

ABSTRACT
In β cells, stimulation by metabolic, hormonal, neuronal, and pharmacological factors is coupled to secretion of insulin through different intracellular signaling pathways. Our knowledge about the molecular machinery supporting these pathways and the patterns of signals it generates comes mostly from rodent models, especially the laboratory mouse. The increased availability of human islets for research during the last few decades has yielded new insights into the specifics in signaling pathways leading to insulin secretion in humans. In this review, we follow the most central triggering pathway to insulin secretion from its very beginning when glucose enters the β cell to the calcium oscillations it produces to trigger fusion of insulin containing granules with the plasma membrane. Along the way, we describe the crucial building blocks that contribute to the flow of information and focus on their functional role in mice and humans and on their translational implications.
KEYWORDS: human, mouse, pancreas, islet of Langerhans, β cell, ion channel, membrane potential, calcium, oscillations, triggering pathway, translation
Introduction
Insulin secreted by pancreatic β cells regulates postprandial storage and interprandial usage of energy rich nutrients. A relative or absolute lack of insulin results in diabetes mellitus, a disease that affects more than 400 million people around the world, and this number is expected to increase to 640 million by 2040.1 The burden of this disease is immense and includes public health costs of treating diabetic patients, as well as personal costs related to serious complications.2 More than 90% of all diabetics have type 2 diabetes mellitus (T2DM), which is characterized by obesity, insulin resistance, and eventually insufficient insulin secretion.3 A genetic susceptibility, age, obesity, dietary habits, and a sedentary lifestyle most importantly contribute to the development of the disease.4 It has been shown that the progressive nature of T2DM can be slowed down upon or reversed by lifestyle interventions.5,6 However, pharmacological agents are still prescribed to practically all patients, especially later during the course of the disease, to increase the deficient secretion of insulin from β cells, to improve its action on target tissues, or promote urinary glucose excretion by inhibiting its renal reuptake.7
β cells couple stimulation by metabolic, hormonal, neuronal, and pharmacological factors to insulin secretion by at least 3 interconnected intracellular signaling pathways (Fig. 1). After more than 4 decades of intensive research, by far the most investigated one is the so-called triggering pathway. It consists of several events starting with influx of glucose through glucose transporters, glucose metabolism increasing intracellular ATP concentration, and triggering closure of ATP-dependent K+ channels (KATP channels). The resulting decrease in K+ efflux causes membrane depolarization, the consequent opening of voltage dependent calcium (Ca2+) channels (VDCC), and influx of Ca2+ ions. The resulting increase in the cytosolic concentration of Ca2+ ([Ca2+]C) is tightly coupled to membrane potential changes and typically follows a characteristic temporal pattern in the form of oscillations, activating the secretory machinery and fusion of insulin-containing vesicles with plasma membrane.8 This triggering pathway is essential, i.e. necessary, but not very efficient without so-called amplifying pathways which mainly affect the sensitivity of the secretory machinery to [Ca2+]C. More specifically, there seem to be a KATP-independent Ca2+-dependent and a KATP-independent Ca2+-independent amplifying pathway.9,10 In addition, non-nutrient secretagogues, i.e., neurotransmitters and hormones, activate membrane receptors and set into motion protein kinase A- and protein kinase C-dependent pathways (Fig. 1). To complicate things further, both glucose- and non-nutrient secretagogues shape the triggering signal by influencing uptake and release of Ca2+ in intracellular stores.11
Figure 1.
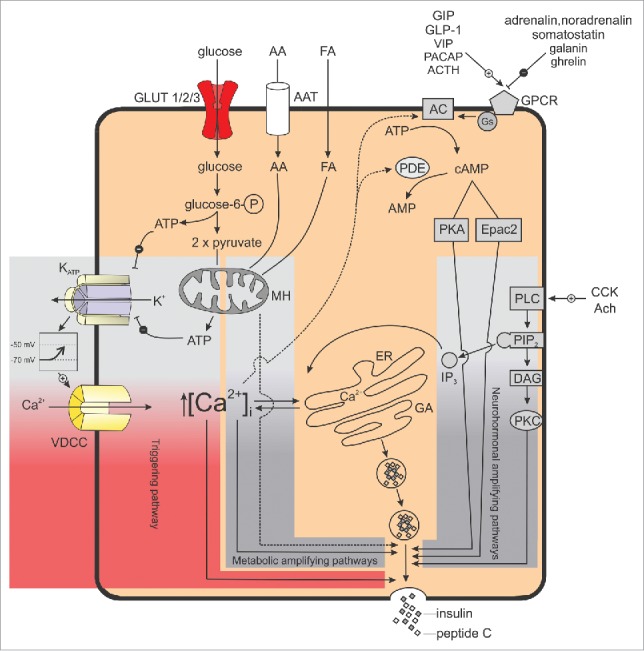
The three interconnected intracellular signaling pathways in pancreatic β cells. The KATP-dependent triggering pathway is indicated in red, the metabolic and neurohormonal amplifying pathways are indicated in gray.
A large proportion of our knowledge about the abovementioned pathways comes from work on rodents, especially mouse models. Although the human and the mouse pancreas and islets of Langerhans share many common anatomic and physiologic characteristics, they are not the same.12,13 During the last decade, islets obtained from humans have been increasingly used and studies suggested many important functional differences between the human and the mouse β cells, so far mostly for the triggering pathway. Validating findings obtained in mice and studying functional similarities and differences between β cells from humans and mice may help us understand why some therapeutic and other interventions do not yield the expected results and to better choose the experimental approach when looking for new therapeutic targets. Thus, in the present review we aim to highlight some similarities and differences between the two species at different stages of the triggering pathway, from the most proximal event, i.e., entry of glucose into the β cell, to the most distal changes in [Ca2+]C (Table 1).
Table 1.
Summary of differences in the triggering pathway between mouse and human β cells.
| Species | Mouse | Human |
|---|---|---|
| Glucose transporter | GLUT2 Km≈17 mM |
GLUT1 and GLUT3 Km≈7 mM and 1.8 mM |
| Hexokinase | Glucokinase | Glucokinase |
| ATP-dependent K+ channels | Kir6.2 and SUR1 | Kir6.2 and SUR1 Kir2.1 |
| Transient receptor potential channels | TRPC1 TRPV2 TRPA1 TRPM2, TRPM3, TRPM4, TRPM5 |
TRPV4 TRPM2, TRPM4, TRPM5 |
| Voltage-dependent Ca2+ channels | CaV1.2 (L-type) - >50% CaV2.3 (R-type) - 25% CaV2.1 (P/Q-type) - 15% of total VDC currents |
CaV2.1 (P/Q-type) - 40 - 45% CaV1.2 and CaV1.3 (L-type) - 40 - 45% CaV3.2 (T-type) - 10 - 20% of total VDC currents |
| Voltage-dependent Na+ channels | NaV1.7 - 85% and NaV1.3 - 15% of total Na+ currents | NaV1.6 - 75% and NaV1.7 - 25% of total Na+ currents |
| Voltage- or calcium-dependent K+ channels | Delayed rectifying K+ channels (KV2.1) Small conductance Ca2+-activated K+ channels (SK4) ERG1 |
BK channels Delayed rectifying K+ channels (KV2.2) Small conductance Ca2+-activated K+ channels (SK3 and SK4) hERG1 |
| Pattern of membrane potential oscillations | Bursts of APs (spikes), glucose dependent, continuous firing of APs observed at higher glucose concentrations (above 16–20 mM) | Continuous or irregularly spaced APs without clear bursts or a more organized oscillatory electrical activity (for details see Table 2). |
| Coupling between β cells | Tight junctions and gap junctions (Cx36) The junctional conductance between cell pairs is estimated at about 200–350 pS. |
Tight junctions and gap junctions (Cx36) A coupling conductance of 100–200 pS is suggested. |
| Pattern of [Ca2+]C oscillations | Globally synchronized [Ca2+]C oscillations. | Globally or locally synchronized [Ca2+]C oscillations or no oscillations observed (for details see Table 3). |
Glucose transporters
Influx of glucose into the β cell through glucose transporters is the first step in GSIS and in the series of signaling events collectively termed stimulus-secretion coupling (SSC). In both mice and humans, glucose enters the β cell cytosol by facilitated diffusion through insulin-independent membrane-bound glucose transporters (GLUTs). Most of the glucose transporters expressed in mouse β cells are GLUT2, the type of carrier proteins with the lowest binding affinity and highest transport capacity for glucose (Km ≈17 mM).14 The glucose transport rate is ≈20-50-fold greater than the rate of glucose phosphorylation by glucokinase.14 Genetic inactivation of GLUT2 suppresses glucose uptake and GSIS in mouse pancreatic β cells.15
In human β cells on the other hand, GLUT2 are almost completely dispensable, since GLUT1 and GLUT3 are far more abundantly expressed.16 Uptake of 3-O-methylglucose was estimated at 2.2 and 2.9 mmol/min per liter intracellular space in humans17 and at 3.0 and 4.8 mmol/min per liter intracellular space in rats at 5 and 10 mM glucose, respectively.18 However, these data can hardly be directly compared since glucose uptake in human β cells was measured at 37°C while in rat β cells, experiments were performed at 15°C to slow down the process sufficiently to allow for accurate calculations. Glucose transport in mouse β cells has been assessed with a different protocol and compared with the rate in human β cells using the fluorescent glucose analog 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxy-D-glucose (2-NBDG). The uptake of 2-NBDG in 10 mM glucose at 37°C was approximately 0.8 nmol/min/5 × 104 cells in mice and approximately 0.9 nmol/min/1 × 105 cells in humans.19
The GLUT1 protein (encoded by SLC2A1 gene) displays 97.2% homology1 between humans and mice and plays a principal role in human islets, since Km in GLUT1 is lower (≈7 mM) compared with GLUT2 (SLC2A2 gene, 82% homology) and more compatible with the dose-response curve for GSIS in human β cells. One of the most striking differences between mouse and human GLUT expression is the distribution of GLUT3 (SLC2A3 gene, 83.5% homology), which has the highest affinity to glucose (Km ≈1,8 mM) among all 3 glucose transporters expressed in pancreatic β cells. In mice, GLUT3 is expressed only at a background level, whereas it is as abundant as GLUT1 in human β cells, suggesting an equally important role for both transporters in the regulation of insulin secretion in humans.20 Although GLUT 1 and GLUT3 are far more abundant in human β cells than GLUT2, the latter still seems necessary for normal GSIS. Namely, mutations in the GLUT2 gene cause the Fanconi-Bickel syndrome manifested by hepatomegaly, growth retardation, and renal syndrome.21 Impaired GSIS in adults has only been reported in a few cases of Fanconi-Bickel syndrome.22 On the other hand, a transient absolute requirement for GLUT2 for the control of insulin secretion in the first months of life exists, since neonates with Fanconi-Bickel syndrome have transient diabetes mellitus that disappears after 18 months of age.23
Cytosolic and mitochondrial metabolism
Rapid entry of glucose through glucose transporters ensures glucose sensing, meaning that the rate of glucose entry is proportional to blood glucose levels and it accounts for fast equilibration of glucose between the extracellular space and the cytosol. This enables glucose unrestrained access to glucokinase (GCK), the hexokinase isozyme found in both mouse and human β cells (GCK gene, 95.9% homology) that catalyzes the rate-controlling step in glucose metabolism.24 GCK has more than a 20-times lower affinity for glucose compared with other hexokinases (Km ≈6–11 mM) and is not inhibited by its product, glucose-6-phosphate.25 Km values in mouse and humans are very similar, reported at 7 mM and 7.9 mM, respectively.26 Its sigmoidal glucose dependency guarantees optimal responsiveness at physiological glucose levels, since the inflection point of the sigmoidal curve (4-5 mM) is close to the physiological threshold for GSIS in β cells. Functionally, this positive cooperativity with glucose allows the enzyme to have increased sensitivity to fluctuations in blood glucose levels.27 The values of Hill coefficients in mice and humans are practically identical (≈1.7).26 At 5 mM and 10 mM glucose glucokinase activity was 0.38 and 0.60 mmol/min per liter intracellular space in nondiabetic human β cells, respectively,17 and similar to that seen in rodent β cells.28 In another study, the rate of glycolysis in 6 and 12 mM glucose was estimated at ≈40 and ≈90 pmol/islet/h in mouse, respectively, and at ≈125 and ≈160 pmol/islet/h in human islets, respectively.29
GCK exerts a unique regulatory role in β cell metabolism because the reaction it catalyzes is virtually irreversible.26,27 The functional reserve of GCK is low in β cells, since a decrease of only ≈50% of enzyme activity is tolerated.30,31 Therefore, GCK expression is indispensable for SSC in β cells. It has been demonstrated that homozygous GCK knockout mice develop lethal hyperglycemia.32 In humans, the majority of approximately 200 known mutations in the GCK gene, including missense, non-sense, and splice site mutations, result in mild hyperglycemia that is usually discovered by routine examination. Based on these characteristics, diabetes associated with GCK mutations was included in the group of maturity onset diabetes of the young (MODY) and named MODY 2.33 These mutations result in a GCK molecule that is less sensitive or less responsive to glucose. β cells in patients with these mutations have a normal ability to produce and secrete insulin, but expectedly a higher threshold (7-8 mM) for GSIS. In rare cases, when infants inherit mutations from 2 heterozygous parents, permanent neonatal diabetes mellitus can occur.34 On the other hand, heterozygous activating mutations can lead to varying degrees of hyperinsulinemia and hypoglycemia.35 These observations are the main reason GCK activators have been studied for more than a decade. Systemic GCK activators alter GCK's affinity to glucose and thereby decrease the threshold for GSIS, but their main drawback is their potential to provoke hypoglycemic episodes. Therefore, hepatoselective GCK activators that can overcome this side effect seem more appropriate candidates for treating T2DM.36,37
Since GCK controls the rate-limiting step in glucose metabolisms, its activity partially determines the pattern of [Ca2+]C oscillations and the consequent pulsatile insulin secretion from β cells. It has been suggested that the slower component of compound oscillations with a period of approximately 5–15 minutes reflects oscillations in glycolysis, whereas the fast component is due to electrical activity in pancreatic β cells (see below). The enzyme downstream from GCK, phosphofructokinase (PFK) with positive feedback regulation by its product fructose bisphosphate, is believed to be directly responsible for oscillatory glycolysis (and slow oscillations). However, if the glucokinase flux rate is too low or too high, glycolysis will be non-oscillatory. In other words, glycolytic oscillations occur only at intermediate levels of GCK flux rate.38,39
Evidence for the molecular events linking glucose-6-phosphate to ATP production in mice and humans is scarce. Glucose-induced hyperpolarization of the mitochondrial inner membrane in mice40 is consistent with the production of pyruvate, its translocation into the mitochondrion, oxidative phosphorylation, activation of the electron transport chain, and translocation of protons across the mitochondrial membrane.41,42 Subsequent transient increase in the mitochondrial [Ca2+] is the consequence of a tightly regulated interplay between Ca2+ entry on one hand and Ca2+ buffering in the matrix and extrusion on the other hand.41-47
Mitochondrial [Ca2+] changes are tightly coupled to [Ca2+]C dynamics, at least in mice.45,48-51 Glucose stimulation evokes an oscillatory behavior of mitochondrial [Ca2+]45,50,51 that is synchronized across cells in an islet.50 The matrix signal seems to be delayed in respect to [Ca2+]C in mice,45,48,51 implying a modulatory role of [Ca2+]C on its matrix counterpart. The latter is corroborated by the observation that the frequency of electrically induced [Ca2+]C modulates the amplitude of matrix [Ca2+], such that matrix [Ca2+] closely follows slow changes in [Ca2+]C (which occur in slow bursting, see below), whereas at high frequencies of cytosolic Ca2+ oscillations, matrix [Ca2+] is unable to follow and the frequency of cytosolic oscillations is decoded as the amplitude of increases in matrix [Ca2+].49,52 Further, there is some evidence that matrix Ca2+ modulates its cytosolic counterpart as impairment of Ca2+ efflux from the mitochondrion reduced glucose-induced cytosolic Ca2+ response in mice.53 On the other hand, impairing Ca2+ entry into the mitochondrion in mice failed to do so.48 A detailed recent study revealing many aspects of coupling between glucose and mitochondrial metabolism in human islets unfortunately did not compare cytosolic and matrix [Ca2+] changes and thus at present, a direct comparison between the 2 species in this regard is impossible. However, the glucose- and calcium- dependent changes in ATP synthesis and insulin release indicate that the functional role of mitochondria in the signaling cascade is comparable in the two species.54 Along this line of reasoning, we wish to point out a recent finding that is of relevance for what was stated above but also for later parts of this paper. Cytosolic ATP concentration ([ATP]C) dynamics induced by the incretin hormone GLP-1 involved changes in mitochondrial potential and were found to be much better correlated between different cells in mouse than in human islets.55 This is in contrast with the degree of GLP-1-induced coupling of ionic events, e.g., [Ca2+]C changes, between β cells in human islets56 (see below) and might also indicate a different degree of coupling between [Ca2+]C and matrix [Ca2+]/mitochondrial metabolism in the two species.
