 |
Annotation of genomic repeat sequences in the gayal genome
To search for the repeated sequences in the gayal genome, including tandem repeats, interspersed repeats, and transposable elements (TE; e.g., LINE, SINE, LTR, DNA transposons), we leveraged both de novo and homolog-based methods as used in previous publications [28, 29]. For the homolog-based methods, we used RepeatMasker (RepeatMasker, RRID:SCR_012954) and RepeatProteinMask [30] to search against the known Repbase TE library (RepBase21.01) [31] and TE protein database, respectively. In the de novo method, Piler [32] and RepeatModeler (RepeatModeler, RRID:SCR_015027) [33] are used to generate a de novo gayal repeat library, which is subsequently used in Repeat-Masker to annotate repeats. TRF [34] is then employed to predict tandem repeats. The combined results show that a total of 1.37 Gb of non-redundant repetitive sequences are identified in the gayal genome, which account for 48.13% of the whole genome. The most predominant repeat is the long interspersed nuclear elements (LINEs), which account for 40.43% (1.15 Gb in total) of the genome (Table 3; Additional file 1: Table S4, Figure S1, Figure S2).
Table 3:
Statistics of repeats in Bos frontalis genome
| Type | Repeat size, bp | % of genome |
|---|---|---|
| Trf | 17 696 175 | 0.62 |
| Repeatmasker | 868 885 926 | 30.50 |
| Proteinmask | 265 003 148 | 9.30 |
| De novo | 917 371 710 | 32.20 |
| Total | 1 371 023 312 | 48.13 |
Gayal genome gene structure prediction
For gene structure prediction, we combined both de novo and homolog-based approaches to predict protein-coding genes in the gayal genome. In homolog-based method, gene sets from Bos taurus [19], Canis familiaris [35], Homo sapiens (ENSEMBL 80), Sus scrofa [36], Rattus norvegicus (ENSEMBL 80), and Ovis aries [37] were used as queries to search against the gayal genome (Additional file 1: Table S5). For the de novo–based method, AUGUSTUS (Augustus: Gene Prediction, RRID:SCR_008417) [38], Genescan (GENSCAN, RRID:SCR_012902) [39], and GlimmerHMM (GlimmerHMM, RRID:SCR_002654) [40] were used as engines to predict gene models. We then merged the gene prediction results derived from both methods using GLEAN [41] to generate a consensus gene set. In total, we have identified 26 667 protein-coding genes with a mean of 3.27 exons per gene (Table 4; Additional file 1: Figure S3). The lengths of genes, coding sequence (CDS), introns, and exons in gayal are comparable to those of the genomes used for homolog-based predictions (Additional file 1: Figure S3). In addition, we predicted non-coding RNA genes in the gayal genome. We used blast to search rRNA against the Human rRNA database, and tRNAscan-SE (tRNAscan-SE, RRID:SCR_010835) [42] to search tRNA in the genome sequences. We also used blast to search miRNA and snRNA via the Rfam database (release 11.0; Rfam, RRID:SCR_007891) [43]. We reveal a total of 2357 ribosomal RNA (rRNA), 29 821 transfer RNA (tRNA), 16 305 microRNAs (miRNA), and 1380 snRNA genes in the gayal genome (Additional file 1: Table S5).
Table 4:
General statistics of predicted protein-coding genes
| Gene set | Total | Exon number | CDS length, bp | mRNA length, bp | Exons per gene | Exon length, bp | Intron length, bp | |
|---|---|---|---|---|---|---|---|---|
| Homolog | Bos taurus | 19 666 | 141 323 | 1325 | 20 618 | 7.19 | 184 | 3118 |
| Canis familiaris | 17 627 | 121 986 | 1323 | 20 802 | 6.92 | 191 | 3290 | |
| Homo sapiens | 24 783 | 146 172 | 1108 | 17 567 | 5.89 | 187 | 3360 | |
| Sus scrofa | 20 283 | 121 282 | 1142 | 16 288 | 5.97 | 191 | 3041 | |
| Rattus norvegicus | 17 988 | 117 965 | 1277 | 19 469 | 6.55 | 194 | 3273 | |
| Ovis aries | 20 947 | 147 367 | 1287 | 20 973 | 7.03 | 183 | 3261 | |
| De novo | AUGUSTUS | 41 227 | 180 664 | 1127 | 22 786 | 4.38 | 257 | 6403 |
| GlimmerHMM | 27 067 | 104 294 | 874 | 5433 | 3.85 | 226 | 1597 | |
| Genescan | 46 598 | 297 828 | 1321 | 36 828 | 6.39 | 206 | 6585 | |
| Glean (final) | 26 667 | 87 392 | 1156 | 4996 | 3.27 | 352 | 1686 | |
Functional annotation of protein-coding genes
Gene functional annotation refers to searching functional motifs, domains, and possible biological processes by aligning translated gene coding sequences to known databases such as SwissProt and TrEMBL [44], NT database (from NCBI), Gene Ontology (GO, RRID:SCR_002811), and Kyoto Encyclopedia of Genes and Genomes (KEGG, RRID:SCR_012773) [45]. We have annotated all the protein-coding genes identified in this study to retrieve functional terms according to InterPro, KEGG, and GO terms. Overall, 81.74% (21 798), 54.56% (14 550), and 66.39% (17 704) genes show enrichment in InterPro, KEGG, and GO, respectively. In total, 25 916 protein-coding genes (97.18%) were successfully annotated for conserved functional motifs and functional terms (Additional file 1: Table S6).
