

Abstract
Background
Chronic fatigue syndrome (CFS) is a prevalent and disabling condition among adolescent. The disease mechanisms are unknown. Previous studies have suggested elevated plasma levels of several cytokines, but a recent meta-analysis of 38 articles found that of 77 different cytokines measured in plasma, transforming growth factor beta (TGF-β) was the only one that was elevated in patients compared to controls in a sufficient number of articles. In the present study we therefore compared the plasma levels of the three TGF-β isoforms in adolescent CFS patients and healthy controls. In addition, the study explored associations between TGF-β levels, neuroendocrine markers, clinical markers and differentially expressed genes within the CFS group.
Methods
CFS patients aged 12–18 years (n = 120) were recruited nation-wide to a single referral center as part of the NorCAPITAL project (ClinicalTrials ID: NCT01040429). A broad case definition of CFS was applied, requiring 3 months of unexplained, disabling chronic/relapsing fatigue of new onset, whereas no accompanying symptoms were necessary. Healthy controls (n = 68) were recruited from local schools. The three isoforms of TGF-β (TGF-β1, TGF-β2, TGF-β3) were assayed using multiplex technology. Neuroendocrine markers encompassed plasma and urine levels of catecholamines and cortisol, as well as heart rate variability indices. Clinical markers consisted of questionnaire scores for symptoms of post-exertional malaise, inflammation, fatigue, depression and trait anxiety, as well as activity recordings. Whole blood gene expression was assessed by RNA sequencing in a subgroup of patients (n = 29) and controls (n = 18).
Results
Plasma levels of all three isoforms of TGF-β were equal in the CFS patients and the healthy controls. Subgrouping according to the Fukuda and Canada 2003 criteria of CFS did not reveal differential results. Within the CFS group, all isoforms of TGF-β were associated with plasma cortisol, urine norepinephrine and urine epinephrine, and this association pattern was related to fatigue score. Also, TGF-β3 was related to expression of the B cell annotated genes TNFRSF13C and CXCR5.
Conclusions
Plasma levels of all TGF-β isoforms were not altered in adolescent CFS. However, the TGF-β isoforms were associated with neuroendocrine markers, an association related to fatigue score. Furthermore, TGF-β3 might partly mediate an association between plasma cortisol and B cell gene expression.
Trial registration Clinical Trials NCT01040429
Electronic supplementary material
The online version of this article (10.1186/s12967-017-1350-1) contains supplementary material, which is available to authorized users.
Keywords: Chronic fatigue syndrome, Adolescent, Inflammation, Transforming growth factor beta
Background
Chronic fatigue syndrome (CFS) is a long-lasting and disabling condition characterized by disproportional fatigue after exertions, musculoskeletal pain, headaches, cognitive impairments, orthostatic intolerance and other symptoms [1, 2]. CFS is a major threat towards adolescent health. The prevalence among teenagers is estimated at 0.1–1.0% [3, 4], and the condition may have detrimental effects on psychosocial and academic development [5], as well as family functioning [6].
Disease mechanisms of CFS remain poorly understood. However, several studies indicate modest immunological alterations, such as enhancement of systemic inflammation [7–9] and attenuation of B cell function [10–12]. A recent study on whole blood gene expression in CFS corroborated a general picture of increased innate immunity and decreased B cell differentiation and survival in CFS [13].
Despite these findings, studies of plasma cytokine levels have been inconclusive; findings include increased levels of interleukin (IL)-1 and tumor necrosis factor (TNF) [14], increased levels of IL-1α and IL-1β but normal levels of TNF [15], and no differences between CFS patients and healthy controls [16, 17]. A recent review including meta-analysis of 38 articles studying all together 77 different cytokines, including interleukins, chemokines and growth factors, found evidence of elevated blood levels only of transforming growth factor beta (TGF-β) in CFS (63% of the studies), whereas other cytokines were not found to be consistently altered in any direction [18]. In a previous study from our laboratory [17], we found that the 27 plasma cytokines measured were identical in the CFS and the control group, without any tendency for differences. This study using modern multiplex technique did not, however, include TGF-β, which here prompted us to investigate our population for this marker, in particular extending inclusion of all three different isoforms (denoted TGF-β1, TGF-β2 and TGF-β3).
