

Abstract
The fruit fly Drosophila melanogaster has emerged as a powerful model for investigating the molecular mechanisms that regulate animal metabolism. However, a major limitation of these studies is that many metabolic assays are tedious, dedicated to analyzing a single molecule, and rely on indirect measurements. As a result, Drosophila geneticists commonly use candidate gene approaches, which, while important, bias studies toward known metabolic regulators. In an effort to expand the scope of Drosophila metabolic studies, we used the classic mutant lysine (lys) to demonstrate how a modern metabolomics approach can be used to conduct forward genetic studies. Using an inexpensive and well-established gas chromatography-mass spectrometry-based method, we genetically mapped and molecularly characterized lys by using free lysine levels as a phenotypic readout. Our efforts revealed that lys encodes the Drosophila homolog of Lysine Ketoglutarate Reductase/Saccharopine Dehydrogenase, which is required for the enzymatic degradation of lysine. Furthermore, this approach also allowed us to simultaneously survey a large swathe of intermediate metabolism, thus demonstrating that Drosophila lysine catabolism is complex and capable of influencing seemingly unrelated metabolic pathways. Overall, our study highlights how a combination of Drosophila forward genetics and metabolomics can be used for unbiased studies of animal metabolism, and demonstrates that a single enzymatic step is intricately connected to diverse aspects of metabolism.
Keywords: Drosophila, metabolomics, lysine, LKRSDH, familial hyperlysinemia
Our modern understanding of Drosophila metabolism is due, in part, to genetic screens conducted during the early 20th century. Many of the classic mutations isolated by Morgan and his colleagues—such as vermillion, rudimentary, cinnabar, ebony, rosy, and Henna—disrupt key metabolic enzymes (Lindsley and Zimm 1992). Subsequent analysis of these mutants helped establish the field of biochemical genetics and provided key insights regarding the in vivo regulation of intermediate metabolism. In contrast to these classic forward genetic studies, modern analyses of Drosophila metabolism primarily rely on reverse genetic approaches, which, while important, inevitably bias our understanding of animal metabolism toward gene families with known roles in human metabolic disease.
The importance of using forward genetics to study fly metabolism is exemplified by the Drosophila gene adipose (adp), which was initially identified in the 1950s as a regulator of the starvation response and triglyceride storage (Doane 1960; Teague et al. 1986). Forty years later, the molecular cloning of adp uncovered a novel, highly conserved gene that regulates triglyceride metabolism in organisms ranging from Caenorhabditis elegans to humans (Hader et al. 2003; Suh et al. 2007). Similarly, forward genetic screens have uncovered dozens of conserved genes that influence triglyceride and carbohydrate metabolism in Drosophila larvae (Pospisilik et al. 2010; Reis et al. 2010; Ugrankar et al. 2015; Song et al. 2017). Despite such successes, the use of forward genetics in metabolic research, and especially in studies of central carbon metabolism, is limited due to the tedious nature of using metabolites as the primary phenotypic readout. However, recent advances in metabolomics have simplified the extraction and measurement of small molecule metabolites from Drosophila tissues (Cox et al. 2017), making forward genetic studies of intermediary metabolism a realistic possibility. To demonstrate the feasibility of using metabolomics to conduct phenotype-driven analysis, we reexamined the classic Drosophila mutation lys1 using gas chromatography-mass spectrometry (GC-MS).
E. H. Grell serendipitously discovered lys1 as a background mutation present within Ed Lewis’ stock collection (Grell 1961). Yet, even though the lys1 mutation induces highly elevated lysine levels, lys1 mutants fail to display visible phenotypes under normal growth conditions (Grell 1961). As a result, this gene has not been studied in over 50 years. Here, we genetically mapped and molecularly characterized lys1 by using a GC-MS-based method to directly measure lysine abundance. Our efforts revealed that lys1 disrupts the Drosophila ortholog of Lysine Ketoglutarate Reductase/Saccharopine Dehydrogenase (LKRSDH), which encodes the first enzyme involved in lysine catabolism and is mutated in humans with familial hyperlysinemia (Markovitz et al. 1984; Cakouros et al. 2008). We then used the same GC-MS-based method to conduct a targeted metabolomic analysis of lys1, thereby allowing us to rapidly characterize the metabolic phenotype of these mutants. This analysis revealed that the lys1 mutant exhibits a metabolic profile reminiscent of patients with familial hyperlysinemia, suggesting that flies and humans catabolize lysine using similar metabolic mechanisms. Furthermore, our metabolomics approach uncovers novel relationships between lysine and other compounds involved in amino acid and carbohydrate metabolism, emphasizing that even a relatively simple enzymatic step can influence seemingly unrelated metabolic processes. Overall, our study demonstrates how metabolomics can simplify forward genetic studies of Drosophila intermediate metabolism, and raises the possibility that a similar method could be used in future genetic screens.
