

Abstract
Objective:
To describe clinical and imaging responses in neurosarcoidosis to infliximab, a monoclonal antibody against tumor necrosis factor–α.
Methods:
Investigators at 6 US centers retrospectively identified patients with CNS sarcoidosis treated with infliximab, including only patients with definite or probable neurosarcoidosis following rigorous exclusion of other causes.
Results:
Of 66 patients with CNS sarcoidosis (27 definite, 39 probable) treated with infliximab for a median of 1.5 years, the mean age was 47.5 years at infliximab initiation (SD 11.7, range 24–71 years); 56.1% were female; 62.1% were white, 37.0% African American, and 3% Hispanic. Sarcoidosis was isolated to the CNS in 19.7%. Using infliximab doses ranging from 3 to 7 mg/kg every 4–8 weeks, MRI evidence of a favorable treatment response was observed in 82.1% of patients with imaging follow-up (n = 56), with complete remission of active disease in 51.8% and partial MRI improvement in 30.1%; MRI worsened in 1 patient (1.8%). There was clinical improvement in 77.3% of patients, with complete neurologic recovery in 28.8%, partial improvement in 48.5%, clinical stability in 18.2%, worsening in 3%, and 1 lost to follow-up. In 16 patients in remission when infliximab was discontinued, the disease recurred in 9 (56%), typically in the same neuroanatomic location.
Conclusions:
Most patients with CNS sarcoidosis treated with infliximab exhibit favorable imaging and clinical treatment responses, including some previously refractory to other immunosuppressive treatments.
Classification of evidence:
This study provides Class IV evidence that for patients with CNS sarcoidosis infliximab is associated with favorable imaging and clinical responses.
Sarcoidosis is an inflammatory disorder characterized by a heightened granulomatous immune response.1 The pathologic hallmarks are compact, well-formed, coalescent non-necrotizing or minimally necrotizing epithelioid granulomas with associated scattered lymphocytes.2 Sarcoidosis exhibits complex genetic inheritance patterns, and nearly all susceptibility genes identified to date are important in immune function.3 The annual age-adjusted incidence of sarcoidosis in the United States ranges from 3 to 10/100,000 among Caucasians to 35–80/100,000 among African Americans.4 Neurosarcoidosis occurs in 5%–15% of patients with sarcoidosis and can cause substantial morbidity.1
While glucocorticoids are a mainstay of treatment for neurosarcoidosis,5 doses required to achieve and sustain an optimal treatment response can be prohibitive due to disease severity or glucocorticoid toxicity.6 Steroid-sparing immunosuppressive therapies have been used to treat neurosarcoidosis with variable success.7–9 There have been no randomized controlled trials focused on CNS sarcoidosis.
Infliximab is a chimeric mouse–human monoclonal antibody against tumor necrosis factor–α (TNF-α) .10 Epithelioid and giant cells in human sarcoidosis granulomas avidly express TNF-α,11 a cytokine critical for granuloma formation and maintenance.12 Infliximab has emerged as a neurosarcoidosis treatment option, including in refractory and steroid-dependent patients, but reports have largely been of single patients or small case series (table e-1 at Neurology.org).
The study of rare neurologic disorders can benefit from collaborative research. An investigator-initiated neurosarcoidosis research consortium was established to advance science and therapeutics in the field.13 In this multi-institutional case series, representing its collaborative effort, we analyze imaging and clinical responses of patients with CNS sarcoidosis treated with infliximab.
METHODS
Patients and study design.
Investigators from 6 US academic medical centers with experience in neurosarcoidosis—University of California, San Francisco (UCSF); Vanderbilt University Medical Center; University of Maryland; Washington University in St. Louis; Harvard (Massachusetts General Hospital); and Kansas University—retrospectively identified patients with CNS sarcoidosis who had been treated with infliximab at their institutions through September 2015 and submitted de-identified data for pooled analysis using a standardized case report form.
We elected to include only those patients who had definite neurosarcoidosis (defined as having a CNS biopsy consistent with sarcoidosis and a neurologic syndrome consistent with granulomatous inflammation) or probable neurosarcoidosis (defined as biopsy-confirmed sarcoidosis in an organ outside the nervous system and a neurologic syndrome consistent with granulomatous inflammation) along with rigorous exclusion of other causes.5,14 Cases of possible or suspected neurosarcoidosis without biopsy confirmation were excluded. We also excluded one patient with combined variable immunodeficiency (CVID)–associated noncaseating granulomatous pulmonary disease who had an inflammatory CNS process (and who notably developed a Nocardia brain abscess on infliximab), as emerging data suggests that CVID-associated granulomatous processes are probably a distinct pathophysiologic entity from classic sarcoidosis.15 Although sarcoidosis can cause peripheral neuropathy and myopathy, in this study of neurosarcoidosis we focus on CNS sarcoidosis.
Standard protocol approvals, registrations, and patient consents.
Institutional review board approval for retrospective chart review was obtained locally at each institution.
Outcome measures.
