

Abstract
Purpose
Studies of Fuchs' dystrophy have largely focused on individuals of European origin. Characterization of disease among African Americans is required to ensure prognostic factors and therapeutic approaches are applicable across diverse patient populations.
Methods
We assessed all self-reported black and white patients aged older than 40 years at a tertiary care institution with a diagnosis of cataract over a 3-year period for concurrent diagnosis of Fuchs' dystrophy. Affected patients in a longitudinal cohort were invited to provide a blood sample from which we extracted genomic DNA. The CTG18.1 trinucleotide repeat length was determined using a two-step, triplet repeat primed PCR protocol. Expansion was defined as >40 CTG repeats. Demographic information, including race, was documented.
Results
Of 59,365 self-reported black and white adults who presented for cataract evaluation, the odds ratio of presenting with Fuchs' dystrophy among black compared to white patients was 0.6992 (95% confidence interval [CI], 0.6210–0.7872). A total of 60 black and 549 white patients with Fuchs' corneal dystrophy enrolled in the longitudinal study, of which 21 (35.0%) black and 343 (62.5%) white patients demonstrated trinucleotide repeat expansion, a significant difference (P = 7.7 × 10−5). In a multivariable linear regression model, repeat expansion but not race was significantly associated with mean clinical grading of severity.
Conclusions
Black patients with Fuchs' dystrophy were less likely than white patients to demonstrate CTG18.1 allele expansion. The data contribute to our understanding of population differences in clinical presentation, and highlight the need for considering diversity of patient populations in clinical research.
Keywords: Fuchs' corneal dystrophy, African-Americans, trinucleotide repeat expansion
Fuchs' corneal dystrophy (FCD) is a hereditary corneal condition resulting in bilateral, progressive vision loss in middle age. The leading indication for corneal transplantation in the United States,1 it is pathologically characterized by formation of guttae, excrescences of Descemet membrane, and associated with corneal endothelial cell loss. Edema begins in early stages2 and progresses to painful bullous keratopathy in late stages of disease.
Differences exist across populations in the presentation of FCD, and worldwide, estimates of FCD prevalence vary across regions when viewed as an indicator for corneal transplantation.3 In the United States, previous studies suggest that African Americans demonstrate similar rates of mild disease but are less likely to progress to a more severe phenotype.4 This is supported by distinctions in corneal transplantation rates between black and white patients.5
To date, the largest investigations recruiting patients with Fuchs' dystrophy have generally drawn from white populations, who comprise 93% or more of participants in such studies,6 although the presence of Fuchs' dystrophy among African Americans has been documented for over a century.7 Research that includes diverse patient populations is important to ensure that conclusions drawn, and potential therapies developed, are broadly applicable. To characterize clinical and genetic features of disease among black and white Americans with Fuchs' dystrophy, we utilized a two-step approach: first, an investigation of our cataract population for an association between Fuchs' dystrophy diagnosis and self-reported race, and second, assessment of CTG18.1 trinucleotide repeat expansion in patients from both populations to determine whether the frequency of trinucleotide repeat expansion may contribute to such a measured association.
Methods
Retrospective Chart Review
To confirm whether FCD is diagnosed at a comparable rate between black and white individuals in our patient population, we retrospectively selected from coding data all patients who presented to a single tertiary care institution (Wilmer Eye Institute) from June 6, 2014 to June 6, 2017, who were 40 to 110 years of age and received an encounter diagnosis of cataract. Among such patients, race was defined by self-assessed categorization at the time of registration, and we assessed the total number of persons who identified as black or white. The number of patients with concurrent diagnosis of Fuchs' dystrophy and cataract in each group was summated. Cataract was chosen as a concurrent diagnosis since it is independent from FCD pathogenesis, evaluation includes testing for Fuchs' dystrophy, and to limit, to the extent possible, confusion between Fuchs' dystrophy and pseudophakic bullous keratopathy, which would be expected to occur in the setting of pseudophakia. Odds ratio and χ2 analyses were employed to assess for an association between race and affection in the cataract cohort.
