

Abstract
PDRG1 is a small oncogenic protein of 133 residues. In normal human tissues, the p53 and DNA damage-regulated gene 1 (PDRG1) gene exhibits maximal expression in the testis and minimal levels in the liver. Increased expression has been detected in several tumor cells and in response to genotoxic stress. High-throughput studies identified the PDRG1 protein in a variety of macromolecular complexes involved in processes that are altered in cancer cells. For example, this oncogene has been found as part of the RNA polymerase II complex, the splicing machinery and nutrient sensing machinery, although its role in these complexes remains unclear. More recently, the PDRG1 protein was found as an interaction target for the catalytic subunits of methionine adenosyltransferases. These enzymes synthesize S-adenosylmethionine, the methyl donor for, among others, epigenetic methylations that occur on the DNA and histones. In fact, downregulation of S-adenosylmethionine synthesis is the first functional effect directly ascribed to PDRG1. The existence of global DNA hypomethylation, together with increased PDRG1 expression, in many tumor cells highlights the importance of this interaction as one of the putative underlying causes for cell transformation. Here, we will review the accumulated knowledge on this oncogene, emphasizing the numerous aspects that remain to be explored.
Keywords: Epigenetic modifications, Glutathione, Methylation, Oncogenes, Intermediary metabolism, p53 and DNA damage-regulated gene 1, Protein complexes, R2TP/prefoldin complex, S-adenosylmethionine synthesis, Redox stress
Core tip: PDRG1 is an understudied protein of the R2TP/prefoldin-like complex that has been found as part of different multiprotein complexes. Increased PDRG1 expression levels have been associated with several types of tumors that concomitantly show global DNA hypomethylation. More recently, this protein has been uncovered as an interaction target for methionine adenosyltransferase catalytic subunits MATα1 and MATα2. Through this interaction, PDRG1 downregulates nuclear S-adenosylmethionine synthesis, hence impacting epigenetic methylations.
INTRODUCTION
Late in the 20th century, global DNA hypomethylation was described as a main characteristic of cancer cells and as the underlying cause of their altered expression pattern[1,2]. Moreover, tumor cell growth was found to be totally dependent on glutamine and methionine, and the latter amino acid could not be replaced by homocysteine[3]. Methionine is one of the substrates required for the synthesis of the main cellular methyl donor, S-adenosylmethionine (AdoMet), which is used for epigenetic methylations occurring at both the DNA and its associated proteins for the regulation of gene expression. These modifications are among the dozens of methylation reactions that occur in a cell using S-adenosylmethionine (AdoMet) as a methyl donor[4,5]. However, the methyltransferases catalyzing these modifications are not the main consumers of AdoMet, despite their importance in regulating cell function. This fact is especially clear in the liver, where three methyltransferases involved in the synthesis of small compounds use the majority of the methyl donor synthesized[6,7]. Those are glycine N-methyltransferase (GNMT), guanidinoacetate N-methyltransferase (GAMT) and phosphatidylethanolamine N-methyltransferase (PEMT), which produce sarcosine, creatine and phosphatidylcholine, respectively[8-11]. Nevertheless, these estimations were made on the basis that AdoMet synthesis occurs exclusively in the cytoplasm and that the methyl donor would be transported to other subcellular localizations as required. This assumption has been challenged lately by reports describing the existence of nuclear pools of several enzymes of the methionine cycle, the pathway where AdoMet is produced, in turn suggesting the existence of two independent AdoMet stocks in the cytoplasm and the nucleus[12-16]. The existence of a nuclear branch of this pathway is supported by the importance of epigenetic methylations and the need to guarantee their AdoMet supply.
To date, information regarding the cytoplasmic and nuclear branches of the methionine cycle has been mainly obtained in liver, and the accumulated knowledge can be summarized as follows. The cytoplasmic methionine cycle involves the following (Figure 1): (1) methionine adenosyltransferases (MATs) that synthesize AdoMet; (2) a large number of methyltransferases, which methylate many types of substrates, also rendering S-adenosylhomocysteine (AdoHcy); (3) AdoHcy hydrolase (AHCY), which eliminates AdoHcy leading to adenosine and homocysteine (Hcy); and (4) betaine homocysteine S-methyltransferases (BHMT and BHMT2) and methionine synthase (MTR), which methylate Hcy to obtain methionine using betaine or S-methylmethionine and 5-methyltetrahydrofolate, respectively[17,18]. In contrast, the nuclear branch of the pathway involves a more reduced set of enzymes as follows (Figure 1): (1) MATs; (2) methyltransferases such as those involved in DNA and histone methylations, GNMT and GAMT; (3) AHCY[12,13,15,16]; and (4) BHMT[19]. Of note, levels of MATs, GNMT and AHCY are markedly higher in the cytoplasm than in the nucleus, but increases in the latter compartment can be observed as a consequence of pathological changes in glutathione concentrations and, precisely, decreases in the GSH/GSSG ratio[14]. More recently, PDRG1 [Consensus nomenclature for the human (PDRG1) and mouse genes (Pdrg1) is used throughout the text. The protein is in all the cases referred to as PDRG1] was added to this nuclear branch of the cycle as an interaction target and regulator of MATs, and hence, of AdoMet synthesis[20,21].
Figure 1.
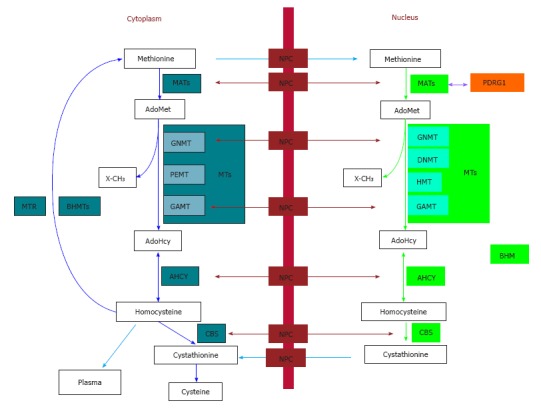
The cytoplasmic and nuclear branches of the hepatic methionine cycle and transsulfuration. Essential components (metabolites and enzymes) of the pathways are shown in the figure. Cytoplasmic enzymes appear in aquamarine boxes, their nuclear equivalents are shown in green boxes, and protein interaction targets are depicted in orange. Only key methyltranferases (MTs) mentioned in the text are illustrated in both subcellular compartments. The nucleocytoplasmic transport of metabolites (blue arrows) and enzymes (maroon arrows) is shown, whereas protein interactions are indicated by violet arrows. AdoHcy: S-adenosylhomocysteine; AdoMet: S-adenosylmethionine; AHCY: S-adenosylhomocysteine hydrolase; BHMT: Betaine homocysteine S-methyltransferase; CBS: Cystathionine β-synthase; DNMT: DNA methyltransferase; GAMT: Guanidinoacetate N-methyltransferase; GNMT: Glycine N-methyltransferase; HMT: Histone methyltransferase; MATs: Methionine adenosyltransferases; MTR: Methionine synthase; NPC: Nuclear pore complex; PDRG1: p53 and DNA damage-regulated gene 1; PEMT: Phosphatidylethanolamine N-methyltransferase; X-CH3: Methylated acceptors (metabolites, proteins, DNA, etc).
PDRG1 stands for p53 and DNA damage-regulated gene 1 as it was discovered during studies devoted to the identification of components of the cellular response to DNA damage, or genotoxic stress, and it is regulated by p53[22]. However, what do we really know about PDRG1? This is the question that will be addressed in the next sections of the present mini-review.
PDRG1 GENE STRUCTURE AND REGULATION
Studies devoted to the identification of components of the cellular response to DNA damage have been carried out for a number of years, and many of them used UV-irradiation as a stressor. Some of these investigations identified several novel UV-induced genes using a UV-treated low-ratio hybridization subtraction-enriched cDNA hamster library, and they were followed by screenings of partial-length expressed sequence tag (EST) clones regulated by this type of stress[22-24]. This latter procedure rendered a UV-induced EST clone that hybridized with a 1.4 kb transcript, which was named PDRG1[22]. Sequencing of the corresponding human and mouse EST clones from different tissues showed a single open reading frame coding for a protein of 133 amino acids.
