

Abstract
FurE, a member of the Nucleobase Cation Symporter 1 transporter family in Aspergillus nidulans, is specific for allantoin, uric acid (UA), uracil, and related analogs. Herein, we show that C- or N-terminally-truncated FurE transporters (FurE-ΔC or FurE-ΔΝ) present increased protein stability, but also an inability for UA transport. To better understand the role of cytoplasmic terminal regions, we characterized genetic suppressors that restore FurE-ΔC-mediated UA transport. Suppressors map in the periphery of the substrate-binding site [Thr133 in transmembrane segment (TMS)3 and Val343 in TMS8], an outward-facing gate (Ser296 in TMS7, Ile371 in TMS9, and Tyr392 and Leu394 in TMS10), or in flexible loops (Asp26 in LN, Gly222 in L5, and Asn308 in L7). Selected suppressors were also shown to restore the wild-type specificity of FurE-ΔΝ, suggesting that both C- and/or N-terminal domains are involved in intramolecular dynamics critical for substrate selection. A direct, substrate-sensitive interaction of C- and/or N-terminal domains was supported by bimolecular fluorescence complementation assays. To our knowledge, this is the first case where not only the function, but also the specificity, of a eukaryotic transporter is regulated by its terminal cytoplasmic regions.
Keywords: nucleobase, purine, allantoin, Mhp1, genetics, gating
Years of research have led to a model where solute transmembrane transporters operate by an alternating access mechanism that exhibits at least two structurally distinct conformations, outward (extracellular)-facing and inward (cytoplasmic)-facing, the alternation of which is promoted by substrate binding and release (Forrest et al. 2011; Kaback et al. 2011). More recent genetic, functional, and structural evidence has supported the idea that during transport catalysis, transporters acquire multiple distinct conformations, a distinction based not only on whether the binding site is facing the extracellular or cytoplasmic side of the cell, but also on whether specific domains of the protein allow or occlude substrate access to, or release from, the binding site. These distinct domains seem to function as gates, gating elements, or selectivity filters (Diallinas 2008, 2016). Thus, current models consider that during substrate translocation, transporters obtain at least four structurally distinct sequential conformations: an outward-facing “open” conformation that provides access to substrates; an outward-facing “closed” conformation, where the substrate is bound in the major substrate-binding site while a domain (e.g., an external gate) moves to occlude further access of substrates from the extracellular side or leakage of the substrate from the binding site; an inward-facing closed conformation, where the substrate is still bound in the major substrate-binding site while a distinct domain (e.g., an internal gate) occludes substrate release into the cytoplasm; and an inward-facing open conformer, where the internal gate is displaced to allow the release of the substrate into the cytoplasm (Shi 2013; Diallinas 2014, 2016; Penmatsa and Gouaux 2014; Colas et al. 2016; Quistgaard et al. 2016).
Interestingly, genetic, functional, and structural approaches have shown that mutations altering the specificity of transporters are preferably located at gating elements or selectivity filters, rather than within the major binding site. Such gating or selectivity elements can be located at the periphery of the major binding site, but also at flexible transmembraneα-helices or hydrophilic loops that act as dynamic hinges (Papageorgiou et al. 2008; Weyand et al. 2008; Kosti et al. 2010; Shimamura et al. 2010; Adelman et al. 2011; Kazmier et al. 2014; Simmons et al. 2014; Alguel et al. 2016a; Diallinas 2016). The independent action of gating or selectivity elements has been further supported by the fact that specific mutations in these elements often do not alter the transport kinetics for physiological substrates, and may lead to additive or synthetic transport activities and specificities when combined with substrate-binding mutations (Papageorgiou et al. 2008; Kosti et al. 2010; Alguel et al. 2016a). Overall, these findings strongly suggest that transporter specificity might be determined through molecular and functional synergy of multiple domains (Diallinas 2014, 2016). Recently, homo-oligomerization was also found to be important for transporter specificity (Alguel et al. 2016a,b; Diallinas 2016).
In this work, we present genetic and biochemical evidence supporting the idea that transporter specificity in the NCS1 (Nucleobase Cation Symporter 1) transporter family (Pantazopoulou and Diallinas 2007; Weyand et al. 2008; Krypotou et al. 2012, 2015; Sioupouli et al. 2017) is determined via dynamic intramolecular interactions of multiple domains. More specifically, we show that cytoplasmic N- and C-terminal segments of FurE, an allantoin–uric acid–uracil transporter of the fungus Aspergillus nidulans (Krypotou et al. 2015), interact with each other and that this interaction is critical for substrate specificity. Our results are discussed in the context of available knowledge from crystallographic and in silico approaches to study NSC1 transporters.
Materials and Methods
Media, strains, and growth conditions
Standard complete (CM) and minimal media (MM) for A. nidulans growth were used. Media and supplemented auxotrophies were used at the concentrations given in the Fungal Genetics Stock Centre (FGSC; http://www.fgsc.net). Glucose 1% (w/v) or Fructose 0.1% (w/v) was used as carbon sources. For nitrogen sources, 10 mM ammonium tartrate (NH4) or sodium nitrate (NO3) was used. Nucleobases and analogs were used at the following final concentrations: 5-fluorouracil (5FU) 100 μΜ, 5-fluorocytosine (5FC) 50 μΜ, 5-fluorouridine (5FUd) 10 μΜ, uric acid (UA) 0.5 mM, xanthine (XAN) 1mM, uracil (URA) 1mM and allantoin (ALL) 1mM. For induction of genes under the alcAp promoter, 50 mM ethanol was also added to the MM. All media and chemical reagents were obtained from Sigma [Sigma Chemical] (St. Louis, MO) or AppliChem (Bioline Scientific SA, Hellas). A ΔuapA ΔuapC::AfpyrG ΔazgA pabaA1 argB2 mutant (Pantazopoulou et al. 2007) was the recipient strain in transformations with plasmids carrying the furE and yfp fusions. Selection was based on complementation of arginine auxotrophy argB2 and p-aminobenzoic acid auxotrophy pabaA1. A ΔfurD::riboB ΔfurA::riboB ΔfcyB::argB ΔazgA ΔuapA ΔuapC::AfpyrG ΔcntA::riboB pabaA1 pantoB100 mutant strain, named Δ7, was the recipient strain in transformations with plasmids carrying fur genes or alleles based on complementation of the pantothenic acid auxotrophy pantoB100 (Krypotou and Diallinas 2014). Derivatives of mutant strains were obtained through genetic crosses based on auxotrophic markers for heterokaryon establishment and, if needed, identified by relevant PCR. A. nidulans protoplast isolation and transformation was performed as previously described (Koukaki et al. 2003). Growth tests were performed at 37° for 48 hr, at pH 6.8.
Standard molecular biology manipulations and plasmid construction
Genomic DNA extraction from A. nidulans was performed as described in FGSC. Plasmids, prepared in Escherichia coli, and DNA restriction or PCR fragments were purified from agarose 1% gels with a Nucleospin Plasmid Kit or Nucleospin ExtractII kit, according to the manufacturer’s instructions (Macherey-Nagel, Lab Supplies Scientific SA, Hellas). Standard PCR reactions were performed using KAPATaq DNA polymerase (Kapa Biosystems). PCR products used for cloning, sequencing, and reintroduction by transformation in A. nidulans were amplified by a high-fidelity KAPA HiFi HotStart Ready Mix (Kapa Biosystems) polymerase. DNA sequences were determined by VBC-Genomics (Vienna, Austria). Site-directed mutagenesis was carried out according to the instructions accompanying the Quik-Change Site-Directed Mutagenesis Kit (Agilent Technologies, Stratagene, La Jolla, CA). The principal vector used for most A. nidulans mutants is a modified pGEM-T-easy vector carrying a version of the gpdA promoter, the trpC 3′ termination region, and the panB selection marker (Krypotou et al. 2015). For bimolecular fluorescence complementation (BiFC) analyses, the N-terminal half of yellow fluorescent protein (YFPn; 154 amino acids of YFP), or the C-terminal half of YFP (YFPc; 86 amino acids of YFP) was amplified from plasmids PDV7 and PDV8 (Takeshita et al. 2008) and cloned into pAN510exp-alcAp or pAN520exp-alcAp (Martzoukou et al. 2015), followed by cloning of the furE ORF. Mutations and segment truncations in Fur transporters were constructed by oligonucleotide-directed mutagenesis or appropriate forward and reverse primers (Supplemental Material, Table S1 in File S1). Transformants arising from single-copy integration events with intact Fur ORFs were identified by Southern and PCR analysis.