Genetic Characterization
To understand the prevalence of CTG18.1 trinucleotide repeat expansion in TCF4 across populations, we explored expansion rate among black and white members of a longitudinal cohort of Fuchs' dystrophy. Patients were recruited after slit-lamp examination revealed clinically significant FCD, defined as at least 12 central guttae in either eye, or 5 central guttae in both eyes. Severity of FCD was graded using the Krachmer scale.8 The participants provided a blood sample from which genomic DNA was extracted using a commercial blood and tissue kit (DNeasy; Qiagen, Hilden, Germany). Race was self-reported by patients.
The protocols in this study were approved by the Institutional Review Board of the Johns Hopkins University School of Medicine and adhere to the tenets of the Declaration of Helsinki. Informed consent was obtained from the subjects after explanation of the nature and possible consequences of the study.
The CTG18.1 trinucleotide repeat length was determined using the two-step, triplet repeat primed PCR (TP-PCR) protocol as described by Vasanth et al.9 The forward primer (P1) was labeled with 6- carboxyfluorescein (5′ CAGATGAGTTTGGTGTAAGATG 3′). The two unlabeled reverse primers were P2 (5′ TACGCATCCCAGTTTGAGACGCAGCAGCAGCAGCAG 3′) and P3 (5′ TACGCATCCCAGTTTGAGACG 3′). We added 1 μL genomic DNA was mixed with 13.5 μL of supermix (Platinum PCR Supermix High Fidelity; Life Technologies), and 0.1 μL of P1 and P2. This was cycled at 95°C for 5 minutes; 95°C for 30 seconds, 58°C for 1 minute, 68°C for 4 minutes × 20 cycles, with an additional 15 seconds for each of the 8th to 20th cycle, 68°C for 10 minutes. We added 2 μL of the PCR products to 13 μL of supermix and 0.1 μL of P1 and P3. This was cycled at 95°C for 5 minutes; 95°C for 30 seconds, 62°C for 1 minute, 68°C for 6 minutes × 25 cycles; 68°C for 10 minutes. The final PCR products were resolved in a DNA Analyzer (ABI3730XL; Applied Biosystems, Inc., Foster City, CA, USA) with a dye size standard (GeneScan 500 Liz; Life Technologies). The results were analyzed using Gene Mapper (Applied Biosystems, Inc.).
Statistical Analysis
Statistical analysis was carried out using statistical software (IBM SPSS, version 24; SPSS, Inc., Chicago, IL, USA). The mean age and mean Krachmer grading of the groups were compared using the independent-samples Mann-Whitney U test. Association between allele expansion, biallelic expansion, and race was examined using one-sided Fisher's exact test. A standard regression analysis was performed to identify the best-fit correlation model between Krachmer grading and age. Stepwise multiple regression analysis, using probability of F as stepping criteria; with F ≤ 0.050 for entry and F ≥ 0.100 for removal, was carried out to evaluate age, sex, race, and allelic expansion as predictors of Krachmer grading. Data from our previously reported cohort was used to compare the association between allele expansion and race. For consistency with previous studies, alleles with >40 CTG repeats were considered expanded, but additional calculations to consider a cutoff of 50 CTG repeats were implemented. Clinical grading using the Krachmer scale8 was documented for each eye. An average Krachmer grading of the two eyes was used. Participants with a history of corneal transplant were given a Krachmer grade of 6 in that eye.
Results
Clinical Cohort
A total of 65,476 individuals presented for a clinical encounter with diagnosis of cataract during the time interval, of whom 59,365 self-reported as black or white. Table 1 reveals classification by race and affection. Relative to white patients, the odds ratio of Fuchs' dystrophy diagnosis among African Americans was 0.6992 (95% CI, 0.6210–0.7872).
Table 1.