TGF-β is excreted by macrophages and other immune cells. All isoforms are involved in a multitude of regulatory processes, including control of inflammation and specific immunity [19]. Notably, in some conditions, TGF-β promotes inflammatory responses combined with an inhibitory effect on several stages in B cell differentiation and survival. Thus, TGF-β is a possible mediator of the subtly skewed immune responses observed in a previous CFS study [13]. Furthermore, TGF-β stimulates the generation of regulatory T cells [20], which has been shown to be increased in CFS by previous reports [21, 22].
In addition to immune changes, CFS disease mechanisms are characterized by neuroendocrine alterations including enhanced sympathetic and attenuated parasympathetic cardiovascular nervous activity [23–26] and attenuation of the hypothalamus–pituitary–adrenal (HPA) axis [27–29]. These phenomena might be causally related, as suggested in the “sustained arousal” model of CFS [30]. For instance, sympathetic activity is shown to enhance TGF-β3 mRNA expression in macrophages [31]. Another study has demonstrated a relationship between HPA dynamics and plasma levels of TGF-β1 [32]. Recently, whole blood gene expression of immune annotated genes in CFS was demonstrated to be strongly associated with neuroendocrine markers of altered HPA-axis and autonomic nervous activity [13]. Thus, a possible relationship between neuroendocrine alterations and TGF-β levels in CFS deserves further attention.
Symptoms that might suggest immune activation, such as chills, sore throat and tender lymphatic nodes, are common complaints among CFS sufferers [1, 33]. A previous study revealed no associations between plasma cytokine levels and symptom scores [17]. Still, a relationship between differential gene expression and symptoms of post-exertional malaise, which is considered a hallmark of the CFS phenotype, has been observed [13]. This finding warrants further exploration of associations between clinical symptoms and immune markers, including TGF-β.
In general, the scientific investigation of adolescent CFS suffers several shortcomings as compared to adult CFS. For instance, to the best of our knowledge, no other studies have specifically addressed circulating TGF-β expression in adolescent CFS patients. Nevertheless, adolescent studies might benefit from low prevalence of comorbidity as well as a limited possibility of confounding effects from aging processes.
Taken together, the main aim of the present study was to compare plasma levels of TGF-β isoforms in adolescent CFS patients and healthy controls. We hypothesized increased levels among the CFS patients based on the previous meta-analysis [18]. In addition, the present study had two exploratory aims: (a) To explore associations between TGF-β levels, neuroendocrine markers and differentially expressed genes in the CFS group, and (b) To explore associations between TGF-β levels and CFS clinical symptoms.
Methods
CFS patients
This study is part of the NorCAPITAL-project (The Norwegian Study of Chronic Fatigue Syndrome in Adolescents: Pathophysiology and Intervention Trial; ClinicalTrials ID: NCT01040429). Details of the recruitment procedure and inclusion/exclusion criteria are described elsewhere [34]. Briefly, all hospital pediatric departments in Norway (n = 20), as well as primary care pediatricians and general practitioners, were invited to refer CFS patients aged 12–18 years consecutively to our study center. A standard form required the referral unit to confirm the result of clinical investigations considered compulsory to diagnose pediatric CFS according to national Norwegian recommendations (pediatric specialist assessment, comprehensive hematology and biochemistry analyses, chest X-ray, abdominal ultrasound, and brain MRI). Also, the referring units were required to confirm that the patient (a) was unable to follow normal school routines due to fatigue; (b) was not permanently bedridden; (c) did not have any concurrent medical or psychiatric disorder that might explain the fatigue; (d) did not experience any concurrent demanding life event (such as parents’ divorce) that might explain the fatigue; (e) did not use pharmaceuticals (including hormone contraceptives) regularly. Patients considered eligible to this study were summoned to a clinical encounter at our study center after which a final decision on inclusion was made. The CFS group comprised 120 patients.
In agreement with clinical guidelines [2] and previous studies from our group [24–26], we applied a ‘broad’ case definition of CFS, requiring 3 months of unexplained, disabling chronic/relapsing fatigue of new onset. We did not require that patients meet any other accompanying symptom criteria. Subgrouping of the participants according to the Fukuda case definition [35] and Canada 2003 case definition [33] was performed post hoc, based on questionnaire results (cf. below).