We defined clinical response as stability, improvement, or worsening of neurologic symptoms and signs as assessed by the treating clinicians and chart review. We defined imaging responses as stability, improvement, or worsening of findings consistent with neurosarcoidosis including contrast enhancement, signal abnormalities on T2-weighted sequences, and other associated features such hydrocephalus, based on review of local case records at last follow-up. We defined a favorable treatment response as either partial or complete imaging improvement in the context of clinical stability, partial clinical improvement, or complete clinical recovery.
Statistical analysis.
Summary statistics were calculated using Stata 12.0 (StataCorp, College Station, TX). Data are expressed in mean and SD and median and interquartile range, unless otherwise specified. Differences in demographic and clinical variables between centers were analyzed using analysis of variance. Multivariable logistic regression was used to evaluate possible predictors of treatment response, including models examining age, sex, race, disease duration before infliximab initiation, duration of infliximab therapy, prior other immunosuppression, and concurrent other immunosuppression.
RESULTS
Patient cohort.
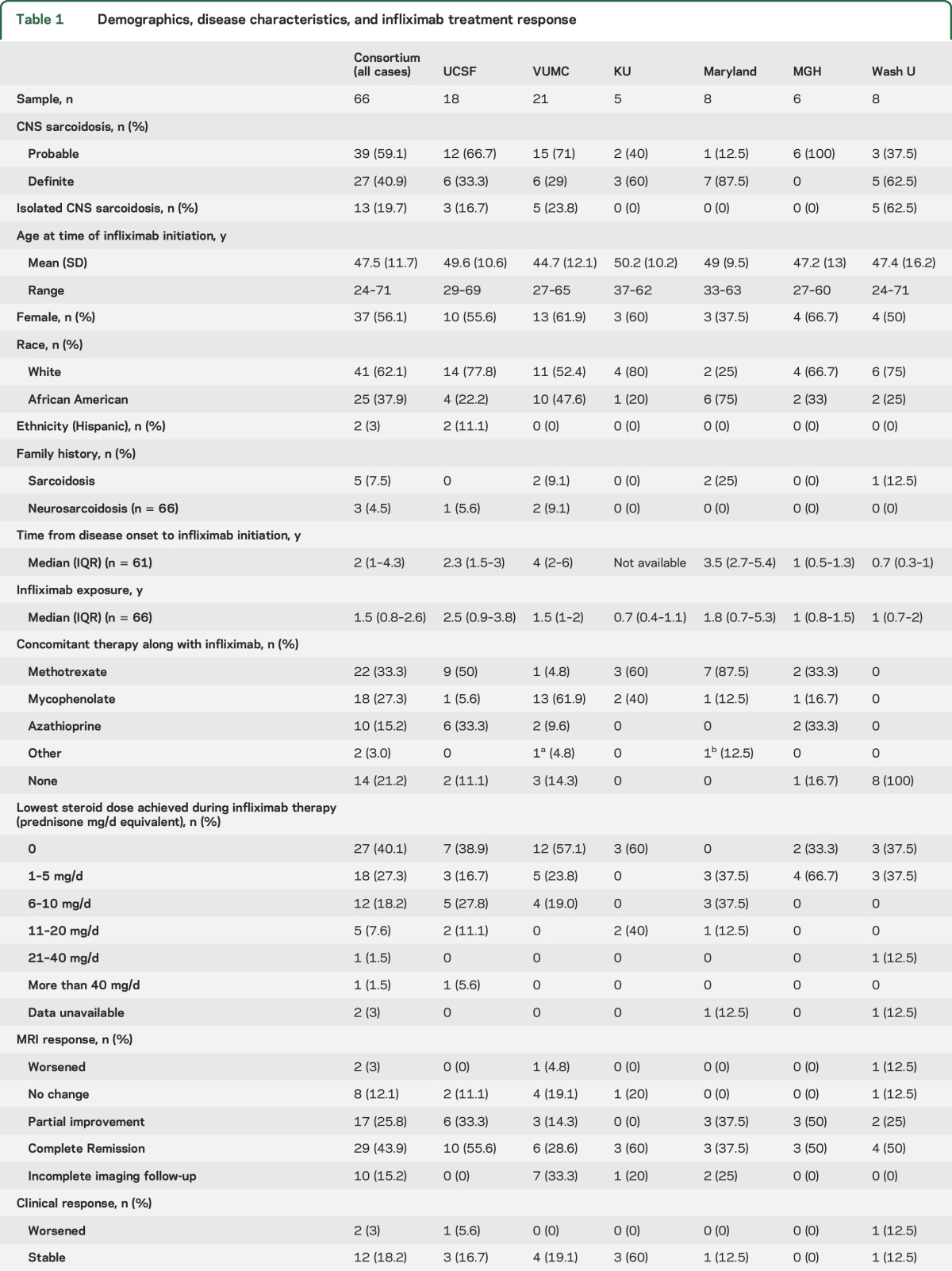
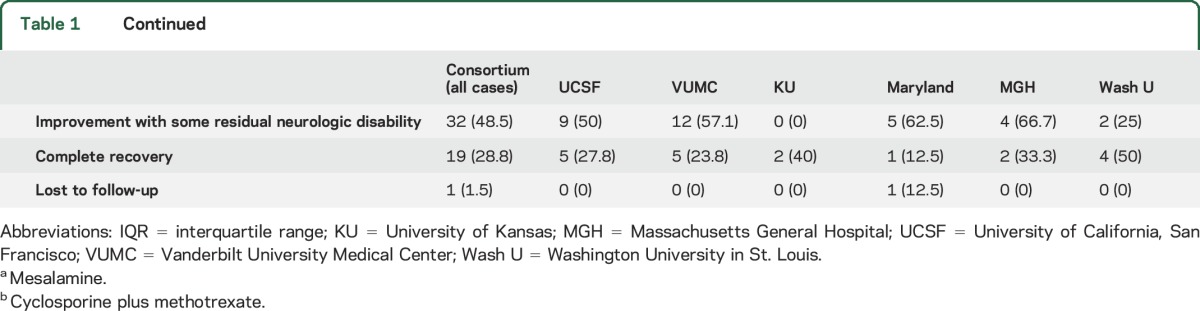
Table 1 summarizes key demographic results and disease characteristics. The clinical syndrome included myelitis or radiculomyelitis in 31 patients (47%), meningitis in 30 (45.5%), pituitary/hypothalamic involvement in 17 (25.8%), and optic neuropathy in 16 (24.2%). Ventriculoperitoneal shunts were present in 8 (12%) patients. Fifty-eight patients had sufficient information for combined clinical and imaging evaluation. There was no statistically significant difference among centers in patient age, sex distribution, or ethnicity, or between relative proportions of clinical and imaging treatment responses.
Table 1.
Demographics, disease characteristics, and infliximab treatment response
Treatment regimens, timing, and duration.
Infliximab dosing regimens varied, and frequency and dose were sometimes adjusted based on treatment response. Loading doses were typically given at weeks 0, 2, and 6. Maintenance dosing ranged from 3 to 7 mg/kg—most commonly 5 mg/kg—and the frequency of infusions ranged from every 4 to every 8 weeks for maintenance infusions. CNS disease was considered active in 100% of treated patients at the time of infliximab initiation.
Previous and concomitant therapies.