Protein extraction and western blotting
Cultures for membrane protein extraction were grown in MM supplemented with NaNO3 at 25° for 14 hr prior to the addition of NH4+ or substrate. Membrane protein extraction was performed as previously described (Evangelinos et al. 2016). Equal sample loading was estimated by Bradford assays. Total proteins (30–50 μg) were separated by SDS-PAGE (8% w/v polyacrylamide gel) and electroblotted (Mini Protean Tetra Cell, Bio-Rad, Hercules, CA) onto PVDF membranes (Macherey-Nagel). Immunodetection was performed with a primary mouse anti-GFP monoclonal antibody (Roche Diagnostics), or a mouse anti-actin monoclonal (C4) antibody (MP Biomedicals Europe) and a secondary goat anti-mouse IgG HRP-linked antibody (Cell Signaling Technology). Blots were developed by the chemiluminescent method using the LumiSensor Chemiluminescent HRP Substrate kit (Genscript USA) and SuperRX Fuji medical X-Ray films (FujiFILM Europe).
Uptake assays
Kinetic analysis of Fur transporter activity was measured by estimating uptake rates of [3H]-uracil (40 Ci mmol−1, Moravek Biochemicals, CA), as previously described in Krypotou and Diallinas (2014). In brief, [3H]-uracil uptake was assayed in A. nidulans conidiospores germinating for 4 hr at 37°, at 140 rpm, in liquid MM (pH 6.8). Initial velocities were measured on 107 conidiospores/100 μl by incubation with concentrations of 0.2–2.0 μΜ of [3H]-uracil at 37°. The time of incubation was defined through time course experiments and the period of time when each transporter showed linear increased activity was chosen, respectively. Km/i values were obtained using radiolabeled uracil at 0.2–0.5 μM in the presence of various concentrations (0.5–2000 μM) of nonlabeled substrates. Ki values were calculated by satisfying the criteria for use of the Cheng and Prusoff equation, Ki = IC50 / [1 + (L / Km)], in which L is the permeant concentration. In our assays, Ki values equal IC50 values as the L is very low (at least 10-fold lower than the Km value). Background counts are subtracted from the values obtained in strains expressing the relevant transporter. Km, Ki, and Vmax determination is carried out using standard Lineweaver–Burk or Eadie–Hofstee equation formulae, or relevant software (e.g., GraphPad Prism). All transport assays were carried out in triplicate. SD was < 20%.
Isolation and characterization of suppressor mutations
Mutagenesis was performed at a standard distance of 20 cm from an Osram HNS30 UV-B/C lamp. Suppressor mutations were obtained after 3 min 45 sec UV mutagenesis of 109 conidiospores of the strain FurE-ΔC and subsequent selection of colonies capable of growing on MM containing UA as a sole nitrogen source, at 25°. Spores from positive colonies were collected after 6–8 days and isolated colonies were obtained by standard purification on the same selective medium that was used to obtain the original colonies. Genomic DNA from 45 purified colonies was isolated and the ORF of FurE-ΔC was amplified and sequenced. In all cases, the amplified fragments contained a new mutation (Table 1). Strains carrying suppressor mutations were outcrossed with the original nonmutagenized strain to show that suppressor phenotypes cosegregate with sequenced FurE mutations.
Table 1. Profile of the mutations suppressing the inability of FurE-ΔC to grow on uric acid. The relevant nucleobase substitution is underlined.
| Mutation | Codon Change | Number of Isolates | Location | Putative Domain |
|---|---|---|---|---|
| D26N | GAC > AAC | 1 | N-tail | Flex loop |
| T133V | ACG > GTG | 4 | TMS3 | Binding site filter |
| G222K | GGG > AAG | 1 | L5 | Flex loop |
| GGG > AAG | 4 | |||
| N308T | AAC > ACC | 7 | L7 | Flex loop |
| AAC > ACA | 1 | |||
| AAC > ACT | 1 | |||
| S296R | AGC >CGC | 4 | TMS7 | Gate |
| V343I | GTC > ATC | 1 | TMS8 | Binding site filter |
| I371L | ATC > CTC | 1 | TMS9 | Gate |
| I371F | ATC > TTC | 3 | TMS9 | Gate |
| I371P | ATC > CCC | 1 | TMS9 | Gate |
| Y392N | TAC > AAC | 6 | TMS10 | Gate |
| Y392C | TAC > TGC | 5 | TMS10 | Gate |
| Y392E | TAC > GAA | 1 | TMS10 | Gate |
| L394P | CTG > CCG | 4 | TMS10 | Gate |
TMS, transmembrane segment.
Epifluorescence microscopy
Samples for inverted epifluorescence microscopy were prepared as previously described (Valdez-Taubas et al. 2004; Gournas et al. 2010; Karachaliou et al. 2013). In brief, sterile 35-mm 1-liter dishes with glass bottoms (Ibidi, Germany), containing liquid minimal media supplemented with NaNO3 and 0.1% glucose or fructose, were inoculated from a spore solution and incubated for 16–22 hr at 25°. The samples were observed on an Axioplan Zeiss ([Carl Zeiss], Thornwood, NY) phase contrast epifluorescent microscope and the resulting images were acquired with a Zeiss-MRC5 digital camera using AxioVs40 V4.40.0 software. FM4-64 terminal vacuolar staining was as described in Evangelinos et al. (2016). Image processing, contrast adjustment, and color combining were made using Adobe Photoshop CS3 software or ImageJ software. To quantify transporter endocytosis, Vacuolar Surface (Total surface of vacuoles containing GFP/hypha) and Vacuolar GFP Fluorescence (Total fluorescence intensity of vacuoles containing GFP/hypha) were measured using the Area Selection Tool of the ICY application (Martzoukou et al. 2017). Tukey’s Multiple Comparison Test (one-way ANOVA) was performed to test the statistical significance of the results for five Regions of Interest using Graphpad Prism3. Images were further processed and annotated in Adobe Photoshop CS3 (Martzoukou et al. 2017).
Data availability
Strains constructed in this work are available upon request.
Results
C-terminally-truncated Fur transporters show modified apparent substrate specificities
Members of the NCS1 transporter family are present in prokaryotes, fungi, and some plants. Fungal, and especially A. nidulans, NCS1 transporters have been extensively studied in respect to their function, specificity, regulation of expression, and evolution, and are classified into two distinct subfamilies: the Fcy- and the Fur-like transporters (de Koning and Diallinas 2000; Pantazopoulou and Diallinas 2007; Krypotou et al. 2015). A. nidulans possesses seven Fur transporters (FurA–G): two function as major uracil (FurD) or allantoin (FurA) transporters (Amillis et al. 2007; Hamari et al. 2009); one is a secondary, low-affinity, promiscuous transporter specific for uracil, allantoin, UA, and related analogs (FurE), and the rest function as cryptic, very low-efficiency uracil transporters (Krypotou et al. 2015).
Using functional GFP-tagged Fur versions, we have previously shown that Fur transporters are differentially sensitive to endocytic turnover in response to two well-characterized endocytic triggers (Krypotou et al. 2015), those elicited by either an excess of substrate or the presence of a primary nitrogen source (Gournas et al. 2010). In particular, FurE is highly sensitive to endocytosis, showing a degree of internalization and turnover even in the absence of endocytic signals (e.g., constitutive endocytosis), FurD is mostly sensitive to ammonium-elicited and less so to substrate-elicited internalization, and FurA is sensitive solely to ammonium-elicited endocytosis. Some of the other Fur transporters (FurC and FurG) do not respond to any known endocytic trigger. In all cases studied, fungal transporter downregulation via endocytosis necessitates prior Lys63-ubiquitylation by Hect-type ubiquitin ligases (Lauwers et al. 2010; Gournas et al. 2016).
To investigate the basis of the differential response of Fur transporters to endocytosis and turnover, and based on the observation that all Fur proteins possess several Lys residues as candidates for ubiquitylation at their C-terminal cytoplasmic regions (Gournas et al. 2010; Karachaliou et al. 2013), we constructed and functionally analyzed strains expressing truncated versions of FurA, FurD, and FurE transporters, all lacking a C-terminal region (CTR) including the three most terminally located Lys residues (Figure S1 in File S1). We also constructed a C-terminally-truncated version of a specific allele of FurE, FurE-K252F, which increases FurE-mediated transport activity and thus permits better assessment of the wild-type FurE function (Krypotou et al. 2015). All truncated fur gene versions were fused C-terminally with the gfp ORF, while the constitutive gpdAp promoter was used to drive their transcription (see Materials and Methods).
The truncated Fur versions (named FurA-ΔC-GFP, FurD-ΔC-GFP, FurE-ΔC-GFP, and FurE-K252F-ΔC-GFP) were introduced by standard genetic transformation in an A. nidulans strain, called Δ7, lacking all seven major transporters specific for nucleobases, allantoin, or nucleosides, as previously performed for the nontruncated FurA-GFP, FurD-GFP, and FurE-GFP transporters (Krypotou et al. 2015). Selected purified transformants arising from intact single-copy plasmid integration events were further analyzed by growth tests, radiolabeled substrate uptake assays, and epifluorescence microscopy.