ATP-dependent potassium channels
Increased β cell glycolytic and tricarboxylic acid (TCA) cycle activity lead to an increased [ATP]C and decreased ADP concentration ([ADP]c). In low glucose, [ATP]C is low and the KATP channel open probability is high, resulting in K+ efflux that keeps the cell membrane hyperpolarized and prevents insulin secretion. At supratreshold glucose, [ATP]C increases due to increased metabolism, thus lowering open probability of the KATP channels. The ensuing decrease in K+ efflux is co-responsible (together with the putatively unchanged leak conductance, see later) for depolarization and initiation of electrical activity.57,58
β cell KATP channels are members of the inwardly rectifying K+ channel family, composed of 4 potassium-selective pore-forming Kir6.2 subunits (KCNJ11 gene, 95.9% homology) and 4 regulatory SUR1 subunits (ABCC8 gene, 95.4% homology).59 The regulatory subunits are termed SURs because they bind sulfonylureas and therefore play a key role in determining the pharmacological regulation of KATP channels. Because KATP channels transport K+ with a greater tendency into a cell then out, they are named inward rectifiers. The assembly and trafficking of the channels has been described to be precisely regulated. Assembly occurs in the endoplasmic reticulum (ER) and only completely assembled and full length complexes are then transported to the plasma membrane and expressed on the surface, while incompletely assembled complexes are retained in the ER.60
KATP channel subunits display differences in their sensitivity to adenosine nucleotides. Intracellular ATP acts on Kir6.2 to decrease channel open probability, while MgADP increases channel open probability through the SUR1 subunit, a member of the ATP-binding cassette superfamily.61 Therefore, changes in [ATP]C and [ATP]C/[ADP]C are believed to determine channel activity.62 However, only fully assembled KATP channels have a normal sensitivity to ATP. The Kir6.2 isoform alone forms a weekly ATP-inhibited KATP channel (IC50 ≈100–200 µM), while reconstitution with SUR1 increases the affinity to inhibitory ATP (IC50 ≈5–10 µM).63 Since KATP channels are not voltage dependent, their current-voltage relation is linear, although the current increases very little at membrane potential above +20 mV. This primarily results from a voltage-dependent block of outward K+ currents by internal Na+ and Mg2+.64 The kinetic studies of KATP channels showed channel openings grouped in bursts. The dominant effect of ATP is to reduce the number of openings per burst and to prolong the time when the channel is closed. Furthermore, high concentrations of phosphatidylinositol phosphate (PIP2) reduce the sensitivity of KATP channels to inhibitory ATP by increasing the number of KATP channel openings.65 The kinetics of the KATP channel in human β cells qualitatively resemble those reported for rodent β cells.58,66
Due to the critical role KATP channels play in insulin secretion, it is not surprising that polymorphisms in KATP subunits alter the biophysical properties of the channel and can lead to both hypo- and hyperglycemia.67 In humans, KATP channel mutations that result in decreased K+ currents cause persistent hyperinsulinemic hypoglycemia of infancy (PHHI).68 Mutations can be found in either of the subunits that make up KATP channels. Mild forms of PHHI associated with mutations in SUR1 produce KATP channels that can be inhibited by ATP, but only poorly stimulated with MgADP. Severe forms of the disease have been associated with mutations that truncate SUR1 and block expression of the KATP channels on the cell membrane. However, remarkable physiologic differences exist between human neonates missing KATP channels and the KATP channel knockout mice (Kir6.2−/− or SUR6.1−/−).61 In both PHHI and KATP channel null mice loss of K+ currents causes an elevated membrane potential and consequently increased [Ca2+]C, leading to an inappropriately high rate of insulin release and producing hypoglycemia. As in PHHI, KIR6.2−/− and SUR1−/− mice display hypoglycemia immediately after birth, however their hypoglycemia is transient and persists only during the first 2–3 days of life.69 It seems that KATP channel null mice become refractory to increased [Ca2+]C and develop a KATP-independent mechanism for regulating insulin release. During adulthood, PHHI patients are hyperglycemic with high prevalence of diabetes, while KATP channel null mice display normal insulin and glucose levels when fed ad libitum, and are hypoglycemic upon fasting and exhibit a mild intolerance toward i.p. injection of glucose. SUR1−/− islets lack the first-phase of insulin secretion and have an attenuated second-phase after stimulation with high glucose, while Kir6.2−/− islets have a small first-phase of insulin secretion with no second phase.61
Conversely, gain-of-function mutations result in neonatal diabetes characterized by an insulin secretory deficit and hyperglycemia. The first indication that overactive KATP channels can produce neonatal diabetes came from transgenic mice expressing a Kir6.2 subunit lacking a segment of its N-terminus responsible for channel gating. Its deletion resulted in nearly constantly open KATP channels that have a reduced sensitivity to ATP and hypoglycemic sulfonylureas.70 Severe hyperglycemia is lethal within first weeks after birth.71 In humans, missense activating mutations associated with neonatal diabetes were also found in the gene encoding the Kir6.2 subunit of the KATP channel (KCNJ11).67 Furthermore, activating mutations in SUR1 in mice and humans directly enhance MgATP activation of KATP channel or indirectly alter channel gating and reduce ATP inhibition at Kir6.2.72,73
Leak channels
The consensus model of SSC predicts that closure of KATP channels triggers membrane depolarization. However, according to the Nernst and Goldman-Hodgkin-Katz equations, closure of KATP channels alone is not sufficient for moving the membrane potential away from the equilibrium potential for K+, as long as the membrane is permeable to K+ only. Therefore, the presence of an additional inward current is needed to achieve depolarization by reducing K+ permeability. Since the input resistance of β cells upon closure of KATP channels is increased, the current needed for depolarization is likely small, however the identity of this current and its properties have not yet been fully elucidated. The most likely ion channel candidates for depolarizing and hypepolarizing currents can be classified in at least 4 different groups, transient receptor potential (TRP) channels, 2-pore domain potassium or K2P channels, NALCN channels and connexins. Unstimulated β cells are to some extent permeable to Na+ and Ca2+ without activation of voltage-dependent Na+ channels and VDCCs.10
TRP channels are candidates for Na+ or Ca2+ influx contributing to the depolarizing current. The number of different TRP channels expressed in β cells is large (TRPC1, TRPC4, TRPV1, TRPV2, TRPV4, TRPA1, TRPM2, TRPM3, and TRPM5)74 and is likely to increase (Fig. 2). The channels are to some extent differentially expressed in β cells of different species. In the following lines, only a few examples will be listed. On the one hand, they translocate to plasma membrane upon glucose stimulation and stimulation with insulin or insulin-like growth factors (TRPV2), resulting in Ca2+ influx and increased insulin secretion.75 This positive feedback to increase insulin secretion may result in hyperinsulinemia, commonly found at early stage of type 2 diabetes. On the other hand, knockdown of a specific insulin receptor attenuated insulin-induced translocation of TRPV2 and knockdown of TRPV2 channels and reduces GSIS.75
Figure 2.
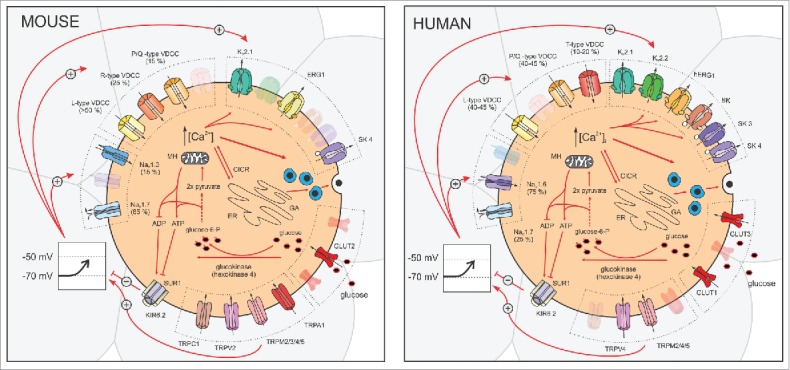
Ion channels involved in the triggering pathway of glucose-induced insulin secretion in mouse (left) and human (right) β cells.
In addition to glucose, other activators like islet amyloid polypeptide (TRPV4),76 inflammatory mediators, glibenclamide (TRPA1),77 pregnenolone sulfate, as well as clotrimazole and tamoxifen and structurally related compounds (TRPM3),78-80 or steviol glycosides (TRPM5)81 can enhance β cell function. Among all TRP channels present in β cells, the TRPM5 seems to play the most important role in insulin secretion since TRPM5 knockdown mice showed significantly reduced Ca2+-activated nonselective cation current. Furthermore, glucose-induced oscillations of membrane potential and [Ca2+]C were reduced, particularly due to a lack of fast Ca2+ oscillations.81 Consequently, glucose-induced insulin secretion from TRPM5 knock-down mice was reduced, resulting in impaired glucose tolerance.81,82
Lately, another group of hyperpolarizing currents have entered the stage as fine tuners of GSIS, namely K2P channels. Inhibition of the 2-pore-domain acid-sensitive potassium channel (TASK-1) significantly stimulates both human and mouse β cells.83 Likewise, ablation of TWIK-related alkaline pH-activated K2P (TALK-1) also seems to result in β cell membrane depolarization.84 These studies suggest that these 2 K2P channels play a role in limiting glucose-stimulated depolarization and insulin secretion. At least in the case of TALK-1, an intracellular binding partner, osteopontin, has recently been described, however its function is not well understood.85 Another candidate responsible for membrane depolarization after closure of KATP channels is the so called voltage-independent non-selective Na+ leak channel (NALCN),86 but its precise role in insulin secretion from mouse and human β cells still has to be elucidated.
Finally, with KATP and other K+ channels closed, the mere presence of electrical coupling between β cells via Cx36 gap junctions can serve as a source of possible electrical interference. More specifically, currents from less or more excitable neighboring cells can respectively hyperpolarize or depolarize a given cell, leading to a complex electrical activity pattern.87-91
Voltage-dependent calcium channels
Since both human and mouse β cells maintain a resting membrane potential at -70 mV or lower, closure of KATP channels and the subsequent decrease in K+ efflux depolarize the membrane. When membrane potential exceeds the threshold potential (≈ -50 mV), the open probability for voltage dependent calcium channels (VDCCs) increases.
A number of differences exist between mouse and human VDCCs (Fig. 2). In mouse β cells, whole-cell Ca2+ currents are activated by depolarization to potentials more positive than -50 mV, increase to a maximum at about -20 mV, and reverse at about +50 mV (in the presence of 2.6 mM Ca2+).92 In human β cells, the Ca2+ current can be measured during depolarization to -50 mV and above with maximal amplitude at 0 mV.93 The major VDCC subtype expressed in mouse β cells are L-type Ca2+ channels (Cav1.2), which account for more than 50% of all Ca2+ currents. Pharmacological inhibition and genetic ablation of the channel result in a severe reduction of GSIS during both first and second phase.94,95 Although L-type Ca2+ channels play a major role in GSIS, this is not the only type of VDCCs expressed in mouse β cells. Around 25% of the Ca2+ current in mouse β cells can be attributed to R-type VDCCs (Cav2.3), which have an important role in GSIS during the second phase of insulin release, since pharmacological inhibition or genetic ablation of R-type VDCCs results in an unaffected first phase but strongly reduced second phase of insulin secretion.96 Besides the aforementioned types of VDCCs, P/Q-type VDCCs (Cav2.1) are responsible for the remaining 15–25% of Ca2+ currents.94,95
Human β cells, on the other hand, do not express R-type VDCCs but T-type VDCCs (Cav3.2), although it was demonstrated that polymorphisms in the gene encoding R-type VDCC (Cav2.3) associate with type 2 diabetes and impaired insulin secretion.97 Furthermore, P/Q-type VDCCs (CACNA1A gene, 92,2% homology) and L-type VDCCs (CACNA1C gene, 95.1% homology) contribute roughly equally (40-45%) to the Ca2+ current in human β cells.98,99 In contrast to mice, human β cells possess both Cav1.2 and Cav1.3 L-type VDCCs. Since L-type VDCCs in human β cells activate quickly during depolarization to membrane potentials above -40 mV, Cav1.3 seems to be the dominant isoform expressed in human β cells.93 L-type VDCCs, but not P/Q-type VDCCs, undergo Ca2+ dependent inactivation.98 T-type VDCCs in human β cells is the only low-voltage activated calcium channel activated already at -60 mV, compared with high voltage-activated L-type and P/Q-type channels. T-type VDCCs reach their peak conductance at -30 mV and undergo fast voltage-dependent inactivation, which is half-maximal at -65 mV.98 Differences between the roles VDCCs play in mouse and human β cells are illustrated in Fig. 2 and 3.
Figure 3.

Ion channels involved in glucose-induced electrical activity in mouse (left) and human (right) β cells. Numbers indicate ion channels responsible for the slow depolarization phase, the upstroke and the repolarization phase of an individual spike (action potential) and for the repolarization phase between bursts.
Voltage-dependent sodium channels
Mouse β cells possess NaV1.3 (Scn3a) and NaV1.7 (Scn9a) channels, with NaV1.7 being the quantitatively more important type. Knockout of Scn9a lowers Na+ current by >85%, disclosing a small Scn3a-dependent component.100 Inactivation of NaV1.7 current is half-maximal at ≈ −105 mV while inactivation of NaV1.3 current is half-maximal at ≈ −50 mV and thus the latter likely contributes to action potential (AP) firing. A contribution of Na+ channels to AP firing in mouse β cells is further corroborated by the finding that the specific sodium channel blocker tetrodotoxin (TTX) reduces the height of APs.101 Scn3a is obviously important in insulin secretion, since genetic ablation of Scn3a reduces insulin secretion, while genetic ablation of Scn9a does not affect insulin secretion.100 Taken together, these data suggest that NaV1.3 represents the functionally more important Na+ channel subtype in mouse β cells.
Human β cells also possess voltage-dependent Na+ channels that are responsible for the AP upstroke102 (Fig. 3). The total Na+ current in human β cells activates instantaneously by depolarization to -30 mV and above, reaches a maximal amplitude at 0 mV and inactivates with a time constant of 2 ms.103 Sodium channels in human β cells undergo voltage-dependent steady-state inactivation that is half-maximal at -40 mV.93,99,101 This is >50 mV more positive than in mouse β cells where the quantitatively more abundant Snc9a sodium channels are fully inactivated already at the resting membrane potential (-80 mV) and therefore, in contrast to humans, cannot effectively contribute to the oscillatory pattern of membrane potential (see below). NaV1.6 channels probably account for the major part of Na+ current in human β cells, and they are important for GSIS since secretion elicited by 6 and 20 mmol/l glucose is reduced by 70% and 55%, respectively, in the presence of TTX.93 Furthermore, TTX reduces the AP amplitude by ∼10% and prolongs their duration.104,105 Like in mice, human β cells also express NaV1.7 channels (SCN9a gene, 92% homology), but they account for only 25% of all Na+ channel transcripts,106 inactivate at hyperpolarized voltage (≈ -105 mV), and are thus not involved in AP firing.100
Voltage- or calcium-dependent potassium channels
In human β cells, the voltage-dependent potassium current consist of at least 2 different components.93 BK channels which show both voltage- and Ca2+-dependence107 are responsible for a transient component activated rapidly upon membrane depolarization above -40 mV and with peak amplitude at +30 mV.93 Above +30 mV, amplitude decreases due to reduced Ca2+ entry. Blockade of BK channels increases AP amplitude in human β cells, while in mouse β cells BK channels do not seem to play a major role in electrical activity and insulin secretion.108 The second component of voltage-dependent potassium current is due to delayed rectifying K+ channels. In mouse β cells mainly KV2.1 are expressed, while in human β cells mRNA expression levels suggest that KV2.2 channels are more abundantly expressed than KV2.193 but latter has not been confirmed by electrophysiological measurements. In KV2.1 null mice, glucose-induced AP duration is prolonged while the firing frequency (see below) is reduced.109 Genetic silencing or pharmacological inhibition of KV2.1 revealed > 70% reduction of KV currents comparing to those from control experiments.109-111 Furthermore, ablation of KV2.1 improves glucose tolerance and enhances insulin secretion in mice.109,110 On the other hand, pharmacological inhibition of KV2.x currents failed to reduce glucose levels in vivo, moreover a significant reduction of KV2.2 mRNA levels were observed and glucose-stimulated somatostatin release was enhanced. The later seems to be a reason for paracrine inhibition of insulin secretion and no significant effect on blood glucose lowering in vivo.110 The same mechanisms might be involved in insulin secretion from human β cells since KV2.x inhibitors, like GxTX-1E and Ry796, profoundly enhance GSIS and somatostatin release in human islets in vitro.110 Furthermore, pharmacological inhibition of KV2.1/2.2 currents using whole-cell patch-clamp suppresses KV currents in human β cells93,110 while has a week effect on electrical activity; it increases the half-width of APs, while it has no effect on action potential amplitude.93 Regarding the effect on insulin secretion, the impact of Kv2.1/2.2 in human β cells is controversial. While previously mentioned study clearly shows enhanced GSIS during pharmacological inhibition of Kv2.1/2.2, no effect on insulin secretion is found in stromatoxin treated islets.93 This different effect on insulin secretion and AP firing between mouse and human β cells may be due to the longer AP duration in mouse compared with human β cells, which results in a greater amount of KV2.1 current being activated.112 In human β cells the time constant of activation measured at -20 mV when BK channels are active and when KV2.1/2.2 channels are active were ≈2 ms and >10 ms, respectively.98 This indicates that KV2.1/2.2 channels do not even activate during the AP upstroke (Fig. 3) and therefore blocking them does not affect electrical activity and insulin secretion.98
In addition to KV2.1 and KV2.2 channels, KV11.1 (ERG channels) are expressed in mouse and human β cells (KCNH2 gene is also named ether-a-go-go-related gene, 96.4% homology). Selective blockage of hERG K+ channels increased firing frequency and insulin secretion in human β cells.113 Similarly, blocking ERG1 increases [Ca2+]C and therefore promotes insulin secretion also in mouse β cells.114 Furthermore, ERG channels are responsible for long-lasting tail currents and may thus influence the intervals between spikes.98 In the heart, hERG1 channels are the molecular basis for the so called long QT-syndrome, and some genetically determined hyperinsulinemias might involve mutations of hERG channels.113
Beside the abovementioned voltage-dependent potassium channels, both mouse and human β cells also possess small conductance (SK) Ca2+-activated K+ channels98,112 (Fig. 2). SK3 and SK4 channels were found in human β cells.98 Their activity is proposed as the underlying mechanism controlling the bursting pattern (see below). The silent phase between subsequent bursts is attributed to activation of potassium permeability, since blocking SK channels stimulates continuous AP firing.115 A similar effect was observed in mouse β cells which express SK4 channels (KACNN4 gene, 87.7% homology).116
The pattern of membrane potential oscillations
Since the first description of membrane potential changes in mouse β cells upon stimulation with glucose,117 numerous studies on mice using different experimental paradigms have confirmed that glucose-induced membrane potential changes in β cells occur in the form of bursts of APs (spikes), which follow a fast, a slow, or a combined pattern coined compound oscillations (Fig. 4B).118-121
Figure 4.
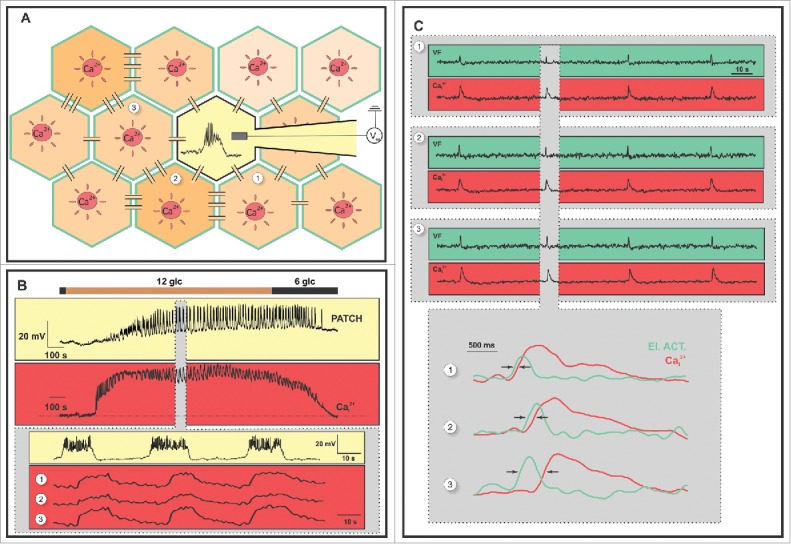
Coupling between membrane potential and [Ca2+]C oscillations in mouse β cells. (A). A schematic representation of 2 experimental methods. The first involves a simultaneous measurement of membrane potential and [Ca2+]C changes, using whole-cell patch-clamp (light yellow) and [Ca2+]C imaging in the neighboring cells (red) with a CCD camera, respectively. The second involves a confocal imaging of membrane potential changes using a voltage sensitive dye (green) and [Ca2+]C changes using a Ca2+ sensitive dye (red). Connections between cells represent gap junctions. More strongly connected cells are represented with a darker cytoplasm while less connected cells are represented with a lighter cytoplasm. (B) The upper trace on the yellow background represents oscillations of membrane potential (bursts) and the upper trace on the red background corresponds to typical [Ca2+]C oscillations in a single β cell during the same glucose protocol: 6 to 12 to 6 mM glucose. The lower traces depict a close-up of the upper traces showing electrical activity from the same cell as in the upper trace and [Ca2+]C from 3 β cells from the same islet. (C) Membrane potential dynamics (green) correlated with simultaneously obtained [Ca2+]C dynamics (red) from 3 β cells of a single islet. The gray rectangle encloses the responses presented below in more detail. Arrows mark the delay between membrane potential and [Ca2+]C oscillations.