Phylogenetic analysis and divergence time estimation
To investigate the phylogenic position of gayal, we retrieved nucleotide and protein data for cattle (Bos taurus) [19], yak (Bos grunniens) [17], wisent (Bison bonasus) [20], bison (Bison bison) [21], zebu (Bos indicus) [22], and buffalo (Bubalus bubalis) [23] from the NCBI database. Gene ortholog relationships of gayal and other bovine species were identified by reciprocal blast searching with an e-value of 1e-7. Genes with alternative splicing variants are represented by the longest transcript. Multiple sequence alignment of the genes within 1 copy gene set were performed using the MUSCLE program (MUSCLE, RRID:SCR_011812) [46]. Aligned sequences were trimmed to remove potentially unreliably aligned regions and gaps using Gblocks [47]. Alignments with lengths shorter than 100 bp were also discarded. Four-fold degenerate sites were extracted and concatenated into a supergene. Modeltest [48] was used to select the best substitution model. MrBayes (MrBayes, RRID:SCR_012067) [49] and RaxML (RAxML, RRID:SCR_006086) [50] software were used to reconstruct the evolutionary relationships between species, and MEGA5 [51] was used to view the tree. From these analyses, gayal clusters with the common ancestor of cattle and zebu (Figure 4).
Figure 4:
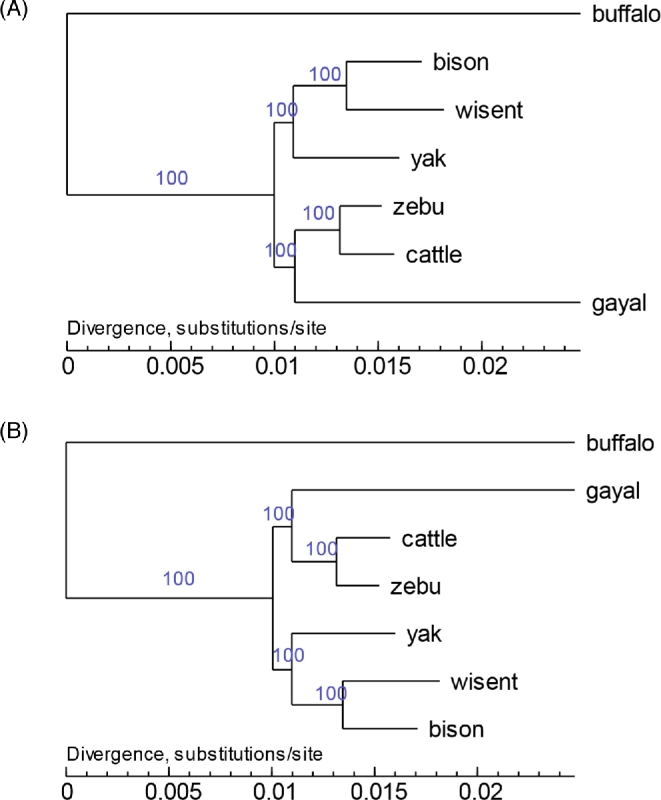
Phylogenetic trees of gayal and other bovine species. (A) Tree constructed based on maximum likelihood method. (B) Tree constructed using Bayesian inference.
