

Abstract
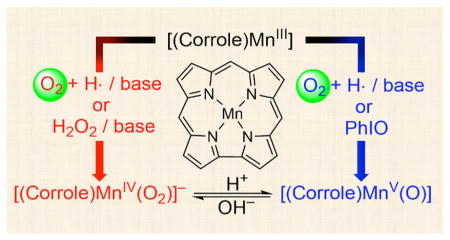
Activation of dioxygen (O2) in enzymatic and biomimetic reactions has been intensively investigated over the past several decades. More recently, O–O bond formation, which is the reverse of the O2-activation reaction, has been the focus of current research. Herein, we report the O2-activation and O–O bond formation reactions by manganese corrole complexes. In the O2-activation reaction, Mn(V)-oxo and Mn(IV)-peroxo intermediates were formed when Mn(III) corroles were exposed to O2 in the presence of base (e.g., OH−) and hydrogen atom (H atom) donor (e.g., THF or cyclic olefins); the O2-activation reaction did not occur in the absence of base and H atom donor. Moreover, formation of the Mn(V)-oxo and Mn(IV)-peroxo species was dependent on the amounts of base present in the reaction solution. The role of the base was proposed to lower the oxidation potential of the Mn(III) corroles, thereby facilitating the binding of O2 and forming a Mn(IV)-superoxo species. The putative Mn(IV)-superoxo species was then converted to the corresponding Mn(IV)-hydroperoxo species by abstracting a H atom from H atom donor, followed by the O–O bond cleavage of the putative Mn(IV)-hydroperoxo species to form a Mn(V)-oxo species. We have also shown that addition of hydroxide ion to the Mn(V)-oxo species afforded the Mn(IV)-peroxo species via O–O bond formation and the resulting Mn(IV)-peroxo species reverted to the Mn(V)-oxo species upon addition of proton, indicating that the O–O bond formation and cleavage reactions between the Mn(V)-oxo and Mn(IV)-peroxo complexes are reversible. The present study reports the first example of using the same manganese complex in both O2-activation and O–O bond formation reactions.
Graphical Abstract
1. INTRODUCTION
Dioxygen (O2) is one of the most important molecules for sustaining aerobic life. In biological reactions, metalloenzymes bind and activate O2, and metal–oxygen intermediates, such as metal-superoxo, -peroxo, -hydroperoxo, and -oxo species, are generated as reactive species in the catalytic cycle of O2-activation.1 One notable example is the O2-activation by iron-containing heme and nonheme enzymes, in which iron–oxygen intermediates have been trapped and characterized using various spectroscopic techniques.2,3 In biomimetic studies, a large number of iron–oxygen intermediates bearing porphyrin and nonporphyrin ligands have been successfully synthesized as the model compounds of the heme and nonheme iron enzymes, and their chemical properties as well as the mechanisms of the formation of the iron–oxygen intermediates have been elucidated using the model compounds.4,5 In most of the model studies, artificial oxidants, such as iodosylbenzene (PhIO), peracids, NaOCl, H2O2, and alkyl hydroperoxides, have been used as surrogates for O2 for the synthesis of the iron–oxygen intermediates.4,5 However, enzymes utilize O2, not the artificial oxidants, in most of their reactions and O2 is an ideal, environmentally benign oxidant in oxidation reactions. Therefore, tremendous efforts have been exerted to use O2 as the primary oxidant in biomimetic oxidation reactions, and the formation of iron–oxygen complexes by activating O2 in the absence or presence of electron and proton donors have been reported recently.6,7
Similar to the iron systems, high-valent manganese-oxo intermediates have been considered as the reactive species in the oxidation of organic substrates as well as in the water oxidation reaction by the oxygen-evolving complex (OEC) in Photosystem II (PS II).8 Therefore, a number of high-valent Mn(IV or V)-oxo complexes have been synthesized in the reactions of heme and nonheme Mn models using artificial oxidants (e.g., PhIO, peracids, and NaOCl).7 In addition to the Mn-oxo species, other Mn-oxygen intermediates, such as Mn-peroxo, -hydroperoxo, and -alkylperoxo species, have been synthesized using H2O2 and alkyl hydroperoxides as the oxidants.9 However, in most of the reactions, artificial oxidants were used for the synthesis of the Mn-oxygen intermediates, and there are only a couple of examples using O2 as the oxidant for the synthesis of Mn(V)-oxo complexes.10 Moreover, to the best of our knowledge, the formation of other Mn-oxygen intermediates (e.g., Mn-peroxo species) has never been observed in the O2-activation reactions.
The reverse of the O2-activation reaction is the O–O bond formation reaction, in which high-valent metal-oxo species have been proposed as the active oxidants responsible for the O–O bond formation (e.g., Mn(V)-oxo species in the water oxidation by OEC).8 Although the water oxidation reaction has been the topic of current research and intense effort has been devoted to the development of highly efficient catalytic systems,8,11 the nature of reactive intermediates and the mechanism(s) of the O–O bond formation still remain elusive. Only a couple of examples showing the O–O bond formation between high-valent Mn(V)-oxo species and hydroxide ion have been reported in biomimetic studies.12 Thus, compared to the O–O bond activation chemistry, the O–O bond formation mechanism is less clearly understood. Taken together, it is indeed a big challenge to achieve both of O–O bond activation and O–O bond formation by employing a single biomimetic complex.
Herein, we report the O2-activation and O–O bond formation reactions by manganese corrole complexes (see Scheme 1). In the O2-activation reaction, the formation of Mn(V)-oxo and Mn(IV)-peroxo complexes was observed when Mn(III) corrole complexes were exposed to O2 in the presence of base and hydrogen atom (H atom) donors. The Mn(V)-oxo and Mn(IV)-peroxo intermediates were characterized spectroscopically by synthesizing authentic compounds. In the O–O bond formation reaction, addition of hydroxide ion to the Mn(V)-oxo complexes yielded the Mn(IV)-peroxo species, and the reversible O–O bond formation and cleavage reactions between the Mn(V)-oxo and Mn(IV)-peroxo complexes were demonstrated. Mechanistic aspects for the O2-activation and O–O bond formation reactions have also been discussed in the present study.
Scheme 1.
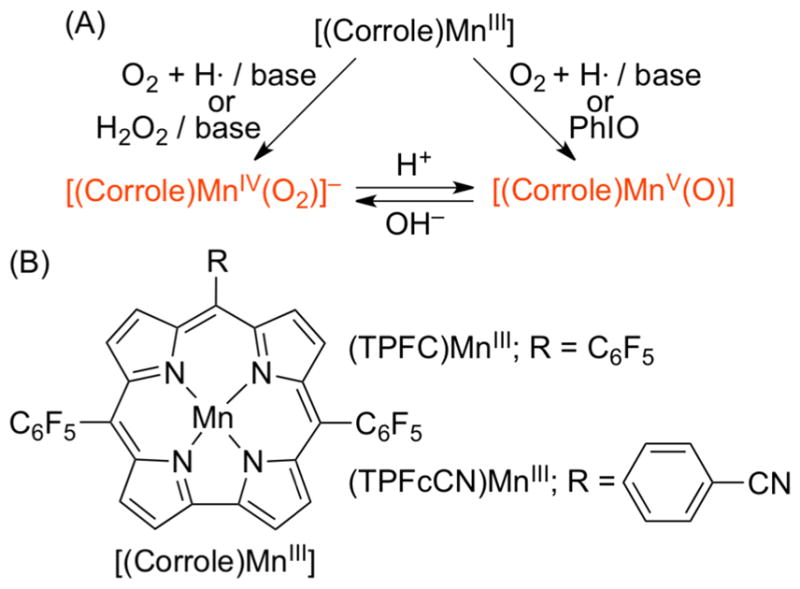
(A) Manganese–Oxygen Intermediates Formed in the O2-Activation and O–O Bond Formation Reactions and (B) Manganese Corroles Used in This Study
2. RESULTS AND DISCUSSION
2.1. Formation of Mn(V)-Oxo and Mn(IV)-Peroxo in O2-Activation Reactions
2.1.1. Generation of Mn(V)-Oxo by Activating O2
The manganese(III) corrole complexes used in this study were [(TPFC)MnIII] (TPFC = 5,10,15-tris-(pentafluorophenyl)corrolato trianion) and [(TPFcCN)MnIII] (TPFcCN = 5,15-bis(pentafluorophenyl)-10-benzonitrilecorro-lato trianion) (see Scheme 1B for structures), which were stable in O2 atmosphere at room temperature in THF. Interestingly, when [(TPFC)MnIII] was exposed to air in the presence of 2 equiv of tetramethylammonium hydroxide (TMAH) in THF at 25 °C,13 the color of the reaction solution changed from green to pink and a new intermediate 1 with absorption bands at 347, 407, and 520 nm was formed within 2 min (Figure 1a); the electronic absorption spectrum of 1 was similar to those of [(Corrole)MnV(O)] complexes reported previously.14 Intermediate 1 was also synthesized as an authentic compound by reacting [(TPFC)MnIII] with 1-(tert-butylsulfonyl)-2-iodosyl-benzene (sPhIO) in CH3CN at 0 °C (Scheme 2, pathway h) (Supporting Information (SI), Figure S2).14 Intermediate 1 was stable (t1/2 ~ 3 h at 25 °C) and was characterized using various spectroscopic methods. The cold-spray ionization mass (CSI-MS) spectrum exhibited a prominent ion peak at a mass-to-charge ratio (m/z) of 864.0 (SI, Figure S3a), which corresponds to the mass and isotope distribution pattern of [(TPFC)Mn-(O)] (calcd m/z = 864.0). When the reaction was carried out with 18O2 gas, the CSI-MS exhibited a peak at m/z 866.0 (SI, Figure S3b), which corresponds to [(TPFC)Mn(18O)] (calcd m/z = 866.0). The observation of the two-mass unit shift in the 18O-labeled experiment suggests that 1 contains one oxygen atom and the oxygen atom in 1 derives from O2. The X-band EPR and 1H NMR spectra of 1 indicate that 1 is diamagnetic with a low-spin (S = 0) state (SI, Figures S4 and S5), as reported in other Mn(V)-oxo complexes.10,12,14,15 The resonance Raman (rRaman) spectrum of 1, measured at –40 °C upon 405 nm excitation, displayed one isotopically sensitive band at 958 cm−1, which shifted to 918 cm−1 upon 18O-substitution (Figure 1b).12b The observed isotopic shift of –40 cm−1 with 18O-substitution was in good agreement with the calculated value for a diatomic Mn–O oscillator (–42 cm−1). The calculated force constant for the 958 cm−1 mode by a simple Hook’s law was 6.7 mdyne/Å, suggesting that Mn–O moiety possesses triple Mn–O bond character as reported in other Mn(V)-oxo complexes.10c,12b,14c,15a,b
Figure 1.
