

Abstract
The dichloromethane extract of the roots of Jatropha dioica afforded riolozatrione (1) and a C-6 epimer of riolozatrione, 6-epi-riolozatrione (2), as a new structure and only the second reported riolozane diterpenoid. The two known diterpenoids jatrophatrione (3) and citlalitrione (4) were also isolated and characterized. Both epimers 1 and 2 are genuine plant constituents, with 2 likely being the biosynthesis precursor of 1 due to the tendency for the quantitative transformation of 2 into 1 under base catalysis. The structural characterization and distinction of the stereoisomers utilized 1H iterative full-spin analysis, yielding complete J-correlation maps that were represented as quantum interaction and linkage tables. The absolute configuration of compounds 1–4 was established by means of vibrational circular dichroism and via X-ray diffraction analysis for 1, 2, and 4. Additionally, the cytotoxic and antiherpetic in vitro activities of the isolates were evaluated.
Graphical abstract
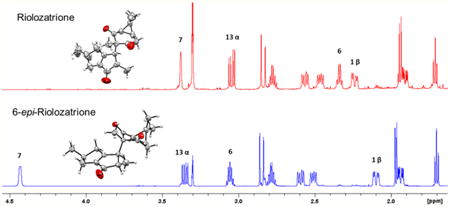
Jatropha dioica, commonly known as “sangre de drago” (“dragon's blood”), has been used in Mexican herbal medicine since the pre-Hispanic era.1 Active compounds reported from J. dioica roots include riolozatrione (1),2 which was hypothesized to arise from the rearrangement of lathyrol or an unknown macrocyclic precursor. To date, the rearranged diterpenoid skeleton (riolozane) of 1 is the only known structure of its kind, and Jatropha species are its sole natural source. Recently, 1 was reported to exhibit a weak in vitro activity against herpes simplex virus (HSV).3 Other Jatropha diterpenoids comprise jatrophatrione (3), a tricyclic compound including a nine-membered ring isolated from the roots of J. microrhiza, which has displayed tumor-inhibitory activity in the P-388 (3PS) lymphocytic leukemia assay.4 A congeneric compound, citlalitrione (4), has subsequently been reported from J. dioica,5 but its bioactivity has not been evaluated. The total synthesis of 3 and 4 has been accomplished, their relative configuration has been confirmed by X-ray crystallography, 4,5 and the identity of synthesized 3 and 4 relative to the isolated natural product has been demonstrated by NMR spectroscopy.6
Ongoing studies regarding the biosynthesis of riolozatrione (1) triggered a search for congeneric compounds and led to the isolation of a new riolozane, 6-epi-riolozatrione (2), from the CH2Cl2 extract of J. dioica roots. Unambiguous establishment of its structure required detailed 1D and 2D NMR studies, including full-spin analysis, and its absolute configuration was determined by means of vibrational circular dichroism (VCD) and X-ray diffraction analysis. Recent insights in the understanding of the terminal steps in the biosynthesis of the two riolozanes are in line with the assignment of the absolute configurations of 1 and 2 established in the present study, as well as with those of other diterpenoids from J. dioica.
While the reported stereochemistry of 1 could be confirmed, its NMR assignments are in need of revision. The close similarity between 1 and 2 necessitated a 1H iterative full-spin analysis (HiFSA). Their full 1H NMR spin parameters include complete J-coupling relationships that were compiled in the form of quantum interaction and linkages tables (QuILTs).7 In addition, the shared absolute configurations of 1–4 and highly congruent structures of 1 and 2 indicate their close biogenetic relationship. While this article was under preparation, the absolute configurations of 3 and 4 were reported by experimental and calculated IR and VCD spectra using DFT B3LYP/DGDZVP level of theory calculations, as well as by single-crystal X-ray diffraction analysis of 4.8 Moreover, this study shows the good agreement between computed VCD spectra that are based on conformational analysis of 3 and 4 at the mPW1B95/DGDZVP and B3PW91/DGDZVP levels and the experimental VCD results. In support of the possible potential of J. dioica diter-penoids as bioactive leads, the present study evaluated the cytotoxicity and anti-HSV activities of 2, 3, and 4, comparing their activities with those reported previously for 1.3
Results and Discussion
As J. dioica is known to produce the structurally unique diterpenoid riolozatrione (1), studies of the biosynthesis pathways of the distinctive riolozane skeleton can benefit from a search of congeneric compounds. Conventional chromatography of the dichloromethane extract of J. dioica roots afforded compounds 1–4 (Chart 1 and Figure S1, Supporting Information). Compound 2 was obtained as colorless crystals and subjected to NMR, IR, and MS analysis. HR-EIMS established its molecular formula as C20H26O3 (Figures S2 and S3, Supporting Information). The UV maximum at 243 nm confirmed the presence of an α,β unsaturated carbonyl moiety, and IR absorptions at 2952 and 2870 cm−1 indicated C–H stretching from alkyl groups, whereas bands at 1722 and 1696 cm−1 were consistent with nonconjugated and conjugated carbonyl groups, respectively (Figure S4, Supporting Information). The 400 MHz 1H NMR spectrum in CDCl3 showed five methyl signals, two doublets at δ 1.15 and 1.07 and three singlets at δ 1.23, 1.08, and 0.85, suggesting the terpenoidal origin of 2. Several highly complex resonance patterns between 3.2 and 1.5 ppm and a methine proton giving rise to a broad doublet at δ 4.22 indicated the aliphatic nature of the structure (Figure S5, Supporting Information). Comparison of the 1H NMR chemical shifts of 2 with those of 1 showed close similarities albeit with specific differences. An array of 1D and 2D NMR experiments supported the structure of 2 as a riolozane-type diterpenoid epimeric with compound 1 (Figures S6, S8, S9, S10, and S11, Supporting Information). Signals ascribed to H-6 and H-7 of 2 appeared at lower field, and the H3-18 and H3-20 methyl protons were shifted upfield relative to 1. Therefore, the structural difference between the compounds had to be in the C-6 and/or C-7 configurations. Such a difference was expected to cause a conformational change in the six-membered ring and, therefore, a change in the spin–spin coupling pattern of H-12 and H-13 (Figure S5, Supporting Information).
Chart 1.
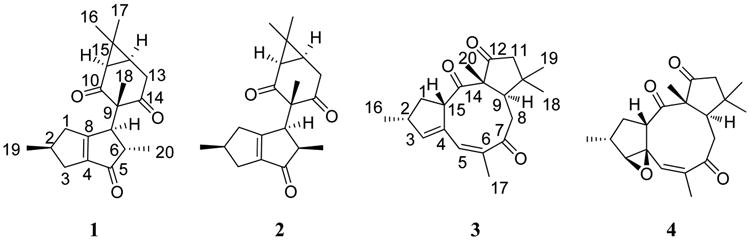
The 13C NMR spectrum exhibited 20 signals, confirming the diterpenoid character of 2. DEPT experiments indicated that these 20 signals corresponded to five methyl, three methylene, five methine, seven nonprotonated, three carbonyl, and two olefinic carbons. Notably, the similarities in the pattern of the olefinic carbons of 1 (180.62 and 149.33 ppm) vs 2 (181.27 and 149.67 ppm) was interpreted to represent a characteristic pattern of a 3,4,5,6-tetrahydropentalen-1(2H)-one system (Figure S5, Supporting Information).
In order to generate unambiguous NMR reference data, the spectra of 1 were re-examined with respect to the original communication.2 The corrected and complete 1H and 13C NMR assignments in CDCl3 and methanol-d4 are shown in Table 1. The relative configuration of 2 was established on the basis of NOESY correlations and compared with those of 1 (Figures 1 and S11, Supporting Information). In both compounds, the dipolar coupling of H3-18 to H-1b and of H3-19 to H-1b and H-3b allowed the assignment of the β-orientations of H-1b and H-3b. Accordingly, H-1a, H-2, and H-3a could be assigned as α-oriented. The NOE correlations between H3-18 and H-6 and between H3-20 and H-7 were observed only in 1, indicating the different chirality of the C-6 and C-7 stereogenic centers in these compounds. Moreover, dipolar couplings between the H3-18 and H3-20 methyl protons and NOE correlations between H-6 and H-7 were observed only in 2. Considering the observed differences in the dipolar couplings of 1 vs 2 showed that 2 is the C-6 epimer of 1. Compared to 1, the 13C NMR resonances of the adjacent C3-18, C-9, and C-7 of 2 show significantly reduced intensities and peak broadening (Figure S5, Supporting Information). Peak intensity increased slightly when longer relaxation delays (20 and 40 s) were applied, indicating the prevalence of dynamic (conformational averaging) over relaxation effects leading to this characteristic behavior of the three carbon resonances. Preliminary evaluation of rotameric populations indicated similar energy barriers for 1 and 2, and this is in line with variable-temperature (−40 to +40 °C) 13C NMR measurements, showing no major effects on molecular dynamics in this temperature range. More detailed dynamic NMR studies and consideration of the keto-enol tautomerism and its impact on both the dynamics and the relative stability of the two epimers are required to fully explain the underlying mechanism, but were beyond the scope of the present study.
