

Abstract
Potassium (K+) is constantly exiting electrically excitable cells during normal and pathophysiological activity. Currently, potassium-sensitive electrodes and electrical measurements are the primary tools to detect K+ efflux even though small molecule, visible-light K+ sensors exist. To utilize state-of-the-art in vivo imaging techniques, a fluorescent sensor should absorb and emit at long wavelengths (> 650 nm) and be easily derivatized for implementation in a variety of biological systems. Here we describe a palladium-mediated synthesis of a near-IR, oxazine fluorescent K+ sensor (KNIR-1) with a dissociation constant that is suited for detecting changes in intracellular and extracellular potassium concentrations. KNIR-1 treatment of cells expressing voltage-gated K+ channels enabled the visualization of intracellular potassium depletion upon K+ channel opening and restoration of cytoplasmic K+ after channel closing. Given the commercial-availability of fluorophores bearing alkynes, the synthetic methodology described herein will facilitate the synthesis of near-IR fluorescent sensors to enable the visualization of intracellular and extracellular K+.
Graphical abstract
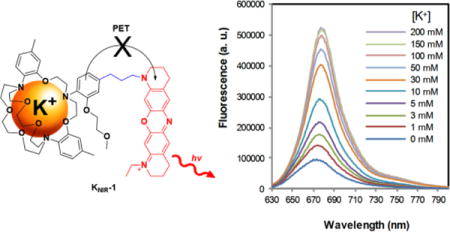
Neuronal and muscle excitability, cardiac rhythmicity, vasodilation, insulin secretion, and renal function all require the spatiotemporal release of potassium (K+) ions from cells and tissues1–6. During normal physical activity, the concentration of extracellular K+ surrounding human muscle nearly triples, rising from 3.5 mM to 10 mM2,3. This substantial change in extracellular K+ is predicted to be even greater in the transverse tubules7–9, though current methods do not allow for accurate measurements in these microscopic invaginations. Regulated K+ efflux is also essential for the developing pancreas, kidney, and vestibular and cochlear endolymphs5,6,10. Accordingly, dysregulated exit of cellular K+ from tissues has been associated with a multitude disease states from epilepsy11, migraine auras12, hypokalemic periodic paralysis13, and cardiac ar-rhythmias14 to neonatal diabetes15, hyperkalemia16 and congenital deafness17. Electrical measurements and potassium-sensitive electrodes provide pinpoint accuracy of K+ efflux, but both approaches are invasive, technically-demanding, and time consuming compared to the small molecule18 and genetically-encodable fluorescent-based sensors19,20 that have revolutionized the intracellular signaling field. Although several small molecule fluorescent K+ sensors have been synthesized21–25, the current cadre of water soluble, visible-light K+ sensors is not routinely used to visualize K+ fluxes from cells, tissues or animal models. Part of the problem is that cells and tissues absorb visible light26, resulting in cellular damage, loss of signal and unwanted autofluo-rescence. However, the major stumbling block has been the inefficient and limited routes to attach fluorophores to the K+ binding domain23–25, which has hampered the application of these sensors for detecting K+ accumulation and depletion.
To overcome these barriers, we sought a synthesis of a near-IR fluorescent K+ sensor where fluorophore attachment to the K+ binding domain was facile. For the potassium-selective binding domain, we chose a triazacryptand (TAC) to detect physiological and pathophysiological K+ concentrations for three reasons (Figure 1): (1) The TAC ion binding site is slightly larger than the K+ ion diameter, yielding an apparent millimolar Kd for potassium23–25. (2) TAC does not measurably bind to the smaller and physiologically abundant extracellular cations23–25: Na+, Ca2+, and Mg2+. (3) The electron-rich aromatic groups of TAC enable K+ sensing via photoinduced electron transfer (PET) with fluorophores. Given this flexibility, we opted to synthesize a K+ sensor with an oxazine dye KNIR-1 (Figure 1) because of its compact structure, photostability27, amenability to PET quenching28, and excitation and fluorescence emission wavelengths in the near-IR region (650 − 900 nm)29.
Figure 1.
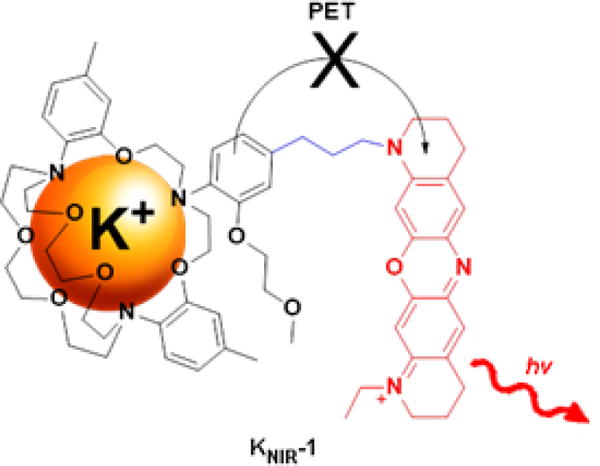
Near-IR K+ sensor KNIR-1 consists of a TAC K+ binding domain covalently linked to an oxazine fluorophore. Binding of K+ to the TAC domain quenches photoinduced electron transfer (PET) from the o-alkoxyaniline moiety to the oxazine fluorophore, resulting in enhanced fluorescence emission.
Previous syntheses of visible-light K+ sensors have directly coupled the fluorophore to the TAC binding domain using aldehyde precursor 1a (TAC-CHO, Scheme 1). Similar to the reported low yields in the literature23–25, our attempts to form a near-IR dye utilizing TAC-CHO were unsuccessful. Stymied by the inability to efficiently derivatize TAC-CHO, we pursued a palladium-mediated aryl coupling approach to append fluorophores to the TAC K+ binding domain. Aryl halogenation of the TAC binding domain (Scheme 1) is ideal for selective modification of the o-alkoxyaniline PET donor because it is the only aromatic ring that is not blocked by a methyl group in the para position. Treatment of 1b with bromine resulted in the selective and near quantitative bromination of the aryl PET donor ring. Iodination of TAC also proceeds quantitatively; however, the iodinated product is more challenging to purify and thus was not pursued further.
Scheme 1.