Materials and Methods
Drosophila husbandry and strain creation
Fly stocks were maintained at 25° on Bloomington Drosophila Stock Center (BDSC) food. Unless noted, all mutations and transgenes were studied in a w1118 background. The chromosome containing lys1 was isolated from BDSC stock #692 (lys1 rc1; ss1) by crossing mutant males with w1118; In(2LR)Gla, wgGla-1/CyO, P{GAL4-twi.G}2.2, P{UAS-2xEGFP}AH2.2 (BDSC stock #6662) virgin females. F1 male progeny of the genotype w1118; lys1 rc1/CyO, P{GAL4-twi.G}2.2, P{UAS-2xEGFP}AH2.2; ss1/+ were again crossed with w1118; In(2LR)Gla, wgGla-1/CyO, P{GAL4-twi.G}2.2, P{UAS-2xEGFP}AH2.2 virgin females. Individual F2 male and virgin female siblings of the genotype w1118; lys1 rc1/CyO, P{GAL4-twi.G}2.2, P{UAS-2xEGFP}AH2.2 flies were intercrossed to generate a homozygous w1118; lys1 rc1 strain that lacked ss1. Rescue experiments were conducted by using da-GAL4 to ubiquitously express a previously described UAS-LKRSDH transgene (Cakouros et al. 2008), which was generously provided by Sharad Kumar (University of South Australia).
The lys1 chromosome also harbored red cell1 (rc1), which is an uncloned recessive mutation that is linked to lys1 on chromosome 2 and located to the right of dachs. Homozygous rc1 mutants display ectopic accumulation of a red pigment in adult fat cells. In nearly all of our experiments, both mutant and control animals harbored a single copy of rc1; however, rc1 heterozygotes failed to display the red cell phenotype and lys1 rc1/+ + animals exhibited lysine levels that were similar to both w1118 controls and LKRSDHMB01942/+ heterozygotes, suggesting that a single copy of rc1 does not influence the lysine phenotype.
Deficiency mapping and complementation tests
The lys1 mutation was mapped by mating males from either w1118 controls or w1118; lys1 rc1 mutants with female flies that harbored molecularly-defined deficiencies (Cook et al. 2012). Adult male F1 progeny with straight wings were analyzed for free lysine levels using GC-MS (see below). Complementation tests were conducted using the Minos insertion Mi{ET1}LKRSDHMB01942 (Bellen et al. 2004), which was isolated from BDSC stock #23382. For all mapping experiments and complementation tests, newly eclosed F1 male offspring were aged for 5 days on BDSC food.
GC-MS analysis
Lysine levels were measured using a previously described GC-MS-based method (Tennessen et al. 2014). Briefly, 25 adult males were placed in a pretared 2 ml screw cap tube containing 1.4 mm ceramic beads (Catalog No. 15-340-153; Fisher Scientific, Pittsburgh, PA), the mass was determined with an analytical balance, and the tube was immediately dropped into liquid nitrogen. Samples were stored at −80° until processing. Metabolite extraction was achieved using prechilled 90% methanol (HPLC grade) containing 2 μg/ml of succinic-d4 acid as an internal standard, and a two-step derivatization was conducted using 40 mg/ml of methoxyamine hydrochloride in pyridine and N-methyl-N-trimethylsilyltrifluoracetamide containing 1% trimethylchlorosilane, respectively. GC-MS analyses for all genetic mapping experiments, complementation tests, and UAS-LKRSDH rescue experiments were performed on an Agilent GC6890-5973i MS equipped with a Gerstel MPS autosampler and a 30 m Phenomex ZB5-5 MSi column. The retention time for lysine in our analysis was 21.6 min, and relative lysine levels were quantified based on the abundance of ions with m/z = 317. Values were normalized based on sample mass and the succinic-d4 acid internal standard. The software package Prism 7 version 7.0b (GraphPad Software) was used to statistically analyze targeted lysine measurements and generate scatter plots. The comparison of lysine levels between w1118 and w1118; lys1 rc1 male flies was conducted using a two-tailed, unpaired Student’s t-test with Welch’s correction. All other genetic experiments were analyzed using one-way ANOVA.