Figure 1A shows a growth test of selected transformants and isogenic controls on MM containing UA or allantoin as sole nitrogen sources, or nitrate plus toxic nucleobase analogs known to be transported by the Fur transporters. A wild-type positive control strain grows on UA or allantoin and is sensitive to all nucleobase/nucleoside toxic analogs tested (e.g., 5FU, 5FC, and 5FUd), because it expresses all relevant transporters. A negative control strain, Δ7, lacking all major nucleobase transporters, does not grow on UA or allantoin, and is resistant to all toxic nucleobase/nucleoside analogs tested. It should be noted that the Δ7 strain carries, among other transporter null mutations, total deletions of furA and furD, but not furE gene. However, furE expression driven by its native promoter is extremely low, so that no FurE-dependent growth on UA or allantoin—or no sensitivity to 5FU, 5FC, or 5FUd—is detected. The transport activity of FurE is only observed when FurE is expressed from the strong gpdAp promoter in Δ7 (Krypotou et al. 2015). Isogenic Δ7 strains expressing the nontruncated GFP-tagged Fur versions from the gpdAp promoter confer distinct growth profiles on UA, allantoin, or toxic analogs as follows. Strains expressing FurA grow on allantoin, albeit the relevant colonies are very compact due to overaccumulation of this toxic metabolite and show sensitivity to all toxic nucleobase/nucleoside analogs. Strains expressing FurD grow weakly on UA and are sensitive to 5FU, 5FC, or 5FUd. Strains expressing FurE grow well on allantoin, weakly on UA, and are sensitive to 5FU, 5FC, or 5FUd. Strains expressing the hyperactive FurE-K252F grow equally well on allantoin or UA, and are sensitive to 5FU or 5FC, but not to 5FUd. These growth phenotypes are in agreement with what has been reported before (Krypotou et al. 2015). Strains expressing the corresponding truncated versions of FurD, FurE, or FurE-K252F (i.e., FurD-ΔC, FurE-ΔC, or FurE-K252F-ΔC) showed distinct growth phenotypes when compared to their nontruncated equivalents. The strain expressing the truncated FurD transporter shows stronger growth on UA and allantoin, but is more resistant to 5FC. More impressively, the strain expressing the truncated FurE transporter could not grow at all on UA, despite retaining full capacity to grow on allantoin or being sensitive to 5FU, 5FC, or 5FUd. The strain expressing the truncated FurE-K252F showed reduced capacity to grow on UA, especially at 37°, but was equally sensitive to all toxic analogs tested with the nontruncated version. In contrast, no apparent growth differences could be detected between strains expressing FurA or FurA-ΔC. Thus, deletion of the C-terminal part of at least FurD and FurE, or FurE-K252F, led to a modification of the apparent substrate specificity of these transporters. The molecular rational behind this rather unexpected observation became the major theme of this work.
Figure 1.

Functional characterization of truncated Fur transporters. (A) C-terminally-truncated Fur transporters show modified apparent substrate specificities. Growth test of strains expressing, from gpdAp promoter, nontruncated (WT) and truncated Fur-GFP transporters. The test is performed at 37° on MM media containing as sole nitrogen source nitrate (control), UA, or ALL, or on nitrate media containing a nucleobase toxic analog (5FU, 5FC, or 5FUd). The growth on UA is recorded at both 37 and 25°. WT stands for a standard A. nidulans wild-type strain. Δ7 is a mutant strain carrying total deletions in all seven major transporters for nucleobases, nucleosides, and ALL (uapAΔ uapCΔ azgAΔ furDΔ furAΔ fcyBΔ cntAΔ). The Δ7 strain has an intact endogenous furE gene transporter, but this is very little expressed under standard conditions and thus does not contribute to detectable transport of its potential substrates (UA, URA, or ALL). All other strains are single-copy isogenic transformants of Δ7 expressing the indicated nontruncated or truncated Fur transporter. K252F is a missense mutation increasing the activity of FurE (Krypotou et al. 2015). (B) C-terminally-truncated Fur transporters do not undergo endocytosis. Subcellular localization of nontruncated (WT) and truncated GFP-tagged Fur transporters, expressed from the gpdAp promoter, as analyzed by in vivo inverted epifluorescence microscopy. Samples are grown for 18 hr at 25° under control (MM with nitrate as N source), or substrate-elicited (ALL or URA) or ammonium-elicited endocytic conditions (MM with nitrate as N source plus addition of substrate or ammonium for the last 2 hr of the culture). Endocytic turnover is identified by the appearance of cytoplasmic structures corresponding to endosomes and vacuoles and the progressive diminution of the peripheral fluorescent signal (Gournas et al. 2010; Krypotou et al. 2015). Notice that different Fur transporters have distinct sensitivities to endocytosis (FurE > FurD, FurA), but truncation of the C-terminus stabilizes the Fur-GFP chimeras in all cases. (C) C-terminally-truncated Fur transporters show dramatically reduced turnover. Western blot analysis of total protein extracts of strains expressing WT and truncated Fur-GFP versions, using anti-GFP (upper panel) or anti-actin (control, lower panel) antibody. Growth conditions are as in (B). Free GFP levels reflect vacuolar degradation of Fur-GFP proteins. (D and E) C-terminally-truncated Fur transporters show modified transport kinetics. (D) Time course of [3H]-URA uptake by truncated (FurD-ΔC and FurE-ΔC) and nontruncated (FurD and FurE) transporters. SD is depicted with error bars. (E) Ki/m values (micromolar) for truncated and nontruncated FurD and FurE transporters determined using [3H]-URA uptake competition. Results are averages of three measurements for each concentration point. SD was < 20%. 5FC, 5-fluorocytosine; 5FU, 5-fluorouracil; 5FUd, 5-fluorouridine; ALL, allantoin; MM, minimal media; n.m., nonmeasurable; URA, uracil; UA, uric acid; WT, wild-type.
C-terminally-truncated Fur transporters show increased protein stability and modified transport kinetics
To investigate the effects of C-terminal truncation in different Fur transporters, we followed the subcellular localization and turnover of the truncated versions by in vivo epifluorescence microscopic and western blot analysis. First, we tested both the rate of constitutive endocytosis and that triggered by either an excess of substrate or the addition of a primary nitrogen source (e.g., ammonium). Figure 1B shows that, in all cases, C-terminal truncation of the Fur proteins stabilizes the Fur transporters in the plasma membrane. As a consequence, very little, if any, fluorescence is associated within vacuoles, unlike what is observed for the nontruncated Fur versions. Vacuolar GFP fluorescence is a standard measure to detect endocytosis of GFP-tagged transporters in fungi and mammalian cells, as transporters, following internalization from the plasma membrane, are sorted into vacuoles via the endosomal/multivesicular bodies (MVB) pathway. Once in the vacuolar lumen, GFP remains rather stable for a sufficient period of time, which permits quantification of fluorescence (Gournas et al. 2010). Figure S2 in File S1 depicts the colocalization of the GFP fluorescent signal, coming from degradation of the Fur-GFP chimaeras, with the endosome/vacuole-specific FM4-64 molecular stain. Quantification and statistical analyses of the surface and intensity of vacuolar GFP fluorescence confirmed the essential role of the CTR in Fur endocytosis (Figure S3 in File S1). Western blot analysis (Figure 1C) confirmed that the steady-state levels of truncated Fur proteins are similar and remain high in all conditions tested, including conditions triggering endocytosis. This contrasts with the picture of the nontruncated Fur proteins, where the relevant steady-state levels show a dramatic reduction in the presence of ammonium and a significant drop in the presence of substrate (allantoin or uracil), when compared to nonendocytic conditions (i.e., NO3−). Notice also that in the strains expressing the truncated versions free GFP polypeptide levels, which arise from the turnover of Fur-GFP, remain low, whereas in strains expressing the nontruncated Fur proteins free GFP polypeptide levels are significantly higher (Figure 1C). In general, the western blot analysis is in perfect line with the results observed from in vivo epifluorescence microscopy.
The increased stability of the truncated versions could, in principle, explain the observation that FurD-ΔC grows better on UA or allantoin compared to the nontruncated FurD, but could not explain the increased resistance to 5FC of the former vs. the latter. More importantly, the increased stability of the truncated FurE-ΔC contrasted with its specific inability to transport UA. Thus, the apparent changes in the specificity of FurD and FurE could not be rationalized on a quantitative basis of increased protein levels.
We also measured directly the transport activity of the truncated vs. the nontruncated versions of FurD and FurE by standard uptake assays using radiolabeled uracil (Krypotou and Diallinas 2014). FurA activity cannot be assayed as there is no commercially available radiolabeled allantoin. Figure 1D shows that truncated FurD and FurE had similar initial uptake rates (see values at 1 min uptake), albeit 40–50% reduced steady-state accumulation of uracil compared to the nontruncated equivalents. This suggests that the absence of the C-tail has a moderate, but detectable, reduction in the steady-state uptake capacity of Fur transporters. We do not have an explanation for this finding, but it seems that stabilization of truncated versions might lead to their overaccumulation in the plasma membrane, and that this in turn might have a negative effect on transport activity.