Additionally, we sequenced the complete mitochondrial DNA (mtDNA, the first complete mtDNA of the gayal submitted to GenBank: MF614103) using the Sanger sequencing method, due to the fact that next-generation sequencing methods have lower ability and accuracy in recovering repeat sequences [28, 52], particularly in regions with rich GC content like the D-loop. We then downloaded mtDNA sequences of gayal and other bovine species from GenBank for phylogenic analysis. As shown in Figure 5 and Figure S4, the gayal we sequenced clusters with gaur (Figure 5; Additional file 1: Figure S4). Our results from both whole-genome and mtDNA data differ from the conclusion made by Mei et al., who mapped gayal genome resequencing data to a bovine reference [5]. Furthermore, the MCMCTREE program, implemented using the PAML (PAML, RRID:SCR_014932) [53] package, was used to estimate divergence times. The JC69 model and correlated molecular clock rates (clock = 3) were used in the calculation. Calibration time for the common ancestor of buffalo and cattle obtained from the TimeTree database [54] was used to calibrate the divergence time. This analysis estimated the divergence time of gayal from cattle and zebu at approximately 5.1 million years ago (Figure 6).
Figure 5:

Maximum likelihood trees of gayal and other bovine species using whole complete mtDNA. IDs in parentheses are GenBank accession number.
Figure 6:
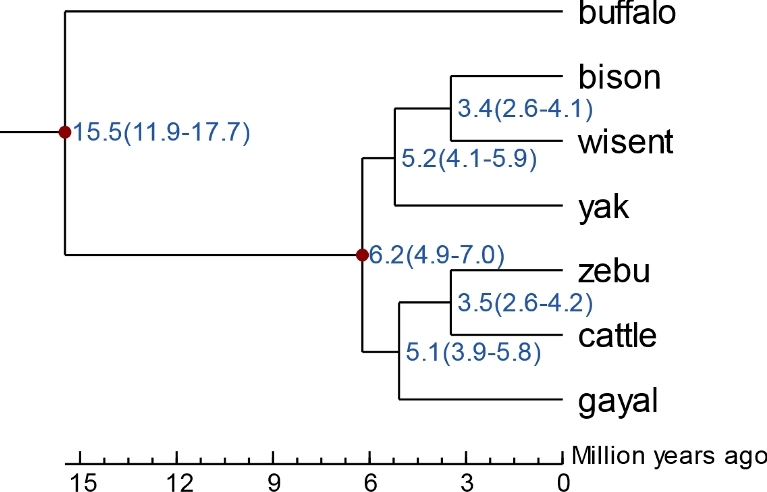
Divergence time estimated between gayal and other bovine species.
In conclusion, we have constructed a de novo assembly of the gayal genome, and we describe its genetic attributes. To our knowledge, this is the first de novo assembled genome for this species. We also demonstrate that together with the genomes of other bovine species, the new gayal genome supports investigations concerning the origin, evolutionary history, and local adaptation of gayal. This resource is also important for the future conservation of this endangered species. In addition, the de novo gayal genome adds to the list of available bovine genomes and has advantages over resequenced genomes in allowing accurate whole-genome alignment and retrieving constraint and/or rapidly evolved elements. It also strengthens the capacity to better assess introgression, incomplete lineage sorting (ILS), and structural variation (SV) among bovine species, as well as inferring their effects on the species tree. The assembled genome could be used as a reference in population genomic studies [55] of the gayal. Furthermore, comprehensive comparative analyses of these genomes will improve understanding of the formation and speciation of bovine species.
Availability of supporting data
The genome sequencing raw reads were deposited in the NCBI SRA database, project ID: PRJNA387130. The assembly and annotation of the gayal genome are available in the GigaScience database, GigaDB [56]. The complete mtDNA for the gayal generated by Sanger sequencing is also available in GenBank under the ID: MF614103. All supplementary figures and tables are provided in Additional file 1.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Y.P.Z., D.D.W., and M.S.W. designed the study. W.W. and Y.D. supervised the analyses. W.H.N., W.T.S., and J.H.W. cultivated the cells. Y.Z. and X.W. performed genome assembly and annotation. M.S.W. extracted genomic DNA and wrote the manuscript with the other authors’ input. M.S.W. and S.Q.Y. sequenced the gayal complete mitochondrial DNA and submitted to GenBank. S.W., Z.J.X., K.X.Q., N.O.O., D.Y., D.D.W., and Y.P.Z. revised the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Acknowledgements
This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences, grant No. XDB13020600, and the Chinese 973 program (2013CB835200, 2013CB835204). D.D.W. was supported by the Youth Innovation Promotion Association, Chinese Academy of Sciences. N.O.O. is thankful for the support of the CAS-TWAS President's Fellowship Program for Doctoral Candidates.