Targeted metabolomic studies were conducted at the University of Utah Metabolomics Core facility, as previously described (Cox et al. 2017). This analysis was focused on a set of ∼150 metabolites for which fragmentation patterns and retention times were initially established by analyzing chemical standards. In addition, the retention times of all metabolites were calibrated using a series of fatty acid methyl ester standards. Processed data were normalized to both sample mass and a succinic-d4 acid internal standard, and statistically analyzed using MetaboAnalyst 3.0 (metaboanalyst.ca; Xia et al. 2015; Xia and Wishart 2016).
PCR amplification of lys1
Tiling fragments of the lys locus were amplified and sequenced using a PCR-based strategy (see Supplemental Material, Figure S1 for oligonucleotide sequences). Long-range PCR amplification of exon 2 in the lys1 mutant was conducted using oligonucleotides 5′-AAGTGGTGTTTACAAGGTGC-3′ and 5′-TGACGACTACCGACCGATATG-3′. Sequencing of the lys1 insertion was conducted using the oligonucleotides 5′-CTGCTTGACCAACTTCTGGAC-3′ and 5′-GATTTACGACTGGGTCCAACTG-3′, which are nested within the PCR product.
Southern blot analysis
Genomic DNA was isolated from w1118 and lys1 adult flies with a Wizard DNA Purification Kit (Promega, Madison, WI) and 5 μg of purified genomic DNA was digested overnight at 37° using PstI (Thermo Fisher Scientific). Southern blot analysis was conducted as previously described (Sullivan et al. 2000). A DNA probe corresponding to exon 2 and the surrounding sequence was generated via PCR amplification of w1118 genomic DNA using the oligonucleotides 5′-TGACGACTACCGACCGATATG-3′ and 5′-AAGTGACAATCACCAGCAGC-3′.
Quantitative (q)RT-PCR
Total RNA was isolated from 5-day-old adult male flies using Tripure Reagent (Roche). cDNA synthesis was conducted using the Thermo Maxima H Minus First Strand cDNA Synthesis Kit with dsDNase (K1681; Thermo Fisher Scientific). cDNAs and the appropriate oligonucleotides (see below) were added to FastStart Essential DNA Green Master (Roche), and a Roche LightCycler 96 was used to quantify the relative abundance of LKRSDH. rp49 was used as an internal reference. The following primer sets were used to measure the relative abundance of LKRSDH mRNA:
Data availability
All strains and reagents are available upon request. Table S1 and Table S2 contain the targeted GC-MS metabolomic data, which has been normalized to both the sample mass and the succinic-d4 acid internal standard.
Results
Genetic mapping of lys1 using GC-MS
To demonstrate how Drosophila intermediate metabolism can be efficiently studied using a combination of metabolomics and forward genetics, we chose to analyze the lys1 mutation, which has no visible phenotype when raised on a standard diet. Since lys1 was last studied in 1961, we used GC-MS to verify that the mutant strain still exhibited abnormally high lysine levels. Our analysis revealed that the levels of lysine are elevated more than fivefold in lys1 mutant males as compared with w1118 controls (Figure 1), thereby confirming that lys is an essential regulator of lysine metabolism and demonstrating that we can reliably quantify this phenotype.
Figure 1.
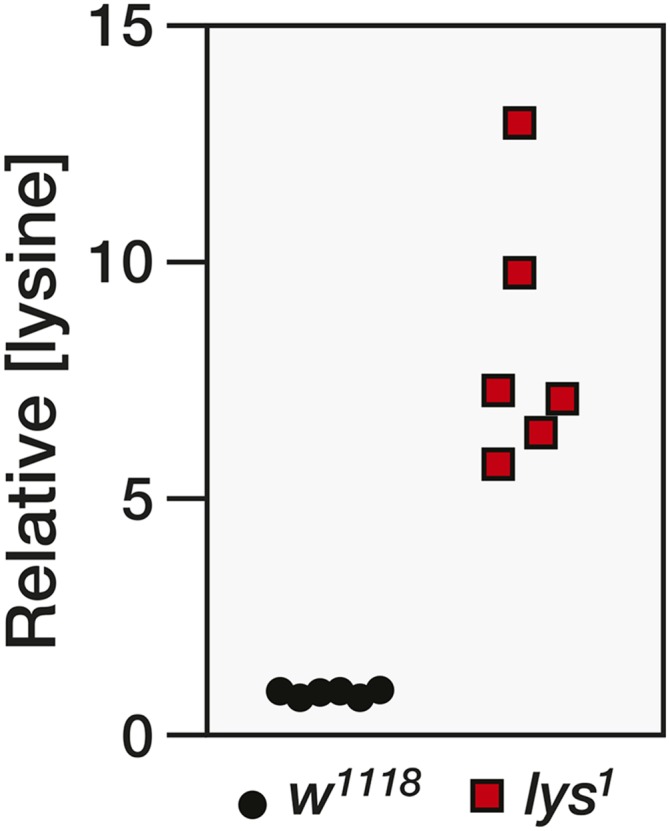
Lysine levels are elevated in lys1 mutants. The relative abundance of lysine was measured in w1118 controls and w1118; lys1 mutants. Each data point represents a single sample that contained 25 adult male flies (n = 6 samples per genotype, P < 0.001).