Healthy controls
A group of healthy controls (n = 68) with a comparable distribution of gender and age were recruited from local schools. Controls were not matched to cases on any variable. No chronic disease and no regular use of pharmaceuticals (including hormone contraceptives) were allowed.
Study design and ethics
A 1-day in-hospital assessment included clinical examination and blood sampling and always commenced between 7.30 and 9.30 a.m. All participants were instructed to fast overnight and abstain from tobacco products and caffeine for at least 48 h. The participants were instructed to apply an ointment containing the local anesthetic lidocaine (Emla®) on the skin in the antecubital area 1 h in advance. After at least 5 min supine rest in calm surroundings, blood samples were obtained in a fixed sequence from antecubital venous puncture. A questionnaire was completed after the clinical encounter and returned in a pre-stamped envelope.
Data were collected in the period from March 2010 until October 2012. The NorCAPITAL project has been approved by the Norwegian National Committee for Ethics in Medical Research (Reference ID: 2009/2541 S-09354c). Written informed consent was obtained from all participants and from parents/next-of-kin if required. CFS patients were randomized to treatment with low-dose clonidine or placebo and followed for a total of 30 weeks; however, only baseline data are used in the present study. Details of the design are reported elsewhere [34].
TGF-β isoforms: TGF-β1, TGF-β2 and TGF-β3
The blood samples for TGF-β analyses were collected in 4 ml EDTA tubes. The samples were placed on ice for approximately 30 min; thereafter, plasma was separated by centrifugation (2500g, 10 min, 4 °C) and frozen at − 80 °C until assayed. The samples were analyzed using Bio-Plex Human TGF-β 3-plex (Bio-Rad Laboratories Inc., Hercules, CA), containing the three isoforms of TGF-β: TGF-β1, TGF-β2 and TGF-β3. The assay was performed using the Bio-Plex 200 system (Bio-Rad Laboratories Inc.) according to the manufactures’ instructions. All samples were obtained, treated stored and analysed in exactly in the same way, using the same personnel at each step, to exclude methodological variation since TGFβ is sensible for sample handling, including release from platelets.
Neuroendocrine markers
Blood samples for analyses of plasma norepinephrine and epinephrine were obtained in vacutainer tubes treated with ethylene glycol tetraacetic acid (EGTA)–glutathione. The samples were placed on ice for approximately 30 min, and thereafter separated by centrifugation (2250×g, 15 min, 4 °C) and assayed by high-performance liquid chromatography (HPLC) with a reversed-phase column and glassy carbon electrochemical detector (Antec, Leyden Deacade II SCC, Zoeterwoude, The Netherlands) as described in detail elsewhere [29]. Blood samples for analyses of plasma cortisol were obtained in 4 ml EDTA vacutainer tubes; plasma cortisol levels were determined by routine assays at the accredited laboratory at Oslo University Hospital, Norway. Morning spot urine samples for norepinephrine and epinephrine analyses were assayed with the same HPLC protocol as for plasma measurements, whereas morning spot urine free cortisol (non-conjugated cortisol) was assayed by solid phase competitive luminescence immunoassay (type Immulite® 2000, Siemens Healthcare Diagnostics, NY, USA). The urine levels of creatinine were analyzed using standard automatic analyzer techniques at the accredited laboratory at Oslo University Hospital, Norway.
Indices of heart rate variability (HRV) were obtained from ECG recordings of participants laying in a horizontal position and connected to the Task Force Monitor (TFM) (Model 3040i, CNSystems Medizintechnik, Graz, Austria). Methodological details are provided elsewhere [36]. Power spectral analysis of HRV was automatically provided by the TFM, returning numerical values for low frequency (LF) power (0.05–0.17 Hz), high frequency (HF) power (0.17–0.4 Hz) and the LF/HF ratio. HF power is considered indicative of parasympathetic heart rate modulation, LF power reflects the combined effect of sympathetic and parasympathetic heart rate control, whereas the LF/HF ratio is an index of sympathetic/parasympathetic balance.
Routine blood analyses
The serum concentration of C-reactive protein (CRP) was analyzed using a highly sensitive assay (Roche Diagnostics, Indianapolis, IN, USA), as described previously [34]. Hematology and biochemistry routine assays were performed at the accredited laboratory at Oslo University Hospital, Norway.
