

Abstract
Detection of nuclear biomarkers such as nucleic acids and nuclear proteins is critical for early-stage cancer diagnosis and prognosis. Conventional methods relying on morphological assessment of cell nuclei in histopathology slides may be subjective, whereas colorimetric immunohistochemical and fluorescence-based imaging are limited by strong light absorption, broad-emission bands and low contrast. Here, we describe the development and use of a scanning laser-emission-based microscope that maps lasing emissions from nuclear biomarkers in human tissues. 41 tissue samples from 35 patients labelled with site-specific and biomarker-specific antibody-conjugated dyes were sandwiched in a Fabry-Pérot microcavity while an excitation laser beam built a laser-emission image. We observed multiple sub-cellular lasing emissions from cancer cell nuclei, with a threshold of tens of μJ/mm2, sub-micron resolution (<700 nm), and a lasing band in the few-nanometre range. Different lasing thresholds of nuclei in cancer and normal tissues enabled the identification and multiplexed detection of nuclear proteomic biomarkers, with a high sensitivity for early-stage cancer diagnosis. Laser-emission-based cancer screening and immunodiagnosis might find use in precision medicine and facilitate research in cell biology.
Keywords: lasers, Fabry-Pérot cavity, microscopy, sub-cellular resolution, tissues, nuclear biomarkers, nucleic acids, cancer, diagnosis, multiplexed detection
Nucleus lies in the heart of every cell and carries abundance of nuclear proteins and the majority of cell’s genetic materials. Molecular changes in the levels of nucleic acids and chromatin and altered expression patterns of nuclear-matrix proteins are highly relevant to malignant progression1. Recent studies have found that a higher level of nucleic acids (aggregates of chromatins) are present in various cancers1–4, indicating the potential use of nucleic acids for cancer screening and monitoring. In addition, overexpression of specific protein biomarkers, such as epidermal growth factor receptor (EGFR)5–7, cellular tumor antigen p538, cell regulator protein Bcl-29, and cell division cycle protein CDK-110, in cell nuclei, is of particular prognostic significance11, as they are highly correlated with patients’ survival rate and their response to chemotherapy and radiotherapy in many types of cancers (e.g., lung cancer6, triple-negative breast cancer12, colorectal cancer13, ameloblastoma14, ovarian cancer15, gastric cancer and colorectal cancers9,16). Detection and evaluation of those nuclear biomarkers (nucleic acids, genetics, and proteins) in human tissues will not only help scientists better understand the signaling pathway in cancers, but more importantly, provide critical information for early stage cancer diagnosis, prognosis, malignant transformation, and the efficacy of anticancer therapies17–21.
To date, evaluation of microscopic histopathology slides by pathologists still remains as the golden standard for cancer diagnosis, which is based mainly on the morphological assessment of cell nuclei in tissues using the Hematoxylin and Eosin (H&E) staining22,23. However, due to the lack of quantitative analysis capability, this method is highly subjective and even the best-characterized histopathological features receive only modest agreement among experienced pathologists, which may result in misdiagnosis and poor treatment management24. This issue becomes particularly serious and challenging when we deal with very early stage cancers25.
Meanwhile, for cancer prognosis and guided therapy, immunohistochemistry (IHC) is widely used to analyze specific antigens, especially those prognostic and predicative biomarkers26,27. IHC relies on colorimetric detection to identify the location of a given proteomic biomarker within a tissue. While simple, IHC performs poorly in distinguishing biomarkers in nuclei from their surrounding background due to the nonlinear optical effect and low dynamic range20 (example given in Supplementary Fig. 1). Consequently, the cells having overexpressed nuclear proteomic biomarkers may be misdiagnosed. This problem exacerbates when multiple targets in a nucleus are to be analyzed. Compared to colorimetric detection, immunofluorescence (IF) uses different fluorescent labels to tag different biomarkers, thus resulting in improved contrast and multiplexing capability over IHC21,28. However, despite elevated biomarker concentrations in the nuclei for the cancers at an advanced stage, the fluorescence signals from the nuclear biomarkers are oftentimes embedded in a large patch of background fluorescence from cytoplasm that may also have the expression of the same biomarkers (Supplementary Fig. 1). In addition, IF suffers significantly from the broad emission spectra of fluorophores, which poses yet another hurdle to distinguish the biomarkers from nearby features with similar emission wavelengths. Therefore, it becomes challenging to accurately identify the nuclei having biomarker expression and further precisely pinpoint the exact biomarker locations inside the nuclei, which may cause significant distortion in and subsequent misinterpretation of the cancer tissue characterization29.
Recently, biolasers are being developed, in which the laser emission is used as the sensing signal30–44. In comparison with fluorescence-based detection, laser emission has narrow linewidth, threshold behavior, high intensity, and high sensitivity to biomolecular/cellular changes. Therefore, biolasers may provide a new bioanalytical tool that complements traditional fluorescent techniques. In one of the biolaser embodiments, bio-species (such as biomolecules, cells, and tissues) are placed inside a Fabry-Pérot (FP) cavity and the laser characteristics such as lasing wavelength, threshold and intensity are modulated passively (i.e., bio-species are not labeled and the gain media are outside those bio-species)30–32 or actively (i.e., bio-species are labeled with fluorophores that serve as the laser gain media)33,45,46. However, to date, no studies have been carried out to use biolasers to identify and detect specific biomarkers within cells/tissues, in particular, using actual patients’ samples. Whether biolasers can truly provide new bioanalytical capability remains a question that has yet to answer.
In this Article, we proposed a scanning “laser-emission-based microscope” (LEM) for improved tumor tissue characterization based on the earlier development of the tissue laser platform45. Successful mapping of the lasing emissions from nuclear biomarkers (such as nucleic acids and/or specific antigens) were achieved in human tissues with a sub-cellular and sub-micron resolution (<700 nm) and a lasing threshold on the order of tens of μJ/mm2. Fig. 1a illustrates the concept of the LEM, in which a tissue labeled with site specific fluorophores (e.g., nucleic acid probes) and/or antibody-conjugated fluorophores is sandwiched inside an FP microcavity formed by two mirrors. The fluorophores serve as the laser gain medium, which are designed to respond to intracellular binding and intratissue activities, thus generating the sensing signal in the form of laser emission. A 2D scanning stage was integrated to map the tissue and construct images by scanning the pump beam across the whole tissue. As illustrated in Fig. 1b, while fluorescence (top illustration) from a nucleus usually provides “spatially-blurred” signal, which covers a large area with a low spatial resolution and a low contrast between the sites with high and low biomarker expressions, laser emission (bottom illustration) offers distinct advantages due to its high intensity/sensitivity34,35,38,39, high fluorescence background suppression (for high contrast imaging)43,45, high spatial resolution35,47, high spectral resolution (for highly multiplexed detection), and unique threshold behavior (to selectively switch on/off specific laser emission signals (Figs. 1c and 1d)48.
Figure 1. Conceptual illustration of the laser-emission based microscope.
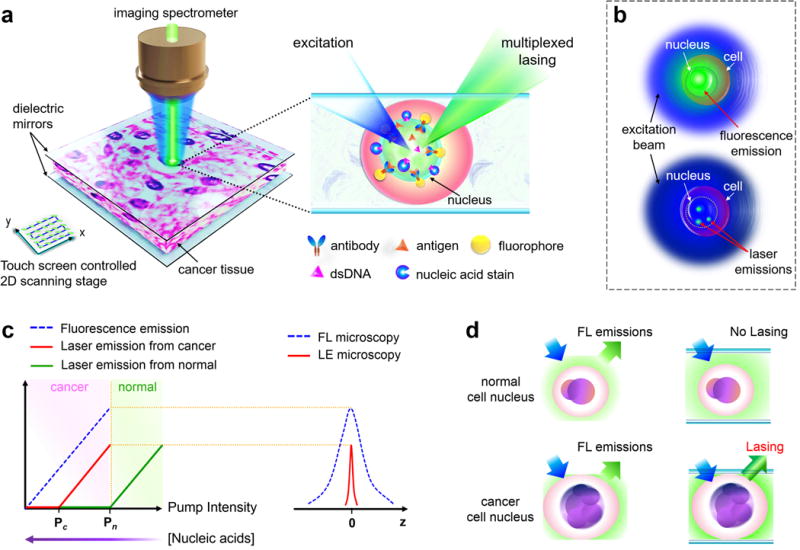
a, Illustration of the laser-emission based microscope (LEM) configuration when a human cancer tissue is sandwiched within a high-Q Fabry-Pérot cavity and integrated with a 2D raster scanning stage. The laser emission from fluorophores is achieved upon external excitation. The inset shows the details of using nucleic acid staining dyes and antibody-conjugated dyes to achieve multiplexed laser emissions from a tissue. Laser emissions are achieved only when probes are bound to the nucleus or targeted nuclear biomarker within the tissue. Here only one antibody is plotted for example; however, multiple targeted antibodies/fluorophores can be used. b, Comparison between the traditional fluorescence emission (top) and “star-like” laser emission (bottom) from a single nucleus. c, (Left) Output intensity of laser emission as a function of pump intensity. Pc, lasing threshold of cancer cell lasing; Pn, lasing threshold of normal cell lasing. A higher/lower nucleic acid concentration leads to a lower/higher lasing threshold. (Right) Laser emission (red solid line) has a much narrower emission profile than traditional fluorescence (blue dashed line) d, Fluorescence emission is detected in both normal and cancer cell nuclei, whereas laser emission can be detected only in cancer cell nuclei when pump energy density is set between Pc and Pn.