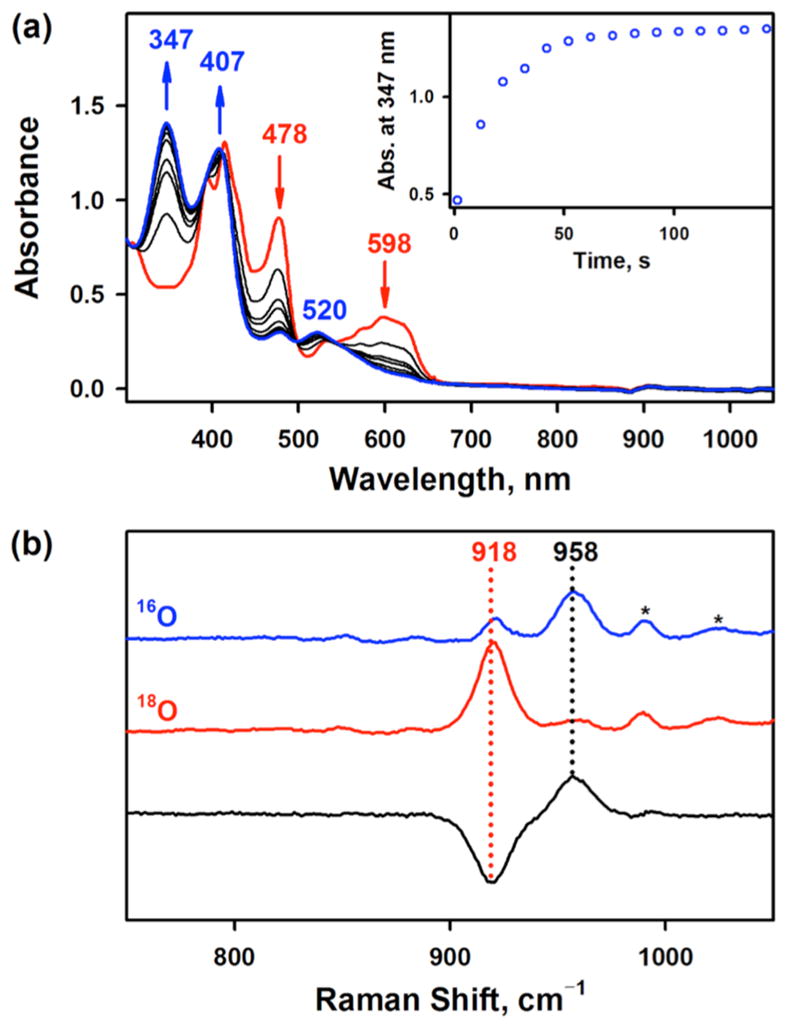
(a) UV–vis spectral changes observed in the O2-activation by [(TPFC)MnIII] (0.030 mM, red line) in the presence of TMAH (0.060 mM, 2 equiv) to form 1 (blue line) in THF at 25 °C. Inset shows the time trace monitored at 347 nm due to 1. (b) Resonance Raman spectra of 1-16O (blue line) and 1-18O (red line) obtained upon excitation at 405 nm. Black line is the difference spectrum of 1-16O and 1-18O.
Scheme 2.
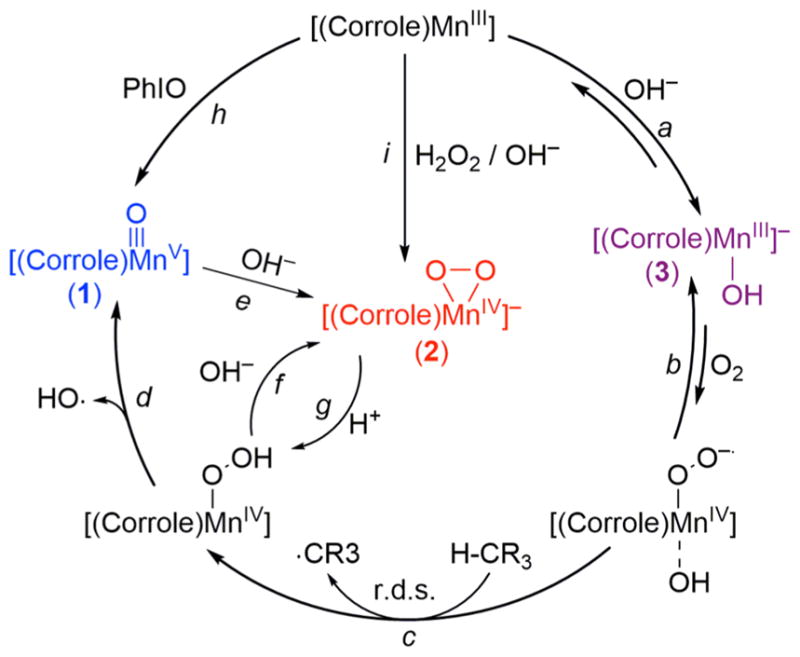
Proposed Mechanisms for the Generation of Mn(V)-Oxo and Mn(IV)-Peroxo Species in O2-Activation and O–O Bond Formation Reactions
Similarly, [(TPFcCN)MnV(O)] was formed when [(TPFcCN)MnIII] was exposed to air in the presence of 2 equiv of TMAH in THF at 25 °C (see the structure of [(TPFcCN)MnIII] in Scheme 1B) (SI, Figure S6a). [(TPFcCN)MnV(O)] was also synthesized by reacting [(TPFcCN)MnIII] with sPhIO in CH3CN at 0 °C (SI, Figure S6b). The [(TPFcCN)MnV(O)] complex was characterized with other spectroscopic methods, such as EPR (data not shown). Taken together, Mn(V)-oxo corrole complexes were formed in the activation of O2 by Mn(III) corroles in the presence of base in THF at 25 °C.
2.1.2. Generation of Mn(IV)-Peroxo by Activating O2
When a THF solution containing [(TPFC)MnIII] (0.030 mM) and large excess TMAH (0.60 mM) was exposed to air at 25 °C, the formation of a new intermediate 2 with absorption bands at 435 and 574 nm in an electronic absorption spectrum was observed within 10 s (Figure 2a, blue line); the color of the reaction solution changed from green to yellowish brown. The X-band EPR spectrum of 2 exhibited a broad signal at g ≈ 5.1, which was assigned to a high-spin S = 3/2 Mn(IV) species (SI, Figure S7, blue line).16 Intermediate 2 was also synthesized by reacting [(TPFC)MnIII] and H2O2 in the presence of TMAH (Scheme 2, pathway i) (SI, Figure S2b).12
Figure 2.
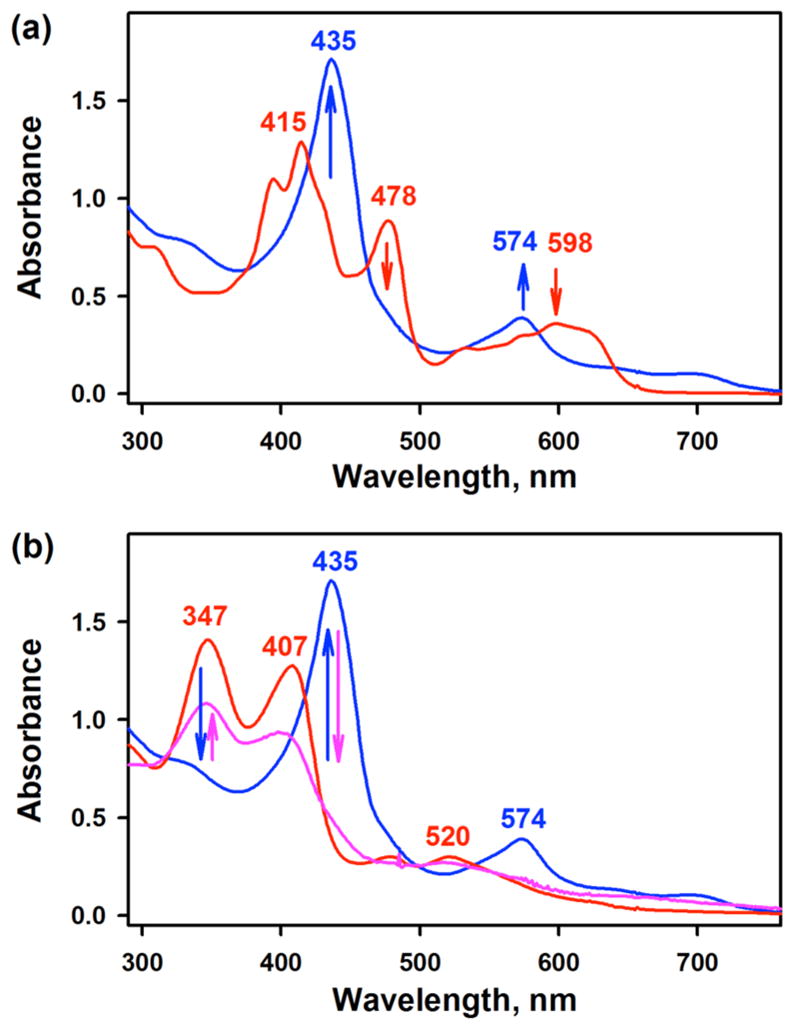
(a) UV–vis spectral changes observed in the O2-activation by [(TPFC)MnIII] (0.030 mM, red line) in the presence of TMAH (0.60 mM, 20 equiv) to form 2 (blue line) in THF at 25 °C. (b) UV–vis spectral changes showing the conversion of 1 (0.030 mM, red line) to 2 (blue line) upon addition of TMAH (0.60 mM, 20 equiv) and the regeneration of 1 (pink line) upon addition of HClO4 (0.60 mM, 20 equiv) to the resulting solution of 2.