E. H. Grell previously mapped the genetic location of lys1 to 2–22.9, which places it to the left of dachs on chromosome 2. To further refine the genomic position of lys, we crossed w1118 and lys1 male flies with virgin females that harbored a series of overlapping deficiencies (Figure 2A). GC-MS analysis of F1 males revealed that Df(2L)ED508, Df(2L)ED12527, and Df(2L)BSC142 failed to complement lys1 (Figure 2), as trans-heterozygous offspring exhibited lysine levels that were 10–50-fold higher than those observed in the heterozygous controls (Figure 2B).
Figure 2.

Deficiency mapping of lys1. (A) A schematic diagram illustrating the size and position of the molecularly-defined deficiencies used to genetically map lys1. Homozygous lys1 females were crossed with deficiencies (Df) that span the genomic region to the left of dachs (d). Deficiencies highlighted in red failed to complement lys1. (B) GC-MS was used to measure relative lysine levels in 3-day-old adult males. Animals that were heterozygous for either lys1 or any of the examined deficiencies exhibited similar lysine levels. In contrast, homozygous lys1 mutants and males that were trans-heterozygous for lys1 and either Df(2L)ED508, Df(2L)ED12527, or Df(2L)BSC142 displayed lysine levels that were up to 50-fold higher than controls. Each data point represents a single sample that contained 25 adult male flies. GC-MS, gas chromatography-mass spectrometry.
Our deficiency mapping results narrowed the location of lys1 to an ∼10 kb region containing the genes LKRSDH and Herp (Figure 3A). Considering that LKRSDH encodes an enzyme that catalyzes the first two steps in lysine degradation, we examined the possibility that lys1 is a mutation in this gene. Homozygous lys1 mutants were mated with flies that harbored a Minos insertion in the third exon of LKRSDH (Mi{ET1}LKRSDHMB01942) and lysine levels were measured in the resulting F1 progeny. Similar to lys1 homozygous mutants, adult male flies that were trans-heterozygous for lys1 and LKRSDHMB01942 accumulated significantly higher lysine levels than lys1 heterozygous controls (Figure 3B). We obtained similar results when either lys1 or LKRSDHMB01942 were placed in trans to the deficiency Df(2L)ED508, suggesting that both lys1 and LKRSDHMB01942 severely reduce LKRSDH enzyme function. Moreover, ubiquitous expression of a UAS-LKRSDH transgene in a lys1/LKRSDHMB01942 trans-heterozygous background completely rescued the elevated lysine phenotype, demonstrating that the lys1 phenotype is due to loss of the LKRSDH enzyme.
Figure 3.

lys1 disrupts LKRSDH. (A) A schematic diagram of the genomic region deleted by both Df(2L)ED508 and Df(2L)ED12527. The only genes present within this region are LKRSDH and Herp. The Minos insertion LKRSDHMB01942 disrupts the third exon of this gene and was used in all subsequent genetic analyses. (B) As compared with heterozygous control strains, lysine levels are elevated between 20- and 40-fold in adult males that harbor a trans-heterozygous combination of lys1 and either LKRSDHMB or Df(2L)ED508 (Df), indicating that lys1 disrupts LKRSDH function. A similar increase in lysine (lys) concentration is observed in males of the genotype w1118; LKRSDHMB01942/Df(2L)ED508. (C) The relative abundance of lysine in adult males of the following genotypes: w1118; lys1/+, w1118; lys1/LKRSDHMB01942, w1118; lys1/LKRSDHMB01942; +/da-GAL4, w1118; lys1/LKRSDHMB01942; UAS-LKRSDH/+, and w1118; lys1/LKRSDHMB01942; UAS-LKRSDH +/+ da-GAL4. (B and C) Each data point represents a single sample that contained 25 adult male flies. n.s, not significant. ** P < 0.01 and *** P < 0.001.
