

Abstract
Pancreatic ductal adenocarcinoma (PDAC) presents at metastatic stage in over 50% of patients. With a survival rate of just 2.7% for patients presenting with distant disease, it is imperative to uncover novel mechanisms capable of suppressing metastasis in PDAC. Previously, we reported that the loss of metastasis suppressor protein 1 (MTSS1) in PDAC cells results in significant increase in cellular migration and invasion. Conversely, we also found that overexpressing MTSS1 in metastatic PDAC cell lines corresponds with not only decreased metastatic phenotype, but also greater overall survival. While it is known that MTSS1 is downregulated in late-stage PDAC, the mechanism behind that loss has not yet been elucidated. Here, we build off our previous findings to present a novel regulatory mechanism for the stabilization of MTSS1 via the tumor suppressor protein phosphatase and tensin homolog (PTEN). We show that PTEN loss in PDAC cells results in a decrease in MTSS1 expression and increased metastatic potential. Additionally, we demonstrate that PTEN forms a complex with MTSS1 in order to stabilize and protect it from proteasomal degradation. Finally, we show that the inflammatory tumor microenvironment, which makes up over 90% of PDAC tumor bulk, is capable of downregulating PTEN expression through secretion of miRNA-23b, potentially uncovering a novel extrinsic mechanism of MTSS1 regulation. Collectively, these data offer new insight into the role and regulation of MTSS1in suppressing tumor cell invasion and migration and help shed light as to what molecular mechanisms could be leading to early cell dissemination in PDAC.
Abbreviations: 3T3, NIH3T3 mouse fibroblast cells; CAF, cancer-associated fibroblast; CAFM, CAF-conditioned media; CHX, cyclohexamide; DMSO, dimethyl sulfoxide; EpM, epithelial-conditioned media; IgG, immunoglobulin G; IP, immunoprecipitate; MOE, MTSS1 overexpression; MTSS1, metastasis suppressor protein 1; PDAC, pancreatic ductal adenocarcinoma; PTEN, phosphatase and tensin homolog; TrCP, transducin repeat containing protein; VEC, vector
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the third leading cause of cancer-related deaths in the United States with a 5-year survival rate of 9% [1], [2]. With research currently struggling to identify new biomarkers and methods for earlier detection, most patients present with advanced disease. Whereas 39% of PDAC patients present with regional or distant disease, an astounding 53% of patients afflicted with PDAC are diagnosed at metastatic stage [3], [4]. This accounts for 92% of all patients diagnosed with PDAC being diagnosed after the primary tumor has already spread. This phenomenon is due in part to the asymptomatic nature of the disease and a lack of reliable early detection methods [5], [6]. Because of this late disease presentation, less than 20% of patients will be viable candidates for surgical resection, the treatment that still has the best outcome for pancreatic cancer patients [7], [8]. These challenges combine to make PDAC an incredibly difficult disease to diagnose at localized stage, thereby highlighting the importance of designing and implementing innovative approaches to harness the disease before it is able to spread. Specifically, elucidation of the molecular mechanisms responsible for the early tumor cell dissemination observed in PDAC that can lead to metastasis is an area that still requires much focus [9].
One of the molecular mechanisms that has recently been shown to be critical for preventing tumor cell dissemination is the regulation of metastasis suppressor protein 1 (MTSS1) [10]. Also known as Missing in Metastasis (MIM), MTSS1 is believed to prevent metastasis by increasing Rac-GTP levels in order to inhibit cell-cell junction disassembly, thereby elevating actin assembly at the cell-cell contacts and preventing cellular dissemination [11]. While very little is known about the mechanism of action and molecular relationships of MTSS1, it has been found to play a role in combatting cellular migration and invasion in PDAC [10]. However, while the functional role of MTSS1 in PDAC has recently been uncovered, the regulation of MTSS1 in PDAC is still an enigma.
Multiple microRNAs have been shown to be capable of regulating MTSS1 expression [12], [13], [14], [15], whereas ubiquitination-driven destruction and DNA methylation have also been found to play a role in MTSS1 downregulation as well [16], [17], [18]. Additionally, MTSS1 has also been shown to have affinity for protein phosphatases, such as PTPδ and PTPN11, in a range of different cancer subtypes [19], [20]. The center of the MTSS1 protein is rich in proline, serine, and threonine residues [21]. These residues, all containing hydroxyl groups, interact and bind with these phosphatases, which then may provide a functional link between the MTSS1 protein and signal transduction pathways that prevent an invasive state from arising. This interaction is intriguing because one of the most commonly implicated tumor suppressors involved in cancer progression is a phosphatase known as phosphatase and tensin homolog (PTEN).
Here, we present evidence of a novel regulatory mechanism for MTSS1 stabilization via the canonical tumor suppressor protein PTEN. PTEN expression has been found to be crucial in minimizing the lethality of PDAC through regulation of the PI3K/AKT pathway [22]; however, its role in PDAC metastasis has only been briefly studied [23], [24], [25]. In this manuscript, we demonstrate that loss of PTEN in PDAC cells not only leads to a more invasive and migratory phenotype, but also results in decreased in MTSS1 expression. We find that PTEN positively regulates the stability and protein level of MTSS1. Furthermore, we demonstrate that PTEN is a protein phosphatase for MTSS1 and inhibits the interaction between MTSS1 and its E3 ligase SCFβ-TRCP to block the proteasome degradation of MTSS1. Finally, we introduce a new, cell-extrinsic mechanism for the downregulation of PTEN via miRNA-23b from the PDAC tumor microenvironment, suggesting a novel role for both the tumor microenvironment and PTEN in PDAC metastatic regulation.
Materials and Methods
Cell Culture and Transfection
Cancer-associated fibroblast (CAF) cell line UH1301-63 was obtained from Melissa L. Fishel, Ph.D. (Department of Pediatrics, IU School of Medicine), and prepared for study as previously described [26]. Fibroblast nomenclature was reduced for purposes of simplicity with “S63” referring to UH1301-63 CAFs. Other commercial cell lines were purchased from ATCC. S63, NIH3T3, PANC-1, MIA PaCa-2, and 293T cells were cultured in DMEM supplemented with 10% FBS, 100 U/ml of penicillin, and 100 μg/ml of streptomycin with 5% CO2 at 37°C. Cells were trypsinized and passaged at approximately 90% confluence. Cell transfection was performed using Lipofectamine 2000 (Invitrogen), unless otherwise noted. Cells were harvested at 24 hours post-transfection for protein analysis, unless otherwise noted.
Cell Authentication
Fibroblast and epithelial cell lines were authenticated by Genetica DNA Laboratories. All cell lines were a 97% to 100% match to the correct cell line in both the ATCC and DSMZ database. The CAFs were previously determined not to have a match in either database, were non-tumorigenic in mice, and did not have a mutation for K-RAS [26].
Virus Production and Transduction
For overexpression of PTEN or MTSS1, 293T cells at 60% confluence were transfected with retroviral plasmid (5 μg) containing vector control, PTEN or MTSS1, VSVG (3 μg), and GAG (2 μg). For knocking down PTEN or MTSS1, 293T cells at 60% confluence were transfected with lentiviral plasmid (5 μg) containing Scramble, shPTEN or shMTSS1, VSVG (3 μg), and GAG (2 μg). Virus was collected 48 hours after transfection. Virus containing media was then added to 70% confluent S63 or NIH3T3 cells supplemented with polybrene (8 μg/ml). Stable cell pools were selected with puromycin for 5 days. Verification was accomplished via Western blot analysis. For shMTSS1, the following target sequence was used: GCTGATGCATTGTGCATAT. The sequence was proven effective for both human and mouse MTSS1 gene as previously described [27]. For shPTEN, the following target sequence was used: CCACAAATGAAGGGATATAAA. The sequence was proven effective for both human and mouse PTEN gene as previously described [28], [29], [30], [31].
For shPTEN + MOE rescue experiments, 1.5 × 105 PANC-1 Scramble and shPTEN cells were plated in a 6-well setting. Twenty-four hours post-plating, shPTEN cells were transfected with MTSS1 Myc WT plasmid (2 μg) using the Lipofectamine 3000 Transfection Reagent (Thermo Fisher, L3000015) according to manufacturer's protocol.
Western Blotting Analysis
Protein lysates were prepared from S63, NIH3T3, PANC-1, MIA PaCa-2, 293T, and stable cell pools in a buffer containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 0.1% SDS, 0.5% deoxycholate, 1% NP-40, and1 mM EDTA supplemented with a protease inhibitor cocktail tablet (Roche 11836153001). Cell lysate (40 μg) was resolved by gel electrophoresis (BioRad Mini Protean TGX Gel 400091313) and transferred to a nitrocellulose membrane. The membranes were blocked in 5% dry milk in TBS-T. Primary antibodies diluted in 5% dry milk and TBS-T include MTSS1 (Cell Signaling 4386S), PTEN (Cell Signaling 9559S), phospho-Akt (Ser473) (Cell Signaling 4060S), and β-actin (Cell Signaling 4970L) at 1:1000. Secondary antibodies used include anti-rabbit (Cell Signaling 7074S). Protein levels were detected by enhanced chemiluminescence (Thermo Scientific 32106) and quantitated with Image Lab software (Bio-Rad). Densitometry analysis was completed using ImageJ64 software.