The PDRG1 gene is organized among five exons and four introns in both humans and mice (Figure 2)[22]. The start codon is located at position 120 in exon 1 of the human gene, whereas the stop codon occurs at position 85 on exon 5. The mouse gene is located on chromosome 2, whereas the human gene appears on the long arm of chromosome 20 at the 20q11.2 region[22]. Interestingly, the human gene localization coincides with a position in which gains of DNA are detected in hepatocellular carcinoma (HCC) and dysplastic nodules, and where alterations in cirrhotic processes are also found[25]. Moreover, these are pathologies in which impairment of the methionine cycle, especially of AdoMet production, have been described[17].
Figure 2.
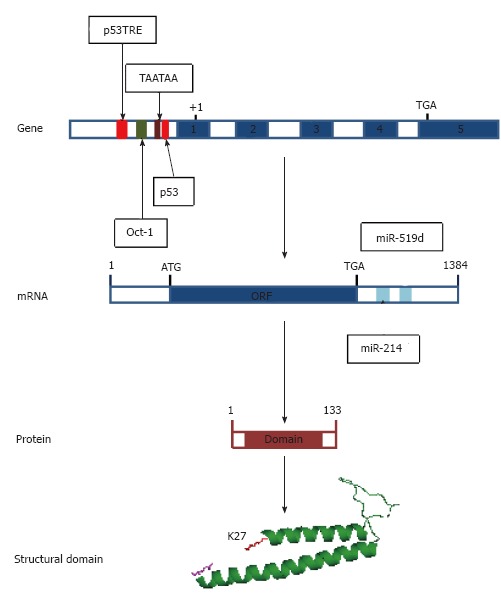
Regulatory elements on the human PDRG1 gene and mRNA and structural features of the protein. From top to bottom, the figure shows first a scheme of the human PDRG1 gene, indicating the regulatory elements on the promoter identified to date as follows: p53 and p53TRE elements (red), an Oct-1 binding element (green) and the TATA-box-like element (crimson). Exons appear as blue boxes. Second, a schematic representation of the mRNA is depicted, including the ORF (blue box) and the miRNA binding sites identified at the 3’-UTR (aquamarine boxes). Third, a scheme of the PDRG1 protein is shown where the sequence encoding the prefoldin-like domain is indicated as a crimson box. Finally, a cartoon of the modeled prefoldin-like domain of rat PDRG1 is included with the N-terminal K27 (red) and the C-terminal Q106 (magenta) depicted as sticks.
Analysis of the human PDRG1 promoter was initially carried out using a 1.7 kb fragment that included the 5’-UTR and part of the non-coding region of exon 1, and it showed high levels of promoter activity in breast cancer MCF-7 cells when fused to the luciferase gene[22]. The gene contains a TATA-like element (TAATAA) localized at -268 from the start codon, but no canonical TATA box was identified[22]. Promoter activity was eliminated by deletion of the -138/+1 region, in turn suggesting the presence of key regulatory elements in this segment[22]. These same authors also detected two putative downregulatory elements in the promoter. The first, a p53 binding element, was identified at position -240/-214 and includes two RRRCWWGYYY (R = purine, Y = pyrimidine, W = A or T, S = G or C, D = A, G or T) repeats arranged head to tail and separated by a 6 bp sequence[26-28]. The second corresponds to a putative p53 transcriptional repressor element (TRE) located at -854/-834[29]. Regulation of the PDRG1 promoter by p53 was further proven when luciferase activity was abolished in the presence of this tumor suppressor[22]. Basal PDRG1 expression in p53 null HCT116 colon cancer cells was in fact higher than that in their wild-type counterparts, results that further supported repression of PDRG1 by p53.
Additionally, the PDRG1 promoter includes a canonical Oct-1 binding element located at -689/-683[22,30]; this factor is known to upregulate expression of a number of genes when activated by UV irradiation[31,32]. Cotransfection of a PDRG1 promoter-luciferase construct and Oct-1 into MCF-7 cells rendered approximately 8-fold induction of the luciferase activity, thus demonstrating the functionality of this Oct-1 binding element[22]. This increased promoter activity was far greater than that caused exclusively by UV irradiation, a fact that Luo et al[22] ascribed putatively to p53 activation in these cells.
More recently, the presence of complementary sequences for miR-214 (position 842-850) and miR-519d (position 1076-1082) in the 3’-UTR of the PDRG1 mRNA was reported[33,34]. Overexpression of miR-214, a miRNA with dual functionality as tumor suppressor or oncogene, in bladder cancer cell lines reduced PDRG1 expression and protein levels[33]. Similarly, overexpression of miR-519d leads to decreased PDRG1 mRNA and protein levels in two nasopharyngeal carcinoma cell lines, which in turn correlates with a higher radiation-induced mortality (Figure 3)[34].
Figure 3.
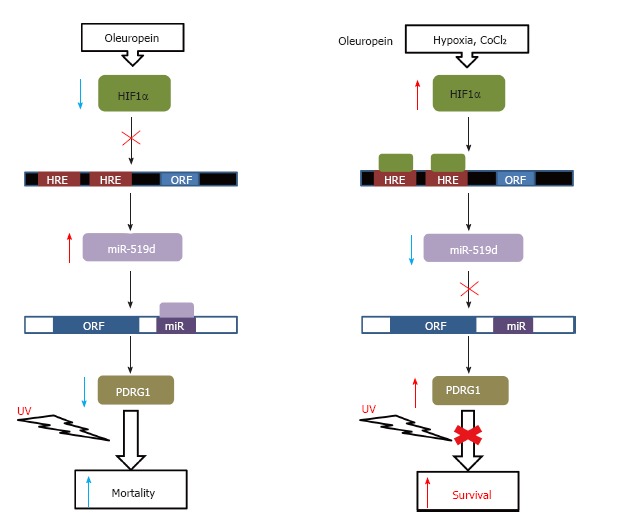
Regulation of cell survival. A schematic representation of the effects on cell survival exerted by PDRG1 regulation through miR-519d expression is shown. Binding sites for HIF1a (HRE) in the promoter of the miR-519d gene (black box) and for this miRNA on the PDRG1 mRNA (white box) are indicated. Agents used for induction or blockade of UV-irradiation effects are indicated on the top.
Regarding the PDRG1 expression profile, this aspect was initially studied using commercial human poly(A)+ Northern blots, where a 1.4 kb transcript was identified in brain, heart, liver, lung, spleen, stomach, testis and skeletal muscle[22]. More recently, real-time RTqPCR of rat tissues also demonstrated wide expression of Pdrg1, further including the cerebellum, intestine, pancreas, kidney and cava vein[20]. In human tissues the highest levels were found in testis, whereas in the rat similar expression levels were detected in brain, cerebellum and testis. Nevertheless, both species showed very low PDRG1 expression in the liver[20,22].
PDRG1 PROTEIN
Mammalian PDRG1 proteins share a high level of conservation, and most of them are over 90% identical (Figure 4). The sequences usually comprise 133 residues (15.5 kDa for the human protein), although larger proteins have been predicted in several marine mammals. In those cases, PDRG1 presents with N-terminal extensions of variable length. The protein’s isoelectric point is 5.81, and several post-translational modifications have been predicted and identified in high-throughput studies. Among them, phosphorylations at human serine residues 3, 52, 54 and mouse serine 120 and ubiquitinations on human lysine residues 27, 108 and 125 have been postulated or found[35-41]. Initial studies suggested association of the protein into homo-oligomers, a fact that was later confirmed by detection of HA-PDRG1 hexamers in embryonic kidney HEK293T cells using analytical gel filtration chromatography[20-22].
Figure 4.