In particular, lasing with lung and colon cancer tissues was demonstrated with a total of 41 samples from 35 patients’ biopsies (N=41 and >100 tissue sections) involved. Nucleic acid dye, YO-PRO-1 Iodide (YOPRO), and several antibodies (e.g., anti-EGFR, mutant-p53, and Bcl-2) conjugated with fluorescein isothiocyanate (FITC) were used to analyze nucleic acids and the proteomic biomarkers within the nuclei, respectively. We demonstrated that the LEM is able to not only clearly distinguish between cancer and normal tissues, but diagnose early stage lung cancer tissues with a high sensitivity of 97.5%. Tumor tissues with and without nuclear proteomic biomarkers can be quantified due to vastly different lasing thresholds resulting from different nuclear expressions of biomarkers. Furthermore, we showed the wavelength-multiplexed immuno-lasing capability of the LEM. Dual lasing emissions were achieved to signify the co-localization of nucleic acid and nuclear proteomic biomarkers. We envision that this study will provide an imaging tool complementary to H&E, IHC and IF for better cancer diagnosis and prognosis, as well as to improve the basic understanding of fundamental cell biology.
Results
The detailed description of mirror fabrication/characterization, FP cavity assembly, and the laser emission-based microscope (LEM) setup can be found in Methods and Supplementary Fig. 2. All the human tissue sections (cancerous and normal) were prepared under the same conditions with a thickness of 15 μm, all of which were labeled with either YOPRO to target nucleic acids inside cells or FITC-conjugated antibodies that bind specifically to EGFR, mutant-p53, or Bcl-2, which can be expressed on the cell membranes, cytoplasmic organelles (endoplasmic reticulum, Golgi, and endosome), and nuclei. It should be noticed that these antibodies are not only specific to the cell nuclei; however, clinical studies have identified the presence of such biomarkers in the nuclei as a critical sign to determine the patients’ survival rate and prognosis13,18. Details of sample preparation and staining procedures can be found in Methods.
Lasing in tissues with nucleic-acid probes
We began with the lung tissues labeled with YOPRO alone. Figs. 2a and b show the lasing spectra of cancerous and normal lung tissues under various pump energy densities when 0.5 mM YOPRO solution was used to label the tissues (H&E images in Supplementary Fig. 3). A sharp lasing peak emerges around 547 nm. As a control experiment (not shown), no lasing was observed with pure YOPRO solution (without any tissue) at 0.5 mM, which is expected, as YOPRO has virtually no emission in the absence of nucleic acids. The spectrally integrated laser emission versus pump energy density extracted from Figs. 2a and b are both presented in Fig. 2c, from which the lasing threshold of cancer and normal tissue are derived to be 21 μJ/mm2 and 32 μJ/mm2, respectively. Furthermore, in Fig. 2d we investigated the dependence of the lasing threshold on the concentration of the YOPRO solution used to stain the tissues at a fixed resonator length (15 μm) for both cancer and normal tissues. When the YOPRO solution concentration decreases from 0.5 mM to 0.05 mM, the effective YOPRO concentration within a nucleus is expected to decrease accordingly, which leads to a drastic increase in the lasing threshold, especially for the YOPRO concentration below 0.25 mM. For all YOPRO concentrations, we find that the lasing thresholds for cancer tissues are consistently lower than those of normal tissues, which is attributed to the higher amount of nuclear chromatin (and hence higher YOPRO concentrations) inside cancer cells (due to higher DNA replication activities)1,49,50. In addition, we notice that in Figs. 2a and b the lasing emissions remain single mode operation even at a pump energy density significantly higher than the respective lasing threshold. The full-width-at-half-maximum (FWHM) of the lasing emission is only 0.16 nm, limited by the spectrometer resolution. Such a single-mode lasing emission band is >100X narrower than that for the corresponding fluorescence, thus enabling highly wavelength-multiplexed detection. Although at an extremely high pump energy density (5X threshold) the YOPRO-stained tissue can support multi-mode lasing operation (see Supplementary Fig. 4), the overall lasing band is still less than 10 nm wide, attesting to the intrinsic narrow gain profile for YOPRO, despite a wide fluorescence band.
Figure 2. Lasing in lung tissue with nucleic acid staining dye.
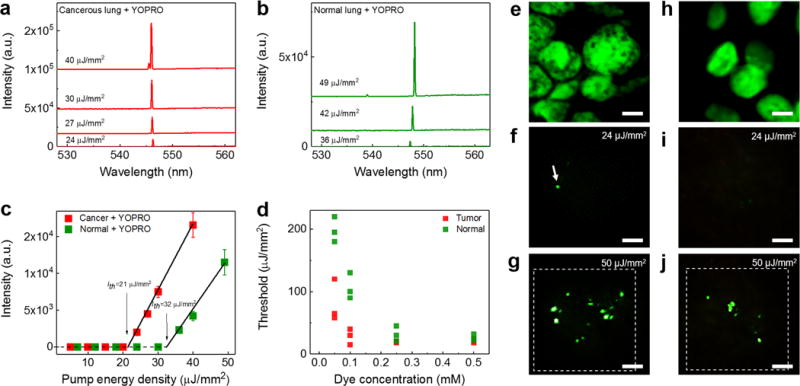
a and b, Examples of lasing spectra of a human lung cancer tissue (a) and normal lung tissue (b), stained with YOPRO under various pump energy densities. Curves are vertically shifted for clarity. c, Comparison of spectrally integrated (540 nm – 550 nm) laser output as a function of pump energy density extracted from the spectra in a and b. The solid lines are the linear fit above the lasing threshold, indicating a lasing threshold of 21 μJ/mm2 for cancer tissue and 32 μJ/mm2 for normal lung tissue. The error bars (s.d.) are defined by considering the pump energy density variation of OPO pulsed laser during the measurements. d, Lasing threshold with different concentrations of YOPRO used to stain the tissue. Three individual measurements were measured for each concentration at different sites, as presented individually in green and red squares. e, Confocal fluorescence image of a lung cancer nucleus (shown in green). f-g, CCD images of the laser output from a nucleus in a lung cancer tissue (f) around (24 μJ/mm2) and (g) far above (50 μJ/mm2) the lasing threshold. The image shows clearly several sharp “lasing stars” within the nucleus, whereas the background fluorescence is significantly suppressed. h, Confocal image of a normal lung nucleus (in green). i-j, CCD images of the laser output from a nucleus in a normal lung tissue (i) below (24 μJ/mm2) and (j) above (50 μJ/mm2) the lasing threshold. Note that e/h and g/j were taken from the same piece of tissue, but does not exactly represent the same cells. All the tissues in a-j were stained with YOPRO (0.5 mM in bulk staining solution) under the same preparation conditions. The dashed squares in g and j show the laser pump beam area in LEM. All scale bars, 5 μm. The corresponding H&E images of the cancer tissues and normal tissues are provided in Supplementary Fig. 3.
Fig. 2e shows the confocal fluorescence microscopy image of a nucleus in a cancer cell from the lung cancer tissue. Due to the low contrast of the fluorescence emission within the nucleus, we are unable to extract the exact locations having high nucleic acid concentrations. However, as visualized by the CCD images in Figs. 2f and g, multiple sharp laser emissions with distinct and strong output against the surrounding background (‘lasing stars’) are visible at specific sites with the highest abundance of nucleic acids. With the increased pump energy density, more sites having slightly lower abundance of nucleic acids start to lase (see Movie 1). Note that there may be fluorescence background emitting from the places that have relatively low concentrations of nucleic acids and therefore are unable to generate laser emission. However, such fluorescence is completely blocked by the top mirror that has >99.8 % reflectivity. Similarly, in Figs. 2h–j we also carried out a series of parallel experiments using a normal lung tissue and observed the same phenomena as in Figs. 2f–g, except that the lasing stars start to emerge at a higher pump energy density and, at a given pump energy density, fewer lasing stars are observed, both of which reflect the lower abundance of nucleic acids in a normal tissue, as discussed previously in Fig. 2d.
Characterization of laser-emission-based microscope
To further characterize those “lasing stars”, in Fig. 3a we plot the laser emission profile of a single lasing star from the lung cancer tissue captured on the CCD. The FWHM is measured to be approximately 678 nm, providing a sub-micron and sub-cellular optical resolution (defined by the FWHM). Fig. 3b illustrates that two adjacent lasing stars separated by only 1.3 μm can be well resolved. Comparison among bright field images, fluorescence images and laser-emission images using the identical cancer cells is given in Supplementary Fig. 5, showing high spatial resolution and high contrast of the LEM, which is due mainly to the background suppression mechanism in the LEM (the background here is defined as the noise or low level signals within the cell nuclei). The insets of Figs. 3c–f illustrate that the sub-cellular lasing stars emerge progressively from a single to multiple lasing stars within the same pumping beam spot when the pump energy density increases gradually. The spectral analysis in Figs. 3c–f suggests that those lasing stars are independent of each other. Each of them is in single lasing mode operation, but may have slightly different lasing wavelengths due possibly to different local environments (such as nucleus thickness, refractive index, and gain distribution, etc.). As exemplified in Figs. 3c–f, at a relatively low pump energy density, only those sites having the highest analyte concentration can lase. With the increased pump energy density, lasing from multiple sites can be observed. Conversely, multiple lasing sites can be “turned-off” down to a single lasing site by decreasing the pump energy density (see Movies 1), signifying the repeatability and controllability of those lasing stars.