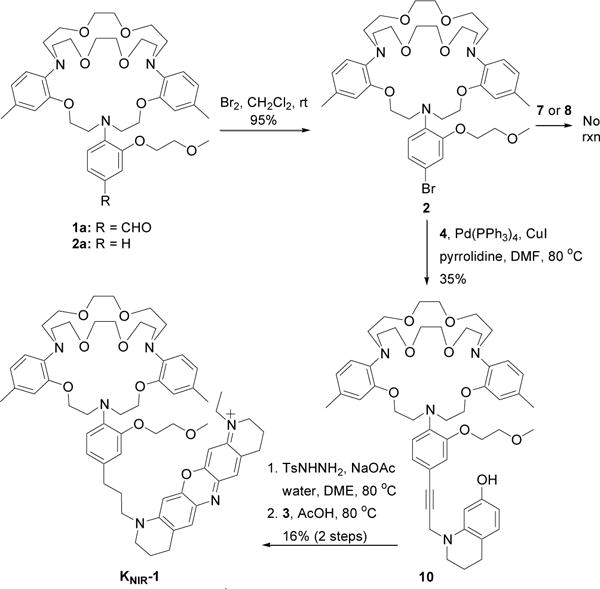
Synthesis of near-IR fluorescent K+ sensor, KNIR-1
With halogenated TAC 2 in hand, we initially envisioned attaching an intact oxazine fluorophore via palladium–catalyzed cross-coupling reactions (e.g. Sonogashira, Suzuki, Stille, etc.). To synthesize the oxazine dye with a pendant propargyl group, aryl azobenzene 3 and propargyl tetrahydroquinoline 4 were prepared separately, and then reacted together in acid (Scheme 2). To prepare the aryl azobenzene 3, 7-methoxytetrahydroquinoline 5 was first N-alkylated with iodoethane, and then subsequently reacted with 4-nitrodiazobenzene. The tetrahydroquinoline 4 was synthesized by N-alkylation of 7-hydroxytetrahydroquinoline 6 with propargyl bromide, followed by acetylation of the 7-hydroxy group with acetic anhydride. Condensation of aryl azobenzene 3 and propargyl tetrahydroquinoline 4 in acetic acid or 1.5% hydrochloric acid yielded the desired oxazine dye 7. Synthesis of the borylated oxazine 8 was accomplished by borylation of tetrahydroquinoline 4 with pinacol borane to generate boronate ester 9, followed by condensation between 9 and arylazobenzene 3 in 1.5% hydrochloric acid.
Scheme 2.
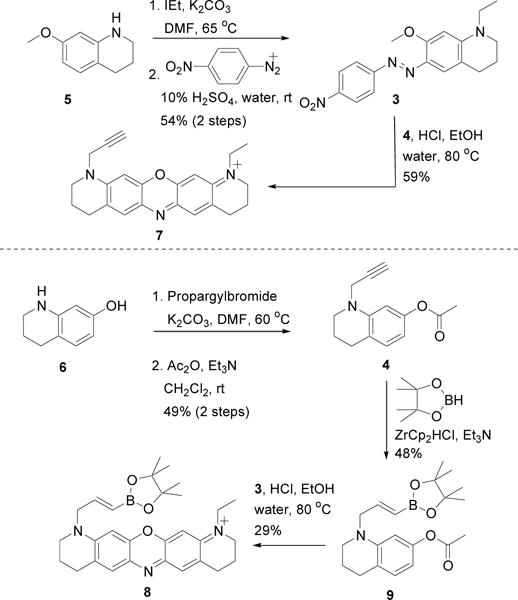
Synthesis of oxazine dyes and precursors for palladium-based coupling reactions
Despite trying several different palladium ligands, co-catalysts, counter anions, and solvents, the Sonogashira reaction between 2 and propargyl oxazine dye 7 did not occur (Scheme 1). Suzuki couplings between 2 and boronate ester oxazine dye 8 were also unsuccessful. To test whether the lack of reactivity was due to the seemingly cantankerous TAC ligand or the oxazine dye, we repeated the palladium-based reactions with the oxazine dye precursors 4 and 9 shown in Scheme 2. Surprisingly, the Sonogashira reaction between 2 and 4 proceeded smoothly, yielding TAC-alkyne 10 (Scheme 1). TAC-Br 2 was also amenable to a Suzuki coupling with boronate ester 9 shown in Scheme 2 (data not shown), suggesting that the positive-charge and/or reduction of the intact dye may impede palladium-catalyzed aryl coupling reactions. Palladium impurities in the resultant alkyne were removed using a palladium scavenger (SiliaMetS® DMT) and the purified alkyne was reduced by tosyl hydrazide hydrogenation. Failure to remove all of the palladium impurities results in cleavage of the tetrahydroquinoline from the TAC K+ binding domain during reduction. Oxazine dye formation between reduced 10 and aryl azobenzene 3 proceeded in acetic acid as previously reported30 and was purified by reverse phase HPLC. Interestingly, dye formation to afford TAC-oxazine KNIR-1 did not proceed in 1.5% hydrochloric, trifluoroacetic, or formic acid, suggesting that protonation of the TAC K+ binding domain hinders oxazine dye formation.
The fluorescence of TAC-oxazine KNIR-1 was initially tested in the presence of various concentrations of physiological extracellular cations: Na+, Ca2+, Mg2+ and K+. Figure 2A shows that KNIR1 is essentially non-responsive to physiological concentrations of extracellular Na+ (150 mM), Ca2+ (1.5 mM), and Mg2+ (1 mM). In contrast, the fluorescence emission of KNIR-1 was highly dependent on K+ concentration in the presence of these ions (Figure 2B) with an apparent Kd of 12.1 ± 0.5 mM (Figure 2C). The dissociation constant of KNIR-1 is ~ 5-fold lower than previous TAC-based K+ sensors where the fluorophore was directly conjugated to the K+ binding domain31. The peak emission wavelength of KNIR-1 is 680 nm (Figure 2B) whereas the peak ab-sorbance was 670 nm with a molar extinction coefficient ε670 = 39,800 ± 100 L·mol-1·cm-1 (Figure 2D), enabling excitation with either visible or near-IR light. K+ binding (200 mM) to KNIR-1 had no discernable effect on absorbance (Figure 2D). Using oxazine-170 as fluorescent reference standard32, the quantum yield for apo KNIR-1: 0.0597 ± 0.0003 and K+ bound (200 mM): Φ = 0.289 ± 0.005 were determined from three experiments ± SEM (supporting information). Much like K+ channels that bind to and permeate Rb+ and Cs+33, KNIR-1 also responds to these larger, but non-physiological cations (Figure 2E). As expected, smaller diameter transition metals (Zn2+ and Fe3+) had no effect on KNIR-1 fluorescence. The fluorescence emission of KNIR-1 is stable at extracellular and intracellular physiological pH 6.8 − 7.8 (Figure 2E), but begins to increase at pH 6.5 (Figure S1) as protonation starts to compete with K+ binding. Thus, under suitably buffered conditions, the fluorescence of KNIR-1 should faithfully report on K+ accumulation and depletion during physiological and pathophysiological activity.