To determine the molecular nature of lys1, we used a PCR-based strategy to sequence LKRSDH in lys1 mutants. During this analysis, we were unable to amplify the second LKRSDH exon from lys1 genomic DNA using standard PCR techniques, suggesting that a large aberration existed in this region (Figure S1, A and B). We tested this possibility using a Southern blot to examine exon 2 of LKRSDH in both w1118 control and lys1 mutants. A probe corresponding to the LKRSDH exon 2 region hybridized with a single ∼1.8 kb DNA fragment in the w1118 control, but hybridized to two fragments in genomic DNA isolated from the lys1 mutant (Figure S1C). Since the mutant bands are collectively larger than the control band, this result suggests that the lys1 mutation arises from either a large insertion or an inversion in LKRSDH. Indeed, when we sequenced a long-range PCR product that amplified from the lys1 chromosome, we discovered a large insertion in the second exon and a small deletion that removes a portion of the 5′-UTR and the first six amino acids of the enzyme (Figure S1D). Consistent with this finding, qRT-PCR analysis reveals that lys1 mutants exhibit an ∼80% decrease in LKRSDH transcript levels as compared with w1118 controls (Figure S1E), indicating that this insertion is a severe loss-of-function allele. Therefore, we will refer to lys1 as LKRSDH1 for the remainder of this study.
Metabolomic analysis of LKRSDH mutants
Although loss-of-function mutations in both the human and Drosophila LKRSDH homologs fail to produce obvious visible phenotypes, recent observations in the fly suggest that both lysine and LKRSDH are key regulators of physiology and behavior (Cakouros et al. 2008; Bjordal et al. 2014). In an effort to better understand the role of LRKSDH in Drosophila metabolism, we used the same GC-MS method that served as the basis of our genetic analysis to conduct a targeted metabolomic study of LKRSDH1/LKRSDHMB01942 mutants and +/LKRSDHMB01942 controls. Two independent analyses of adult male samples detected between 85 and 100 metabolites (Table S1 and Table S2). A principle component analysis of the resulting data revealed that the mutant strains exhibited a metabolomic profile that was significantly different from controls (Figure 4A and Figure S2A). Moreover, relative lysine concentrations were increased by more than fivefold in LKRSDH mutant samples, and lysine was the most significantly altered metabolite in our analysis (Figure 4, B and C and Figure S2B).
Figure 4.

Metabolomic analysis of LKRSDH mutants. w1118; LKRSDHMB/+ and w1118; LKRSDH1/MB01942 adult males were analyzed using a targeted gas chromatography-mass spectrometry (GC-MS) approach. (A) A comparison of the metabolomic data from control and mutant samples using principle component (PC) analysis. (B) Differences in metabolite abundance between control and mutant samples are represented as a volcano plot. Dashed vertical line represents a fold change (FC) of > 1.5. Dashed horizontal line represents P < 0.01. (C–E) The relative abundance of lysine (Lys), 2-aminoadipate (2-Aad), pipecolic acid (Pip), 5-aminopentanoate (5-Apt), histidine (His), and sorbitol (Sorb). Each data point represents a single sample that contained 25 adult male flies. P < 0.001 for all panels.
In addition to lysine, we observed an unexpected increase in 2-aminoadipate (2Aad; Figure 4, B and D, Figure 5, and Figure S2B), which is a downstream product of lysine catabolism. Since loss of LKRSDH activity should inhibit lysine catabolism, we anticipated that 2Aad levels would be decreased in LKRSDH mutants. This observed increase in 2Aad suggests that, in the absence of LKRSDH, flies produce 2Aad using alternative metabolic pathways. In this regard, our metabolomic data present two possible mechanisms that could bypass the requirement for LKRSDH in 2Aad synthesis (Figure 5). First, some animals are thought to be capable of converting lysine into 2Aad via the poorly understood and somewhat controversial pipecolic acid pathway (Figure 5; Broquist 1991; Struys and Jakobs 2010). Furthermore, human patients with familial hyperlysinemia, which is caused by mutations in LKRSDH, exhibit elevated pipecolic acid levels (Markovitz et al. 1984). Since pipecolic acid was not measured during our initial targeted analysis, we reanalyzed our mutants for the presence of this compound and found that pipecolic acid levels were also significantly elevated in LKRSDH mutants as compared with controls (Figure 4D). This result indicates that Drosophila synthesizes pipecolic acid and suggests that flux through this pathway is increased in LKRSDH mutants. Second, LKRSDH mutants exhibit elevated 5-aminopentanoate levels (5Apt; Figure 4D, Figure 5, and Figure S2B), which can be produced when lysine is catabolized via a cadaverine intermediate (Fothergill and Guest 1977). While there is some evidence that animals can directly convert lysine to cadaverine by an unknown mechanism, the cadaverine pathway is most commonly associated with bacterial and plant metabolism (Miller-Fleming et al. 2015). Therefore, these results hint at the possibility that either the fly microbial community is contributing to lysine catabolism in LKRSDH mutants or that Drosophila produces this compound by an uncharacterized metabolic mechanism.