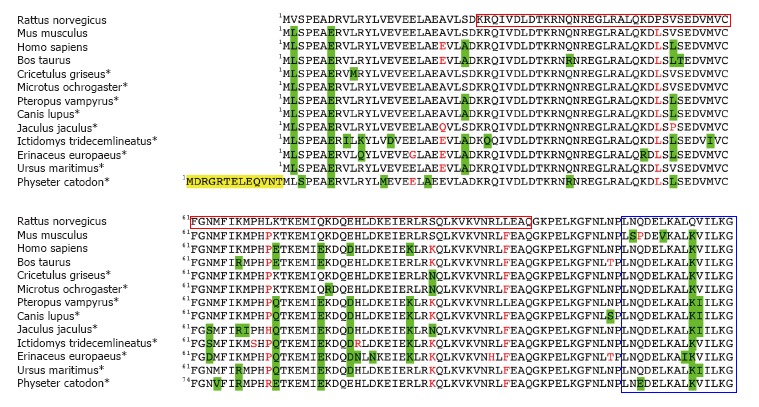
Alignment of mammalian PDRG1 proteins. The figure shows the BLASTP alignment of thirteen representative mammalian PDRG1 sequences using that of Rattus norvegicus as a reference. Most of the data correspond to predicted sequences (*). Conservative changes are highlighted in green, non-conservative substitutions appear in red, and N-terminal extensions are indicated in yellow. The C-terminal helix-turn-helix motif and the prefoldin-like domain are indicated in blue and crimson boxes, respectively.
Motif searches identified a helix-turn-helix (LNQDEL KALKVILKG) at the C-terminal end of the human and mouse protein sequences[22]. These motifs are normally involved in interactions with either the DNA or other proteins. Moreover, human and mouse PDRG1 are characterized by the presence of a β-prefoldin-like domain that is also recognized by the Protein Homology/analogY Recognition Engine (PHYRE) in the rat counterpart. PHYRE2 identified prefoldin as the closest homologue to rat PDRG1 during the preparation of a structural model. This model included residues K27-Q106 (Figure 2), thus excluding approximately 25 residues from each protein end. Elimination of the C-terminal end has divergent effects on the interaction of PDRG1 with MAT catalytic subunits MATα1 and MATα2[20,21]. Additionally, motif searches carried out with the rat PDRG1 sequence predicted nucleocytoplasmic localization (PSORT II) and a nuclear export signal (NES) involving serine 93; the NetNES score increases between residues E89-L95.
Subcellular localization was initially examined by fluorescence microscopy using PDRG1-GFP and GFP-PDRG1 tagged forms in fixed NIH 3T3 fibroblasts and HCT116 colon cancer cells. In both cases, cytoplasmic aggregates of the protein were described, whereas no localization within subcellular compartments such as the mitochondria, endoplasmic reticulum and nucleus was observed using specific dyes for these organelles[22]. Moreover, these authors indicated that this distribution was not affected by UV irradiation, and hence, a putative PDRG1-DNA interaction was excluded. In contrast, PDRG1 was found in the nuclear fractions used to isolate protein complexes from LNCaP prostate cancer cells, hence suggesting that the protein may be distributed between subcellular compartments[42]. This aspect was reexamined using confocal microscopy and several cell lines in which HA-PDRG1 or PDRG1-EGFP was overexpressed. The results of direct fluorescence and immunofluorescence experiments demonstrated that, in fact, PDRG1 is a nucleocytoplasmic protein in ovary (CHO), kidney (COS-7), neuroblastoma (N2a) and hepatoma (H35) cell lines[20,21]. Moreover, quantification of the fluorescence signals indicated a stronger protein level in the nuclear compartment, where PDRG1 also colocalized with nuclear speckles containing SC-35.
PROTEIN-PROTEIN INTERACTIONS INVOLVING PDRG1
Lately, several high-throughput studies have been carried out with the goal of identifying components of several large protein complexes (Figure 5). For this purpose, the following two main techniques were used: (1) affinity purification of subunits including single (AP) or tandem tags (TAP) followed by mass spectrometry (MS); and (2) yeast two-hybrid (YTH). During some of these studies PDRG1 was identified either as a subunit or interaction target of several complexes, and interestingly, methylation plays a direct or indirect role in many of the processes involving these multimers (e.g., nutrient signaling and mRNA capping).
Figure 5.
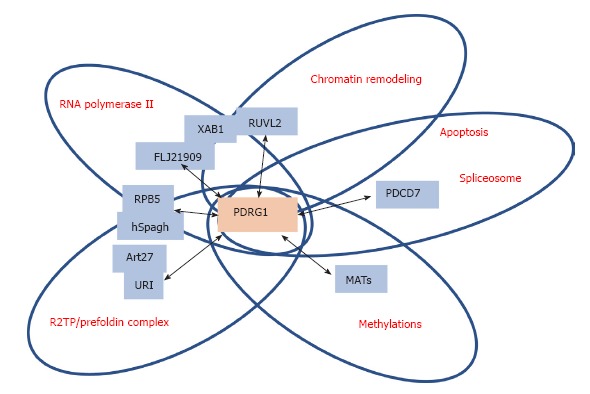
Schematic representation of the main PDRG1 interaction targets. The figure highlights the main targets of interaction with PDRG1 identified using either affinity purification/mass spectrometry or yeast two-hybrid screening. The proteins involved are shown in blue boxes within circles, according to the function or multiprotein complex in which they are involved. PDRG1 appears at the intersection of these circles.
PDRG1 is indirectly linked to the RNA polymerase II machinery
MS, together with a computer method to minimize the contribution of false positives, was used to identify the interaction network of human RNA polymerase II using tagged subunits that allowed TAP purification from kidney HEK293 cells[43]. Some key proteins were selected for additional TAP-MS experiments, and among them are XAB1, which appears placed at the interface with members of chaperone complexes (C19orf2/URI, PFDN6, PFDN2 and UXT), and the AAA+ chaperone-like ATPases RUVBL1/TIP49a and RUVBL2/TIP49b. These experiments showed TAP-XAB1 copurification with FLJ21909 and subsequent TAP-FLJ21909 copurification with PDRG1[43]. Moreover, this same work also demonstrated copurification of PDRG1 with TAP-RUVBL2, C1orf82/RPAP2 and FLJ21908/RPAP3, although no further validation of the interactions using additional techniques was carried out[43].
Additional studies to analyze the cytoplasmic assembly of RNA polymerase II were later performed by Boulon et al[44]. These MS-proteomics studies showed association of unassembled RPB1, the largest subunit of this transcription machinery, with the R2TP/prefoldin-like complex[44]. Moreover, in this same study the interactions of a central component of the R2TP/prefoldin-like complex, hSpagh that binds HSP90 as a cofactor, were further explored. Independent interactions of hSpagh with RPB1 and RPB5 subcomplexes were then identified, which also involved other members of the R2TP/prefoldin-like complex, such as PDRG1. However, YTH only confirmed the implication of PDRG1 in the interaction with RBP5, although this binding seemed indirect through its association with hSpagh[44].
PDRG1 may have a role in chromatin remodeling, splicing and apoptosis
AP-MS data also suggested that PDRG1 might be linked to the SWR-C and INO80 chromatin remodeling complexes. Such a relationship could be established by the identification of its interaction targets, RUVBL1 and RUVBL2, in both complexes in experiments carried out in nuclear fractions of human cells[45,46]. Additionally, YTH screens identified PDRG1 interacting with programmed cell death 7 (PDCD7), Cip1 zinc interacting finger (CIZ1) and microtubule-associated protein 1S (MAP1S) in human testis, although immunoprecipitation only confirmed the PDRG1-PDCD7 interaction[47]. These results linked PDRG1 with the modulation of apoptosis and the U12-type spliceosome, a process and a complex that involve PDCD7[48].