Figure 2.
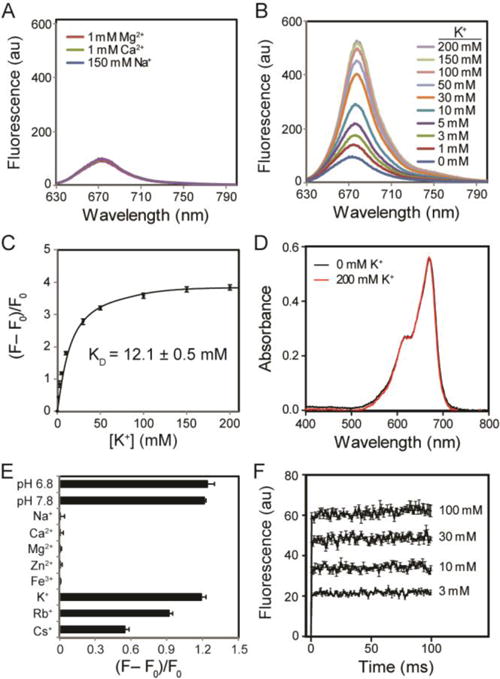
Fluorescence emission (Ex: 620 nm) and absorbance of KNIR-1 in 10 mM HEPES. (A) Fluorescence emission in the presence of Mg2+, Ca2+ and Na+. (B) Fluorescence emission at various K+ concentrations in the presence of Mg2+ (1 mM), Ca2+ (1.5 mM) and Na+ (150 mM). (C) Integrated fluorescence intensities at different K+ concentrations. F is the integrated emission intensity between 630 and 800 nm; F0 is the integrated baseline fluorescence intensity (with 0 mM K+). (D) Absorbance spectra at 0 and 200 mM K+. (E) Fluorescence intensity (F − F0)/F0 as determined in (C) in the presence of various cations [Na+ (150 mM), Ca2+ (1.5 mM), Mg2+ (1 mM), Zn2+ (0.3 mM), Fe3+ (50 μM), K+ (5 mM), Rb+ (5 mM) and Cs+ (5 mM)] and at different pH values with 5 mM K+ present. Unless indicated, the pH was 7.4. (F) Stopped-flow experiments at various K+ concentrations; emission was collected at all wavelengths above 645 nm. Data were averaged from three experiments; error bars are ± SEM. Data could not be fittted because the equilibrium rate was faster than the mixing time (1.4 ms).
We next used stopped-flow experiments to determine the K+ binding kinetics to TAC-oxazine KNIR-1. Figure 2F shows the fluorescence responses from rapid mixing (1.4 ms dead time) of KNIR-1 with different millimolar solutions of K+. Even with rapid mixing, the binding between K+ and KNIR-1 at all K+ concentrations reached equilibrium within the dead time of the stopped-flow instrument. The magnitude of the K+-dependent fluorescent response confirmed the Kd of KNIR-1 (Figure 2C) and that the equilibrium between K+ and KNIR-1 was reached within ~ 1 ms, which is faster than previously reported for TAC-based K+ sensors using fluorescence correlation spectroscopy31. Thus, the sub-millisecond K+ association and disassociation kinetics of KNIR-1 are sufficient to rapidly report on changes in K+ concentration due to ion channel activity at the plasma membrane.
Although the dissociation constant of KNIR-1 is somewhat better suited for extracellular K+ detection, we determined whether the highly membrane permeant KNIR-1 could detect intracellular K+ depletion upon K+ channel opening (Figure 3). Chinese hamster ovary (CHO) cells transiently transfected with empty vector (pCDNA3.1) or a voltage-gated K+ channel (Shaker-IR) were treated with 50 μM KNIR-1 for 3 min and the excess reagent removed before forming a gigaseal with a single cell (Figure 3A, dotted circles). Whole cell patch clamp fluorometry (Figure 3B) with untransfected cells (−) showed no voltage-dependent currents (I) or change in fluorescence (ΔF/F0). In contrast, Shaker-IR expressing cells showed robust outward currents with depolarizing voltages ≥ 0 mV (Figure 3C). Simultaneous fluorescent imaging (635 nm excitation) of the whole cell at 10 Hz revealed a concomitant loss in fluorescence that was commensurate with the outward K+ currents during the 2-s depolarization. The kinetics of the fluorescence signals were substantially slower than the currents, which was consistent with cytoplasmic K+ depletion at the Shaker-IR intracellular vestibule. Upon channel closing by repolarization to − 80 mV, the current rapidly returned to zero (< 100 ms) whereas it required seconds for the fluorescent signal to return to the pre-depolarization values. Based on the sub-millisecond association and disassociation kinetics between K+ and KNIR-1 in stopped-flow experiments (Figure 2F), the ΔF/F0 kinetics in Figure 3C are consistent with intracellular K+ depletion during channel opening followed by reestablishment of the cytoplasmic K+ concentration after channel closing. The extremely long time to restore cellular K+ to steady state concentrations highlights the importance of the Na+/K+-ATPase in maintaining the cellular K+ and Na+ gradients in electrically excitable cells.
Figure 3.
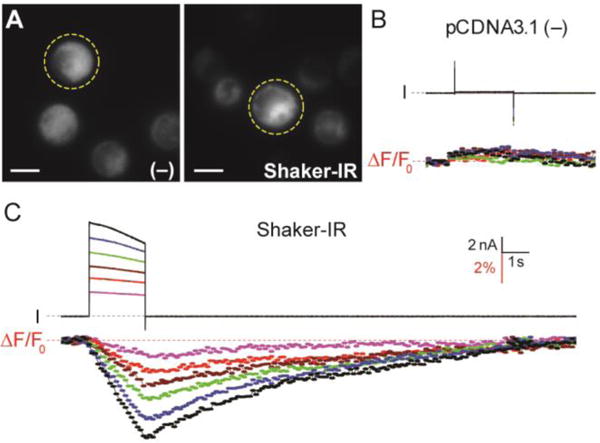
Fluorescent detection of K+ depletion in cells. (A) CHO cells transiently transfected with empty pCDNA3.1 plasmid (−) or Shaker-IR DNA labeled with 50 μM KNIR-1. Yellow dotted circles indicate the patch clamped cell in the field of view; fluorescent image scale bars represents 10 μm. Voltage-clamp fluorometry traces for (B) pCDNA3.1 (−) and (C) Shaker-IR: cells were held at −80 mV and currents (I) were elicited from 2-s command voltages from 0 to 100 mV in 20-mV increments. Total cellular fluorescence was simultaneously collected at 10 Hz by 10-ms excitation at 635 nm; F0 is the average fluorescence intensity of the first ten data points before the test depolarization. Voltage clamp fluorometry scale bars are for both (B) and (C); dotted line indicates zero current or change in fluorescence.