A comparison of the Mn K-edge XAS data for [(TPFC)MnIII] and 2 is shown in Figure 3a. The data show that on going from the starting MnIII species to 2, the rising-edge inflection point shifts by 2.2 eV to higher energy (from 6550.0 to 6552.2 eV). Because the rising-edge energy position shifts to higher energies with an increase in the effective nuclear charge at the absorbing Mn center,16c,17 the data indicate a clear increase in oxidation state at the Mn site and show that 2 is a Mn(IV) species. The Mn K-pre-edge spectrum, which arises due to Mn 1s → 3d electric dipole forbidden, quadrupole allowed transitions, is strongly affected by changes in the ligand-field strength and bonding at the Mn center.18,19 On going from [(TPFC)MnIII] to 2, the pre-edge shifts to higher energy by 0.6 eV (6540.5 to 6541.1 eV). In addition, the pre-edge intensity pattern changes from a single sharp feature in [(TPFC)MnIII] to a broad, weaker feature in 2. Together, these data indicate an increase in ligand-field and a more symmetrical structure in 2 relative to [MnIII(TPFC)]. This qualitative structural assessment from the Mn K-pre-edge data was augmented with Mn K-edge EXAFS data for [(TPFC)MnIII] and 2. Figure 3b shows a comparison of the nonphase shift corrected Fourier transform data for the two species (see Figure 3b, inset for the EXAFS comparison). The data reveal a significant increase in the first shell intensity indicating an increase in coordination at the Mn. FEFF best fits to the data for [(TPFC)MnIII] are consistent with a first shell with 4 Mn–N at 1.91 Å,20 while for 2, a split first shell is required with 2 Mn–O contributions at 1.83 Å and 4 Mn–N at 1.96 Å. For both [(TPFC)MnIII] and 2, the second and third shells were fit with single and multiple scattering contributions from the TPFC ligand (SI, Figure S8 and Table S1). These data are in good agreement with the Mn K-pre-edge intensities and together the Mn K-edge XAS and EXAFS data are consistent with a side-on bound peroxo Mn(IV) description for 2.
Figure 3.
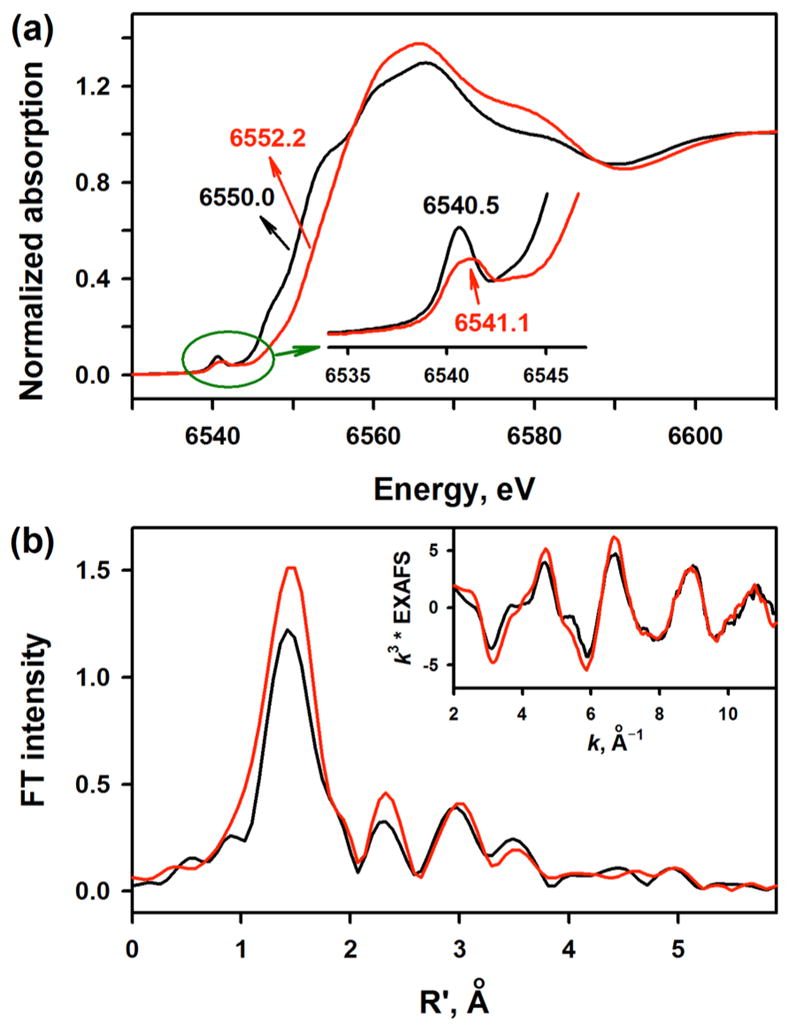
(a) Normalized Mn K-edge XAS data for [(TPFC)MnIII] (black line) and 2 (red line). Inset shows the expanded pre-edge region. (b) Non-phase-shift corrected Fourier Transform data for [(TPFC)-MnIII] (black line) and 2 (red line). Inset shows the Mn K-edge EXAFS comparison.
Similarly, we observed the formation of [(TPFcCN)-MnIV(O2)]− when [(TPFcCN)MnIII] was exposed to air in the presence of 20 equiv or TMAH in THF at 25 °C (SI, Figure S9a). The [(TPFcCN)MnIV(O2)]− complex was also synthesized as an authentic compound by reacting [(TPFcCN)MnIII] with H2O2 in the presence of TMAH in CH3CN at 0 °C (SI, Figure S9b). On the basis of the spectroscopic data presented in this study and the previous reports,12 we conclude that Mn(IV)- peroxo complexes were formed by activating O2 in the presence of a large excess amount of base in THF at 25 °C.
2.1.3. Reversible O–O Bond Formation and Cleavage between Mn(V)-Oxo and Mn(IV)-Peroxo Complexes
We have shown above that Mn(V)-oxo and Mn(IV)-peroxo complexes can be formed in the O2-activation reaction, depending on the amounts of TMAH present in the reaction solution; Mn(V)-oxo and Mn(IV)-peroxo species were formed in the presence of 2 and 20 equiv of TMAH, respectively. We propose the following mechanisms for the formation of different Mn-oxygen intermediates depending on the amounts of TMAH: In the reaction using an excess amount of TMAH (i.e., 20 equiv), 1, which was generated in the O2-activation reaction by [(TPFC)-MnIII] initially, reacted further with OH− at a fast rate, thereby affording the formation of 2 (Scheme 2, pathway e). Indeed, the fast conversion of 1 to 2 was confirmed by carrying out a control reaction, in which addition of 20 equiv of TMAH to the solution of 1, which was generated either in the O2-activation reaction (Figure 2) or in the reaction of [(TPFC)MnIII] and sPhIO (SI, Figure S10), resulted in the immediate conversion of 1 to 2. Thus, 2 can be generated via the O–O bond formation process between Mn(V)-oxo and OH−, as reported previously.12 An alternative mechanism proposing the formation of 2 in the presence of an excess amount of OH− is the fast deprotonation of the putative Mn(IV)-OOH species generated in the catalytic cycle (Scheme 2, pathway f). Thus, there are two competing pathways for the reaction of the Mn(IV)-OOH species, such as the O–O bond cleavage of Mn(IV)-OOH to form 1 (Scheme 2, pathway d), followed by the O–O bond formation between 1 and OH− (Scheme 2, pathway e) or the deprotonation of Mn(IV)-OOH by OH− to form 2 (Scheme 2, pathway f). Because the rates of the O–O bond cleavage and deprotonation steps of the Mn(IV)-OOH species cannot be determined without capturing the Mn(IV)-OOH species, we cannot distinguish the formation mechanism of 2 from the putative Mn(IV)-OOH species at this moment (e.g., Scheme 2, pathways d versus f).
The reverse reaction of the O–O bond formation between Mn(V)-oxo and OH− (Scheme 2, pathway e) is the O–O bond cleavage reaction upon addition of H+ to the Mn(IV)-peroxo species (Scheme 2, pathway g, followed by pathway d). Indeed, we observed the immediate formation of 1 upon addition of H+ to 2 (Figure 2b), as reported previously.12,21 For the latter reaction, we propose that addition of H+ to 2 forms the putative Mn(IV)-OOH species (Scheme 2, pathway g), followed by the O–O bond cleavage of the Mn(IV)-OOH species to form 1 (Scheme 2, pathway d).12
2.2. Mechanistic Investigations
2.2.1. Role of Base in O2-Activation Reaction
The O2-activation by Mn(III) corroles occurs only in the presence of base (e.g., TMAH), indicating that base is an essential component in the O2-activation reaction. We therefore investigated the role of base in the O2-activation reaction by [(TPFC)MnIII]. Because it is difficult to control the fast reaction of [(TPFC)MnIII] with O2 in THF, mechanistic studies were performed in CH3CN; [(TPFC)MnIII] does not react with O2 in CH3CN even in the presence of base when there is no hydrogen atom (H atom) donor in reaction solution (e.g., THF).
First, addition of TMAH to [(TPFC)MnIII] in CH3CN at 25 °C afforded the formation of a new species 3 with an absorption band at 471 nm (SI, Figure S12); this species was stable under an air atmosphere. 3 was EPR silent (SI, Figure S13a), and CSI-MS of 3 exhibited an ion peak at m/z of 947.2, corresponding to the mass and isotope distribution pattern of [(TPFC)Mn-(OH)(CH3CN)2]− (calcd m/z = 947.0) (SI, Figure S13b). The spectroscopic data and the previous examples of Mn(III) corroles binding hydroxide, [(Corrole)MnIII(OH)]−,22 led us to assign 3 as [(TPFC)MnIII(OH)]−.
Then, electrochemical measurements of [(TPFC)MnIII] were carried out in the absence and presence of OH− (Figure 4). The cyclic voltammogram (CV) of [(TPFC)MnIII] in CH3CN revealed reversible anodic and cathodic peaks centered at 0.74 V vs SCE, which is assigned as a MnIV/III redox couple (Figure 4, red line). This value was similar to the previously reported redox potential of [(TPFC)MnIII] in CH2Cl2 (0.71 V vs SCE).23 Interestingly, when [(TPFC)MnIII] was converted to [(TPFC)-MnIII(OH)]− by adding TMAH (2.0 mM) to the solution of [(TPFC)MnIII] (2.0 mM), the MnIV/III redox potential exhibited a large negative shift from 0.74 V vs SCE in the absence of TMAH to 0.18 V vs SCE in the presence of TMAH (Figure 4, blue line). Because of the large negative shift of the first oxidation potential of [(TPFC)MnIII(OH)]−, the second and third redox couples were observed at 0.45 and 0.71 V vs SCE, respectively. Thus, within the same region of the potential between +1.0 and –0.4 V for the oxidation of [(TPFC)MnIII] in the absence of TMAH, up to three-electron oxidation of [(TPFC)MnIII(OH)]− becomes possible, although whether the second oxidation occurs at the metal center to afford the MnV complex or at the ligand to give the corrole radical cation has yet to be determined. Similar negative shift of the MnIV/III redox couple was obtained under an Ar atmosphere in THF. The redox potential was 0.89 V vs SCE in the absence of TMAH, which shifted negatively to 0.50 V vs SCE in the presence of TMAH (SI, Figure S14). The smaller negative shift in THF than that in CH3CN may result from the weaker binding of OH− of TMAH in THF.
Figure 4.