PDRG1 is a member of the URI/prefoldin complex involved in nutrient signaling
PDRG1 was identified, together with HKE2, BC014022, POL3A, FLJ21908 and FLJ20643, as a new member of the URI/prefoldin complex with abundance similar to that exhibited by the consensus subunits known at that time[49]. URI stands for unconventional prefoldin RPB5 interactor, also known as RMP, and some of the interaction studies described in this section showed this interaction. Additional data from this AP-MS study identified PDRG1 interacting with TIP49a/b in HEK293/FRT and HeLa cells, as well as bound to BC014022, DPCD, FLJ20643, NUFIP, RPB5MP and UXT1[49]. Reciprocal studies using FLAG-tagged PDRG1 identified interactors with moderate (POL3A, PPP1CA, PPP1CC, PFDN2, HKE2, RPB5, PPP1CB, NPM1, UXT1, H2BS, H2BR, RPB5MP, FLJ21908, BC014022, H2AZ, NOP5/NOP58, FLJ20643) or low probability scores (TUB6, HSP70/1, HSP70/1B, BAF53, TIP49a, TIP49b)[49]. However, none of these PDRG1 interactions were confirmed by immunoprecipitation. More recently, MS analysis of immunoprecipitates from nuclear fractions of LNCaP prostate cells stably transfected with FLAG-URI also showed the presence of PDRG1 in the R2TP/prefoldin-like complex, and the interaction was confirmed by detection of URI and PDRG1 in anti-Art27 immunoprecipitates[42]. Both studies considered the PDRG1-URI interaction abundant, according to the number of peptides retrieved[42,49].
PDRG1 overexpression did not modify URI or Art27 levels, whereas overexpression of URI upregulates PDRG1 protein levels, an effect that was ascribed to protein stabilization[42]. The PDRG1-URI interaction seems to involve the hook-shaped β-strands of the URI N-terminal prefoldin-like domain, through which interaction with Art27 is also established. In fact, these authors suggested that all prefoldin-like proteins of the R2TP/prefoldin-like complex (URI, Art27, PDRG1, PFD2, PFD6) use the hook to interact with each other[42].
AP-MS experiments carried out in HeLa cells identified URI, TIP49/RUVL, RMP and RPB5 in association with STAP-1, and hence potentially linking PDRG1 with signal transduction and the TOR complex[50]. These interactions were further confirmed by immunoprecipitation, where anti-URI binds RPB5 and STAP-1 and anti-TIP48 rendered TIP49, URI, STAP1 and SKP2. Similarly, PDRG1 could be indirectly implicated in the roles of URI as a transcriptional repressor and as a regulator of apoptosis that involve interactions with the RPB5/POLR2E subunit of the three RNA polymerases and PPIγ[51,52]. In the latter case, phosphorylation of mitochondrial URI by S6K1 allows dissociation of the complex. Moreover, the URI complex is targeted by nutrient signaling and, in turn, participates in the control of gene expression downstream of the TOR kinase, a process that is regulated by methylation of the protein phosphatase PP2A[53].
PDRG1 controls production of S-adenosylmethionine
In yeast, URI deletion alters expression of genes involved mainly in amino acid metabolism, although none of the key genes in the methionine cycle seems affected[50]. On the other hand, YTH screening of a rat liver library identified PDRG1 interacting with MATα1, a result that was further confirmed by immunoprecipitation and pull-down[20]. These results linked PDRG1 with methionine metabolism and AdoMet-dependent methylations[20], which are altered in tumor cells.
MATα1 is the catalytic subunit of homotetrameric MAT I and homodimeric MAT III, the isoenzymes synthesizing AdoMet in normal liver, and accumulates in the cell nucleus in liver disease and extrahepatic cells[12,14]. Despite the nucleocytoplasmic localization of both MATα1 and PDRG1, their interaction was restricted to the nuclear compartment, where large association states (approximately 360 kDa) could be detected[20,21]. Taking into account the size of each protein and their individual oligomeric states, this means binding of two PDRG1 hexamers per nuclear MAT I homotetramer. Moreover, pull-down assays also showed the ability of PDRG1 to interact with the MATα2 catalytic subunit of heterotrimeric MAT II, the main isoenzyme found in extrahepatic tissues, fetal liver and hepatic diseases[20,21]. This fact could be expected from the high percentage of identity (84%) between the MATα1 and MATα2 subunits[54]. Additionally, the PDRG1-MATα2 interaction interface seems to overlap with the binding site for the regulatory MATβ subunit that is displaced from the oligomer upon PDRG1 attachment[20]. Further insights into the putative areas of interaction were obtained through the use of truncated PDRG1 forms. These protein variants were designed according to information obtained from the available structural model and lack the N-terminus, the C-terminus or both protein ends[20]. Analysis of their interaction with MATα1 and MATα2 indicated a role for the C-terminal end of PDRG1 in the interaction, an area that is not involved in the interaction with other members of the R2TP/prefoldin-like complex, which seem to use the hook for this purpose[42].
Tissue Pdrg1 expression mimics the expression profile of Mat2a (the MATα2 gene), while following the opposite pattern from that exhibited by Mat1a (the MATα1 gene)[20]. This fact coincides with the preferential nuclear localization of MATα1 in those cell types/tissues where Mat1a expression is low, thus increasing the probability of interaction with PDRG1 in those settings. This interaction impairs AdoMet production by approximately 50%, as demonstrated in vitro using purified MATα1 or MATα2 homo-oligomers or MAT II hetero-oligomers and PDRG1. Similarly, decreased global DNA methylation levels were detected in cultured cells upon overexpression of PDRG1 or coexpression of PDRG1 and MATα1[20,21]. Altogether, these results provide the first evidence for PDRG1 function as a modulator of methyl donor production.
ROLE OF PDRG1 IN DISEASE
To date, there is no disease directly linked to alterations in PDRG1, although the OMIM 610789 code has been associated with this gene. In fact, the NCBI database already includes descriptions of more than 700 SNPs in this gene, including point mutations in the ORF, frameshifts and early stop codons (Table 1). However, the impact of these alterations remains to be studied.
Table 1.
SNPs for the human PDRG1 gene listed in the NCBI database leading to mutated protein forms
| SNP code1 | Base change | Mutated residue | Type of mutation |
| rs777897331 | 1A>C | M1L | Missense |
| rs748585638 | 19G>A | E7K | Missense |
| rs867545839 | 23G>T | R8L | Missense |
| rs779425406 | 29T>C | L10P | Missense |
| rs755286962 | 32G>A | R11Q | Missense |
| rs150958567 | 34T>C | Y12H | Missense |
| rs142496377 | 35A>G | Y12C | Missense |
| rs375829085 | 46G>A | V16M | Missense |
| rs760411451 | 59C>T | A20V | Missense |
| rs763777690 | 57_68delCGCCG- AGGAGGT | A20_V23del | Indel |
| rs765247648 | 67G>T | V23L | Missense |
| rs759432900 | 83G>A | R28Q | Missense |
| rs772260316 | 91G>A | V31M | Missense |
| rs139473332 | 92T>C | V31A | Missense |
| rs147359616 | 110G>T | R37M | Missense |
| rs201481003 | 113A>C | N38T | Missense |
| rs749744129 | 119A>C | N40T | Missense |
| rs142890852 | 121C>G, 121C>T | R41G, R41stop | Missense, stop gained |
| rs539323269 | 122G>A | R41Q | Missense |
| rs746198655 | 134G>A | R45K | Missense |
| rs370045763 | 155G>C | S52T | Missense |
| rs758120112 | 157C>G | L53V | Missense |
| rs748723095 | 175G>T | V59F | Missense |
| rs775880591 | 184G>A | G62R | Missense |
| rs200644098 | 188A>G | N63S | Missense |
| rs767238840 | 190A>G | M64V | Missense |
| rs764202244 | 208C>T | H70Y | Missense |
| rs777531663 | 210_211insAA | P71Nfs | Frameshift |
| rs566583428 | 211C>T | P71S | Missense |
| rs768828941 | 223_224ins2 | E75Vfs | Frameshift |
| rs750997250 | 251_252delTG | L84Rfs | Frameshift |
| rs745401995 | 264A>G | I88M | Missense |
| rs780722451 | 265G>C | E89Q | Missense |
| rs201754951 | 274C>T | R92W | Missense |
| rs186921219 | 275G>A | R92Q | Missense |
| rs140650422 | 295G>A | V99I | Missense |
| rs200378363 | 299A>T | N100I | Missense |
| rs565384763 | 301C>T | R101C | Missense |
| rs779220725 | 302G>A | R101H | Missense |
| rs753937019 | 317A>G | Q106R | Missense |
| rs143995288 | 319G>A | G107S | Missense |
| rs768088765 | 323A>G | K108R | Missense |
| rs200900452 | 326C>T | P109L | Missense |
| rs759134978 | 328G>A | E110K | Missense |
| rs776274160 | 332T>C | L111P | Missense |
| rs370586022 | 340T>C | F114L | Missense |
| rs764544630 | 344_345insA | N115Kfs | Frameshift |
| rs760255766 | 359A>G | N120S | Missense |
| rs772012403 | 361C>T | Q121stop | Stop gained |
| rs149510387 | 365A>G | D122G | Missense |
| rs774273098 | 366T>G | D122E | Missense |
| rs548757558 | 377C>T | A126V | Missense |
Information collected in January 2017;
Inserted sequence TGAGCT TTTCTTTTTTACATGCCCTGCAATAGAGACTGGGTTGA.