For immunoprecipitation experiments, 500 μg of cell lysate was incubated with IgG and Protein A–Agarose (SIGMA P2545) for 3 hours at 4°C. Beads were washed three times with lysis buffer and centrifuged at 5000g for 5 minutes between each wash. Protein was eluted from beads with 50 μl of Laemmli sample buffer (Bio-Rad). Lysates were resolved on SDS-PAGE gels and transferred onto nitrocellulose for Western blotting.
For determining the phosphorylation status of MTSS1, the phos-tag gel was used in which the phosphorylated MTSS1 ran slower than the unphosphorylated MTSS1, as previously described [32].
Conditioned Media Collection
Media from cancer-associated fibroblasts (CAFM) or PANC-1 cells [epithelial-conditioned media (EpM)] were collected after 3 to 4 days at 80% to 90% confluence and centrifuged at 2500 RPM for 5 minutes in order to pellet any debris. Media were stored at 4°C until needed.
Scratch Assay Analysis
Cells were seeded at 1.5 × 105 density in 6-well plates and allowed to grow to 90% confluence. At 90% confluence, a scratch was made down the center of the well using a P10 pipet tip. The cells were washed with 1× PBS (Sigma) and then placed in the appropriate media for 48 hours. Images were taken at 0, 12, 24, and 48 hours posts-cratch using the AMG EVOS FL Cell Imaging System microscope (AMEX4300). Analysis was completed in triplicate using ImageJ64 software.
Transwell Assay Analysis
A total of 4 × 104 PDAC cells were plated on a 24-well Transwell polycarbonate membrane with 8.0-μm pore size (Corning 3422). Wells were coated with 50 μl of 3 mg/ml Matrigel (Corning 354230) and kept at 37°C for 24 hours before plating to ensure solidification. Cells plated in the upper chamber, unless treated with conditioned media, were placed in 150 μl of the appropriate serum-free media, whereas the bottom chamber contained 700 μl of the appropriate serum-containing media to act as the chemoattractant in the study. Cells were collected at 48 hours and gently rinsed with ddH2O and then fixed for 20 minutes with 10% formalin (Azer Scientific). After fixation, the cells were placed in hematoxylin (Sigma) for 2 minutes, rinsed in ddH2O, quickly dipped in 1% acid alcohol (Sigma), and rinsed a final time in ddH2O. Membranes were then imaged, and individual cells were counted manually using an AMG EVOS XL Core Cell Imaging System microscope (AMEX1000).
miRNA-23b Baseline and Inhibition Studies
PDAC cellular RNA was harvested using Trizol Reagent (Ambion 15596026) according to standard protocol. For formation of cDNA, QIAGEN miScript II RT Kit (QIAGEN 218161) was used according to manufacturer's protocol. Quantification of miRNA was completed using the QIAGEN miScript SYBR Green PCR Kit (QIAGEN 218073) according to manufacturer's protocol on the Bio-Rad CFX Connect Real-Time PCR Detection System (Bio-Rad). The following miRNA primer assays from QIAGEN were purchased and utilized: Hs_RNU6-2_11 (QIAGEN MS00033740), Hs_miR23b_2 (QIAGEN MS00031647), and Hs_miR23a_2 (QIAGEN MS00031633). miRNA was normalized to RNU6.
miR-23b inhibition studies were completed according to QIAGEN miScript miRNA Inhibitor protocol. Briefly, 1.0 × 105 PANC-1 cells were plated and incubated while the inhibitor cocktails were prepared. The following miRNA inhibitors were added to the appropriate serum-free culture medium at a final concentration of 50 nM: Negative Control siRNA (QIAGEN SI03650325) and Anti–hsa-miR-23b-3p (QIAGEN MIN0000418). HiPerfect Transfection Reagent (QIAGEN 301707) was used to complete the transfection cocktail. Cells were harvested and/or experimented with at 48 hours post-transfection.
Patient Dataset Analysis
The prognostic value of PTEN and MTSS1 loss in PDAC patients was analyzed in a publically available dataset, GSE21501 [33], using PROGgene gene expression–based survival analysis web application [34].
RNA Collection RT-PCR Analysis
RNA was harvested with Trizol (Life Technologies), resuspended in RNAse-free water, and analyzed for purity by 260/280 absorbance via Nanodrop. RT-PCR was conducted on the BioRad-CFX-Connect cycler using Qiagen QuantiFast SYBR Green RT-PCR kit (Qiagen 204154) and the following primers: Human GAPDH (Qiagen PPH00150F) and Human MTSS1 (Qiagen PPH10073B).
Statistical Analysis
Experiments were performed with a minimum of three biological replicates. Data are presented as the mean ± standard deviation. Statistical significance was calculated via Microsoft Excel using a Student t test. “*” denotes P value < .05, “**” denotes P value < .001, and “***” denotes P value < .0001.
Results
Loss of PTEN In Vitro Results in Increased PDAC Cellular Invasion and Migration
In order to investigate our hypothesis that the phosphatase activity of PTEN stabilizes MTSS1 expression in PDAC, we first set out to define the importance of PTEN itself in pancreatic cancer metastatic progression. While there is strong supporting evidence that PTEN plays a large role in suppressing tumor formation and growth in PDAC [35], [36], [37], its role in metastasis is less well known. Utilizing a stably transduced PANC-1 PDAC cell line to knock down PTEN expression, we were able to run a number of assays to determine if PTEN played a role in PDAC invasion and migration. PANC-1 shPTEN cells display 84% knockdown of PTEN compared to PANC-1 Scramble control (Figure 1A, Supplemental Figure 1). PANC-1 shPTEN cells exhibit significantly increased migration and invasion capabilities when compared to control in both serum-free and serum-containing scratch assay settings (Figure 1B). In serum-free conditions, where cells are not able to proliferate but rather must rely on migratory properties to travel across the wound, PANC-1 shPTEN cells were able to migrate approximately 100 μm farther than Scramble control. Similarly, in serum-containing conditions, PANC-1 shPTEN cells were able to invade across the wound approximately 200 μm farther than Scramble control over a 48-hour period.
Figure 1.

Loss of PTEN results in increased invasion and migration. (A) Western blot confirmation of stable PANC-1 shPTEN cell pool as compared to Scramble control. (B) PANC-1 Scramble and shPTEN cells were plated, and a scratch assay was performed in both serum-free (top) and serum-containing (bottom) conditions. PANC-1 shPTEN cells were able to move across the scratch significantly farther than PANC-1 Scramble cells in both conditions. (C) PANC-1 cells were cultured in PANC-1 EpM or CAFM and harvested at various timepoints. Lysates were analyzed via Western blot, and it was seen that PANC-1 cells cultured in CAFM expressed less PTEN as time went on, which was accompanied by a robust upregulation of P-AKT. (D) PANC-1 Scramble and shPTEN cells were plated, and a scratch assay was performed in either EpM or CAFM conditions. PANC-1 shPTEN cells traveled significantly farther across the scratch, and that phenomenon was augmented in the presence of CAFM, in which the PANC-1 shPTEN cells traveled more than two-fold farther than PANC-1 Scramble control cells. (E) PANC-1 Scramble and shPTEN cells were plated, and a Transwell invasion assay was performed in either EpM or CAFM conditions. PANC-1 shPTEN cells invaded significantly more through the Transwell membrane; however, that significance was increased in shPTEN cells cultured in CAFM. (F) Western blot analysis of PANC-1 Scramble and shPTEN cells shows that MTSS1 expression decreases in PANC-1 shPTEN cells. (G) Western blot analysis of both metastatic (metPDAC.1) and primary (primPDAC.1) PDAC patient cell lysates shows metastatic patient sample containing decreased PTEN and MTSS1 expression. *P value < .05, **P value < .001, ***P value < .0001.
Supplemental Figure 1.
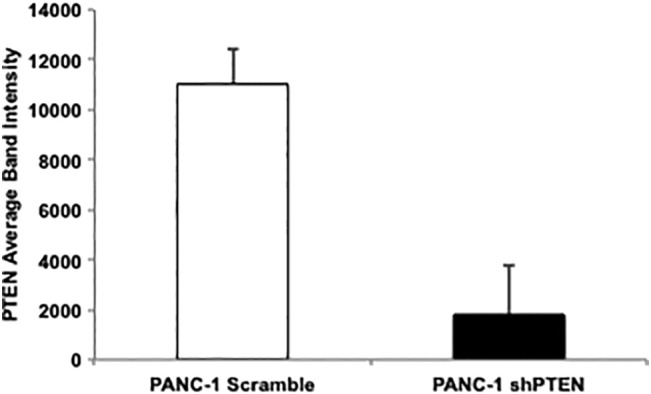
Quantitation of Figure 1A Western blot confirming the loss of PTEN in stable PANC-1 shPTEN cell pool as compared to Scramble control.