All but 1 patient (98.5% of patients) had been treated with glucocorticoids prior to infliximab. In total, 52 (78.8%) patients received at least one steroid-sparing therapy prior to infliximab, including mycophenolate mofetil in 30 patients (45.5%), methotrexate in 24 (36.4%), azathioprine in 9 (13.6%), plasma exchange in 5 (7.5%), hydroxychloroquine in 2 (3%), rituximab in 2 (3%), alemtuzumab in 1 (1.5%), mitoxantrone in 1 (1.5%), IV immunoglobulin in 1 (1.5%), and adalimumab in 1 (1.5%). In total, 49 patients (74.3%) were on another steroid-sparing, nonbiologic, immunosuppressant concurrent with infliximab (table 1). At the time of infliximab initiation, 52 (80%) patients were on concurrent glucocorticoids. During infliximab therapy, 27 patients (40%) were able to discontinue steroids, while another 18 (27%) were able to be maintained on 5 mg/d or less of prednisone or prednisone equivalent (table 1).
Treatment outcome and predictors.
With infliximab treatment, there was a favorable imaging response in 82.1% of patients with adequate MRI follow-up (n = 56), with complete remission of active disease in 29 (51.8%) and partial improvement in 17 (30.1%); MRI findings worsened in 1 patient (1.8%) (table 1). Figures 1 and 2 (and figures e-1 and e-2) provide several examples of observed patterns of infliximab treatment response. With infliximab treatment, there was clinical improvement in 77.3% of patients, with complete recovery in 28.8% and partial improvement in 48.5%; an additional 18.2% of patients exhibited clinical stability; 3% worsened; and 1 was lost to clinical follow-up (table 1). A favorable treatment response was observed in 45 (80.4%) of 58 patients with sufficient information for combined clinical and imaging evaluation. The longer neurosarcoidosis had been present before infliximab initiation, the lower the odds of a favorable treatment response (odds ratio [OR] 0.82, 95% confidence interval [CI] 0.72–0.92, p = 0.02, in a logistic regression model; OR 0.79, 95% CI 0.64–0.97, p = 0.02, in a logistic regression model adjusting for age and sex). The effect size was similar when analyzing by a purely favorable clinical response (OR 0.86, 95% CI 0.75–0.98, p = 0.03) or a purely favorable imaging response (OR 0.83, 95% CI 0.71–0.96, p = 0.01). Combination therapy of infliximab with another steroid-sparing agent was associated with a favorable treatment response (OR 4.8, 95% CI 1.2–19.3, p = 0.03, in a logistic regression model; OR 6.9, 95% CI 1.2–41.3, p = 0.03, in a logistic regression model adjusting for age, sex, and time from disease onset to infliximab initiation). However, combination therapy was not significant as a predictor of a purely imaging response (OR 3.6, 95% CI 0.87–14.6, p = 0.08), complete clinical responses, or complete or partial clinical response. There was no association between infliximab treatment response and age, sex, race, or exposure to prior immunosuppression.