Figure 5.

A schematic diagram illustrating the metabolic pathways that catabolize lysine. Metabolites highlighted in red text were measured using a targeted metabolomics approach. Solid black arrows represent the LKRSDH-dependent pathway. Blue solid arrows indicate the postulated pipecolic acid pathway. Dashed arrows indicate those metabolic reactions are catalyzed by bacterial enzymes. *, saccharopine was undetectable in both control and mutant samples.
In addition to the metabolic changes associated with lysine catabolism, our metabolomic analysis of LKRSDH mutants also revealed reproducible changes in histidine, sorbitol, and sarcosine. The decrease in histidine concentration is particularly notable, as after lysine, histidine is the most altered metabolite in LKRSDH mutants. However, the metabolic relationships between lysine and these three compounds remain unknown and represent novel metabolic links that could only be discovered using a metabolomics approach.
Discussion
Here, we demonstrate how a combination of forward genetics and metabolomics can be used to rapidly identify and characterize defects in Drosophila intermediate metabolism. In our study, we genetically mapped and characterized defects associated with lys1, a mutant with no obvious morphological defects. The fact that we could conduct a metabolomic analysis of our mutants with the same method used for mapping LKRSDH1 also allowed us to rapidly identify metabolic phenotypes caused by this mutation. Our efforts revealed that LKRSDH mutants appear to compensate for loss of LKRSDH activity by utilizing the poorly understood pipecolic acid pathway, which is also active in human patients with familial hyperlysinemia (Markovitz et al. 1984). Furthermore, the elevated levels of 5Apt in LKRSDH mutants indicate that either flies are capable of synthesizing 5Apt via an unknown metabolic pathway or that lysine levels are controlled, in part, by metabolic cross talk between somatic tissues and the fly microbiome. This latter possibility is supported by a recent study that demonstrated that the fly bacterial community is intimately associated with host amino acid metabolism (Leitao-Goncalves et al. 2017). Overall, our findings demonstrate that both flies and humans use similar metabolic mechanisms to catabolize lysine, thereby establishing Drosophila as a model for both investigating mechanisms of pipecolic acid synthesis and studying potential cross talk between this poorly understood pathway, bacterial metabolism, and LKRSDH.
While our analysis provides an initial metabolic characterization of LKRSDH, lysine was previously shown to regulate larval feeding behavior, and LKRSDH is also known to moonlight as a regulator of ecdysone signaling (Cakouros et al. 2008; Bjordal et al. 2014). Yet, despite these roles for lysine metabolism in development and physiology, LKRSDH mutations were never isolated in genetic screens for visible phenotypes. The fact that LKRSDH was overlooked by genetic studies is consistent with the fact that Drosophila development can tolerate severe metabolic disruptions. For example, Mitochondrial Pyruvate Carrier 1 mutants are viable despite being unable to transport pyruvate into their mitochondria (Bricker et al. 2012), the oxidative branch of the pentose phosphate pathway is dispensable under standard culture conditions (Hughes and Lucchesi 1977), and Malate Dehydrogenase mutants, which lack a functional citric acid cycle, grow at a normal rate during larval development (Wang et al. 2010). If major disruptions of intermediate metabolism fail to elicit easily recognizable phenotypes, then subtle metabolic regulators will be nearly impossible to identify and study based on morphological or behavioral defects. In contrast, a metabolomic approach that uses metabolites as a primary phenotypic readout would quickly identify any of the mutants described above and provide a rapid and reliable means to characterize their metabolic functions.