In human β cells, glucose-induced electrical activity is much more variable compared with mice, with continuous or irregularly spaced APs without any clear bursts reported in the majority of studies93,104,105,122 (also termed the canine-like pattern, Fig. 5 B, upper trace), or a more organized oscillatory electrical activity with Ca2+ -dependent (rodent-like) or Na+- and Ca2+- dependent (distinctly human) bursts being observed in other preparations from the same studies or in other studies.104,122,123 Of note, the bursts of APs are typically much shorter in human β cells than in mice and this pattern has also been termed complex APs or rapid bursts (Fig. 5A, upper trace).104,118,124 In high concentrations of glucose, small amplitude oscillations of membrane potential without any clear APs (the so called wobbly pattern) have also been described (see Table 2 for details).
Figure 5.
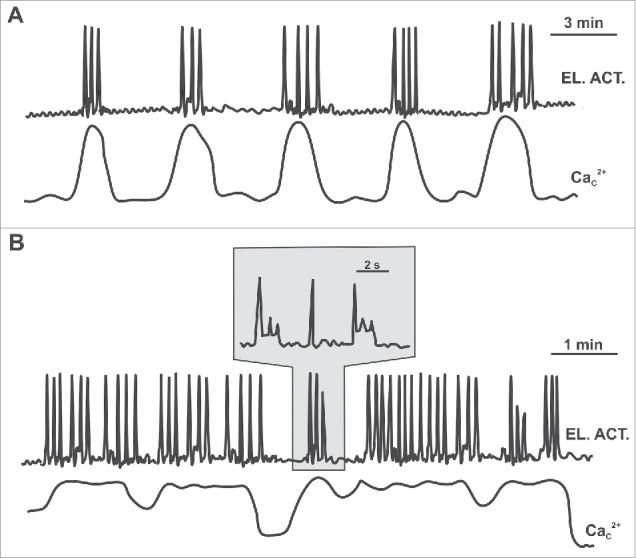
A schematic representation of possible scenarios of coupling between membrane potential and [Ca2+]C oscillations in human β cells. (A) β cells that respond to glucose with an organized oscillatory electrical activity (bursts) exhibit phase-locked [Ca2+]C oscillations. (B) β cells that respond to glucose with continuous or irregular spikes do not show any clear [Ca2+]C oscillations.
Table 2.
Patterns of membrane potential oscillations in human β cells.
| Study | Tissue | Recording | Stimulus | Response | Synchronicity |
|---|---|---|---|---|---|
| Falke et al., FEBS Lett 1989 123 | Isolated β cells from non-diabetic organ donors. Cultured in M199 (7-10 days). | Perforated-patch clamp recordings (0,15 mg/ml amphotericin B), room temperature. | Glc (15), tolbutamide (50 μM), arginine (20 mM) | Bursting (rodent-like) in 7/11 cells (fr not assessed, ampl≈35-55 mV); the provided recordings are not long enough to reliably assess slow oscillations. | n.a. |
| Pressel et al., J Membr Biol 1990 102 | Surface β cells in islet fragments. Islets isolated from cadaveric donors, cultured 2–14 d as described in Misler et al. Diabetes 1989). 221 | Perforated-patch clamp recordings (0,15 mg/ml amphotericin B), room temperature. | Glc (3-10). | Continuous spiking with some rapid bursts (fr not assessed, ampl≈30 mV in 15 glc) in one third to one half of all cells; the provided recordings are not long enough to reliably assess slow oscillations. | n.a. |
| Misler et al., Diabetes 1992 105 | Details not provided. | Perforated-patch clamp recordings (nystatin, further details not provided). | Glc (5,6,8,16) | Continuous spiking (canine-like), rapid bursting (distinctly human), or a rodent-like pattern (mentioned but not shown, fr not quantified, ampl≈30-40 mV in 5–8 glc); the provided recordings are not long enough to reliably assess slow oscillations. | n.a. |
| Misler et al., Diabetes 1992 122 | Isolated islets from cadaveric donors, incubated 1–7 d in HEPES-buffered CMRL 1066 (5,5 mM glc). Used as fragments/cells. | Perforated-patch clamp recordings (further details not provided, reference to Pressel et al, see above). | Glc (0, 3, 5,10,15), tolbutamide (20 μM) | In 0 glc, resting Vm of -60 mV (6/9), in response to elevated glucose (> 5), > 20 mV depolarization with a variable configuration of APs (4/6, not further described) or without APs (2/6); or a resting Vm of -40 mV and depolarization by 5–10 mV in high glucose (3/9). All cells responded to tolbutamide with at least some APs (not further quantified). The provided recordings are not long enough to reliably assess slow oscillations. |
n.a. |
| Barnett et al. Eur J Physiol 1995 99 | Isolated islets from 20 cadaveric donors, incubated 1–4 d at 22–24°C in HEPES-buffered CMRL 1066, then brought to 37°C. Used as islets or fragments/cells (dispersion by dispase 0,33 mg/ml). | Perforated patch on isolated cells/clusters of cells using nystatin (0,1-0,2 mg/ml) Insulin secretion from batches of islets using RIA |
Glc (3, 5, 10) Glc (3 →6/10, +1μM TTX) |
In 3 glc, a resting Vm of -60-(-55) mV; in 5 glc (at background Vm < -45 mV) i) trains of single high amplitude rapid APs (8/23 cells, 4 isolated, 4 in clusters), ii) complex-shaped APs/short bursts (7/23 cells), or iii) wobbly plateau depolarizations without APs (8/23 cells). In 10 glc, membrane potential progressively rised (to a background Vm of -35 mV) and the phenotype changed from i) to iii); the provided recordings are not long enough to reliably assess slow oscillations. TTX nearly abolished rise in insulin secretion in 6, but had no effect in 10 glc. |
n.a. |
| Rosati et al., FASEB J 2000 113 | Surface β cells in islets and islet fragments. Islets isolated from cadaveric donors, cultured 3–15 d in Hepes-buffered CMRL-1066 (7-10 days). | Perforated-patch clamp recordings (0,15 mg/ml amphotericin B), room temperature. | Glc (15), tolbutamide (50 μM), arginine (20 mM) | Continuous spiking (fr≈3 Hz, ampl≈30 mV in 15 glc); the provided recordings are not long enough to reliably assess slow oscillations. | n.a. |
| Misler et al., Eur J Physiol 2005 222 | Cryopreserved-thawed islets from non-diabetic (life-supported) cadavers. Cultured 1–2 d at 37°C, or up to 10 d at RT and then 1–2 d at 37°C. Single cells obtained by incubation in Verseneanddispase or in trypsin-EDTAandmechanical disruption. Single cells cultured up to 5 d in HEPES-buffered CMRL-1066 (5 mM glc) | Perforated patch-clamp | Glc (2, 5, 6, 15) + TTX (500 nM) | In 2 glc, Vm was stable at -60 mV, in 15 glc, background Vm was unstable at - 40 mV, with intermittent large amplitude APs (approaching 0 mV) in 7/7 cells. APs were solitary or grouped. In 6 glc, 3/4 cells showed a similar response as in 15 glc, 1/4 showed no clear response. In 5 glc (to mimick normal postprandial levels), 10/18 cells fired APs (from a background Vm of -55-(-50) mV (fr≈1/4s - 2/s). In 6/10 cells APs were affected by TTX and in 4/10 they peaked at -25-(-20) mV and were not affected by TTX. Slow oscillations n.a. | n.a. |
| Manning Fox et al., Endocrinology 2006 127 | β cells within isolated islets, obtained from dr. Jonathan Lakey and the JDRF Human Islet Distribution Program. Isolated using the Edmonton protocol, cultured in RPMI (5,5 mM glc), culture duration unknown. | Perforated patch using amphotericin B (0,1 mg/ml), performed at 34°C. |
Glc (11,1 → 2,8) | Individual APs in 11,1 (-40-(-10/0) mV, complex patterns (-50/-40-(-15/-10) mV, similar to rapid rodent oscillations during washout in 2,8 mM. A single recording was made. | n.a. |
| Braun et al., Diabetes 2008 93 | Isolated β cells obtained by dispersing islets from non-diabetic organ donors, using a Ca2+ free buffer. Cultured in RPMI 1640 (10 glc+2 L-gln, culture duration unknown). | Standard and perforated-patch clamp recordings, 32–33°C. | Glc (6, 10, 20), tolbutamide (100 μM) | Continuous spiking (fr not quantified, ampl≈20-40 mV in 6 glc); the provided recordings are not long enough to assess slow oscillations and the provided temporal resolution does not allow to assess rapid bursting. | n.a. |
| Rorsman et al., Annu Rev Physiol 2013 98 | β cells in intact isolated islets. Further details not provided. | Perforated patch-clamp. Further details not provided. | Glc (6 mM), +adrenaline (5 μM), +carbachol (20 μM) | Single APs from a background Vm of approx. -50 mV (58% of cells); or oscillatory electrical activity (suggested to underlie [Ca2+]C oscillations) with periods from 1–10 seconds (26% of cells), in both cases large amplitudes (up to 20 mV). The phenotype in the remaining cells (16%) not further specified. Hyperpolarization (by 16 ± 4 mV) in adrenaline and inhibiton of AP firing; depolarization (by 5 ± 3 mV) and stimulation of AP firing in carbachol. | n.a. |
| Fridyland et al., Islets 2013 223 | β cells within isolated islets from cadaveric donors. Further details not provided. | Perforated-patch clamp recordings using amphotericin B as described in Jacobson et al., J Physiol 2010 112 | Glc (14 mM), + TTX (500 nM) | Simple spikes and complex spikes (amplitude approx (-60/-50)-(-20/0) mV, further quantification not provided. Addition of TTX did not abolish AP firing but decreased the maxima of spikes by approx. 10 mV. | n.a. |
| Riz et al., PLoS Comput Biol 2014 104 | Isolated β cells obtained by dispersing islets from non-diabetic organ donors, using a Ca2+ free buffer. Cultured >24 h in RPMI 1640 (7,5 glc). | Perforated-patch clamp recordings (0,24 mg/ml amphotericin B), 31–33°C. | Glc (6 mM), +various channel blockers (TTX, ω-agatoxin IVA, UCL-1684, TRAM-34) | In control conditions (6 mM glucose), 3 main patterns of activity were observed: i) continuous spiking; ii) rapid bursting (1 burst/1-2 s); and iii) slow oscillations in AP firing (period≈5 min). | n.a. |
| Loppini et al., Phys Biol 2015 135 | β cell clusters obtained by dispersing islets from non-diabetic organ donors, using a Ca2+ free buffer. Cultured >24 h in RPMI 1640 (7,5 glc). | Perforated-patch clamp recordings (0,25 mg/ml amphotericin B), 31–33°C. | Glc (6 mM), +TTX (0,1 μg/ml) | 3/10 cells: occasional rapid APs (40 mV) + small oscillations (10 mV) around the resting MP; only small oscillations in TTX 2/10 cells: only rapid Aps, slow oscillations in TTX; 4/10 cells: rapid APs, not responsive to TTX; 1/10: silent in both control cond. and in TTX |
Suggested that active and non-active cells are coupled, and that coupling serves to synchronize active cells, not to activate inactive cells. Junctional conductance between 2 cells estimated to be 100–200 pS (0,01-0,02 nS/pF). Electrical coupling sufficient to synchronize spiking and bursting cells, also to partly synchronize slow electrical oscillations. |
Irrespective of the species, oscillations in membrane potential reflect a balance between activation of Na+ channels or VDCCs and K+ channel activity. The shape of APs triggered in mouse β cells is largely determined by L-type Ca2+ channels responsible for the upstroke phase, and voltage-dependent potassium current through Kv2.1 channels responsible for the repolarization. In human β cells, AP upstroke is mediated by voltage-dependent sodium channels and L-type Ca2+ channels102 while BK channels are mainly responsible for the repolarization phase.93,98,104 Because of the kinetic differences between VDCCs and voltage-dependent sodium currents, APs in mouse β cells are much longer compared with human β cells. The silent phase between subsequent bursts is attributed to SK channels for mouse, and to HERG,103,113 and SK channels for human β cells.104 Additionally, it has been suggested that KATP and T-type Ca2+ channels might play a role in grouping of APs into rapid bursts in human β cells.98 Importantly, in human β cells that do not display bursting and continuously spike, the role of SK channels seems to depend on excitability of the cell, with less excitable cells showing an increase in AP firing frequency and more excitable cells not being affected upon SK channel block.104
In non-stimulatory glucose, β cells are electrically silent. While in mice membrane potential oscillations are observed at 7 mM and above,120 human β cells oscillate already at 5 mM glucose (seeTable 2) where resting K+ conductance maintains the membrane potential at around -70 mV while still allowing occasional openings of VDCCs, mainly the T-type Ca2+ channels. This results in membrane depolarization, the subsequent opening of additional T-type channels and further depolarization which can then trigger opening of L-type Ca2+ channels and VD Na+ channels. In mouse β cells, the glucose concentration needed for triggering membrane potential oscillations is higher, i.e., > 6 mM, since lower glucose does not provide enough energy to increase [ATP]C high enough to decrease K+ conductance to such an extent that L-type VDCCs would open.98
In mice (Fig. 4), increasing glucose concentrations changes burst duration from 3 seconds and a frequency of 2–5 oscillations/min (i.e., period of 12–30 seconds) at the threshold concentration to a continuous spiking activity in 20 mM glucose98,119,121,125,126 As already mentioned, in humans, the responses take several different forms at a given concentration of glucose and are much harder to predict (Fig. 5 and Table 2). For a comprehensive summary of possible patterns of membrane potential changes and factors that might influence them, in Table 2 we briefly review the main findings of all studies that have, to the best of our knowledge, described or even quantified the electrophysiological responses of human β cells to glucose and some other secretagogues (that act via the triggering pathway). All electrophysiological studies on human β cells taken together suggest that with increasing glucose, there is progressive depolarization also in human β cells and that there is at least circumstantial evidence of a trend of patterns switching from no APs to bursts of APs to continuous bursting.99,127
Finally, slow or the so-called glycolytic oscillations in membrane potential with a frequency of 0.2-0.4 oscillations/min are observed in mouse β cells under intermediate GCK flux rates and they are not glucose-sensitive.38 In almost all studies on human β cells, the electrical activity was observed over periods that were too short to enable a reliable assessment of slow oscillations. However, a recent study by Riz et al. provided evidence that slow membrane potential oscillations with a period of 3–5 minutes can be present in human β cells and that they can be accounted for in the model by a glycolytic component.104
Coupling between β cells
Within areas of plasma membrane delimited by tight junctions, both mouse,128,129 and human130 β cells possess gap junctions. In addition to connexin 30.2,131 the connexin 36 (Cx36) is considered the major form expressed in β cells in mice,128,132 and humans133 and is believed to be the mechanical substrate for β cell functional coupling. Functional coupling describes the finding that changes in metabolism, membrane potential, and [Ca2+]C, are synchronized between different cells by means of depolarization and [Ca2+]C waves spreading between cells which we describe in more detail in the following chapter, and putatively also by diffusion of small signaling molecules and metabolic intermediates.
Electrophysiological approaches estimated the gap junctional conductance between 2 β cells at about 200–350 pS in mice,128,129,134 and at about 100–200 pS in humans.135 Taking into account that each β cell is connected to 6 - 8 neighbors,136 the conductance between a cell and all of its neighboring cells was estimated at about 2.5-3.5 nS in mice.137 Recently, gap junctional coupling between β cells has been explored by using fluorescence recovery after photobleaching (FRAP) and this approach suggested a quantitatively similar degree of coupling between β cells in human and mouse islets.138
Gap junctions are permeable to a variety of ions and metabolites.139 The most probable candidate for gap-junctional currents in mice are Ca2+ ions, as their flow through gap junctions is belived to be able to spread depolarization from a cell to its neighbors,134,140,141 although this view has been challenged.142 Conceivably, in humans both Na+ and Ca2+ ions could be implicated in waves of depolarization, but this remains to be demonstrated. Noteworthy, gap junctions are permeable to glycolytic intermediates and their diffusion could account for synchronization of metabolic oscillations in different cells, although this is not necessary and electrical coupling alone can synchronize metabolic oscillations.143
In addition to gap junctions, other means of cell- cell communication, may help control insulin release from β cells. First, ephrin-A-EphA ligand-receptor signaling between β cells has a role in insulin secretion as glucose stimulation enhances insulin secretion via dephosphorylation of the Eph receptor in both mice and humans.144 In addition, disruption of neural cell adhesion molecules (NCAM) seems to influence regulation of insulin release in mice.145 Further, primary cilia of β cells may be involved in the control of insulin release in both mice and humans.146,147 Moreover, there are several possible auto- and paracrine interactions in both human and mouse islets.148 Insulin secreted by β cells binds to insulin receptors on plasma membrane and acts in an auto- or paracrine manner by upregulating insulin gene transcription149 and triggering exocytosis.150 Further, ATP co-secreted with insulin,151-153 binds to membrane-bound purinergic P2X and P2Y (for a review of the expression in mice and humans, see ref. 154). In mice, purinergic stimulation seems to have either an excitatory effect via P2X1 and P2X3155 or an inhibitory effect via P2Y1 receptors.156,157 ATP was also suggested as an agent synchronizing β cell activity.158 In fact, brief (15 seconds-long) stimulations by ATP were able to synchronize Ca2+ oscillations in isolated individual islets that were not in direct physically contact.158 In humans, purinergic stimulation evokes a positive feedforward mechanism,155,159 most probably via P2X3 receptors160 and a transient increase in [Ca2+]C.161 Dopamine, yet another molecule co-secreted with insulin, was shown to inhibit GSIS in both humans and mice.162,163 In contrast, γ-Aminobutyric acid (GABA), which is also co-secreted with insulin, was ascribed a positive feedback role in humans.164,165 Finally, glucagon secreted by α cells was found to synchronize oscillatory Ca2+ activity of β cells in mice, however the exact role of glucagon as a synchronizing factor needs to be explored into more detail as glucagon exhibits out-of-phase oscillations with insulin.148,166,167 At the level of an islet, things are further complicated by the fact that somatostatin released from delta cells might critically influence the function of both β and α cells.11 Finally, coupling between β cells from different islets might conceivably play a role in vivo and be brought about by means of neurotransmitters, such as acetylcholine,168-170 or via metabolic feedback from the liver.143
The relationship between membrane potential and [Ca2+]C changes, waves, and functional connectivity
Twenty years after the first description of glucose-induced membrane potential changes in microdissected mouse islets,117 oscillatory [Ca2+]C changes were recorded in dispersed mouse β cells,171 but their temporal characteristics were difficult to reconcile. More specifically, dispersed β cells showed widely heterogeneous and much slower [Ca2+]C oscillations compared with typical frequencies of bursts. In the early nineties, detailed studies on coupling between membrane potential and [Ca2+]C, as well as [Ca2+]C and insulin secretion on isolated mouse islets showed that in intact islets [Ca2+]C changes closely resemble and are temporally tightly coupled to membrane potential changes and that depolarization and [Ca2+]C waves are probably the synchronizing mechanism.125,172,173 In addition, the observed [Ca2+]C oscillations are closely matched by pulses of insulin secretion.174-177
A large number of studies using different electrophysiological and imaging approaches support the view that membrane potential and [Ca2+]C changes in different β cells in mouse islets are not completely in phase, but phase-locked with a temporal delay and this delay can be explained by depolarization and [Ca2+]C waves spreading from cell to cell via gap junctions.134,140,141,172,177-182 Further, both waves spread in the same direction and with the same velocity and in every β cell [Ca2+]C oscillations closely follow the bursts of electrical activity but are of longer durations than the bursts (Fig. 4C).141,183,184
All studies that have quantified the direction and velocity of [Ca2+]C waves have reported comparable values of wave velocities and also agree in that at least over the time frame of a few minutes, the direction of the waves remains rather constant.140,141,182,185 Moreover, experiments and existing mathematical models suggest that the origin of the wave is usually a single cell or a small cluster of cells at islet periphery, also called the pacemaker region, that heterogeneity in β cell excitability and gap junction coupling is needed to fully reproduce the experimentally observed properties of synchronicity and [Ca2+]C waves, that the cells in the pacemaker region are more excitable than others are, that they determine the [Ca2+]C oscillation period for the rest of the islet, and that such regions might arise by chance due to heterogeneity.182,185 As to why [Ca2+]C waves usually originate at islet periphery, considering that cells with elevated excitability can probably be found in all regions of an islet, we wish to suggest that this might be due to a lower number of neighboring cells and thus a weaker hyperpolarizing contribution from less excitable cells.