Self-Reported Black and White Patients at a Tertiary Care Institution

Genetic Characterization
A total of 609 participants, 60 black and 549 white, were selected from a longitudinal study of FCD. We found CTG18.1 trinucleotide repeat expansion in 35.0% (21/60) of black and in 62.5% (343/549) white individuals, a significant association between race and CTG18.1 repeat expansion (P = 7.7 × 10−5, two-sided Fisher's exact test). A cutoff of 50 rather than 40 repeats, as has been reported in some studies, resulted in one black and four white individuals considered as nonexpanded, but the results remained significant (P = 2.8 × 10−5). Biallelic expansion was seen in 1.7% (1/60) of black and 2.4% (13/549) of white cases, using >40 repeats as the threshold. No significant difference was demonstrated between the proportion of biallelic expansions between the two groups (P = 0.572, two-sided Fisher's exact test). Within each group, the distribution of copy number was comparable among black and white individuals, with median copy length of 18 among black persons without expansion and 91 repeats among those with the expanded allele, compared to 17 and 90.5 in white persons, respectively.
In black patients with Fuchs' dystrophy, the mean Krachmer grade without repeat expansion was 2.72 (median 2+), while with repeat expansion it was 3.44 (median 4+). In white patients, the mean Krachmer grades were 2.55 (median 2+) and 3.22 (median 3+), respectively, for the associated genotypes. To explore contributors to this apparent worsening of severity with repeat expansion, a multivariable linear regression model with variables of age, race, sex, and repeat expansion status was produced. Notably, repeat expansion, but not race, nor other variables, were significantly associated with worsening of the disease phenotype. Details are included in Table 3.
Table 3.
Multivariable Linear Regression Model of Mean Clinical Severity on the Krachmer Scale
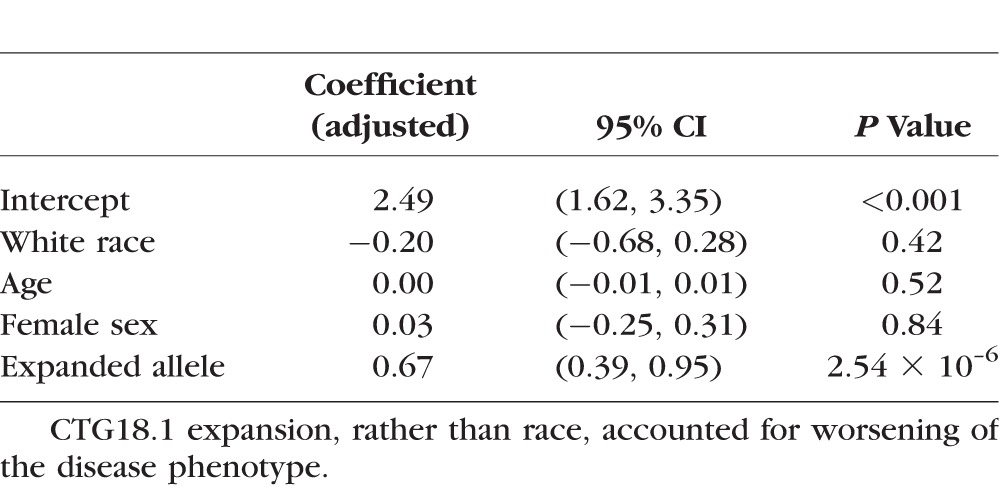
Table 2.
Characteristics of Participants With Fuchs' Dystrophy, by Self-Reported Race

Discussion
In this study, we demonstrate decreased odds of presenting with Fuchs' dystrophy in a large sample of African Americans presenting to a hospital system in the United States, and that self-reported black patients with Fuchs' dystrophy are less likely to harbor expansion of the CTG18.1 trinucleotide repeat in TCF4 compared to white affected individuals.