Whole blood gene expression
In a subgroup of 29 CFS patients and 18 healthy controls, RNA was extracted from whole blood as described in details elsewhere [13]. RNA samples with RNA integrity number value ≥ 7 were used for gene expression characterization by RNA sequencing (RNA-Seq) (Run by the Genomics Core Facilities at the Oslo University Hospital Radiumhospitalet). RNA reads were mapped to the human genome version GRCh38.p2 by STAR [37]. Gene expression abundance was quantified as reads mapped per exon by the Subread package [38]. Differentially expressed genes (DEG) between CFS patients and controls were identified using DESeq2 package [39]. In order to correct for possible confounding background factors, age groups as scaling factor and gender input were included in the design model of DESeq2. Functional annotation of genes obtained from DESeq2 was done by uploading all DEGs into HumanMine [40 ].
Clinical markers
A CFS symptom inventory for adolescents assesses the frequency of 24 common symptoms during the preceding month, as has been described elsewhere [34]. Briefly, each symptom is rated on a 5-point Likert scale, ranging from ‘never/rarely present’ to ‘present all the time’. The inventory includes the accompanying symptom of the Fukuda and Canada 2003 case definitions, facilitating post hoc subgrouping of CFS patients (Additional file 1). A composite score reflecting inflammatory symptoms was generated by taking the arithmetic mean across three single items (fever/chills, sore throat, and tender lymphatic nodes) and a composite score reflecting symptoms of post-exertional malaise was generated by taking the arithmetic mean across two single items (post-exertional fatigue and non-refreshing sleep). For both variables, the total range is from 0 to 5; higher scores imply more severe symptom burden.
The Chalder Fatigue Questionnaire (CFQ) total sum score is applied in the present study [41]; total range is from 0 to 33, where higher scores imply more severe fatigue. The mood and feelings questionnaire (MFQ) consists of 34 items, each scored on a 0–2 Likert scale; thus, the total sum score is from 0 to 68 [42]. The Spielberger State-Trait Anxiety Inventory subscore reflecting trait anxiety is derived from the sum across 20 items; total range is from 20 to 80 [43]. The activPAL accelerometer device (PAL Technologies Ltd, Glasgow, Scotland) was used for monitoring of daily physical activity during seven consecutive days [44], as described elsewhere [34].
Statistical analyses
All statistical analyses were carried out in SPSS (SPSS Inc., Chicago, Illinois, USA). A total of 9 individuals (6 CFS patients, 3 healthy controls) had missing data for TGF-β analyses. As the CFS patients were included in a randomised controlled trial, individual data from follow-up consultations (when available) were used for imputation of missing data at baseline. The remaining missing data (3 CFS patients, 3 healthy controls) were imputed using the mean values within each group. Missing data in other variables were not imputed.
Patients with CFS were compared with healthy controls by applying independent sample t tests, Mann–Whitney tests, Pearson Chi Square tests, or Fisher exact tests as appropriate. CFS patients adhering to the Fukuda criteria and the Canada 2003-criteria were compared to the healthy controls in the same way. Associations between TGF-β and immune markers, neuroendocrine markers, single gene transcriptional counts and clinical markers within each group (CFS patients and healthy controls, respectively) were performed using correlation and linear regression analyses. A p < 0.05 was regarded as statistically significant. The correlation and linear regression analyses involved a multitude of statistical tests; however, as all these tests were considered exploratory, the p values were not adjusted.
Results
Demographic data
The 120 CFS patients fulfilling the inclusion criteria in the NorCAPITAL trial were included as the CFS patients group and 68 healthy individuals were included as control group [34]. The whole blood gene expression analysis (RNA-seq) was carried out in a random subgroup of 29 CFS patients and 18 healthy controls [13]. Gender, age and body mass index were equally distributed in the two groups (Table 1). In the CFS group, 98% were of Scandinavian ethnicity; 73% adhered to the Fukuda case definition and 38% to the Canada 2003-definition. With the exception of 1 patient, all belonging to the Canada 2003 subgroup also belonged to the Fukuda subgroup. A total of 2 CFS patients were on thyroid hormone supplement therapy, 1 used melatonin and 1 used bronchodilators occasionally; no other pharmaceuticals were used. No CFS patients and 3 healthy controls reported occasional use of illicit drugs; 78% in both groups reported no consume of alcoholic beverages. CFS patient had higher levels of serum CRP, plasma norepinephrine and plasma epinephrine, and lower levels of heart rate variability (HRV) LF-power and urine cortisol-to-creatinine ratio (Table 1). Also, CFS patients scored significantly poorer on all clinical markers (Table 1).