Figure 3. Optical resolution of sub-cellular lasers under LEM.
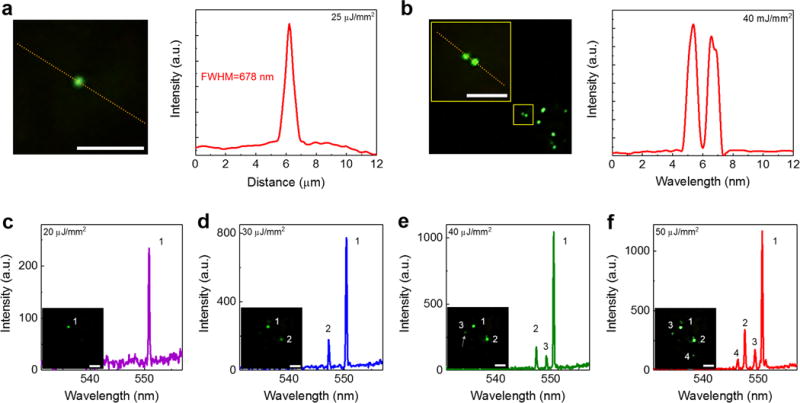
a, Enlarged CCD image (left) of a single laser emission star from a human lung tissue stained with YOPRO. The intensity profile along the yellow dotted line (right) shows the FWHM of 678 nm. b, Enlarged CCD image (left) of two adjacent lasing stars. The yellow square identifies the location of two lasing stars within the tissue. The intensity profile along the yellow dotted line (right) shows two well-resolved peaks. The smallest resolvable distance between two laser emissions is estimated to be better than 1 μm. c-f, Lasing spectra of independent sub-cellular lasers within the same focal beam spot by increasing the pump energy density from (c) 20 μJ/mm2, (d) 30 μJ/mm2, (e) 40 μJ/mm2, to (f) 50 μJ/mm2. The insets show the CCD images of corresponding laser emissions, in which c is an example of a single lasing star, d is an example of two independent lasing stars with different lasing thresholds, e is an example of three independent lasing stars with different lasing thresholds, and f is an example of multiple independent lasing stars emerging simultaneously at a high pump energy density. Note that the slight increase in the background emission beyond 560 nm in c-e is due to the fluorescence leaking out of the FP cavity caused by the reduced reflectivity of the dielectric mirror (see Supplementary Fig. 2 for details). NA= 0.42. All scale bars, 5 μm.
Furthermore, spatial analysis shows that those lasing stars are the lowest order (0,0) Ince-Gaussian mode51,52, which is due largely to the localization of nucleic acids (and hence the YOPRO). In order to validate this, we conducted a series of experiments by staining lung normal/cancer tissues with FITC (non-specific dye) for comparison (Supplementary Fig. 6). Despite the refractive index differences, similar lasing modes (generally higher order Ince-Gaussian modes) were observed for both normal and cancer cells (Supplementary Fig. 6d and e), suggesting that FITC is equally distributed throughout the cell. In contrast, multiple independent lasing modes (lasing stars) can be observed in cells when labeled with YOPRO. The significant difference between FITC and YOPRO in Supplementary Fig. 6 supports the hypothesis that the lasing star is caused by the localization (concentration) effect of nucleic acids (and hence dyes). Consequently, the results in Fig. 3 provide an alternative method to quantify the analyte concentration in tissues (or cells) with a sub-micron spatial resolution by ramping the pump energy density. The image for each the pump energy density can be recorded so that the distribution of analyte relative concentration can be mapped and the histogram of the sites having different levels of analyte concentrations can be built, thus enabling more detailed characterization of tissues and cells in the future.
Lasing thresholds of cancer/normal tissue biopsy
According to the results in Fig. 2d, the lasing thresholds for the cancer cell nuclei are generally lower than those for the normal cell nuclei. In particular, Fig. 2d shows that the binding of YOPRO to nuclear acids starts to saturate beyond 0.1 mM in cancer tissues. Thus, the largest difference in lasing threshold is achieved at 0.1 mM, which can be explored to distinguish between the cancer and normal tissues. In Fig. 4 we systemically investigated the lasing thresholds of 14 patients’ biopsies (including 10 sets of normal/cancer lung tissue, H&E images are given in Supplementary Fig. 7), all of which were stained with 0.1 mM YOPRO. First, we present the statistics of lasing thresholds based on the 6 pairs of lung cancer tissues and normal lung tissues from the same patients (P1–P6) in Figs. 4a and b, respectively. In particular, two types of non-small cell lung cancer, andenocarcinoma and squamous cell carcinoma (Fig. 4c for example), were investigated. At least 20 cells were randomly selected and measured within each tumor/normal tissue region for each patient. Interestingly, all cancer cells exhibit consistently a narrow lasing threshold range around 20 μJ/mm2. In contrast, the normal cells have a much wider threshold range, varying from 30 μJ/mm2 to 180 μJ/mm2. Next, we investigated the lasing thresholds based on 4 pairs of cancer and normal tissue from 8 different patients (4 normal: P7-P10 and 4 cancer: P11-P14) in order to examine the sample-to-sample variations. Similar statistic results of lasing thresholds were observed in Figs. 4d and e. Both the p values of two sample t-Test between normal and tumor cells in both cases (same patient/different patient) are < 0.005.
Figure 4. Statistics of cancer/normal cell lasing thresholds.
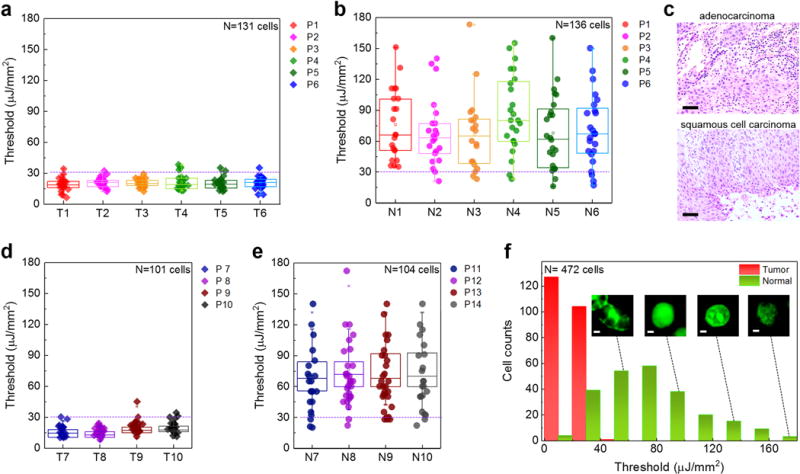
a, Statistics of tumor cell lasing thresholds from six individual lung cancer patients (P1, P2, P3, P4, P5, P6), labeled as Tumor tissue, T1-T6. For each patient, at least 20 cells were randomly selected and measured. b, Statistics of normal cell lasing thresholds of normal lung tissues from the same six patients (P1-P6) in a, labeled as Normal tissue N1-N6. c, Exemplary H&E microscopic images of the two major types of non-small lung cancers used in this work, including andenocarcinoma (top) for P1-P3 and squamous cell carcinoma (bottom) for P4-P6. Scale bars, 100 μm. d, Statistics of tumor cell lasing thresholds from four different lung cancer patients (P7, P8, P9, P10), labeled as Tumor tissue, T7- T10. e, Statistics of normal cell lasing thresholds of normal lung tissues from four different control patients (P11-P14), labeled as Normal tissue N7-N10. For each patient, at least 20 cells were randomly selected and measured. The error bars (s.d.) in a, b, d and e are defined by the lasing threshold variation of 20 cells measured from each patients, respectively. The statistical box plots are also shown in the same figure in a, b, d and e. The dashed purple lines in a, b, d, and e indicate the cutoff threshold of 30 μJ/mm2. f, Histogram of all cancer/normal cell lasing thresholds (N=472) extracted from a, b, d and e. The insets show the confocal fluorescence images of normal cells at different cell phases. Scale bars, 1 μm. The H&E images of the cancer tissues and normal tissues of entire 14 patients, P1-P14, are provided in Supplementary Fig. 7.
The histogram of the lasing thresholds (N=472 cells) in Fig. 4f extracted from Figs. 4a, b, d, and e shows clearly a cutoff threshold around 30 μJ/mm2 between the normal and cancer cells. As discussed previously, the stark difference in the lasing threshold is attributable to the different nucleic acid concentrations within cell nuclei53. As shown in the fluorescence images in the inset of Fig. 4f, normal cells undergo regular cell cycles/stages, whereas cancer cells are highly active abnormal cells that undergo endless divisions and DNA replication. The high proliferation of DNA (chromatins) in cancer cell nuclei will result in high nucleic acid concentrations, thus lower lasing thresholds.