To further dissect the role of the CTR of FurD and FurE in transport, we determined the Km or Ki values of FurD-ΔC and FurE-K252F-ΔC for uracil, UA, or allantoin, and compared them with those of the nontruncated proteins (the low transport activity of FurE-ΔC does not allow rigorous kinetic analysis). Figure 1E shows that truncation of FurD reduces the affinity for uracil (4.6-fold) and UA (> 10-fold), but seemingly does not affect the very low affinity for allantoin. Truncation of FurE-K252F did not affect the affinity for uracil and UA, but led to a moderate 2.5-fold reduction in allantoin binding. Overall, these results show that truncation of the C-tail of Fur transporters not only increases their stability in the plasma membrane (PM), but also leads to differential modification of the relevant transport kinetics, in addition to changes in specificity.
Genetic suppressors of the C-terminal truncation restore substrate specificity in FurE
To better understand how the CTR might affect FurE specificity, we selected genetic suppressors of the C-terminal truncation that restore substrate specificity in FurE by direct selection of revertants on media containing UA as the sole nitrogen source, after standard UV mutagenesis of the strain expressing FurE-ΔC (see Materials and Methods). We obtained > 50 revertants able to grow on UA, and the furE-ΔC ORF of 45 of them was amplified by PCR and sequenced. In nearly all cases, we detected a single-codon change, while in a single case two nearby codons were modified. Table 1 summarizes the profile of the suppressor mutations obtained. Selected strains carrying suppressors corresponding to different mutations were outcrossed with the original nonmutagenized strain, and the progeny analyzed showed that the suppressor phenotype cosegregated with the sequenced FurE mutation. Overall, suppressor mutations mapped in several transmembrane segments (TMS) 3, 7, 8, 9 and 10, but also in external loops (L5 or L7), and in the N-terminal protein region. Growth tests of relevant mutant strains showed that they are all able to grow on UA and allantoin, and that all are sensitive to relevant toxic nucleobase analogs (Figure 2A, upper panel). Importantly, T133V, and to a lesser degree some of the other mutants (e.g., Y392N, Y392E, V343I, or L349P), could also grow moderately on xanthine, which is not a physiological substrate of either FurE or FurE-ΔC. Νone of the mutants acquired the capacity to grow on other purines that are not substrates of Fur transporters, such as adenine, hypoxanthine, or guanine (data not shown). Thus, all suppressor mutations restored the ability of FurE-ΔC to transport UA, but additionally, some specific mutations enlarged the FurE or FurE-ΔC specificity profile to include xanthine. The suppressor mutations did not significantly affect the stable localization of FurE-ΔC in the plasma membrane (Figure 2A, lower panel), despite some retention in the ER or very moderately increased turnover in specific cases, suggesting that the suppressor mutations do not affect the folding of the transporters.
Figure 2.

Functional characterization of FurE-ΔC suppressors. (A) Upper panel: growth tests of FurE-ΔC suppressors. FurE-ΔC suppressors and control strains (WT, Δ7, FurE, and FurE-ΔC) were grown in MM containing nitrate (control), UA, ALL, or XAN as N sources, or on nitrate media containing 5FU or 5FC. All growth tests shown were performed at 37°. Lower panel: subcellular localization of FurE-ΔC suppressors. FurE-ΔC suppressors and control strains were analyzed by in vivo epifluorescence microscopy. Notice that some suppressor mutations lead to partial retention of FurE-ΔC in internal structures resembling the ER (open rings), or to moderate sorting in the vacuole, but still the majority of FurE is sorted in the PM. Cultures are grown in MM containing nitrate as nitrogen source for 18 hr at 25°. (B) Transport kinetics of FurE-ΔC suppressors. Comparative [3H]-URA accumulation (4 min) in strains expressing FurE-ΔC suppressor mutations and control strains (FurE and FurE-ΔC). SD is depicted with error bars. (C) Steady-state protein levels of selected FurE-ΔC suppressors showing increased apparent transport activity. Western blot analysis of total protein extracts of strains expressing truncated FurE-ΔC (the same as that shown in Figure 1), FurE-ΔC-T133V, or FurE-ΔC-Y392N, using anti-GFP (upper panel) or anti-actin (control, lower panel) antibody. Growth conditions are as for microscopy. Free GFP levels reflect vacuolar degradation of the intact Fur-GFP proteins. (D) Transport kinetics of FurE-T133V and FurE-T133V-ΔC. Dose response curve of [3H]-URA uptake by FurE-T133V or FurE-T133V-ΔC in the presence of increasing concentration of nonradiolabeled URA or UA, respectively. The IC50 (equal to Ki/m) values measured are depicted as inserts (for details see Materials and Methods). Results are averages of three measurements for each concentration point. SD was < 20%. 5FC, 5-fluorocytosine; 5FU, 5-fluorouracil; ALL, allantoin; MM, minimal media; PM, plasma membrane; URA, uracil; UA, uric acid; WT, wild-type; XAN, xanthine.
Comparative uptake assays further showed that most suppressors have a moderate positive effect on the generally low rate of accumulation of radioactive uracil mediated by FurE or FurE-ΔC (Figure 2B). The most prominent case was mutation T133V, which led to more than a sevenfold increase in uracil uptake, and to lower degree mutations V343I, L349P, I371P, Y392C, Y392N, or Y392E, which led to 3–4.5-fold increases. Western blot analysis of two selected suppressor mutants showing increased transport capacity, namely T133V and Y392N, showed that this is not justified by an analogous increase in FurE protein steady-state levels (Figure 2C). In fact, these mutations slightly increase the turnover of FurE-ΔC (notice a two- to fourfold increase in the accumulation of free GFP). Thus, the results of uptake and western blot analyses together confirm that mutations T133V and Y392N do lead to a significant increase in FurE-ΔC-mediated transport capacity per se.
The generally low uracil uptake rates of most suppressors did not permit a rigorous estimation of affinity constants for FurE substrates, and thus we could not test whether suppressors generally reconferred UA transport by increasing UA binding. However, the Ki for UA could be measured in mutant FurE-T133V-ΔC, which is the only suppressor that showed sufficient transport activity for the performance of rigorous kinetic analysis. Figure 2D shows that FurE-T133V-ΔC has an affinity constant of 32 µM for UA and 57 µM for uracil. These values cannot be directly compared to the original FurE-ΔC, as this showed accumulation of these radiolabeled substrates that was too low to be analyzed kinetically. However, similar affinity constants for UA (29 µM) and uracil (49 µM) have been obtained in a strain expressing a nontruncated version carrying the suppressor mutation FurE-T133V, suggesting that the effect of mutation T133V on transport kinetics is independent of the truncated C-tail segment. Thus, comparing wild-type FurE (Krypotou et al. 2015 and Figure 1D) to FurE-T133V we detect a significant increase (∼20-fold) in the affinity for uracil (Km from 1000 to 49 μM), which could explain the apparent sevenfold increase in uracil accumulation, but practically no change in the affinity for UA (Ki from 20 to 29 μΜ). In other words, the restoration of UA transport in FurE-T133V-ΔC is, in principle, not due to an increase in the binding affinity for UA, but rather to a change related to the dynamics of transport catalysis.
Overall, the functional analysis of FurE suppressors showed that the relevant mutations do not affect the expression, folding, and turnover of FurE, but that they differentially modify the transport kinetics and specificity of the transporter.
N-terminal truncation of FurE leads to growth phenotypes mimicking those of C-terminal truncation
We also constructed and studied an N-terminally-truncated version of the FurE transporter (Figure S4 in File S1). This includes a deletion of the first 21 amino acid residues of the cytoplasmic N-terminal region of FurE (FurE-ΔN). The deleted segment does not show any sequence conservation in members of the NCS1 family and is thus not expected to affect the proper ER-exit and further exocytosis of the transporter. FurE-ΔN led to specific loss of apparent FurE-mediated transport of UA, and to a very moderate increase in 5FC resistance, a phenotype similar to the one obtained with a strain expressing the C-terminally-truncated version of FurE. Similarly to FurE-ΔC, FurE-ΔN did not affect the ability of FurE to transport allantoin or 5FU (Figure 3A). Transport assays further showed that FurE-ΔN confers a detectable rate of uracil accumulation (Figure 3B), confirming that this truncation has not abolished the transport of substrates other than UA or 5FC. Microscopic analysis showed that the FurE-ΔN version is stably localized in the PM and remains integrated in it, even after imposing conditions triggering wild-type FurE endocytosis, exactly as is the case for the C-terminally-truncated FurE version (Figure 3C, upper panel). The observation that truncation of the N-terminal region stabilizes FurE under endocytic conditions was confirmed by western blot analysis (Figure 3D, left panel). Thus, both N- and C-terminal truncations of FurE lead to specific loss of UA transport and a block of endocytosis.
Figure 3.