The concept of depolarization and [Ca2+]C waves being the synchronizing mechanism seems to be in conflict with more recent reports showing that functionally, islets behave as small world networks where signals of widely spaced β cells may be more similar to each other than to nearby cells. More specifically, if [Ca2+]C oscillations are compared for every cell pair and a so called functional connection is drawn between cells whose signals are similar enough as to exceed a certain threshold, it turns out that a relatively few cells harbor a large proportion of all functional connections.56,88 It is not intuitively clear how a spreading [Ca2+]C wave could allow for such behavior. This conflict has received yet another layer of complexity with the recent report by Johnston and colleagues suggesting that hub cells are in fact bona fide pacemakers that convey signals to other cells called followers.186 In comparison with the more traditional view of synchronization via waves, according to the functional coupling analysis, there seems to be more pacemakers (1-10% of all β cells) and they are not limited to the periphery but are distributed across the whole cross section of an islet.
In our opinion, the above 2 ideas are not mutually exclusive. Namely, it is possible that there is a temporal sequence of activation within the population of hubs, such that they are activated one after another and before other cells and that the depolarization and [Ca2+]C wave spread to follower cells from them. A study seems to support the idea presented above.187 Namely, cellular heterogeneity and nearest neighbor coupling, without any additional direct long range physical connections (via nerves or cilia, see above), are sufficient to account for both the properties of experimentally observed membrane potential and [Ca2+]C waves and the degree of synchronization they enable,140,141,172,178,180,182-185 and the small-world character of the functional connectivity pattern with hubs and followers.56,88,186,188-190 In this model, in 3D there are strings of more strongly coupled direct neighbors that enable propagation of fast [Ca2+]C waves over long distances, and enable a high degree of synchronicity between widely separated cells that thus assume the role of hubs when studied from the functional network perspective. The remaining cells are less well coupled to hubs and to each other and thus their signals correlate with each other to a lesser extent, making them less well connected in the functional network.187 This model is also consistent with our report of segregated communities of nearby cells that become more and more interconnected in higher glucose, when gap junctional connectivity is believed to increase,188 and also with reports of decreased connectivity when gap junction coupling is reduced (see below for details).56,186
There are certainly several open questions regarding connectivity in general and the nature of hub cells in specific and they have been recently summarized in a comprehensive review.190 Here we only wish to point out that in accordance with predictions,182,185 evidence is accumulating that the hub or pacemaker cells are metabolically highly active,186,191 and display some phenotypic characteristics of immature cells, such as high levels of glucokinase, a low insulin content and a low expression of pancreatic duodenum homeobox-1 (Pdx1) and NK6 homeobox 1 (Nkx6.1). A less mature phenotype is also consistent with a lower threshold for insulin secretion.192 Notably, immature cells can indeed be found in varying numbers in adult islets of Langerhans.193 However, considering the role of gap junctions in β cell connectivity and the finding that adult β cells express more Gjd2 (the gene encoding Cx36) and that the expression of this gene has been linked with an increased expression of transcription factors determining a mature β cell phenotype,194 hub cells are probably not phenotypically immature in all regards.
For several reasons, much less is known about normal patterns of [Ca2+]C changes, waves, their relationship with depolarization, and also about functional connectivity in human islets.45,195,196 First, human pancreatic tissue is more difficult to obtain than mouse tissue, and in a large proportion of the few existing studies, it has been collected from patients with pancreatic disease. Second, islets have been isolated in different ways and the culture duration and conditions have varied widely in studies on human islets, which is a possible source of phenotypic variability in their responses to glucose, as has been demonstrated convincingly for mouse islets.197 Finally, [Ca2+]C changes have been recorded by different methods and different stimulation protocols have been used in studies on human islets. Table 3 provides a comprehensive summary of the various aspects discussed above in more detail. In brief, at the one end of the spectrum of possible scenarios lies the view that human islets do not display organized fast [Ca2+]C oscillations and that even if they occasionally occur, they are synchronized only locally.198 Putatively, the structural substrate of this behavior is the unique cytoarchitecture of human islets, where β cells are believed to be less orderly distributed within islets and make a larger number of heterotypic contacts with α and delta cells than in mouse islets.198,199 At the other end, we have the possibility that human islets display orderly fast [Ca2+]C oscillations (albeit with a lower frequency) that are at least partially200 or even completely synchronized between different cells.201 These 2 types of responses seem to be supported by morphological studies showing that in human islets, β cells are organized in clusters202 or ribbon-like patterns,203 and that in 3 dimensions they probably form uninterrupted syncytia and sometimes even islets that are similar to the stereotypical mouse mantle-core islets.12,204,205 Between these 2 extremes lie the reports of [Ca2+]C oscillations that were slower, less orderly, or were not observed in all islets included in a study (Table 3). Noteworthy, in a few recent studies synchronization of [Ca2+]C signals in different cells has been analyzed in human islets and compared with mouse islets. The percentage of area synchronized206 was similar in mouse and human islets138 and human islets showed similar but more clustered functional connectivity.56,186 It remains to be determined whether these different phenotypes reflect true biological variability or are at least partly due to methodological factors. Additionally, the presence of waves, the coupling between membrane potential and [Ca2+]C changes, as well as between [Ca2+]C and insulin secretion, remain to be investigated into more detail. Recently, evidence has been provided that β cells in human islets electrically communicate with each other similarly as in mice, but it remains to be explained how exactly the observed extracellular slow potentials relate to membrane potential oscillations.207 Furthermore, it is difficult to understand how the described properties of [Ca2+]C oscillations can be reconciled with patterns of electrical activity in human islets. The rapid bursts are too rapid to explain the observed periods of [Ca2+]C oscillations and it is also unclear whether they are synchronized between different cells. The recently observed slow membrane potential oscillations seem to be a better candidate for explaining [Ca2+]C oscillations observed in human islets in one study,104,200 although they are too slow to explain the faster [Ca2+]C oscillations observed in another.201 Moreover, the release of insulin from isolated human islets occurs in a fairly regular pulsatile manner, with a period between approximately 4 and 10 minutes,208,209 and it remains to be clarified how the irregular patterns of membrane potential and [Ca2+]C are compatible with this finding. Noteworthy, the recently reported period of slow glycolytic membrane potential oscillations in β cells104 could explain the period of insulin secretory pulse at the lower bound of the reported range.209. Fig. 5 illustrates the 2 abovementioned extremes of possible scenarios.
Table 3.
Patterns of [Ca2+]C oscillations in human β cells.
| Study | Tissue | Recording | Stimulus | Response | Synchronicity |
|---|---|---|---|---|---|
| Kindmark et al., FEBS Lett 1991 161 | i) isolated islets (3 patients with normal glc tolerance, Whipple´s resection, minced tissue, collagenase, cultured 1–4 d in RPMI 1640 (11,1 mM glc) ii) insulinoma cells, (1 patient) |
Fura-2 (340/380 - 515) Spectrofluoro-meter (fr = 1 Hz) |
glc (3→11; 0→10 / 20) K+ (25 mM) tolbutamide ATP Diazoxide |
Only islets with an increase in Ca2+ considered successful (> 50% of all): i) decrease, then increase with stable (1 islet) or »fluctuating« (1 islet) plateau; ii) no initial decrease, plateau with irregular oscillations (period 70–80 s) (1 islet). | n.a. |
| Misler et al., Diabetes 1992 122 | Isolated islets (non-diabetic life supported cadavers, age and gender unknown (collagenase + Ficoll gradient), cultured 1–7 d in 5,5 mM glc |
Fura-2 (340/380 - 505) spectrofluorometry (fr unknown) perofrated patch-clamp |
glc (3→10→20) | i) no response to glc with subsequent response to tolbutamide (4/11 islets); ii) oscillations superimposed on a plateau (4/11); iii) slow rise to a plateau without oscillations (2/11); iv) short transients on an unchanging baseline, coalescing to a spike. | n.a. |
| Kindmark et al., Diabetologia 1994 224 | Isolated islets (9 patients, normal, impaired, and diabetic tolerance, f and m, 57–83 y (mean 65,0 yr) minced pancreas tissue, collagenase, cultured 1–4 d (in 11 mM glc) |
Fura-2 (340/380 - > 510) spectrofluorometry (fr = 1 Hz) perforated patch-clamp |
glc (3→10 / 12; 15 mM for electrophysiology, tolbutamide (100 μM) |
i) fairly regular slow oscillations in both glc and tolbutamide (period = 2–3 min); ii) in some a monophasic increase continuous firing of APs (5-6 Hz), no bursts. | n.a. |
| Hellman et al., Diabetologia 1994 225 |
i) isolated cells, ii) clusters, iii) isolated islets (8 cadaveric donors, 39 ± 5 yr, gender unknown, ductal distension and Ficoll gradient, cultured 3–7 d in 6,1 mM glc, sent by air and stored for another 1–3 d in 5,6 mM glc) |
Fura-2, Indo-1 PMT or video camera, fr unknown |
glc (3→20 glc, 3→3+1mM tolbutamide, 1 mM tolbutamide+20 mM glc, addition of glucagon (10 nM or glycine (10 mM) in isolated islets glc 3→20 |
In isolated cells and clusters large amplitude oscillations from the basal level, fr = 0,1-0,5/min (type a) and superimposed on an elevated Ca (type b), slow oscillations were also present in tolbutamide, glucagon and glycine transformed type a to a plateau. In islets plateau with superimposed oscillations (period 2–5 min). |
Analyzed in clusters, well synchronized slow oscillations, no pacemakers. In isolated islets well synchronized, different amplitudes, no quantification of synchronization. Good overlap with type a oscillations and insulin release. Higher glucose affected amplitude, not frequencies. In isolated islets, tybe b pulses of insulin wered described (but not Ca). |
| Martin et al., Cell Calcium 1996 201 |
Isolated islets 2 cadaveric donors (age unknown) and 2 patients (44 and 80 yr), Tissue minced and collagenase, hand picking, cultured overnight in 5,5 mM glc |
Fura-2 (340/380-510) CCD camera, fr. = 0,167 Hz) |
glc 3→11, 11→16,7; tolbutamide 50 μM |
A triphasic response (31/42 islets), decrease, a rapid transient increase, then oscillations (fr. = 1 ± 0,3/min), or (11/41 islets), slow rise to a plateau w.o. osc.; Upon elevating glc, prolonged duration or slow oscillations (0,15 ± 0,2/min) (14/17) or plateau (3/17). Tol: reversible plateau-like increase (7/7). |
Synchrony in all 3 phases in all regions of an islet (13/15 islets) or regions 10–15 s out of phase (2/15). |
| Cabrera et al., PNAS 2006 198 | Isolated islets (5 cadaveric donors, 48 ± 7 y of age), liberase + purification, cultured in CMRL (5,5 mM glc) |
Fura-2 (340/380-510) CCD camera, fr. unknown. |
glc 3→11 mM | No general oscillatory responses (> 70 islets from 10 preparations), only localized oscillations (single cells or clusters). In dispersed cells, slow oscillations were observed, period = 5min (not further quantified). | Not quantified in detail, qualitatively assessed that there is no sync. |
| Quesada et al., Diabetes 2006 200 | Isolated islets (5 cadaveric donors (22,6 ± 5,3 yr, gender unknown; minced pancreas tissue, then collagenase, recorded acutely) |
Fluo-3 (488/505-530) Ca2+ Confocal microscopy at the equatorial plane (t = 8 μm, fr = 0,5-0,25 Hz |
glc via perifusion, 3→11, 11→16,7 | 6/41 β cells (10 islets): sustained increase without oscillations 35/41 β cells: oscillations in 11 mM (0,45 ± 0,02 /min) |
Assessed qualitatively, in clusters only, sometimes more widely distributed after several minutes (4/7 islets) or a more general but weak coupling (3/7 islets). Heterogeneity in the degree of coupling, even from the same preparation. |
| Hodson et al. J Clin Invest 2013 56 | Isolated islets (21 normoglycemic donors, isolation centers (UK, I, CH), cultured in RPMI in 5,5 mM glc | Spinning-disk confocal; Fluo-2 (491-525/25) CCD, fr = 0,5 Hz |
glc 3→11 GLP-1 (20 nM) |
Stochastic fast oscillations, large deflections in a subpopulation of β cells upon GLP-1. | Moderate correlation in 11 mM glc, large synchronous deflections in Ca upon GLP-1. |
| De Marchi et al., J Biol Chem 2014 54 | Isolated islets (2 donors, purchased (Tebu-bio), cultured in RPMI in 5,5 mM glc | Genetically encoded cameleon sensor YC3.6cyto (430 - 480/40 and 535/30), CCD, fr = 0,5 Hz | glc via perifusion, 1→16,7 mM, inhibition by oligomycin (2,5 μg/ml) and diazoxide (100 μM) | Transient increase, then stable plateau with occasional superimposed »spikes." | n.a. |
| Johnston et al. Cell Metab 2016 186 | Isolated islets (from distant isolation centers (I, CH), cultured in RPMI in 5,5 mM glc | Spinning-disk confocal; Fluo-2 (491-525/25) CCD, fr = 2-8 Hz | glc 3→11 | Not shown. | Complex network theory analyses. Similar topological characteristics as mouse islets, more clustered/compartmentalized synchrony. |
| Farnsworth et al. J Biol Chem 2016 138 | Isolated islets (from distant isolation centers (I, CH), cultured in RPMI in 5,5 mM glc | Wide field microscope; Fluo-4 (490/40-525/36) CCD, fr = 1 Hz | glc 11 | Not shown. | Percentage area synchronized ≈90% in controls. |
Changes in coupling, membrane potential and Ca oscillations, as well as functional connectivity in type 2 diabetes
There is currently no direct evidence for an involvement of disrupted coupling in pathogenesis of T2DM in humans. Yet, a body of circumstantial evidence suggests that disrupted coupling could play an important role. On the one hand, diminishing or abolishing gap junctional coupling by pharmacological or genetic tools in mice leads to a disrupted pattern of membrane potential and [Ca2+]C oscillations, as well as to an absence of [Ca2+]C waves.87,182,185,210 Further, some studies have found an increased basal insulin secretion and a diminished amplitude of the first phase, as well as absence of oscillations in secretion during the second phase of stimulated insulin secretion from islets lacking gap junctions.210,211 These alterations in insulin secretion from isolated islets have not been supported by all studies.210,212 However, measurements in Cx36 knockout mice in vivo strongly support the view that first phase insulin secretion is reduced and the second phase is non-oscillatory in these animals, which could account for the observed glucose intolerance.211 On the other hand, chronically increased glucose has been shown to decrease expression of Gjd2 and this may contribute to the effect of glucotoxicity.213 Similarly, lipotoxicity has been shown to involve Cx36 downregulation.56 In addition, cytokines released from the adipose tissue and in islets under hyperglycemia and hyperlipidemia are able to significantly decrease Cx36 gap junction coupling, disrupt the synchronicity and pattern of [Ca2+]C oscillations, and reduce the number of hubs and correlated links.138,186,214 Most importantly, it has been demonstrated in high-fat diet fed mice that a prediabetic milieu leads to an increase in basal insulin secretion, an impairment in Cx36 mediated intercellular coupling,215 as well as to disorganized [Ca2+]C oscillations in islets isolated from these mice.56 Similar findings have been obtained on islets from ob/ob 216 and db/db 217 mice. In sum, it seems that the series of events connecting altered gap junctional coupling with impaired glucose tolerance via a disrupted pattern of synchronization of membrane potential and [Ca2+]C oscillations operate both ways in mice. In other words, diminished coupling can result in glucose intolerance and vice versa.
Regarding the relevance of this series of events for humans, disruptions to insulin dynamics similar to the ones described above are observed in humans with prediabetes and T2DMs.218,219 Moreover, chronic exposure of human islets to elevated free fatty acids reduces Cx36 expression, disrupts the synchronicity of the [Ca2+]C response, functional connectivity, and insulin release in response to GLP-1 added to stimulatory glucose (but not to stimulatory glucose alone). Importantly, donor BMI seems to be negatively correlated with islet responses to GLP-1.56 It was further demonstrated that proinflammatory cytokines decrease Cx36 gap junction coupling in isolated human islets, albeit in a spatially more uniform manner that in mouse islets,138 and that glucolipotoxicity induces hub failure in isolated human islets, i.e., it reduces the number of hubs and functional links between β cells.186 The above recent lines of evidence suggest that similar although not same pathophysiological mechanisms linking glucose intolerance and gap junctional coupling might be at work in humans. It has been proposed that inherent structural differences between human and mouse islets12 might explain the differences in the spatial pattern of cytokine-induced Cx36 downregulation,138 and that in addition to the inherent differences in islets, patterns of meal consumption might account for the differential responsiveness of human and mouse islets to glucose and incretins.56 To get a more complete picture of β cell connectivity as a target of various pathological processes and future therapies, see some resourceful recent reviews.91,189,190,196,220
Conclusions
At all levels of the triggering pathway to insulin secretion, from the entry of glucose into β cells to the pattern of [Ca2+]C changes that are believed to trigger insulin release, there are important functional differences between mice and humans. At present, our knowledge about these differences is largely limited to the types and functional properties of transporter and channel proteins being expressed in the 2 species, whereas there is still much confusion regarding the normal or prevailing patterns of membrane potential and [Ca2+]C changes and intraislet synchronization of these 2 crucial signals in the signaling cascade, especially in human islets. Even less is known about the mechanisms of interislet synchronization in mouse and human islets that are believed to govern the in vivo oscillations in plasma insulin concentration. In addition, we deliberately limited ourselves to functional aspects in adults and developmental facets were only touched upon. The accumulating evidence regarding interspecies similarities and differences in other pathways to insulin secretion and the role of neurotransmitters, paracrine factors from other islet cells, and hormones, such as incretins, deserves separate reviews.