Lung cancer screening with scanning LEM
Based on the statistic results in Fig. 4f, in Fig. 5 we employed the LEM to distinguish the cancer and normal tissues by mapping the laser emissions from nuclei in both cancer and normal tissues from 3 individual patients (P15, P16, P17, see Supplementary Fig. 8 for H&E images). For each patient, 5 tissue sections (for both cancer and normal tissues) were scanned with the LEM over a field-of-view of 150 μm × 150 μm under a fixed pump energy density of 30 μJ/mm2, as shown in Figs. 5a–c, respectively. It is obvious that no or only a few lasing stars appeared in all normal tissue sections. In contrast, a large number of lasing stars were observed in cancer tissue sections, where isolated individual lasing stars and clusters of lasing stars were both present, since the pump energy density is above the lasing threshold for cancer cells. Quantitative analyses of the number of lasing cells using the LEM results in Figs. 5a–c are plotted in Figs. 5d–f (note: a lasing cell is defined as the cell that has one or more lasing stars). Statistically significant differences between the cancer and normal tissues were achieved (p < 0.005 with two-sample t-Test), demonstrating the superior contrast and screening capability of the LEM. As comparison, in Supplementary Fig. 9, we scanned the same tissues with confocal fluorescence microscopy. Supplementary Fig. 10 further analyzes the same confocal fluorescence images and shows that the fluorescence of the cancer and normal cells is very similar in intensity and spatial distribution. Those results in Supplementary Figs. 9 and 10 suggest that confocal fluorescence microscopy is unable to distinguish/quantify cancer and normal tissues by nucleic acid expression by simply using fluorescence intensity distribution (Note that in Supplementary Figs. 9 and 10, cancer and normal tissues can still be distinguished by the cell sizes/morphologies. However, for morphology based tissue evaluation, H&E (rather than fluorescence microscopy) is the golden standard. Later in Fig. 7 and Supplementary 11, we tested the LEM with early stage cancer tissues, where cancer/normal tissues cannot be distinguished by the cell sizes/morphologies). In comparison with histopathology, which is based on morphological assessment of cells and tissues, the LEM method that maps the lasing emissions from nuclei may provide a simpler, more systematic, and more quantitative tool to complement the traditional H&E method in tissue characterization.
Figure 5. Comparison and statistics of laser-emission microscopic images of normal/cancer tissues.
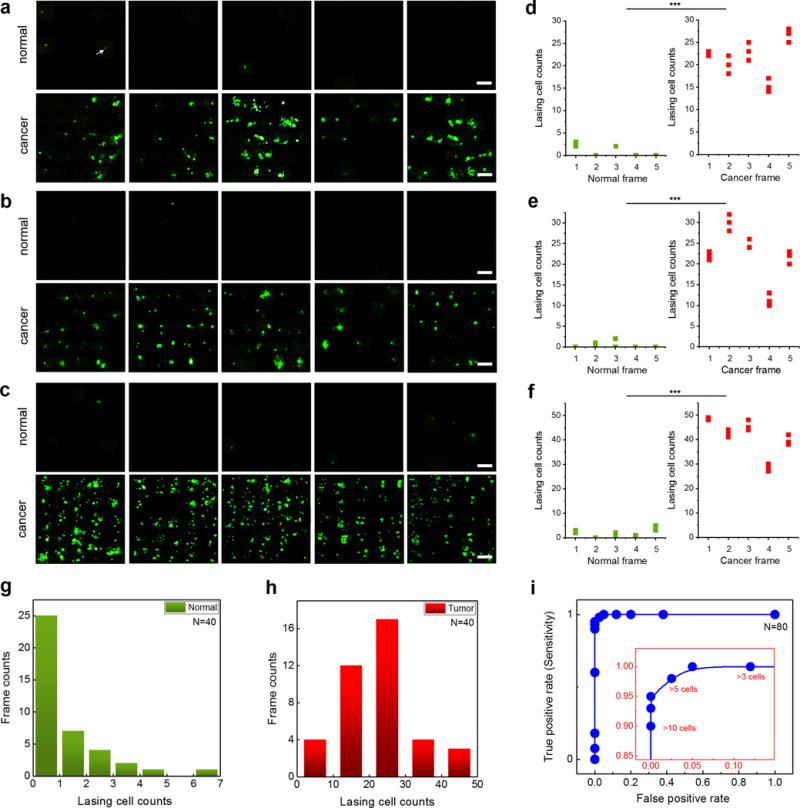
a–c, LEM images by mapping the nucleic acids in normal and lung cancer tissues of (a) Patient 15, (b) Patient 16, and (c) Patient 17. For each patient, five normal/cancer tissue sections (frames) were scanned under a fixed pump energy density of 30 μJ/mm2. The white arrow in a points an example of a single lasing star in a normal tissue of Patient 1. NA= 0.42. All scale bars, 20 μm. Each frame is 150 μm × 150 μm. The corresponding H&E images of the cancer tissues and normal tissues of the three patients are provided in Supplementary Fig. 8. All three patients’ tissues were examined by pathologists and diagnosed as lung cancer. (P15: Stage II, P16: Stage I, P17: Stage I lung cancer). d-f, Statistics of the number of cells per frame that have laser emission from their respective nuclei for (d) Patient 15, (e) Patient 16, and (f) Patient 17 extracted from the LEM images in a-c. Green/red dots represent for the normal/cancer tissues, respectively. The lasing cell counts were calculated three times for each tissue frame as plotted in d-f. g, Histogram of frame counts based on the number of the lasing cells per frame in normal tissues from 8 different patients (P1, P3, P5, P6, P10, P15, P16, P17). For each patient, five frames (150 μm × 150 μm) were scanned under a fixed pump energy density of 30 μJ/mm2. h, Histogram of frame counts based on the number of the lasing cells per frame in cancer tissues from 8 different patients (P1, P3, P5, P6, P10, P15, P16, P17). For each patient, five frames (150 μm × 150 μm) were scanned under a fixed pump energy density of 30 μJ/mm2. i, Receiver Operating Characteristics (ROC) curve based on the 80 frames (40 normal tissue sections, 40 tumor tissue sections) in g and h. The ROC curve is plotted by using the different lasing cell counts per frame. The area under the curve is 0.998. The inset shows the enlarged part of the ROC curve, in which the sensitivity of 97.5% is obtained based on the criterion of >5 lasing cells per LEM frame.
Figure 7. Lasing in lung cancer tissue with anti-EGFR-FITC.
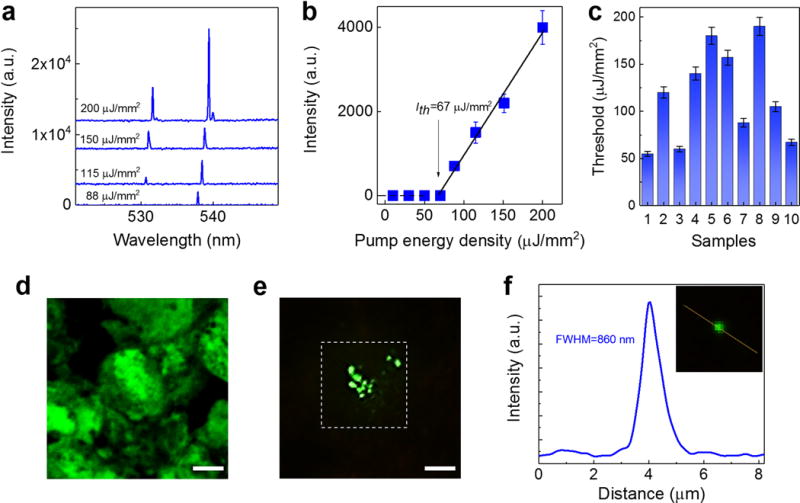
a, Examples of lasing spectra of a human lung cancer tissue with n-EGFR expression stained with anti-EGFR-FITC under various pump energy densities. Curves are vertically shifted for clarity. b, Spectrally integrated (530 nm – 540 nm) laser output as a function of pump energy density extracted from the spectra in a. The solid lines are the linear fit above the lasing threshold, indicating a lasing threshold of 67 μJ/mm2. c, Histogram of n-EGFR-FITC lasing thresholds measured from 10 cells (samples) out of 5 lung cancer patients (patients P21-P25: including squamous cell carcinoma and andenocarcinoma tissues). The error bars (s.d.) in both b and c are defined by considering the pump energy density variation of OPO pulsed laser during the measurements. d, Confocal microscopic image of a cell with n-EGFR expression in the lung cancer tissue. e, CCD images of the laser output from the same lung cancer tissue above the lasing threshold (125 μJ/mm2). The image shows clearly several “lasing stars” corresponding to the highest concentrated EGFR locations within the nuclei. The dashed square shows the laser pump beam area in LEM, which is focused on only one of the cells in the tissue. Note that d and e are not from the identical cells, but from the same piece of tissue. f, The intensity profile along the yellow dotted line (inset) shows the FWHM is measured to be 860 nm. All the tissues in a-e were stained with anti-EGFR-FITC (0.5 mM in bulk staining solution) under the same preparation conditions. NA=0.42. All scale bars, 10 μm.
In order to to establish the sensitivity and specificity of the LEM technique, we randomly selected 8 cancer patients and performed the LEM on both of their normal and cancer tissues. By using the same experimental condition and quantification methods as in Fig. 5, five tissue sections (frames) were scanned for each patient for both cancer tissue and normal tissue (a total of 80 frames, N=80). The histogram of the number of the lasing cells per frame for normal tissues and cancer tissuess are plotted in Figs. 5g and h, respectively. Based on the number of lasing cells per frame, we generate the Receiver Operation Characteristic (ROC) curve in Fig. 5i, which has the area under the curve of 0.998. We found that the optimal threshold to identify a cancer tissue is “5 lasing cells per frame”, which corresponds to a sensitivity of 97.5%. Practically, the threshold can be adjusted for higher sensitivity or higher specificity.