Rates of CTG18.1 trinucleotide repeat expansion have been shown to vary across populations globally, with 25 of 57 affected Chinese patients from Singapore10 and 15 of 44 Indian patients11 demonstrating the expanded repeat. These stand in contrast to data from the United States, in which a majority of white patients with Fuchs' dystrophy harbor CTG18.1 expansion.12
Given that a significant proportion of Fuchs' dystrophy among African Americans is likely related to factors beyond trinucleotide repeat expansion, alternative factors contributing to the disease state must be identified. These data suggest a role for additional variants in the causal pathway; previous research has found that mutations in SLC4A11, TCF8 and COL8A2 do not appear to the predominant contributors to disease in this population.13
Despite distinctions in the predominant genotype, both African American and white groups in this study were composed predominantly of female participants. These data are consistent with a cohort of patients with Fuchs' dystrophy previously reported that included a subset of 14 African-Americans; all were women, 2 of whom harbored repeat expansion.14
Although we found decreased odds of presenting with Fuchs' dystrophy in a tertiary care center among African American patients, both a lesser population prevalence or decreased severity may affect rates of presentation. While an analysis of Medicare codes has shown that FCD appears to be diagnosed less often among African-Americans, with the diagnosis of FCD attributed to 1.55% of white and 1.38% of black patients,5 Lorenzetti4 found a similar rate of black and white individuals with any degree of guttae but worse disease among white persons. We and others have previously shown that CTG18.1 expansion may affect clinical severity,8,14 including an increase in the need for cornea transplantation.15 Therefore, the data reported here may help clarify reports of decreased transplantation rates among African Americans with Fuchs' dystrophy.5
In both groups, we utilized triplet-primed PCR, a technique that has been shown to increase the accuracy of detecting large expanded alleles.16 However, given that some extremely large expansions may avoid detection even with this method, the proportions described here err on the side of a conservative estimate of rates of repeat expansion in each group. There was no substantial difference in rates of repeat expansion between cutoff values of 40 or 50 repeats.
Given that race is a social construct, it is important to clarify interpretability of data from this study. The information presented here reflects population samples and its application for counseling of any one individual reflects probability. The terms “black” and “white” were used to describe categories from clinical registration data; however, a broad set of ethnic groups identify as white or black, and significant genetic variability exists across populations within and between continents.17–19 Differences in the frequency of alleles in self-reported racial or ethnic groups reflect history and movement of populations18; prevalence rates will vary across subsequent years and generations. We use the term “African American” to describe this sample that is generated from an American population, with the understanding that allele frequency will likely vary if tested among self-identified black individuals across countries and continents.
Nevertheless, it is important to ensure that research is inclusive of diverse segments of the population. Given the promise of gene therapy in medicine, building an understanding of disease-associated genetic variants across populations allows scientists to ensure the broad applicability of future therapies; otherwise, benefits may be limited to the specific segment of the population previously studied.
In summary, African-Americans with Fuchs' dystrophy were less likely to demonstrate expansion of the CTG18.1 trinucleotide repeat in TCF4 relative to white participants in this study. The findings carry implications for understanding perceived population differences in severity and prevalence.
Acknowledgments
Supported by National Institutes of Health K12 EY015025-10 (AOE), R01 EY016835 (JDG), and L30 EY024746 (AOE).
Disclosure: A.O. Eghrari, None; S. Vahedi, None; N.A. Afshari, None; S.A. Riazuddin, None; J.D. Gottsch, None
References
- 1. Eye Bank Association of America. 2015. Eye Banking Statistical Report. Available at: http://restoresight.org/wp-content/uploads/2016/03/2015-Statistical-Report.pdf.