Footnotes
Values that determine the homology of proteins are taken from the HomoloGene.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This paper is dedicated to the memory of Matthias Braun (1966-2013) who crucially contributed to our understanding of human β cell physiology and whose work is a continuing source of inspiration.
Funding
The authors acknowledge the financial support from the Slovenian Research Agency (research core funding, Nos. P3-0396 and I0-0029-0552, as well as research projects, Nos. N3-0048, J3-7177, J1-7009, and J7-7226).
References
- [1].Federation ID IDF Diabetes Atlas. Brussels, Belgium: International Diabetes Federation, 2015. [Google Scholar]
- [2].Stratton IM, Adler AI, Neil HAW, Matthews DR, Manley SE, Cull CA, Hadden D, Turner RC, Holman RR. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): Prospective observational study. BMJ 2000; 321:405-12; PMID:10938048; https://doi.org/ 10.1136/bmj.321.7258.405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Weyer C, Bogardus C, Mott DM, Pratley RE. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J Clin Invest 1999; 104:787-94; PMID:10491414; https://doi.org/ 10.1172/JCI7231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Danaei G, Finucane MM, Lu Y, Singh GM, Cowan MJ, Paciorek CJ, Lin JK, Farzadfar F, Khang YH, Stevens GA, et al.. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: Systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2·7 million participants. Lancet 2011; 378:31-40; PMID:21705069; https://doi.org/ 10.1016/S0140-6736(11)60679-X [DOI] [PubMed] [Google Scholar]
- [5].Gregg EW, Chen H, Wagenknecht LE, Clark JM, Delahanty LM, Bantle J, Pownall HJ, Johnson KC, Safford MM, Kitabchi AE, et al.. Association of an intensive lifestyle intervention with remission of type 2 diabetes. JAMA 2012; 308:2489-96; PMID:23288372; https://doi.org/ 10.1001/jama.2012.67929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Steven S, Hollingsworth KG, Al-Mrabeh A, Avery L, Aribisala B, Caslake M, Taylor R. Very low-calorie diet and 6 months of weight stability in type 2 diabetes: Pathophysiological changes in responders and nonresponders. Diabetes Care 2016; 39:808-15; PMID:27002059; https://doi.org/ 10.2337/dc15-1942 [DOI] [PubMed] [Google Scholar]
- [7].Wright JJ, Tylee TS. Pharmacologic therapy of type 2 diabetes. Med Clin North Am 2016; 100:647-63; PMID:27235609; https://doi.org/ 10.1016/j.mcna.2016.03.014 [DOI] [PubMed] [Google Scholar]
- [8].Rorsman P, Renström E. Insulin granule dynamics in pancreatic beta cells. Diabetologia 2003; 46:1029-45; PMID:12879249; https://doi.org/ 10.1007/s00125-003-1153-1 [DOI] [PubMed] [Google Scholar]
- [9].Bratanova-Tochkova TK, Cheng H, Daniel S, Gunawardana S, Liu Y-J, Mulvaney-Musa J, Schermerhorn T, Straub SG, Yajima H, Sharp GW. Triggering and augmentation mechanisms, granule pools, and biphasic insulin secretion. Diabetes 2002; 51:S83-S90; PMID:11815463; https://doi.org/ 10.2337/diabetes.51.2007.S83 [DOI] [PubMed] [Google Scholar]
- [10].Henquin JC, Nenquin M, Ravier MA, Szollosi A. Shortcomings of current models of glucose-induced insulin secretion. Diabetes Obes Metab 2009; 11:168-79; PMID:19817799; https://doi.org/ 10.1111/j.1463-1326.2009.01109.x [DOI] [PubMed] [Google Scholar]
- [11].Tengholm A, Gylfe E. Oscillatory control of insulin secretion. Mol Cell Endocrinol 2009; 297:58-72; PMID:18706473; https://doi.org/ 10.1016/j.mce.2008.07.009 [DOI] [PubMed] [Google Scholar]
- [12].Dolensek J, Rupnik MS, Stozer A. Structural similarities and differences between the human and the mouse pancreas. Islets 2015; 7:e1024405; PMID:26030186; https://doi.org/ 10.1080/19382014.2015.1024405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Levetan CS, Pierce SM. Distinctions between the islets of mice and men: Implications for new therapies for type 1 and 2 diabetes. Endocr Pract 2013; 19:301-12; PMID:23186955; https://doi.org/ 10.4158/EP12138.RA [DOI] [PubMed] [Google Scholar]
- [14].Mueckler M, Thorens B. The SLC2 (GLUT) family of membrane transporters. Mol Aspects Med 2013; 34:121-38; PMID:23506862; https://doi.org/ 10.1016/j.mam.2012.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Guillam MT, Hummler E, Schaerer E, Yeh JI, Birnbaum MJ, Beermann F, Schmidt A, Dériaz N, Thorens B. Early diabetes and abnormal postnatal pancreatic islet development in mice lacking Glut-2. Nat Genet 1997; 17:327-30; PMID:9354799; https://doi.org/ 10.1038/ng1197-327 [DOI] [PubMed] [Google Scholar]
- [16].Thorens B. GLUT2, glucose sensing and glucose homeostasis. Diabetologia 2015; 58:221-32; PMID:25421524; https://doi.org/ 10.1007/s00125-014-3451-1 [DOI] [PubMed] [Google Scholar]
- [17].De Vos A, Heimberg H, Quartier E, Huypens P, Bouwens L, Pipeleers D, Schuit F. Human and rat beta cells differ in glucose transporter but not in glucokinase gene expression. J Clin Invest 1995; 96:2489-95; PMID:7593639; https://doi.org/ 10.1172/JCI118308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Heimberg H, De Vos A, Pipeleers D, Thorens B, Schuit F. Differences in glucose transporter gene expression between rat pancreatic alpha- and beta-cells are correlated to differences in glucose transport but not in glucose utilization. J Biol Chem 1995; 270:8971-5; PMID:7721807; https://doi.org/ 10.1074/jbc.270.15.8971 [DOI] [PubMed] [Google Scholar]
- [19].Ohtsubo K, Chen MZ, Olefsky JM, Marth JD. Pathway to diabetes through attenuation of pancreatic beta cell glycosylation and glucose transport. Nat Med 2011; 17:1067-75; PMID:21841783; https://doi.org/ 10.1038/nm.2414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].McCulloch LJ, van de Bunt M, Braun M, Frayn KN, Clark A, Gloyn AL. GLUT2 (SLC2A2) is not the principal glucose transporter in human pancreatic beta cells: Implications for understanding genetic association signals at this locus. Mol Genet Metab 2011; 104:648-53; PMID:21920790; https://doi.org/ 10.1016/j.ymgme.2011.08.026 [DOI] [PubMed] [Google Scholar]
- [21].Manz F, Bickel H, Brodehl J, Feist D, Gellissen K, Gescholl-Bauer B, Gilli G, Harms E, Helwig H, Nützenadel W. Fanconi-Bickel syndrome. Pediatr Nephrol 1987; 1:509-18; PMID:3153325; https://doi.org/ 10.1007/BF00849262 [DOI] [PubMed] [Google Scholar]
- [22].Chesney RW, Kaplan BS, Colle E, Scriver CR, McInnes RR, Dupont CH, Drummond KN. Abnormalities of carbohydrate metabolism in idiopathic Fanconi syndrome. Pediatr Res 1980; 14:209-15; PMID:7383741; https://doi.org/ 10.1203/00006450-198003000-00006 [DOI] [PubMed] [Google Scholar]
- [23].Sansbury FH, Flanagan SE, Houghton JAL, Shuixian Shen FL, Al-Senani AMS, Habeb AM, Abdullah M, Kariminejad A, Ellard S, Hattersley AT. SLC2A2 mutations can cause neonatal diabetes, suggesting GLUT2 may have a role in human insulin secretion. Diabetologia 2012; 55:2381-5; PMID:22660720; https://doi.org/ 10.1007/s00125-012-2595-0 [DOI] [PubMed] [Google Scholar]
- [24].Matschinsky FM. A lesson in metabolic regulation inspired by the glucokinase glucose sensor paradigm. Diabetes 1996; 45:223; PMID:8549869; https://doi.org/ 10.2337/diab.45.2.223 [DOI] [PubMed] [Google Scholar]
- [25].Iynedjian PB. Molecular physiology of mammalian glucokinase. Cell Mol Life Sci 2008; 66:27-42; PMID:18726182; https://doi.org/ 10.1007/s00018-008-8322-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Matschinsky FM. Regulation of pancreatic beta-cell glucokinase: From basics to therapeutics. Diabetes 2002; 51(Suppl 3):S394-404; PMID:12475782; https://doi.org/ 10.2337/diabetes.51.2007.S394 [DOI] [PubMed] [Google Scholar]
- [27].Matschinsky FM, Glaser B, Magnuson MA. Pancreatic beta-cell glucokinase: Closing the gap between theoretical concepts and experimental realities. Diabetes 1998; 47:307-15; PMID:9519733; https://doi.org/ 10.2337/diabetes.47.3.307 [DOI] [PubMed] [Google Scholar]
- [28].Heimberg H, Devos A, Vandercammen A, Vanschaftingen E, Pipeleers D, Schuit F. Heterogeneity in glucose sensitivity among pancreatic beta-cells is correlated to differences in glucose phosphorylation rather than glucose-transport. EMBO J 1993; 12:2873-9; PMID:8335003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Doliba NM, Qin W, Najafi H, Liu C, Buettger CW, Sotiris J, Collins HW, Li C, Stanley CA, Wilson DF, et al.. Glucokinase activation repairs defective bioenergetics of islets of Langerhans isolated from type 2 diabetics. Am J Physiol Endocrinol Metab 2012; 302:E87-E102; PMID:21952036; https://doi.org/ 10.1152/ajpendo.00218.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Matschinsky FM. Banting lecture 1995. A lesson in metabolic regulation inspired by the glucokinase glucose sensor paradigm. Diabetes 1996; 45:223-41; PMID:8549869; https://doi.org/ 10.2337/diab.45.2.223 [DOI] [PubMed] [Google Scholar]
- [31].Gloyn AL. Glucokinase (GCK) mutations in hyper- and hypoglycemia: Maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemia of infancy. Hum Mutat 2003; 22:353-62; PMID:14517946; https://doi.org/ 10.1002/humu.10277 [DOI] [PubMed] [Google Scholar]
- [32].Grupe A, Hultgren B, Ryan A, Ma YH, Bauer M, Stewart TA. Transgenic knockouts reveal a critical requirement for pancreatic beta cell glucokinase in maintaining glucose homeostasis. Cell 1995; 83:69-78; PMID:7553875; https://doi.org/ 10.1016/0092-8674(95)90235-X [DOI] [PubMed] [Google Scholar]
- [33].Stoffel M, Froguel P, Takeda J, Zouali H, Vionnet N, Nishi S, Weber IT, Harrison RW, Pilkis SJ, Lesage S, et al.. Human glucokinase gene: Isolation, characterization, and identification of two missense mutations linked to early-onset non-insulin-dependent (type 2) diabetes mellitus. Proc Natl Acad Sci U S A 1992; 89:7698-702; PMID:1502186; https://doi.org/ 10.1073/pnas.89.16.7698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Njolstad PR, Sagen JV, Bjorkhaug L, Odili S, Shehadeh N, Bakry D, Sarici SU, Alpay F, Molnes J, Molven A, et al.. Permanent neonatal diabetes caused by glucokinase deficiency: Inborn error of the glucose-insulin signaling pathway. Diabetes 2003; 52:2854-60; PMID:14578306; https://doi.org/ 10.2337/diabetes.52.11.2854 [DOI] [PubMed] [Google Scholar]
- [35].Glaser B, Kesavan P, Heyman M, Davis E, Cuesta A, Buchs A, Stanley CA, Thornton PS, Permutt MA, Matschinsky FM, et al.. Familial hyperinsulinism caused by an activating glucokinase mutation. N Engl J Med 1998; 338:226-30; PMID:9435328; https://doi.org/ 10.1056/NEJM199801223380404 [DOI] [PubMed] [Google Scholar]
- [36].Brouwers MCGJ, Jacobs C, Bast A, Stehouwer CDA, Schaper NC. Modulation of glucokinase regulatory protein: A double-edged sword? Trends Mol Med 2015; 21:583-94; PMID:26432016; https://doi.org/ 10.1016/j.molmed.2015.08.004 [DOI] [PubMed] [Google Scholar]
- [37].Matschinsky FM. Assessing the potential of glucokinase activators in diabetes therapy. Nat Rev Drug Discov 2009; 8:399-416; PMID:19373249; https://doi.org/ 10.1038/nrd2850 [DOI] [PubMed] [Google Scholar]
- [38].Bertram R, Sherman A, Satin LS. Metabolic and electrical oscillations: partners in controlling pulsatile insulin secretion. Am J Physiol Endocrinol Metab 2007; 293:E890-E900; PMID:17666486; https://doi.org/ 10.1152/ajpendo.00359.2007 [DOI] [PubMed] [Google Scholar]
- [39].Bertram R, Sherman A, Satin LS. Electrical bursting, calcium oscillations, and synchronization of pancreatic islets. The islets of langerhans. In: Islam MS, ed.: Springer; Netherlands, 2010:261-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Luciani DS, Misler S, Polonsky KS. Ca2+ controls slow NAD(P)H oscillations in glucose-stimulated mouse pancreatic islets. J Physiol 2006; 572:379-92; PMID:16455690; https://doi.org/ 10.1113/jphysiol.2005.101766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wiederkehr A, Wollheim CB. Mitochondrial signals drive insulin secretion in the pancreatic beta-cell. Mol Cell Endocrinol 2012; 353:128-37; PMID:21784130; https://doi.org/ 10.1016/j.mce.2011.07.016 [DOI] [PubMed] [Google Scholar]
- [42].Wiederkehr A, Wollheim CB. Impact of mitochondrial calcium on the coupling of metabolism to insulin secretion in the pancreatic beta-cell. Cell Calcium 2008; 44:64-76; PMID:18191448; https://doi.org/ 10.1016/j.ceca.2007.11.004 [DOI] [PubMed] [Google Scholar]
- [43].Maechler P. Mitochondrial function and insulin secretion. Mol Cell Endocrinol 2013; 379:12-8; PMID:23792187; https://doi.org/ 10.1016/j.mce.2013.06.019 [DOI] [PubMed] [Google Scholar]
- [44].Pendin D, Greotti E, Pozzan T. The elusive importance of being a mitochondrial Ca(2+) uniporter. Cell Calcium 2014; 55:139-45; PMID:24631327; https://doi.org/ 10.1016/j.ceca.2014.02.008 [DOI] [PubMed] [Google Scholar]
- [45].Gilon P, Chae HY, Rutter GA, Ravier MA. Calcium signaling in pancreatic beta-cells in health and in Type 2 diabetes. Cell Calcium 2014; 56:340-61; PMID:25239387; https://doi.org/ 10.1016/j.ceca.2014.09.001 [DOI] [PubMed] [Google Scholar]
- [46].Nicholls DG. The pancreatic beta-cell: A bioenergetic perspective. Physiol Rev 2016; 96:1385-447; PMID:27582250; https://doi.org/ 10.1152/physrev.00009.2016 [DOI] [PubMed] [Google Scholar]
- [47].Rutter GA, Pullen TJ, Hodson DJ, Martinez-Sanchez A. Pancreatic beta-cell identity, glucose sensing and the control of insulin secretion. Biochem J 2015; 466:203-18; PMID:25697093; https://doi.org/ 10.1042/BJ20141384 [DOI] [PubMed] [Google Scholar]
- [48].Tarasov AI, Semplici F, Ravier MA, Bellomo EA, Pullen TJ, Gilon P, Sekler I, Rizzuto R, Rutter GA. The mitochondrial Ca2+uniporter MCU is essential for glucose-induced ATP increases in pancreatic β-Cells. PLoS ONE 2012; 7:e39722; PMID:22829870; https://doi.org/ 10.1371/journal.pone.0039722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Tarasov A, Semplici F, Li D, Rizzuto R, Ravier M, Gilon P, Rutter GA. Frequency-dependent mitochondrial Ca2+ accumulation regulates ATP synthesis in pancreatic β cells. Pflugers Arch 2013; 465:543-54; PMID:23149488; https://doi.org/ 10.1007/s00424-012-1177-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Quesada I, Villalobos C, Nunez L, Chamero P, Alonso MT, Nadal A, García-Sancho J. Glucose induces synchronous mitochondrial calcium oscillations in intact pancreatic islets. Cell Calcium 2008; 43:39-47; PMID:17499355; https://doi.org/ 10.1016/j.ceca.2007.03.001 [DOI] [PubMed] [Google Scholar]
- [51].Rutter GA, Tsuboi T, Ravier MA. Ca2+ microdomains and the control of insulin secretion. Cell Calcium 2006; 40:539-51; PMID:17030367; https://doi.org/ 10.1016/j.ceca.2006.08.015 [DOI] [PubMed] [Google Scholar]
- [52].Tarasov AI, Griffiths EJ, Rutter GA. Regulation of ATP production by mitochondrial Ca2+. Cell Calcium 2012; 52:28-35; PMID:22502861; https://doi.org/ 10.1016/j.ceca.2012.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Nita II, Hershfinkel M, Fishman D, Ozeri E, Rutter GA, Sensi SL, Khananshvili D, Lewis EC, Sekler I. The mitochondrial Na+/Ca2+ exchanger upregulates glucose dependent Ca2+ signalling linked to insulin secretion. PLoS ONE 2012; 7:e46649; PMID:23056385; https://doi.org/ 10.1371/journal.pone.0046649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].De Marchi U, Thevenet J, Hermant A, Dioum E, Wiederkehr A. Calcium co-regulates oxidative metabolism and ATP-synthase dependent respiration in pancreatic beta-cells. J Biol Chem 2014; 289(13):9182-94; PMID:24554722; https://doi.org/ 10.1074/jbc.M113.513184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Hodson DJ, Tarasov AI, Gimeno Brias S, Mitchell RK, Johnston NR, Haghollahi S, Cane MC, Bugliani M, Marchetti P, Bosco D, et al.. Incretin-modulated beta cell energetics in intact islets of Langerhans. Mol Endocrinol 2014; 28(6):860-71; PMID:24766140; https://doi.org/ 10.1210/me.2014-1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Hodson DJ, Mitchell RK, Bellomo EA, Sun G, Vinet L, Meda P, Li D, Li WH, Bugliani M, Marchetti P, et al.. Lipotoxicity disrupts incretin-regulated human beta cell connectivity. J Clin Invest 2013; 123:4182-94; PMID:24018562; https://doi.org/ 10.1172/JCI68459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kanno T, Rorsman P, Göpel SO. Glucose-dependent regulation of rhythmic action potential firing in pancreatic β-cells by kATP-channel modulation. J Physiol 2002; 545:501-7; PMID:12456829; https://doi.org/ 10.1113/jphysiol.2002.031344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ashcroft FM, Harrison DE, Ashcroft SJH. Glucose induces closure of single potassium channels in isolated rat pancreatic [beta]-cells. Nature 1984; 312:446-8; PMID:6095103; https://doi.org/ 10.1038/312446a0 [DOI] [PubMed] [Google Scholar]