Using the threshold established above, we further demonstrated an important application of the LEM by examining early stage lung cancer tissues, which is regarded as the most critical yet challenging task in clinical histopathology. For this study, we used three samples from three patients, who were diagnosed as early stage lung cancer (in-progress lung cancer, see Figs. 6a–c for the H&E images). The confocal fluorescence images are provided in the Supplementary Fig. 11. It is obvious that the normal cells, in-progress cancer cells, and cancer cells are completely mixed within the whole tissue, making it very difficult to identify/differentiate according to cell morphology and/or fluorescence intensity. In contrast, by using the LEM, cancer cells can be unambiguously identified within the tissue with a high contrast. For each patient, 5 sections were scanned with the LEM under a fixed pump energy density of 30 μJ/mm2, as shown in Figs. 6d–f. Quantitative analyses of the number of lasing cells using the LEM are also plotted in Figs. 6g–i. All three tissues are identified as cancer tissues since all frames have more than 5 lasing cells.
Figure 6. Laser-emission microscopic images of early stage lung cancer tissues.
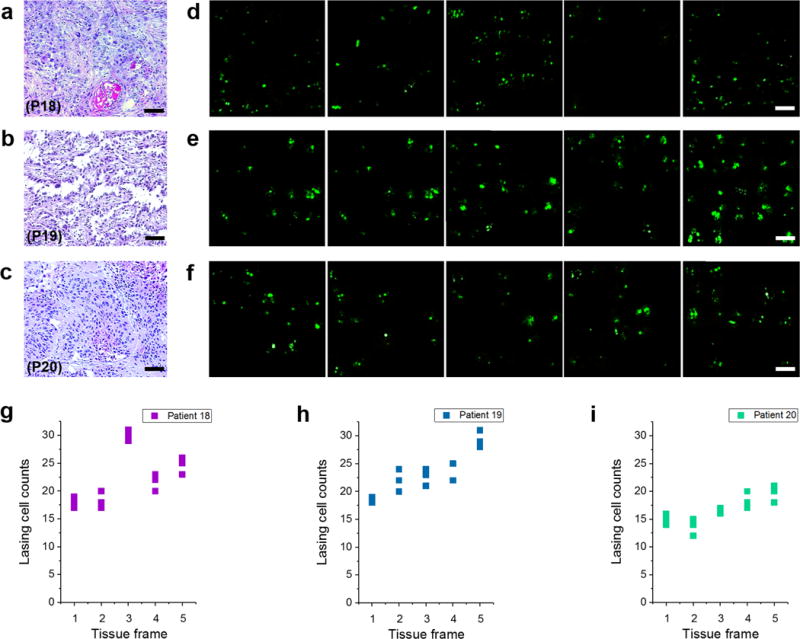
a–c, H&E images of (a) Patient 18 (P18), (b) Patient 19 (P19), and (c) Patient 20 (P20), who are identified as in-progress or early stage lung cancer. d-f, The corresponding LEM images by scanning the nucleic acids of the tissues from the same three patients. For each patient, five tissue sections (frames) were scanned under a fixed pump energy density of 30 μJ/mm2. g-i, Statistics of the number of cells having lasing emission from nuclei extracted from the LEM images. The lasing cell counts were calculated three times for each tissue frame as plotted in g-i. NA= 0.42. Scale bars for a-c, 100 μm; d-f, 20 μm.
Multiplexed detection with immuno-lasing in lung cancer tissues
Moving a step forward, we aim to improve the prognostic prediction of cancer patients by demonstrating immunodiagnosis capability of the LEM. Here in Fig. 7 we studied the lasing emission using EGFR antibody conjugated with FITC (anti-EGFR-FITC) to target nuclear EGFR (n-EGFR) expressed in the same lung cancer tissue used in Fig. 2. Similar to the YOPRO case in Fig. 2a, Fig. 7a shows single-mode lasing emission with an FWHM of about 0.16 nm when the pump energy density is slightly above the threshold. Although with the increased pump energy density the second mode emerges, the lasing band is only about 8 nm, much narrower than the corresponding fluorescence band (>60 nm. See Supplementary Fig. 12). The spectrally integrated laser emission versus pump energy density extracted from Fig. 7a is presented in Fig. 7b, showing a lasing threshold of approximately 67 μJ/mm2. We further investigated 10 samples from 5 lung cancer patients with nuclear EGFR positive response (Patients: P21-P25. Details are given in Supplementary Fig. 13). The measured lasing thresholds is plotted in Fig. 7c. The variation in the lasing threshold is caused by the different degrees of EGFR expression (hence FITC concentration) in each cell nucleus. For comparison, it is difficult for conventional IF microscopy (Fig. 7d) to pinpoint the exact locations within the nucleus that have high EGFR concentrations. Similar to the YOPRO lasing case studied previously, with the laser emission significant improvement is achieved in the imaging contrast and the capability to locate the spots of high EGFR concentrations with a sub-micron resolution, as shown in Figs. 7e and f.
With the sub-cellular lasing from n-EGFR achieved in Fig. 7, we now applied the LEM to distinguish the lung tissues with and without n-EGFR in Fig. 8. We prepared two types of lung cancer tissues, adenocarcinoma with n-EGFR (Tissue type #1) and adenocarcinoma without n-EGFR (Tissue type #2). Both type of tissues are well characterized and verified by pathologists (see Methods and Supplementary Fig. 13). The exemplary lasing spectra of Type #1 (red curve) and Tissue #2 (blue curve) tissues when stained with anti-EGFR-FITC are shown in Fig. 8a. As expected, laser emission at 537 nm is obtained in Tissue #1 with a pump energy density of 80 μJ/mm2; however, no lasing from Tissue #2 can be observed from the entire tissue when pumped with the same energy density. This result indicates that cells having EGFR expression only on the membranes or other cytoplasmic organelles are not capable to provide sufficient gain to lase due to relatively low abundance of EGFR (and hence FITC). To validate the above experiments, we examined a total of 12 human patients with 3 tissue sections from each patient, including 6 lung cancer patients with n-EGFR and 6 patients without n-EGFR (3 normal and 3 lung cancer diagnosed without n-EGFR). Details of the 12 patients are provided in Supplementary Fig. 13, labeled as patient P21-P32. Among all the 6 patients with n-EGFR, 5 of them showed positive lasing emissions (P21-P25). For all normal patients (P30-P32) and those having lung cancer but without n-EGFR (P27-P29), none of them showed lasing emissions from anti-EGFR-FITC. The statistic results are plotted in Fig. 8b. The corresponding IHC image of both types of tissues in Fig. 8b (right column) confirm that Type #1 tissue has a significant amount of EGFR within most cell nuclei, whereas Type #2 tissue does not have any EGFR expression within the cell nuclei (but does have EGFR expressed on the cell membrane).
Figure 8. Multiplexed lasing in lung cancer tissues.
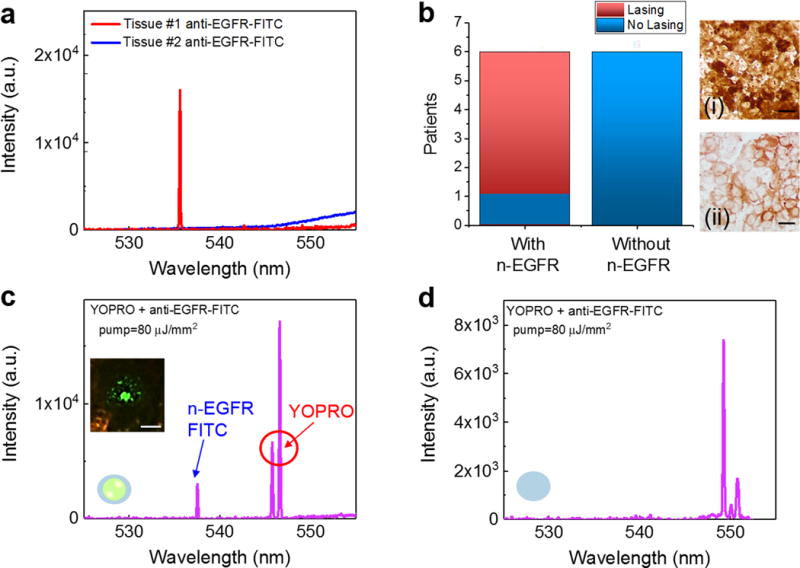
a, Lasing spectra of Type #1 (red curve) and Type #2 (blue curve) tissues stained with anti-EGFR-FITC. Pump energy density=80 μJ/mm2. b, Statistics of the EGFR lasing results for positive and negative n-EGFR lasing from 12 patients (P21-P32). Details and H&E images are provided Supplementary Fig. 13. (i) Brightfield IHC image of a human lung cancer tissue with n-EGFR (Tissue type #1). (ii) Brightfield IHC of a human lung cancer tissue without n-EGFR overexpression (Tissue type #2). c, Lasing spectra of a Type #1 tissue dual-stained with YOPRO and EGFR-anti-FITC. The laser was focused on a single nucleus within the lung cancer tissue. The pump energy density was set above the threshold for both YOPRO and FITC under single excitation wavelength. The inset CCD image is the demonstration of a n-EGFR laser emission, which indicates that EGFR co-localizes with the nucleus. d, Lasing spectra of a Type #2 tissue dual-stained with YOPRO and anti-EGFR-FITC. The laser was focused on a single nucleus within the lung cancer tissue. Note that the slight increase in the background emission beyond 550 nm in a and c is due to the fluorescence leaking out of the FP cavity caused by the reduced reflectivity of the dielectric mirror (see Supplementary Fig. 2). Scale bars, 20 μm.