Figure 1. Progression of CNS sarcoidosis over 10 years despite conventional immunosuppressive therapy with complete remission on infliximab.

At age 37, this previously healthy African American man developed bilateral sequential optic neuropathy. Over the next 3 years, he progressed to blindness with no light perception bilaterally, despite aggressive glucocorticoid therapy. Chest CT at age 39 showed mild bilateral hilar lymphadenopathy, and biopsy of nasal mucosa at age 42 was consistent with sarcoidosis. Over the next decade, he went on to develop hypopituitarism, cognitive impairment (correlating with further subcortical involvement on neuroimaging), and intractable hiccups despite treatment with oral glucocorticoids and azathioprine. At age 50, brain biopsy of an enhancing lesion in the right thalamus showed non-necrotizing granulomatous inflammation consistent with CNS sarcoidosis. The top and bottom rows show serial T1 postgadolinium MRIs from 2 different anatomic levels with the x-axis aligned by time. The MRIs show that there is persistent waxing and waning abnormal nodular enhancement (arrows show some examples) in the same neuroanatomic distribution over 10 years, which finally remitted following treatment with infliximab, initially at an escalated dose of 7 mg/kg IV every 6 weeks and since maintained on infliximab 5 mg/kg Q8 weeks together with azathioprine 50 mg/d and replacement doses of glucocorticoids. Remission of the CNS process has been maintained for over 4 years. The patient had breakthrough sarcoidal granulomatous dermatitis on this maintenance regimen 2.5 years into his CNS remission, and the facial lesions subsequently resolved. His hiccups resolved and cognition returned to baseline; as expected, there has been no improvement in visual function. ACE = angiotensin-converting enzyme; IgG = immunoglobulin G; OCB = oligoclonal band; WBC = white blood cell.
Figure 2. Improvement of leptomeningeal, pituitary/hypothalamic, and optic chiasm involvement of neurosarcoidosis following infliximab and worsening upon infliximab discontinuation.

A 32-year-old white man with definite neurosarcoidosis proven by biopsy of a hypothalamic lesion progressed despite treatment with glucocorticoids, azathioprine, and methotrexate. (A) Coronal and midsagittal T1-weighted MRI brain with gadolinium contrast obtained after treatment with the aforementioned for 2 years demonstrated significant, progressing nodular leptomeningeal enhancement along the interhemispheric fissure (small arrow), surrounding the optic chiasm and pituitary stalk (large arrow), and brainstem (arrowhead) including the cerebellopontine angle and upper cervical spinal cord. (B) Coronal and midsagittal T1-weighted MRI brain with gadolinium contrast after 2 months of infliximab demonstrated near-complete resolution of previously active disease with only a small amount of possible enhancement along the optic chiasm (arrowhead). The patient was maintained in remission while on infliximab for 5 years. (C) Upon cessation of infliximab, the patient had recurrence of disease within 8 months. Coronal and midsagittal T1-weighted MRI brain with gadolinium contrast demonstrated nodular leptomeningeal enhancement at previous sites of active disease including the optic chiasm (arrow) and medullopontine angle (arrowhead). He was retreated with infliximab with clinical improvement.
Safety and tolerability of infliximab.
Seven patients (10.1%) had infections that the investigators considered to be possibly related to infliximab or the cumulative immunosuppressive regimen.
Another patient discontinued infliximab after the first dose due to myositis, which was thought to be medication-related. There were no deaths during the period under study. Notably, one patient (the same patient presented in figure 1) subsequently experienced breakthrough cutaneous sarcoidosis on the face while otherwise demonstrating sustained remission of previously highly active neurosarcoidosis on infliximab; the cutaneous sarcoidosis was managed topically with complete remission of cutaneous involvement.
Treatment discontinuation of infliximab.
Of 16 patients in remission from neurosarcoidosis on treatment when infliximab was discontinued, active CNS sarcoidosis recurred in 9 patients (56%) within a mean of 5.7 months (SD 3) following infliximab discontinuation. The disease reoccurred in the same neuroanatomical location of previous disease activity in 60% of these patients (figure 2). There was no significant association in a logistic regression model between total infliximab treatment duration and time to relapse upon infliximab cessation.
DISCUSSION
Our real-world results report cumulative experience from our institutions of infliximab treatment for neurosarcoidosis that is informative for clinical care and future investigations in the field. In this retrospective analysis of 66 patients with definite or probable CNS sarcoidosis from a diverse demographic and geographic distribution from 6 US centers, over three-quarters of patients treated with infliximab exhibited favorable imaging and clinical treatment responses.