Table 1.
Demographic characteristics
| CFS patients (n = 120) | Healthy controls (n = 68) | p valuec | |||
|---|---|---|---|---|---|
| Background | |||||
| Female gender Number, % |
88 | 73 | 46 | 68 | 0.563 |
| Age (years) Mean, SD |
15.4 | 1.6 | 15.1 | 1.6 | 0.179 |
| Body mass index (kg/m2) Mean, SD |
21.5 | 4.2 | 20.6 | 3.7 | 0.119 |
| Disease duration (months) Median, range |
18 | 4–104 | n/a | ||
| Adheres to the Fukuda criteria of CFSa
Number, % |
88 | 73 | n/a | ||
| Adheres to the Canada 2003-criteria of CFSb
Number, % |
46 | 38 | n/a | ||
| Immune, autonomic and neuroendocrine markers | |||||
| Serum C-reactive protein (mg/l) Median, IQR |
0.43 | 0.96 | 0.35 | 0.46 | 0.049 |
| Heart rate variability, LF power (abs) Median, IQR |
481 | 772 | 682 | 1514 | 0.015 |
| Heart rate variability, HF power (abs) Median, IQR |
788 | 1835 | 1058 | 1837 | 0.070 |
| Heart rate variability, LF/HF-ratio Median, IQR |
0.69 | 0.64 | 0.74 | 0.38 | 0.792 |
| Plasma norepinephrine (pmol/l) Mean, SD |
1991 | 783 | 1483 | 422 | < 0.001 |
| Plasma epinephrine (pmol/l) Median, IQR |
308 | 131 | 265 | 95 | 0.001 |
| Plasma cortisol (nmol/l) Mean, SD |
367 | 143 | 351 | 150 | 0.460 |
| Urine norepinephrine/creatinine ratio (nmol/mmol) Median, IQR |
12.3 | 5.8 | 10.8 | 5.8 | 0.102 |
| Urine epinephrine/creatinine ratio (nmol/mmol) Median, IQR |
1.3 | 1.2 | 1.5 | 1.0 | 0.649 |
| Urine cortisol/creatinine ratio (nmol/mmol) Median, IQR |
3.4 | 3.1 | 5.3 | 6.0 | 0.001 |
| Clinical makers | |||||
| Symptoms of post-exertional malaise (total score) Mean, SD |
4.0 | 1.0 | 1.4 | 0.5 | < 0.001 |
| Inflammatory symptoms (total score) Mean, SD |
2.1 | 0.9 | 1.3 | 0.5 | < 0.001 |
| Chalder Fatigue Questionnaire (total score) Mean, SD |
19.3 | 6.1 | 8.7 | 4.6 | < 0.001 |
| Moods and Feelings Questionnaire (total score) Mean, SD |
17.4 | 10.0 | 6.4 | 7.8 | < 0.001 |
| Spielberger state-trait anxiety questionnaire (trait subscore) Mean, SD |
43.2 | 8.9 | 32.3 | 7.2 | < 0.001 |
| Steps per day (number) Mean, SD |
4662 | 2386 | 10,293 | 3716 | < 0.001 |
P values in italics indicate statistical significance
n/a not applicable, SD standard deviation, IQR interquartile range, LF low-frequency power of heart rate variability, HF high-frequency power of heart rate variability
aCf. Ref. [35]
bCf. Ref. [33]
cStatistical comparison across CFS group and HC group are based upon Independent sample t test, Mann–Whitney test or Fisher exact test as appropriate
Transforming growth factor-β (TGF-β)
Plasma levels of all three isoforms of TGF-β (TGF-β1, TGF-β2 and TGF-β3) were equal in CFS patients and healthy controls (Table 2, Additional file 2: Table S1). Subgroup analyses revealed no differential results, neither for the Fukuda case definition nor for the Canada 2003-definition. Within the CFS group, TGF-β was not associated with clinical markers (Additional file 3: Table S2). Also, whole blood transcriptional counts from the TGFB1 gene in the subgroup subjected to RNA-seq were equal among CFS patients and healthy controls (1184 vs 1208, p = 0.712).