PDRG1 and cancer
The chromosomal location of PDRG1 coincides with a region with high frequency of sequence gains in several types of human cancers, including breast, liver, lung, colon and rectum tumors[25,55-59]. Moreover, p53 is downregulated in several human cancers, and hence, its regulation of the PDRG1 gene is expected to be impaired. The same is true for miR-214, which is also downregulated in cervical, breast, HCC and bladder tumors[33,60-62]. Using commercial cancer profiling arrays, 60%-85% upregulation of PDRG1 was detected in a variety of tumor types with impaired p53 expression, confirming this hypothesis[47]. This same study also showed that the percentage of patients with increased PDRG1 levels was dependent on the type of tumor analyzed. In bladder cancer, miR-214 levels were associated with the tumor stage and recurrence, whereas an inverse correlation with PDRG1 expression was detected[33]. Moreover, this tumor suppressor role was found to be exerted through miR-214 binding to the 3’-UTR of the PDRG1 mRNA, in turn leading to a reduction in its levels.
Correlation of elevated PDRG1 and GLUT1 expression with poor pathological response has been reported in residual rectal cancer cells after pre-operative chemotherapy[63]. However, PDRG1 expression levels were not associated with the overall survival of the patients despite its proposed roles in DNA damage regulation and tumor growth. Furthermore, a role for PDRG1 in cancer can also be inferred from the reports linking its interaction target URI to the development of ovarian cancer and HCC[64,65]. In this same line, the reduced miR-214 levels found in breast cancer cells lead to the accumulation of the enhancer of Zeste homologue 2 (EZH2) methyltransferase. EZH2 is a component of the polycomb repressive complex 2 (PRC2) and catalyzes the inclusion of the repressive epigenetic mark H3K27me3[61]. Levels of this repression mark are enhanced in HCC[66], where nuclear accumulation of MATα1 is induced, putatively favoring its interaction with PDRG1. This apparent contradictory effect could be explained by the high affinity of EZH2 for AdoMet, which putatively makes this particular methyltransferase insensitive to the decreased methyl donor concentrations resulting from the nuclear MATα1/PDRG1 interaction.
The upregulated expression of PDRG1 correlates with enhanced PDRG1 protein levels, for example in colon cancer samples (mainly in epithelial cells) and in some lung cancer cell lines (A549 and H446)[47,67]. Tumor cell lines have been used for stable or transient overexpression and downregulation of PDRG1 to study effects on proliferation, apoptosis or the response to UV irradiation. PDRG1 overexpression in A549 and H446 lung cancer cells enhanced proliferation, whereas no morphological changes in NIH 3T3 fibroblasts, HCT116 colon cancer or H35 hepatoma cells were observed[20,22,67]. Regarding apoptosis, no signs of this process were detected in NIH 3T3 or HCT116 cells after overexpression[22]. In contrast, downregulation of PDRG1 in T24 and 5637 bladder cancer cell lines by overexpression of miR-214 led to decreased proliferation, enhanced apoptosis and reduced invasiveness[33]. PDRG1 knockdown in RKO colon and A549 and H446 lung cancer cells also caused reduction in cell growth[47,67]. However, stable silencing in H35 hepatoma cells did not induce significant changes in growth of any of the clones analyzed or in their morphology[20]. This downregulation was never above 70%, suggesting that further suppression of Pdrg1 expression in hepatoma cells could be lethal. Additionally, the differences observed may arise not only from the downregulation extent achieved in each tumor cell but also from the diverse procedures used for silencing and the distinct basal levels in the corresponding normal cells. Altogether, these results suggest a putative role for PDRG1 in apoptosis and proliferation, which might be exerted through its interaction with different targets. Among those identified to date, PDCD7 contributes to the modulation of apoptosis, MAP1S participates in cell cycle progression through its interaction with RASSF1A, and CIZ interacts with p21 for their shuttling from the nucleus to the cytoplasm.
Focusing our attention on stably silenced H35 hepatoma cells, the maximum downregulation achieved is still well above the levels detected in normal liver[20,21]. Nevertheless, important changes in the expression profile were found using microarrays, these alterations affecting a large variety of pathways[20,21]. A total of 114 genes consistently changed their pattern in different clones, of which only 80% could be ascribed to GO pathways. Among upregulated genes, Lpin1 was found. This is an essential gene in fat metabolism that is regulated by p53 and induced by DNA damage and glucose deprivation[68]. Lpin1 codes for a bifunctional protein that catalyzes diacylglycerol synthesis and acts as a transcriptional coactivator in hepatocytes to regulate fatty acid oxidation[68]. In contrast, expression of neither the Tp53 nor the Mat genes were significantly altered in the silenced cells, but many of the routes affected are implicated in pathological processes in which the pattern of Mat gene expression is modified[20,21].
As mentioned in the initial sections of this mini-review, PDRG1 was identified when the effects of UV irradiation were analyzed. Therefore, it was not surprising that stable HCT116 clones overexpressing HA-PDRG1 displayed increased sensitivity to UV irradiation or that this agent upregulated PDRG1 in several human cell lines, including some lacking p53 or carrying mutant forms of this protein[22]. In fact, higher PDRG1 mRNA levels were detected after irradiation upon TP53 repression by doxycycline in Tet-inducible DLD1 colon cancer cells as well as in TP53-/- cells. These data suggested an inverse correlation of PDRG1 and TP53 expression but also with that of Cip1 (the p21 gene, also named CDKN1), in turn indicating involvement of a p53-independent mechanism[22]. Additional studies carried out by Jiang et al[47] demonstrated that only agents inducing genotoxic stress (adriamycin, etoposide, camptothecin and UV) upregulated PDRG1 expression.
The link between PDRG1 and radioresistance was further supported when an inverse correlation between PDRG1 and ATM levels was detected in lung cancer cells after irradiation[67]. In fact, PDRG1-silenced cells exhibited high ATM levels together with increased p53 phosphorylation on serine 15, and tumors generated in nude mice using these silenced clones were more susceptible to irradiation[67]. Oleuropein treatment of nasopharyngeal cancer cell lines reduced PDRG1 and HIF1α expression and protein levels, while inducing miR-519d levels[34]. Further examination of the effects of the treatment revealed that oleuropein avoids HIF1α downregulation of miR-519, in turn decreasing PDRG1 levels in the cells and favoring sensitivity to irradiation both in the cells and in tumor xenografts.
PDRG1 in liver disease
Pdrg1 expression has also been analyzed in models of liver disease, where the Mat1a to Mat2a expression switch was detected together with oxidative stress. The results reported to date indicate no significant alteration of hepatic Pdrg1 levels in early stages of a model of Wilson disease, the Long Evans Cinnamon rats, despite the evident accumulation of copper[20]. However, acute liver injury induced by D-galactosamine intoxication induced the Mat expression switch, nuclear accumulation of MATα1 and the upregulation of Pdrg1 expression and its nuclear levels[20]. Of note, the upregulation of Pdrg1 reported in this model was more modest than that measured in hepatoma cells.
PDRG1 in T cell selection
Autoimmune diseases present altered T cell selection mechanisms that have been explored using microarrays and animal models such as the non-obese diabetic (NOD) mice[69]. Thymocytes obtained from this model of human autoimmune diabetes have a reduced number of genes allowing distinction between positive and negative T cell selection compared to cells from a control mouse strain. This fact was especially important in the negative selection gene set, which included Pdgr1. Basal Pdrg1 expression was reduced in NOD thymocytes, and this gene was also identified among the high-quality candidates for D2mit490-linked defective thymic deletion, D2mit490 being a marker for the loci contributing to the defective negative selection detected in NOD mice[69].