Limitations of this study include potential biases inherent in any retrospective case series, including the absence of a comparison of outcomes to a non-infliximab-treated group. However, the return of disease activity in 56% of patients who achieved remission on infliximab at a mean of 5.7 months after infliximab discontinuation provides additional support for the presumed benefit of infliximab in neurosarcoidosis. In addition, the favorable treatment response to infliximab in some patients who failed to respond to other immunosuppressive agents is supportive of an infliximab benefit. Another limitation of this study is that infliximab treatment protocols were not standardized within or across the centers, and variability in infusion protocols and use of concurrent immunotherapies could have had some influence on efficacy and safety. In our series, infliximab was most commonly used as combination therapy, rather than monotherapy, and some of the treatment benefit could relate to synergistic effects of combination immunosuppression. Variability in practice patterns related to timing of clinical and imaging follow-up in this retrospective case series precluded more specific time to event analyses, and future studies with standardized clinical and imaging follow-up will be helpful to clarify the specifics of timing of treatment of response with various dosing regimens. As our centers have a special interest in caring for patients with neurosarcoidosis, the patient sample could reflect referral bias. Despite these limitations, our study is important in reporting real-world, clinically relevant outcomes following infliximab treatment in 66 patients with probable or definite CNS neurosarcoidosis. Furthermore, rates of clinical and imaging responses to infliximab were remarkably consistent across study sites.
Treatment of neurosarcoidosis is currently based on expert opinion, observations from case reports, and relatively small case series, and by inferring efficacy from treatment of sarcoidosis in other organ systems. In one series, as many as 79% of patients with neurosarcoidosis achieved clinical remission with oral glucocorticoids,16 although in many others, a substantial proportion of patients with neurosarcoidosis progressed despite glucocorticoids or relapsed when glucocorticoid doses were tapered to more tolerable, less toxic levels.5,8,9,17 Steroid-sparing immunosuppressive agents used in neurosarcoidosis have included methotrexate, azathioprine, mycophenolate mofetil, hydroxychloroquine, cyclophosphamide, and, increasingly, infliximab.7,18–20 Very few comparative studies of neurosarcoidosis treatments exist, but one retrospective analysis of 40 patients with CNS and peripheral nervous system sarcoidosis suggested greater efficacy of methotrexate over mycophenolate (but with more adverse effects with methotrexate) and relatively high relapse rates with either therapy.21 There are incomplete data about the possible efficacy of anti-CD20 B-cell-depleting treatment in neurosarcoidosis22 and sarcoidosis more broadly,23 but it is notable that 2 patients included in our analysis proceeded to infliximab treatment despite prior treatment with rituximab, with partial clinical improvement on infliximab in one patient and complete clinical and imaging improvement in the other.
The scientific rationale for TNF-α-inhibiting therapy in sarcoidosis is the central role TNF-α appears to play in the formation and maintenance of granulomas.24 These observations led to early adoption of treatments with target effects inhibiting TNF-α, including pentoxifylline25 and thalidomide.26 Two double-blind placebo-controlled randomized trials of infliximab for pulmonary sarcoidosis demonstrated a statistically significant, albeit clinically modest, improvement in forced vital capacity.27,28 Post hoc analysis of one of these trials27 analyzing effects of infliximab on extrapulmonary sarcoidosis showed improvements in a novel extrapulmonary sarcoidosis severity score based in part on the number of organ systems involved by sarcoidosis and included a small subset with neurologic involvement.29 Our dataset, in the context of other published series in neurosarcoidosis (table e-1), suggests that infliximab may be particularly effective in treating CNS sarcoidosis. CNS involvement by sarcoidosis may also exhibit patterns of disease activity and treatment responses different from what is sometimes observed in the lungs, skin, or other affected organ systems, including the particular susceptibility of CNS tissue to permanent injury. These data illustrate the importance of studying organ-specific treatment responses in sarcoidosis whenever possible. Organ-specific treatment responses can sometimes even differ within the same patient, as in the example of the patient in our series with sustained remission of CNS sarcoidosis on infliximab but transient breakthrough cutaneous sarcoidosis.
Previous studies reporting infliximab efficacy in neurosarcoidosis were limited to single case reports and smaller case series (table e-1). An earlier review of the literature identified 35 reported cases of neurosarcoidosis treated with infliximab distributed through 18 articles—improvement was reported in all of those cases, suggesting the potential for publication bias in earlier literature.30 Our study adds substantially to the literature by reporting the range of experiences with infliximab treatment of neurosarcoidosis at 6 US specialty centers, including instances of partial or incomplete (though often still meaningful) imaging and clinical responses, as well as patients who did not respond. Relapse after discontinuation of infliximab occurred in 56% of our patients who were in clinical and imaging remission on infliximab, most commonly in the same neuroanatomic distribution. The relapse rates following infliximab discontinuation observed in our series are consistent with the 50% relapse rate following infliximab discontinuation reported in another series published this year.31 When considering discontinuation of infliximab for patients in remission, these data support proactive clinical and imaging surveillance strategies to monitor for evidence of relapse.