Table 1. NMR Spectroscopic Data of Riolozatrione (1) and 6-epi-Riolozatrione (2).
| pos. | type | CDCl3-d at 700 MHz (1H) and 100 MHz (13C) | 1 | 2 | methanol-d4 at 700 MHz (1H) and 100 MHz (13C) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||||||||||
| 1 | 2 | 1 | 2 | ||||||||||||||
|
|
|
|
|
|
|||||||||||||
|
δH [ppm], mult |
J [Hz] | δC |
δH [ppm], mult |
J [Hz] | δC | aΔδH | aΔδC | aΔδH | aΔδC |
δH [ppm], mult |
J [Hz] | δC |
δH [ppm], mult |
J [Hz] | δC | ||
| 1α | CH2 | 2.4853 dddddd | -18.56, 8.28, 2.14, 1.59, 1.02, 0.63 | 40.46 | 2.4794 dddddd | -18.47, 8.24, 2.20, 1.51, 1.12, 0.87 | 40.35 | -0.0784 | -1.15 | -0.1128 | -1.16 | 2.5637 dddddd | -18.72, 8.21, 2.07, 1.69, 1.10, 0.75 | 41.61 | 2.5922 dddddd | -18.58, 8.22, 2.07, 1.61, 1.17, 0.94 | 41.51 |
| 1β | 2.3103 dddddd | -18.56, 6.65, 2.05, 2.04, 1.38, 0.76 | 2.1745 dddddd | -18.47, 6.51, 2.14, 2.05, 1.50, 0.83 | 0.0725 | 0.00 | 0.0764 | 0.00 | 2.2378 dddddd | -18.72, 6.50, 2.04, 2.03, 1.36, 0.78 | 2.0981 dddddd | -18.58, 6.34, 2.00, 1.95, 1.31, 1.00 | |||||
| 2 | CH | 2.7534 ddqdd | 8.41, 8.28, 6.97, 6.65, 6.60 | 37.99 | 2.7580 ddqdd | 8.34, 8.24, 6.95, 6.51, 6.49 | 37.83 | -0.0231 | -1.35 | -0.0270 | -1.27 | 2.7765 ddqdd | 8.40, 8.21, 6.95, 6.50, 6.40 | 39.34 | 2.7850 ddqdd | 8.32, 8.22, 6.96, 6.34, 6.32, 0.50 | 39.10 |
| 3α | CH2 | 2.4949 ddddd | -16.04, 8.40, 2.84, 2.05, 1.02 | 32.58 | 2.5406 ddddd | -16.00, 8.34, 2.82, 2.05, 1.12 | 32.70 | 0.0322 | -0.91 | 0.0317 | -0.92 | 2.4627 ddddd | -15.90, 8.40, 2.85, 2.03, 1.10 | 33.49 | 2.5089 ddddd | -15.85, 8.32, 2.76, 2.00, 1.17, 0.27 | 33.62 |
| 3β | 1.9619 ddddd | -16.04, 6.60, 2.29, 2.14, 2.04 | 2.0021 ddddd | -16.00, 6.48, 2.20, 2.18, 2.14 | 0.0544 | 0.00 | 0.0658 | 0.00 | 1.9075 dddddd | -15.90, 6.40, 2.24, 2.07, 2.04, 0.20 | 1.9363 ddddd | -15.85, 6.32, 2.22, 2.07, 1.95 | |||||
| 4 | C | 149.67 | 149.33 | -0.63 | -0.55 | 150.30 | 149.88 | ||||||||||
| 5 | C | 203.44 | 203.30 | -2.87 | -3.27 | 206.31 | 206.57 | ||||||||||
| 6 | CH | 2.4564 br qddd | 7.46, 2.01, 0.76, 0.63 | 47.94 | 2.9213 qddd | 7.75, 6.32, 0.87, 0.83 | 50.29 | 0.1193 | -1.57 | -0.1346 | -1.51 | 2.3371 qdddd | 7.46, 1.90, 0.78, 0.75, 0.20 | 49.51 | 3.0559 qddd | 7.71, 6.14, 1.00, 0.94, 0.27 | 51.80 |
| 7 | CH | 3.1265 ddddd | 2.84, 2.29, 2.01, 1.59, 1.38 | 52.63 | 4.2193 dddddd | 6.32, 2.82, 2.18, 1.51, 1.50, -0.35 | 45.61 | -0.2517 | -0.30 | -0.2126 | -1.30 | 3.3782 ddddd | 2.85, 2.24, 1.90, 1.69, 1.36 | 52.93 | 4.4319 dddddq | 6.14, 2.76, 2.22, 1.61, 1.31, 0.50, -0.41 | 46.91 |
| 8 | C | 181.27 | 180.62 | -3.50 | -3.48 | 184.77 | 184.10 | ||||||||||
| 9 | C | 65.54 | 68.59 | -1.78 | -1.63 | 67.32 | 70.22 | ||||||||||
| 10 | C | 206.79 | 205.27 | -2.01 | -2.21 | 208.80 | 207.48 | ||||||||||
| 11 | CH | 1.8832 br dd | 7.68, -0.47, -0.12 | 33.16 | 1.8648 d | 7.81 | 33.16 | -0.0557 | -0.92 | -0.1029 | -1.31 | 1.9389 ddd | 7.79, -0.50, -0.20 | 34.08 | 1.9677 d | 7.94 | 34.47 |
| 12 | CH | 1.6141 ddd | 8.01, 7.68, 0.64 | 22.73 | 1.6079 ddd | 8.38, 7.81, 0.64 | 21.82 | -0.0960 | -1.00 | -0.0931 | -1.43 | 1.7101 ddd | 7.93, 7.79, 0.96 | 23.73 | 1.7010 ddd | 8.44, 7.94, 0.64 | 23.25 |
| 13α | CH2 | 2.8822 ddd [ABX] | -18.16, 8.01, -0.47 | 35.85 | 3.1215 dd | -16.95, 8.37 | 36.76 | -0.1615 | -0.90 | -0.2285 | -1.26 | 3.0437 ddd | -18.51, 7.93, -0.20 | 36.75 | 3.3500 dd | -16.99, 8.44 | 38.02 |
| 13β | 2.8848 br dd [ABX] | -18.16, 0.64, -0.12 | 2.9313 dd | -16.95, 0.64 | 0.0473 | 0.0836 | 2.8375 ddd | -18.51, 0.96, -0.50 | 2.8477 dd | -16.99, 0.64 | |||||||
| 14 | C | 207.14 | 206.83 | -2.03 | -2.64 | 209.17 | 209.47 | ||||||||||
| 15 | C | 26.18 | 26.29 | -0.94 | -1.13 | 27.12 | 27.42 | ||||||||||
| 16 | CH3 | 1.2359 s | 28.31 | 1.2265 s | 28.43 | 0.0137 | -0.11 | 0.0194 | -0.17 | 1.2222 s | 28.42 | 1.2071 s | 28.60 | ||||
| 17 | CH3 | 0.8529 s | 16.74 | 0.8041 s | 16.45 | 0.0635 | -0.44 | 0.0704 | -0.44 | 0.7894 s | 17.18 | 0.7337 s | 16.89 | ||||
| 18 | CH3 | 1.0881 s | 12.42 | 1.0196 d | -0.35 | 10.70 | 0.1207 | 0.56 | 0.1135 | -0.24 | 0.9674 s | 11.86 | 0.9061 d | -0.41 | 10.94 | ||
| 19 | CH3 | 1.1535 d | 6.97 | 21.48 | 1.1560 d | 6.95 | 21.51 | 0.0128 | -0.54 | 0.0175 | -0.54 | 1.1407 d | 6.95 | 22.02 | 1.1385 d | 6.95 | 22.05 |
| 20 | CH3 | 1.1571 d | 7.46 | 17.62 | 1.0702 d | 7.75 | 11.84 | 0.0300 | -0.14 | 0.0812 | -0.25 | 1.1271 d | 7.46 | 17.76 | 0.9890 d | 7.71 | 12.09 |
Δδ = difference between CDCl3-d and Methanol-d4 values.
Figure 1.