Next, knowing the importance of the tumor microenvironment on PDAC metastatic progression [9], [38], [39], we investigated how treatment with media taken from CAFs, which are the main component of the PDAC tumor microenvironment, affected PTEN expression in PANC-1 cells. Western blot analysis revealed that PANC-1 cells treated with CAFM at various lysate collection points express diminished levels of PTEN expression and increased levels of P-AKT (Figure 1C). At the 24-hour lysate collection time point, PTEN expression in CAFM-treated PANC-1 cells was reduced by approximately 35%, whereas at the 48-hour collection time point, that reduction in PTEN expression in CAFM-treated cells rose to approximately 45% (Supplemental Figure 1). CAFM treatment also resulted in an increase in P-AKT expression. At the 24-hour lysate collection time point, CAFM-treated PANC-1 cells express 86% more P-AKT than EpM-treated PANC-1 cells. Furthermore, at the 48-hour collection time point, that increase rose to 95% (Supplemental Figure 2).
Supplemental Figure 2.
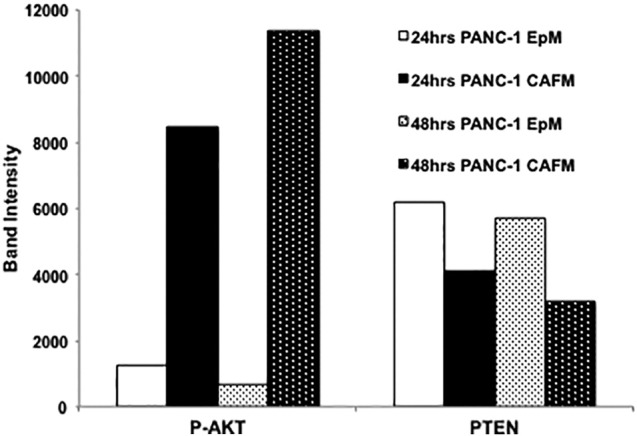
Quantitation of Figure 1C Western blot denoting the loss of PTEN expression and the increase in P-AKT expression in CAFM conditions at both 24- and 48-hour time points.
In order to test if CAFM could further augment the shPTEN cells' ability to increase invasion and migration, PANC-1 shPTEN cells were incubated in either EpM or CAFM and subjected to scratch assay analysis. PANC-1 shPTEN cells travel approximately 50 μm farther than Scramble control in EpM conditions; however, when incubated in CAFM conditions, PANC-1 shPTEN cells travel greater than 150 μm farther than Scramble control (Figure 1D). Additionally, the PANC-1 shPTEN cells were plated in a Matrigel-coated Transwell membrane and incubated in either EpM or CAFM. PANC-1 shPTEN cells incubated in EpM were 1.64-fold more successful at invading through the Matrigel-coated membrane than Scramble control, whereas PANC-1 shPTEN cells incubated in CAFM were 2.8-fold more successful at invading through the Matrigel-coated membrane than PANC-1 Scramble control (Figure 1E).
Having established that loss of PTEN in PDAC epithelial cells results in increased metastatic potential, we next investigated if loss of PTEN affected MTSS1 expression. PANC-1 shPTEN cells exhibit approximately 80% decrease in MTSS1 expression when compared to Scramble control (Figure 1F). Additionally, we performed Western blot analyses on both primary (primPDAC.1) and metastatic (metPDAC.1) PDAC patient cell lysates. metPDAC.1 cells displayed a nearly two-fold decrease in PTEN expression levels when compared to primPDAC.1 cells (Figure 1G). The metastatic patient cells also showed a robust 9.9-fold decrease in MTSS1 expression as compared to the primary patient cells (Figure 1G). Collectively, these results indicate that PTEN loss significantly increases PDAC cell migration and invasion, potentially due to the downregulation of MTSS1 expression. To provide additional validation of this finding, we examined a publically available PDAC patient dataset, GSE21501, to determine if loss of both PTEN and MTSS1 expression correlated with patient outcome. We found that decreased expression levels of both PTEN and MTSS1 were a significant predictor of poorer overall patient survival (HR: 0.59, P = .0240028; Supplemental Figure 3).
Supplemental Figure 3.

Kaplan-Meier survival curve of patient dataset GSE21501 indicating that loss of both PTEN and MTSS1 in patients leads to significantly worse overall patient survival (P = .024).
PTEN Stabilizes MTSS1
Having previously established that stable MTSS1 expression levels are correlated with decreased metastatic potential in PDAC [10] and now having uncovered a potential novel connection between the PTEN tumor suppressor and MTSS1 status, we next investigated the potential molecular mechanism that could be responsible for the loss of PTEN correlating to loss of MTSS1. We hypothesized that PTEN might regulate both the MTSS1 protein level and stability. To test this hypothesis, we manipulated PTEN expression levels by transfecting S63 PDAC fibroblast cells with either a plasmid that produces overexpression of PTEN (PTEN) or a plasmid that knocks down PTEN expression (shPTEN). We observed that overexpression of PTEN significantly increases MTSS1 expression, whereas knockdown of PTEN significantly decreases MTSS1 expression in S63 cells compared to a vector only control (Figure 2A).
Figure 2.

PTEN stabilizes MTSS1 expression levels. (A) MTSS1 protein level is positively regulated by PTEN in S63 fibroblast cells. PTEN overexpression increases MTSS1 protein level, and PTEN knockdown decreases MTSS1 protein level. Cell lysates of S63 cells, which were transfected with control, PTEN, or shPTEN, were analyzed by Western blot. MTSS1 levels were normalized against β-actin. (B) PTEN increases MTSS1 stability in S63 cells. S63 cells stably expressing vector or PTEN were treated with CHX (20 μg/ml) for indicated times. Endogenous MTSS1 protein levels were determined. Relative MTSS1 levels were normalized against β-actin. In (C) and (D), the same assays were carried out in NIH3T3 fibroblast cells and showed similar results. Data are represented as mean ± SEM. *P value < .05, **P value < .001.
Next, we used retroviruses to generate S63 cell pools that either stably overexpressed PTEN or a vector-only control (Supplemental Figure 4). When the stability of MTSS1 was compared between these cell pools, we observed that overexpression of PTEN significantly increases MTSS1 stability in S63 cells treated with the protein synthesis inhibitor cyclohexamide (CHX) in media with serum (Figure 2B). To further confirm our findings in a general fibroblast cell line, the same assays were performed in NIH3T3 cells. We observed that overexpression of PTEN significantly increases MTSS1 protein level in NIH3T3 cells, while PTEN knockdown significantly decreases MTSS1 protein levels (Figure 2C). Finally, we observed that PTEN overexpression significantly increases MTSS1 stability in NIH3T3 cells, matching what we had observed in S63 cells (Figure 2D). Collectively, our data show that MTSS1 stability—and thereby protein level—is positively regulated by PTEN.
Supplemental Figure 4.
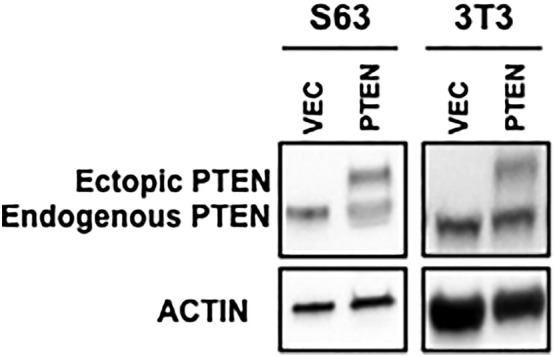
Western blot confirmation of endogenous and ectopic PTEN after transfection in S63 (left) and NIH3T3 cells (right).
PTEN Is a Protein Phosphatase for MTSS1
Thus far, our data show that PTEN regulates MTSS1 in human pancreatic cancer cells as well as mouse fibroblasts. We next sought to elucidate which of PTEN's many functions is responsible for this regulation. PTEN is a tumor suppressor that acts as both a lipid phosphatase and a protein phosphatase [40]. As part of its role as a lipid phosphatase, PTEN negatively regulates the intracellular levels of phosphatidylinositol-3,4,5-trisphosphate in cells and functions as a tumor suppressor by negatively regulating the PI3K/AKT signaling pathway [40], [41]. Thus, we hypothesized that the PI3K/AKT pathway may negatively regulate MTSS1 protein expression.
To test this hypothesis, we treated S63 and NIH3T3 cells with LY294002, a PI3K inhibitor that specifically mimics the lipid phosphatase activity of PTEN [42]. Surprisingly, we found that while LY294002 treatment decreased P-AKT levels, it did not significantly increase MTSS1 levels in either S63 or NIH3T3 cells (Figure 3A). However, when we transfected PPP577 cells, which are a murine cell line that are Pten−/−, with a plasmid that mutated only the lipid phosphatase activity of Pten (PtenG129E), leaving only the protein phosphatase activity as viable, we found that the cells with Pten protein phosphatase activity were able to not only stabilize Mtss1 protein levels but in fact moderately increase Mtss1 expression by approximately 30%, unlike what we saw in Figure 3A (Supplemental Figure 5, lanes 1 and 3). These results suggest that PTEN regulates MTSS1 specifically through its protein phosphatase activity and not its lipid phosphatase activity.
Figure 3.