- [59].Inagaki N, Gonoi T, Clement JP 4th, Namba N, Inazawa J, Gonzalez G, Aguilar-Bryan L, Seino S, Bryan J. Reconstitution of IKATP: An inward rectifier subunit plus the sulfonylurea receptor. Science 1995; 270:1166-70; PMID:7502040; https://doi.org/ 10.1126/science.270.5239.1166 [DOI] [PubMed] [Google Scholar]
- [60].Zerangue N, Schwappach B, Jan YN, Jan LY. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron 1999; 22:537-48; PMID:10197533; https://doi.org/ 10.1016/S0896-6273(00)80708-4 [DOI] [PubMed] [Google Scholar]
- [61].Aguilar-Bryan L, Bryan J, Nakazaki M. Of mice and men: KATP channels and insulin secretion. Recent Prog Horm Res 2001; 56:47-68; PMID:11237225; https://doi.org/ 10.1210/rp.56.1.47 [DOI] [PubMed] [Google Scholar]
- [62].Speier S, Yang SB, Sroka K, Rose T, Rupnik M. KATP-channels in beta-cells in tissue slices are directly modulated by millimolar ATP. Mol Cell Endocrinol 2005; 230:51-8; PMID:15664451; https://doi.org/ 10.1016/j.mce.2004.11.002 [DOI] [PubMed] [Google Scholar]
- [63].Koster JC, Remedi MS, Dao C, Nichols CG. ATP and sulfonylurea sensitivity of mutant ATP-sensitive K+ channels in neonatal diabetes: Implications for pharmacogenomic therapy. Diabetes 2005; 54:2645-54; PMID:16123353; https://doi.org/ 10.2337/diabetes.54.9.2645 [DOI] [PubMed] [Google Scholar]
- [64].Ciani S, Ribalet B. Ion permeation and rectification in ATP-sensitive channels from insulin-secreting cells (RINm5F): effects of K+, Na+ and Mg2+. J Membr Biol 1988; 103:171-80; PMID:2846847; https://doi.org/ 10.1007/BF01870947 [DOI] [PubMed] [Google Scholar]
- [65].Fan Z, Makielski JC. Anionic phospholipids activate ATP-sensitive potassium channels. J Biol Chem 1997; 272:5388-95; PMID:9038137; https://doi.org/ 10.1074/jbc.272.9.5388 [DOI] [PubMed] [Google Scholar]
- [66].Ashcroft FM, Kakei M, Kelly RP, Sutton R. ATP-sensitive K+ channels in human isolated pancreatic B-cells. FEBS Lett 1987; 215:9-12; PMID:2436949; https://doi.org/ 10.1016/0014-5793(87)80103-5 [DOI] [PubMed] [Google Scholar]
- [67].Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining GJ, Slingerland AS, Howard N, Srinivasan S, Silva JM, Molnes J, et al.. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med 2004; 350:1838-49; PMID:15115830; https://doi.org/ 10.1056/NEJMoa032922 [DOI] [PubMed] [Google Scholar]
- [68].Dunne MJ, Cosgrove KE, Shepherd RM, Aynsley-Green A, Lindley KJ. Hyperinsulinism in infancy: From basic science to clinical disease. Physiol Rev 2004; 84:239-75; PMID:14715916; https://doi.org/ 10.1152/physrev.00022.2003 [DOI] [PubMed] [Google Scholar]
- [69].Beaudelaire Tsiaze E, Huang Y, Bombek L, Yang S, Jevšek M, Seino S, Slak Rupnik M. Age-dependent changes in the exocytotic efficacy in Kir6.2 ablated mouse pancreatic β-cells. Open J Mol Integr Physiol 2012; 2:51-60; https://doi.org/ 10.4236/ojmip.2012.23008 [DOI] [Google Scholar]
- [70].Koster JC, Marshall BA, Ensor N, Corbett JA, Nichols CG. Targeted overactivity of beta cell K(ATP) channels induces profound neonatal diabetes. Cell 2000; 100:645-54; PMID:10761930; https://doi.org/ 10.1016/S0092-8674(00)80701-1 [DOI] [PubMed] [Google Scholar]
- [71].Aguilar-Bryan L, Bryan J. Neonatal diabetes mellitus. Endocr Rev 2008; 29:265-91; PMID:18436707; https://doi.org/ 10.1210/er.2007-0029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Babenko AP, Polak M, Cave H, Busiah K, Czernichow P, Scharfmann R, Bryan J, Aguilar-Bryan L, Vaxillaire M, Froguel P. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N Engl J Med 2006; 355:456-66; PMID:16885549; https://doi.org/ 10.1056/NEJMoa055068 [DOI] [PubMed] [Google Scholar]
- [73].Ellard S, Flanagan Sarah E, Girard Christophe A, Patch A-M, Harries Lorna W, Parrish A, Edghill EL, Mackay DJ, Proks P, Shimomura K, et al.. Permanent neonatal diabetes caused by dominant, recessive, or compound heterozygous SUR1 mutations with opposite functional effects. Am J Hum Genet 2007; 81:375-82; PMID:17668386; https://doi.org/ 10.1086/519174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Islam MS. Calcium signaling in the islets the islets of langerhans. In: Islam MS, ed.: Springer; Netherlands, 2010:235-59. [DOI] [PubMed] [Google Scholar]
- [75].Hisanaga E, Nagasawa M, Ueki K, Kulkarni RN, Mori M, Kojima I. Regulation of calcium-permeable TRPV2 channel by insulin in pancreatic beta-cells. Diabetes 2009; 58:174-84; PMID:18984736; https://doi.org/ 10.2337/db08-0862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Casas S, Novials A, Reimann F, Gomis R, Gribble FM. Calcium elevation in mouse pancreatic beta cells evoked by extracellular human islet amyloid polypeptide involves activation of the mechanosensitive ion channel TRPV4. Diabetologia 2008; 51:2252-62; PMID:18751967; https://doi.org/ 10.1007/s00125-008-1111-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Babes A, Fischer MJ, Filipovic M, Engel MA, Flonta ML, Reeh PW. The anti-diabetic drug glibenclamide is an agonist of the transient receptor potential Ankyrin 1 (TRPA1) ion channel. Eur J Pharmacol 2013; 704:15-22; PMID:23461853; https://doi.org/ 10.1016/j.ejphar.2013.02.018 [DOI] [PubMed] [Google Scholar]
- [78].Colsoul B, Vennekens R, Nilius B. Transient receptor potential cation channels in pancreatic β Cells In: Amara GS, Bamberg E, Fleischmann KB, Gudermann T, Jahn R, Lederer JW, et al., eds. Reviews of physiology, biochemistry and pharmacology 161. Berlin, Heidelberg: Springer Berlin Heidelberg, 2011:87-110. [DOI] [PubMed] [Google Scholar]
- [79].Oberwinkler J, Philipp SE. TRPM3 In: Nilius B, Flockerzi V, eds. Mammalian transient receptor potential (TRP) cation channels: Volume I. Berlin, Heidelberg: Springer Berlin Heidelberg, 2014:427-59. [Google Scholar]
- [80].Vriens J, Held K, Janssens A, Tóth BI, Kerselaers S, Nilius B, Vennekens R, Voets T. Opening of an alternative ion permeation pathway in a nociceptor TRP channel. Nat Chem Biol 2014; 10:188-95; PMID:24390427; https://doi.org/ 10.1038/nchembio.1428 [DOI] [PubMed] [Google Scholar]
- [81].Philippaert K, Pironet A, Mesuere M, Sones W, Vermeiren L, Kerselaers S, Pinto S, Segal A, Antoine N, Gysemans C, et al.. Steviol glycosides enhance pancreatic beta-cell function and taste sensation by potentiation of TRPM5 channel activity. Nat Commun 2017; 8:14733; PMID:28361903; https://doi.org/ 10.1038/ncomms14733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Colsoul B, Schraenen A, Lemaire K, Quintens R, Van Lommel L, Segal A, Owsianik G, Talavera K, Voets T, Margolskee RF, et al.. Loss of high-frequency glucose-induced Ca2+ oscillations in pancreatic islets correlates with impaired glucose tolerance in Trpm5-/- mice. Proc Natl Acad Sci U S A 2010; 107:5208-13; PMID:20194741; https://doi.org/ 10.1073/pnas.0913107107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Dadi PK, Vierra NC, Jacobson DA. Pancreatic beta-cell-specific ablation of TASK-1 channels augments glucose-stimulated calcium entry and insulin secretion, improving glucose tolerance. Endocrinology 2014; 155:3757-68; PMID:24932805; https://doi.org/ 10.1210/en.2013-2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Vierra NC, Dadi PK, Jeong I, Dickerson M, Powell DR, Jacobson DA. Type 2 Diabetes–Associated K(+) Channel TALK-1 modulates β-cell electrical excitability, second-phase insulin secretion, and glucose homeostasis. Diabetes 2015; 64:3818-28; PMID:26239056; https://doi.org/ 10.2337/db15-0280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Dickerson MT, Vierra NC, Milian SC, Dadi PK, Jacobson DA. Osteopontin activates the diabetes-associated potassium channel TALK-1 in pancreatic β-cells. PLoS ONE 2017; 12:e0175069; PMID:28403169; https://doi.org/ 10.1371/journal.pone.0175069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Swayne LA, Mezghrani A, Lory P, Nargeot J, Monteil A. The NALCN ion channel is a new actor in pancreatic β-cell physiology. Islets 2010; 2:54-6; PMID:21099296; https://doi.org/ 10.4161/isl.2.1.10522 [DOI] [PubMed] [Google Scholar]
- [87].Speier S, Gjinovci A, Charollais A, Meda P, Rupnik M. Cx36-mediated coupling reduces beta-cell heterogeneity, confines the stimulating glucose concentration range, and affects insulin release kinetics. Diabetes 2007; 56:1078-86; PMID:17395748; https://doi.org/ 10.2337/db06-0232 [DOI] [PubMed] [Google Scholar]
- [88].Stožer A, Gosak M, Dolenšek J, Perc M, Marhl M, Rupnik MS, Korošak D. Functional connectivity in islets of langerhans from mouse pancreas tissue slices. PLoS Comput Biol 2013; 9:e1002923; PMID:23468610; https://doi.org/ 10.1371/journal.pcbi.1002923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Notary AM, Westacott MJ, Hraha TH, Pozzoli M, Benninger RK. Decreases in gap junction coupling recovers Ca2+ and insulin secretion in neonatal diabetes mellitus, dependent on beta cell heterogeneity and noise. PLoS Comput Biol 2016; 12:e1005116; PMID:27681078; https://doi.org/ 10.1371/journal.pcbi.1005116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Nguyen LM, Pozzoli M, Hraha TH, Benninger RK. Decreasing cx36 gap junction coupling compensates for overactive KATP channels to restore insulin secretion and prevent hyperglycemia in a mouse model of neonatal diabetes. Diabetes 2014; 63:1685-97; PMID:24458355; https://doi.org/ 10.2337/db13-1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Farnsworth NL, Benninger RK. New insights into the role of connexins in pancreatic islet function and diabetes. FEBS Lett 2014; 588:1278-87; PMID:24583073; https://doi.org/ 10.1016/j.febslet.2014.02.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Ashcroft FM, Rorsman P. Electrophysiology of the pancreatic beta-cell. Prog Biophys Mol Biol 1989; 54:87-143; PMID:2484976; https://doi.org/ 10.1016/0079-6107(89)90013-8 [DOI] [PubMed] [Google Scholar]
- [93].Braun M, Ramracheya R, Bengtsson M, Zhang Q, Karanauskaite J, Partridge C, Johnson PR, Rorsman P. Voltage-gated ion channels in human pancreatic beta-cells: Electrophysiological characterization and role in insulin secretion. Diabetes 2008; 57:1618-28; PMID:18390794; https://doi.org/ 10.2337/db07-0991 [DOI] [PubMed] [Google Scholar]
- [94].Rorsman P, Braun M, Zhang Q. Regulation of calcium in pancreatic alpha- and beta-cells in health and disease. Cell Calcium 2012; 51:300-8; PMID:22177710; https://doi.org/ 10.1016/j.ceca.2011.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Schulla V, Renstrom E, Feil R, Feil S, Franklin I, Gjinovci A, Jing XJ, Laux D, Lundquist I, Magnuson MA, et al.. Impaired insulin secretion and glucose tolerance in beta cell-selective Ca(v)1.2 Ca2+ channel null mice. EMBO J 2003; 22:3844-54; PMID:12881419; https://doi.org/ 10.1093/emboj/cdg389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Jing X, Li DQ, Olofsson CS, Salehi A, Surve VV, Caballero J, Ivarsson R, Lundquist I, Pereverzev A, Schneider T, et al.. CaV2.3 calcium channels control second-phase insulin release. J Clin Invest 2005; 115:146-54; PMID:15630454; https://doi.org/ 10.1172/JCI200522518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Holmkvist J, Tojjar D, Almgren P, Lyssenko V, Lindgren CM, Isomaa B, Tuomi T, Berglund G, Renström E, Groop L. Polymorphisms in the gene encoding the voltage-dependent Ca(2+) channel Ca (V)2.3 (CACNA1E) are associated with type 2 diabetes and impaired insulin secretion. Diabetologia 2007; 50:2467-75; PMID:17934712; https://doi.org/ 10.1007/s00125-007-0846-2 [DOI] [PubMed] [Google Scholar]
- [98].Rorsman P, Braun M. Regulation of insulin secretion in human pancreatic islets. Annu Rev Physiol 2013; 75:155-79; PMID:22974438; https://doi.org/ 10.1146/annurev-physiol-030212-183754 [DOI] [PubMed] [Google Scholar]
- [99].Barnett DW, Pressel DM, Misler S. Voltage-dependent Na+ and Ca2+ currents in human pancreatic islet beta-cells: evidence for roles in the generation of action potentials and insulin secretion. Pflugers Arch 1995; 431:272-82; PMID:9026789; https://doi.org/ 10.1007/BF00410201 [DOI] [PubMed] [Google Scholar]
- [100].Zhang Q, Chibalina MV, Bengtsson M, Groschner LN, Ramracheya R, Rorsman NJG, Leiss V, Nassar MA, Welling A, Gribble FM, et al.. Na(+) current properties in islet α- and β-cells reflect cell-specific Scn3a and Scn9a expression. J Physiol 2014; 592:4677-96; PMID:25172946; https://doi.org/ 10.1113/jphysiol.2014.274209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Göpel S, Kanno T, Barg S, Galvanovskis J, Rorsman P. Voltage-gated and resting membrane currents recorded from B-cells in intact mouse pancreatic islets. J Physiol 1999; 521:717-28; PMID:10601501; https://doi.org/ 10.1111/j.1469-7793.1999.00717.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Pressel DM, Misler S. Sodium channels contribute to action potential generation in canine and human pancreatic islet B cells. J Membr Biol 1990; 116:273-80; PMID:2167377; https://doi.org/ 10.1007/BF01868466 [DOI] [PubMed] [Google Scholar]
- [103].Pedersen MG. A biophysical model of electrical activity in human beta-cells. Biophys J 2010; 99:3200-7; PMID:21081067; https://doi.org/ 10.1016/j.bpj.2010.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Riz M, Braun M, Pedersen MG. Mathematical modeling of heterogeneous electrophysiological responses in human β-Cells. PLoS Comput Biol 2014; 10:e1003389; PMID:24391482; https://doi.org/ 10.1371/journal.pcbi.1003389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Misler S, Barnett DW, Gillis KD, Pressel DM. Electrophysiology of stimulus-secretion coupling in human beta-cells. Diabetes 1992; 41:1221-8; PMID:1397696; https://doi.org/ 10.2337/diab.41.10.1221 [DOI] [PubMed] [Google Scholar]
- [106].Nica AC, Ongen H, Irminger JC, Bosco D, Berney T, Antonarakis SE, Halban PA, Dermitzakis ET. Cell-type, allelic, and genetic signatures in the human pancreatic beta cell transcriptome. Genome Res 2013; 23:1554-62; PMID:23716500; https://doi.org/ 10.1101/gr.150706.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Fakler B, Adelman JP. Control of K(Ca) channels by calcium nano/microdomains. Neuron 2008; 59:873-81; PMID:18817728; https://doi.org/ 10.1016/j.neuron.2008.09.001 [DOI] [PubMed] [Google Scholar]
- [108].Kukuljan M, Goncalves AA, Atwater I. Charybdotoxin-sensitive K(Ca) channel is not involved in glucose-induced electrical activity in pancreatic beta-cells. J Membr Biol 1991; 119:187-95; PMID:1710672; https://doi.org/ 10.1007/BF01871418 [DOI] [PubMed] [Google Scholar]
- [109].Jacobson DA, Kuznetsov A, Lopez JP, Kash S, Ammala CE, Philipson LH. Kv2.1 ablation alters glucose-induced islet electrical activity, enhancing insulin secretion. Cell Metab 2007; 6:229-35; PMID:17767909; https://doi.org/ 10.1016/j.cmet.2007.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Li X, Herrington J, Petrov A, Ge L, Eiermann G, Xiong Y, Jensen MV, Hohmeier HE, Newgard CB, Garcia ML, et al.. The role of voltage-gated potassium channels Kv2.1 and Kv2.2 in the regulation of insulin and somatostatin release from pancreatic islets. J Pharmacol Exp Ther 2013; 344:407; PMID:23161216; https://doi.org/ 10.1124/jpet.112.199083 [DOI] [PubMed] [Google Scholar]
- [111].MacDonald PE, Sewing S, Wang J, Joseph JW, Smukler SR, Sakellaropoulos G, Wang J, Saleh MC, Chan CB, Tsushima RG, et al.. Inhibition of Kv2.1 Vage-dependent K+channels in pancreatic β-cells enhances glucose-dependent insulin secretion. J Biol Chem 2002; 277:44938-45; PMID:12270920; https://doi.org/ 10.1074/jbc.M205532200 [DOI] [PubMed] [Google Scholar]
- [112].Jacobson DA, Mendez F, Thompson M, Torres J, Cochet O, Philipson LH. Calcium-activated and voltage-gated potassium channels of the pancreatic islet impart distinct and complementary roles during secretagogue induced electrical responses. J Physiol 2010; 588:3525-37; PMID:20643768; https://doi.org/ 10.1113/jphysiol.2010.190207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Rosati B, Marchetti P, Crociani O, Lecchi M, Lupi R, Arcangeli A, Olivotto M, Wanke E. Glucose- and arginine-induced insulin secretion by human pancreatic beta-cells: the role of HERG K(+) channels in firing and release. FASEB J 2000; 14:2601-10; PMID:11099479; https://doi.org/ 10.1096/fj.00-0077com [DOI] [PubMed] [Google Scholar]