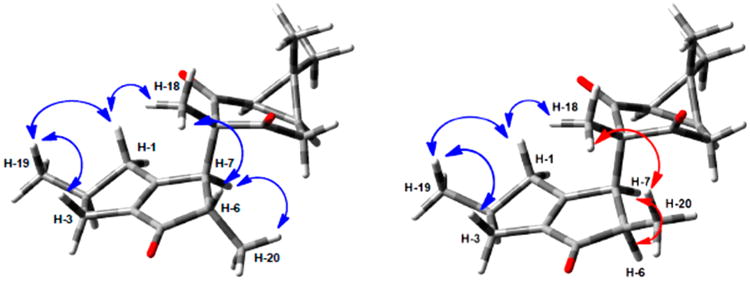
Observed NOE correlations for compounds 1 (left) and 2 (right).
In order to establish the precise difference between 1 and 2, a full determination of the 1H,1H coupling network in both molecules was undertaken using the 1D 1H NMR spectra acquired in CDCl3 at 400 and 700 MHz. Processing the FIDs with Lorentzian–Gaussian apodization permitted the resolution of coupling constants as small as ∼0.5 Hz as line splittings in all signals. In both compounds, several 1H resonances were overlapped at 400 MHz (Figure S5, Supporting Information), making it difficult to extract coupling constants for H-6, H-3α, and H-1α of 2 as well as H-6, H-13α and β, H-3α, and H-1α of 1 by manual analysis, even with resolution enhancement processing. While the 700 MHz spectra of 1 and 2 in CDCl3 exhibited improved resolution (Figure 2), some overlap remained for the resonances of H-6, H-3α, and H-1α of 1, as well as H-6 and H-13β of 2. One remarkable detail of the 1D 1H NMR behavior of 1 vs 2 is that their identical coupling pattern in the six-membered ring gives rise to disparate H-12 resonances with very different apparent multiplicities. This is due to a higher order effect caused by the close AB-pattern of the H-13 methylene protons in 1, which yields an unexpected ddd-like resonance pattern for H-12 that is prone to misinterpretation (Figure S7, Supporting Information).
Figure 2.
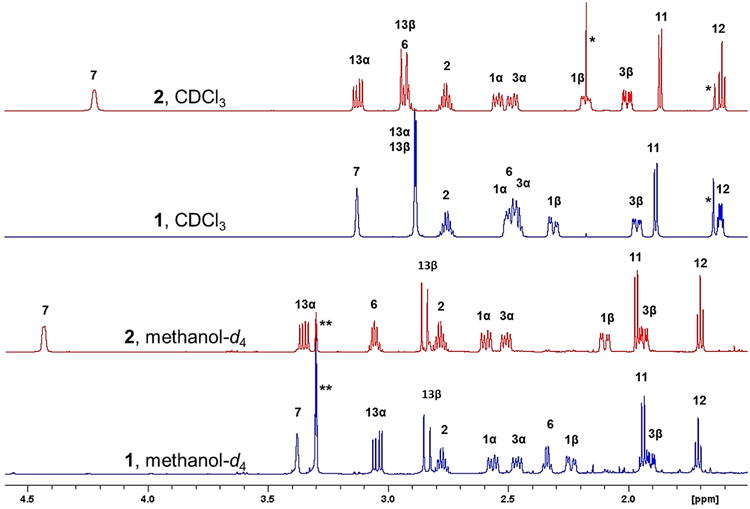
Experimental 1H NMR spectra at 700 MHz of 1 (blue) and 2 (red). Signals marked with (*) and (**) denote impurity and solvent signals, respectively.
Switching to methanol-d4, as a less obvious but still suitable solvent, led to the complete resolution of all proton signals at both 400 and 700 MHz (Figures 2 and S12, Supporting Information). The change of solvent is an important but almost forgotten strategy in 1H NMR analysis.9 One notable detail of the spectra is that H-7, H-6, and H-13α of 1 are shielded relative to their counterparts in 2. This can be explained by the influence of the Me-20 being close to both H-7 and H-13α, and Me-18 being in close proximity to H-6 in 1. In contrast, the Me-20 and Me-18 protons experience a subtle but opposing shielding effect in 2 (Table 1 and Figures 1 and 2). These effects are diagnostic but remain empirical until confirmed by quantum mechanical shift calculations.
For unambiguous extraction of the 1H spin parameters in methanol-d4, the NMR data at 700 MHz of both compounds were analyzed using HiFSA (Figures S13 and S14, Supporting Information).10 The resulting coupling constants and multiplicities (Table 1) showed a relatively complex network of spins. By constructing a QuILTs, a comprehensive visualization of the J-coupling relationships was achieved, allowing for ready distinction between the δ/J patterns of 1 and 2 (Figure 3). The J value of 6.1 Hz representing the coupling of H-6 with H-7 in 2 was in particular agreement with the conformationally averaged 20° dihedral angle calculated from a molecular model. Similarly, the observed 1.9 Hz coupling between H-6 and H-7 in 1 was consistent with a 120° dihedral angle calculated for H-6–C-6–C-7–H-7 in 1.
Figure 3.
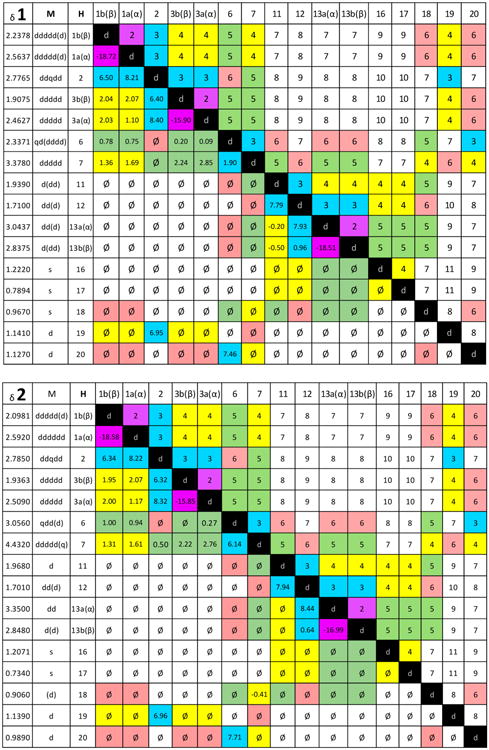
Full H NMR δ and J-correlation maps, termed quantum interaction and linkage tables (QuILTs), of riolozatrione (1) and 6-epi-riolozatrione (2) were achieved by HiFSA processing of the 700 MHz spectra in methanol-d4. Multiplicities within parentheses are due to couplings of ≤1 Hz. Couplings with absolute values of ≤0.10 Hz are given as “ø”.
The spectroscopic parameters found and calculated from methanol-d4 at 700 MHz were fitted to the 1D NMR spectra in CDCl3 at 700 MHz and iterated, thereby creating a 1H fingerprint of compounds 1 and 2 despite the overlapping of several signals. Therefore, HiFSA profiles and the automated consistency analysis (ACA) could be completed, and QuILTs for CDCl3 data were also constructed (Figures S15–S18, Supporting Information).
In order to explain why 2 had not been detected in earlier studies, the reported extraction methodology of riolozatrione was re-examined.2 Initially, 1 was obtained from a petroleum ether extract after refluxing for 1 h in MeOH and chromatographic separation on silica gel. A more recent study3 used an aqueous MeOH extraction at room temperature (RT) for 12 h, evaporation at reduced pressure at 40 °C, and subsequent partitioning to obtain n-hexane, EtOAc, and n-BuOH fractions. The n-hexane fraction afforded 1 after chromatographic separation on silica gel. The present work added the direct extraction of the ground J. dioica roots with dichloromethane for 2 h at RT to the methods. Comparing a hexane partition obtained as reported3 with direct CH2Cl2 extraction using an HPLC method established in our laboratory (Figure S1, Supporting Information) indicated that 2 was present in both extracts but was extracted more efficiently by the direct CH2Cl2 method.
However, as the different abundance of 2 in the two extracts could also be the result of chemical interconversion between 1 and 2, the following two-prong approach was taken to investigate this possibility: (a) determination of the energy of the most stable conformers of both epimers, using density functional theory (DFT) calculations; (b) study of the interconversion induced by NaOMe. According to the energy calculated at the mPW1B95/DGDZVP level of theory, the most stable conformer of compound 1 (−1003.15004 hartree) is 3.7 kcal/mol more stable than the most stable conformer of 2 (−1003.14245 hartree). This difference in energy explains why only compound 1 is observed in the extract. On the other hand, treatment of 10 mg of 1 with 1 equiv of NaOMe in MeOH at RT for 30 min yielded no product. Surprisingly, performing the same procedure with 2 led to the quantitative formation of 1. These results indicated that 2 is more likely to serve as a reactive biosynthetic precursor of 1, in a yet to-be-determined process. A subsequent experiment exposing 2 to the isolation conditions described previously3 demonstrated that 2 is stable in MeOH solution (for 4 months). In contrast, by adding a catalytic amount of HCl, and acquiring 1H NMR spectra over time, the gradual formation of 1 and an unidentified product was observed. Compound 1 does not produce 2 under the same conditions, confirming that 1 is not an artifact from the extraction or the separation procedures. Moreover, over several years we have prepared extracts from ground roots collected in different seasons. Both compounds 1 and 2 were detected in different proportions in all these extracts, and we consider this additional evidence that both compounds are present in the plant rather than being artifacts.