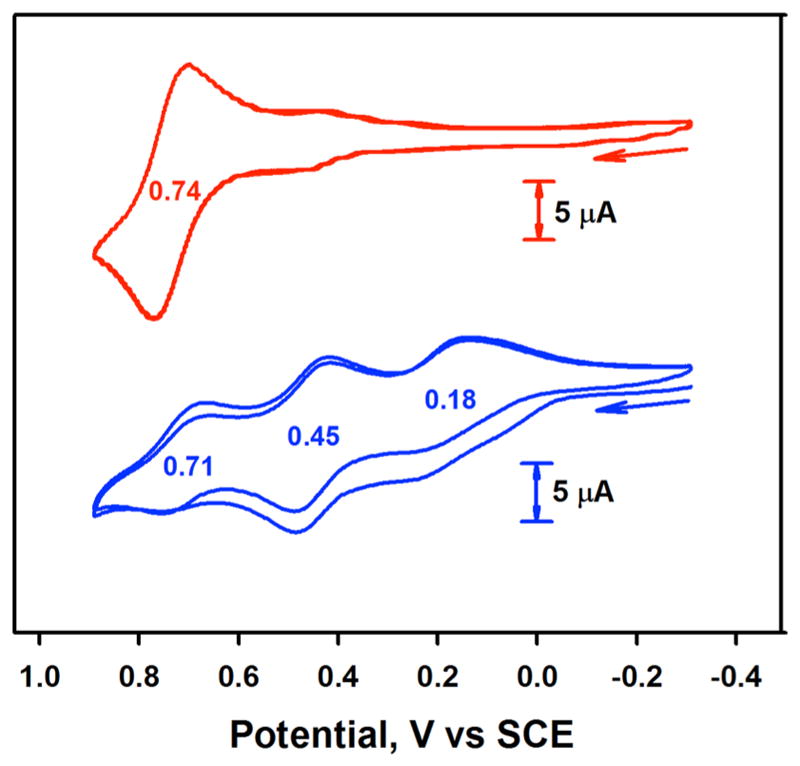
Cyclic voltammograms of [(TPFC)MnIII] (2.0 mM) in the absence (red line) and presence (blue line) of TMAH (2.0 mM) in CH3CN at 25 °C.
The MnIV/III redox couple of [(TPFC)MnIII] was also shifted to the negative direction to 0.46 V vs SCE in the presence of excess Cl− anion (SI, Figure S15, blue line). Such an anion effect on the redox potential of Mn corroles was reported by Kadish and co-workers;22 it has been demonstrated that Mn(III) corroles binding anions, such as Cl−, CN−, OAc−, F−, and OH−, changed their redox potentials with the negative shift of the redox couple of MnIV/III.22 Thus, Mn(III) corroles binding anionic axial ligands can be oxidized more easily as compared with those without axial ligands.
The cyclic voltammetry results corroborate the results of the O2-activation reaction by [(TPFC)MnIII]; the O2-activation reaction by [(TPFC)MnIII] occurs when the oxidation potential of [(TPFC)MnIII] is low, such as in the case of OH− binding (i.e., 0.18 V vs SCE). In other words, [(TPFC)MnIII] binding OH− can bind O2 due to its low oxidation potential. In contrast, when the oxidation potential of [(TPFC)MnIII] is relatively high, such as in the cases of no anionic axial ligand or with Cl− anionic axial ligand, the Mn(III) corroles cannot bind O2 and O2-activation does not occur in these systems. On the basis of the electrochemical study of [(TPFC)MnIII] in the absence and presence of anionic axial ligands, we conclude that the role of OH− is to lower the oxidation potential of [(TPFC)MnIII] and the [(TPFC)MnIII] complex with an axially coordinated OH− can bind O2 to form a putative Mn(IV)-superoxo complex for further reactions (Scheme 2, pathways a and b). Indeed, it has been shown previously that the binding of O2 becomes favorable when cobalt or manganese porphyrins bind imidazole or pyridine as an axial ligand24 and the oxidation potential of a nonheme iron(II) complex becomes lower by changing a solvent system.25
2.2.2. Role of THF and Cyclic Olefins in O2-Activation Reaction
We have shown above that Mn-oxygen intermediates, 1 and 2, are formed when Mn(III) corroles are reacted with O2 in the presence of base in THF, but not in CH3CN (vide supra). Interestingly, in the latter reaction, addition of a small amount of THF to a CH3CN solution containing [(TPFC)MnIII] and TMAH (20 equiv) afforded the immediate formation of 2 at 25 °C (SI, Figure S16a). Similarly, addition of cyclic olefins (e.g., cyclopentene, cyclohexene, and cycloheptene) to a CH3CN solution containing [(TPFC)MnIII] (0.030 mM) and a large excess of TMAH (20 equiv, 0.60 mM) generated 2 immediately (SI, Figure S16b). We therefore investigated the role of THF and cyclic olefins in O2-activation reaction, by carrying out the reaction of [(TPFC)MnIII] with O2 in CH3CN at a lower temperature (e.g., at –20 °C) and by adding different amounts of H atom donors (e.g., THF and cyclic olefins) for kinetic studies. As described above, the mechanistic studies were performed using 20 equiv of TMAH, which was the condition for the generation of 2 with a clean kinetic behavior.
Addition of THF (0.12 M) to a reaction solution containing [(TPFC)MnIII] (0.030 mM) and TMAH (20 equiv, 0.60 mM) at –20 °C afforded clean spectral changes with isosbestic points at 370, 407, 451, 498, 586, 666, and 740 nm for the formation of 2 (Figure 5a). The formation rate of 2 was dependent on the amounts of THF added; the first-order rate constants, determined by pseudo-first-order fitting of the kinetic data, increased linearly with the increase of the concentration of THF, affording a second-order rate constant of 6.1(5) × 10−2 M−1 s−1 at –20 °C (SI, Figure S17). Similarly, when cyclic olefins, such as cyclohexene, cycloheptene and cyclopentene, were added to a CH3CN solution containing [(TPFC)MnIII] (0.030 mM) and TMAH (20 equiv, 0.60 mM) at –20 °C, we observed the formation of 2 with clean isosbestic points (Figure 5b). The formation of 2 was dependent on the concentration of olefins, and the second-order rate constants of 8.0(7) × 10−2, 3.8(4) × 10−2 and 1.9(2) × 10−2 M−1 s−1 were determined in the reactions of cyclohexene, cycloheptene, and cyclopentene, respectively (Figure 5c; SI, Figures S18 and S19). However, no reaction occurred when cyclooctene was used. In addition, the formation of 2 was slower in the reaction of cyclohexene-d10, giving a kinetic isotope effect (KIE) value of 5.0. We have also examined other substrates with weaker C–H bond other than cycloalkenes, such as fluorene. The second-order rate constant of fluorene was determined to be 2.9(3) × 10−1 M−1 s−1 (SI, Figure S20), which was approximately 4 times larger than that of cyclohexene. These results indicate that the formation rate was correlated with the C–H bond dissociation energies (BDEs) of substrates (fluorene, BDE = 79.5 kcal mol−1; cyclohexene, BDE = 81.0 kcal mol−1; cycloheptene, BDE = 82.8 kcal mol−1; cyclopentene, BDE = 83.3 kcal mol−1 and cyclooctene, BDE = 85.4 kcal mol−1), as observed in other metal complex-initiated O2-activation reactions in the presence of cyclic olefins.10d,26 No reaction with cyclooctene suggests that the BDE of the O–H bond of the Mn(IV)-hydroperoxo complex may be smaller than that of the C–H bond of cyclooctene (85.4 kcal mol−1), when the H atom abstraction from cyclooctene by the Mn(IV)-hydroperoxo complex is uphill. The weaker O–H bond of the Mn(IV)-hydroperoxo complex than that of H2O2 (87.49 ± 0.07 kcal mol−1)27 may result from the conjugation of the Mn(IV)-superoxo radical complex, since it is known that the smaller the spin density of the terminal oxygen, the smaller the BDH of the O–H bond.28 The linear correlation of log kH, where the kH values were obtained by dividing the second-order rate constants (k2) by the numbers of equivalent target C–H bonds in the substrates (i.e., 4 for cycloalkenes and 2 for fiuorine), vs BDE is shown in Figure S21 in SI, where the Bell–Evans–Polanyi α value is determined as 0.42 ± 0.05.
Figure 5.

(a) UV–vis spectral changes for the formation of 2 (blue line) and the disappearance of 3 (red line) upon addition of THF (0.12 M) to a CH3CN solution of [(TPFC)MnIII(OH)]− (3) (0.030 mM, red line) at –20 °C; 3 was prepared by adding TMAH (0.60 mM) to a CH3CN solution of [(TPFC)MnIII] (0.030 mM). Inset shows time course monitored at 431 nm due to 2. (b) UV–vis spectral changes for the formation of 2 (blue line) and the disappearance of 3 (red line) upon addition of cyclohexene (0.20 M) to a CH3CN solution of [(TPFC)MnIII(OH)]− (3) (0.030 mM, red line) at –20 °C; 3 was prepared by adding TMAH (0.60 mM) to a CH3CN solution of [(TPFC)MnIII] (0.030 mM). Inset shows time course monitored at 431 nm due to 2. (c) Plots of kobs against concentration of cyclohexene (black circles) and cyclohexene-d10 (red rectangles) to determine the second-order rate constants for the formation of 2 in the O2-activation by [(TPFC)MnIII] (0.030 mM) in the presence of TMAH (0.60 mM) in air-saturated CH3CN at –20 °C.