- [114].Hardy AB, Fox JEM, Giglou PR, Wijesekara N, Bhattacharjee A, Sultan S, Gyulkhandanyan AV, Gaisano HY, MacDonald PE, Wheeler MB. Characterization of Erg K+ Channels in α- and β-Cells of mouse and human islets. J Biol Chem 2009; 284:30441-52; PMID:19690348; https://doi.org/ 10.1074/jbc.M109.040659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Atwater I, Rosario L, Rojas E. Properties of the Ca-Activated K+ channel in pancreatic beta-cells. Cell Calcium 1983; 4:451-61; PMID:6323007; https://doi.org/ 10.1016/0143-4160(83)90021-0 [DOI] [PubMed] [Google Scholar]
- [116].Dufer M, Gier B, Wolpers D, Krippeit-Drews P, Ruth P, Drews G. Enhanced glucose tolerance by SK4 channel inhibition in pancreatic beta-cells. Diabetes 2009; 58:1835-43; PMID:19401418; https://doi.org/ 10.2337/db08-1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Dean PM, Matthews EK. Electrical activity in pancreatic islet cells. Nature 1968; 219:389-90; PMID:4873864; https://doi.org/ 10.1038/219389a0 [DOI] [PubMed] [Google Scholar]
- [118].Felix-Martinez GJ, Godinez-Fernandez JR. Mathematical models of electrical activity of the pancreatic beta-cell: A physiological review. Islets 2014; 6:e949195; PMID:25322829; https://doi.org/ 10.4161/19382014.2014.949195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Henquin JC, Meissner HP, Schmeer W. Cyclic variations of glucose-induced electrical activity in pancreatic B cells. Pflügers Arch 1982; 393:322-7; PMID:6750552; https://doi.org/ 10.1007/BF00581418 [DOI] [PubMed] [Google Scholar]
- [120].Rorsman P, Eliasson L, Kanno T, Zhang Q, Gopel S. Electrophysiology of pancreatic β-cells in intact mouse islets of Langerhans. Prog Biophys Mol Biol 2011; 107:224-35; PMID:21762719; https://doi.org/ 10.1016/j.pbiomolbio.2011.06.009 [DOI] [PubMed] [Google Scholar]
- [121].Sanchez-Andres JV, Gomis A, Valdeolmillos M. The electrical activity of mouse pancreatic beta-cells recorded in vivo shows glucose-dependent oscillations. J Physiol 1995; 486(Pt 1):223-8; PMID:7562637; https://doi.org/ 10.1113/jphysiol.1995.sp020804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Misler S, Barnett DW, Pressel DM, Gillis KD, Scharp DW, Falke LC. Stimulus-secretion coupling in beta-cells of transplantable human islets of Langerhans. Evidence for a critical role for Ca2+ entry. Diabetes 1992; 41:662-70; PMID:1375175; https://doi.org/ 10.2337/diab.41.6.662 [DOI] [PubMed] [Google Scholar]
- [123].Falke LC, Gillis KD, Pressel DM, Misler S. ‘Perforated patch recording’ allows long-term monitoring of metabolite-induced electrical activity and voltage-dependent Ca2+ currents in pancreatic islet B cells. FEBS Lett 1989; 251:167-72; PMID:2473925; https://doi.org/ 10.1016/0014-5793(89)81448-6 [DOI] [PubMed] [Google Scholar]
- [124].Braun M, Ramracheya R, Johnson PR, Rorsman P. Exocytotic properties of human pancreatic β-cells. Ann N Y Acad Sci 2009; 1152:187-93; PMID:19161389; https://doi.org/ 10.1111/j.1749-6632.2008.03992.x [DOI] [PubMed] [Google Scholar]
- [125].Valdeolmillos M, Santos RM, Contreras D, Soria B, Rosario LM. Glucose-induced oscillations of intracellular Ca2+ concentration resembling bursting electrical activity in single mouse islets of Langerhans. FEBS Lett 1989; 259:19-23; PMID:2689228; https://doi.org/ 10.1016/0014-5793(89)81484-X [DOI] [PubMed] [Google Scholar]
- [126].Valdeolmillos M, Gomis A, Sánchez-Andrés JV. In vivo synchronous membrane potential oscillations in mouse pancreatic beta-cells: Lack of co-ordination between islets. J Physiol 1996; 493:9-18; PMID:8735691; https://doi.org/ 10.1113/jphysiol.1996.sp021361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Manning Fox JE, Gyulkhandanyan AV, Satin LS, Wheeler MB. Oscillatory membrane potential response to glucose in islet β-cells: A comparison of islet-cell electrical activity in mouse and Rat. Endocrinology 2006; 147:4655-63; PMID:16857746; https://doi.org/ 10.1210/en.2006-0424 [DOI] [PubMed] [Google Scholar]
- [128].Moreno AP, Berthoud VM, Pérez-Palacios G, Pérez-Armendariz EM. Biophysical evidence that connexin-36 forms functional gap junction channels between pancreatic mouse β-cells. Am J Physiol Endocrinol Metab 2005; 288:E948-E56; PMID:15625088; https://doi.org/ 10.1152/ajpendo.00216.2004 [DOI] [PubMed] [Google Scholar]
- [129].Pérez-Armendariz M, Roy C, Spray DC, Bennett MV. Biophysical properties of gap junctions between freshly dispersed pairs of mouse pancreatic beta cells. Biophys J 1991; 59:76-92; PMID:2015391; https://doi.org/ 10.1016/S0006-3495(91)82200-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Orci L, Malaisse-Lagae F, Amherdt M, Ravazzola M, Weisswange A, Dobbs R, Perrelet A, Unger R. Cell contacts in human islets of langerhans. J Clin Endocrinol Metab 1975; 41:841-4; PMID:1102552; https://doi.org/ 10.1210/jcem-41-5-841 [DOI] [PubMed] [Google Scholar]
- [131].Coronel-Cruz C, Hernandez-Tellez B, Lopez-Vancell R, Lopez-Vidal Y, Berumen J, Castell A, Pérez-Armendariz EM. Connexin 30.2 is expressed in mouse pancreatic beta cells. Biochem Biophys Res Commun 2013; 438:772-7; PMID:23831630; https://doi.org/ 10.1016/j.bbrc.2013.06.100 [DOI] [PubMed] [Google Scholar]
- [132].Serre-Beinier V, Le Gurun S, Belluardo N, Trovato-Salinaro A, Charollais A, Haefliger JA, Condorelli DF, Meda P. Cx36 preferentially connects beta-cells within pancreatic islets. Diabetes 2000; 49:727-34; PMID:10905480; https://doi.org/ 10.2337/diabetes.49.5.727 [DOI] [PubMed] [Google Scholar]
- [133].Serre-Beinier V, Bosco D, Zulianello L, Charollais A, Caille D, Charpantier E, Gauthier BR, Diaferia GR, Giepmans BN, Lupi R, et al.. Cx36 makes channels coupling human pancreatic β-cells, and correlates with insulin expression. Hum Mol Genet 2009; 18:428-39; PMID:19000992; https://doi.org/ 10.1093/hmg/ddn370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Zhang Q, Galvanovskis J, Abdulkader F, Partridge CJ, Gopel SO, Eliasson L, Rorsman P. Cell coupling in mouse pancreatic beta-cells measured in intact islets of Langerhans. Philos Trans A Math Phys Eng Sci 2008; 366:3503-23; PMID:18632454; https://doi.org/ 10.1098/rsta.2008.0110 [DOI] [PubMed] [Google Scholar]
- [135].Loppini A, Braun M, Filippi S, Pedersen MG. Mathematical modeling of gap junction coupling and electrical activity in human beta-cells. Phys Biol 2015; 12:066002; PMID:26403477; https://doi.org/ 10.1088/1478-3975/12/6/066002 [DOI] [PubMed] [Google Scholar]
- [136].Nittala A, Ghosh S, Wang X. Investigating the role of islet cytoarchitecture in its oscillation using a new β-cell cluster model. PLoS ONE 2007; 2:e983; PMID:17912360; https://doi.org/ 10.1371/journal.pone.0000983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Mears D, Sheppard NF, Atwater I, Rojas E. Magnitude and modulation of pancreatic beta-cell gap junction electrical conductance In-Situ. J Membr Biol 1995; 146:163-76; PMID:7473686; https://doi.org/ 10.1007/BF00238006 [DOI] [PubMed] [Google Scholar]
- [138].Farnsworth NL, Walter RL, Hemmati A, Westacott MJ, Benninger RK. Low level pro-inflammatory cytokines decrease connexin36 gap junction coupling in mouse and human islets through nitric oxide-mediated protein kinase Cdelta. J Biol Chem 2016; 291:3184-96; PMID:26668311; https://doi.org/ 10.1074/jbc.M115.679506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Cigliola V, Chellakudam V, Arabieter W, Meda P. Connexins and β-cell functions. Diabetes Res Clin Pract 2013; 99:250-9; PMID:23176806; https://doi.org/ 10.1016/j.diabres.2012.10.016 [DOI] [PubMed] [Google Scholar]
- [140].Stožer A, Dolenšek J, Rupnik MS. Glucose-stimulated calcium dynamics in islets of langerhans in acute mouse pancreas tissue slices. PLoS ONE 2013; 8:e54638; PMID:23358454; https://doi.org/ 10.1371/journal.pone.0054638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Dolenšek J, Stožer A, Skelin Klemen M, Miller EW, Slak Rupnik M. The relationship between membrane potential and calcium dynamics in glucose-stimulated beta cell syncytium in acute mouse pancreas tissue slices. PLoS ONE 2013; 8:e82374; PMID:24324777; https://doi.org/ 10.1371/journal.pone.0082374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Tsaneva-Atanasova K, Zimliki CL, Bertram R, Sherman A. Diffusion of calcium and metabolites in pancreatic islets: Killing oscillations with a pitchfork. Biophys J 2006; 90:3434-46; PMID:16500973; https://doi.org/ 10.1529/biophysj.105.078360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Pedersen MG, Bertram R, Sherman A. Intra- and inter-islet synchronization of metabolically driven insulin secretion. Biophys J 2005; 89:107-19; PMID:15834002; https://doi.org/ 10.1529/biophysj.104.055681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Konstantinova I, Nikolova G, Ohara-Imaizumi M, Meda P, Kucera T, Zarbalis K, Wurst W, Nagamatsu S, Lammert E. EphA-Ephrin-A-mediated beta cell communication regulates insulin secretion from pancreatic islets. Cell 2007; 129:359-70; PMID:17448994; https://doi.org/ 10.1016/j.cell.2007.02.044 [DOI] [PubMed] [Google Scholar]
- [145].Olofsson CS, Hakansson J, Salehi A, Bengtsson M, Galvanovskis J, Partridge C, SörhedeWinzell M, Xian X, Eliasson L, Lundquist I, et al.. Impaired insulin exocytosis in neural cell adhesion molecule-/- mice due to defective reorganization of the submembrane F-actin network. Endocrinology 2009; 150:3067-75; PMID:19213846; https://doi.org/ 10.1210/en.2008-0475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Gerdes JM, Christou-Savina S, Xiong Y, Moede T, Moruzzi N, Karlsson-Edlund P, Leibiger B, Leibiger IB, Östenson CG, Beales PL, et al.. Ciliary dysfunction impairs beta-cell insulin secretion and promotes development of type 2 diabetes in rodents. Nat Commun 2014; 5:5308; PMID:25374274; https://doi.org/ 10.1038/ncomms6308 [DOI] [PubMed] [Google Scholar]
- [147].Gan WJ, Zavortink M, Ludick C, Templin R, Webb R, Ma W, Poronnik P, Parton RG, Gaisano HY, Shewan AM, et al.. Cell polarity defines three distinct domains in pancreatic beta-cells. J Cell Sci 2017; 130:143-51; PMID:26919978; https://doi.org/ 10.1242/jcs.185116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Gylfe E, Tengholm A. Neurotransmitter control of islet hormone pulsatility. Diabetes Obes Metab 2014; 16(Suppl 1):102-10; PMID:25200303; https://doi.org/ 10.1111/dom.12345 [DOI] [PubMed] [Google Scholar]
- [149].Rhodes CJ, White MF, Leahy JL, Kahn SE. Direct autocrine action of insulin on beta-cells: does it make physiological sense? Diabetes 2013; 62:2157-63; PMID:23801714; https://doi.org/ 10.2337/db13-0246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Aspinwall CA, Lakey JR, Kennedy RT. Insulin-stimulated insulin secretion in single pancreatic beta cells. J Biol Chem 1999; 274:6360-5; PMID:10037726; https://doi.org/ 10.1074/jbc.274.10.6360 [DOI] [PubMed] [Google Scholar]
- [151].Hutton JC, Penn EJ, Peshavaria M. Low-molecular-weight constituents of isolated insulin-secretory granules. Bivalent cations, adenine nucleotides and inorganic phosphate. Biochem J 1983; 210:297-305; PMID:6344863; https://doi.org/ 10.1042/bj2100297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Leitner JW, Sussman KE, Vatter AE, Schneider FH. Adenine nucleotides in the secretory granule fraction of rat islets. Endocrinology 1975; 96:662-77; PMID:163731; https://doi.org/ 10.1210/endo-96-3-662 [DOI] [PubMed] [Google Scholar]
- [153].Dyachok O, Gylfe E. Ca2+-induced Ca2+ release via inositol 1,4,5-trisphosphate receptors is amplified by protein kinase a and triggers exocytosis in pancreatic β-Cells. J Biol Chem 2004; 279:45455-61; PMID:15316011; https://doi.org/ 10.1074/jbc.M407673200 [DOI] [PubMed] [Google Scholar]
- [154].Petit P, Lajoix AD, Gross R. P2 purinergic signalling in the pancreatic beta-cell: Control of insulin secretion and pharmacology. Eur J Pharm Sci 2009; 37:67-75; PMID:19429412; https://doi.org/ 10.1016/j.ejps.2009.01.007 [DOI] [PubMed] [Google Scholar]
- [155].Silva AM, Rodrigues RJ, Tome AR, Cunha RA, Misler S, Rosario LM, Santos RM. Electrophysiological and immunocytochemical evidence for P2X purinergic receptors in pancreatic beta cells. Pancreas 2008; 36:279-83; PMID:18362842; https://doi.org/ 10.1097/MPA.0b013e31815a8473 [DOI] [PubMed] [Google Scholar]
- [156].Poulsen CR, Bokvist K, Olsen HL, Hoy M, Capito K, Gilon P, Gromada J. Multiple sites of purinergic control of insulin secretion in mouse pancreatic beta-cells. Diabetes 1999; 48:2171-81; PMID:10535451; https://doi.org/ 10.2337/diabetes.48.11.2171 [DOI] [PubMed] [Google Scholar]
- [157].Leon C, Freund M, Latchoumanin O, Farret A, Petit P, Cazenave JP, Gachet C. The P2Y(1) receptor is involved in the maintenance of glucose homeostasis and in insulin secretion in mice. Purinergic Signal 2005; 1:145-51; PMID:18404499; https://doi.org/ 10.1007/s11302-005-6209-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Gylfe E, Grapengiesser E, Dansk H, Hellman B. The neurotransmitter ATP triggers Ca2+ responses promoting coordination of pancreatic islet oscillations. Pancreas 2012; 41:258-63; PMID:22076565; https://doi.org/ 10.1097/MPA.0b013e3182240586 [DOI] [PubMed] [Google Scholar]
- [159].Fernandez-Alvarez J, Hillaire-Buys D, Loubatieres-Mariani MM, Gomis R, Petit P. P2 receptor agonists stimulate insulin release from human pancreatic islets. Pancreas 2001; 22:69-71; PMID:11138974; https://doi.org/ 10.1097/00006676-200101000-00012 [DOI] [PubMed] [Google Scholar]
- [160].Jacques-Silva MC, Correa-Medina M, Cabrera O, Rodriguez-Diaz R, Makeeva N, Fachado A, Diez J, Berman DM, Kenyon NS, Ricordi C, et al.. ATP-gated P2X3 receptors constitute a positive autocrine signal for insulin release in the human pancreatic beta cell. Proc Natl Acad Sci U S A 2010; 107:6465-70; PMID:20308565; https://doi.org/ 10.1073/pnas.0908935107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Kindmark H, Köhler M, Nilsson T, Arkhammar P, Wiechel K-L, Rorsman P, Efendić S, Berggren PO. Measurements of cytoplasmic free Ca2+ concentration in human pancreatic islets and insulinoma cells. FEBS Lett 1991; 291:310-4; PMID:1936280; https://doi.org/ 10.1016/0014-5793(91)81309-V [DOI] [PubMed] [Google Scholar]
- [162].Simpson N, Maffei A, Freeby M, Burroughs S, Freyberg Z, Javitch J, Leibel RL, Harris PE. Dopamine-mediated autocrine inhibitory circuit regulating human insulin secretion in vitro. Mol Endocrinol 2012; 26:1757-72; PMID:22915827; https://doi.org/ 10.1210/me.2012-1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Ustione A, Piston DW, Harris PE. Minireview: Dopaminergic regulation of insulin secretion from the pancreatic islet. Mol Endocrinol 2013; 27:1198-207; PMID:23744894; https://doi.org/ 10.1210/me.2013-1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Braun M, Ramracheya R, Bengtsson M, Clark A, Walker JN, Johnson PR, Rorsman P. Gamma-aminobutyric acid (GABA) is an autocrine excitatory transmitter in human pancreatic beta-cells. Diabetes 2010; 59:1694-701; PMID:20413510; https://doi.org/ 10.2337/db09-0797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Taneera J, Jin Z, Jin Y, Muhammed SJ, Zhang E, Lang S, Salehi A, Korsgren O, Renström E, Groop L, et al.. gamma-Aminobutyric acid (GABA) signalling in human pancreatic islets is altered in type 2 diabetes. Diabetologia 2012; 55:1985-94; PMID:22538358; https://doi.org/ 10.1007/s00125-012-2548-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Hellman B, Salehi A, Grapengiesser E, Gylfe E. Isolated mouse islets respond to glucose with an initial peak of glucagon release followed by pulses of insulin and somatostatin in antisynchrony with glucagon. Biochem Biophys Res Commun 2012; 417:1219-23; PMID:22227186; https://doi.org/ 10.1016/j.bbrc.2011.12.113 [DOI] [PubMed] [Google Scholar]
- [167].Hellman B, Salehi A, Gylfe E, Dansk H, Grapengiesser E. Glucose generates coincident insulin and somatostatin pulses and antisynchronous glucagon pulses from human pancreatic islets. Endocrinology 2009; 150:5334-40; PMID:19819962; https://doi.org/ 10.1210/en.2009-0600 [DOI] [PubMed] [Google Scholar]