Interpretation of the NMR spectra of 3 and 4 was established previously without details about stereochemical properties of the compounds.6 Although the 13C and 1H NMR data of 3 were obtained in CDCl3, and those of 4 in benzene-d6 and acetone-d6, the 13C and 1H NMR data obtained here were consistent with reported data,6 and 2D experiments permitted the unequivocal assignment of the 1H and 13C NMR resonances (Table 2). The relative configuration of 3 was confirmed via NOESY experiments. NOE correlations between H3-16 and the proton resonating at 1.35 ppm identified this signal as belonging to H-1α. This led to assignment of H-1β as the signal at 1.95 ppm, which was consistent with its NOE with H3-20. In the same way, the NOE association between H3-20 and the Me signal at δ 0.97 showed the β-orientation of H-19. Correlations between H3-19 and H-8b (δ 2.58) led to the assignment of H-8b as being β, placing H-8a in an α position. As most of the NOE interactions observed in 3 were similar to those in 4 (Figure 4), both compounds were concluded to have congruent configurations. Accordingly, assignments of the α- vs β-orientation of the methylene protons at C-1, C-8, and C-11 were done by analogy.
Table 2. NMR Data of Compounds 3 and 4 in CDCl3 at 400 MHz (1H) and 100 MHz (13C).
| position | 3 | 4 | ||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| δH, mult. (J in Hz) | δC (type) | δH, mult. (J in Hz) | δC (type) | HMBCa | ||
| 1 | α | 1.453, ddd (−13.1, 5.2, 5.1) | 37.95 (CH2) | 1.351, ddd (−13.7, 1.5, 1.3) | 34.90 (CH2) | 2, 3, 4, 16 |
| β | 2.303, ddd (−13.1, 9.1, 9.0) | 1.945, ddd (−13.7, 9.5, 8.8) | 2, 14, 15, 16 | |||
| 2 | 2.801–2.873, m | 39.82 (CH) | 2.364 dqd (8.8, 7.5, 1.3) | 33.80 (CH) | 3, 4, 15, 16 | |
| 3 | 5.887, s br | 144.53 (CH) | 3.304, s | 72.93 (CH) | 2, 5 | |
| 4 | 138.38 (C) | 67.58 (C) | ||||
| 5 | 6.147, s br | 128.11 (CH) | 5.513, q (1.6) | 128.35 (CH) | 3, 4, 7, 15, 17 | |
| 6 | 135.91 (C) | 145.55 (C) | ||||
| 7 | 210.63 (C) | 208.41 (C) | ||||
| 8 | α | 2.896, dd (−13.0, 2.1) | 38.30 (CH2) | 2.945, dd (−12.7, 1.8) | 38.01 (CH2) | 7, 9 10, 13 |
| β | 2.519, dd (−13.1, 13.0) | 2.577, dd (−13.3, 12.7) | 6, 7, 9 | |||
| 9 | 2.430 dd (13.1, 2.1) | 51.34 (CH) | 2.508, dd (13.3, 1.8) | 52.84 (CH) | 10, 13, 18, 19, 20 | |
| 10 | 37.44 (C) | 37.57 (C) | ||||
| 11 | α | 2.462 d (−16.7) | 55.46 (CH2) | 2.424, d (−12.0) | 55.52 CH2 | 9, 10, 12, 19 |
| β | 2.326 d (−16.7) | 2.424, d (−12.0) | ||||
| 12 | 218.01 (C) | 217.23 (C) | ||||
| 13 | 64.55 (C) | 65.72 (C) | ||||
| 14 | 215.66 (C) | 215.52 (C) | ||||
| 15 | 4.123, dddd (9.1, 5.1, 2.9, 1.5) | 51.02 (CH) | 3.656, dd (9.5, 1.5) | 46.65 (CH) | 2, 3, 13, 14 | |
| 16 | 1.126, d (7.0) | 20.54 (CH3) | 1.156, d (7.5) | 16.12 (CH3) | 1, 2, 3 | |
| 17 | 1.947, s broad | 20.68 (CH3) | 1.945, d (1.6) | 20.54 (CH3) | 5, 6, 7 | |
| 18 | 1.238, s | 27.92 (CH3) | 1.277, s | 28.29 (CH3) | 9, 10, 11, 19 | |
| 19 | 0.921, s | 23.59 (CH3) | 0.969, s | 23.67 (CH3) | 9, 10, 18 | |
| 20 | 1.479, s | 14.04 (CH3) | 1.441, s | 14.60 (CH3) | 9, 13 | |
Indicates 2J and 3J interactions.
Figure 4.
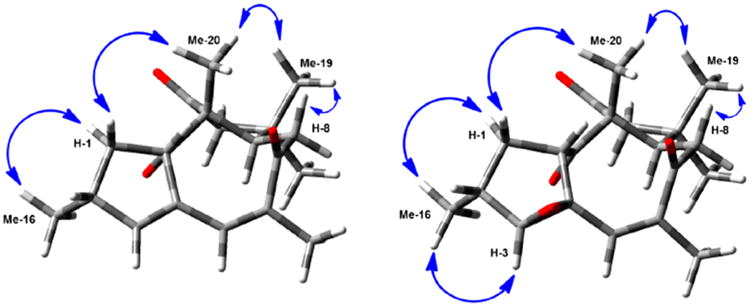
Observed NOE correlations for 3 (left) and 4 (right).
The absolute configurations of compounds 1–4 were initially obtained by comparison of the calculated and experimental VCD spectra. In the first step, a Monte Carlo conformational search was performed using a 10 kcal/mol energy window, yielding 15, eight, three, and four conformers for 1–4, respectively. These structures were submitted to DFT geometry optimizations using the DGDZVP basis set and the mPW1B95 functional, chosen to more accurately predict the thermochemical data than the B3LYP functional.11 The relative energy values of the four most stable conformers of 1 and 2 represented 98% and 97% of the conformational population, respectively. The selected conformers of the four compounds were considered to compute the IR and VCD spectra using the values of the dipole transition moments and rotational strengths. Individual spectra were processed using Lorentzian functions with a half-width of 6 cm−1. For 1 and 2, the final spectra were computed based on the Boltzmann population shown in Table 3. For 3 and 4, the relative energies of the conformers were ≥4 kcal/mol greater than those of the most stable structure; therefore, only one conformer was considered for each compound in the next steps.
Table 3. DFT Relative Energies and Conformational Populations of Riolozatrione (1) and 6-epi-Riolozatrione (2).
| conformer | ΔEmPW1B95 | %OPT | ΔGmPW1B95 | %mPW1B95 | ΔEB3PW91 | %OPT | ΔGB3PW91 | %B3PW91 |
|---|---|---|---|---|---|---|---|---|
| 1a | 0.36 | 25.54 | 0.00 | 56.92 | 0.00 | 43.24 | 0.00 | 51.17 |
| 1b | 1.02 | 8.31 | 1.03 | 10.05 | 0.94 | 8.80 | 0.40 | 25.92 |
| 1c | 0.00 | 46.49 | 0.62 | 20.10 | 0.05 | 39.94 | 0.63 | 17.61 |
| 1d | 0.51 | 19.66 | 0.88 | 12.93 | 1.00 | 8.02 | 1.34 | 5.29 |
| 2a | 0.00 | 69.23 | 0.00 | 86.00 | 0.00 | 61.42 | 0.00 | 56.84 |
| 2b | 0.84 | 16.88 | 1.17 | 11.06 | 0.94 | 12.78 | 0.57 | 21.75 |
| 2c | 1.45 | 6.03 | 2.44 | 1.33 | 0.91 | 13.22 | 0.77 | 15.41 |
| 2d | 1.29 | 7.86 | 1.59 | 5.57 | 0.94 | 12.57 | 1.33 | 5.98 |
The confidence-level data for the IR and VCD spectra comparisons of 1–4 are given in Table 4. For 1 and 3, the IR and VCD spectra were similar according to the SIR and SE index values,12 with a 99% confidence level, which permitted assignment of their absolute configurations as (2S, 6S, 7S, 9S, 11R, 12S) for 1 and (2R, 9R, 14S, 15S) for 3 (Chart 1). The IR and VCD similarity indices for 2 and 4 were sufficiently high, but only with confidence levels of 86% and 87%, respectively. This result is interesting because for the calculation of the IR and VCD spectra of 3 and 4, only one conformer was considered. The structure of each conformer differs only in the epoxide functional group. This result shows the limitations of the mPW1B95 functional to predict the VCD spectra of 2 and 4.