References
- 1. Payne WJA, Hodges J. Tropical Cattle: Origins, Breeds and Breeding Policies. Oxford: Blackwell Science; 1997. [Google Scholar]
- 2. Uzzaman MR, Bhuiyan MS, Edea Z et al. Semi-domesticated and Irreplaceable genetic resource gayal (Bos frontalis) needs effective genetic conservation in Bangladesh: a review. Mol Biol Evol 2014;27:1586–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Miao YW, Ha F, Gao HS et al. Polymorphisms of inhibin α gene exon 1 in buffalo (Bubalus bubalis), gayal (Bos frontalis) and yak (Bos grunniens). Zool Res 2012;33:402–8. [DOI] [PubMed] [Google Scholar]
- 4. Payne WJA. Cattle production in the tropics. In: General Introduction and Breeds and Breeding, vol. 1. London: Longman Group Ltd.; 1970. pp.xv + 336 pp. [Google Scholar]
- 5. Mei C, Wang H, Zhu W et al. Whole-genome sequencing of the endangered bovine species gayal (Bos frontalis) provides new insights into its genetic features. Sci Rep 2016;6:19787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Winter H, Mayr B, Schleger W et al. Karyotyping, red blood cell and haemoglobin typing of the mithun (Bos frontalis), its wild ancestor and its hybrids. Res Vet Sci 1984;36:276–83. [PubMed] [Google Scholar]
- 7. Gallagher DS Jr., Womack JE. Chromosome conservation in the Bovidae. J Hered 1992;83:287–98. [DOI] [PubMed] [Google Scholar]
- 8. Shan XN, Chen YF, Luo LH et al. Comparative studies on the chromosomes of five species of catties of the genus Bos in China. Zool Res 1980;1:75–81. [Google Scholar]
- 9. Adbullah MH, Idris I, Hilmi M. Karyotype of malayan gaur (Bos gaurus hubbacki), Sahiwal-Friesian cattle and gaur x cattle hybrid backcrosses. Pak J Biol Sci 2009;12:896–901. [DOI] [PubMed] [Google Scholar]
- 10. Qu KX, He ZX, Nie WH et al. Karyotype analysis of mithun (Bos frontalis) and mithun bull x Brahman cow hybrids. Genet Mol Res 2012;11:131–40. [DOI] [PubMed] [Google Scholar]
- 11. Gou X, Wang Y, Yang S et al. Genetic diversity and origin of gayal and cattle in Yunnan revealed by mtDNA control region and SRY gene sequence variation. J Anim Breed Genet 2010;127:154–60. [DOI] [PubMed] [Google Scholar]
- 12. Li SP, Chang H, Ma GL et al. Molecular phylogeny of the gayal inferred from the analysis of cytochrome b gene entire sequences. Yi Chuan 2008;30:65–70. [DOI] [PubMed] [Google Scholar]
- 13. Nijman IJ, van Boxtel DCJ, van Cann LM et al. Phylogeny of Y chromosomes from bovine species. Cladistics 2008;24:723–6. [Google Scholar]
- 14. Dorji T, Mannen H, Namikawa T et al. Diversity and phylogeny of mitochondrial DNA isolated from mithun Bos frontalis located in Bhutan. Anim Genet 2010;41:554–6. [DOI] [PubMed] [Google Scholar]
- 15. Tanaka K, Takizawa T, Murakoshi H et al. Molecular phylogeny and diversity of Myanmar and Bhutan mithun based on mtDNA sequences. Anim Sci J 2011;82:52–56. [DOI] [PubMed] [Google Scholar]
- 16. Baig M, Mitra B, Qu KX et al. Mitochondrial DNA diversity and origin of Bos frontalis. Curr Sci 2013;104:115–20. [Google Scholar]
- 17. Qiu Q, Zhang G, Ma T et al. The yak genome and adaptation to life at high altitude. Nat Genet 2012;44:946–9. [DOI] [PubMed] [Google Scholar]
- 18. Porto-Neto LR, Sonstegard TS, Liu GE et al. Genomic divergence of zebu and taurine cattle identified through high-density SNP genotyping. BMC Genomics 2013;14:876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zimin AV, Delcher AL, Florea L et al. A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol 2009;10:R42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang K, Wang L, Lenstra JA et al. The genome sequence of the wisent (Bison bonasus). Gigascience 2017.doi: 10.1093/gigascience/gix016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. American bison (Bison bison bison) genome assembly. https://www.ncbi.nlm.nih.gov/assembly/GCF_000754665.1/. Accessed 1 November 2017.