Functional characterization of the FurE-ΔN truncation. (A) Truncation of the N-terminal region confers growth phenotype mimicking C-terminal truncation. Growth tests of strains expressing FurE-ΔN, selected FurE-ΔC suppressors (T133V, Y392N), versions of FurE-ΔN carrying the selected suppressor mutations (FurE-T133V-ΔN and FurE-Y392N-ΔN), and control strains. Growth conditions and labeling are as in the legend of Figure 1A. (B) Transport kinetics of different versions of FurE-ΔN. Left panel: comparative radiolabeled URA accumulation rates in strains expressing the different versions of FurE and control strains, which were tested in (A and B). Right panel: dose response curve of [3H]-URA uptake in the presence of increasing concentration of nonradiolabeled URA or UA, respectively, in strains expressing FurE-T133V or FurE-T133V-ΔN. The IC50 (equal to Ki/m) values measured are depicted as inserts (for details see Materials and Methods). Results are averages of three measurements for each concentration point. SD was < 20%. (C) Subcellular localization of different versions of FurE-ΔN analyzed by in vivo epifluorescence microscopy. Details are as in Figure 1B. Notice that FurE-ΔN does not undergo endocytosis in response to either ammonium, ALL or URA, whereas FurE-T133V-ΔN and FurE-Y392N-ΔN are normally endocytosed in response to ammonium, but less in response to ALL or URA. Nontruncated versions, FurE-ΔN, FurE-T133V, and FurE-Y392N-ΔN, included as controls, are also normally endocytosed. (D) Steady-state protein levels of different versions of FurE-ΔN. Western blot analysis of total protein extracts of strains expressing FurE, FurE-T133V, or FurE-Y392N, as well as truncated FurE-ΔN, FurE-ΔN-T133V, or FurE-ΔN-Y392N, using anti-GFP (upper panel) or anti-actin (control, lower panel) antibody. Growth conditions are as in (C). Notice that both mutations studied reduce the steady-state levels of FurE, but truncation of the N-terminal region partially counteracts this effect. Free GFP levels reflect vacuolar degradation of the intact Fur-GFP proteins. 5FC, 5-fluorocytosine; 5FU, 5-fluorouracil; ALL, allantoin; MM, minimal media; URA, uracil; UA, uric acid; wt, wild-type; XAN, xanthine.
Given the similarity of phenotypes caused by the N- and C-truncations, we next examined whether selected suppressors of the C-terminal truncation might also suppress the N-terminal truncation phenotypes. The mutations selected, T133V and Y392N, were examined in the context of either FurE-ΔΝ or nontruncated FurE. When these mutations were included in a nontruncated FurE, both conferred growth on allantoin, UA, or xanthine, sensitivity to relevant toxic substrates, and increased FurE endocytosis very slightly, judging from an increased number of fluorescent vacuoles (Figure 3, A and C, middle and lower panels). Most importantly, both suppressors restored growth in UA in strains expressing the truncated FurE-ΔN version, and in addition, T133V also conferred growth on xanthine of a FurE-ΔN strain (Figure 3A). Somewhat surprisingly, mutations T133V and Y392N also increased FurE-ΔN endocytosis, more evident in response to the presence of ammonium (Figure 3C), in contrast to what was observed when these mutations were present in the FurE-ΔC-truncated version (as shown in Figure 2A, lower panel). Relevant western blot analysis (Figure 3D) showed that mutations T133V or Y392N do increase the turnover of the nontruncated protein under all conditions tested, but that the N-terminal truncation partially counteracts this effect, especially when cells grow under nonendocytic conditions (i.e., NO3−).
Overall, our results suggested that both the C- and N-terminally-truncated segments of FurE contain cis-acting elements that are critical for endocytosis. This further suggested that the two cytoplasmic terminal regions might synergize in determining a FurE topology necessary for recruiting the endocytic machinery. This hypothesis gained further support though the independent experiments described in the next section.
The N- and C-terminal cytoplasmic segments of FurE come into close proximity in the absence of substrate
The similarity of phenotypes arising from truncations of either the N-terminal regions or CTRs prompted us to test whether these two domains interact, and whether this interaction is part of a mechanism regulating FurE function and stability. To test this interaction, we employed intramolecular BiFC (see Materials and Methods). Figure 4A shows that expression of YFPn-FurE-YFPc reconstitutes YFP fluorescence at the plasma membrane, whereas expression of each chimera by itself (FurE-YFPc or FurE-YFPn) does not produce any detectable fluorescent signal. Importantly, when the same assay was performed in the presence of substrates (e.g., allantoin) added for increasing time periods, the fluorescent signal was practically lost in a time-dependent manner (Figure 4B). The latter observation suggests that when FurE is actively transporting its substrates, its two terminal cytoplasmic tails do not stably reconstitute split-YFP, unlike what is observed in the absence of transport activity (i.e., the absence of substrates).
Figure 4.
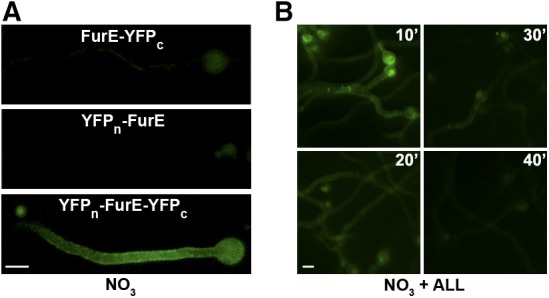
The N- and C-terminal cytoplasmic segments of FurE come into close proximity. (A) Isogenic strains expressing YFPn-FurE-YFPc, FurE-YFPc, or YFPn-FurE were analyzed by in vivo epifluorescence microscopy (for details see Materials and Methods). Notice that that expression of FurE-YFPc or YFPn-FurE does not lead to a detectable fluorescent signal, whereas expression of YFPn-FurE-YFPc leads to clear cortical fluorescence, compatible with reconstitution of the two parts of YFP in the chimeric transporter, apparently expressed in the PM. (B) In the strain expressing YFPn-FurE-YFPc, notice that addition of substrate (ALL), prior to microscopic examination, reduces and eventually turns off the fluorescent signal in a time-dependent manner. ALL, allantoin; PM, plasma membrane; YFP, yellow fluorescent protein.
Of note, strains expressing FurE-YFPn or/and FurE-YFPc grow on allantoin and are sensitive to 5FU, but do not grow on UA (results not shown). This suggests that FurE molecules tagged at their termini with either of the two parts of YFP are generally functional, but have lost their ability to specifically transport UA, similarly to the truncated versions. This observation further supports the idea that modifications of the two cytoplasmic terminal regions of FurE are critical for UA transport and FurE specificity in general.
A doubly N- and C-terminally-truncated version of FurE does not respond to endocytosis and shows wild-type function and specificity
We also constructed and tested a doubly truncated FurE version (FurE-ΔN/ΔC). Surprisingly, a strain expressing FurE-ΔN/ΔC could grow on UA in addition to allantoin, and was sensitive to toxic analogs, an apparent specificity profile that is similar to wild-type FurE (Figure S5A in File S1). Transport assays confirmed that FurE-ΔN/ΔC was capable of uracil accumulation, with an apparent measured rate of uracil accumulation very similar to that of a wild-type nontruncated FurE (data not shown). FurE-ΔN/ΔC was stably localized in the PM and did not respond to endocytosis (Figure S5, B and C in File S1). In other words, simultaneous truncation of both cytoplasmic terminal regions, which when truncated individually lead to functional modification of FurE, seems to restore the wild-type FurE transport function. In the absence of relevant structural data, this result is not easy to explain. However, it is an important observation as it strongly suggests that the basic mechanism of transport in NCS1 transporters does not require the extended N- and C-terminal domains that are uniquely present in eukaryotic homologs, which in the context of the present work further establishes that the acquisition of specific cytoplasmic termini in eukaryotic NCS1 members serves a fine regulatory role in determining transport kinetics and specificity.
Suppressors reveal a critical functional role of distinct gating or selectivity elements in FurE
Fur transporters are homologous to the bacterial Mhp1 benzyl-hydantoin transporter, which also belongs to the NCS1 family. We have previously constructed and validated structural models of FurA, FurD, and FurE based on the crystal structure of Mhp1. Validation included substrate docking, molecular dynamics, and an analysis of mutations that alter the function or specificity of Fur transporters. This work has identified the putative binding site of relative substrates and revealed the importance of TMS10, acting as an external gate critical for the high specificity of FurD (Krypotou et al. 2015). Herein, we mapped all characterized residues concerning suppressor mutations in the 3D-modeled structure of FurE (Figure 5). Based on this, we could classify suppressors as one of three types. Type I suppressors concern amino acids proximal to substrate-binding residues, as these were defined genetically, functionally, and by in silico docking (Krypotou et al. 2015). These are suppressors T133V (TMS3) and V343I (TMS8), which are very close to the major substrate-binding residues Trp130 and Gln134, or Asn341, respectively. Thr133 seems to be an essential element of the major binding site, whereas, Val343 lies “one-step down” in the theoretical trajectory from the binding site toward the cytoplasm. These residues can act as selectivity filters. Type II suppressors concern amino acids Ser296 in TMS7, Ile371 in TMS9, and Tyr392 and Leu394 in TMS10. TMS10 is a flexible transmembrane segment that has been shown to act as an outward-facing gate not only in FurD, but also in Mhp1 (Kazmier et al. 2014; Simmons et al. 2014). Molecular dynamics have indeed shown that TMS10 participates in an occlusion mechanism via its dramatic movement over the major substrate-biding site, while specific mutations in this segment modify (mostly enlarge) the specificity of the transporters, apparently by compromising the occlusion mechanism (Shimamura et al. 2010; Adelman et al. 2011; Kazmier et al. 2014; Simmons et al. 2014). In FurD, mutation M389A (in TMS10) converts this uracil transporter to a transporter capable of recognizing all purines and pyrimidines (Krypotou et al. 2015). Mutations in Leu363 (in TMS10) also affect the substrate specificity of Mhp1 (Simmons et al. 2014). The evidence from molecular dynamics in Mhp1 further suggests that TMS10 acts as a dynamic outward gate in concert with TMS9 and the extracellular hydrophilic segment linking TMS7 and TMS8 (EL4 in Mhp1) (Kazmier et al. 2014; Simmons et al. 2014). Thus, the FurE-ΔC suppressors related to residues Ser296 in TMS7 and Ile371 in TMS9 might also affect the dynamics of the outward-facing gate in FurE. Type III suppressors concern amino acids located in flexible loops, distantly located from the substrate-binding site or the translocation trajectory. Asp26 is at the border of TMS1 with the N-terminal cytoplasmic loop, Gly222 is in the extracellular loop L5, and Asn308 is in extracellular loop L7. Although there are no genetic or functional data related to these residues, molecular dynamics in Mhp1 have shown that substrate binding triggers the movement of TMS1, TMS5, and TMS7, consistent with the closing or opening of the intracellular (TMS1 and TMS5) and extracellular (TMS7) vestibules that lead to the major substrate-binding site (Kazmier et al. 2014; Simmons et al. 2014).