Table 4. Confidence Data for IR and VCD Spectra Comparisons for Compounds 1–4.
| compound | level of theory | anHa | SIRb | SEc | S−Ed | ESIe | C (%)f |
|---|---|---|---|---|---|---|---|
| 1 | mPW1B95/DGDZVP | 0.960 | 91.9 | 72.5 | 9.6 | 62.9 | 99 |
| B3PW91/DGDZVP | 0.971 | 92.2 | 75.9 | 6.5 | 69.4 | 99 | |
| 2 | mPW1B95/DGDZVP | 0.960 | 90.4 | 57.6 | 20.9 | 36.7 | 86 |
| B3PW91/DGDZVP | 0.971 | 93.3 | 72.2 | 4.9 | 67.3 | 99 | |
| 3 | mPW1B95/DGDZVP | 0.974 | 90.4 | 72.4 | 3.9 | 68.5 | 99 |
| B3PW91/DGDZVP | 0.972 | 90.5 | 73.8 | 4.5 | 69.3 | 99 | |
| 4 | mPW1B95/DGDZVP | 0.960 | 92.4 | 67.4 | 18.5 | 48.9 | 87 |
| B3PW91/DGDZVP | 0.976 | 91.8 | 80.1 | 6.8 | 73.3 | 99 | |
| 1 exp vs 2 calc | 0.971 | 90.2 | 65.6 | 9.1 | 56.5 | 93 | |
| 2 exp vs 1 calc | 0.970 | 92.3 | 49.8 | 19.9 | 29.9 | 69 |
Scaling frequencies factor.
IR spectral similarity.
VCD spectral similarity for the correct enantiomer.
VCD spectral similarity for the incorrect enantiomer.
Enantiomer similarity index calculated as SE – S−E.
Confidence level for the stereochemical assignment.
Because the B3PW91 functional has been used successfully to compute the IR and VCD spectra of a great number of diterpenoids,13 the quantum mechanics (QM) geometry optimization for the conformers was performed at the B3PW91/DGDZVP level of theory. A single conformer represented more than 99% of the conformational population of 3 and 4. For 1 and 2, the relative energy values of the most likely structures are displayed in Table 3. These conformers were submitted to Gaussian 0914 to calculate the dipole transition moments and rotational strengths affording the calculated IR and VCD spectra shown in Figure 5 along with the experimental spectra. The comparison indices presented in Table 4, SIR, SE, and ESI, increased with respect to those obtained with the mPW1B95 functional, which confirms the proposed absolute configuration. With the B3PW91 functional, the SE spectral similarity values of 2 and 4 were 72.2 and 80.1, respectively, with a 99% confidence level (Table 4). Cross comparisons between IR and VCD spectra of 1 and 2 showed a poor agreement, with a confidence level below 95% in both cases (Figure 5 and Table 4). This confirmed the (2S, 6S, 7S, 9S, 11R, 12S) and (2S, 6R, 7S, 9S, 11R, 12S) absolute configurations for 1 and 2, respectively (Chart 1), and also demonstrated that 3 and 4 share (2R, 9R, 14S, 15S) absolute configurations.
Figure 5.
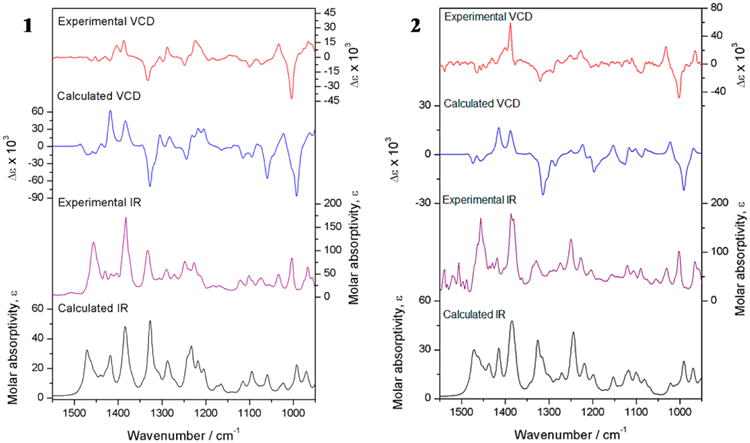
Experimental and calculated IR and VCD spectra at the B3PW91/DGDZVP level of theory for 1 and 2.
The absolute configurations of compounds 3 and 4 were recently published via computed data generated at the B3LYP/6-31G(d) single-point and B3LYP/DGDZVP full geometry optimization levels.8 In the current study the conformational populations were estimated with two calculation models11 with different capabilities: mPW1B95/DGDZVP and B3PW91/DGDZVP. As expected, the nature of the conformers is different when calculated at different levels of theory; nevertheless results are quite similar (Figure S19, Supporting Information).
In order to confirm the absolute configurations of 1, 2, and 4 (Figure 6), crystals of those compounds were subjected to crystallographic analysis by the anomalous X-ray scattering from the oxygen atoms,15 asserting that the isolated compounds crystallized as enantiopure compounds. Compound 2 shows two isoforms described in the Experimental Section. The Flack parameter refined to those values indicated in Table 5 for the “hole-in-one” fit and for the selected quotients from Parsons' method.16,17 These assessment factors suggest that the resonant-scattering contributions to the observed intensities are weak, but still significant enough to assign the absolute stereostructures correctly.
Figure 6.
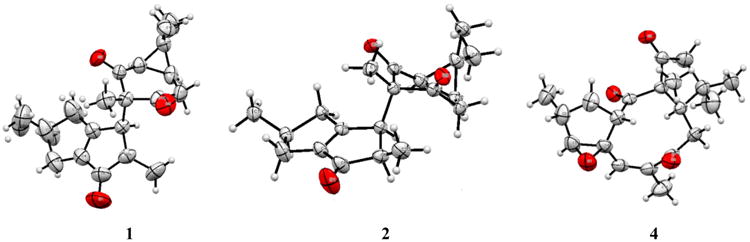
ORTEP drawings of the X-ray structures of 1, 2, and 4.
Table 5. Flack, Parsons, and Hooft Parameters for Absolute Configuration Determination.
| 1 | 2a | 4 | |
|---|---|---|---|
| space group | P21 21 21 | P21 | P212121 |
| wavelength | 1.54178 | 1.54178 | 1.54178 |
| Flack param x | −0.01(5) | 0.04(6) | 0.03(4) |
| Parsons z | 0.00(5) | 0.03(6) | 0.05(4) |
| StudentT ν | 99 | 47 | 99 |
| select pairs | 1567 | 3509 | 1648 |
| θmin | 7.37 | 5.06 | 8.02 |
| θmax | 74.64 | 82.71 | 77.49 |
| P2 (true) | 1.000 | 1.000 | 1.000 |
| P3 (true) | 1.000 | 1.000 | 1.000 |
| P3 (rac-twin) | 0.1 × 10−17 | 0.2 × 10−12 | 0.2 × 10−23 |
| P3 (false) | 0.2 × 10−68 | 0.3 × 10−52 | 0.4 × 10−98 |
| G | 0.9896 | 0.9535 | 0.9192 |
| G (su) | 0.1074 | 0.1239 | 0.0865 |
| Hooft y | 0.01(5) | 0.02(6) | 0.04(4) |
Calculated values for isoform B.
To further increase the confidence of the absolute structure determination, a Bayesian analysis as implemented in PLATON18 was used to analyze the Bijvoet differences19 and suggested that compounds 1, 2, and 4 are enantiopure: the probability P2 (true) = P3 (true) = 1.000 with P3 (racemate-twin), P3 (false), G, and the Hooft parameter y for the 3R enantiomer of 4. However, prior to and after Bayesian refinement, the Flack parameter and its standard deviation as well as the excellent figures of merit clearly indicate the reliability of X-ray analyses and also verify the absolute configurations of 1, 2, and 4.2,4,5,8
Finally, 1–4 were assessed biologically for their toxicity against Vero cells using the Mossman method and for in vitro anti-HSV activity using the plaque reduction assay with HSV-1- and HSV-2-infected Vero cells (Table 6). Compared to 1, compound 2 exhibited higher nonspecific cytotoxicity and also displayed lower antiviral activity against both viruses, leading to SI values around 3. Both 1 and 3 showed similar activity against HSV-1, while 4 was inactive against either virus at the tested concentrations.