In order to further confirm that FITC lasing is only from the expression of EGFR in the nucleus, co-localization of EGFR and nucleus lasing signals from sub-cellular locations is critical, which not only verifies the presence of n-EGFR in a nucleus but also makes it easier for computer-aided imaging processing in the future. To demonstrate co-localization, both Type #1 and Type #2 tissues were dual-stained with YOPRO and anti-EGFR-FITC (see Methods). It is apparent that we are unable to clearly determine the sub-cellular locations by their fluorescence, or distinguish the EGFR expressions in nucleus and cytoplasm, as a result of the huge spectral overlap between YOPRO and FITC (Supplementary Fig. 12). In contrast, lasing signals of the two dyes can be observed and distinguished spectrally for Type #1 tissue. As shown in Fig. 8c, two sharp lasing peaks appear at 537 nm and 547 nm when the tissue was pumped at the nucleus, which is the evidence of dual lasing from both EGFR and nucleic acids. The inset of Fig. 8c shows a CCD image, which confirms the presence of EGFR in the nucleus. Note that since the lasing threshold of FITC is much higher than that of for YOPRO, the lasing intensity from FITC is lower than that from YOPRO under the same pump energy density. Also note that even at higher pump energy densities at which higher order modes may emerge, the lasing spectra of YOPRO and FITC can still be distinguished due to their very narrow emission bands (Supplementary Fig. 4). As a negative control, for Type #2 tissue (Fig. 8d) only lasing spectrum from YOPRO was obtained when the tissue was pumped at the nucleus, no lasing signal from EGFR was observed, indicating that no EGFR or very low EGFR exists in the nucleus.
Immuno-lasing for nuclear biomarker detection in colorectal cancer tissues
Finally, we applied the LEM to other types of tissues and proteomic biomarkers to validate its broad utility. In Supplementary Fig. 14 we examined three different important cancer biomarkers (EGFR, p53 and Bcl-2) in human colon cancer tissues. For the purpose of tissue characterization, Supplementary Figs. 14a–c present the IHC and confocal IF images of a colorectal cancer tissues labeled with the corresponding antibodies (i.e., anti-EGFR, anti-mutant p53, and anti-Bcl-2), showing a high amount of EGFR, p53 and Bcl-2 in the nuclei of the respective tissues. Similar to the procedures used previously, the lasing emissions from those colon tissues labeled with anti-EGFR-FITC, anti-mutant p53-FITC, and anti-Bcl-2-FITC were achieved in Supplementary Figs. 14d–f, when the nuclei were pumped. Our results show that lasing is achieved when there exist a high amount of biomarkers within the nuclei, which can be used for immunodiagnosis of various nuclear biomarkers. Despite the existence of multiple lasing peaks at the high pump energy density, the laser emission band is still approximately only 5 nm, showing the potential capability of multiplexed detection. The corresponding lasing threshold curves are presented in Supplementary Figs. 15a–c, showing that the lasing threshold is on the order of 200 μJ/mm2.
Outlook
In this work, we have developed a laser-emission based imaging tool (LEM) that takes advantage of the high intensity, high background suppression, and high spectral/spatial resolution of the laser emission. We have further tested its validity in early stage cancer diagnosis and evaluation of various nuclear biomarkers (nucleic acids, EGFR, p53, and Bcl-2) in human cancer tissues, which may significantly benefit the current clinical practice by providing complementary information that the conventional H&E, IHC, and IF are unable or difficult to obtain. As an outlook, below we discuss a few possible areas that deserve further exploration in the near future.
Conventional methods (H&E, IHC and IF) are known to be more subjective and may cause significant variations in tissue evaluation from one pathologist to another22–24,54. The LEM can potentially provide a means to quantify the number of sites within a cell that have different levels of biomarker expression and the number of cells that express high nuclear biomarkers. In Fig. 5, each lasing cell was counted only once even though there might be multiple lasing stars present inside the lasing cell. Despite this significantly simplified way of categorization, the LEM is already able to distinguish cancer and normal tissues. On the other hand, we have discussed that the lasing stars in cancer tissues may have different lasing thresholds due to different biomarker concentrations. By ramping the pump energy density, the histogram of the lasing stars with different thresholds can be constructed, which may allow us to sub-categorize the cancer tissue (e.g., in terms of severity and sensitivity to therapies). We will work closely with pathologists to develop algorithms that can make best use of this new information for more objective evaluation of cancer tissues.
Fundamentally, through the LEM we have found that there exist multiple sites within a nucleus that have high biomarker expressions. Further interdisciplinary collaboration is needed to elucidate mechanisms of the occurrence of those spots and how to make use of them in cancer diagnosis/prognosis.
Besides cancer cells, the same technology can be extended to stroma cells (cancer-associated fibroblasts) that have overexpressed biomarkers. For example, Supplementary Fig. 16 gives a lasing example using a colon tissue expressed with Bcl-2. Recent reports have revealed that the interaction between stromal cells and tumor cells plays a major role in cancer growth and progression55. Moreover, stromal gene expression have also demonstrated to define poor-prognosis subtypes in several cancers56,57. Therefore the capability to observe tumor stroma lasing will be of potential significance to predict clinical outcomes and strengthen clinical prognostic factors.
Other potential applications will also be explored, including, but are not limited to, Pap smear stain examination, intraoperative image-guided neurosurgery and tumor resection, and Fluorescence In-Site Hybridization (FISH).
While dual detection (2-plexed detection) was achieved in this work, there is no doubt that the LEM platform is capable to accommodate more fluorophores. For instance, in Supplementary Fig. 17a lasing emission from a third dye (high-affinity nucleic acid dye - SYTOX Blue) was achieved, which is centered on 500 nm with a band width less than 5 nm. Furthermore, we demonstrate that at least four lasing emission wavelengths can be clearly distinguished within the range of 500 nm – 550 nm in Supplementary Fig. 17b. In the future, high wavelength multiplexicity of the lasing emission will be explored for better characterization of cells and tissues.
The laser output from tissues provides a number of optical parameters unseen in fluorescence, such as the lasing threshold of various biomarkers, lasing mode spatial distribution, lasing mode competition, and lasing gain clamping. We will conduct further investigations to understand how those parameters reflect the underlying biological and physiological processes of cancerous tissues as well as to achieve better spectral and spatial resolution.
Methods and Materials
Tissue and device preparation
In this study, a total of 41 tissue samples (N=41) from 35 patients’ tissue were used, including human lung tissues (N=38, labeled as P1-P32) and human colon tissues (N=3, P33-P35). In particular, P18-P20 are identified as early stage lung cancer samples. All the human lung and colon tissues were purchased from OriGene Technologies in the form of OCT (Optimal Cutting Temperature) frozen tissue blocks. Both male and female patients diagnosed with stage I/II cancer, aging from 46–78 were examined. All human subjects are fully informed and are explicitly asked for their consent to future research use of their samples. Samples are collected from a diverse set of medical institutions throughout the United States in order to maximize patient diversity. Those tissues were verified at the Company by pathologists with full pathological evaluation data, clinical annotation (including patient age, gender, and minimum stage grouping), abstracted pathology reports to ensure the accuracy of the sample level diagnosis (i.e., a normal adjacent sample collected from a cancer patient) and detail the sample’s cellularity (% normal cells, % lesion cells, % tumor cells, % stroma and % necrosis). For the exemplary tissues demonstrated used in Fig. 8, both tissues were examined by pathologists and verified with lung adenocarcinoma and EGFR.
Upon receiving, all OCT tissue blocks were sliced into 15 μm thick sections by using a cryostat (Leica 3050S). The selected tissue section was picked up and placed on the top of a poly-L–lysine (Sigma-Aldrich #P8920) coated dielectric mirror, which was first cleaned and rinsed with lysine for better tissue adhesion. The tissue were then rinsed with PBS (phosphate buffered solution, ThermoFisher # 10010023) and air dried before staining/labeling (see staining/labeling details in the next section). Finally, the tissue was covered by the top dielectric mirror. For confocal IF microscopy, the tissue was first deposited on the top of a superfrost glass slide (ThermoFisher #3021–002), followed by the same staining process, and then mounted with Fluoromount (Sigma-Aldrich #4680) and covered with a coverslip before scanning.
For experiments in Fig. 4, all tissues were first examined with H&E histopathology slides to select the area that consists of all tumor cells or all normal cells (as shown in Supplementary Fig. 7). All of the tissues were sectioned into 4 slices and at least 5 cells within the tumor/normal region from each tissue section were randomly selected and measured.
Staining and labeling
For IHC staining, the tissue was fixed on a superfrost glass slide (ThermoScientific #15–188-48) by immersing in pre-cooled acetone (−20 °C) for 3 minutes and dried off at room temperature. The slide was then rinsed with PBS twice. Next, the tissue was first blocked with BSA buffer for 10 minutes to prevent non-specific binding and rinsed with PBS thoroughly. Then the tissue was incubated with 200 μl of diluted primary antibody (anti-human-EGFR antibody (abcam #52894) overnight at 4 °C. The primary antibody was prepared with 1:50 solution, with a final concentration of 0.005 mg/ml. After incubation of primary antibodies, the tissue was rinsed with PBS, followed by 30 minutes of staining with HRP conjugated anti-Rb IgG secondary antibody (abcam #6721) at room temperature with a dilution of 1:200 (final concentration=0.01 mg/ml). Then DAB substrate solution (abcam #64238) was applied to the tissue for 5 minutes to reveal the color of the antibody staining. After rinsing 5 times, the tissue was dehydrated through pure alcohol, then mounted with mounting medium (abcam #ab64230), and finally covered with a coverslip. The same procedure was applied to both lung and colon tissues. For colon tissues, two additional primary antibodies, anti-mutant p53 (abcam #32049) and anti-Bcl-2 (abcam #182858), were used.