The results presented above demonstrate unambiguously that the rate of the formation of 2 depends on the BDEs of C–H bonds of cyclic olefins. However, it is of interest to note that although BDE of the C–H bond on α-position of THF is ~92 kcal mol−1,29 which is much higher than the BDE of the allylic C–H bonds of cyclohexene (81.0 kcal mol−1), the rates of the formation of 2 in the reactions of THF (6.1 × 10−2 M−1 s−1 at –20 °C) and cyclohexene (8.0 × 10−2 M−1 s−1 at –20 °C) are similar. Indeed, such a unusual reactivity in the reaction of THF was previously reported in the H atom transfer reactions by tert-butoxyl radical30 and a (μ- ν2: ν2-peroxo)dicopper(II) complex.31 In the latter case, the reactivity of THF was similar to the substrate with a BDE of ~82 kcal mol−1, which is close to that of cyclohexene.31 The easy oxidation of THF by C–H bond activation was explained previously with a pronounced stereo-electronic effect on the H atom abstraction from those C–H bonds adjacent to the oxygen atom in THF.31,32
On the basis of the experimental results presented above, it is evident that THF and cyclic olefins function as a H atom donor. In addition, no reaction occurred between 3 and THF or cycloalkenes under anaerobic conditions. Therefore, the H atom abstraction from the H atom donors (e.g., THF or cyclic olefins) by the putative Mn-superoxo species is the rate-determining step (r.d.s.) for the formation of Mn-oxygen intermediates in the O2-activation reaction (Scheme 2, pathway c). It has been demonstrated recently that metal-superoxo species are capable of abstracting H atom from substrates to form metal-hydroperoxo species in enzymatic and biomimetic reactions.33
2.2.3. Kinetic Studies of O2-Binding and O–O Bond Formation
The concentration effect of O2 in the dioxygen activation reaction was also investigated. The rate constant of the generation of 2 from 3 with cyclohexene in an O2-saturated CH3CN was determined to be 3.9(4) × 10 1 M−1 s−1 (SI, Figure S22), which is approximately 5 times larger than that in an air-saturated CH3CN (8.0(7) × 10−2 M−1 s−1). Thus, the equilibrium for formation of the Mn(IV)-superoxo complex lies far to the left-hand side, when the concentration of the Mn(IV)-superoxo complex is proportional to concentration of O2 (Scheme 2, pathway b). As indicated by the deuterium kinetic isotope effect in the generation of 2 with cyclohexene-d10 (KIE = 5.0), hydrogen atom transfer from cyclohexene to the Mn(IV)-superoxo species is the rate-determining step, which competes with the back reaction of the Mn(IV)-superoxo species to regenerate the Mn(III) complex. In such a case, the observed rate constant corresponds to kHK, in which the equilibrium constant for the generation of the Mn(IV)-superoxo complex cannot be determined separately from the rate constant of hydrogen transfer (kH).
Kinetic studies on the O–O bond formation between 1 and OH− to produce 2 were examined as well. These experiments were performed at –40 °C, due to its fast conversion (SI, Figure S23). The rate of formation of 2 obeyed first-order kinetics and the pseudo-first-order rate constants were proportional to concentration of OH−. The second-order rate constant was determined from the slope of the linear plot to be 5.6(5) × 10 M−1 s−1. The linear dependence of the rate constant on OH−concentration also indicates that the nucleophilic attack of OH−to the Mn(V)-oxo species is the rate-determining step in the O–O bond formation, as proposed in literatures.8a,b,34 Moreover, this rate constant (5.6(5) × 10 M−1 s−1 at –40 °C) is much larger than those of cycloalkenes in the O2 activation to generate 2 (8.0(7) × 10−2 M−1 s−1 for cyclohexene at –20 °C). This is fully consistent with the result that the rate-determining step in the O2 activation to generate 2 is the H atom transfer from H atom donors to the Mn(IV)-superoxo species, followed by fast O–O bond formation to generate 2.
2.3. Proposed Mechanisms and Conclusions
The overall reaction mechanism for the formation of 1 and 2 in the O2-activation reaction by [(TPFC)MnIII] is depicted in Scheme 2, which shows (i) the formation of [(TPFC)-MnIII(OH)]− (3) upon addition of base to [(TPFC)MnIII] (pathway a), (ii) the binding of O2 by 3 to form a putative MnIV-superoxo species, [(TPFC)MnIV(O2.−)] (pathway b), (iii) the H atom abstraction from H atom donor by [(TPFC)MnIV(O2.−)] to form a putative MnIV-hydroperoxo species, [(TPFC)-MnIV(OOH)] (pathway c), (iv) the conversion of [(TPFC)-MnIV(OOH)] to [(TPFC)MnV(O)] via O–O bond homolysis (pathway d) or to [(TPFC)MnIV(O2)]− by deprotonation of the hydroperoxide ligand (pathway f), and (v) the conversion of 1 to 2 upon addition of OH− to 1 (pathway e). We have also shown that (vi) addition of proton to 2 afforded 1 via the formation of [(TPFC)MnIV(OOH)] (pathway g), followed by the hydroperoxide O–O bond cleavage of the latter species (pathway d). The Mn(V)-oxo and Mn(IV)-peroxo intermediates were also synthesized independently by reacting [(TPFC)-MnIII] with PhIO (pathway h) and H2O2/OH− (pathway i), respectively, and were used for spectroscopic characterization and mechanistic studies. Detailed discussions for the reactions i–vi are presented below.
In the first step of the reaction cycle (Scheme 2, pathway a), the oxidation potential of [(TPFC)MnIII] is shifted negatively by 0.56 V upon binding of OH−. Thus, the role of OH− is to lower the oxidation potential of the Mn(III) corrole, which is then oxidized easily for further reaction.
The Mn(III) corrole (3) with a low oxidation potential can bind O2 readily, thereby forming a putative Mn(IV)-superoxo species (Scheme 2, pathway b) via electron transfer from 3 to O2, followed by binding of O2 •− to the Mn(IV) center. Although electron transfer from 3 (Eox = 0.18 V vs SCE) to O2 (Ered = –0.87 V vs SCE) is still endergonic,35 the binding of O2 to the Mn(III) center may produce Mn(IV)-superoxo species via an inner-sphere electron transfer process, which is thermodynamically feasible. This step is probably in equilibrium, as shown in the binding of O2 by many metal complexes (e.g., Co(II) complexes).24,36 If there are no H atom donors, such as THF and cyclic olefins, no further reaction occurs and 3 is the only detectable species in reaction solution because the equilibrium is toward the dissociation of O2 from the Mn(IV)-superoxo species.
In the presence of H atom donors, such as THF or cyclic olefins, the putative Mn(IV)-superoxo species abstracts a H atom from H atom donors, thereby forming a putative Mn(IV)-hydroperoxo species (Scheme 2, pathway c). We have verified that THF and cyclic olefins function as a H atom donor. In addition, based on the large KIE value determined in the reactions of cyclohexene and deuterated cyclohexene and the linear relationship between the BDEs of cyclic olefins and the formation rate constants of Mn-oxygen intermediates, H atom abstraction by the Mn(IV)-superoxo species was proposed to be the r.d.s.
In the reaction of using a smaller amount of base (e.g., 2 equiv to Mn corrole), Mn(V)-oxo species (1) was the product formed in the O2-activation by [(TPFC)MnIII] in the presence of a H atom donor (e.g., THF or cyclohexene). A proposed mechanism is the conversion of the putative Mn(IV)-hydroperoxo species to 1 via O–O bond homolysis (Scheme 2, pathway d). An alternative mechanism that can be considered is the O–O bond heterolysis of the Mn(IV)-hydroperoxo species to yield a highly unstable Mn(VI)-oxo species and OH−, followed by a fast one-electron reduction of the Mn(VI)-oxo species to form the Mn(V)-oxo species. The mechanisms of the hydroperoxide O–O bond cleavage of metal-hydroperoxo species has been extensively investigated in biomimetic studies.5b,37
-
In the case of using a larger amount of base (e.g., 20 equiv to Mn corrole), Mn(IV)-peroxo species (2) instead of Mn(V)-oxo (1) was formed as the product in the O2-activation reaction by [(TPFC)MnIII]. As proposed in Scheme 2, 2 can be formed either from the reaction of 1 and OH− (pathway e) or from the deprotonation of the Mn(IV)-hydroperoxo species (pathway f). The former pathway, which shows the O–O bond formation between Mn(V)-oxo and OH−, has been the topic of current research for the O2-evolution process in water oxidation by PS II. It is of interest to note that despite the intense research with a large number of high-valent metal-oxo species aimed at understanding the O–O bond formation step in model studies of PS II, such an O–O bond formation has only been directly observed with Mn(V)–O corrole species.12 The nucleophilic attack of OH− to 1 might produce [(TPFC)MnIII(OOH)]− complex and the latter species was then oxidized by 1 to form [(TPFC)-MnIV(OOH)]− intermediate, followed by the deprotona-tion by OH− to afford 2 as the final product. However, detailed mechanism for the O–O bond formation between high-valent metal-oxo species and OH− remains elusive.
Another plausible pathway for the formation of 2 is the deprotonation of the putative Mn(IV)-hydroperoxo species (pathway f), which has been well documented in the acid–base chemistry between metal-hydroperoxo and -peroxo species in biomimetic reactions.17c,37c,38 As mentioned above, the formation mechanism of 2, such as the O–O bond formation between 1 and OH− or the deprotonation of the Mn(IV)-hydroperoxo species (pathway f), cannot be distinguished without capturing the Mn(IV)-OOH species and determining the rates of the O–O bond cleavage and deprotonation steps of the putative Mn(IV)-OOH species.
The reverse reaction of the O–O bond formation between 1 and OH− is the protonation of 2 that leads to the formation of the Mn(IV)-hydroperoxo species (pathway g), followed by the O–O bond cleavage of the Mn(IV)-hydroperoxo species to form 1 (pathway d). It has been well documented in biomimetic studies that protonation of metal-peroxo species generates high-valent metal-oxo species via the formation of metal-hydroperoxo species, followed by either homolysis or heterolysis of the hydroperoxide O–O bond of the metal-hydroperoxo species.5b,37
In summary, we have shown that Mn(III) corroles are able to activate O2 in the presence of base and H atom donor, thereby forming Mn(V)-oxo and Mn(IV)-peroxo species. We have also shown the reversibility of the O–O bond formation and cleavage reactions between Mn(V)-oxo and Mn(IV)-peroxo species. To the best of our knowledge, the present study reports the first example of using the same manganese complex in both O2-activation and O–O bond formation reactions.
3. EXPERIMENTAL SECTION
3.1. Materials
Commercially available chemicals were used without further purification unless otherwise indicated. Solvents were dried according to the literature procedures and redistilled under Ar before use.39 1-(tert-butylsulfonyl)-2-iodosylbenzene (sPhIO) was synthesized by a literature method.40 The purity of the oxidant was determined by iodometric titration. [(TPFC)MnIII] and [(TPFcCN)MnIII] were synthesized by following the literature.41 18O2 (98% 18O-enriched) and H218O (95% 18O-enriched) were purchased from ICON Services Inc. (Summit, NJ, USA).