Table 6. Cytotoxic Effect against Vero Cell Lines and Antiherpetic Activity of Compounds 1–4.
| compound | Vero cells | HSV-1 | HSV-2 | SIb |
|---|---|---|---|---|
|
|
|
|
|
|
| CC50, μMa | IC50, μMa | IC50, μMa | CC50/IC50 | |
| 1c | 1222.9 ± 1.9 | 210.2 ± 8.6 | 210.2 ± 5.9 | 5.8 |
| 2 | 584.2 ± 17 | 179.8 ± 4.1 | 172.4 ± 4.3 | 3.2/3.4 |
| 3 | 1523.0 ± 39.5 | 292.9 ± 9.4 | >318.5 | 5.2 |
| 4 | >1515.1 | >303.0 | >303.0 | ND |
| acyclovird | NDe | 4.75 × 10−6 ± 0.31 | 3.11 × 10−6 ± 0.10 | ND |
Results are expressed as a mean (n = 3) ± SD.
SI is the selective index, SI = CC50/IC50.
See ref 3.
Positive control.
Not determined.
Experimental Section
General Experimental Procedures
Melting points were measured on an electrothermal apparatus. IR and VCD spectra were recorded on a BioTools ChiralIR 2× VCD spectrometer (BioTools, Inc., Jupiter, FL, USA). X-ray data were obtained on a Bruker D8 Venture Geometry diffractometer. The 1D and 2D NMR experiments were performed on a Bruker AVANCE III HD 400 MHz and a Bruker AVANCE III HD 700 MHz. Chemical shifts were referenced to tetramethylsilane, and J values are given in Hz. The HRESIMS data were acquired on a Jeol AccuTOF JMS T100LC spectrometer. Silica gel 40–63 μm (Aldrich) and LiChroprep RP-18 40–63 μm (Merck) were used for column chromatography (CC). Thin-layer chromatography (TLC) was carried out on silica gel 60 F254 (Merck).
Acquisition of NMR Spectra
1H NMR data on a Bruker AVANCE III HD 700 MHz were acquired under the following conditions: temperature 298 K, probe 5 mm BBO GRD Z120187/0029, T1 1.0000 s, pulse width 6.76, AQ time 2.3243 s, spectral width 14097.7 Hz, acquired size 32 768, spectral size 65 536. 1H NMR data on a Bruker AVANCE III HD 400 MHz were acquired under the following conditions: temperature 298 K, probe 5 mm PABBO GRD Z116098/0245, T1 1.0000 s, pulse width 10.00, AQ time 4.0894 s, spectral width 8012.8 Hz, acquired size 32 768, spectral size 65 536.
General NMR Data Processing
The 1D 1H NMR data were processed with NUTS software (v.201004, Acorn NMR, Inc., Las Positas, CA, USA) using Lorentzian-to-Gaussian apodization for resolution enhancement (line broadening = −1.0 Hz, Gaussian factor = 0.10), followed by zero filling to 256 K data points prior to Fourier transformation. The resulting NMR spectra were subjected to manual phase adjustment and baseline correction using fifth-order polynomial functions. PERCH NMR software (v.2014.1, PERCH Solutions Ltd.) was used for all QM-based NMR spectroscopic analysis including iteration, simulation, and HiFSA, as described before.20,21
Plant Material
J. dioica var. sessiliflora (Hook) was collected from Villaldama Municipality of Nuevo León, Mexico, and authenticated at the Institutional Herbarium of the Biological Sciences School at Universidad Autónoma de Nuevo León (UANL). Voucher specimens (UAN-24077) have been deposited.
Extraction and Isolation
Dried and powdered roots of J. dioica (250 g) were extracted with CH2Cl2 (3 × 1 L) at room temperature to give 3.5 g of crude extract upon evaporation in vacuo. The extract was fractionated on silica gel by low-pressure CC using CH2Cl2/acetone (19:1) to give four fractions (A–D). Fraction B was further fractionated by silica gel CC using n-hexane/EtOAc (6:4) to give six fractions (B1–B6) based on TLC monitoring results. Fractions B2 and B3 were separated by CC on RP-18 silica gel using an isocratic mode with MeOH/H2O (7:3 v/v) to give 4 (30 mg) and 3 (50 mg), respectively. Compounds 1 (180 mg) and 2 (90 mg) were purified from fraction C using silica gel CC eluted with n-hexane/EtOAc (6:4), followed by RP-18 silica gel CC with MeOH/H2O (7:3 v/v). The HPLC analysis was performed on a Waters liquid chromatograph 1525 linked to a Waters diode array detector 2996, using a Waters AccQ-Tag column and isocratic elution with acetonitrile/water (50:50).
Riolozatrione (1)
colorless crystals (Et2O/petroleum ether); mp 116–118 °C (118.6 °C); [α]25D +49.2 (c 0.4, CHCl3); IR (KBr) νmax 2955, 2929, 2871, 1688, 1625, 1454, 1380, 729 cm−1; 1H NMR (CDCl3 and methanol-d4, 400 MHz) and 13C NMR (CDCl3 and methanol-d4, 100 MHz), see Table 1; ESIMS m/z [M + H]+ 315.19420 (calcd for C20H26O3 315.19385).
6-epi-Riolozatrione (2)
colorless crystals (Et2O/petroleum ether); mp 126–128 °C; [α]25D +14.2 (c 0.2, CHCl3); IR (KBr) νmax 2952, 2870, 1722, 1696, 1632, 1454, 1383, cm−1; 1H NMR (CDCl3 and methanol-d4, 400 MHz) and 13C NMR (CDCl3 and methanol-d4, 100 MHz), see Table 1; ESIMS m/z [M + H]+ 315.19650 (calcd for C20H26O3 315.19602).
Jatrophatrione (3)
colorless solid; mp 147–149 °C (148–150 °C);4 1H NMR (CDCl3, 400 MHz) and 13C NMR (CDCl3, 100 MHz), see Table 2.
Citlalitrione (4)
colorless crystals (MeOH/n-hexane); mp 196–198 °C (194–196 °C);5 1H NMR (CDCl3, 400 MHz) and 13C NMR (CDCl3, 100 MHz), see Table 2.
IR and VCD Analysis
Compounds 1–4 were dissolved in 100% CDCl3 (5 mg/0.15 mL, 8.8 mg/0.2 mL, 7.5 mg/0.21 mL, and 5 mg/0.15 mL, respectively) and placed in a 100 μm path-length cell with BaF2 windows. IR and VCD spectra were collected with a 4 cm−1 resolution over 8 h for 1, 19 h for 2, 21 h for 3, and 5 h for 4. In all collections, the instrument was optimized at 1400 cm−1. The blank solvent spectra measured under the same conditions were subtracted from the spectra of the molecules.
Computational Methods
A molecular mechanics conformational search was performed for 1–4 using ComputeVOA software with the MMFF94 force field. The conformations obtained for each compound were subjected to geometry optimization using the mPW1B95 calculation models and the DGDZVP basis set. For 3 and 4, the respective single most stable conformer represented 99.99% of the conformational populations, whereas the four conformers of 1 and 2 listed in Table 3 accounted for 97% of the conformational space of each of these molecules. All these structures were considered to obtain the harmonic frequencies, dipole transition moments, and rotational strengths that were subsequently used to compute the IR and VCD spectra of each conformer. The final spectra of 1 and 2 were obtained considering a Boltzmann distribution with the ΔG values of Table 3. Comparisons of experimental and calculated spectra were performed with the CompareVOA software.22 The same procedure was performed at the B3PW91/DGDZVP level of theory. After DFT optimization, one single conformer respectively for 3 and 4 and four conformers of each of 1 and 2 represented at least 98% of the conformational populations.
X-ray Diffraction Analysis
Crystals of 1, 2, and 4 were mounted on the goniometer of a Bruker D8 Venture geometry diffractometer operating with Cu Kα radiation (λ = 1.541 78 Å). Data collection, unit-cell refinement, and data processing were carried out with the APEX2.41 program. The structures were solved using SHELXS and refined using SHELXL-2014/7.42. The absolute configurations were established from the anomalous dispersion effects.17
Crystal data of riolozatrione (1, CCDC 1543367)
colorless prisms, C20H26O3, M = 314.41, orthorhombic, crystal size = 0.351 × 0.248 × 0.142 mm, a = 7.51070(10) Å, b = 11.4024(2) Å, c = 21.1329(4) Å, α = 90°, β = 90°, γ = 90°, V = 1809.82(5) Å3, T = 298(2) K, space group P212121, Z = 4, Dcald = 1.154 mg/m3, λ(Cu Kα) = 1.541 78 Å, reflections collected = 68 276, independent reflections = 3702 [R(int) = 0.0382]. Final R indices for I > 2σ(I): R1 = 0.0387, wR2 = 0.1035. R indices for all data: R1 = 0.0430, wR2 = 0.1082. Flack parameter = −0.01(5).