The difference between clinical and imaging outcomes in some patients in our dataset is likely attributable to sustained (and possibly irreversible) neurologic deficits incurred following CNS injury from sarcoidosis even when inflammation subsequently remitted on effective therapy. These data highlight the need for developing better disease-specific clinical, imaging, and biological metrics to improve the responsiveness and granularity of outcome data in neurosarcoidosis. The findings from our study also lend support for a randomized controlled trial of infliximab in neurosarcoidosis, which would necessarily need to be multicenter. While a standard parallel-group placebo-controlled design could be considered, the data presented in our study would also support a placebo-controlled randomized-withdrawal trial design for neurosarcoidosis in which all patients would be treated with infliximab (or another therapy under investigation). Infliximab nonresponders would be censored and responders randomized to either continue with active therapy or switch to placebo in a randomized double-blinded fashion, with the primary outcome being a return of disease activity. A potential advantage of a randomized withdrawal approach is that previously effective treatment could be immediately reinitiated upon meeting the trial endpoint in order to minimize suffering and reduce the potential for irreversible neurologic injury; the precedent for this approach is a registration study of etanercept for polyarticular juvenile idiopathic arthritis.32
Although practice patterns varied in our series and across centers, we typically reserve infliximab for more severe cases of CNS sarcoidosis, and tend to start with a dose of 5 mg/kg IV loading at weeks 0, 2, and 6 and maintenance dosing every 4–8 weeks based on estimation of disease severity and risk of neurologic compromise. In the majority of patients (78.8%) in our analysis, infliximab was used in combination with an oral immunosuppressant agent, typically mycophenolate, methotrexate, or azathioprine. Combination therapy may reduce the risk of developing anti-infliximab antibodies that can neutralize therapeutic effects and other adverse events (for a recent review of anti-infliximab antibodies, see Murdaca et al.33). Neutralizing antibodies and infliximab levels can be checked via clinical laboratory studies if warranted. Combination therapy may also have synergistic disease-modifying effects.
Further investigation is needed to determine how combination therapy compares with monotherapy in neurosarcoidosis and how various combinations compare in terms of efficacy, safety, and tolerability. The optimal duration of therapy also remains to be determined, but instances of relapse after long-term infliximab treatment together with the observation that over half of patients in remission on infliximab relapse after infliximab discontinuation supports the use of prolonged infliximab therapy in some patients and the need for close clinical and imaging follow-up if electing to discontinue infliximab. Many questions remain regarding longer-term management strategies and maintenance therapy. For example, after achieving remission, should the total dose or frequency of infliximab infusions be decreased? And when using combination regimens, what is the best strategy for tapering?
The expanding number of biologic therapies that target the TNF-α pathway are not identical in terms of their antigenicity and treatment effects, and the benefit observed with infliximab in sarcoidosis could be target, antibody, or molecule-specific. Adalimumab, a fully humanized monoclonal TNF-α antibody that has also been used in cutaneous and systemic sarcoidosis,34 has been reported to be effective in a few cases of neurosarcoidosis.35 Etanercept, a fusion protein that binds to TNF-α and inhibits binding to the TNF-α receptor, appeared promising in some patients with refractory systemic sarcoidosis.36 However, an open-label trial and a randomized controlled trial of etanercept found no benefit in pulmonary sarcoidosis37 or in systemic sarcoidosis with uveitis.38 A randomized study of golimumab in chronic pulmonary or skin sarcoidosis failed to meet its primary outcome.39
Infliximab has several well-recognized risks, including black box warnings related to infection and malignancy, and risks related to infusion and hypersensitivity reactions. Rarely, paradoxical development of granulomatous inflammation has also been reported on anti-TNF-α therapy.40 While our observational study was not focused on the full range of potential adverse event reporting, over 10% of patients developed infections on infliximab that in the opinion of the investigator could have been related to treatment. Prospective studies will be needed to determine the comparative risk of infliximab to other disease-modifying agents in neurosarcoidosis and what dose, frequency, and treatment combinations are optimal for neurosarcoidosis.
These data, based on the largest cohort reported to date, provide Class IV evidence (because of the absence of a comparison of outcomes to a non-infliximab-treated group) that for patients with CNS sarcoidosis, infliximab is associated with favorable imaging and clinical responses, including in patients who have failed other therapies.
GLOSSARY
- CI
confidence interval
- CVID
combined variable immunodeficiency
- OR
odds ratio
- TNF-α
tumor necrosis factor–α
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Jeffrey M. Gelfand: study concept/design, data collection, interpretation, manuscript preparation, critical review. Michael J. Bradshaw: study concept/design, site recruitment, data collection, interpretation, manuscript preparation, critical review. Barney J. Stern: data contribution, critical review. David B. Clifford: data contribution, critical review, site recruitment. Yunxia Wang: data contribution, critical review. Tracey A. Cho: data contribution, critical review. Laura L. Koth: data contribution, critical review. Stephen L. Hauser: data contribution, critical review. Jason Dierkhising: data contribution, critical review. NgocHanh Vu: data contribution, critical review. Subramaniam Sriram: data contribution, critical review. Harold Moses: data contribution, critical review. Francesca Bagnato: data contribution, critical review. Jeffrey Kaufmann: data contribution, critical review. Deidre Ammah: data contribution, critical review. Tsion H. Yohannes: data contribution, critical review. Mark J. Hamblin: data contribution, critical review. Nagagopal Venna: data contribution, critical review. Ari J. Green: data contribution, critical review. Siddharama Pawate: study concept/design, data contribution, site recruitment, manuscript editing and critical review.
STUDY FUNDING
Research reported in this publication was supported, in part, by the National Center for Advancing Translational Sciences of the NIH under award number KL2TR000143. Dr. Green received a Research to Prevent Blindness grant.