Our synthesis of TAC-oxazine KNIR-1 has several advantages over previous syntheses of visible-light K+ sensors. First, selective halogenation of the PET donor greatly expands the repertoire of chemistries to attach a fluorophore to the TAC ion binding domain. For example, the Sonogashira coupling described herein should enable the synthesis of a wide palette of fluorescent K+ sensors using alkynyl-derivatized fluorophores that have become commercially-available due to the emergence of bioorthogonal Huisgen 1,3-dipolar cycloadditions (click chemistry). Second, the absorption and emission spectra of TAC-oxazine KNIR-1 enable K+ detection using near-IR light, which has better tissue penetration and reduced autofluorescence compared to visible light26. In addition, TAC-oxazine KNIR-1 can be utilized with the standard repertoire of visible-light calcium sensors18–20 and channelrhodopsins34,35 − negating the need for electrodes and enabling the visualization of multiple ion activities over the landscape of a cell, neuronal circuit, or brain slice. Lastly, the modular synthesis of the oxazine dye will enable the synthesis of chemically-reactive derivatives of KNIR-1 that can be conjugated to membrane proteins36 and/or the cell surface37 to detect extracellular K+ accumulation and depletion.
EXPERIMENTAL SECTION
General experimental procedures
Materials were obtained from commercial suppliers and were used without further purification. The final oxazine compound was purified by RP HPLC. NMR spectra were recorded in CDCl3, CD3OD or DMSO-d6 on a 400 MHz spectrometer. HRMS were obtained on a Q-TOF mass spectrometer equipped with an ESI source.
UV-visible and fluorescence experiments
Unless otherwise stated, all UV-visible and fluorescence experiments were performed in 10 mM HEPES solutions (pH 7.4) using acetonitrile as a vehicle (0.5% final concentration) in 3 mL quartz cuvettes (1 cm optical path length). Excitation wavelength (λex) was 620 nm and slit width was 5 nm. Fluorescence measurements were corrected for dilution when aliquots of the different test cation solutions significantly changed the total volume.
TAC-Br (2)
Bromination of triazacryptand 1 was carried out according to a previously reported aryl bromination procedure38. Triazacryptand 123 (220 mg, 0.32 mmol, 1.0 eq) was dissolved in CH2Cl2 (5 mL) and cooled to 0°C in an ice bath. A 0.55 M stock solution of bromine in dry CH2Cl2 was added dropwise by syringe (0.63 mL, 0.35 mmol, 1.1 eq) to above solution over a 5 min period. After 10 minutes of stirring, the ice bath was removed and the mixture was stirred at room temperature for an additional 1.25 h. The reaction was cooled again to 0°C in an ice bath and another aliquot of the 0.55 M stock solution of bromine in dry CH2Cl2 was added via syringe (0.63 mL, 0.35 mmol, 1.1 eq). After 10 minutes of stirring, the ice bath was removed and the mixture was stirred at ambient temperature for an additional 1.25 h. The reaction was quenched by adding 10% aqueous NaOH (10 mL) and saturated aqueous Na2S2O3 solution (10 mL). The organic layer was washed with water (10 mL), dried with Na2SO4, filtered, and concentrated under reduced pressure to give 2 as a brown solid (237 mg, 95%) which was used in the next step without further purification. 1H NMR (CDCl3, 400 MHz) δ: 6.96–7.01 (m, 3H), 6.87 (d, J = 7.8, 2H), 6.65 (d, J = 7.8, 2H), 6.56 (s, 2H) 4.12–4.18 (m, 2H), 3.98–4.01 (m, 4H), 3.89–3.92 (m, 4H), 3.78–3.80 (m, 2H) 3.59–3.73 (m, 16H), 3.46 (s, 3H), 3.40–3.45 (m, 4H), 3.26–3.33 (m, 4H), 2.24 (s, 6H). 13C NMR (CDCl3, 100 MHz): δ 153.3, 153.1, 138.3, 137.7, 132.8, 124.6, 123.0, 121.6, 121.2, 117.4, 114.5, 114.1, 71.4, 71.2, 70.2, 68.3, 67.2, 59.3, 54.1, 53.2, 21.3. HRMS (ESI): m/z calculated for C39H55BrN3O8 ([M+H]+): 772.3173; found: 772.3169.
1-Ethyl-7-methoxy-1,2,3,4-tetrahydroquinoline (11)
Potassium carbonate (1.2 g, 8.4 mmol, 1.5 eq) was added to a solution of 1-ethyl-7-methoxy-1,2,3,4-tetrahydroxyquinoline (5)30 (0.91 g, 5.6 mmol, 1.0 eq) in DMF (5 mL). Ethyl iodide (0.54 mL, 6.8 mmol, 1.2 eq) was added and the reaction was stirred at 65°C for 18 hrs. The mixture was allowed to cool to room temperature, diluted with water (80 mL), and acidified to pH 3 with 1 M HCl. The aqueous phase was extracted with ethyl acetate (1 × 80 mL, 5 × 50 mL) and the combined organic phases were washed with water (3 × 60 mL) and brine (80 mL) and dried over Na2SO4. The solvent was evaporated under vacuum and the crude product was purified by flash column chromatography on silica (100% hexanes to acetone/hexanes 5:95) to yield 11 as a yellow oil (0.91 g, 85%). Rf = 0.58 (silica/EtOAc/hexanes 1:19). 1H-NMR (CDCl3): δ 6.84 (d, 1H, J = 8.1 Hz), 6.18 (d, 1H, J = 2.1 Hz), 6.13 (dd, 1H, J = 2.4 Hz, 8.1 Hz), 3.78 (s, 3H), 3.32 (q, 2H, J = 7.1 Hz), 3.24 (t, 2H, J = 5.6 Hz), 2.68 (t, 2H, J = 6.4 Hz), 1.96–1.9 (m, 2H), 1.14 (t, 3H, J = 7.1Hz). 13C-NMR (CDCl3): δ 159.6, 146.1, 129.7, 115.6, 99.8, 97.6, 55.4, 48.6, 45.7, 27.8, 22.8, 11.1. HRMS (ESI) m/z calculated for C12H18NO2: 192.1388 ([M+H+]); found: 192.1379.