Figure 5.

Location of suppressor mutations in the modeled FurE structure. (A) Model structure of FurE highlighting the residues altered in suppressor mutations (orange spheres) in relation to the substrate-binding site residues (blue spheres), and a critical residue in the putative outer gate (green sphere) (Krypotou et al. 2015). (B) Topology of amino acids (Asp26 in LN, Asn308 in L7, and Gly222 in L5) modified in type III suppressors, located in loops that may function as flexible hinges (see text). (C) Topology of amino acids [Thr133 in transmembrane segment (TMS)3 and Val343 in TMS8] modified in type I suppressors (see text), located proximal to residues shown to interact with substrates in the major substrate-binding pocket of FurE (Ser54 and Ser56 in TMS1, Gln134 and Trp130 in TMS3, Pro251 and Lys252 in TMS6, and Asn342 in TMS8). (D) Topology of amino acids (Ser296 in TMS7, Ile371 in TMS9, and Tyr392 and Leu394 in TMS10) modified in type II suppressors, located next to or in the periphery of a major residue of the outer gate (Leu389 in TMS10).
Overall, the suppressors isolated concern residues mapping either in or close to the major substrate-binding site (TMS3 and TMS8; type I), in an outward-facing gate (TMS7, TMS9, and TMS10; type II), or in dynamic loops that might act as hinges to control gating or the alteration from inward to outward and vice versa (N-terminal-TMS1, L5, and L7; type III).
Discussion
The N- and C-terminal cytoplasmic segments of FurE proved to be important in determining transport kinetics and substrate specificity. A critical but less pronounced role in transport activity and specificity of the CTR of FurD was also implied by growth tests, but this was not analyzed further. No apparent functional modification was detected in the version lacking the CTR of FurA. Thus, the specificity of Fur transporters, and possibly other eukaryotic NCS1 transporters, seems to be differentially dependent on molecular interactions involving domains located at their cytoplasmic N- or CTRs. Although truncations of N- and/or C-terminal segments led to blocks in endocytosis, this could not explain the observed changes in transport kinetics and specificity. Furthermore, transport kinetic analysis has shown that specificity changes cannot always be directly related to analogous modifications in substrate-binding affinities or apparent transport rates. This is best exemplified in the case of FurE-K252F-ΔC, where the specific reduction of UA transport cannot be assigned to a change in the affinity for UA, as this remains practically identical to that of the nontruncated FurE-K252F version. Finally, the cytoplasmic location of the truncated regions makes it rather improbable that the segments deleted are parts of the substrate-binding site or even of the substrate translocation trajectory, as these were previously defined by genetic, in silico, and structural analyses in NCS1 transporters. Of note, the deleted segments in the Fur proteins are “naturally” absent in the prokaryotic members of the NCS1 family. These findings indicate that the cytoplasmic terminal truncations affect the specificity of certain Fur transporters without modifying their bona fide substrate-binding site or translocation trajectory. In a most rational scenario, terminal truncations modify the transport dynamics of FurE due to the role of the deleted segments in critical intramolecular interactions affecting distinct gating or selectivity elements (Shimamura et al. 2010; Adelman et al. 2011; Kazmier et al. 2014; Simmons et al. 2014; Krypotou et al. 2015; Sioupouli et al. 2017).
Using an unbiased genetic approach, we fortified the above conclusion by selecting suppressors “restoring” the loss of UA transport in the truncated FurE-ΔC transporter. We obtained several suppressor mutations in residues located at the periphery of the substrate-binding site, in a tentative external gate, and in flexible loops that might act as hinges during conformational changes associated with transport catalysis. Notably, we did not obtain any mutations affecting residues that bind substrates directly, which supports our previous notion that specificity in transporters is principally determined by gating elements or peripheral selectivity filters, rather than the substrate-binding site (Amillis et al. 2001; Papageorgiou et al. 2008; Diallinas 2014, 2016; Krypotou et al. 2015; Alguel et al. 2016a).
BiFC analysis strongly suggested that the cytoplasmic N- and C-termini of FurE interact physically in the absence of substrates, but that this interaction is practically lost when the transporter is actively transporting substrates. These findings further suggest that, in the absence of substrates, the cytoplasmic N- and C-termini are in close contact, compatible with a rather “immobilized” outward-facing conformation. This idea is in agreement with reports stating that once the two parts of split-YFP interact, they tend to remain associated (Horstman et al. 2014). In the presence of substrates the transporter becomes active, so that upon substrate entry into the binding cavity, exposed in the outward-facing conformation, induced-fit phenomena will force the alteration of the transporter to an inward-facing conformation, and thus the loss of interaction of the split-YFP parts attached to the two cytoplasmic termini. Subsequent dynamic cycles of transport will hinder the stable association of the two cytoplasmic termini, compatible with lack of detection of YFP fluorescent signal.
The interaction of the N- and C-termini, detected genetically and by BiFC for FurE, is apparently not essential for the alternation of outward- and inward-facing conformations that accompany transport catalysis. This conclusion arises from the observation that the singly-truncated versions do transport substrates other than UA, and mostly because the doubly-truncated FurE behaves, unexpectedly, similarly to wild-type FurE. Thus, it seems more probable that the interaction of the N- and C-termini regulates the fine functioning of gating or selectivity elements along the substrate translocation trajectory. In a speculative model for the functioning of FurE, the order of events might be as follows (Figure 6). In the nontruncated version found in the outward-facing conformation, an interaction of the N- and C-termini opens the external gate “fully” and thus all physiological substrates (UA, allantoin, or uracil) fit in the major substrate-binding site. Proper substrate binding triggers occlusion of the outward-facing gate. Closure of the outward-facing gate elicits a major conformational change leading to the inward-closed structure. This will subsequently disrupt the interaction of the N- and C-termini, followed by opening of the internal gate and the release of substrate into the cytoplasm. Lack of the C- or N-terminal region alleviates the full opening of the external gate, in a way that only smaller or specific substrates (e.g., uracil and allantoin) can have access to the binding site. Thus, UA is excluded either because the outer gate is not sufficiently open or because the closure of the gate has not occurred properly. Suppressor mutations modify the action of gating elements so that all substrates regain access to the binding site and are transported.
Figure 6.

Highly speculative scheme of the role of the interaction of the N- and C-terminal regions in FurE functioning. (A) In the nontruncated version found in the outward-facing conformation, an interaction of the N- and C-termini opens “fully” the external gate and thus all physiological substrates [uric acid (UA), allantoin (ALL), or uracil: red rhomboid] fit within the major substrate-binding site. Proper substrate binding triggers occlusion of the outward-facing gate (in orange). Closure of the outward-facing gate elicits a gross conformational change leading to the inward-closed structure. This will subsequently disrupt of the N- and C-termini interaction, open the internal gate, and lead to release of substrate into the cytoplasm. (B) Lack of the C- (or N-terminal) region restricts the full opening of the outer gate, in a way that allows only smaller or specific substrates (e.g., ALL or uracil) to have access to the binding site. Thus, UA is excluded either because the outer gate is not sufficiently open or because the closure of the gate does not occur properly. (C) Suppressor mutations indirectly modify the outer gate action, or other filtering or gating elements (in green), so that UA regains access to the binding site and is transported.