DISCLOSURE
J. Gelfand reports consulting for Genentech and medical legal consulting and research support to UCSF from Genentech, MedDay, and Quest Diagnostics. M. Bradshaw and B. Stern report no disclosures relevant to the manuscript. D. Clifford reports consulting with Biogen, Genentech, Inhibikase, and Takeda, and serves on data safety or adjudication committees for Amgen, Biogen, Takeda, Sanofi-Genzyme, Shire, Genentech, and Protagonist. Y. Wang, T. Cho, L. Koth, S. Hauser, J. Dierkhising, N. Vu, S. Sriram, H. Moses, F. Bagnato, J. Kaufmann, D. Ammah, T. Yohannes, M. Hamblin, N. Venna, A. Green, and S. Pawate report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med 2007;357:2153–2165. [DOI] [PubMed] [Google Scholar]
- 2.Rossi G, Cavazza A, Colby TV. Pathology of sarcoidosis. Clin Rev Allergy Immunol 2015;49:36–44. [DOI] [PubMed] [Google Scholar]
- 3.Fingerlin TE, Hamzeh N, Maier LA. Genetics of sarcoidosis. Clin Chest Med 2015;36:569–584. [DOI] [PubMed] [Google Scholar]
- 4.Rybicki BA, Iannuzzi MC. Epidemiology of sarcoidosis: recent advances and future prospects. Semin Respir Crit Care Med 2007;28:22–35. [DOI] [PubMed] [Google Scholar]
- 5.Zajicek JP, Scolding NJ, Foster O, et al. Central nervous system sarcoidosis: diagnosis and management. QJM 1999;92:103–117. [DOI] [PubMed] [Google Scholar]
- 6.Miloslavsky EM, Naden RP, Bijlsma JW, et al. Development of a glucocorticoid toxicity index (GTI) using multicriteria decision analysis. Ann Rheum Dis 2016;76:543–546. [DOI] [PubMed] [Google Scholar]
- 7.Doty JD, Mazur JE, Judson MA. Treatment of corticosteroid-resistant neurosarcoidosis with a short-course cyclophosphamide regimen. Chest 2003;124:2023–2026. [DOI] [PubMed] [Google Scholar]
- 8.Pawate S, Moses H, Sriram S. Presentations and outcomes of neurosarcoidosis: a study of 54 cases. QJM 2009;102:449–460. [DOI] [PubMed] [Google Scholar]
- 9.Scott TF, Yandora K, Valeri A, Chieffe C, Schramke C. Aggressive therapy for neurosarcoidosis: long-term follow-up of 48 treated patients. Arch Neurol 2007;64:691–696. [DOI] [PubMed] [Google Scholar]
- 10.Horiuchi T, Mitoma H, Harashima S, Tsukamoto H, Shimoda T. Transmembrane TNF-alpha: structure, function and interaction with anti-TNF agents. Rheumatology 2010;49:1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baughman RP, Strohofer SA, Buchsbaum J, Lower EE. Release of tumor necrosis factor by alveolar macrophages of patients with sarcoidosis. J Lab Clin Med 1990;115:36–42. [PubMed] [Google Scholar]
- 12.Kindler V, Sappino AP, Grau GE, Piguet PF, Vassalli P. The inducing role of tumor necrosis factor in the development of bactericidal granulomas during BCG infection. Cell 1989;56:731–740. [DOI] [PubMed] [Google Scholar]
- 13.Clifford DB. Nemesis of neglected neurosarcoidosis. Ann Clin Transl Neurol 2015;2:947–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Judson MA, Costabel U, Drent M, et al. The WASOG sarcoidosis organ assessment instrument: an update of a previous clinical tool. Sarcoidosis Vasc Diffuse Lung Dis 2014;31:19–27. [PubMed] [Google Scholar]
- 15.Bouvry D, Mouthon L, Brillet PY, et al. Granulomatosis-associated common variable immunodeficiency disorder: a case-control study versus sarcoidosis. Eur Respir J 2013;41:115–122. [DOI] [PubMed] [Google Scholar]
- 16.Stern BJ, Aksamit A, Clifford D, Scott TF, Neurosarcoidosis Study Group. Neurologic presentations of sarcoidosis. Neurol Clin 2010;28:185–198. [DOI] [PubMed] [Google Scholar]
- 17.Agbogu BN, Stern BJ, Sewell C, Yang G. Therapeutic considerations in patients with refractory neurosarcoidosis. Arch Neurol 1995;52:875–879. [DOI] [PubMed] [Google Scholar]
- 18.Lower EE, Broderick JP, Brott TG, Baughman RP. Diagnosis and management of neurological sarcoidosis. Arch Intern Med 1997;157:1864–1868. [PubMed] [Google Scholar]
- 19.Pettersen JA, Zochodne DW, Bell RB, Martin L, Hill MD. Refractory neurosarcoidosis responding to infliximab. Neurology 2002;59:1660–1661. [DOI] [PubMed] [Google Scholar]