PTEN is a protein phosphatase for MTSS1. (A) In both S63 and NIH3T3 fibroblast cells, inhibition of PI3K does not increase MTSS1 protein level. Cells were treated with PI3K inhibitor LY294002 (30 μM) for 6 hours. MG132 (10 μM for 6 hours) treatment was included as indicator of increased MTSS1. S473 (pAKT) was detected as positive control for LY294002 treatment. Cell lysates were analyzed by Western blot. Panels (B) and (C) show that PTEN and MTSS1 are in the same protein complex. (B) PTEN was co-transfected with MTSS1 into 293T cells as indicated. (C) Myc-MTSS1 was transfected in 293T cells. PTEN and MTSS1 associations were examined by reciprocal co-IP as indicated. “^” indicates the presence of the heavy chain of rabbit IgG. (D) PTEN decreases the phosphorylation level of MTSS1. PTEN was co-transfected with MTSS1 or MTSS1S322A into 293T cells as indicated. The phosphorylation level of MTSS1 was determined by the phos-tag gel. Data are represented as mean ± SEM.
Supplemental Figure 5.

Western blot showing implications of mutating PTEN lipid phosphatase activity, leaving only its protein phosphatase activity as viable. As can be seen in lanes 1 and 3, cells with mutated lipid phosphatase activity (PTENG129E) not only were able to maintain Mtss1 levels but in fact were able to moderately increase them when compared to the control P577 Pten−/− cells in lane 1.
Given that it is not uncommon for dephosphorylation events between a phosphatase and its target protein to occur via physical interaction, we hypothesized that PTEN was exerting its phosphatase activity on MTSS1 through a direct physical interaction. Hence, we performed co-immunoprecipitation (co-IP) experiments with ectopically expressed proteins to determine whether PTEN could bind with MTSS1. We found that ectopic expression of Flag-tagged PTEN could be readily pulled down by Myc-MTSS1 (Figure 3B). Similarly, reciprocal co-IP experiments also showed that Myc-MTSS1 co-precipitated with endogenous PTEN (Figure 3C). Taken together, these results show that MTSS1 and PTEN are, at the very least, in the same protein complex, consistent with our hypothesis that MTSS1 is a substrate for PTEN's phosphatase activity.
To further test this hypothesis, we next wanted to investigate whether PTEN actually alters the phosphorylation level of MTSS1 via Western blot analysis. However, due to the lack of an antibody that can detect phosphorylated MTSS1, we could not evaluate the phosphorylation level of MTSS1 directly. Therefore, we used Phos-tag SDS-PAGE analysis to detect the phosphorylation change of MTSS1. In a Phos-tag gel, phosphorylated proteins run slower than unphosphorylated proteins. Thus, if two bands are observed in a sample, the lower band corresponds to the unphosphorylated form of the protein, and the upper band corresponds to the phosphorylated form of the protein. When we ectopically expressed MTSS1 alone in 293T cells, the lower, unphosphorylated band for MTSS1 was faint, but upon co-expression of MTSS1 and PTEN, the lower band for unphosphorylated MTSS1 was seen clearly (arrow in Figure 3D, left panel). This suggests that PTEN can decrease MTSS1 phosphorylation levels.
Finally, we sought to identify a phosphorylation site on MTSS1 that would mediate its interaction with PTEN. Previous literature has shown not only that the serine 322 residue (S322) on MTSS1 is phosphorylated by kinase CKIδ, but that phosphorylated S322 is necessary for MTSS1 proteasome degradation [16]. We therefore hypothesized that PTEN might be responsible for dephosphorylating the S322 residue of MTSS1, thereby protecting it from proteasome degradation and thus stabilizing its expression. To test this hypothesis, we ectopically expressed a form of MTSS1 that harbors a point mutation at residue 322 from serine to alanine (MTSS1S322A) in 293T cells. These cells were co-transfected with or without PTEN in order to see whether PTEN could still alter the MTSS1 phosphorylation level despite a mutation at the S322 site. The data show that there is no significant difference between the upper, phosphorylated band and lower, unphosphorylated band with or without overexpression of PTEN (Figure 3D, right panel). This suggests that the S322 residue could be the dephosphorylation site through which PTEN stabilizes MTSS1.
PTEN Blocks Proteasome Degradation of MTSS1
While MTSS1 has been linked to the regulation of metastasis in various cancer types, most of the published studies have focused on the transcriptional regulation of MTSS1 [43], [44]. The only previous study that examined posttranslational regulation of MTSS1 found that ubiquitination of MTSS1 by SCFβ-TRCP promotes its proteasome degradation [16]. Our data showing that PTEN stabilizes MTSS1 levels in cells exposed to CHX indicate that PTEN regulates MTSS1 in a posttranslational manner because CHX interferes with translational elongation. Additionally, we found that while our shPTEN cells did in fact express decreased levels of MTSS1, the difference was not significant, further indicating a need to investigate the novel PTEN/MTSS1 relationship at the translational level (Supplemental Figure 6). Thus, to elucidate whether PTEN regulates MTSS1 through proteasome degradation, we treated S63 and NIH3T3 cells with MG132, a proteasome inhibitor, combined with knockdown of PTEN. In both S63 and NIH3T3 cells, the decrease of MTSS1 protein levels induced by knockdown of PTEN is significantly blocked by MG132 (Figure 4, A and B). These results suggest that PTEN has the same effect on MTSS1 protein level as MG132 in that they both prevent the proteasome degradation of MTSS1.
Supplemental Figure 6.
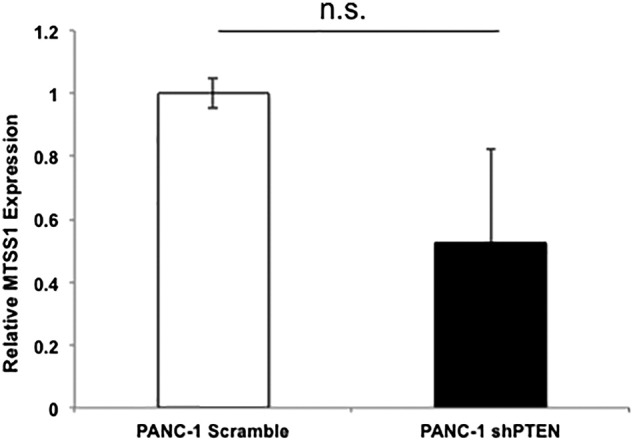
RT-PCR data indicating no significant difference in MTSS1 mRNA expression levels between PANC-1 Scramble and shPTEN cell lines. (P value = .336168).
Figure 4.

PTEN blocks proteasome degradation of MTSS1. (A and B) MG132 blocks PTEN knockdown-induced decrease of MTSS1 protein levels in both S63 and NIH3T3 fibroblast cells. S63 and NIH3T3 fibroblast cells stably expressing Scramble or shPTEN were treated with MG132 (10 μM) for 6 hours. Endogenous MTSS1 protein levels were determined along with the β-actin control. (C and D) The indicated plasmids were transfected into 293T cells. Co-IP was performed to determine the interaction between MTSS1 and β-TrCP. When overexpressing PTEN, the interaction between MTSS1 and β-TrCP decreased in reciprocal co-IP. Overexpression of PTEN does not affect the interaction between MTSS1S322A and β-TrCP. (E) MG132 blocks PTEN knockdown-induced decrease of MTSS1 protein levels in PDAC cell lines. PANC-1 and MIA PaCa-2 epithelial cells transfected with Scramble or shPTEN were treated with MG132 (10 μM) for 6 hours. Endogenous MTSS1 protein levels were determined along with the β-actin control. Data are represented as mean ± SEM. **P value < .001.
The proteasome degradation of MTSS1 requires an interaction with the E3 ligase SCFβ-TRCP and phosphorylation at the S322 residue [16]. Having earlier shown that PTEN is a phosphatase for MTSS1 and that the S322 residue may be the dephosphorylation site for PTEN, we next sought to show that MTSS1 protein level change occurs through SCFβ-TRCP. Our results show that overexpression of PTEN decreased the binding between β-TrCP and MTSS1 but did not change the binding between β-TrCP and MTSS1S322A (Figure 4, C and D). This is consistent with our earlier results showing that PTEN decreases the phosphorylation level of MTSS1 but not the phosphorylation level of MTSS1S322A.
Until this point, our data on MTSS1 regulation via PTEN had been completed in 293T cells and multiple fibroblast cell lines. However, we still wanted to determine whether PTEN stabilizes MTSS1 in a similar way in epithelial PDAC cells [45]. Thus, we performed the same experiments with PANC-1 and MIA PaCa-2 cells, two epithelial PDAC cells lines that have been previously used by our laboratory [10]. We observed that PDAC cells with PTEN knockdown showed similar results when they were treated with MG132 (Figure 4E), indicating that this novel regulation of MTSS1 by PTEN happens in both CAFs and PDAC cells.