3.2. Instrumentation
UV–vis spectra were recorded on a Hewlett-Packard Agilent 8453 UV–vis spectrophotometer equipped with an UNISOKU cryostat system (USP-203; UNISOKU, Japan). Coldspray ionization time-of-flight mass (CSI-MS) spectral data were collected on a JMS-T100CS (JEOL) mass spectrometer equipped with a CSI source. Typical measurement conditions were as follows: needle voltage, 2.2 kV; orifice 1 current, 50–500 nA; orifice 1 voltage, 0 to 20 V; ring lens voltage, 10 V; ion source temperature, 5 °C; spray temperature, –40 °C. Resonance Raman scattering was dispersed by a single polychromator (Ritsu Oyo Kogaku, MC-100DG) and was detected by a liquid-nitrogen-cooled CCD detector (HORIBA JOBIN YVON, Symphony 1024 × 128 Cryogenic Front Illuminated CCD Detector). Raman spectra were collected with backscattering geometry at an excitation wavelength (λex) of 405 nm (LM-405-PLR-40-2, ONDAX) using a spinning sample cell (NMR tube with 5 mm OD), which was placed in a thermostated quarts Dewar at –40 °C by flashing cold nitrogen gas. The laser power at a measuring point in front of a quartz Dewar was adjusted to 20 mW. Raman shifts were calibrated using indene (accurate to within ±1 cm−1). X-band electron paramagnetic resonance (EPR) spectra were recorded at 5 K using an X-band Bruker EMX-plus spectrometer equipped with a dual mode cavity (ER 4116DM). Low temperatures were achieved and controlled with an Oxford Instruments ESR900 liquid Helium quartz cryostat fitted with an Oxford Instruments ITC503 temperature and gas flow controller. The experimental parameters for EPR spectra were as follows: Microwave frequency = 9.647 GHz, microwave power = 1.0 mW, modulation amplitude = 10 G, gain = 1 × 104, modulation frequency = 100 kHz, time constant = 40.96 ms, and conversion time = 81.00 ms. Electrochemical measurements were performed on a CHI630B electrochemical analyzer (CH Instruments, Inc.) in deaerated CH3CN containing 0.10 M Bu4NPF6 (TBAPF6) as a supporting electrolyte at 25 °C. A conventional three-electrode cell was used with a platinum working electrode (surface area of 0.30 mm2), a platinum wire as a counter electrode, and an Ag/Ag+ electrode as a reference electrode. The platinum working electrode was routinely polished with BAS polishing alumina suspension and rinsed with acetone and acetonitrile before use. The measured potentials were recorded with respect to an Ag/Ag+ (0.010 M) reference electrode. All potentials (vs Ag/Ag+) were converted to values vs SCE by adding 0.29 V.
3.3. X-ray Absorption Spectroscopy
The Mn K-edge X-ray absorption spectra of [(TPFC)MnIII] and 2 were measured at the Stanford Synchrotron Radiation Lightsource (SSRL) on the wiggler BL9-3 under standard ring conditions of 3 GeV and ~500 mA. A Si(220) double crystal monochromator was used for energy selection. A Rh-coated harmonic rejection mirror was used to reject components of higher harmonics. During data collection, the samples were maintained at a constant temperature of ~10 K using an Oxford Instruments CF 1208 liquid helium cryostat. A 100-element Ge monolith fluorescence detector from Canberra Industries was employed for fluorescence data measurement to k = 12 Å−1. Solution samples for [(TPFC)MnIII] and intermediate 2 were transferred into 2 mm delrin XAS cells with 70 m Kapton tape windows under synthesis conditions and were immediately frozen after preparation and stored under liquid N2 conditions. Internal energy calibration was accomplished by simultaneous measurement of the absorption of a Mn-foil placed between two ionization chambers situated after the sample. The first inflection point of the foil spectrum was set at 6539.0 eV. Data presented here are 8-scan average spectra for [(TPFC)MnIII] and 16-scan average spectrum for 2. To avoid potential photodamage, at most two scans were obtained on a fresh sample. Significant photoreduction was not observed (judging by comparison of scan 1 and scan 2). Data were processed by fitting a second-order polynomial to the pre-edge region and then subtracting this from the entire spectrum as background. A three-region spline of orders 2, 3, and 3 was used to model the smoothly decaying postedge region. The data were normalized by subtracting the cubic spline and assigning the edge jump to 1.0 at 6555 eV using the Pyspline42 program. Data were then renormalized in Kaleidagraph for comparison and quantitation purposes. Theoretical EXAFS signals χ (k) were calculated by using FEFF (Macintosh version)43–45 using geometry optimized preliminary structures obtained from DFT calculations. The input structure was improved based on preliminary EXAFS fit parameters to generate more accurate theoretical EXAFS signals. Data fitting was performed in EXAFSPAK.46 The structural parameters varied during the fitting process were the bond distance (R) and the bond variance σ2, which is related to the Debye–Waller factor resulting from thermal motion, and static disorder of the absorbing and scattering atoms. The nonstructural parameter E0 (the energy at which k = 0) was also allowed to vary but was restricted to a common value for every component in a given fit. Coordination numbers was systematically varied in the course of the fit but were fixed within a given fit.
3.4. Generation of Intermediate 1
[(TPFC)MnV(O)] (1) intermediate was generated by adding tetramethylammonium hydroxide (TMAH, 2 equiv, 0.060 mM) into a UV–vis cuvette containing a THF solution of [(TPFC)MnIII] (0.030 mM) under air atmosphere at 25 °C. Formation of 1 was confirmed by monitoring UV–vis spectral changes at 478 and 598 nm due to the decay of [(TPFC)MnIII] and at 347 and 407 nm due to the formation of 1. The 18O-labeled complex [(TPFC)MnV(18O)] (1-18O) was also generated by adding tetrame-thylammonium hydroxide (TMAH, 2 equiv, 0.060 mM) into a UV–vis cuvette containing a THF solution of [(TPFC)MnIII] (0.030 mM) under 18O2 atmosphere at 25 °C.
1 can also be generated by adding sPhIO (2 equiv) into a CH3CN solution of [(TPFC)MnIII] (0.030 mM) at 0 °C. 1 was generated within 1 min. [(TPFcCN)MnV(O)] intermediate was generated by the same procedure for the generation of 1.
UV–vis of 1 in THF, λmax (nm) (ε, M−1 cm−1): 347 (4.5 × 104), 407 (4.0 × 104), 520 (7.6 × 103). UV–vis of 1 in CH3CN: 347 (4.6 × 104), 405 (4.2 × 104), 518 (8.4 × 103).
3.5. Generation of Intermediate 2
[(TPFC)MnIV(O2)]− (2) complex was generated by adding tetramethylammonium hydroxide (TMAH, 20 equiv, 0.60 mM) into a UV–vis cuvette containing a THF solution of [(TPFC)MnIII] (0.030 mM) under air atmosphere at 25 °C. 2 was also generated by adding tetramethylammonium hydroxide (TMAH, 20 equiv, 0.60 mM) into a UV–vis cuvette containing a CH3CN solution of [(TPFC)MnIII] (0.030 mM) in the presence of THF or cyclic olefins, such as cyclohexene and cyclopentene, under air atmosphere at 25 °C.
Alternatively, 2 can be prepared by reacting [(TPFC)MnIII] (0.030 mM) with H2O2 solution (0.12 mM) in the presence of TMAH (0.60 mM, 20 equiv) in CH3CN at 0 °C. [(TPFcCN)MnIV(O2)]− was generated by the same procedure for the generation of 2.
UV–vis of 2 in THF, λmax (nm) (ε, M−1 cm−1): 435 (5.7 × 104), 574 (1.3 × 104), 696 (3.5 × 103). UV–vis of 2 in CH3CN: 431 (5.3 × 104), 571 (1.3 × 104), 701 (3.2 × 103).
3.6. Reversible Transformation between 1 and 2. 1
(0.030 mM), which was generated by dioxygen activation from [(TPFC)-MnIII] and TMAH under air atmosphere, was converted to 2 immediately by adding TMAH (20 equiv) into a THF solution of 1 at 25 °C. The same conversion was also obtained by using 1 produced in the reaction of [(TPFC)MnIII] and sPhIO.
To the opposite, 2 (0.030 mM), which was generated by dioxygen activation from [(TPFC)MnIII] and TMAH, was also reversibly converted to 1 by adding HClO4 (0.60 mM, 20 equiv) into a THF solution of 2 at 25 °C. The same conversion was also obtained by using 2 produced in the reaction of [(TPFC)MnIII] and H2O2 in the presence of TMAH.
3.7. Generation of Intermediate 3
[(TPFC)MnIII(OH)]− was produced by adding TMAH (20 equiv) into a CH3CN solution of [(TPFC)MnIII] (0.030 mM) under air at 25 °C. No further spectral change was observed upon addition of more than 20 equiv of TMAH.
UV–vis of 3 (CH3CN), λmax (nm) (ε, M−1 cm−1): 401 (3.7 × 104), 428 (4.0 × 104), 471 (4.7 × 103), 562 (9.5 × 103), 595 (9.0 × 103), 636 (1.1 × 104).
3.8. Kinetic Studies
All reactions were investigated in a 1.0 cm quartz cuvette and followed by monitoring UV–vis spectral changes of the generation of 2 in CH3CN/THF or CH3CN/cyclic olefins at –20 °C. The kinetic experiments were run at least in triplicate, and the data reported here represented the average of these reactions. Rate constants were determined under pseudo-first-order conditions (i.e., [substrate]/ [intermediate] > 10) by fitting the changes in absorbance at 431 nm due to the formation of 2. Kinetic isotope efiect was determined by comparing the formation rates of 2 in the presence of cyclohexene and cyclohexene-d10.
Supplementary Material
Acknowledgments
This work is dedicated to the memory of Professor Takashi Ogura. This work was supported by the NRF of Korea through CRI (NRF-2012R1A3A2048842 to W.N.), GRL (NRF-2010-00353 to W.N.) and Basic Science Research Program (2017R1D1A1B03029982 t o Y.- M.L. and 2017R1D1A1B03032615 to S.F.), and by a Grant-in-Aid (no. 16H02268 to S.F.) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), a SENTAN project from the Japan Science and Technology Agency (JST) to S.F., and National Natural Science Foundation of China (No. 21371059 and No. 21671068). The SSRL SMB resource was supported by the NIH National Institute of General Medical Sciences (NIGMS) through a Biomedical Technology Research Resource P41 grant (P41GM103393) and by the DOE Office of Biological and Environmental Research.
Footnotes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b08678.