- 20.Sharma OP. Effectiveness of chloroquine and hydroxychloroquine in treating selected patients with sarcoidosis with neurological involvement. Arch Neurol 1998;55:1248–1254. [DOI] [PubMed] [Google Scholar]
- 21.Bitoun S, Bouvry D, Borie R, et al. Treatment of neurosarcoidosis: a comparative study of methotrexate and mycophenolate mofetil. Neurology 2016;87:2517–2521. [DOI] [PubMed] [Google Scholar]
- 22.Bomprezzi R, Pati S, Chansakul C, Vollmer T. A case of neurosarcoidosis successfully treated with rituximab. Neurology 2010;75:568–570. [DOI] [PubMed] [Google Scholar]
- 23.Cinetto F, Compagno N, Scarpa R, Malipiero G, Agostini C. Rituximab in refractory sarcoidosis: a single centre experience. Clin Mol Allergy 2015;13:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iannuzzi MC, Fontana JR. Sarcoidosis: clinical presentation, immunopathogenesis, and therapeutics. Jama 2011;305:391–399. [DOI] [PubMed] [Google Scholar]
- 25.Zabel P, Entzian P, Dalhoff K, Schlaak M. Pentoxifylline in treatment of sarcoidosis. Am J Respir Crit Care Med 1997;155:1665–1669. [DOI] [PubMed] [Google Scholar]
- 26.Hammond ER, Kaplin AI, Kerr DA. Thalidomide for acute treatment of neurosarcoidosis. Spinal Cord 2007;45:802–803. [DOI] [PubMed] [Google Scholar]
- 27.Baughman RP, Drent M, Kavuru M, et al. Infliximab therapy in patients with chronic sarcoidosis and pulmonary involvement. Am J Respir Crit Care Med 2006;174:795–802. [DOI] [PubMed] [Google Scholar]
- 28.Rossman MD, Newman LS, Baughman RP, et al. A double-blinded, randomized, placebo-controlled trial of infliximab in subjects with active pulmonary sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2006;23:201–208. [PubMed] [Google Scholar]
- 29.Judson MA, Baughman RP, Costabel U, et al. Efficacy of infliximab in extrapulmonary sarcoidosis: results from a randomised trial. Eur Respir J 2008;31:1189–1196. [DOI] [PubMed] [Google Scholar]
- 30.Lorentzen AO, Sveberg L, Midtvedt O, Kerty E, Heuser K. Overnight response to infliximab in neurosarcoidosis: a case report and review of infliximab treatment practice. Clin Neuropharmacology 2014;37:142–148. [DOI] [PubMed] [Google Scholar]
- 31.Cohen Aubart F, Bouvry D, Galanaud D, et al. Long-term outcomes of refractory neurosarcoidosis treated with infliximab. J Neurol 2017;264:891–897. [DOI] [PubMed] [Google Scholar]
- 32.Lovell DJ, Giannini EH, Reiff A, et al. Etanercept in children with polyarticular juvenile rheumatoid arthritis: Pediatric Rheumatology Collaborative Study Group. N Engl J Med 2000;342:763–769. [DOI] [PubMed] [Google Scholar]
- 33.Murdaca G, Spano F, Contatore M, et al. Immunogenicity of infliximab and adalimumab: what is its role in hypersensitivity and modulation of therapeutic efficacy and safety? Expert Opin Drug Saf 2016;15:43–52. [DOI] [PubMed] [Google Scholar]
- 34.Lahmer T, Knopf A, Lanzl I, Heemann U, Thuermel K. Using TNF-alpha antagonist adalimumab for treatment for multisystem sarcoidosis: a case study. Rheumatol Int 2012;32:2367–2370. [DOI] [PubMed] [Google Scholar]
- 35.Marnane M, Lynch T, Scott J, Stack J, Kelly PJ. Steroid-unresponsive neurosarcoidosis successfully treated with adalimumab. J Neurol 2009;256:139–140. [DOI] [PubMed] [Google Scholar]
- 36.Khanna D, Liebling MR, Louie JS. Etanercept ameliorates sarcoidosis arthritis and skin disease. J Rheumatol 2003;30:1864–1867. [PubMed] [Google Scholar]
- 37.Utz JP, Limper AH, Kalra S, et al. Etanercept for the treatment of stage II and III progressive pulmonary sarcoidosis. Chest 2003;124:177–185. [DOI] [PubMed] [Google Scholar]
- 38.Baughman RP, Lower EE, Bradley DA, Raymond LA, Kaufman A. Etanercept for refractory ocular sarcoidosis: results of a double-blind randomized trial. Chest 2005;128:1062–1047. [DOI] [PubMed] [Google Scholar]
- 39.Judson MA, Baughman RP, Costabel U, et al. Safety and efficacy of ustekinumab or golimumab in patients with chronic sarcoidosis. Eur Respir J 2014;44:1296–1307. [DOI] [PubMed] [Google Scholar]
- 40.Tong D, Manolios N, Howe G, Spencer D. New onset sarcoid-like granulomatosis developing during anti-TNF therapy: an under-recognised complication. Intern Med J 2012;42:89–94. [DOI] [PubMed] [Google Scholar]