The power of using metabolomics in forward genetics studies was recently demonstrated by an analysis of the Saccharomyces cerevisiae gene knockout collection (Mulleder et al. 2016). This study revealed that one-third of coding genes influence the concentration of at least one amino acid, and of the ∼1000 unstudied genes affecting amino acid metabolism, 440 have human homologs, thereby revealing our surprising lack of knowledge regarding the regulation of eukaryotic intermediate metabolism. Previous metabolomic studies in flies suggest that the regulation of animal metabolism is similarly complex (Cox et al. 2017), indicating that a large-scale metabolomic analysis of the available Drosophila mutant and RNA interference collections has the potential to dramatically expand our understanding of eukaryotic metabolism.
Finally, our study highlights the work of E. H. Grell, who discovered the lys1 mutation as the result of a series of serendipitous observations (Grell 1958). At the time, the lys1 phenotype could be scored only by chromatography or based on its ability to enhance the phenotype of red cell mutations. The fact that Grell identified and mapped lys1 is a testament to the tenacity of Drosophila geneticists. In addition, Grell conducted a series of unpublished experiments suggesting that lys1 mutants are able to catabolize both pipecolic acid and 2Aad (Grell 1958). While unknown at the time, this observation pinpointed LKRSDH as the origin of the lys1 phenotype (see Figure 5). Fifty years later, our analysis of lys1 both provides closure to Grell’s observations and demonstrates how emerging metabolomic technologies can be used in forward genetic studies.
Supplementary Material
Supplemental material is available online at www.genetics.org/lookup/suppl/doi:10.1534/genetics.117.300201/-/DC1.
Acknowledgments
We thank the Bloomington Drosophila Stock Center (BDSC) for providing fly strains, the Indiana University Mass Spectrometry Facility for technical assistance, and J. Cox at the University of Utah Metabolomics Core Facility for conducting the targeted metabolomics analysis. We also thank L. Golden, K. Cook, and other members of the BDSC for helpful discussions and reagents. Metabolomics analysis was performed at the Metabolomics Core Facility at the University of Utah, which is supported by National Institute of Health grants 1S10 OD-016232-01, 1S10 OD-021505-01, and 1U54 DK-110858-01. S.L.S.C. was supported by a Victoria Finnerty Undergraduate Travel Award from the Genetics Society of America. J.M.T. is supported by a National Institutes of Health R35 Maximizing Investigators’ Research Award (1R35 GM-119557) from the National Institute of General Medical Sciences.
Footnotes
Communicating editor: N. Perrimon
Literature Cited
- Bellen H. J., Levis R. W., Liao G., He Y., Carlson J. W., et al. , 2004. The BDGP gene disruption project: single transposon insertions associated with 40% of Drosophila genes. Genetics 167: 761–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjordal M., Arquier N., Kniazeff J., Pin J. P., Leopold P., 2014. Sensing of amino acids in a dopaminergic circuitry promotes rejection of an incomplete diet in Drosophila. Cell 156: 510–521. [DOI] [PubMed] [Google Scholar]
- Bricker D. K., Taylor E. B., Schell J. C., Orsak T., Boutron A., et al. , 2012. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 337: 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broquist H. P., 1991. Lysine-pipecolic acid metabolic relationships in microbes and mammals. Annu. Rev. Nutr. 11: 435–448. [DOI] [PubMed] [Google Scholar]
- Cakouros D., Mills K., Denton D., Paterson A., Daish T., et al. , 2008. dLKR/SDH regulates hormone-mediated histone arginine methylation and transcription of cell death genes. J. Cell Biol. 182: 481–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook R. K., Christensen S. J., Deal J. A., Coburn R. A., Deal M. E., et al. , 2012. The generation of chromosomal deletions to provide extensive coverage and subdivision of the Drosophila melanogaster genome. Genome Biol. 13: R21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J. E., Thummel C. S., Tennessen J. M., 2017. Metabolomic studies in Drosophila. Genetics 206: 1169–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doane W. W., 1960. Developmental physiology of the mutant female sterile(2)adipose of Drosophila melanogaster. I. Adult morphology, longevity, egg production, and egg lethality. J. Exp. Zool. 145: 1–21. [DOI] [PubMed] [Google Scholar]
- Fothergill J. C., Guest J. R., 1977. Catabolism of L-lysine by Pseudomonas aeruginosa. J. Gen. Microbiol. 99: 139–155. [DOI] [PubMed] [Google Scholar]