Increase in MTSS1 Expression Results in Decreased Metastatic Phenotype in PTEN-Deficient Cells
Thus far, our data show that PTEN status in PDAC cells correlates with MTSS1 expression levels. We have identified that loss of PTEN has not only significant functional consequences through increasing cellular migration and invasion capabilities but also significant molecular consequences in that its loss allows for MTSS1 to be tagged for proteasomal degradation. However, we wanted to further test our hypothesis that MTSS1 is a target of PTEN through functional rescue experiments. Utilizing the same PANC-1 shPTEN cell lines from our previous experiments, we transiently transfected Myc-MTSS1 plasmid into the cells in order to generate a cell line that was deficient in PTEN yet still expressed MTSS1 (Figure 5A). We then took these cells and performed a scratch assay in either the presence or absence of serum in order to functionally define the importance of MTSS1 in a PTEN-deficient setting. In serum-free conditions, PANC-1 shPTEN cells were still significantly more able to migrate across the scratch by approximately 50 μm farther than either Scramble control or shPTEN + MOE cells (Figure 5B). However, upon rescue with MTSS1, shPTEN + MOE cells displayed a significantly less migratory phenotype even when compared to PANC-1 Scramble, indicating a reversion to a more stationary phenotype. The same results were seen in serum-containing conditions, where PANC-1 shPTEN cells invaded approximately 150 μm farther than both Scramble and shPTEN + MOE cells (Figure 5B). Still, the rescue shPTEN + MOE cells once again reverted to a more stationary phenotype, in which there was no significant difference seen between the Scramble and shPTEN + MOE cell lines.
Figure 5.

Increase in MTSS1 expression results in decreased invasion and migration in PTEN-deficient cells. (A) PANC-1 cells were stably transduced with either Scramble control vector or shPTEN knockdown plasmid. PANC-1 shPTEN cells were then transiently transfected with Myc-MTSS1 plasmid, and MTSS1 overexpression was confirmed via Western blot. (B) PANC-1 Scramble, shPTEN, and shPTEN + MOE cells were plated, and a scratch assay was performed in both serum-free (top) and serum-containing (bottom) conditions. Cells that were deficient in PTEN expression moved significantly further across the wound in both conditions, but upon overexpression of MTSS1 in those same cells, there was no significant difference denoted when compared to Scramble control. (C) PANC-1 Scramble, shPTEN, and shPTEN + MOE cells were plated, and a Transwell invasion assay was performed in the presence of Matrigel. PANC-1 shPTEN cells invaded through the membrane significantly more often than Scramble control until MTSS1 was overexpressed, and then no significant difference was seen. *P value < .05, **P value < .001.
Additionally, these cell lines were plated in Matrigel-coated wells for Transwell invasion analysis. PANC-1 shPTEN cells invaded through the Transwell membrane significantly more often than either Scramble or shPTEN + MOE cells (Figure 5C). However, even in the absence of PTEN, the PANC-1 shPTEN + MOE cells were able to return to a similar invading cell number as PANC-1 Scramble control cells, as there was no significant difference seen between the two cell lines. These data indicate the crucial presence of MTSS1 in PDAC progression. We show that even if an important tumor suppressor, such as PTEN, is lost, as long as MTSS1 is still present, the ability for a cell to migrate and invade is diminished significantly.
Inhibition of miRNA-23b Leads to Decreased Metastatic Phenotype and Increased PTEN and MTSS1 Expression
While all of our data thus far have been focused on the cell-intrinsic regulation of MTSS1 through the tumor suppressor PTEN, we have also presented evidence indicating a potential cell-extrinsic role for the CAFs found in the tumor microenvironment (Figure 1, D and E). One of the avenues that we hypothesized may play a role in the downregulation of MTSS1 via PTEN loss was miRNA regulation. It is well documented that MTSS1 is a target for downregulation of many miRNAs [12], [13], [14], [15]. However, we chose to investigate miRNAs that target PTEN in order to determine if miRNA regulation of PTEN played any role on MTSS1 expression. One such miRNA cluster, the miR-23 cluster, has been reported to inhibit PTEN gene expression in a number of different cancer models [46], [47], [48]. Upon further investigation, we found that one specific member of the miR-23 cluster, namely, miR-23b, is predicted to target the 3′ UTR of PTEN with an miTG score of 0.824 (Figure 6, A and B).
Figure 6.

Manipulation of miRNA-23b leads to altered cellular invasion, migration, and PTEN and MTSS1 expression. (A) Schematic of miR-23b binding site in PTEN mRNA 3′ UTR sequence at nucleotide position 1608 to 1615. (B) DIANA LAB miTG prediction score of the probability that miR-23b will target PTEN. The closer to 1.0 the miTG score, the higher the probability of targeting. (C) qPCR analysis of relative miR-23b expression levels in PDAC epithelial cells or PDAC CAFs. (D) PANC-1 cells were transiently transfected with miR-23b27b cluster plasmid. These cells showed robust loss of PTEN expression upon Western blot examination. (E) qPCR confirmation of PANC-1 cells transiently transfected with anti–miR-23b inhibitor. (F) PANC-1 cells treated with anti–miR-23b were significantly less able to travel across the wound in a scratch assay setting as compared to (−) control. (G) PANC-1 cells treated with anti–miR-23b were significantly less able to travel through a Matrigel-coated membrane in a Transwell invasion assay setting as compared to (−) control. (H) Treatment of PANC-1 cells with anti–miR-23b resulted in an increase in expression levels of both PTEN and MTSS1 as confirmed via Western blot analysis. (I) Schematic for the hypothesized mechanism of action for the downregulation of MTSS1 via PTEN loss. The dense tumor microenvironment, majorly composed of CAFs, contains higher amounts of miRNA-23b. miRNA-23b is then secreted to the PDAC tumor epithelium, where it targets PTEN, downregulating it. This downregulation leaves MTSS1 unstabilized from proteasomal degradation and thus results in loss of MTSS1 and thereby increased metastatic potential. *P value < .05, **P value < .001, ***P value < .0001.
Having determined that miR-23b would be an appropriate upstream inhibitor of PTEN, we next sought out to define the relative expression level of miR-23b in a PDAC epithelial cell line versus a PDAC CAF cell line. qPCR analysis revealed that PDAC CAFs have a nearly 12-fold higher miR-23b expression level than epithelial PDAC cells (Figure 6C). We also transiently transfected a plasmid containing two members of the miR-23 cluster, namely, miR-23b and miR-27b, into PANC-1 cells and performed a Western blot on their lysates. In PANC-1 cells transfected with miR-23b27b, PTEN expression was dramatically decreased when compared to MiArrest control (Figure 6D).
Upon uncovering a potential link between miR-23b expression and PTEN status, we next sought to investigate the functional and genomic repercussions of inhibiting miR-23b. Utilizing transiently transduced PANC-1 cell lines that were treated with anti–miR-23b (Figure 6E), we performed a scratch assay to determine the effects of diminishing miR-23b expression levels. PANC-1 anti–miR-23b cells were significantly less able to invade across the wound, traveling approximately 100 μm less than (−) control (Figure 6F). Additionally, these PANC-1 anti–miR-23b cells were 1.7-fold less able to travel through a Matrigel-coated Transwell membrane when compared to (−) control, indicating miR-23b's importance in promoting cellular invasion and migration (Figure 6G). In order to investigate the genomic consequences of miR-23b loss, we treated PANC-1 cells with either (−) control or anti–miR-23b inhibitor and found that inhibition of miR-23b results in a moderate increase in both MTSS1 and PTEN status (Figure 6H).
Together, these data suggest a novel, cell-intrinsic mechanism for the regulation of MTSS1 via the protein phosphatase activity of PTEN. However, we hypothesize that there is also a cell-extrinsic component to this regulation, specifically through the transfer of miR-23b from the PDAC CAFs of the tumor microenvironment to the tumor epithelium, where miR-23b targets PTEN for downregulation, leading to the lack of protection for MTSS1 from proteasomal degradation, thus causing an increase in cellular invasion and migration (Figure 6I).
Discussion
In this report, we uncover a new mechanism of regulating MTSS1 stability via the protein phosphatase activity of PTEN. This regulation is essential for suppressing pancreatic cancer cell invasion and migration. We previously identified MTSS1 as a metastasis suppressor protein crucial for keeping cells from further disseminating from the primary tumor [10]. Upon validating its functional role in PDAC, we sought to determine novel ways in which MTSS1 could be regulated in pancreatic cancer. Knowing the affinity that the MTSS1 protein has for protein phosphatases, we began our investigation looking at the role that PTEN plays in PDAC metastatic progression. We showed that loss of PTEN significantly increases PDAC cell migration and invasion, while at the same time resulting in decreased expression of MTSS1. Next, we demonstrated that CAF-conditioned medium has the ability to increase the migratory and invasive properties of PTEN-deficient cells. We then provided evidence to support our hypothesis that PTEN stabilizes MTSS1 via its protein phosphatase activity. Through dephosphorylation of the S322 residue of MTSS1, PTEN decreases the binding between MTSS1 and its E3 ligase SCFβ-TRCP to prevent the proteasome degradation of MTSS1. We also showed that, even in the absence of PTEN in PDAC cells, ectopic expression of MTSS1 was able to revert cells to a more stationary phenotype. Finally, we offered a novel, cell-extrinsic mechanism of how PTEN downregulation occurs via miR-23b in the PDAC tumor microenvironment, leading to eventual loss of MTSS1 expression.