A similar mechanism, proposing that intramolecular interactions of cytoplasmic domains are critical for proper transporter functioning, has been described in a number of recently reported cases. One such case concerns the Saccharomyces cerevisiae Fur4p uracil transporter, which is homologous to the A. nidulans Fur transporters studied herein. In Fur4p, an N-terminal domain called LID (Loop Interaction Domain) has been shown to regulate ubiquitylation and endocytic turnover in response to transport activity or stress conditions that promote unfolding (Keener and Babst 2013). It seems that this domain senses conformational alterations in the transporter, in a way that renders a region adjacent to it accessible for ubiquitylation, a prerequisite for Fur4p endocytosis and turnover. Evidence based on crystal structures of the prokaryotic homolog Mhp1 has shown that in an outward-facing conformation, the LID interacts tightly with all of the cytoplasmic loops and the C-terminus of the transporter via a series of hydrogen bonds. In contrast, when the transporter acquires an inward-facing conformation, the interaction of LID with the other cytoplasmic domains is loosened. The LID–loop interactions also seem to play an important role in stabilizing the basic fold of Fur4p, as N-terminal deletions that remove it cause ER retention and degradation (Keener and Babst 2013). However, in the case of Fur4, cytoplasmic interactions affect the turnover of the protein, rather than its transport function directly.
In the case of the fungal Mep2 ammonium transporters, which also act as receptors and are thus called transceptors (Diallinas 2017), a CTR has been shown, using both structural and genetic evidence, to act as a domain that dynamically interacts with the main body of the transporter, and is thus directly involved in a mechanism that regulates the opening and closing of the substrate translocation pathway (van den Berg et al. 2015). Under nitrogen-sufficient conditions, where Mep2 is not phosphorylated and transport is inactive, the CTR makes relatively few contacts with the main body of the transporter. Under nitrogen-limited conditions, Mep2 is activated via Npr1 kinase-dependent phosphorylation of Ser457 in the CTR. Interestingly, in the Arabidopsis thaliana homologous Amt-1 ammonium transporters, phosphorylation of the CTR under conditions of high ammonium inhibits transport activity; that is, the nonphosphorylated state of the plant transporter is active (Loqué et al. 2007; Lanquar and Frommer 2010; Boeckstaens et al. 2014). This means that phosphorylation can either lead to channel closing (in the case of ammonium transporters) or channel opening (in the case of Mep2). Overall, the results on Mep2/Amt1 lead to a model for the regulation of transition between closed and open states of eukaryotic ammonium transporters involving the phosphorylation-dependent dynamic interaction of CTRs with specific cytoplasmic loops (e.g., ICL1/ICL3) (Boeckstaens et al. 2014). However, how exactly the substrate translocation pathway in Mep2/Amt1 opens and closes remains elusive.
Another interesting case where cytoplasmic regions control transport functioning directly occurs in prokaryotic ATP-binding cassette (ABC) transporters or the homologous human cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel involved in cystic fibrosis [Gadsby et al. 2006; Mihályi et al. 2016, for a review see Locher (2016)]. In particular, in CFTR and other ABC transporters, ATP-binding-dependent dimerization of two cytosolic nucleotide-binding domains (NBDs) opens the pore, whereas dimer disruption following ATP hydrolysis closes it. Spontaneous openings without ATP are rare in wild-type, but not in mutant versions of CFTR, but are still strictly coupled to NBD dimerization. Coordinated NBD/pore movements are therefore intrinsic to CFTR, suggesting that ATP alters the stability, but not the fundamental structural architecture, of open- and closed-pore conformations. The apparently cyclic, dynamic restructuring of the intramolecular domain made of two NBD domains might be mechanistically analogous to the N- and C-tail interaction, which we propose to be the basis of the gating cycle in FurE.
Phosphorylation is likely to be a common mechanism for the regulation of eukaryotic transporters and channels in response to environmental or stress signals, or transport activity. Prominent examples, besides Fur4p and Mep2/Amt-1 discussed above, include transporters specific for urea (Klein 2014), nitrate (Parker and Newstead 2014; Jacquot et al. 2017), amino acids (Gournas et al. 2016), or aquaporins (Törnroth-Horsefield et al. 2010). Our work has not investigated a possible functional role of phosphorylation of cytoplasmic terminal regions in FurE or other Fur transporters. The only Fur-like transporter studied with respect to phosphorylation is the Fur4p of S. cerevisiae. In this transporter, an N-terminal cytoplasmic PEST-like sequence seems to be essential for transporter turnover, via ubiquitylation and endocytosis (Marchal et al. 1998, 2000). However, no direct effect of N-terminal phosphorylation on the function of Fur4p has been identified.
In conclusion, the present work adds another transporter family to a handful of eukaryotic transporters that have been shown to be regulated by dynamic interactions of their cytoplasmic tails. What is novel in our work is that cytoplasmic terminal regions affect not simply transporter activity or turnover, but can finely regulate substrate specificity. This, in turn, allows us to propose that transporter-specific interactions of terminal cytoplasmic regions provide a flexible mechanism for the generation of novel specificities over the course of evolution. Given that most prokaryotic homologs of eukaryotic transporters do not possess long cytoplasmic terminal regions, it seems that the acquisition of such extended terminal cytoplasmic regions served as a molecular novelty that allowed fine transporter neo-functionalization. Additionally, given that terminal regions of transporters are little conserved, even among close eukaryotic homologs, it is becoming evident that we cannot predict substrate specificities a priori simply by comparing similarities in substrate-binding sites. Thus, after the recent discovery of dynamic gating elements and oligomerization (Diallinas 2017), terminal cytoplasmic regions, acting as allosteric switches, have come to add an extra level of complexity regarding what determines transporter function and specificity.
Supplementary Material
Supplemental material is available online at www.genetics.org/lookup/suppl/doi:10.1534/genetics.117.300327/-/DC1.
Acknowledgments
We are grateful to George Lambrinidis for his help in making Figure 6. This work was supported by a Stavros S. Niarchos Foundation for Charity grant.
Footnotes
Communicating editor: J. Heitman
Literature Cited
- Adelman J. L., Dale A. L., Zwier M. C., Bhatt D., Chong L. T., et al. , 2011. Simulations of the alternating access mechanism of the sodium symporter Mhp1. Biophys. J. 101: 2399–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alguel Y., Cameron A. D., Diallinas G., Byrne B., 2016a Transporter oligomerization: form and function. Biochem. Soc. Trans. 44: 1737–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alguel Y., Amillis S., Leung J., Lambrinidis G., Capaldi S., et al. , 2016b Structure of eukaryotic purine/H (+) symporter UapA suggests a role for homodimerization in transport activity. Nat. Commun. 7: 11336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amillis S., Koukaki M., Diallinas G., 2001. Substitution F569S converts UapA, a specific uric acid-xanthine transporter, into a broad specificity transporter for purine-related solutes. J. Mol. Biol. 313: 765–774. [DOI] [PubMed] [Google Scholar]
- Amillis S., Hamari Z., Roumelioti K., Scazzocchio C., Diallinas G., 2007. Regulation of expression and kinetic modeling of substrate interactions of a uracil transporter in Aspergillus nidulans. Mol. Membr. Biol. 24: 206–214. [DOI] [PubMed] [Google Scholar]
- Boeckstaens M., Llinares E., Van Vooren P., Marini A. M., 2014. The TORC1 effector kinase Npr1 fine tunes the inherent activity of the Mep2 ammonium transport protein. Nat. Commun. 5: 3101. [DOI] [PubMed] [Google Scholar]
- Colas C., Ung P. M., Schlessinger A., 2016. SLC transporters: structure, function, and drug discovery. Medchemcomm 7: 1069–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Koning H., Diallinas G., 2000. Nucleobase transporters. Mol. Membr. Biol. 17: 75–94. [DOI] [PubMed] [Google Scholar]
- Diallinas G., 2008. Biochemistry. An almost-complete movie. Science 322: 1644–1645. [DOI] [PubMed] [Google Scholar]
- Diallinas G., 2014. Understanding transporter specificity and the discrete appearance of channel-like gating domains in transporters. Front. Pharmacol. 5: 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diallinas G., 2016. Dissection of transporter function: from genetics to structure. Trends Genet. 32: 576–590. [DOI] [PubMed] [Google Scholar]
- Diallinas G., 2017. Transceptors as a functional link of transporters and receptors. Microb. Cell 4: 69–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evangelinos M., Martzoukou O., Chorozian K., Amillis S., Diallinas G., 2016. BsdA(Bsd2)-dependent vacuolar turnover of a misfolded version of the UapA transporter along the secretory pathway: prominent role of selective autophagy. Mol. Microbiol. 100: 893–911. [DOI] [PubMed] [Google Scholar]