Crystal data of 6-epi-riolozatrione (2, CCDC 1543373)
isoform A colorless prism, C20H26O3, M = 314.41, monoclinic, crystal size = 0.394 × 0.222 × 0.094 mm, a = 10.7807(12) Å, b = 14.2755(16) Å, c = 11.7322(13) Å, α = 90°, β = 105.260°, γ = 90°, V = 1741.9(3) Å3, T = 150(2) K, space group P212121, Z = 4, Dcald = 1.199 mg/m3, λ(Cu Kα) = 1.541 78 Å, reflections collected = 72 830, independent reflections = 7410 [R(int) = 0.0457]. Final R indices for I > 2σ(I): R1 = 0.0374, wR2 = 0.0901. R indices for all data: R1 = 0.0410, wR2 = 0.0930. Flack parameter = 0.04(5). CCDC 1544841; isoform B: colorless prism, C20H26O3, M = 314.41, monoclinic, crystal size = 0.374 × 0.362 × 0.098 mm, a = 10.6742(13) Å, b = 14.1451(16) Å, c = 11.5436(16) Å, α = 90°, β = 105.595(12)°, γ = 90°, V = 1678.8(4) Å3, T = 298(2) K, space group P21, Z = 4, Dcald = 1.244 mg/m3, λ(Cu Kα) = 1.541 78 Å, reflections collected = 62 416, independent reflections = 7333 [R(int) = 0.0483]. Final R indices for I > 2σ(I):R1 = 0.0427, wR2 = 0.1084. R indices for all data: R1 = 0.0499, wR2 = 0.1163. Flack parameter = 0.04(6).
Crystal data of citlalitrione (4, CCDC 1543369)
colorless prism, C20H26O4, M = 330.41, orthorhombic, crystal size = 0.380 × 0.235 × 0.112 mm, a = 6.57050(1) Å, b = 11.1729(2) Å, c = 25.1590(5) Å, α = 90°, β = 90°, γ = 90°, V = 1846.96(6) Å3, T = 298(2) K, space group P212121, Z = 4, Dcald = 1.188 mg/m3, λ(Cu Kα) = 1.541 78 Å, reflections collected = 71 131, independent reflections = 3927 [R(int) = 0.0384]. Final R indices for I > 2σ(I): R1 = 0.0369, wR2 = 0.0949. R indices for all data: R1 = 0.0404, wR2 = 0.0978. Flack parameter = 0.03(4).
Crystallographic data for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre. Copies of the data can be obtained, free of charge, upon application to the Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: +44-(0)1223-336033 or deposit@ccdc.cam.ac.uk).
Cell Culture and Viral Particles
Mammalian Vero cells ATCC CRL-1586 were used for the cytotoxicity and antiviral assays. Cells were grown in advanced DMEM media that was supplemented with 2% fetal bovine serum with glutamine, essential amino acids, streptomycin, and 1% penicillin. Cells were maintained at 37 °C in a 5% CO2 atmosphere to reach 80–90% confluence. HSV-1 was obtained from a clinical isolate of an infected patient at the Department of Dermatology, UANL. HSV-2 was obtained from a clinical isolate from patients attending the Dental School. Both isolates were found to be positive for the herpes simplex virus thymidine kinase gene by the polymerase chain reaction (PCR) assay and positive for the cytopathic effect of HSV infection by Vero cell culture.
Cytotoxicity Assay
Cell viability was determined according to the MTT method.23 The compounds were further examined for toxicity in a Vero cell line at concentrations of 100, 200, 400, 800, and 1600 μM. After 3 days of incubation, cell viability was assessed by adding 10 μL of a solution of 5 mg/mL of 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenylte-trazolium bromide (MTT). The CC50 was determined as the concentration of the compound required to reduce cell viability by 50%, taking as much as 100% of the untreated cells. The experiments were performed in triplicate for each compound.
Antiherpetic Assay
The antiherpetic effects on HSV-1 and HSV-2 in vitro were evaluated using the reduction plaque assay.24 Briefly, 5 × 105 Vero cells were seeded onto six-well culture plates and then incubated with 100 plaque forming units of HSV-1 or HSV-2 for 1 h at 37 °C. Supernatant was discarded, fresh medium was supplemented with 1% DMSO, and 0.32% IgG was added. Concentrations of 80, 160, and 320 μM of each compound were tested. Cells were incubated for 72 h for HSV-1 and HSV-2. Finally, the cells were fixed with MeOH and stained with Giemsa reagent. Negative (mock) and positive (acyclovir) controls were used for each assay. All assays were carried out in triplicate.
Supplementary Material
Figure S1. Chromatograms obtained from HPLC analysis for hexane fraction (A) and methylene chloride extract (B) from J. dioica roots.
Figure S2. Elemental analysis of 6-epi-Riolozatrione (2)
Figure S3. HRESIMS spectrum of 6-epi-Riolozatrione (2)
Figure S4. Infrared spectrum of 6-epi-Riolozatrione (2)
Figure S5. Experimental 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra of 1 and 2 in CDCl3.
Figure S6. Comparison of the 1D 1H-NMR spectra of compounds 1 and 2 in CD3OD at 700 MHz, FIDs processed with Lorentzian-Gaussian apodization.
Figure S7. Comparison of the 1H-NMR signals of the H-12 proton in compound 1 at 400 and 750 MHz and compound 2 at 750 MHz. Only in 1, the highly coupled vicinal methylene protons, 2H-13, form an AB pair with very close chemical shifts (Δδ=0.0014ppm). Therefore, H-12 in 1 gives rise to a higher order resonance pattern with a ddd-like multiplicity that can be readily misinterpreted when working under first-order assumptions. In contrast, as the methylene protons, 2H-13, resonate as relatively distant AM/AX nuclei in 2, H-12 gives rise to an apparent triplet (pseudo triplet; with two additional long-range couplings). In fact, as the 6-epi relationship between 1 and 2 does essentially not affect the geometry of the six-membered ring, its coupling patters have to be identical in both molecules – only the resonances are not, due to the higher order effects caused by the differences in relative chemical shifts.
Figure S8. Comparison of the 100 MHz 1D 13C-NMR spectra of compounds 1 and 2 in CD3OD.
Figure S9. HSQC spectrum (400 MHz, CD3OD) of 6-epi-Riolozatrione (2).
Figure S10. HMBC spectrum (400 MHz, CD3OD) of 6-epi-Riolozatrione (2).
Figure S11. NOESY spectrum (400 MHz, CD3OD) of 6-epi-Riolozatrione (2).
Figure S12. Calculated (red) vs. experimental (blue) and difference (green) 1D 1H-NMR signal of H-1b in compound 1 in CD3OD at 700 MHz.
Figure S13. 1H NMR HiFSA data for Riolozatrione (1) in CD3OD (PERCH .pms file format)
Figure S14. 1H NMR HiFSA data for 6-epi-Riolozatrione (2) in CD3OD (PERCH .pms file format)
Figure S15. 1H NMR HiFSA data for Riolozatrione (1) in CDCl3 (PERCH .pms file format)
Figure S16. 1H NMR HiFSA data for 6-epi-Riolozatrione (2) in CDCl3 (PERCH .pms file format)
Figure S17. Full 1H NMR δ and J-correlation maps, termed Quantum Interaction and Linkage Tables (QuILTs), of riolozatrione (1) and 6-epi-riolozatrione (2) were achieved by HiFSA processing of the 700 MHz spectra in CDCl3. Multiplicities within parentheses are due to couplings of ≤1 Hz. Couplings with absolute value of ≤0.10 Hz are given as “ø”.
Figure S18. Calculated (red) vs. experimental (blue) and difference (green) 1D 1H-NMR signals in compounds 1 and 2 in CDCl3 at 700 MHz.
Figure S19. Experimental and calculated IR and VCD spectra at the B3PW91/DGDZVP level of theory for 3 (left) and 4 (right).
Acknowledgments
We are grateful to I. Carrera (FM-UANL) for technical assistance with the extraction procedures, to Dra. Beatriz Quiróz García, Q. Luis Velasco-Ibarra, Dr. Javier Pérez-Flores, Q. María de la Paz Orta Pérez (IQ-UNAM), and Dra. Karla Ramírez-Gualito (Centro de Nanociencias y Nanotecnología-IPN) for technical assistance. We also acknowledge financial support by the Dirección de Cómputo y de Tecnologías de Información y Comunicación de la Universidad Nacional Autónoma de México, via Grant SC16-1-IG-105; the Universidad Autónoma de Nuevo León via Grant PAICYT-CS657-11; the Programa para el Desarrollo Profesional Docente (PRODEP) from the Secretaría de Educación Pública (SEP) via Grant 103.5/15/14156; and the Consejo Nacional de Ciencia y Tecnología (CONACYT-México) via Grant 252589. E.M.M.M. acknowledges CONACYT for scholarship number 208998. G.F.P. and S.N.C. acknowledge support by Grant U41 AT008706 from NCCIH and ODS.
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jnatprod.7b00193.