Metastasis is a significant contributor to mortality in cancer patients, especially in PDAC due to its late diagnosis [49]. MTSS1 is a metastasis suppressor gene originally identified as a protein downregulated in metastatic bladder carcinoma cell lines [50]. Growing evidence exists showing that downregulation of MTSS1 expression correlates with metastatic progression in a number of cancers, including bladder, prostate, breast, and pancreatic cancer [43], [44], [51], [52]. In our previous study, we have shown that loss of MTSS1 results in increased metastatic potential in pancreatic cancer [10]. Maintaining robust MTSS1 expression levels is difficult, especially in late-stage cancers, where it has been found that MTSS1 is frequently downregulated [50], [51], [52], [53], [54]. Therefore, uncovering the molecular mechanisms responsible for regulating MTSS1 expression is crucial for combatting cancer cell dissemination.
A number of different studies have been completed looking at the different regulatory mechanisms of action for manipulating MTSS1 expression levels. Most commonly, MTSS1 is found to be the target of a small number of miRNAs that are capable of downregulating its expression. In squamous cellular carcinoma, miR-96 was found to target and downregulate MTSS1 expression, leading to increased cellular proliferation and metastasis [55]. Similarly, miR-135b in colorectal cancer [13], miR-182 in ovarian cancer [56], and miR-135a in hepatocellular carcinoma [57] have all been shown to target and downregulate MTSS1 expression, leading to increased metastatic potential. Additionally, MTSS1 has been found to be a target of methylation-induced silencing [18], [58]. Interestingly, all of these aforementioned studies looking into the regulation of MTSS1 occur during pre-translational phases, that is, at the transcriptional, mRNA level. Instead of taking that canonical approach to this particular regulatory situation, we hypothesized that the protein structure of MTSS1 may lead to novel regulatory opportunities.
MTSS1 is a multidomain, scaffold protein that normally regulates cytoskeletal dynamics and actin polymerization of the cell [21], [59]. As a multidomain protein, MTSS1 contains a WH2 [WASP (Wiskott-Aldrich syndrome protein) homology 2] domain and an IMD [IRSp53 (insulin receptor substrate protein of 53 kDa) and MIM homology] domain to help develop the cytoskeletal interactions necessary to promote cellular adhesion [59]. However, the relevant portion of the MTSS1 protein to this study is the center due to its wealth of proline, serine, and threonine residues. These residues, all containing hydroxyl groups, are prime targets for interactions with a number of different proteins, specifically including phosphatases like PTEN.
PTEN is a well-studied tumor suppressor that negatively regulates the PI3K/AKT pathway [40], [41]. Many publications have shown that PTEN inhibits tumor cell proliferation, migration, and invasion [60], [61], [62], [63], [64], [65]. While many of these studies have focused on how PTEN's regulation of AKT signaling limits oncogenic potential, no publication to date has established a novel connection between the PTEN tumor suppressor and the MTSS1 metastasis suppressor. Our data define a functional relevance for PTEN loss in the metastatic progression of PDAC, as PTEN-deficient cells were more invasive and migratory in nature. Additionally, the PTEN-deficient cells also harbor decreased levels of MTSS1. Our data show that despite its more traditional role as a lipid phosphatase, in this context, PTEN's protein phosphatase activity is responsible for stabilizing MTSS1.
Previous data have shown MTSS1 to be vulnerable to proteasome degradation via phosphorylation of its Ser322 residue [16]. The hydroxyl group of the Ser322 residue undergoes a phosphorylation event by casein kinase 1δ. That phos-tagged Ser322 essentially becomes a beacon for ubiquitination via the 26S proteasome. Zhong et al. noted in their study that phosphatase interaction could disrupt this ubiquitination event, but they did not stipulate further as to which phosphatase, if any, may be critical to this stabilization of MTSS1. Here, for the first time, we provide data showing that PTEN can protect MTSS1 from proteasome degradation via dephosphorylation of this S322 residue, thus stabilizing its expression levels. Our data do not show PTEN or MG132 having the ability to significantly increase MTSS1 expression levels, although they are both acting in ways that are meant to disrupt the proteasomal degradation of MTSS1. This correlates with our initial hypothesis that PTEN plays a role in simply stabilizing MTSS1 expression, not necessarily increasing it. Through the dephosphorylation activity of PTEN or through proteasomal inhibition via MG132, MTSS1 is protected from degradation, and simply by maintaining normal levels of MTSS1 expression, we showed that cellular invasion and migration are significantly diminished.
While the majority of the data presented here strongly supports our hypothesis that PTEN stabilizes MTSS1 in a cell-intrinsic fashion, we also present results that suggest that the dense tumor microenvironment that is characteristic of PDAC also plays a cell-extrinsic role in regulating MTSS1 expression. CAFs make up the majority of cells that comprise the tumor microenvironment in PDAC [38], [66], [67]. Based on our prior MTSS1 studies, we know that CAFM leads to a robust downregulation of MTSS1 expression [10]. In this study, we found that CAFM is capable of downregulating PTEN expression as well. Thus, we hypothesized that something secreted by the CAFs into their media was responsible for the downregulation of both PTEN and MTSS1. Since we found PTEN to be the protein responsible for the stabilization of the expression level of MTSS1, we focused our attention on potential regulators of the phosphatase protein that were found in the CAFM. One of the better-studied inhibitors of PTEN in prior reports has been the miRNA-23 cluster [46], [47], [48]. We found that one specific member of this miRNA cluster, namely miR-23b, was significantly upregulated in CAFs as compared to PDAC epithelial cells. Upon overexpression of this miRNA in PANC-1 PDAC epithelial cells, we noticed complete ablation of PTEN expression. Conversely, when we inhibited miR-23b, we found that not only did cellular migration and invasion decrease, but also both PTEN and MTSS1 expression increased moderately.
These data suggested to us that the CAFs found in the PDAC tumor microenvironment may be secreting miR-23b, which is finding its way to the tumor epithelium, where it is targeting PTEN for downregulation. This downregulation of PTEN causes the destabilization of MTSS1, allowing its Ser322 residue to be phosphorylated by casein kinase 1δ, thus tagging it for proteasomal degradation. This novel, cell-extrinsic mechanism compiled with our original finding of a regulatory relationship between the canonical tumor suppressor PTEN and the metastasis suppressor MTSS1 offers new insight into one of the controlling mechanisms at hand in PDAC metastatic progression. Future studies will focus further on how other members of the miRNA-23 cluster may impact PDAC metastasis as well as uncovering other factors in the tumor microenvironment that may be responsible for perpetuating increased metastatic characteristics in PDAC. Additionally, in vivo analyses showing that PTEN inhibits tumor growth and invasion via the MTSS1 pathway will be critical in further supporting these findings.
In conclusion, our findings support our central hypothesis that MTSS1 is stabilized by the protein phosphatase activity of the tumor suppressor PTEN. Our data show that PTEN loss in PDAC cells results in both increased metastatic potential and decreased MTSS1 expression. Furthermore, we show that ectopic MTSS1 expression rescues this effect. Additionally, we demonstrate that PTEN forms a complex with MTSS1 in order to stabilize it from proteasomal degradation. Finally, we show that the tumor microenvironment, which makes up over 90% of PDAC tumor bulk, is capable of downregulating PTEN expression, potentially uncovering a novel extrinsic, upstream mechanism of MTSS1 regulation. Collectively, these data not only offer new insight into the role and regulation of MTSS1 in suppressing tumor cell invasion and migration but also uncover novel mechanisms of action of both PTEN and the PDAC tumor microenvironment.
The following are the supplementary data related to this article.
Author Contributions
The project was conceived by W. H., A. E. Z., and R. H.; experiments were performed by A. E. Z. and W. H., who share first authorship; M. L. F. and R. H. contributed expertise; the paper was written by A. E. Z., W. H., and R. H. and approved by all the authors.
Funding
R. Hill was awarded financial support from the Walther Cancer Foundation and the Joseph D. Boyle Memorial Fund. W. Huang was supported by the Walther Cancer Foundation Engineering Novel Solutions to Cancer's Challenges at the Interdisciplinary Interface (ENSCCII) Training Project. A. E. Zeleniak was supported by Walther Cancer Foundation Interdisciplinary Interface Training Program (IITP) Award and by the University of Notre Dame via funding raised for the Harper Cancer Research Institute on Notre Dame Day. Work by M. L. Fishel was supported by grants from the National Institutes of Health, NCI CA167291, with additional support from the Biomedical Research Grant and in part by Jeff Gordon Children's Foundation. We thank the Notre Dame Men's and Women's Rowing Teams for their fundraising support.
Disclosure of Potential Conflicts of Interest
The authors declare no conflict of interest.
Acknowledgments
Acknowledgements
We thank all our colleagues who generously provided reagents and our laboratory colleagues for their support, advice, and good cheer. We thank Dr. Rich Dahl and his laboratory for their gracious sharing of miR-23 cluster plasmids. We also thank Dr. Wenyi Wei and his laboratory for their generous sharing of the Myc-tagged MTSS1 plasmid used for overexpression analysis.