(1-Ethyl-7-methoxy-1,2,3,4-tetrahydro-quinolin-6-yl)-(4-nitro-phenyl)-diazene (3)
4-Nitrobenzenediazonium tetrafluoroborate (0.57 g, 2.4 mmol, 1.0 eq) was suspended in 10% aqueous H2SO4 (1.6 mL) and added dropwise to a solution of 11 (0.46 g, 2.4 mmol, 1.0 eq) in methanol (2 mL). The resulting dark purple solution was stirred at room temperature for 1 hour and cooled in an ice bath. The reaction was neutralized with aqueous NH4OH (28–30%) and the resulting precipitate was isolated by filtration and washed with water (300 mL) to give a dark red solid. The crude product was recrystallized from n-butanol/water to yield 3 as dark purple crystalline solid (0.52 g, 64%). Rf = 0.14 (silica/EtOAc/hexanes 1:19). 1H-NMR (CDCl3): δ 8.26 (d, 2H, J = 8.9 Hz), 7.84 (d, 2H, J = 8.9 Hz), 7.58 (s, 1H), 6.11 (s, 1H), 4.00 (s, 3H), 3.46 (q, 2H, J = 7.1 Hz), 3.38 (t, 2H, J = 5.6 Hz), 2.71 (t, 2H, J = 6.0 Hz), 1.95(p, 2H, J = 5.6 Hz, 6.1 Hz), 1.26 (t, 3H, J = 7.1Hz). 13C-NMR (CDCl3): δ 160.3, 158, 151.4, 146.7, 133.6, 124.9, 122.5, 117.7, 116.5, 92.8, 56.5, 49.2, 46.3, 27.5, 22.2, 11.5. HRMS (ESI) m/z calculated for C18H21N4O3: 341.1614 ([M+H]+); found: 341.1625.
1-(prop-2-yn-1-yl)-1,2,3,4-tetrahydroquinolin-7-ol (12)
7-Hydroxy-1,2,3,4-tetrahydroquinoline (6)30 (0.60 g, 4.0 mmol, 1.0 eq) and K2CO3 (0.66 g, 4.8 mmol, 1.2 eq) were combined in DMF (5 mL). Propargyl bromide (0.9 mL of an 80 wt% solution in toluene, 6.0 mmol, 1.5 eq) was added, and the reaction was stirred at 60°C for 1.5 hrs. The reaction was cooled, and partitioned between EtOAc (100 mL) and water (20 mL). The aqueous layer was washed with EtOAc (3 × 20 mL), organic layers were combined, dried over Na2SO4, filtered, and concentrated under vacuum. The crude product was purified by flash chromatography on silica (EtOAc/hexanes 1:4) to yield 12 as a brown oil which acquired a light pink color upon standing (0.52 g, 69%). Rf = 0.35 (silica/EtOAc/hexanes 1:4). 1H NMR (CDCl3, 400 MHz) δ: 6.82 (d, J = 8.0 Hz, 1H), 6.24 (d, J = 3.6 Hz), 6.16 (dd, J = 8.0 Hz, J = 3.6 Hz, 1H), 4.68 (s, 1H), 3.97 (d, J = 2.4 Hz, 2H), 3.27 (t, J = 5.6 Hz, 2 H), 2.69 (t, J = 6.4 Hz, 2H), 2.17 (t, J = 2.4 Hz, 1H), 2.01 − 1.94 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ 154.7, 145.5, 129.8, 116.4, 104.2, 99.3, 79.5, 71.8, 49.1, 40.8, 26.9, 22.5. HRMS (ESI): m/z calculated for C12H14NO ([M+H]+): 188.1070; found 188.1065.
1-(prop-2-yn-1-yl)-1,2,3,4-tetrahydroquinolin-7-yl acetate (4)
The propargylamine 12 (200 mg, 1.1 mmol, 1.0 eq), acetic anhydride (402 μL, 4.4 mmol, 4.0 eq) and triethylamine (306 μL, 2.2 mmol, 2.0 eq) were dissolved in CH2Cl2 (5 mL), and the reaction was stirred at room temperature for 18 hrs. The reaction was washed with saturated aqueous Na2CO3 (20 mL) and 0.5 M HCl (20 mL), dried over MgSO4, filtered and solvent was removed. Purification by flash chromatography on silica (EtOAC/hexanes 5:95) yielded 4 as a yellow oil which became a white solid upon standing (192 mg, 71%). Rf = 0.33 (silica/EtOAc/hexanes 1:19). 1H NMR (CDCl3, 400 MHz) δ: 6.97 (d, J = 8.0 Hz, 1H), 6.42 (d, J = 2.0 Hz, 1H), 6.40 (dd, J = 8.0 Hz, J = 2.0 Hz), 3.97 (d, J = 2.4 Hz, 2H), 3.29 (t, J = 5.6 Hz, 2 H), 2.74 (t, J = 6.4 Hz, 2H), 2.29 (s, 3H), 2.20 (t, J = 2.4 Hz, 1H), 2.02 − 1.96 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ 169.9, 149.9, 145.4, 129.5, 121.4, 110.0, 105.1, 79.3, 72.0, 48.9, 40.7, 27.2, 22.2, 21.2. HRMS (ESI): m/z calculated for C14H16NO2 ([M+H]+): 230.1181; found: 230.1171.
11-ethyl-1-(prop-2-yn-1-yl)-2,3,4,8,9,10-hexahydro-1H-dipyrido[3,2-b:2′,3′-i]phenoxazin-11-ium chloride (7)
The aryl azobenzene 3 (20 mg, 0.59 mmol, 1.0 eq) and the propargylamine 4 (13 mg, 0.59 mmol, 1.0 eq) were combined in 0.5 mL of 1.5% HCl (37% HCl/EtOH/water 0.5 mL:10 mL:1 mL) and stirred at 80°C for 4 hrs. The resulting dark green reaction was concentrated under vacuum. Purification by column chromatography on silica (MeOH/CH2Cl2 1:10) yielded the chloride salt of 7 as a dark blue solid (13 mg, 59%). Rf = 0.59 (silica/MeOH/CH2Cl2 1:9). 1H NMR (CDCl3, 400 MHz) δ: 7.47 (s, 1H), 7.36 (s, 1H), 7.29 (s, 1H), 6.51 (s, 1H), 3.72 –3.68 (m, 2H), 3.55 (t, J = 6.8 Hz, 4H), 3.49 (s, 2H), 2.87 (t, J = 6.0 Hz, 4H), 2.17 (s, 1H), 2.08 − 1.97 (m, 4H), 1.34 (t, J = 7.2 Hz, 3H). 13C NMR (CDCl3, 100 MHz): δ 155.1, 153.4, 146.3, 132.8, 130.7, 130.4, 130.3, 130.2, 128.5, 95.6, 94.9, 76.5, 73.7, 60.9, 57.8, 50.1, 26.9, 26.8, 20.7, 20.4, 10.4. HRMS (ESI): m/z calculated for C23H24N3O (M+): 358.1914; found: 358.1917.