For nucleic acid labeling, YOPRO solution (ThermoFisher #Y3603) was dissolved in PBS at a concentration of 0.5 mM, then diluted to lower concentrations with PBS. The prepared YOPRO solution was then applied to the tissue sections for 10 minutes and rinsed with PBS solution three times before measurements. The above process is the same for both IF and laser measurements.
For antibody-fluorophore labeling of lung tissues, the glass slide was first rinsed with PBS twice and blocked with BSA buffer for 10 minutes to prevent non-specific binding, then rinsed again with PBS. Next, the tissue was incubated with 200 μl of diluted primary antibody (anti-human-EGFR antibody (abcam #52894)) overnight at 4 °C. The primary antibody was prepared with 1:50 solution, with a final concentration at 0.01 mg/ml. After incubation with primary antibodies, the tissue was rinsed with PBS thoroughly, followed by FITC conjugated anti-Rb IgG secondary antibody (stained for 2 hours at room temperature). The secondary antibody (ThermoFisher #65-6111) was prepared with 1:5 dilution to reach the final concentration of 0.2 mg/ml. Finally, the tissue was rinsed again with PBS before laser experiment. The same procedure was applied to colon tissues, in which the primary antibodies, (anti-EGFR antibody (abcam #52894), anti-mutant p53 antibody (abcam #32049) and anti-Bcl-2 antibody (abcam#182858), were used, followed by FITC conjugated anti-Rb IgG secondary antibody (stained for 2 hours at room temperature). The above process is the same for both IF and laser measurements.
For dual staining of anti-EGFR-FITC and YOPRO, we first applied anti-EGFR-FITC (the same procedure as described for primary and secondary antibody labeling) to the lung tissue sections, then 0.1 mM YOPRO solution was applied for 20 minutes to the same tissue and rinsed with PBS solution twice before measurement. For staining of SYTOX Blue (ThermoFisher #S11348) and BOBO-1 Iodide (ThermoFisher #B3582) in Supplementary Fig. 17, SYTOX and BOBO-1 Iodide were dissolved in PBS at a concentration of 0.2 mM, following by the same staining procedures as for YOPRO.
Optical imaging techniques and LEM setup
The bright field IHC images were taken with a Nikon-E800 Wide field Microscope. The confocal fluorescence microscopic images were taken by using Nikon A1 Spectral Confocal Microscope with an excitation of 488 nm laser source. The bright field images as well as the movie (Movie 1) of the laser emissions (“lasing stars”) in tissues were captured by using a CCD (Thorlabs #DCU223C) integrated directly on top of the objective in our experimental setup (see Supplementary Fig. 2a for details). In particular, the fluorescence images in Supplementary Fig. 5 were first captured with a wide-field fluorescence microscope (without the top mirror), then immediately the corresponding lasing measurements were performed by covering the tissue samples with the top mirror.
A typical confocal setup was used to excite the sample and collect emission light from the FP cavity (see the optical setup in Supplementary Fig. 2a). A pulsed OPO laser (pulse width: 5 ns, repetition rate: 20 Hz) at 465 nm was used to excite the stained tissues. The FWHM of the focused laser beam size was ~ 30 μm in diameter, in which the spatial sampling area is slightly larger than the focal beam size. The pump energy density was adjusted by a continuously variable neutral density filter, normally in the range of 1 μJ/mm2 −200 μJ/mm2. The emission light was collected through the same objective then separated by a beam splitter to the spectrometer (Horiba iHR550, spectral resolution ~0.2 nm) and CCD for spectral and image analysis.
The laser-emission scanned images were collected through the same optical setup, in which the images were taken by the CCD (10 fps, Thorlabs #DCU223C) mounted on top of the objective (NA 0.42, 20X). The raster scanning stage was home-built using two linear actuators with electric controllers (Newport #CONEX TRA25CC) and integrated with a raspberry PI/touchscreen (Digikey #8997466) for operation. The LEM images shown in Figs. 5–7 (FOV of 150 μm × 150 μm per frame in this work) was formed by mapping and integration of 25 individual CCD images (30 μm × 30 μm). However, larger mapping area up to 1mm × 1mm is achievable with the aid of algorithms. Currently the accuracy of the stage is limited by 3 μm during each displacement.
FP microcavities
The FP microcavity was formed by two customized dielectric mirrors (Supplementary Fig. 2). The top mirror (made by Qingdao Novel Beam Technology Co. Ltd, China) had a high reflectivity in the spectral range of 520 nm – 555 nm to provide optical feedback and high transmission around 465 nm for the pump light to pass through, whereas the bottom mirror (made by Evaporated Coating INC, USA) had a slightly wider reflection band. The respective reflectivity for the top mirror and bottom mirror at the lasing wavelength (535–555 nm) is approximately 99.8% and 99.9%, while the transmission of the top mirror at pump wavelength (465 nm) is ~ 90.2%. The Q-factor for the FP cavity was on the order of 104, at a cavity length of 15 μm (without tissues). Details of the fabrication and characterization of the FP cavities are described in the reference58.
Data availability
All raw images and data generated in this work, including the representative images provided in the manuscript, are available from the corresponding author upon request. The authors declare that all data supporting the findings of this study are available within the paper and its supplementary information.
Supplementary Material
Acknowledgments
We acknowledge the support from National Science Foundation (ECCS-1607250), National Institutes of Health (NIBIB-1R21EB016783), and National Natural Science Foundation of China (Grant No. 61471254). We also thank the support of the Molecular Imaging Analysis Lab at the University of Michigan for cryostats and confocal microscopy. We would also like to thank Dr. Wei-Hung Weng from Harvard Medical School for pathology assistance.
Footnotes
Author contributions. Y.C.C. and X.F. conceived the research; Y.C.C, X.T, and X.F. designed the experiments; Y.C.C. and Q.C. performed the experiments; Q.S. developed the coding for 2D scanning system; W.W. and X.F. designed, fabricated, and characterized the optical mirrors and cavities. Y.C.C. and X.F. analyzed data; and Y.C.C. and X.F. wrote the paper.
Additional Information. The authors declare no competing financial interests.
References
- 1.Zink D, Fischer AH, Nickerson JA. Nuclear structure in cancer cells. Nat Rev Cancer. 2004;4:677–687. doi: 10.1038/nrc1430. [DOI] [PubMed] [Google Scholar]
- 2.Swarup V, Rajeswari M. Circulating (cell-free) nucleic acids–a promising, non-invasive tool for early detection of several human diseases. FEBS Lett. 2007;581:795–799. doi: 10.1016/j.febslet.2007.01.051. [DOI] [PubMed] [Google Scholar]
- 3.Schwarzenbach H, Hoon DS, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. 2011;11:426–437. doi: 10.1038/nrc3066. [DOI] [PubMed] [Google Scholar]
- 4.Morgan MA, Shilatifard A. Chromatin signatures of cancer. Genes & Dev. 2015;29:238–249. doi: 10.1101/gad.255182.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lo H, Hung M. Nuclear EGFR signalling network in cancers: linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Br J Cancer. 2006;94:184–188. doi: 10.1038/sj.bjc.6602941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Traynor AM, et al. Nuclear EGFR protein expression predicts poor survival in early stage non-small cell lung cancer. Lung Cancer. 2013;81:138–141. doi: 10.1016/j.lungcan.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang L, et al. Evaluation of Raman spectroscopy for diagnosing EGFR mutation status in lung adenocarcinoma. Analyst. 2014;139:455–463. doi: 10.1039/c3an01381b. [DOI] [PubMed] [Google Scholar]
- 8.Sarkis AS, et al. Prognostic value of p53 nuclear overexpression in patients with invasive bladder cancer treated with neoadjuvant MVAC. J Clin Oncol. 1995;13:1384–1390. doi: 10.1200/JCO.1995.13.6.1384. [DOI] [PubMed] [Google Scholar]
- 9.Manne U, et al. Prognostic significance of Bcl-2 expression and p53 nuclear accumulation in colorectal adenocarcinoma. Int J Cancer. 1997;74:346–358. doi: 10.1002/(sici)1097-0215(19970620)74:3<346::aid-ijc19>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 10.Porter LA, Donoghue DJ. Cyclin B1 and CDK1: nuclear localization and upstream regulators. Prog Cell Cycle Res. 2003;5:335–348. [PubMed] [Google Scholar]
- 11.Konety BR, Getzenberg RH. Nuclear structural proteins as biomarkers of cancer. J Cell Biochem. 1999;75:183–191. doi: 10.1002/(sici)1097-4644(1999)75:32+<183::aid-jcb22>3.3.co;2-1. [DOI] [PubMed] [Google Scholar]
- 12.Rakha EA, et al. Prognostic markers in triple‐negative breast cancer. Cancer. 2007;109:25–32. doi: 10.1002/cncr.22381. [DOI] [PubMed] [Google Scholar]