References
- 1.Society. AC . The Society; Atlanta, GA: 2010. Cancer Facts and Figures; p. v. [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics. CA Cancer J Clin. 2016;66(1):7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 3.Loncle C, Bonjoch L, Folch-Puy E, Lopez-Millan MB, Lac S, Molejon MI, Chuluyan E, Cordelier P, Dubus P, Lomberk G. IL17 Functions through the Novel REG3beta-JAK2-STAT3 Inflammatory Pathway to Promote the Transition from Chronic Pancreatitis to Pancreatic Cancer. Cancer Res. 2015;75(22):4852–4862. doi: 10.1158/0008-5472.CAN-15-0896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loncle C, Molejon MI, Lac S, Tellechea JI, Lomberk G, Gramatica L, Fernandez Zapico MF, Dusetti N, Urrutia R, Iovanna JL. The pancreatitis-associated protein VMP1, a key regulator of inducible autophagy, promotes Kras(G12D)-mediated pancreatic cancer initiation. Cell Death Dis. 2016;7:e2295. doi: 10.1038/cddis.2016.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steeg PS. Targeting metastasis. Nat Rev Cancer. 2016;16(4):201–218. doi: 10.1038/nrc.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mettu NB, Abbruzzese JL. Clinical insights into the biology and treatment of pancreatic cancer. J Oncol Pract. 2016;12(1):17–23. doi: 10.1200/JOP.2015.009092. [DOI] [PubMed] [Google Scholar]
- 7.Yeo CJ, Cameron JL. Prognostic factors in ductal pancreatic cancer. Langenbecks Arch Surg. 1998;383(2):129–133. doi: 10.1007/s004230050104. [DOI] [PubMed] [Google Scholar]
- 8.Gebhardt C, Meyer W, Reichel M, Wunsch PH. Prognostic factors in the operative treatment of ductal pancreatic carcinoma. Langenbeck's Arch Surg. 2000;385(1):14–20. doi: 10.1007/s004230050004. [DOI] [PubMed] [Google Scholar]
- 9.Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148(1-2):349–361. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeleniak AE, Huang W, Brinkman MK, Fishel ML, Hill R. Loss of MTSS1 results in increased metastatic potential in pancreatic cancer. Oncotarget. 2017;8(10):16473–16487. doi: 10.18632/oncotarget.14869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dawson JC, Bruche S, Spence HJ, Braga VM, Machesky LM. Mtss1 promotes cell-cell junction assembly and stability through the small GTPase Rac1. PLoS One. 2012;7(3):e31141. doi: 10.1371/journal.pone.0031141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kedmi M, Ben-Chetrit N, Korner C, Mancini M, Ben-Moshe NB, Lauriola M, Lavi S, Biagioni F, Carvalho S, Cohen-Dvashi H. EGF induces microRNAs that target suppressors of cell migration: miR-15b targets MTSS1 in breast cancer. Sci Signal. 2015;8(368):ra29. doi: 10.1126/scisignal.2005866. [DOI] [PubMed] [Google Scholar]
- 13.Wu W, Wang Z, Yang P, Yang J, Liang J, Chen Y, Wang H, Wei G, Ye S, Zhou Y. MicroRNA-135b regulates metastasis suppressor 1 expression and promotes migration and invasion in colorectal cancer. Mol Cell Biochem. 2014;388(1-2):249–259. doi: 10.1007/s11010-013-1916-z. [DOI] [PubMed] [Google Scholar]
- 14.Jahid S, Sun J, Edwards RA, Dizon D, Panarelli NC, Milsom JW, Sikandar SS, Gumus ZH, Lipkin SM. miR-23a promotes the transition from indolent to invasive colorectal cancer. Cancer Discov. 2012;2(6):540–553. doi: 10.1158/2159-8290.CD-11-0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J, Li J, Shen J, Wang C, Yang L, Zhang X. MicroRNA-182 downregulates metastasis suppressor 1 and contributes to metastasis of hepatocellular carcinoma. BMC Cancer. 2012;12:227. doi: 10.1186/1471-2407-12-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhong J, Shaik S, Wan L, Tron AE, Wang Z, Sun L, Inuzuka H, Wei W. SCF beta-TRCP targets MTSS1 for ubiquitination-mediated destruction to regulate cancer cell proliferation and migration. Oncotarget. 2013;4(12):2339–2353. doi: 10.18632/oncotarget.1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fan H, Chen L, Zhang F, Quan Y, Su X, Qiu X, Zhao Z, Kong KL, Dong S, Song Y. MTSS1, a novel target of DNA methyltransferase 3B, functions as a tumor suppressor in hepatocellular carcinoma. Oncogene. 2012;31(18):2298–2308. doi: 10.1038/onc.2011.411. [DOI] [PubMed] [Google Scholar]
- 18.Utikal J, Gratchev A, Muller-Molinet I, Oerther S, Kzhyshkowska J, Arens N, Grobholz R, Kannookadan S, Goerdt S. The expression of metastasis suppressor MIM/MTSS1 is regulated by DNA methylation. Int J Cancer. 2006;119(10):2287–2293. doi: 10.1002/ijc.22106. [DOI] [PubMed] [Google Scholar]
- 19.Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, van der Burgt I, Crosby AH, Ion A, Jeffery S. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001;29(4):465–468. doi: 10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- 20.Woodings JA, Sharp SJ, Machesky LM. MIM-B, a putative metastasis suppressor protein, binds to actin and to protein tyrosine phosphatase delta. Biochem J. 2003;371(Pt 2):463–471. doi: 10.1042/BJ20021962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chaudhary F, Lucito R, Tonks NK. Missing-in-Metastasis regulates cell motility and invasion via PTPdelta-mediated changes in SRC activity. Biochem J. 2015;465(1):89–101. doi: 10.1042/BJ20140573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A. 1999;96(8):4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wartenberg M, Centeno I, Haemmig S, Vassella E, Zlobec I, Galvan JA, Neuenschwander M, Schlup C, Gloor B, Lugli A. PTEN alterations of the stromal cells characterise an aggressive subpopulation of pancreatic cancer with enhanced metastatic potential. Eur J Cancer. 2016;65:80–90. doi: 10.1016/j.ejca.2016.06.013. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Zhang DW, Lin DQ, Cao LQ. Peroxisome proliferator-activated receptor-gamma inhibits pancreatic cancer cell invasion and metastasis via regulating MMP-2 expression through PTEN. Mol Med Rep. 2015;12(4):6255–6260. doi: 10.3892/mmr.2015.4224. [DOI] [PubMed] [Google Scholar]
- 25.Wang MC, Jiao M, Wu T, Jing L, Cui J, Guo H, Tian T, Ruan ZP, Wei YC, Jiang LL. Polycomb complex protein BMI-1 promotes invasion and metastasis of pancreatic cancer stem cells by activating PI3K/AKT signaling, an ex vivo, in vitro, and in vivo study. Oncotarget. 2016;7(8):9586–9599. doi: 10.18632/oncotarget.7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richards KE, Zeleniak AE, Fishel ML, Wu J, Littlepage LE, Hill R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene. 2017;36(13):1770–1778. doi: 10.1038/onc.2016.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lei R, Tang J, Zhuang X, Deng R, Li G, Yu J, Liang Y, Xiao J, Wang HY, Yang Q. Suppression of MIM by microRNA-182 activates RhoA and promotes breast cancer metastasis. Oncogene. 2014;33(10):1287–1296. doi: 10.1038/onc.2013.65. [DOI] [PubMed] [Google Scholar]
- 28.Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124(6):1283–1298. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 29.Kim JS, Lee C, Bonifant CL, Ressom H, Waldman T. Activation of p53-dependent growth suppression in human cells by mutations in PTEN or PIK3CA. Mol Cell Biol. 2007;27(2):662–677. doi: 10.1128/MCB.00537-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang J, Zhang P, Wei Y, Piao HL, Wang W, Maddika S, Wang M, Chen D, Sun Y, Hung MC. Deubiquitylation and stabilization of PTEN by USP13. Nat Cell Biol. 2013;15(12):1486–1494. doi: 10.1038/ncb2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang G, Redelman-Sidi G, Rosen N, Glickman MS, Jiang X. Inhibition of mycobacterial infection by the tumor suppressor PTEN. J Biol Chem. 2012;287(27):23196–23202. doi: 10.1074/jbc.M112.351940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteomics. 2006;5(4):749–757. doi: 10.1074/mcp.T500024-MCP200. [DOI] [PubMed] [Google Scholar]