CONCLUSION
PDRG1 is a member of the R2TP/prefoldin-like complex with nucleocytoplasmic distribution. In acute liver injury and a variety of tumors, high levels of PDRG1 expression have been detected. Several protein-protein interactions involving PDRG1 have been found while exploring the components of multiprotein complexes, putatively linking this protein to processes such as splicing, apoptosis and nutrient signaling. However, the role of these interactions and how and when do they occur remains unclear. Increased PDRG1 expression correlates with decreased levels of AdoMet, the main cellular methyl donor. Moreover, this increased expression also correlates with nuclear accumulation of MATα1 and with changes in epigenetic methylations that may be of a different sign depending on the AdoMet affinity of each specific methyltransferase. Such behavior can be explained by the decreased capacity for AdoMet synthesis exhibited by the MAT isoenzymes upon their interaction with PDRG1. This modulatory role of AdoMet levels is the first function ascribed to PDRG1. The fact that this interaction is restricted to the nucleus may exert a counteracting effect on liver pathology, precluding the objectives pursued by the nuclear accumulation of MATα1. Moreover, the interest and impact of this nuclear interaction could be of major importance for most cells, where PDRG1 expression is higher and MATα1 localization is restricted to this compartment. Altogether, the aspects that deserve further attention include the identification of the interaction surfaces in complexes involving PDRG1, how the interactions are regulated, and the structure of the oncogene or its regulation. Knowledge of structural features may be of use for the design of new therapeutic options that, for example, interfere with pathological protein-protein interactions involving PDRG1.
ACKNOWLEDGMENTS
The author wishes to thank Drs. Juliana Pérez-Miguelsanz and Dolores Pérez-Sala for their thoughtful comments on the manuscript and former members of her group at the Instituto de Investigaciones Biomédicas Alberto Sols (CSIC-UAM) for their contributions through the years. The author also wishes to acknowledge the long-standing support by the Ministerio Educación y Ciencia and Ministerio de Economía y Competitividad of Spain (until June 2013).
Footnotes
Conflict-of-interest statement: The author declares no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Manuscript source: Unsolicited manuscript
Specialty type: Biochemistry and molecular biology
Country of origin: Spain
Peer-review report classification
Grade A (Excellent): A, A
Grade B (Very good): B, B
Grade C (Good): 0
Grade D (Fair): D
Grade E (Poor): 0
Peer-review started: October 6, 2017
First decision: October 30, 2017
Article in press: November 20, 2017
P- Reviewer: Chandra D, Chui YL, Miloso M, Ocker M, Rangel-Corona R S- Editor: Ji FF L- Editor: A E- Editor: Lu YJ
References
- 1.Diala ES, Hoffman RM. Hypomethylation of HeLa cell DNA and the absence of 5-methylcytosine in SV40 and adenovirus (type 2) DNA: analysis by HPLC. Biochem Biophys Res Commun. 1982;107:19–26. doi: 10.1016/0006-291x(82)91663-1. [DOI] [PubMed] [Google Scholar]
- 2.Laird PW, Jaenisch R. DNA methylation and cancer. Hum Mol Genet. 1994;3 Spec No:1487–1495. doi: 10.1093/hmg/3.suppl_1.1487. [DOI] [PubMed] [Google Scholar]
- 3.Mecham JO, Rowitch D, Wallace CD, Stern PH, Hoffman RM. The metabolic defect of methionine dependence occurs frequently in human tumor cell lines. Biochem Biophys Res Commun. 1983;117:429–434. doi: 10.1016/0006-291x(83)91218-4. [DOI] [PubMed] [Google Scholar]
- 4.Cantoni GL. Biological methylation: selected aspects. Annu Rev Biochem. 1975;44:435–451. doi: 10.1146/annurev.bi.44.070175.002251. [DOI] [PubMed] [Google Scholar]
- 5.Petrossian TC, Clarke SG. Uncovering the human methyltransferasome. Mol Cell Proteomics. 2011;10:M110.000976. doi: 10.1074/mcp.M110.000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stead LM, Brosnan JT, Brosnan ME, Vance DE, Jacobs RL. Is it time to reevaluate methyl balance in humans? Am J Clin Nutr. 2006;83:5–10. doi: 10.1093/ajcn/83.1.5. [DOI] [PubMed] [Google Scholar]
- 7.Mudd SH, Brosnan JT, Brosnan ME, Jacobs RL, Stabler SP, Allen RH, Vance DE, Wagner C. Methyl balance and transmethylation fluxes in humans. Am J Clin Nutr. 2007;85:19–25. doi: 10.1093/ajcn/85.1.19. [DOI] [PubMed] [Google Scholar]
- 8.Luka Z, Mudd SH, Wagner C. Glycine N-methyltransferase and regulation of S-adenosylmethionine levels. J Biol Chem. 2009;284:22507–22511. doi: 10.1074/jbc.R109.019273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brosnan JT, da Silva RP, Brosnan ME. The metabolic burden of creatine synthesis. Amino Acids. 2011;40:1325–1331. doi: 10.1007/s00726-011-0853-y. [DOI] [PubMed] [Google Scholar]
- 10.Vance DE. Phospholipid methylation in mammals: from biochemistry to physiological function. Biochim Biophys Acta. 2014;1838:1477–1487. doi: 10.1016/j.bbamem.2013.10.018. [DOI] [PubMed] [Google Scholar]
- 11.Mato JM, Pajares MA, Varela I. How many phopsholipid methyltransferases are there in mammalian cells? Trends Biochem Sci. 1984;9:471–472. [Google Scholar]
- 12.Reytor E, Pérez-Miguelsanz J, Alvarez L, Pérez-Sala D, Pajares MA. Conformational signals in the C-terminal domain of methionine adenosyltransferase I/III determine its nucleocytoplasmic distribution. FASEB J. 2009;23:3347–3360. doi: 10.1096/fj.09-130187. [DOI] [PubMed] [Google Scholar]
- 13.Katoh Y, Ikura T, Hoshikawa Y, Tashiro S, Ito T, Ohta M, Kera Y, Noda T, Igarashi K. Methionine adenosyltransferase II serves as a transcriptional corepressor of Maf oncoprotein. Mol Cell. 2011;41:554–566. doi: 10.1016/j.molcel.2011.02.018. [DOI] [PubMed] [Google Scholar]
- 14.Delgado M, Garrido F, Pérez-Miguelsanz J, Pacheco M, Partearroyo T, Pérez-Sala D, Pajares MA. Acute liver injury induces nucleocytoplasmic redistribution of hepatic methionine metabolism enzymes. Antioxid Redox Signal. 2014;20:2541–2554. doi: 10.1089/ars.2013.5342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krupenko NI, Wagner C. Transport of rat liver glycine N-methyltransferase into rat liver nuclei. J Biol Chem. 1997;272:27140–27146. doi: 10.1074/jbc.272.43.27140. [DOI] [PubMed] [Google Scholar]
- 16.Radomski N, Kaufmann C, Dreyer C. Nuclear accumulation of S-adenosylhomocysteine hydrolase in transcriptionally active cells during development of Xenopus laevis. Mol Biol Cell. 1999;10:4283–4298. doi: 10.1091/mbc.10.12.4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pajares MA, Markham GD. Methionine adenosyltransferase (s-adenosylmethionine synthetase) Adv Enzymol Relat Areas Mol Biol. 2011;78:449–521. doi: 10.1002/9781118105771.ch11. [DOI] [PubMed] [Google Scholar]