(E)-1-(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)allyl)-1,2,3,4-tetrahydroquinolin-7-yl acetate (9)
The propargylamine 4 (220 mg, 0.74 mmol, 1.0 eq), pinacolborane (110 mg, 0.89 mmol, 1.2 eq) and Et3N (8.6 μL, 0.060 mmol, 0.086 eq) were combined in a sealable reaction vessel and the vessel was purged with Ar for 10 min. ZrCp2HCl (8.1 mg, 0.031 mmol, 0.042 eq) was quickly added, vessel was sealed and the reaction was stirred at 60°C for 15 hrs under Ar. Reaction contents were dissolved in EtOAc/hexanes (1:9) and the product was purified by flash chromatography on silica (EtOAc/hexanes 1:99 to 3:97) to yield 7 as a yellow oil (127 mg, 48%). Rf = 0.15 (silica/EtOAc/hexanes 1:19). 1H NMR (CD3OD, 400 MHz) δ: 6.79 (dd, J = 12 Hz, J = 8 Hz, 1H), 6.09 − 6.01 (m, 2H), 5.62 (d, J = 18 Hz, 1H), 4.86 (m, 2H), 3.20 − 3.18 (m, 2H), 2.64 − 2.61 (m, 2H), 2.06 (s, 3H), 1.94 − 1.86 (m, 2H), 1.24 (d, 12H).13C NMR (CD3OD, 100 MHz): δ 167.9, 155, 145.7, 136.2, 129.5, 124.7, 113.1, 104.3, 100.9, 82.4, 74.5, 71.8, 50.9, 41.4, 23.4, 23.5, 22.3, 17.0. HRMS (ESI): m/z calculated for C20 H29BNO4 ([M+H]+): 358.2190; found: 358.2186.
(E)-11-ethyl-1-(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)allyl)-2,3,4,8,9,10-hexahydro-1H-dipyrido[3,2-b:2′,3′-i]phenoxazin-11-ium chloride salt (8)
The aryl azobenzene 3 (28 mg, 0.095 mmol, 1.0 eq) and the borane ester 9 (40 mg, 0.095 mmol, 1.0 eq) were combined in 0.5 mL of 1.5% HCl (37% HCl/EtOH/water 0.5 mL:10 mL:1 mL) and stirred at 80°C for 4 hrs. The resulting dark green reaction was concentrated under vacuum. Purification by column chromatography on silica (MeOH/CH2Cl2 1:10) yielded 8 as a dark blue solid (14 mg, 29%). Rf = 0.57 (silica/MeOH/CH2Cl2 3:17).1H NMR (CDCl3, 400 MHz) δ: 6.96 (s, 1H), 6.94 (s, 1H), 6.41 − 6.37 (m, 4H), 3.97 (d, J = 2.7 Hz, 2H), 3.29 (m, 4H), 2.72 (t, J = 6.8 Hz, 4H), 2.28 (s, 12H). 2.17 (m, 3H), 2.00 − 1.97 (m, 4H). 13C NMR (CDCl3, 100 MHz): δ 155.2, 153.5, 148.0, 130.8, 130.3, 128.7, 121.3, 111.1, 74.8, 73.8, 61.1, 57.9, 50.2, 27.1, 26.9, 20.8, 20.6, 10.5. HRMS (ESI): m/z calculated for C29H37BN3O3 (M+): 484.9222; found: 484.9227.
TAC-N-propargyl-1,2,3,4-tetrahydroquinolin-7-ol (10)
A reaction vessel containing TAC-Br (271 mg, 0.35 mmol, 1.0 eq) and propargylamine 4 (250 mg, 1.1 mmol, 3.1 eq) was purged with Ar for 10 min and the Ar was removed by applying high vacuum. The above procedure was repeated two more times. DMF (1.0 mL) and pyrrolidine (1.0 mL) were injected into the reaction vessel and Ar was bubbled through the resulting solution for 10 min. Pd(PPh3)4 (66 mg, 0.058 mmol, 0.17 eq) and Cu(I)I (9.5 mg, 0.050 mmol, 0.014 eq) were quickly added, reaction was purged with Ar for 5 min and stirred at 80°C for 18 hrs. Solvents were removed under high vacuum and the resulting brown residue was purified by flash chromatography on neutral alumina (CH2Cl2 to MeOH/CH2Cl2 1:9). The purified product was redissolved in 1:2 MeOH/CH2Cl2 (6 mL), SiliaMet® DMT (450 mg of silica with 0.60 mmol/g scavenger loading, 5 eq) was added and the reaction was refluxed at 80°C for 4 hrs. The silica was filtered off and the solvent removed to yield 10 as a brown oil (108 mg, 35%). A 30 mg aliquot of 10 was purified by preparative HPLC according to the conditions below to obtain a sample with sufficient purity for spectroscopic characterization. Method started at 30% solvent B (0.1% trifluoroacetic acid in acetonitrile) and 70% solvent A (0.1% trifluoroacetic acid in water), then 30–60% solvent B over 15 min, then 60–100% solvent B over 1 min, then 100% solvent B for 5 min. HPLC absorbance detector was set at 254 nm. The product eluted at 15.0 min. HPLC fractions were combined and lyophilized to yield 10 as a brown oil. Rf = 0.30 (neutral alumina/methanol/CH2Cl2 1:49).1 H NMR (CDCl3, 400 MHz) δ: 7.08 (d, J = 8.0 Hz, 1H), 6.96 − 6.82 (m, 6H), 6.75 (m, 2H), 6.20 (dd, J = 6.0 Hz, J = 2.4 Hz, 1H), 6.03 (d, J = 2.4 Hz, 1H), 4.21 (t, J = 4.2 Hz, 4H), 4.10 (t, J = 4.4 Hz, 2H), 3.79 − 3.61 (m, 27H), 3.40 (t, J = 4.8 Hz, 1H), 3.35 (s, 3H), 3.29 − 3.26 (m, 8H), 2.69 (t, J = 6.4 Hz, 4H), 2.36 (s, 6H), 1.94 − 1.88 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ 153.3, 150.4, 148.8, 131.8, 129.5, 127.4, 121.6, 118.6, 112.0, 109.5, 102.3, 98.5, 81.9, 68.2, 68.1, 67.6, 65.3, 64.2, 51.0, 50.5, 49.9, 39.2, 28.5, 23.7, 19.9, 18.5, 18.2. HRMS (ESI): m/z calculated for C51H66N4O9Na ([M+Na]+): 901.4727; found: 901.4726.