- Grell E. H., 1958. Genetics and biochemistry of “red cell”, pp. 58 in Drosophila melanogaster. California Institute of Technology, Pasadena, CA. [Google Scholar]
- Grell E. H., 1961. The genetics and biochemistry of red fat cells in Drosophila melanogaster. Genetics 46: 925–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hader T., Muller S., Aguilera M., Eulenberg K. G., Steuernagel A., et al. , 2003. Control of triglyceride storage by a WD40/TPR-domain protein. EMBO Rep. 4: 511–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes M. B., Lucchesi J. C., 1977. Genetic rescue of a lethal “null” activity allele of 6-phosphogluconate dehydrogenase in Drosophila melanogaster. Science 196: 1114–1115. [DOI] [PubMed] [Google Scholar]
- Leitao-Goncalves R., Carvalho-Santos Z., Francisco A. P., Fioreze G. T., Anjos M., et al. , 2017. Commensal bacteria and essential amino acids control food choice behavior and reproduction. PLoS Biol. 15: e2000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley D., Zimm G., 1992. The Genome of Drosophila melanogaster. Academic Press, Inc., San Diego, CA. [Google Scholar]
- Markovitz P. J., Chuang D. T., Cox R. P., 1984. Familial hyperlysinemias. Purification and characterization of the bifunctional aminoadipic semialdehyde synthase with lysine-ketoglutarate reductase and saccharopine dehydrogenase activities. J. Biol. Chem. 259: 11643–11646. [PubMed] [Google Scholar]
- Miller-Fleming L., Olin-Sandoval V., Campbell K., Ralser M., 2015. Remaining mysteries of molecular biology: the role of polyamines in the cell. J. Mol. Biol. 427: 3389–3406. [DOI] [PubMed] [Google Scholar]
- Mulleder M., Calvani E., Alam M. T., Wang R. K., Eckerstorfer F., et al. , 2016. Functional metabolomics describes the yeast biosynthetic regulome. Cell 167: 553–565.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pospisilik J. A., Schramek D., Schnidar H., Cronin S. J., Nehme N. T., et al. , 2010. Drosophila genome-wide obesity screen reveals hedgehog as a determinant of brown vs. white adipose cell fate. Cell 140: 148–160. [DOI] [PubMed] [Google Scholar]
- Reis T., Van Gilst M. R., Hariharan I. K., 2010. A buoyancy-based screen of Drosophila larvae for fat-storage mutants reveals a role for Sir2 in coupling fat storage to nutrient availability. PLoS Genet. 6: e1001206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W., Cheng D., Hong S., Sappe B., Hu Y., et al. , 2017. Midgut-derived activin regulates glucagon-like action in the fat body and glycemic control. Cell Metab. 25: 386–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struys E. A., Jakobs C., 2010. Metabolism of lysine in alpha-aminoadipic semialdehyde dehydrogenase-deficient fibroblasts: evidence for an alternative pathway of pipecolic acid formation. FEBS Lett. 584: 181–186. [DOI] [PubMed] [Google Scholar]
- Suh J. M., Zeve D., McKay R., Seo J., Salo Z., et al. , 2007. Adipose is a conserved dosage-sensitive antiobesity gene. Cell Metab. 6: 195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan W., Ashburner M., Hawley R., 2000. Laboratory Culture of Drosophila pp. 589–590 in Drosophila Protocols. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Teague B. D., Clark A. G., Doane W. W., 1986. Developmental analysis of lipids from wild-type and adipose60 mutants of Drosophila melanogaster. J. Exp. Zool. 240: 95–104. [DOI] [PubMed] [Google Scholar]
- Tennessen J. M., Barry W. E., Cox J., Thummel C. S., 2014. Methods for studying metabolism in Drosophila. Methods 68: 105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugrankar R., Berglund E., Akdemir F., Tran C., Kim M. S., et al. , 2015. Drosophila glucome screening identifies Ck1alpha as a regulator of mammalian glucose metabolism. Nat. Commun. 6: 7102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Lam G., Thummel C. S., 2010. Med24 and Mdh2 are required for Drosophila larval salivary gland cell death. Dev. Dyn. 239: 954–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J., Wishart D. S., 2016. Using MetaboAnalyst 3.0 for comprehensive metabolomics data analysis. Curr. Protoc. Bioinformatics 55: 14.10.1–14.10.91. [DOI] [PubMed] [Google Scholar]
- Xia J., Sinelnikov I. V., Han B., Wishart D. S., 2015. MetaboAnalyst 3.0–making metabolomics more meaningful. Nucleic Acids Res. 43: W251–W257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All strains and reagents are available upon request. Table S1 and Table S2 contain the targeted GC-MS metabolomic data, which has been normalized to both the sample mass and the succinic-d4 acid internal standard.