- 18.Pajares MA, Pérez-Sala D. Betaine homocysteine S-methyltransferase: just a regulator of homocysteine metabolism? Cell Mol Life Sci. 2006;63:2792–2803. doi: 10.1007/s00018-006-6249-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pérez-Miguelsanz J, Vallecillo N, Garrido F, Reytor E, Pérez-Sala D, Pajares MA. Betaine homocysteine S-methyltransferase emerges as a new player of the nuclear methionine cycle. Biochim Biophys Acta. 2017;1864:1165–1182. doi: 10.1016/j.bbamcr.2017.03.004. [DOI] [PubMed] [Google Scholar]
- 20.Pérez C, Pérez-Zúñiga FJ, Garrido F, Reytor E, Portillo F, Pajares MA. The Oncogene PDRG1 Is an Interaction Target of Methionine Adenosyltransferases. PLoS One. 2016;11:e0161672. doi: 10.1371/journal.pone.0161672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pérez C, Pérez-Zúñiga FJ, Garrido F, Reytor E, Portillo F, Pajares MA. Correction: The Oncogene PDRG1 Is an Interaction Target of Methionine Adenosyltransferases. PLoS One. 2016;11:e0163761. doi: 10.1371/journal.pone.0161672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luo X, Huang Y, Sheikh MS. Cloning and characterization of a novel gene PDRG that is differentially regulated by p53 and ultraviolet radiation. Oncogene. 2003;22:7247–7257. doi: 10.1038/sj.onc.1207010. [DOI] [PubMed] [Google Scholar]
- 23.Sheikh MS, Chen YQ, Smith ML, Fornace AJ Jr. Role of p21Waf1/Cip1/Sdi1 in cell death and DNA repair as studied using a tetracycline-inducible system in p53-deficient cells. Oncogene. 1997;14:1875–1882. doi: 10.1038/sj.onc.1201004. [DOI] [PubMed] [Google Scholar]
- 24.Sheikh MS, Fernandez-Salas E, Yu M, Hussain A, Dinman JD, Peltz SW, Huang Y, Fornace AJ Jr. Cloning and characterization of a human genotoxic and endoplasmic reticulum stress-inducible cDNA that encodes translation initiation factor 1(eIF1(A121/SUI1)) J Biol Chem. 1999;274:16487–16493. doi: 10.1074/jbc.274.23.16487. [DOI] [PubMed] [Google Scholar]
- 25.Raidl M, Pirker C, Schulte-Hermann R, Aubele M, Kandioler-Eckersberger D, Wrba F, Micksche M, Berger W, Grasl-Kraupp B. Multiple chromosomal abnormalities in human liver (pre)neoplasia. J Hepatol. 2004;40:660–668. doi: 10.1016/j.jhep.2003.12.020. [DOI] [PubMed] [Google Scholar]
- 26.Johnson RA, Ince TA, Scotto KW. Transcriptional repression by p53 through direct binding to a novel DNA element. J Biol Chem. 2001;276:27716–27720. doi: 10.1074/jbc.C100121200. [DOI] [PubMed] [Google Scholar]
- 27.Resnick-Silverman L, St Clair S, Maurer M, Zhao K, Manfredi JJ. Identification of a novel class of genomic DNA-binding sites suggests a mechanism for selectivity in target gene activation by the tumor suppressor protein p53. Genes Dev. 1998;12:2102–2107. doi: 10.1101/gad.12.14.2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nat Genet. 1992;1:45–49. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- 29.Wong J, Li PX, Klamut HJ. A novel p53 transcriptional repressor element (p53TRE) and the asymmetrical contribution of two p53 binding sites modulate the response of the placental transforming growth factor-beta promoter to p53. J Biol Chem. 2002;277:26699–26707. doi: 10.1074/jbc.M203020200. [DOI] [PubMed] [Google Scholar]
- 30.Latchman DS. POU family transcription factors in the nervous system. J Cell Physiol. 1999;179:126–133. doi: 10.1002/(SICI)1097-4652(199905)179:2<126::AID-JCP2>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 31.Takahashi S, Saito S, Ohtani N, Sakai T. Involvement of the Oct-1 regulatory element of the gadd45 promoter in the p53-independent response to ultraviolet irradiation. Cancer Res. 2001;61:1187–1195. [PubMed] [Google Scholar]
- 32.Zhao H, Jin S, Fan F, Fan W, Tong T, Zhan Q. Activation of the transcription factor Oct-1 in response to DNA damage. Cancer Res. 2000;60:6276–6280. [PubMed] [Google Scholar]
- 33.Wang J, Zhang X, Wang L, Yang Y, Dong Z, Wang H, Du L, Wang C. MicroRNA-214 suppresses oncogenesis and exerts impact on prognosis by targeting PDRG1 in bladder cancer. PLoS One. 2015;10:e0118086. doi: 10.1371/journal.pone.0118086. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Xu T, Xiao D. Oleuropein enhances radiation sensitivity of nasopharyngeal carcinoma by downregulating PDRG1 through HIF1α-repressed microRNA-519d. J Exp Clin Cancer Res. 2017;36:3. doi: 10.1186/s13046-016-0480-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharma K, D’Souza RC, Tyanova S, Schaab C, Wiśniewski JR, Cox J, Mann M. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 2014;8:1583–1594. doi: 10.1016/j.celrep.2014.07.036. [DOI] [PubMed] [Google Scholar]
- 36.Mertins P, Yang F, Liu T, Mani DR, Petyuk VA, Gillette MA, Clauser KR, Qiao JW, Gritsenko MA, Moore RJ, et al. Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol Cell Proteomics. 2014;13:1690–1704. doi: 10.1074/mcp.M113.036392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou H, Di Palma S, Preisinger C, Peng M, Polat AN, Heck AJ, Mohammed S. Toward a comprehensive characterization of a human cancer cell phosphoproteome. J Proteome Res. 2013;12:260–271. doi: 10.1021/pr300630k. [DOI] [PubMed] [Google Scholar]
- 38.Olsen CM, Carroll HJ, Whiteman DC. Estimating the attributable fraction for cancer: A meta-analysis of nevi and melanoma. Cancer Prev Res (Phila) 2010;3:233–245. doi: 10.1158/1940-6207.CAPR-09-0108. [DOI] [PubMed] [Google Scholar]
- 39.Wu X, Tian L, Li J, Zhang Y, Han V, Li Y, Xu X, Li H, Chen X, Chen J, et al. Investigation of receptor interacting protein (RIP3)-dependent protein phosphorylation by quantitative phosphoproteomics. Mol Cell Proteomics. 2012;11:1640–1651. doi: 10.1074/mcp.M112.019091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wiśniewski JR, Nagaraj N, Zougman A, Gnad F, Mann M. Brain phosphoproteome obtained by a FASP-based method reveals plasma membrane protein topology. J Proteome Res. 2010;9:3280–3289. doi: 10.1021/pr1002214. [DOI] [PubMed] [Google Scholar]
- 41.Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2011;44:325–340. doi: 10.1016/j.molcel.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mita P, Savas JN, Ha S, Djouder N, Yates JR 3rd, Logan SK. Analysis of URI nuclear interaction with RPB5 and components of the R2TP/prefoldin-like complex. PLoS One. 2013;8:e63879. doi: 10.1371/journal.pone.0063879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jeronimo C, Forget D, Bouchard A, Li Q, Chua G, Poitras C, Thérien C, Bergeron D, Bourassa S, Greenblatt J, et al. Systematic analysis of the protein interaction network for the human transcription machinery reveals the identity of the 7SK capping enzyme. Mol Cell. 2007;27:262–274. doi: 10.1016/j.molcel.2007.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boulon S, Pradet-Balade B, Verheggen C, Molle D, Boireau S, Georgieva M, Azzag K, Robert MC, Ahmad Y, Neel H, et al. HSP90 and its R2TP/Prefoldin-like cochaperone are involved in the cytoplasmic assembly of RNA polymerase II. Mol Cell. 2010;39:912–924. doi: 10.1016/j.molcel.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruhl DD, Jin J, Cai Y, Swanson S, Florens L, Washburn MP, Conaway RC, Conaway JW, Chrivia JC. Purification of a human SRCAP complex that remodels chromatin by incorporating the histone variant H2A.Z into nucleosomes. Biochemistry. 2006;45:5671–5677. doi: 10.1021/bi060043d. [DOI] [PubMed] [Google Scholar]