- 22. Canavez FC, Luche DD, Stothard P et al. Genome sequence and assembly of Bos indicus. J Hered 2012;103:342–8. [DOI] [PubMed] [Google Scholar]
- 23. Water buffalo (Bubalus bubalis) genome assembly https://www.ncbi.nlm.nih.gov/assembly/GCA_000471725.1#/st. Accessed 1 November 2017.
- 24. Luo R, Liu B, Xie Y, Li Z et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 2012;1:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kajitani R, Toshimoto K, Noguchi H et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res 2014;24:1384–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bovine Genome S, Analysis C, Elsik CG et al. The genome sequence of taurine cattle: a window to ruminant biology and evolution. Science 2009;324:522–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Simao FA, Waterhouse RM, Ioannidis P et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015;31:3210–2. [DOI] [PubMed] [Google Scholar]
- 28. Wang MS, Li Y, Peng MS et al. Genomic analyses reveal potential independent adaptation to high altitude in Tibetan chickens. Mol Biol Evol 2015;32:1880–9. [DOI] [PubMed] [Google Scholar]
- 29. Xiong Z, Li F, Li Q et al. Draft genome of the leopard gecko, Eublepharis macularius. Gigascience 2016;5:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. RepeatMasker http://repeatmasker.org/. Accessed 1 November 2017.
- 31. Kapitonov VV, Jurka J. A universal classification of eukaryotic transposable elements implemented in Repbase. Nat Rev Genet 2008;9:411–2. [DOI] [PubMed] [Google Scholar]
- 32. Edgar RC, Myers EW. PILER: identification and classification of genomic repeats. Bioinformatics 2005;21(Suppl 1):152–8. [DOI] [PubMed] [Google Scholar]
- 33.RepeatModeler http://www.repeatmasker.org/. Accessed 1 November 2017.
- 34. Benson G. Tansdem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 1999;27:573–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lindblad-Toh K, Wade CM, Mikkelsen TS et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 2005;438:803–19. [DOI] [PubMed] [Google Scholar]
- 36. Groenen MA, Archibald AL, Uenishi H et al. Analyses of pig genomes provide insight into porcine demography and evolution. Nature 2012;491:393–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jiang Y, Xie M, Chen W et al. The sheep genome illuminates biology of the rumen and lipid metabolism. Science 2014;344:1168–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stanke M, Keller O, Gunduz I et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 2006;34:W435–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cai Y, Gonzalez JV, Liu Z et al. Computational systems biology methods in molecular biology, chemistry biology, molecular biomedicine, and biopharmacy. Biomed Res Int 2014;2014:746814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Majoros WH, Pertea M, Salzberg SL. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 2004;20:2878–9. [DOI] [PubMed] [Google Scholar]
- 41. Elsik CG, Mackey AJ, Reese JT et al. Creating a honey bee consensus gene set. Genome Biol 2007;8:R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 1997;25:955–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gardner PP, Daub J, Tate J et al. Rfam: Wikipedia, clans and the “decimal” release. Nucleic Acids Res 2011;39:D141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. UniProt C. UniProt: a hub for protein information. Nucleic Acids Res 2015;43:D204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kanehisa M, Goto S, Sato Y et al. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res 2014;42:D199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 2004;5:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol 2007;56:564–77. [DOI] [PubMed] [Google Scholar]
- 48. Posada D. Using MODELTEST and PAUP* to select a model of nucleotide substitution. Curr Protoc Bioinformatics 2003;Chapter 6:Unit 6 5. [DOI] [PubMed] [Google Scholar]
- 49. Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001;17:754–5. [DOI] [PubMed] [Google Scholar]
- 50. Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006;22:2688–90. [DOI] [PubMed] [Google Scholar]
- 51. Tamura K, Peterson D, Peterson N et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 2011;28:2731–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang MS, Yang HC, Otecko NO et al. Olfactory genes in Tibetan wild boar. Nat Genet 2016;48:972–3. [DOI] [PubMed] [Google Scholar]
- 53. Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 2007;24:1586–91. [DOI] [PubMed] [Google Scholar]
- 54.TimeTree http://www.timetree.org/. Accessed 1 November 2017.
- 55. Chen H. Population genetic studies in the genomic sequencing era. Zool Res 2015;36: 223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang MS, Zeng Y, Wang X et al. Supporting data for “Draft genome of the gayal, Bos frontalis.” GigaScience Database. 2017. http://dx.doi.org/10.5524/100354. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.