TAC-N-propan-1,2,3,4-tetrahydroquinolin-7-ol (13)
The TAC-alkyne 10 (104 mg, 0.12 mmol, 1.0 eq) and tosylhydrazide (440 mg, 2.4 mmol, 20 eq) were dissolved in DME (1.0 mL) and stirred at 80°C. A solution of NaOAc (190 mg, 2.4 mmol, 20 eq) in water (1.0 mL) was added dropwise to the above solution over a period of 6 hrs. The resulting mixture was stirred at 80°C for 18 hrs and then stirred at room temperature for another 4 hrs. The reaction was partitioned between EtOAc (10 mL) and water (5 mL), and the aqueous phase was extracted with EtOAc (3 × 10 mL). The organic layers were combined, dried over MgSO4, filtered and solvent was removed. Purification by flash chromatography on neutral alumina (CH2Cl2 to MeOH/CH2Cl2 1:9) yielded 13 as a brown oil (76 mg, 72%). A 30 mg aliquot of 13 was purified by preparative HPLC according to the conditions below to obtain a sample with sufficient purity for spectroscopic characterization. Method started at 30% solvent B (0.1% trifluoroacetic acid in acetonitrile) and 70% solvent A (0.1% trifluoroacetic acid in water), then 30–60% solvent B over 15 min, then 60–100% solvent B over 1 min, then 100% solvent B for 5 min. HPLC absorbance detector was set at 254 nm. The product eluted at 15.7 min. HPLC fractions were combined and lyophilized to yield 13 as a brown oil. Rf = 0.32 (neutral alumina/methanol/CH2Cl2 1:49). 1H NMR (CDCl3, 400 MHz) δ: 7.12 (dd, J = Hz, J = Hz, 1H), (m, 7H), 6.74 (m, 2H), 6.20 (dd, J = 2.8 Hz, J = 8.4 Hz, 1H), 6.03 (d, J = 2.8 Hz, 1H), 4.23 − 4.20 (m, 4H), 4.12 − 4.09 (m, 2H), 3.79 − 3.61 (m, 26H), 3.43 − 3.40 (m, 4H), 3.35 (s, 3H), 3.29 − 3.26 (m, 8H), 2.69 (t, J = 6.4 Hz, 2H), 2.36 (s, 6H), 1.94 − 1.84 (m, 4H), 1.49 − 1.45 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ 162.7, 152.7, 146.5, 139.5, 134.2, 127.7, 121.3, 115.3, 113.6, 113.2, 71.2, 67.5 67.0, 55.1, 58.9, 52.4, 50.1, 36.4, 31.3, 21.1. HRMS (ESI): m/z calculated for C51H70N4O9Na ([M+Na]+): 905.5040; found: 905.5042.
KNIR-1
The aryl azobenzene 3 (28 mg, 0.095 mmol, 1.0 eq) and TAC-alkane 13 (40 mg, 0.095 mmol, 1.0 eq) were combined in AcOH (0.5 mL) and stirred at 80°C for 18 hrs. The resulting dark green reaction was concentrated under vacuum, and purified by preparative HPLC according to the conditions. Method started at 30% solvent B (0.1% trifluoroacetic acid in acetonitrile) and 70% solvent A (0.1% trifluoroacetic acid in water), then 30–60% solvent B over 15 min, then 60–100% solvent B over 1 min, then 100% solvent B for 5 min. HPLC absorbance detector was set at 254 nm. The product eluted at 15.7 min. HPLC fractions were combined and lyophilized to yield the trifluoroacetate salt of KNIR-1 as a blue solid (mg, 22%), which was first converted to the hydroxide salt and then to the acetate salt by passing through DOWEX® 1×8 200–400 hydroxide and acetate anion exchange columns. Rf = 0.20 (silica/methanol/CH2Cl2 1:9). 1H NMR (CD3OD, 400 MHz) δ: 7.58 (d, J = 9.6 Hz, 2H), 7.45 (d, J = 14.4 Hz, 2H), 7.22 (d, 8.0 Hz, 1H), 7.03 − 6.98 (m, 5H), 6.92 (s, 2H), 6.85 (d, J = 8.0 Hz, 1H), 6.60 (s, 1H), 4.35 − 4.20 (m, 6H), 4.25 − 3.85 (m, 12H), 3.85 − 3.75 (m, 5H), 3.75 − 3.50 (m, 17H), 3.50 − 3.35 (m, 5H), 3.31 (s, 3H), 3.85 − 3.75 (m, 4H), 2.70 (t, J = 7.2 Hz, 2H), 2.30 (s, 6H), 2.20 − 1.80 (m, 6H), 1.38 (t, J = 7.2 Hz, 3H). 13C NMR (DMSO-d6, 100 MHz): δ 172.5, 163.5, 156.0, 152.9, 152.8, 151.3, 151.1, 147.4, 131.9, 123.9, 122.2, 121.6, 121.5, 121.4, 116.7, 114.5, 114.4, 96.9, 95.3, 70.7, 70.5, 70.3, 67.9, 58.5, 52.6, 52.5, 52.5, 35.5, 32.5, 27.1, 21.5, 14.5, 11.8. HRMS (ESI): m/z calculated for C62H81N6O9Na ([M+Na]2+): 527.3069; found: 527.3068.
Supplementary Material
Acknowledgments
This work was supported by a grant to W.R.K. from the National Institutes of Health (GM-070650). S.C.M. acknowledges support from the NIH (GM-087460), and S.M.P. received fellowship support from the American Heart Association.
Footnotes
Supporting Information
Experimental procedures, compound characterization, and supplementary figures are available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interests.