- 33.Stratford JK, Bentrem DJ, Anderson JM, Fan C, Volmar KA, Marron JS, Routh ED, Caskey LS, Samuel JC, Der CJ. A six-gene signature predicts survival of patients with localized pancreatic ductal adenocarcinoma. PLoS Med. 2010;7(7):e1000307. doi: 10.1371/journal.pmed.1000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goswami CP, Nakshatri H. PROGgene: gene expression based survival analysis web application for multiple cancers. J Clin Bioinforma. 2013;3(1):22. doi: 10.1186/2043-9113-3-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hill R, Calvopina JH, Kim C, Wang Y, Dawson DW, Donahue TR, Dry S, Wu H. PTEN loss accelerates KrasG12D-induced pancreatic cancer development. Cancer Res. 2010;70(18):7114–7124. doi: 10.1158/0008-5472.CAN-10-1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Asano T, Yao Y, Zhu J, Li D, Abbruzzese JL, Reddy SA. The PI 3-kinase/Akt signaling pathway is activated due to aberrant Pten expression and targets transcription factors NF-kappaB and c-Myc in pancreatic cancer cells. Oncogene. 2004;23(53):8571–8580. doi: 10.1038/sj.onc.1207902. [DOI] [PubMed] [Google Scholar]
- 37.Ebert MP, Fei G, Schandl L, Mawrin C, Dietzman K, Herrera P, Friess H, Gress TM, Malfertheiner P. Reduced PTEN expression in the pancreas overexpressing transforming growth factor-beta 1. Br J Cancer. 2002;86(2):257–262. doi: 10.1038/sj.bjc.6600031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25(6):735–747. doi: 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee JJ, Perera RM, Wang H, Wu DC, Liu XS, Han S, Fitamant J, Jones PD, Ghanta KS, Kawano S. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc Natl Acad Sci U S A. 2014;111(30):E3091–E3100. doi: 10.1073/pnas.1411679111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13(5):283–296. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 41.Leslie NR, Downes CP. PTEN: The down side of PI 3-kinase signalling. Cell Signal. 2002;14(4):285–295. doi: 10.1016/s0898-6568(01)00234-0. [DOI] [PubMed] [Google Scholar]
- 42.Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) J Biol Chem. 1994;269(7):5241–5248. [PubMed] [Google Scholar]
- 43.Zhou L, Li J, Shao QQ, Guo JC, Liang ZY, Zhou WX, Zhang TP, You L, Zhao YP. Expression and Significances of MTSS1 in Pancreatic Cancer. Pathol Oncol Res. 2016;22(1):7–14. doi: 10.1007/s12253-015-9963-2. [DOI] [PubMed] [Google Scholar]
- 44.Du P, Ye L, Ruge F, Yang Y, Jiang WG. Metastasis suppressor-1, MTSS1, acts as a putative tumour suppressor in human bladder cancer. Anticancer Res. 2011;31(10):3205–3212. [PubMed] [Google Scholar]
- 45.Seeley ES, Carriere C, Goetze T, Longnecker DS, Korc M. Pancreatic cancer and precursor pancreatic intraepithelial neoplasia lesions are devoid of primary cilia. Cancer Res. 2009;69(2):422–430. doi: 10.1158/0008-5472.CAN-08-1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tan X, Wang S, Zhu L, Wu C, Yin B, Zhao J, Yuan J, Qiang B, Peng X. cAMP response element-binding protein promotes gliomagenesis by modulating the expression of oncogenic microRNA-23a. Proc Natl Acad Sci U S A. 2012;109(39):15805–15810. doi: 10.1073/pnas.1207787109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tian L, Fang YX, Xue JL, Chen JZ. Four microRNAs promote prostate cell proliferation with regulation of PTEN and its downstream signals in vitro. PLoS One. 2013;8(9):e75885. doi: 10.1371/journal.pone.0075885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zaman MS, Thamminana S, Shahryari V, Chiyomaru T, Deng G, Saini S, Majid S, Fukuhara S, Chang I, Arora S. Inhibition of PTEN gene expression by oncogenic miR-23b-3p in renal cancer. PLoS One. 2012;7(11):e50203. doi: 10.1371/journal.pone.0050203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stathis A, Moore MJ. Advanced pancreatic carcinoma: current treatment and future challenges. Nat Rev Clin Oncol. 2010;7(3):163–172. doi: 10.1038/nrclinonc.2009.236. [DOI] [PubMed] [Google Scholar]
- 50.Lee YG, Macoska JA, Korenchuk S, Pienta KJ. MIM, a potential metastasis suppressor gene in bladder cancer. Neoplasia. 2002;4(4):291–294. doi: 10.1038/sj.neo.7900231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Loberg RD, Neeley CK, Adam-Day LL, Fridman Y, St John LN, Nixdorf S, Jackson P, Kalikin LM, Pienta KJ. Differential expression analysis of MIM (MTSS1) splice variants and a functional role of MIM in prostate cancer cell biology. Int J Oncol. 2005;26(6):1699–1705. [PubMed] [Google Scholar]
- 52.Parr C, Jiang WG. Metastasis suppressor 1 (MTSS1) demonstrates prognostic value and anti-metastatic properties in breast cancer. Eur J Cancer. 2009;45(9):1673–1683. doi: 10.1016/j.ejca.2009.02.019. [DOI] [PubMed] [Google Scholar]
- 53.Liu K, Wang G, Ding H, Chen Y, Yu G, Wang J. Downregulation of metastasis suppressor 1(MTSS1) is associated with nodal metastasis and poor outcome in Chinese patients with gastric cancer. BMC Cancer. 2010;10:428. doi: 10.1186/1471-2407-10-428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Callahan CA, Ofstad T, Horng L, Wang JK, Zhen HH, Coulombe PA, Oro AE. MIM/BEG4, a Sonic hedgehog-responsive gene that potentiates Gli-dependent transcription. Genes Dev. 2004;18(22):2724–2729. doi: 10.1101/gad.1221804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guo Y, Ren MS, Shang C, Zhu L, Zhong M. MTSS1 gene regulated by miR-96 inhibits cell proliferation and metastasis in tongue squamous cellular carcinoma Tca8113 cell line. Int J Clin Exp Med. 2015;8(9):15441–15449. [PMC free article] [PubMed] [Google Scholar]
- 56.Liu Z, Liu J, Segura MF, Shao C, Lee P, Gong Y, Hernando E, Wei JJ. MiR-182 overexpression in tumourigenesis of high-grade serous ovarian carcinoma. J Pathol. 2012;228(2):204–215. doi: 10.1002/path.4000. [DOI] [PubMed] [Google Scholar]
- 57.Liu S, Guo W, Shi J, Li N, Yu X, Xue J, Fu X, Chu K, Lu C, Zhao J. MicroRNA-135a contributes to the development of portal vein tumor thrombus by promoting metastasis in hepatocellular carcinoma. J Hepatol. 2012;56(2):389–396. doi: 10.1016/j.jhep.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 58.Yamashita S, Tsujino Y, Moriguchi K, Tatematsu M, Ushijima T. Chemical genomic screening for methylation-silenced genes in gastric cancer cell lines using 5-aza-2'-deoxycytidine treatment and oligonucleotide microarray. Cancer Sci. 2006;97(1):64–71. doi: 10.1111/j.1349-7006.2006.00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Machesky LM, Johnston SA. MIM: a multifunctional scaffold protein. J Mol Med. 2007;85(6):569–576. doi: 10.1007/s00109-007-0207-0. [DOI] [PubMed] [Google Scholar]
- 60.Chen JS, Wang Q, Fu XH, Huang XH, Chen XL, Cao LQ, Chen LZ, Tan HX, Li W, Bi J. Involvement of PI3K/PTEN/AKT/mTOR pathway in invasion and metastasis in hepatocellular carcinoma: Association with MMP-9. Hepatol Res. 2009;39(2):177–186. doi: 10.1111/j.1872-034X.2008.00449.x. [DOI] [PubMed] [Google Scholar]
- 61.Dupont J, Renou JP, Shani M, Hennighausen L, LeRoith D. PTEN overexpression suppresses proliferation and differentiation and enhances apoptosis of the mouse mammary epithelium. J Clin Invest. 2002;110(6):815–825. doi: 10.1172/JCI13829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lacalle RA, Gomez-Mouton C, Barber DF, Jimenez-Baranda S, Mira E, Martinez AC, Carrera AC, Manes S. PTEN regulates motility but not directionality during leukocyte chemotaxis. J Cell Sci. 2004;117(Pt 25):6207–6215. doi: 10.1242/jcs.01545. [DOI] [PubMed] [Google Scholar]
- 63.Leslie NR, Yang X, Downes CP, Weijer CJ. The regulation of cell migration by PTEN. Biochem Soc Trans. 2005;33(Pt 6):1507–1508. doi: 10.1042/BST0331507. [DOI] [PubMed] [Google Scholar]
- 64.Zhang LL, Liu J, Lei S, Zhang J, Zhou W, Yu HG. PTEN inhibits the invasion and metastasis of gastric cancer via downregulation of FAK expression. Cell Signal. 2014;26(5):1011–1020. doi: 10.1016/j.cellsig.2014.01.025. [DOI] [PubMed] [Google Scholar]
- 65.Zhao H, Dupont J, Yakar S, Karas M, LeRoith D. PTEN inhibits cell proliferation and induces apoptosis by downregulating cell surface IGF-IR expression in prostate cancer cells. Oncogene. 2004;23(3):786–794. doi: 10.1038/sj.onc.1207162. [DOI] [PubMed] [Google Scholar]
- 66.Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, Ji B, Evans DB, Logsdon CD. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68(3):918–926. doi: 10.1158/0008-5472.CAN-07-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]