- 2. Kopplin LJ, Przepyszny K, Schmotzer B,et al. Relationship of Fuchs' endothelial corneal dystrophy severity to central corneal thickness. Arch Ophthalmol. 2012; 130: 433– 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Eghrari AO, Gottsch JD. . Fuchs' corneal dystrophy. Expert Rev Ophthalmol. 2010; 5: 147– 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lorenzetti DW, Uotila MH, Parikh N, Kaufman HE. . Central cornea guttata. Incidence in the general population. Am J Ophthalmol. 1967; 64: 1155– 1158. [PubMed] [Google Scholar]
- 5. Mahr MA, Baratz KH, Hodge DO, Erie JC. . Racial/ethnic differences in rates of penetrating or endothelial keratoplasty for Fuchs' endothelial corneal dystrophy among US Medicare beneficiaries. JAMA Ophthalmol. 2016; 1134: 1178– 1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sugar A, Tanner JP, Dontchev M,et al. Recipient risk factors for graft failure in the cornea donor study. Ophthalmology. 2009; 116: 1023– 1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Whitham LB. . Clinical contribution to the etiology of dystrophia epithelialis corneae. Trans Am Ophthalmol Soc. 1924; 22: 290– 299. [PMC free article] [PubMed] [Google Scholar]
- 8. Krachmer JH, Purcell JJ Jr, Young CW, Bucher KD. . Corneal endothelial dystrophy. A study of 64 families. Arch Ophthalmol. 1978; 96: 2036– 2039. [DOI] [PubMed] [Google Scholar]
- 9. Vasanth S, Eghrari AO, Gapsis BC,et al. Expansion of CTG18.1 Trinucleotide repeat in TCF4 is a potent driver of Fuchs' corneal dystrophy. Invest Ophthalmol Vis Sci. 2015; 56: 4531– 4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xing C, Gong X, Hussain I,et al. Transethnic replication of association of CTG18.1 repeat expansion of TCF4 gene with Fuchs' corneal dystrophy in Chinese implies common causal variant. Invest Ophthalmol Vis Sci. 2014; 55: 7073– 7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nanda GG, Padhy B, Samal S, Das S, Alone DP. . Genetic association of TCF4 intronic polymorphisms, CTG18.1 and rs17089887, with Fuchs' endothelial corneal dystrophy in an Indian population. Invest Ophthalmol Vis Sci. 2014; 55: 7674– 7680. [DOI] [PubMed] [Google Scholar]
- 12. Wieben ED, Aleff RA, Tosakulwong N,et al. A common trinucleotide repeat expansion within the transcription factor 4 (TCF4, E2-2) gene predicts Fuchs' corneal dystrophy. PLoS One. 2012; 7: e49083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Minear MA, Li YJ, Rimmler J,et al. Genetic screen of African Americans with Fuchs' endothelial corneal dystrophy. Mol Vis. 2013; 19: 2508– 2516. [PMC free article] [PubMed] [Google Scholar]
- 14. Soliman AZ, Xing C, Radwan SH, Gong X, Mootha VV. . Correlation of severity of Fuchs' endothelial corneal dystrophy with triplet repeat expansion in TCF4. JAMA Ophthalmol. 2015; 133: 1386– 1391. [DOI] [PubMed] [Google Scholar]
- 15. Eghrari AO, Vasanth S, Wang J, Vahedi F, Riazuddin SA, Gottsch JD. . CTG18.1 expansion in TCF4 increases likelihood of transplantation in Fuchs' corneal dystrophy. Cornea. 2017; 36: 40– 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Singh S, Zhang A, Dlouhy S, Bai S. . Detection of large expansions in myotonic dystrophy type 1 using triplet primed PCR. Front Genet. 2014; 5: 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tishkoff SA, Reed FA, Friedlaender FR,et al. The genetic structure and history of Africans and African-Americans. Science. 2009; 324: 1035– 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. O'Dushlaine C, McQuillan R, Weale ME,et al. Genes predict village of origin in rural Europe. Eur J Hum Genet. 2010; 18: 1269– 1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McEvoy BP, Lind JM, Wang ET. . Whole-genome genetic diversity in a sample of Australians with deep aboriginal ancestry. Am J Hum Genet. 2010; 87: 297– 305. [DOI] [PMC free article] [PubMed] [Google Scholar]