References
- 1.Hounsgaard J, Nicholson C. J Physiol. 1983;340:359. doi: 10.1113/jphysiol.1983.sp014767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martin G, Morad M. J Physiol. 1982;328:205. doi: 10.1113/jphysiol.1982.sp014260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zifarelli G, Pusch M. J Gen Physiol. 2010;136:593. doi: 10.1085/jgp.201010567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dunn KM, Nelson MT. Circulation journal: official journal of the Japanese Circulation Society. 2010;74:608. doi: 10.1253/circj.cj-10-0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCrossan ZA, Abbott GW. Neuropharmacology. 2004;47:787. doi: 10.1016/j.neuropharm.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 6.Nichols CG. Nature. 2006;440:470. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- 7.Wallinga W, Vliek M, Wienk ED, J AM, Ypey DL. Proceedings of the 18th Annual International Conference of the IEEE. 1996;5:1909. [Google Scholar]
- 8.Shepherd N, McDonough HB. Am J Physiol. 1998;275:H852. doi: 10.1152/ajpheart.1998.275.3.H852. [DOI] [PubMed] [Google Scholar]
- 9.Swift F, Stromme TA, Amundsen B, Sejersted OM, Sjaastad I. J Appl Physiol. 2006;101:1170. doi: 10.1152/japplphysiol.00297.2006. [DOI] [PubMed] [Google Scholar]
- 10.Wangemann P. Hear Res. 2002;165:1. doi: 10.1016/s0378-5955(02)00279-4. [DOI] [PubMed] [Google Scholar]
- 11.Frohlich F, Bazhenov M, Iragui-Madoz V, Sejnowski TJ. Neuroscientist. 2008;14:422. doi: 10.1177/1073858408317955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Costa C, Tozzi A, Rainero I, Cupini LM, Calabresi P, Ayata C, Sarchielli P. The journal of headache and pain. 2013;14:62. doi: 10.1186/1129-2377-14-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cannon SC. J Physiol. 2010;588:1887. doi: 10.1113/jphysiol.2010.186874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marban E. Nature. 2002;415:213. doi: 10.1038/415213a. [DOI] [PubMed] [Google Scholar]
- 15.Ashcroft FM. J Clin Invest. 2005;115:2047. doi: 10.1172/JCI25495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiss J, Shine KI. Am J Physiol. 1982;243:H318. doi: 10.1152/ajpheart.1982.243.2.H318. [DOI] [PubMed] [Google Scholar]
- 17.Lee MP, Ravenel JD, Hu RJ, Lustig LR, Tomaselli G, Berger RD, Brandenburg SA, Litzi TJ, Bunton TE, Limb C, Francis H, Gorelikow M, Gu H, Washington K, Argani P, Goldenring JR, Coffey RJ, Feinberg AP. J Clin Invest. 2000;106:1447. doi: 10.1172/JCI10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paredes RM, Etzler JC, Watts LT, Zheng W, Lechleiter JD. Methods. 2008;46:143. doi: 10.1016/j.ymeth.2008.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, Looger LL, Svoboda K, Kim DS. Nature. 2013;499:295. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tian L, Hires SA, Mao T, Huber D, Chiappe ME, Chalasani SH, Petreanu L, Akerboom J, McKinney SA, Schreiter ER, Bargmann CI, Jayaraman V, Svoboda K, Looger LL. Nat Methods. 2009;6:875. doi: 10.1038/nmeth.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carpenter RD, Verkman AS. Organic letters. 2010;12:1160. doi: 10.1021/ol902836c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou X, Su F, Tian Y, Youngbull C, Johnson RH, Meldrum DR. J Am Chem Soc. 2011;133:18530. doi: 10.1021/ja207345s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He H, Mortellaro MA, Leiner MJ, Fraatz RJ, Tusa JK. J Am Chem Soc. 2003;125:1468. doi: 10.1021/ja0284761. [DOI] [PubMed] [Google Scholar]
- 24.Padmawar P, Yao X, Bloch O, Manley GT, Verkman AS. Nat Methods. 2005;2:825. doi: 10.1038/nmeth801. [DOI] [PubMed] [Google Scholar]
- 25.Hirata T, Terai T, Komatsu T, Hanaoka K, Nagano T. Bioorg Med Chem Lett. 2011;21:6090. doi: 10.1016/j.bmcl.2011.08.056. [DOI] [PubMed] [Google Scholar]
- 26.Weissleder R. Nature biotechnology. 2001;19:316. doi: 10.1038/86684. [DOI] [PubMed] [Google Scholar]
- 27.Eggeling C, Widengren J, Brand L, Schaffer J, Felekyan S, Seidel CA. Thejournal of physical chemistry A. 2006;110:2979. doi: 10.1021/jp054581w. [DOI] [PubMed] [Google Scholar]
- 28.Doose S, Neuweiler H, Sauer M. Chemphyschem: a European journal of chemical physics and physical chemistry. 2009;10:1389. doi: 10.1002/cphc.200900238. [DOI] [PubMed] [Google Scholar]
- 29.Hintersteiner M, Enz A, Frey P, Jaton AL, Kinzy W, Kneuer R, Neumann U, Rudin M, Staufenbiel M, Stoeckli M, Wiederhold KH, Gremlich HU. Nature biotechnology. 2005;23:577. doi: 10.1038/nbt1085. [DOI] [PubMed] [Google Scholar]
- 30.Pauff SM, Miller SC. Organic letters. 2011;13:6196. doi: 10.1021/ol202619f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Magzoub M, Padmawar P, Dix JA, Verkman AS. The journal of physical chemistry B. 2006;110:21216. doi: 10.1021/jp0633392. [DOI] [PubMed] [Google Scholar]
- 32.Rurack K, Spieles M. Analytical chemistry. 2011;83:1232. doi: 10.1021/ac101329h. [DOI] [PubMed] [Google Scholar]
- 33.Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. Science. 1998;280:69. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 34.Kleinlogel S, Feldbauer K, Dempski RE, Fotis H, Wood PG, Bamann C, Bamberg E. Nat Neurosci. 2011;14:513. doi: 10.1038/nn.2776. [DOI] [PubMed] [Google Scholar]
- 35.Caldwell JH, Herin GA, Nagel G, Bamberg E, Scheschonka A, Betz H. J Biol Chem. 2008;283:24300. doi: 10.1074/jbc.M802593200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hirata T, Terai T, Yamamura H, Shimonishi M, Komatsu T, Hanaoka K, Ueno T, Imaizumi Y, Nagano T, Urano Y. Analytical chemistry. 2016;88:2693. doi: 10.1021/acs.analchem.5b03970. [DOI] [PubMed] [Google Scholar]
- 37.Zhang L, Bellve K, Fogarty K, Kobertz WR. Cell chemical biology. 2016;23:1449. doi: 10.1016/j.chembiol.2016.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Metten B, Smet M, Boens N, Dehaen W. Synthesis. 2005;11:1838. doi: 10.1039/b505969k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.