- [168].Rodriguez-Diaz R, Menegaz D, Caicedo A. Neurotransmitters act as paracrine signals to regulate insulin secretion from the human pancreatic islet. J Physiol 2014; 592(16):3413-7; PMID:24591573; https://doi.org/ 10.1113/jphysiol.2013.269910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Rodriguez-Diaz R, Speier S, Molano RD, Formoso A, Gans I, Abdulreda MH, Cabrera O, Molina J, Fachado A, Ricordi C, et al.. Noninvasive in vivo model demonstrating the effects of autonomic innervation on pancreatic islet function. Proc Natl Acad Sci U S A 2012; 109:21456-61; PMID:23236142; https://doi.org/ 10.1073/pnas.1211659110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Molina J, Rodriguez-Diaz R, Fachado A, Jacques-Silva MC, Berggren PO, Caicedo A. Control of insulin secretion by cholinergic signaling in the human pancreatic islet. Diabetes 2014; 63:2714-26; PMID:24658304; https://doi.org/ 10.2337/db13-1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Grapengiesser E, Gylfe E, Hellman B. Glucose-induced oscillations of cytoplasmic Ca2+ in the pancreatic β-cell. Biochem Biophys Res Commun 1988; 151:1299-304; PMID:3281672; https://doi.org/ 10.1016/S0006-291X(88)80503-5 [DOI] [PubMed] [Google Scholar]
- [172].Santos RM, Rosario LM, Nadal A, Garcia-Sancho J, Soria B, Valdeolmillos M. Widespread synchronous [Ca2+]i oscillations due to bursting electrical activity in single pancreatic islets. Pflugers Arch 1991; 418:417-22; PMID:1876486; https://doi.org/ 10.1007/BF00550880 [DOI] [PubMed] [Google Scholar]
- [173].Gilon P, Henquin JC. Influence of membrane potential changes on cytoplasmic Ca2+ concentration in an electrically excitable cell, the insulin-secreting pancreatic B-cell. J Biol Chem 1992; 267:20713-20; PMID:1400388 [PubMed] [Google Scholar]
- [174].Gilon P, Shepherd RM, Henquin JC. Oscillations of secretion driven by oscillations of cytoplasmic Ca2+ as evidenced in single pancreatic islets. J Biol Chem 1993; 268:22265-8; PMID:8226733 [PubMed] [Google Scholar]
- [175].Bergsten P, Grapengiesser E, Gylfe E, Tengholm A, Hellman B. Synchronous oscillations of cytoplasmic Ca2+ and insulin release in glucose-stimulated pancreatic islets. J Biol Chem 1994; 269:8749-53; PMID:8132606 [PubMed] [Google Scholar]
- [176].Bergsten P. Slow and fast oscillations of cytoplasmic Ca2+ in pancreatic islets correspond to pulsatile insulin release. Am J Physiol 1995; 268:E282-E7; PMID:7864105 [DOI] [PubMed] [Google Scholar]
- [177].Gilon P, Henquin JC. Distinct effects of glucose on the synchronous oscillations of insulin release and cytoplasmic Ca2+ concentration measured simultaneously in single mouse islets. Endocrinology 1995; 136:5725-30; PMID:7588329; https://doi.org/ 10.1210/endo.136.12.7588329 [DOI] [PubMed] [Google Scholar]
- [178].Aslanidi OV, Mornev OA, Skyggebjerg O, Arkhammar P, Thastrup O, Sørensen MP, Christiansen PL, Conradsen K, Scott AC. Excitation wave propagation as a possible mechanism for signal transmission in pancreatic islets of langerhans. Biophys J 2001; 80:1195-209; PMID:11222284; https://doi.org/ 10.1016/S0006-3495(01)76096-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Meda P, Orci L, Atwater I, Bangham A, Rojas E, Gonçalves A. The topography of electrical synchrony among β-cells in the mouse islet of langerhans. Q J Exp Physiol 1984; 69:716-35; PMID:6440208; https://doi.org/ 10.1113/expphysiol.1984.sp002864 [DOI] [PubMed] [Google Scholar]
- [180].Palti Y, BenDavid G, Lachov E, Mika YH, Omri G, Schatzberger R. Islets of Langerhans generate wavelike electric activity modulated by glucose concentration. Diabetes 1996; 45:595-601; PMID:8621009; https://doi.org/ 10.2337/diab.45.5.595 [DOI] [PubMed] [Google Scholar]
- [181].Valdeolmillos M, Nadal A, Soria B, Garciasancho J. Fluorescence digital image-analysis of glucose-induced Ca2+ oscillations in mouse pancreatic islets of langerhans. Diabetes 1993; 42:1210-4; PMID:8325454; https://doi.org/ 10.2337/diab.42.8.1210 [DOI] [PubMed] [Google Scholar]
- [182].Benninger RK, Zhang M, Head WS, Satin LS, Piston DW. Gap junction coupling and calcium waves in the pancreatic islet. Biophys J 2008; 95:5048-61; PMID:18805925; https://doi.org/ 10.1529/biophysj.108.140863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Dolensek J, Spelic D, Klemen MS, Zalik B, Gosak M, Rupnik MS, Stožer A. Membrane potential and calcium dynamics in beta cells from mouse pancreas tissue slices: Theory, experimentation, and analysis. Sensors (Basel) 2015; 15:27393-419; PMID:26516866; https://doi.org/ 10.3390/s151127393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Gosak M, Dolenšek J, Markovič R, Slak Rupnik M, Marhl M, Stožer A. Multilayer network representation of membrane potential and cytosolic calcium concentration dynamics in beta cells. Chaos Solitons Fractals 2015; 80:76-82; https://doi.org/ 10.1016/j.chaos.2015.06.009 [DOI] [Google Scholar]
- [185].Benninger RK, Hutchens T, Head WS, McCaughey MJ, Zhang M, Le Marchand SJ, Satin LS, Piston DW. Intrinsic islet heterogeneity and gap junction coupling determine spatiotemporal Ca(2)(+) wave dynamics. Biophys J 2014; 107:2723-33; PMID:25468351; https://doi.org/ 10.1016/j.bpj.2014.10.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Johnston NR, Mitchell RK, Haythorne E, Pessoa MP, Semplici F, Ferrer J, Piemonti L, Marchetti P, Bugliani M, Bosco D, et al.. Beta cell hubs dictate pancreatic islet responses to glucose. Cell Metab 2016; 24:389-401; PMID:27452146; https://doi.org/ 10.1016/j.cmet.2016.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Cappon G, Pedersen MG. Heterogeneity and nearest-neighbor coupling can explain small-worldness and wave properties in pancreatic islets. Chaos 2016; 26:053103; PMID:27249943; https://doi.org/ 10.1063/1.4949020 [DOI] [PubMed] [Google Scholar]
- [188].Markovic R, Stozer A, Gosak M, Dolensek J, Marhl M, Rupnik MS. Progressive glucose stimulation of islet beta cells reveals a transition from segregated to integrated modular functional connectivity patterns. Sci Rep 2015; 5:7845; PMID:25598507; https://doi.org/ 10.1038/srep07845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Rutter GA, Hodson DJ. Beta cell connectivity in pancreatic islets: a type 2 diabetes target? Cell Mol Life Sci 2014; 72(3):453-67; PMID:25323131; https://doi.org/ 10.1007/s00018-014-1755-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Rutter GA, Hodson DJ, Chabosseau P, Haythorne E, Pullen TJ, Leclerc I. Local and regional control of calcium dynamics in the pancreatic islet. Diabetes Obes Metab 2017; PMID:28466490; https://doi.org/ 10.1111/dom.12990 [DOI] [PubMed] [Google Scholar]
- [191].Gosak M, Stozer A, Markovic R, Dolensek J, Marhl M, Rupnik MS, Perc M. The relationship between node degree and dissipation rate in networks of diffusively coupled oscillators and its significance for pancreatic beta cells. Chaos 2015; 25:073115; PMID:26232966; https://doi.org/ 10.1063/1.4926673 [DOI] [PubMed] [Google Scholar]
- [192].Blum B, Hrvatin S, Schuetz C, Bonal C, Rezania A, Melton DA. Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nat Biotechnol 2012; 30:261-4; PMID:22371083; https://doi.org/ 10.1038/nbt.2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Szabat M, Lynn FC, Hoffman BG, Kieffer TJ, Allan DW, Johnson JD. Maintenance of β-cell maturity and plasticity in the adult pancreas: Developmental biology concepts in adult physiology. Diabetes 2012; 61:1365-71; PMID:22618775; https://doi.org/ 10.2337/db11-1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Nlend RN, Ait-Lounis A, Allagnat F, Cigliola V, Charollais A, Reith W, Haefliger JA, Meda P. Cx36 is a target of Beta2/NeuroD1, which associates with prenatal differentiation of insulin-producing beta cells. J Membr Biol 2012; 245:263-73; PMID:22729650; https://doi.org/ 10.1007/s00232-012-9447-1 [DOI] [PubMed] [Google Scholar]
- [195].Satin LS, Butler PC, Ha J, Sherman AS. Pulsatile insulin secretion, impaired glucose tolerance and type 2 diabetes. Mol Aspects Med 2015; 42:61-77; PMID:25637831; https://doi.org/ 10.1016/j.mam.2015.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Benninger RK, Piston DW. Cellular communication and heterogeneity in pancreatic islet insulin secretion dynamics. Trends Endocrinol Metab 2014; 25(8):399-406; PMID:24679927; https://doi.org/ 10.1016/j.tem.2014.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Gilon P, Jonas J, Henquin J. Culture duration and conditions affect the oscillations of cytoplasmic calcium concentration induced by glucose in mouse pancreatic islets. Diabetologia 1994; 37:1007-14; PMID:7851679; https://doi.org/ 10.1007/BF00400464 [DOI] [PubMed] [Google Scholar]
- [198].Cabrera O, Berman DM, Kenyon NS, Ricordi C, Berggren P-O, Caicedo A. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc Natl Acad Sci U S A 2006; 103:2334-9; PMID:16461897; https://doi.org/ 10.1073/pnas.0510790103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Brissova M, Fowler MJ, Nicholson WE, Chu A, Hirshberg B, Harlan DM, Powers AC. Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. J Histochem Cytochem 2005; 53:1087-97; PMID:15923354; https://doi.org/ 10.1369/jhc.5C6684.2005 [DOI] [PubMed] [Google Scholar]
- [200].Quesada I, Todorova MG, Alonso-Magdalena P, Beltrá M, Carneiro EM, Martin F, Nadal A, Soria B. Glucose induces opposite intracellular Ca2+ Concentration oscillatory patterns in identified α- and β-cells within intact human islets of langerhans. Diabetes 2006; 55:2463-9; PMID:16936194; https://doi.org/ 10.2337/db06-0272 [DOI] [PubMed] [Google Scholar]
- [201].Martin F, Soria B. Glucose-induced [Ca2+]i oscillations in single human pancreatic islets. Cell Calcium 1996; 20:409-14; PMID:8955555; https://doi.org/ 10.1016/S0143-4160(96)90003-2 [DOI] [PubMed] [Google Scholar]
- [202].Erlandsen SL, Hegre OD, Parsons JA, McEvoy RC, Elde RP. Pancreatic islet cell hormones distribution of cell types in the islet and evidence for the presence of somatostatin and gastrin within the D cell. J Histochem Cytochem 1976; 24:883-97; PMID:60437; https://doi.org/ 10.1177/24.7.60437 [DOI] [PubMed] [Google Scholar]
- [203].Grube D, Bohn R. The microanatomy of human islets of langerhans, with special reference to somatostatin (D-) Cells. Arch Histol Jpn 1983; 46:327-53; PMID:6139102; https://doi.org/ 10.1679/aohc.46.327 [DOI] [PubMed] [Google Scholar]
- [204].Orci L, Unger RH. Functional subdivision of islets of langerhans and possible role of D cells. Lancet 1975; 2:1243-4; PMID:53729; https://doi.org/ 10.1016/S0140-6736(75)92078-4 [DOI] [PubMed] [Google Scholar]
- [205].Bosco D, Armanet M, Morel P, Niclauss N, Sgroi A, Muller YD, Giovannoni L, Parnaud G, Berney T. Unique arrangement of α- and β-cells in human islets of langerhans. Diabetes 2010; 59:1202-10; PMID:20185817; https://doi.org/ 10.2337/db09-1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Hraha TH, Bernard AB, Nguyen LM, Anseth KS, Benninger RK. Dimensionality and size scaling of coordinated Ca(2+) dynamics in MIN6 beta-cell Clusters. Biophys J 2014; 106:299-309; PMID:24411262; https://doi.org/ 10.1016/j.bpj.2013.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Lebreton F, Pirog A, Belouah I, Bosco D, Berney T, Meda P, Bornat Y, Catargi B, Renaud S, Raoux M, et al.. Slow potentials encode intercellular coupling and insulin demand in pancreatic beta cells. Diabetologia 2015; 58:1291-9; PMID:25788295; https://doi.org/ 10.1007/s00125-015-3558-z [DOI] [PubMed] [Google Scholar]
- [208].Marchetti P, Scharp DW, McLear M, Gingerich R, Finke E, Olack B, Swanson C, Giannarelli R, Navalesi R, Lacy PE. Pulsatile insulin secretion from isolated human pancreatic islets. Diabetes 1994; 43:827-30; PMID:8194670; https://doi.org/ 10.2337/diab.43.6.827 [DOI] [PubMed] [Google Scholar]
- [209].Ritzel RA, Veldhuis JD, Butler PC. Glucose stimulates pulsatile insulin secretion from human pancreatic islets by increasing secretory burst mass: dose-response relationships. J Clin Endocrinol Metab 2003; 88:742-7; PMID:12574208; https://doi.org/ 10.1210/jc.2002-021250 [DOI] [PubMed] [Google Scholar]
- [210].Ravier MA, Güldenagel M, Charollais A, Gjinovci A, Caille D, Söhl G, Wollheim CB, Willecke K, Henquin JC, Meda P. Loss of connexin36 channels alters β-cell coupling, islet synchronization of glucose-induced Ca2+ and insulin oscillations, and basal insulin release. Diabetes 2005; 54:1798-807; PMID:15919802; https://doi.org/ 10.2337/diabetes.54.6.1798 [DOI] [PubMed] [Google Scholar]
- [211].Head WS, Orseth ML, Nunemaker CS, Satin LS, Piston DW, Benninger RK. Connexin-36 gap junctions regulate in vivo first- and second-phase insulin secretion dynamics and glucose tolerance in the conscious mouse. Diabetes 2012; 61:1700-7; PMID:22511206; https://doi.org/ 10.2337/db11-1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Benninger RKP, Head WS, Zhang M, Satin LS, Piston DW. Gap junctions and other mechanisms of cell–cell communication regulate basal insulin secretion in the pancreatic islet. J Physiol 2011; 589:5453-66; PMID:21930600; https://doi.org/ 10.1113/jphysiol.2011.218909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Haefliger JA, Rohner-Jeanrenaud F, Caille D, Charollais A, Meda P, Allagnat F. Hyperglycemia downregulates Connexin36 in pancreatic islets via the upregulation of ICER-1/ICER-1 gamma. J Mol Endocrinol 2013; 51:49-58; PMID:23613279; https://doi.org/ 10.1530/JME-13-0054 [DOI] [PubMed] [Google Scholar]
- [214].O'Neill CM, Lu C, Corbin KL, Sharma PR, Dula SB, Carter JD, Ramadan JW, Xin W, Lee JK, Nunemaker CS. Circulating levels of IL-1B+IL-6 cause ER stress and dysfunction in islets from prediabetic male mice. Endocrinology 2013; 154:3077-88; PMID:23836031; https://doi.org/ 10.1210/en.2012-2138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Carvalho CPF, Oliveira RB, Britan A, Santos-Silva JC, Boschero AC, Meda P, Collares-Buzato CB. Impaired β-cell-β-cell coupling mediated by Cx36 gap junctions in prediabetic mice. Am J Physiol Endocrinol Metab 2012; 303:E144-E51; PMID:22569071; https://doi.org/ 10.1152/ajpendo.00489.2011 [DOI] [PubMed] [Google Scholar]
- [216].Ravier MR, Sehlin JS, Henquin JH. Disorganization of cytoplasmic Ca2+ oscillations and pulsatile insulin secretion in islets from ob/ob mice. Diabetologia 2002; 45:1154-63; PMID:12189446; https://doi.org/ 10.1007/s00125-002-0883-9 [DOI] [PubMed] [Google Scholar]
- [217].Do OH, Low JT, Gaisano HY, Thorn P. The secretory deficit in islets from db/db mice is mainly due to a loss of responding beta cells. Diabetologia 2014; 57:1400-9; PMID:24705605; https://doi.org/ 10.1007/s00125-014-3226-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Kanatsuka A, Makino H, Sakurada M, Hashimoto N, Iwaoka H, Yamaguchi T, Taira M, Yoshida S, Yoshida A. First-phase insulin response to glucose in nonobese or obese subjects with glucose intolerance: analysis by C-peptide secretion rate. Metabolism 1988; 37:878-84; PMID:3047522; https://doi.org/ 10.1016/0026-0495(88)90123-0 [DOI] [PubMed] [Google Scholar]
- [219].O'Rahilly S, Turner RC, Matthews DR. Impaired pulsatile secretion of insulin in relatives of patients with non-insulin-dependent diabetes. N Engl J Med 1988; 318:1225-30; PMID:3283553; https://doi.org/ 10.1056/NEJM198805123181902 [DOI] [PubMed] [Google Scholar]
- [220].Rutter GA, Hodson DJ. Minireview: Intraislet regulation of insulin secretion in humans. Mol Endocrinol 2013; 27:1984-95; PMID:24243488; https://doi.org/ 10.1210/me.2013-1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Misler S, Gee WM, Gillis KD, Scharp DW, Falke LC. Metabolite-regulated ATP-sensitive K+ channel in human pancreatic islet cells. Diabetes 1989; 38:422-7; PMID:2647551; https://doi.org/ 10.2337/diab.38.4.422 [DOI] [PubMed] [Google Scholar]
- [222].Misler S, Dickey A, Barnett DW. Maintenance of stimulus-secretion coupling and single beta-cell function in cryopreserved-thawed human islets of Langerhans. Pflügers Arch 2005; 450:395-404; PMID:15988591; https://doi.org/ 10.1007/s00424-005-1401-y [DOI] [PubMed] [Google Scholar]
- [223].Fridlyand LE, Jacobson DA, Philipson LH. Ion channels and regulation of insulin secretion in human beta-cells: a computational systems analysis. Islets 2013; 5:1-15; PMID:23624892; https://doi.org/ 10.4161/isl.24166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Kindmark H, Kohler M, Arkhammar P, Efendic S, Larsson O, Linder S, Nilsson T, Berggren PO. Oscillations in cytoplasmic free calcium concentration in human pancreatic islets from subjects with normal and impaired glucose tolerance. Diabetologia 1994; 37:1121-31; PMID:7867884; https://doi.org/ 10.1007/BF00418376 [DOI] [PubMed] [Google Scholar]
- [225].Hellman B, Gylfe E, Bergsten P, Grapengiesser E, Lund P, Berts A, Tengholm A, Pipeleers DG, Ling Z. Glucose induces oscillatory Ca signalling and insulin release in human pancreatic beta cells. Diabetologia 1994; 37:S11-S20; PMID:7821725; https://doi.org/ 10.1007/BF00400821 [DOI] [PubMed] [Google Scholar]