HPLC analysis, elemental analysis, IR, MS, NMR spectra, and HiFSA profiles of compounds 1 and 2 (PDF)
ORCID: Guido F. Pauli: 0000-0003-1022-4326
Shao-Nong Chen: 0000-0003-0748-0863
Verónica M. Rivas-Galindo: 0000-0002-7981-3674
Notes: The authors declare no competing financial interest.
The original NMR data (FIDs) are made available at DOI: http://dx.doi.org/10.7910/DVN/QHYRAM.
References
- 1.Artschwager KM. Healing with Plants in the American and Mexican West. University of Arizona Press; USA: 1996. p. 168. [Google Scholar]
- 2.Domínguez XA, Cano G, Franco R, Villarreal AM, Watson WH, Zabel V. Phytochemistry. 1980;19:2478. [Google Scholar]
- 3.Silva-Mares D, Torres-López E, Rivas-Estilla AM, Cordero-Pérez P, Waksman-Minsky N, Rivas-Galindo VM. Nat Prod Commun. 2013;8:297–298. [PubMed] [Google Scholar]
- 4.Torrance SJ, Wiedhopf RM, Cole JR, Arora SK, Bates RB, Beavers WA, Cutler RS. J Org Chem. 1976;41:1855–1857. doi: 10.1021/jo00872a038. [DOI] [PubMed] [Google Scholar]
- 5.Villarreal AM, Dominguez XA, Williams HJ, Scott AI, Reibenspies J. J Nat Prod. 1988;51:749–753. doi: 10.1021/np50058a014. [DOI] [PubMed] [Google Scholar]
- 6.Yang J, Long YO, Paquette LA. J Am Chem Soc. 2003;125:1567–1574. doi: 10.1021/ja021177r. [DOI] [PubMed] [Google Scholar]
- 7.Pauli GF, Niemitz M, Bisson J, Lodewyk MW, Soldi C, Shaw JT, Tantillo DJ, Saya JM, Vos K, Kleinnijenhuis RA, Hiemstra H, Chen SN, McAlpine JB, Lankin DC, Friesen JB. J Org Chem. 2016;81:878–889. doi: 10.1021/acs.joc.5b02456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burgueño-Tapia E, Chávez-Castellanos K, Cedillo-Portugal E, Joseph-Nathan P. Tetrahedron: Asymmetry. 2017;28:166–174. [Google Scholar]
- 9.(a) Freeman R, Bhacca N. J Chem Phys. 1966;45:3795–3805. [Google Scholar]; (b) Bowie J, Cameron D, Schütz P, Williams D, Bhacca N. Tetrahedron. 1966;22:1771–1775. [Google Scholar]
- 10.Laatikainen R, Niemitz M, Weber U, Sundelin J, Hassinen T, Vepsäläinen J. J Magn Reson, Ser A. 1996;A120:1–10. [Google Scholar]
- 11.Barquera-Lozada JE, Quiroz-García B, Quijano L, Cuevas G. J Org Chem. 2010;75:2139–2146. doi: 10.1021/jo902170w. [DOI] [PubMed] [Google Scholar]
- 12.(a) Burgueño-Tapia E, Zepeda LG, Joseph-Nathan P. Phytochemistry. 2010;71:1158–1161. doi: 10.1016/j.phytochem.2010.04.005. [DOI] [PubMed] [Google Scholar]; (b) Penicooke N, Walford K, Badal S, Delgoda R, Williams LAD, Joseph-Nathan P, Gordillo-Román B, Gallimore W. Phytochemistry. 2013;87:96–101. doi: 10.1016/j.phytochem.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 13.Joseph-Nathan P, Gordillo-Román B. In: Progress in the Chemistry of Organic Natural Products. Kinghorn AD, Falk H, Kobayashi J, editors. Vol. 100. Springer International Publishing; Switzerland: 2015. pp. 311–452. Chapter 4. [DOI] [PubMed] [Google Scholar]
- 14.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision E.01. Gaussian, Inc; Wallingford, CT: 2009. [Google Scholar]
- 15.Flack HD, Bernardinelli G. J Appl Crystallogr. 2000;33:1143–1148. [Google Scholar]
- 16.Flack HD. Acta Crystallogr, Sect A: Found Crystallogr. 1983;39:876–881. [Google Scholar]
- 17.Parsons S, Flack HD, Wagner T. Acta Crystallogr, Sect B: Struct Sci, Cryst Eng Mater. 2013;B69:249–259. doi: 10.1107/S2052519213010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spek AL. Acta Crystallogr, Sect D: Biol Crystallogr. 2009;D65:148–155. doi: 10.1107/S090744490804362X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hooft RWW, Straver LH, Spek AL. J Appl Crystallogr. 2008;41:96–103. doi: 10.1107/S0021889807059870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Napolitano JG, Lankin DC, McAlpine JB, Niemitz M, Korhonen SP, Chen SN, Pauli GF. J Org Chem. 2013;78:9963–9968. doi: 10.1021/jo4011624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Napolitano JG, Gödecke T, Rodriguez Brasco MF, Jaki BU, Chen SN, Lankin DC, Pauli GF. J Nat Prod. 2012;75:238–248. doi: 10.1021/np200949v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Debie E, De Gussem E, Dukor RK, Herrebout W, Nafie LA, Bultinck P. ChemPhysChem. 2011;12:1542–1549. doi: 10.1002/cphc.201100050. [DOI] [PubMed] [Google Scholar]
- 23.Mosmann T. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 24.Russell WC. Nature. 1962;195:1028–1029. doi: 10.1038/1951028a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Chromatograms obtained from HPLC analysis for hexane fraction (A) and methylene chloride extract (B) from J. dioica roots.
Figure S2. Elemental analysis of 6-epi-Riolozatrione (2)
Figure S3. HRESIMS spectrum of 6-epi-Riolozatrione (2)
Figure S4. Infrared spectrum of 6-epi-Riolozatrione (2)
Figure S5. Experimental 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra of 1 and 2 in CDCl3.
Figure S6. Comparison of the 1D 1H-NMR spectra of compounds 1 and 2 in CD3OD at 700 MHz, FIDs processed with Lorentzian-Gaussian apodization.
Figure S7. Comparison of the 1H-NMR signals of the H-12 proton in compound 1 at 400 and 750 MHz and compound 2 at 750 MHz. Only in 1, the highly coupled vicinal methylene protons, 2H-13, form an AB pair with very close chemical shifts (Δδ=0.0014ppm). Therefore, H-12 in 1 gives rise to a higher order resonance pattern with a ddd-like multiplicity that can be readily misinterpreted when working under first-order assumptions. In contrast, as the methylene protons, 2H-13, resonate as relatively distant AM/AX nuclei in 2, H-12 gives rise to an apparent triplet (pseudo triplet; with two additional long-range couplings). In fact, as the 6-epi relationship between 1 and 2 does essentially not affect the geometry of the six-membered ring, its coupling patters have to be identical in both molecules – only the resonances are not, due to the higher order effects caused by the differences in relative chemical shifts.
Figure S8. Comparison of the 100 MHz 1D 13C-NMR spectra of compounds 1 and 2 in CD3OD.
Figure S9. HSQC spectrum (400 MHz, CD3OD) of 6-epi-Riolozatrione (2).
Figure S10. HMBC spectrum (400 MHz, CD3OD) of 6-epi-Riolozatrione (2).
Figure S11. NOESY spectrum (400 MHz, CD3OD) of 6-epi-Riolozatrione (2).
Figure S12. Calculated (red) vs. experimental (blue) and difference (green) 1D 1H-NMR signal of H-1b in compound 1 in CD3OD at 700 MHz.
Figure S13. 1H NMR HiFSA data for Riolozatrione (1) in CD3OD (PERCH .pms file format)
Figure S14. 1H NMR HiFSA data for 6-epi-Riolozatrione (2) in CD3OD (PERCH .pms file format)
Figure S15. 1H NMR HiFSA data for Riolozatrione (1) in CDCl3 (PERCH .pms file format)
Figure S16. 1H NMR HiFSA data for 6-epi-Riolozatrione (2) in CDCl3 (PERCH .pms file format)
Figure S17. Full 1H NMR δ and J-correlation maps, termed Quantum Interaction and Linkage Tables (QuILTs), of riolozatrione (1) and 6-epi-riolozatrione (2) were achieved by HiFSA processing of the 700 MHz spectra in CDCl3. Multiplicities within parentheses are due to couplings of ≤1 Hz. Couplings with absolute value of ≤0.10 Hz are given as “ø”.
Figure S18. Calculated (red) vs. experimental (blue) and difference (green) 1D 1H-NMR signals in compounds 1 and 2 in CDCl3 at 700 MHz.
Figure S19. Experimental and calculated IR and VCD spectra at the B3PW91/DGDZVP level of theory for 3 (left) and 4 (right).