- 13.Dekanić A, et al. Strong nuclear EGFR expression in colorectal carcinomas is associated with cyclin-D1 but not with gene EGFR amplification. Diagn Pathol. 2011;6:1. doi: 10.1186/1746-1596-6-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pereira NB, et al. Nuclear localization of epidermal growth factor receptor (EGFR) in ameloblastomas. Oncotarget. 2015;6:9679. doi: 10.18632/oncotarget.3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xia W, et al. Nuclear expression of epidermal growth factor receptor is a novel prognostic value in patients with ovarian cancer. Mol Carcinog. 2009;48:610–617. doi: 10.1002/mc.20504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Müller W, Schneiders A, Hommel G, Gabbert H. Prognostic value of bcl-2 expression in gastric cancer. Anticancer Res. 1997;18:4699–4704. [PubMed] [Google Scholar]
- 17.Beer DG, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;8:816–824. doi: 10.1038/nm733. [DOI] [PubMed] [Google Scholar]
- 18.Ludwig JA, Weinstein JN. Biomarkers in cancer staging, prognosis and treatment selection. Nat Rev Cancer. 2005;5:845–856. doi: 10.1038/nrc1739. [DOI] [PubMed] [Google Scholar]
- 19.Irish JM, Kotecha N, Nolan GP. Mapping normal and cancer cell signalling networks: towards single-cell proteomics. Nat Rev Cancer. 2006;6:146–155. doi: 10.1038/nrc1804. [DOI] [PubMed] [Google Scholar]
- 20.Levenson RM, Borowsky AD, Angelo M. Immunohistochemistry and mass spectrometry for highly multiplexed cellular molecular imaging. Lab Invest. 2015;95:397–405. doi: 10.1038/labinvest.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou L, et al. Single-band upconversion nanoprobes for multiplexed simultaneous in situ molecular mapping of cancer biomarkers. Nature Commun. 2015;6 doi: 10.1038/ncomms7938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Collins LG, Haines C, Perkel R, Enck RE. Lung cancer: diagnosis and management. Am Fam Physician. 2007;75:56–63. [PubMed] [Google Scholar]
- 23.Silvestri GA, et al. Noninvasive staging of non-small cell lung cancer: ACCP evidenced-based clinical practice guidelines. Chest. (2nd) 2007;132:178S–201S. doi: 10.1378/chest.07-1360. [DOI] [PubMed] [Google Scholar]
- 24.Yu KH, et al. Predicting non-small cell lung cancer prognosis by fully automated microscopic pathology image features. Nat Commun. 2016;7 doi: 10.1038/ncomms12474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vansteenkiste J, Dooms C, De Leyn P. Early stage non-small-cell lung cancer: challenges in staging and adjuvant treatment: evidence-based staging. Ann Oncol. 2010;21:vii189–vii195. doi: 10.1093/annonc/mdq424. [DOI] [PubMed] [Google Scholar]
- 26.Shi SR, Cote RJ, Taylor CR. Antigen retrieval immunohistochemistry: past, present, and future. J Histochem Cytochem. 1997;45:327–343. doi: 10.1177/002215549704500301. [DOI] [PubMed] [Google Scholar]
- 27.Arnould L, et al. Trastuzumab-based treatment of HER2-positive breast cancer: an antibody-dependent cellular cytotoxicity mechanism. Brit J Cancer. 2006;94:259–267. doi: 10.1038/sj.bjc.6602930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gerdes MJ, et al. Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc Natl Sci Acad USA. 2013;110:11982–11987. doi: 10.1073/pnas.1300136110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu L, Qu X. Cancer biomarker detection: recent achievements and challenges. Chem Soc Rev. 2015;44:2963–2997. doi: 10.1039/c4cs00370e. [DOI] [PubMed] [Google Scholar]
- 30.Gourley P. Semiconductor microlasers: A new approach to cell–structure analysis. Nat Med. 1996;2:942–944. doi: 10.1038/nm0896-942. [DOI] [PubMed] [Google Scholar]
- 31.Gourley PL. Biocavity laser for high-speed cell and tumour biology. J Phys D: Appl Phys. 2003;36:R228. [Google Scholar]
- 32.Gourley P, et al. Ultrafast Nanolaser Flow Device for Detecting Cancer in Single Cells. Biomed Microdevices. 2005;7:331–339. doi: 10.1007/s10544-005-6075-x. [DOI] [PubMed] [Google Scholar]
- 33.Gather MC, Yun SH. Single-cell biological lasers. Nature Photon. 2011;5:406–410. [Google Scholar]
- 34.Sun Y, Fan X. Distinguishing DNA by Analog-to-Digital-like Conversion by Using Optofluidic Lasers. Angew Chem Int Ed. 2012;51:1236–1239. doi: 10.1002/anie.201107381. [DOI] [PubMed] [Google Scholar]
- 35.Fan X, Yun SH. The potential of optofluidic biolasers. Nat Methods. 2014;11:141–147. doi: 10.1038/nmeth.2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nizamoglu S, Gather MC, Yun SH. All-Biomaterial Laser using Vitamin and Biopolymers. Adv Mater. 2013;25:5943–5947. doi: 10.1002/adma201300818. [DOI] [PubMed] [Google Scholar]
- 37.Humar M, Gather MC, Yun SH. Cellular dye lasers: lasing thresholds and sensing in a planar resonator. Opt Express. 2015;23:27865–27879. doi: 10.1364/OE.23.027865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Humar M, Yun SH. Intracellular microlasers. Nature Photon. 2015;9:572–576. doi: 10.1038/nphoton.2015.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schubert M, et al. Lasing within live cells containing intracellular optical micro-resonators for barcode-type cell tagging and tracking. Nano Lett. 2015;15:5647–5652. doi: 10.1021/acs.nanolett.5b02491. [DOI] [PubMed] [Google Scholar]
- 40.Wei Y, et al. Starch-Based Biological Microla sers. ACS nano. 2016 [Google Scholar]
- 41.Chen YC, Chen Q, Fan X. Optofluidic chlorophyll lasers. Lab Chip. 2016;16:2228–2235. doi: 10.1039/c6lc00512h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caixeiro S, Gaio M, Marelli B, Omenetto FG, Sapienza R. Silk-Based Biocompatible Random Lasing. Adv Opt Mater. 2016;4:998–1003. [Google Scholar]
- 43.Chen YC, Chen Q, Fan X. Lasing in blood. Optica. 2016;3:809–815. doi: 10.1364/OPTICA.3.000809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aas M, Chen Q, Jonáš A, Kiraz A, Fan X. Optofluidic FRET lasers and their applications in novel photonic devices and biochemical sensing. IEEE J Sel Top Quantum Electron. 2016;22:1–15. [Google Scholar]
- 45.Chen YC, Chen Q, Zhang T, Wang W, Fan X. Versatile tissue lasers based on high-Q Fabry–Pérot microcavities. Lab Chip. 2017;17:538–548. doi: 10.1039/c6lc01457g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nizamoglu S, et al. A Simple Approach to Biological Single-Cell Lasers Via Intracellular Dyes. Adv Opt Mater. 2015;3:1197–1200. [Google Scholar]
- 47.Cho S, Humar M, Martino N, Yun SH. Laser Particle Stimulated Emission Microscopy. Phys Rev Lett. 2016;117:193902. doi: 10.1103/PhysRevLett.117.193902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen Q, et al. Highly sensitive fluorescent protein FRET detection using optofluidic lasers. Lab Chip. 2013;13:2679–2681. doi: 10.1039/c3lc50207d. [DOI] [PubMed] [Google Scholar]
- 49.Beljanski M. The Regulation of DNA Replication and Transcription. Demos Medical Publishing; 2013. [Google Scholar]
- 50.Lane AN, Fan TWM. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015;43:2466–2485. doi: 10.1093/nar/gkv047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bandres MA, Gutiérrez-Vega JC. Ince–Gaussian beams. Opt Lett. 2004;29:144–146. doi: 10.1364/ol.29.000144. [DOI] [PubMed] [Google Scholar]
- 52.Schwarz UT, Bandres MA, Gutiérrez-Vega JC. Observation of Ince–Gaussian modes in stable resonators. Opt Lett. 2004;29:1870–1872. doi: 10.1364/ol.29.001870. [DOI] [PubMed] [Google Scholar]
- 53.Pliss A, Kuzmin AN, Kachynski AV, Prasad PN. Nonlinear optical imaging and Raman microspectrometry of the cell nucleus throughout the cell cycle. Biophys J. 2010;99:3483–3491. doi: 10.1016/j.bpj.2010.06.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grilley-Olson JE, et al. Validation of interobserver agreement in lung cancer assessment: hematoxylin-eosin diagnostic reproducibility for non-small cell lung cancer: the 2004 World Health Organization classification and therapeutically relevant subsets. Arch Pathol Lab Med. 2012;137:32–40. doi: 10.5858/arpa.2012-0033-OA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bremnes RM, et al. The role of tumor stroma in cancer progression and prognosis: emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J Thorac Oncol. 2011;6:209–217. doi: 10.1097/JTO.0b013e3181f8a1bd. [DOI] [PubMed] [Google Scholar]
- 56.Finak G, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14:518–527. doi: 10.1038/nm1764. [DOI] [PubMed] [Google Scholar]
- 57.Calon A, et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat Genet. 2015;47:320–329. doi: 10.1038/ng.3225. [DOI] [PubMed] [Google Scholar]
- 58.Wang W, et al. Optofluidic laser array based on stable high-Q Fabry-Perot microcavities. Lab Chip. 2015;15:3862–3869. doi: 10.1039/c5lc00847f. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw images and data generated in this work, including the representative images provided in the manuscript, are available from the corresponding author upon request. The authors declare that all data supporting the findings of this study are available within the paper and its supplementary information.