EXAFS data and spectroscopic characterization and kinetic data (PDF)
References
- 1.(a) Nam W. Acc Chem Res. 2007;40:465. doi: 10.1021/ar700027f. [DOI] [PubMed] [Google Scholar]; (b) Que L., Jr J Biol Inorg Chem. 2017;22:171. doi: 10.1007/s00775-017-1443-6. [DOI] [PubMed] [Google Scholar]
- 2.(a) Krebs C, Galonić Fujimori D, Walsh CT, Bollinger JM., Jr Acc Chem Res. 2007;40:484. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kovaleva EG, Lipscomb JD. Nat Chem Biol. 2008;4:186. doi: 10.1038/nchembio.71. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Solomon EI, Goudarzi S, Sutherlin KD. Biochemistry. 2016;55:6363. doi: 10.1021/acs.biochem.6b00635. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Proshlyakov DA, McCracken J, Hausinger RP. J Biol Inorg Chem. 2017;22:367. doi: 10.1007/s00775-016-1406-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Ortiz de Montellano PR. Chem Rev. 2010;110:932. doi: 10.1021/cr9002193. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shaik S, Cohen S, Wang Y, Chen H, Kumar D, Thiel W. Chem Rev. 2010;110:949. doi: 10.1021/cr900121s. [DOI] [PubMed] [Google Scholar]; (c) Rittle J, Green MT. Science. 2010;330:933. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]; (d) Poulos TL. Chem Rev. 2014;114:3919. doi: 10.1021/cr400415k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Yosca TH, Ledray AP, Ngo J, Green MT. J Biol Inorg Chem. 2017;22:209. doi: 10.1007/s00775-016-1430-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Ray K, Pfaff FF, Wang B, Nam W. J Am Chem Soc. 2014;136:13942. doi: 10.1021/ja507807v. [DOI] [PubMed] [Google Scholar]; (b) Cook SA, Borovik AS. Acc Chem Res. 2015;48:2407. doi: 10.1021/acs.accounts.5b00212. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Nam W. Acc Chem Res. 2015;48:2415. [Google Scholar]; (d) Puri M, Que L., Jr Acc Chem Res. 2015;48:2443. doi: 10.1021/acs.accounts.5b00244. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Engelmann X, Monte-Pérez I, Ray K. Angew Chem, Int Ed. 2016;55:7632. doi: 10.1002/anie.201600507. [DOI] [PubMed] [Google Scholar]
- 5.(a) Nam W. Acc Chem Res. 2007;40:522. doi: 10.1021/ar700027f. [DOI] [PubMed] [Google Scholar]; (b) Oszajca M, Franke A, Brindell M, Stochel G, van Eldik R. Coord Chem Rev. 2016;306:483. [Google Scholar]; (c) Huang X, Groves JT. J Biol Inorg Chem. 2017;22:185. doi: 10.1007/s00775-016-1414-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Sahu S, Goldberg DP. J Am Chem Soc. 2016;138:11410. doi: 10.1021/jacs.6b05251. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hong S, Lee YM, Ray K, Nam W. Coord Chem Rev. 2017;334:25. [Google Scholar]; (c) Bruijnincx PCA, van Koten G, Klein Gebbink RJM. Chem Soc Rev. 2008;37:2716. doi: 10.1039/b707179p. [DOI] [PubMed] [Google Scholar]; (d) Korendovych IV, Kryatov SV, Rybak-Akimova EV. Acc Chem Res. 2007;40:510. doi: 10.1021/ar600041x. [DOI] [PubMed] [Google Scholar]; (e) Borovik AS. Acc Chem Res. 2005;38:54. doi: 10.1021/ar030160q. [DOI] [PubMed] [Google Scholar]
- 7.(a) Yin G. Acc Chem Res. 2013;46:483. doi: 10.1021/ar300208z. [DOI] [PubMed] [Google Scholar]; (b) Liu W, Groves JT. Acc Chem Res. 2015;48:1727. doi: 10.1021/acs.accounts.5b00062. [DOI] [PubMed] [Google Scholar]; (c) Neu HM, Baglia RA, Goldberg DP. Acc Chem Res. 2015;48:2754. doi: 10.1021/acs.accounts.5b00273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Nocera DG. Acc Chem Res. 2012;45:767. doi: 10.1021/ar2003013. [DOI] [PubMed] [Google Scholar]; (b) Yano J, Yachandra V. Chem Rev. 2014;114:4175. doi: 10.1021/cr4004874. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bang S, Lee YM, Hong S, Cho KB, Nishida Y, Seo MS, Sarangi R, Fukuzumi S, Nam W. Nat Chem. 2014;6:934. doi: 10.1038/nchem.2055. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Young KJ, Brennan BJ, Tagore R, Brudvig GW. Acc Chem Res. 2015;48:567. doi: 10.1021/ar5004175. [DOI] [PubMed] [Google Scholar]; (e) Blakemore JD, Crabtree RH, Brudvig GW. Chem Rev. 2015;115:12974. doi: 10.1021/acs.chemrev.5b00122. [DOI] [PubMed] [Google Scholar]
- 9.(a) Cho J, Sarangi R, Nam W. Acc Chem Res. 2012;45:1321. doi: 10.1021/ar3000019. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Leto DF, Jackson TA. J Biol Inorg Chem. 2014;19:1. doi: 10.1007/s00775-013-1067-4. [DOI] [PubMed] [Google Scholar]; (c) Kovacs JA. Acc Chem Res. 2015;48:2744. doi: 10.1021/acs.accounts.5b00260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Prokop KA, Goldberg DP. J Am Chem Soc. 2012;134:8014. doi: 10.1021/ja300888t. [DOI] [PubMed] [Google Scholar]; (b) Jung J, Ohkubo K, Prokop-Prigge KA, Neu HM, Goldberg DP, Fukuzumi S. Inorg Chem. 2013;52:13594. doi: 10.1021/ic402121j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Neu HM, Jung J, Baglia RA, Siegler MA, Ohkubo K, Fukuzumi S, Goldberg DP. J Am Chem Soc. 2015;137:4614. doi: 10.1021/jacs.5b00816. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hong S, Lee YM, Sankaralingam M, Vardhaman AK, Park YJ, Cho KB, Ogura T, Sarangi R, Fukuzumi S, Nam W. J Am Chem Soc. 2016;138:8523. doi: 10.1021/jacs.6b03874. [DOI] [PubMed] [Google Scholar]
- 11.(a) Weinberg DR, Gagliardi CJ, Hull JF, Murphy CF, Kent CA, Westlake BC, Paul A, Ess DH, McCafferty DG, Meyer TJ. Chem Rev. 2012;112:4016. doi: 10.1021/cr200177j. [DOI] [PubMed] [Google Scholar]; (b) Sala X, Maji S, Bofill R, García-Antón J, Escriche L, Llobet A. Acc Chem Res. 2014;47:504. doi: 10.1021/ar400169p. [DOI] [PubMed] [Google Scholar]; (c) Duan L, Wang L, Li F, Li F, Sun L. Acc Chem Res. 2015;48:2084. doi: 10.1021/acs.accounts.5b00149. [DOI] [PubMed] [Google Scholar]; (d) Askerka M, Brudvig GW, Batista VS. Acc Chem Res. 2017;50:41. doi: 10.1021/acs.accounts.6b00405. [DOI] [PubMed] [Google Scholar]
- 12.(a) Gao Y, Åkermark T, Liu J, Sun L, Åkermark B. J Am Chem Soc. 2009;131:8726. doi: 10.1021/ja901139r. [DOI] [PubMed] [Google Scholar]; (b) Kim SH, Park H, Seo MS, Kubo M, Ogura T, Klajn J, Gryko DT, Valentine JS, Nam W. J Am Chem Soc. 2010;132:14030. doi: 10.1021/ja1066465. [DOI] [PubMed] [Google Scholar]
- 13.1 could be generated in the dioxygen activation by [(TPFC) MnIII] in THF with 1 equiv of TMAH. However, the rate of formation of 1 was quite slow for the full generation (see SI, Figure S1). Thus, 2 equiv of TMAH was used in the present study.
- 14.(a) Bougher CJ, Liu S, Hicks SD, Abu-Omar MM. J Am Chem Soc. 2015;137:14481. doi: 10.1021/jacs.5b09759. [DOI] [PubMed] [Google Scholar]; (b) Kumar A, Goldberg I, Botoshansky M, Buchman Y, Gross Z. J Am Chem Soc. 2010;132:15233. doi: 10.1021/ja1050296. [DOI] [PubMed] [Google Scholar]; (c) Liu HY, Yam F, Xie YT, Li XY, Chang CK. J Am Chem Soc. 2009;131:12890. doi: 10.1021/ja905153r. [DOI] [PubMed] [Google Scholar]; (d) Gross Z, Golubkov G, Simkhovich L. Angew Chem, Int Ed. 2000;39:4045. doi: 10.1002/1521-3773(20001117)39:22<4045::aid-anie4045>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 15.(a) Collins TJ, Powell RD, Slebodnick C, Uffelman ES. J Am Chem Soc. 1990;112:899. [Google Scholar]; (b) Mandimutsira BS, Ramdhanie B, Todd RC, Wang H, Zareba AA, Czernuszewicz RS, Goldberg DP. J Am Chem Soc. 2002;124:15170. doi: 10.1021/ja028651d. [DOI] [PubMed] [Google Scholar]; (c) Lansky DE, Mandimutsira B, Ramdhanie B, Clausén M, Penner-Hahn J, Zvyagin SA, Telser J, Krzystek J, Zhan R, Ou Z, Kadish KM, Zakharov L, Rheingold AL, Goldberg DP. Inorg Chem. 2005;44:4485. doi: 10.1021/ic0503636. [DOI] [PubMed] [Google Scholar]; (d) Popescu DL, Chanda A, Stadler M, de Oliveira FT, Ryabov AD, Münck E, Bominaar EL, Collins TJ. Coord Chem Rev. 2008;252:2050. [Google Scholar]
- 16.(a) Parsell TH, Behan RK, Green MT, Hendrich MP, Borovik AS. J Am Chem Soc. 2006;128:8728. doi: 10.1021/ja062332v. [DOI] [PubMed] [Google Scholar]; (b) Garcia-Bosch I, Company A, Cady CW, Styring S, Browne WR, Ribas X, Costas M. Angew Chem, Int Ed. 2011;50:5648. doi: 10.1002/anie.201100907. [DOI] [PubMed] [Google Scholar]; (c) Wu X, Seo MS, Davis KM, Lee YM, Chen J, Cho KB, Pushkar YN, Nam W. J Am Chem Soc. 2011;133:20088. doi: 10.1021/ja208523u. [DOI] [PubMed] [Google Scholar]
- 17.(a) Jensen SC, Davis KM, Sullivan B, Hartzler DA, Seidler GT, Casa DM, Kasman E, Colmer HE, Massie AA, Jackson TA, Pushkar Y. J Phys Chem Lett. 2017;8:2584. doi: 10.1021/acs.jpclett.7b01209. [DOI] [PubMed] [Google Scholar]; (b) Leto DF, Jackson TA. Inorg Chem. 2014;53:6179. doi: 10.1021/ic5006902. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) So H, Park YJ, Cho KB, Lee YM, Seo MS, Cho J, Sarangi R, Nam W. J Am Chem Soc. 2014;136:12229. doi: 10.1021/ja506275q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chen J, Lee YM, Davis KM, Wu X, Seo MS, Cho KB, Yoon H, Park YJ, Fukuzumi S, Pushkar YN, Nam W. J Am Chem Soc. 2013;135:6388. doi: 10.1021/ja312113p. [DOI] [PubMed] [Google Scholar]
- 18.Sarangi R. Coord Chem Rev. 2013;257:459. doi: 10.1016/j.ccr.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Westre TE, Kennepohl P, DeWitt JG, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 1997;119:6297. [Google Scholar]
- 20.The Mn K-pre-edge intensity for [(TPFC)MnIII] indicates that the species is not symmetric. Thus, although the EXAFS data are consistent with 4 Mn-N, it is likely that a solvent molecule coordinates axially to the Mn center in [(TPFC)MnIII], which increases the pre-edge intensity. This solvent interaction is not detected in the EXAFS data due to the known error in coordination number determination.