- Forrest L. R., Krämer R., Ziegler C., 2011. The structural basis of secondary active transport mechanisms. Biochim. Biophys. Acta 1807: 167–188. [DOI] [PubMed] [Google Scholar]
- Gadsby D. C., Vergani P., Csanády L., 2006. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature 440: 477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gournas C., Amillis S., Vlanti A., Diallinas G., 2010. Transport-dependent endocytosis and turnover of a uric acid-xanthine permease. Mol. Microbiol. 75: 246–260. [DOI] [PubMed] [Google Scholar]
- Gournas C., Prévost M., Krammer E. M., André B., 2016. Function and regulation of fungal amino acid transporters: insights from predicted structure. Adv. Exp. Med. Biol. 892: 69–106. [DOI] [PubMed] [Google Scholar]
- Hamari Z., Amillis S., Drevet C., Apostolaki A., Vágvölgyi C., et al. , 2009. Convergent evolution and orphan genes in the Fur4p-like family and characterization of a general nucleoside transporter in Aspergillus nidulans. Mol. Microbiol. 73: 43–57. [DOI] [PubMed] [Google Scholar]
- Horstman A., Tonaco I. A., Boutilier K., Immink R. G., 2014. A cautionary note on the use of split-YFP/BiFC in plant protein-protein interaction studies. Int. J. Mol. Sci. 15: 9628–9643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquot A., Li Z., Gojon A., Schulze W., Lejay L., 2017. Post-translational regulation of nitrogen transporters in plants and microorganisms. J. Exp. Bot. 68: 2567–2580. [DOI] [PubMed] [Google Scholar]
- Kaback H. R., Smirnova I., Kasho V., Nie Y., Zhou Y., 2011. The alternating access transport mechanism in LacY. J. Membr. Biol. 239: 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karachaliou M., Amillis S., Evangelinos M., Kokotos A. C., Yalelis V., et al. , 2013. The arrestin-like protein ArtA is essential for ubiquitination and endocytosis of the UapA transporter in response to both broad-range and specific signals. Mol. Microbiol. 88: 301–317. [DOI] [PubMed] [Google Scholar]
- Kazmier K., Sharma S., Islam S. M., Roux B., Mchaourab H. S., 2014. Conformational cycle and ion-coupling mechanism of the Na+/hydantoin transporter Mhp1. Proc. Natl. Acad. Sci. USA 111: 14752–14757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keener J. M., Babst M., 2013. Quality control and substrate-dependent downregulation of the nutrient transporter Fur4. Traffic 14: 412–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein J. D., 2014. Expression of urea transporters and their regulation. Subcell. Biochem. 73: 79–107. [DOI] [PubMed] [Google Scholar]
- Kosti V., Papageorgiou I., Diallinas G., 2010. Dynamic elements at both cytoplasmically and extracellularly facing sides of the UapA transporter selectively control the accessibility of substrates to their translocation pathway. J. Mol. Biol. 397: 1132–1143. [DOI] [PubMed] [Google Scholar]
- Koukaki M., Giannoutsou E., Karagouni A., Diallinas G., 2003. A novel improved method for Aspergillus nidulans transformation. J. Microbiol. Methods 55: 687–695. [DOI] [PubMed] [Google Scholar]
- Krypotou E., Diallinas G., 2014. Transport assays in filamentous fungi: kinetic characterization of the UapC purine transporter of Aspergillus nidulans. Fungal Genet. Biol. 63: 1–8. [DOI] [PubMed] [Google Scholar]
- Krypotou E., Kosti V., Amillis S., Myrianthopoulos V., Mikros E., et al. , 2012. Modeling, substrate docking, and mutational analysis identify residues essential for the function and specificity of a eukaryotic purine-cytosine NCS1 transporter. J. Biol. Chem. 287: 36792–36803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krypotou E., Evangelidis T., Bobonis J., Pittis A. A., Gabaldón T., et al. , 2015. Origin, diversification and substrate specificity in the family of NCS1/FUR transporters. Mol. Microbiol. 96: 927–950. [DOI] [PubMed] [Google Scholar]
- Lanquar V., Frommer W. B., 2010. Adjusting ammonium uptake via phosphorylation. Plant Signal. Behav. 5: 736–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauwers E., Erpapazoglou Z., Haguenauer-Tsapis R., André B., 2010. The ubiquitin code of yeast permease trafficking. Trends Cell Biol. 20: 196–204. [DOI] [PubMed] [Google Scholar]
- Locher K. P., 2016. Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nat. Struct. Mol. Biol. 23: 487–493. [DOI] [PubMed] [Google Scholar]
- Loqué D., Lalonde S., Looger L. L., von Wirén N., Frommer W. B., 2007. A cytosolic trans-activation domain essential for ammonium uptake. Nature 446: 195–198. [DOI] [PubMed] [Google Scholar]
- Martzoukou O., Karachaliou M., Yalelis V., Leung J., Byrne B., et al. , 2015. Oligomerization of the UapA purine transporter is critical for ER-exit, plasma membrane localization and turnover. J. Mol. Biol. 427: 2679–2696. [DOI] [PubMed] [Google Scholar]
- Martzoukou O., Amillis S., Zervakou A., Christoforidis S., Diallinas G., 2017. The AP-2 complex has a specialized clathrin-independent role in apical endocytosis and polar growth in fungi. Elife 21: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchal C., Haguenauer-Tsapis R., Urban-Grimal D., 1998. A PEST-like sequence mediates phosphorylation and efficient ubiquitination of yeast uracil permease. Mol. Cell. Biol. 18: 314–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchal C., Haguenauer-Tsapis R., Urban-Grimal D., 2000. Casein kinase I-dependent phosphorylation within a PEST sequence and ubiquitination at nearby lysines signal endocytosis of yeast uracil permease. J. Biol. Chem. 275: 23608–23614. [DOI] [PubMed] [Google Scholar]
- Mihályi C., Töröcsik B., Csanády L., 2016. Obligate coupling of CFTR pore opening to tight nucleotide-binding domain dimerization. Elife 5: e18164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantazopoulou A., Diallinas G., 2007. Fungal nucleobase transporters. FEMS Microbiol. Rev. 31: 657–675. [DOI] [PubMed] [Google Scholar]
- Pantazopoulou A., Lemuh N. D., Hatzinikolaou D. G., Drevet C., Cecchetto G., et al. , 2007. Differential physiological and developmental expression of the UapA and AzgA purine transporters in Aspergillus nidulans. Fungal Genet. Biol. 44: 627–640. [DOI] [PubMed] [Google Scholar]
- Papageorgiou I., Gournas C., Vlanti A., Amillis S., Pantazopoulou A., et al. , 2008. Specific interdomain synergy in the UapA transporter determines its unique specificity for uric acid among NAT carriers. J. Mol. Biol. 382: 1121–1135. [DOI] [PubMed] [Google Scholar]
- Parker J. L., Newstead S., 2014. Molecular basis of nitrate uptake by the plant nitrate transporter NRT1.1. Nature 507: 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penmatsa A., Gouaux E., 2014. How LeuT shapes our understanding of the mechanisms of sodium-coupled neurotransmitter transporters. J. Physiol. 592: 863–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quistgaard E. M., Löw C., Guettou F., Nordlund P., 2016. Understanding transport by the major facilitator superfamily (MFS): structures pave the way. Nat. Rev. Mol. Cell Biol. 17: 123–132. [DOI] [PubMed] [Google Scholar]
- Shi Y., 2013. Common folds and transport mechanisms of secondary active transporters. Annu. Rev. Biophys. 42: 51–72. [DOI] [PubMed] [Google Scholar]
- Shimamura T., Weyand S., Beckstein O., Rutherford N. G., Hadden J. M., et al. , 2010. Molecular basis of alternating access membrane transport by the sodium-hydantoin transporter Mhp1. Science 328: 470–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons K. J., Jackson S. M., Brueckner F., Patching S. G., Beckstein O., et al. , 2014. Molecular mechanism of ligand recognition by membrane transport protein, Mhp1. EMBO J. 33: 1831–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sioupouli G., Lambrinidis G., Mikros E., Amillis S., Diallinas G., 2017. Cryptic purine transporters in Aspergillus nidulans reveal the role of specific residues in the evolution of specificity in the NCS1 family. Mol. Microbiol. 103: 319–332. [DOI] [PubMed] [Google Scholar]
- Takeshita N., Higashitsuji Y., Konzack S., Fischer R., 2008. Apical sterol-rich membranes are essential for localizing cell end markers that determine growth directionality in the filamentous fungus Aspergillus nidulans. Mol. Biol. Cell 19: 339–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Törnroth-Horsefield S., Hedfalk K., Fischer G., Lindkvist-Petersson K., Neutze R., 2010. Structural insights into eukaryotic aquaporin regulation. FEBS Lett. 584: 2580–2588. [DOI] [PubMed] [Google Scholar]
- Valdez-Taubas J., Harispe L., Scazzocchio C., Gorfinkiel L., Rosa A. L., 2004. Ammonium-induced internalisation of UapC, the general purine permease from Aspergillus nidulans. Fungal Genet. Biol. 41: 42–51. [DOI] [PubMed] [Google Scholar]
- van den Berg B., Chembath A., Jefferies D., Basle A., Khalid S., et al. , 2015. Structural basis for Mep2 ammonium transceptor activation by phosphorylation. Nat. Commun. 7: 11337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyand S., Shimamura T., Yajima S., Suzuki S., Mirza O., et al. , 2008. Structure and molecular mechanism of a nucleobase-cation-symport-1 family transporter. Science 322: 709–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Strains constructed in this work are available upon request.