- 46.Jin J, Cai Y, Yao T, Gottschalk AJ, Florens L, Swanson SK, Gutiérrez JL, Coleman MK, Workman JL, Mushegian A, et al. A mammalian chromatin remodeling complex with similarities to the yeast INO80 complex. J Biol Chem. 2005;280:41207–41212. doi: 10.1074/jbc.M509128200. [DOI] [PubMed] [Google Scholar]
- 47.Jiang L, Luo X, Shi J, Sun H, Sun Q, Sheikh MS, Huang Y. PDRG1, a novel tumor marker for multiple malignancies that is selectively regulated by genotoxic stress. Cancer Biol Ther. 2011;11:567–573. doi: 10.4161/cbt.11.6.14412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Will CL, Schneider C, Hossbach M, Urlaub H, Rauhut R, Elbashir S, Tuschl T, Lührmann R. The human 18S U11/U12 snRNP contains a set of novel proteins not found in the U2-dependent spliceosome. RNA. 2004;10:929–941. doi: 10.1261/rna.7320604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sardiu ME, Cai Y, Jin J, Swanson SK, Conaway RC, Conaway JW, Florens L, Washburn MP. Probabilistic assembly of human protein interaction networks from label-free quantitative proteomics. Proc Natl Acad Sci USA. 2008;105:1454–1459. doi: 10.1073/pnas.0706983105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gstaiger M, Luke B, Hess D, Oakeley EJ, Wirbelauer C, Blondel M, Vigneron M, Peter M, Krek W. Control of nutrient-sensitive transcription programs by the unconventional prefoldin URI. Science. 2003;302:1208–1212. doi: 10.1126/science.1088401. [DOI] [PubMed] [Google Scholar]
- 51.Dorjsuren D, Lin Y, Wei W, Yamashita T, Nomura T, Hayashi N, Murakami S. RMP, a novel RNA polymerase II subunit 5-interacting protein, counteracts transactivation by hepatitis B virus X protein. Mol Cell Biol. 1998;18:7546–7555. doi: 10.1128/mcb.18.12.7546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Djouder N, Metzler SC, Schmidt A, Wirbelauer C, Gstaiger M, Aebersold R, Hess D, Krek W. S6K1-mediated disassembly of mitochondrial URI/PP1gamma complexes activates a negative feedback program that counters S6K1 survival signaling. Mol Cell. 2007;28:28–40. doi: 10.1016/j.molcel.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 53.Sutter BM, Wu X, Laxman S, Tu BP. Methionine inhibits autophagy and promotes growth by inducing the SAM-responsive methylation of PP2A. Cell. 2013;154:403–415. doi: 10.1016/j.cell.2013.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sánchez-Pérez GF, Bautista JM, Pajares MA. Methionine adenosyltransferase as a useful molecular systematics tool revealed by phylogenetic and structural analyses. J Mol Biol. 2004;335:693–706. doi: 10.1016/j.jmb.2003.11.022. [DOI] [PubMed] [Google Scholar]
- 55.Hodgson JG, Chin K, Collins C, Gray JW. Genome amplification of chromosome 20 in breast cancer. Breast Cancer Res Treat. 2003;78:337–345. doi: 10.1023/a:1023085825042. [DOI] [PubMed] [Google Scholar]
- 56.Wong MP, Fung LF, Wang E, Chow WS, Chiu SW, Lam WK, Ho KK, Ma ES, Wan TS, Chung LP. Chromosomal aberrations of primary lung adenocarcinomas in nonsmokers. Cancer. 2003;97:1263–1270. doi: 10.1002/cncr.11183. [DOI] [PubMed] [Google Scholar]
- 57.Schlegel J, Stumm G, Scherthan H, Bocker T, Zirngibl H, Rüschoff J, Hofstädter F. Comparative genomic in situ hybridization of colon carcinomas with replication error. Cancer Res. 1995;55:6002–6005. [PubMed] [Google Scholar]
- 58.Lukásová E, Kozubek S, Falk M, Kozubek M, Zaloudík J, Vagunda V, Pavlovský Z. Topography of genetic loci in the nuclei of cells of colorectal carcinoma and adjacent tissue of colonic epithelium. Chromosoma. 2004;112:221–230. doi: 10.1007/s00412-003-0263-3. [DOI] [PubMed] [Google Scholar]
- 59.Verbeek W, Schulten HJ, Sperling M, Tiesmeier J, Stoop H, Dinjens W, Looijenga L, Wörmann B, Füzesi L, Donhuijsen K. Rectal adenocarcinoma with choriocarcinomatous differentiation: clinical and genetic aspects. Hum Pathol. 2004;35:1427–1430. doi: 10.1016/j.humpath.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 60.Peng RQ, Wan HY, Li HF, Liu M, Li X, Tang H. MicroRNA-214 suppresses growth and invasiveness of cervical cancer cells by targeting UDP-N-acetyl-α-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 7. J Biol Chem. 2012;287:14301–14309. doi: 10.1074/jbc.M111.337642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Derfoul A, Juan AH, Difilippantonio MJ, Palanisamy N, Ried T, Sartorelli V. Decreased microRNA-214 levels in breast cancer cells coincides with increased cell proliferation, invasion and accumulation of the Polycomb Ezh2 methyltransferase. Carcinogenesis. 2011;32:1607–1614. doi: 10.1093/carcin/bgr184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shih TC, Tien YJ, Wen CJ, Yeh TS, Yu MC, Huang CH, Lee YS, Yen TC, Hsieh SY. MicroRNA-214 downregulation contributes to tumor angiogenesis by inducing secretion of the hepatoma-derived growth factor in human hepatoma. J Hepatol. 2012;57:584–591. doi: 10.1016/j.jhep.2012.04.031. [DOI] [PubMed] [Google Scholar]
- 63.Saigusa S, Tanaka K, Toiyama Y, Matsushita K, Kawamura M, Okugawa Y, Hiro J, Inoue Y, Uchida K, Mohri Y, et al. Gene expression profiles of tumor regression grade in locally advanced rectal cancer after neoadjuvant chemoradiotherapy. Oncol Rep. 2012;28:855–861. doi: 10.3892/or.2012.1863. [DOI] [PubMed] [Google Scholar]
- 64.Theurillat JP, Metzler SC, Henzi N, Djouder N, Helbling M, Zimmermann AK, Jacob F, Soltermann A, Caduff R, Heinzelmann-Schwarz V, et al. URI is an oncogene amplified in ovarian cancer cells and is required for their survival. Cancer Cell. 2011;19:317–332. doi: 10.1016/j.ccr.2011.01.019. [DOI] [PubMed] [Google Scholar]
- 65.Yang Y, Zheng L, Chen Y. [Study of HBV X protein and RMP, an RPB5 mediate protein competitively interacting with general transcription factor TF2B] Zhonghua Gan Zang Bing Za Zhi. 2000;8:15–17. [PubMed] [Google Scholar]
- 66.Yang H, Cho ME, Li TW, Peng H, Ko KS, Mato JM, Lu SC. MicroRNAs regulate methionine adenosyltransferase 1A expression in hepatocellular carcinoma. J Clin Invest. 2013;123:285–298. doi: 10.1172/JCI63861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tao Z, Chen S, Mao G, Xia H, Huang H, Ma H. The PDRG1 is an oncogene in lung cancer cells, promoting radioresistance via the ATM-P53 signaling pathway. Biomed Pharmacother. 2016;83:1471–1477. doi: 10.1016/j.biopha.2016.08.034. [DOI] [PubMed] [Google Scholar]
- 68.Assaily W, Rubinger DA, Wheaton K, Lin Y, Ma W, Xuan W, Brown-Endres L, Tsuchihara K, Mak TW, Benchimol S. ROS-mediated p53 induction of Lpin1 regulates fatty acid oxidation in response to nutritional stress. Mol Cell. 2011;44:491–501. doi: 10.1016/j.molcel.2011.08.038. [DOI] [PubMed] [Google Scholar]
- 69.Liston A, Hardy K, Pittelkow Y, Wilson SR, Makaroff LE, Fahrer AM, Goodnow CC. Impairment of organ-specific T cell negative selection by diabetes susceptibility genes: genomic analysis by mRNA profiling. Genome Biol. 2007;8:R12. doi: 10.1186/gb-2007-8-1-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]