- 21.The low absorbance of 1, which was regenerated from 2 by adding acid, was due to the instability of 1 in the presence of acid (SI, Figure S11), as reported previously (see reference 14a).
- 22.(a) Shen J, El Ojaimi M, Chkounda M, Gros CP, Barbe J-M, Shao J, Guilard R, Kadish KM. Inorg Chem. 2008;47:7717. doi: 10.1021/ic8007415. [DOI] [PubMed] [Google Scholar]; (b) Fang Y, Ou Z, Kadish KM. Chem Rev. 2017;117:3377. doi: 10.1021/acs.chemrev.6b00546. [DOI] [PubMed] [Google Scholar]
- 23.Golubkov G, Bendix J, Gray HB, Mahammed A, Goldberg I, DiBilio AJ, Gross Z. Angew Chem, Int Ed. 2001;40:2132. doi: 10.1002/1521-3773(20010601)40:11<2132::AID-ANIE2132>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 24.(a) Walker FA. J Am Chem Soc. 1973;95:1154. doi: 10.1021/ja00785a026. [DOI] [PubMed] [Google Scholar]; (b) Weschler CJ, Hoffman BM, Basolo F. J Am Chem Soc. 1975;97:5278. doi: 10.1021/ja00851a043. [DOI] [PubMed] [Google Scholar]; (c) Hoffman BM, Szymanski T, Brown TG, Basolo F. J Am Chem Soc. 1978;100:7253. [Google Scholar]
- 25.Kim SO, Sastri CV, Seo MS, Kim J, Nam W. J Am Chem Soc. 2005;127:4178. doi: 10.1021/ja043083i. [DOI] [PubMed] [Google Scholar]
- 26.Lee YM, Hong S, Morimoto Y, Shin W, Fukuzumi S, Nam W. J Am Chem Soc. 2010;132:10668. doi: 10.1021/ja103903c. [DOI] [PubMed] [Google Scholar]
- 27.Ruscic B, Boggs JE, Burcat A, Csaszar AG, Demaison J, Janoschek R, Martin JML, Morton ML, Rossi MJ, Stanton JF, Szalay PG, Westmoreland PR, Zabel F, Berces T. J Phys Chem Ref Data. 2005;34:573. [Google Scholar]
- 28.Wiberg KB, Ellison GB, McBride JM, Petersson GA. J Phys Chem A. 2013;117:213. doi: 10.1021/jp310510y. [DOI] [PubMed] [Google Scholar]
- 29.(a) Luo YR. Comprehensive Handbook of Chemical Bond Energies. CRC Press, Taylor & Francis Group; Boca Raton: 2007. [Google Scholar]; (b) Xue X-S, Ji P, Zhou B, Cheng J-P. Chem Rev. 2017;117:8622. doi: 10.1021/acs.chemrev.6b00664. [DOI] [PubMed] [Google Scholar]
- 30.Finn M, Friedline R, Suleman NK, Wohl CJ, Tanko JM. J Am Chem Soc. 2004;126:7578. doi: 10.1021/ja0493493. [DOI] [PubMed] [Google Scholar]
- 31.Matsumoto T, Ohkubo K, Honda K, Yazawa A, Furutachi H, Fujinami S, Fukuzumi S, Suzuki M. J Am Chem Soc. 2009;131:9258. doi: 10.1021/ja809822c. [DOI] [PubMed] [Google Scholar]
- 32.(a) Malatesta V, Scaiano JC. J Org Chem. 1982;47:1455. [Google Scholar]; (b) Malatesta V, Ingold KU. J Am Chem Soc. 1981;103:609. [Google Scholar]
- 33.(a) Maiti D, Fry HC, Woertink JS, Vance MA, Solomon EI, Karlin KD. J Am Chem Soc. 2007;129:264. doi: 10.1021/ja067411l. [DOI] [PubMed] [Google Scholar]; (b) Xing G, Diao Y, Hoffart LM, Barr EW, Prabhu KS, Arner RJ, Reddy CC, Krebs C, Bollinger JM. Proc Natl Acad Sci U S A. 2006;103:6130. doi: 10.1073/pnas.0508473103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Maiti D, Lee D-H, Gaoutchenova K, Würtele C, Holthausen MC, Narducci Sarjeant AA, Sundermeyer J, Schindler S, Karlin KD. Angew Chem, Int Ed. 2008;47:82. doi: 10.1002/anie.200704389. [DOI] [PubMed] [Google Scholar]; (d) Cho J, Woo J, Nam W. J Am Chem Soc. 2010;132:5958. doi: 10.1021/ja1015926. [DOI] [PubMed] [Google Scholar]; (e) Hong S, Sutherlin KD, Park J, Kwon E, Siegler MA, Solomon EI, Nam W. Nat Commun. 2014;5:5440. doi: 10.1038/ncomms6440. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Chiang CW, Kleespies ST, Stout HD, Meier KK, Li PY, Bominaar EL, Que L, Jr, Münck E, Lee WZ. J Am Chem Soc. 2014;136:10846. doi: 10.1021/ja504410s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Wang CC, Chang HC, Lai YC, Fang H, Li CC, Hsu HK, Li ZY, Lin TS, Kuo TS, Neese F, Ye S, Chiang YW, Tsai ML, Liaw WF, Lee WZ. J Am Chem Soc. 2016;138:14186. doi: 10.1021/jacs.6b08642. [DOI] [PubMed] [Google Scholar]
- 34.(a) Siegbahn PEM. Inorg Chem. 2008;47:1779. doi: 10.1021/ic7012057. [DOI] [PubMed] [Google Scholar]; (b) Siegbahn PEM. Chem - Eur J. 2006;12:9217. [Google Scholar]
- 35.Sawyer DT, Chiericato G, Jr, Angelis CT, Nanni EJ, Jr, Tsuchiya T. Anal Chem. 1982;54:1720. [Google Scholar]
- 36.(a) Basolo F, Hoffman BM, Ibers JA. Acc Chem Res. 1975;8:384. [Google Scholar]; (b) Hoffman BM, Diemente DL, Basolo F. J Am Chem Soc. 1970;92:61. [Google Scholar]
- 37.(a) Meunier B, de Visser SP, Shaik S. Chem Rev. 2004;104:3947. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]; (b) van Eldik R, Hubbard CD. Coord Chem Rev. 2010;254:297. [Google Scholar]; (c) Cho J, Jeon S, Wilson SA, Liu LV, Kang EA, Braymer JJ, Lim MH, Hedman B, Hodgson KO, Valentine JS, Solomon EI, Nam W. Nature. 2011;478:502. doi: 10.1038/nature10535. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Liu LV, Hong S, Cho J, Nam W, Solomon EI. J Am Chem Soc. 2013;135:3286. doi: 10.1021/ja400183g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Oszajca M, Drzewiecka-Matuszek A, Franke A, Rutkowska-Zbik D, Brindell M, Witko M, Stochel G, van Eldik R. Chem - Eur J. 2014;20:2328. doi: 10.1002/chem.201303694. [DOI] [PubMed] [Google Scholar]; (f) Bang S, Park S, Lee YM, Hong S, Cho KB, Nam W. Angew Chem, Int Ed. 2014;53:7843. doi: 10.1002/anie.201404556. [DOI] [PubMed] [Google Scholar]; (g) Gamba I, Codolà Z, Lloret-Fillol J, Costas M. Coord Chem Rev. 2017;334:2. [Google Scholar]
- 38.(a) Jensen BK, McKenzie CJ, Nielsen LP, Pedersen JZ, Svendsen HM. Chem Commun. 1999;1313 [Google Scholar]; (b) Ho RYN, Que L, Jr, Roelfes G, Feringa BL, Hermant R, Hage R. Chem Commun. 1999:2161. [Google Scholar]; (c) Kim YM, Cho KB, Cho J, Wang B, Li C, Shaik S, Nam W. J Am Chem Soc. 2013;135:8838. doi: 10.1021/ja404152q. [DOI] [PubMed] [Google Scholar]; (d) Tcho WY, Wang B, Lee YM, Cho KB, Shearer J, Nam W. Dalton Trans. 2016;45:14511. doi: 10.1039/c6dt01194b. [DOI] [PubMed] [Google Scholar]
- 39.Armarego WLF, Chai CLL. Purification of Laboratory Chemicals. 6. Pergamon Press; Oxford: 2009. [Google Scholar]
- 40.Guo M, Dong H, Li J, Cheng B, Huang Y-q, Feng Y-q, Lei A. Nat Commun. 2012;3:1190. doi: 10.1038/ncomms2196. [DOI] [PubMed] [Google Scholar]
- 41.Orlowski R, Gryko D, Gryko DT. Chem Rev. 2017;117:3102. doi: 10.1021/acs.chemrev.6b00434. [DOI] [PubMed] [Google Scholar]
- 42.Tenderholt A, Hedman B, Hodgson KO. X-ray Absorption Fine Structure - XAFS13. In: Hedman B, Pianetta P, editors. AIP Conference Proceedings. Vol. 882. American Institute of Physics; College Park, MD: 2007. p. 105. [Google Scholar]
- 43.Zabinsky SI, Rehr JJ, Ankudinov A, Albers RC, Eller MJ. Phys Rev B: Condens Matter Mater Phys. 1995;52:2995. doi: 10.1103/physrevb.52.2995. [DOI] [PubMed] [Google Scholar]
- 44.Mustre de Leon J, Rehr JJ, Zabinsky SI, Albers RC. Phys Rev B: Condens Matter Mater Phys. 1991;44:4146. doi: 10.1103/physrevb.44.4146. [DOI] [PubMed] [Google Scholar]
- 45.Rehr JJ, Mustre de Leon J, Zabinsky SI, Albers RC. J Am Chem Soc. 1991;113:5135. [Google Scholar]
- 46.George GN. EXAFSPAK and EDG-FIT. Stanford Synchrotron Radiation Laboratory, Stanford Linear Accelerator Center; Stanford, CA: 2000. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.