Table 2.
TGF-β isoforms across CFS patients (including Fukuda and Canada 2003-subgroups) and healthy controls
| CFS patients (n = 120) | p valuea | Healthy controls (n = 68) | CFS, Fukuda subgroup (n = 88) | p valuea | CFS, Canada 2003 subgroup (n = 46) | p valuea | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Plasma TGF-β1 (pg/ml) Median, IQR |
4927 | 5157 | 0.482 | 5692 | 6350 | 4944 | 4276 | 0.599 | 4880 | 6247 | 0.831 |
| Plasma TGF-β2 (pg/ml) Mean, SD |
758 | 262 | 0.928 | 754 | 278 | 712 | 246 | 0.809 | 780 | 252 | 0.621 |
| Plasma TGF-β3 (pg/ml) Median, IQR |
230 | 227 | 0.901 | 251 | 250 | 230 | 208 | 0.990 | 243 | 254 | 0.760 |
TGF transforming growth factor beta, SD standard deviation, IQR interquartile range
aCompared with healthy controls. Statistical comparisons across groups are based upon Independent sample t test or Mann–Whitney test as appropriate
TGF-β, neuroendocrine markers and fatigue score
Plasma levels of TFG-β were associated with neuroendocrine markers, in particular plasma cortisol as well as plasma and urine catecholamines. Multiple linear regression analyses revealed that these associations differed in CFS patients and healthy controls (Fig. 1), despite similar levels of TFG-β in the two groups. In the CFS patients, all TGF-β isoforms were significantly and independently associated with plasma cortisol, urine norepinephrine-to-creatinine ratio and urine epinephrine-to-creatinine ratio, whereas no significant associations to these markers were observed among healthy control. This differential association pattern was related to symptom severity (Table 3). When divided according to fatigue severity (Chalder Fatigue Questionnaire), CFS patients with the highest fatigue score (≥ 20) displayed the strongest associations between TGF-β and neuroendocrine markers, whereas CFS patients with the lowest fatigue score (< 20) had a pattern of associations that was more similar to the one observed in healthy controls.
Fig. 1.
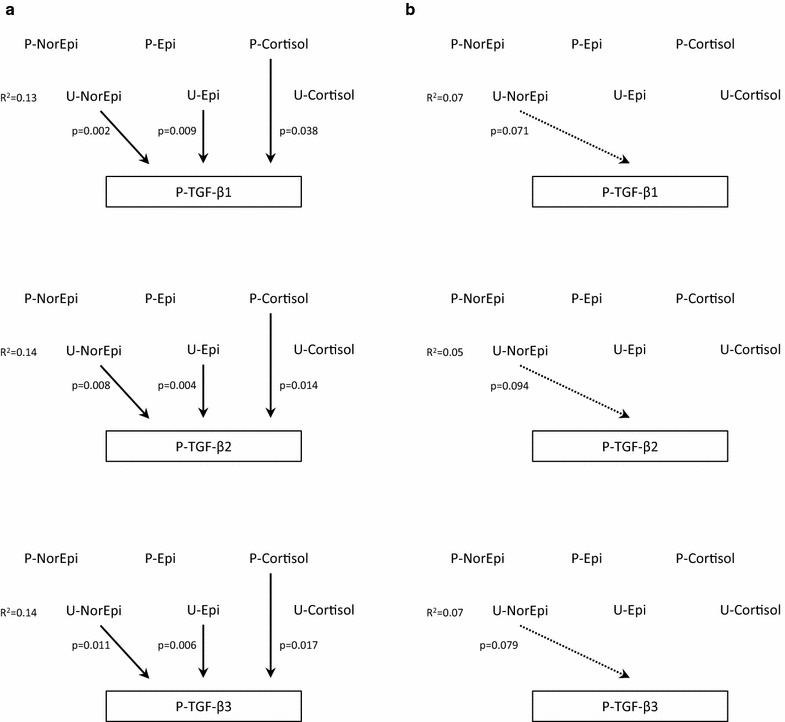
Multiple linear regression models exploring associations between neuroendocrine markers and isoforms of TGF-β in CFS patients (a) and healthy controls (b). Significant associations are indicated with a continuous arrow, whereas associations close to the level of significance are indicated with a dotted arrow. P-NorEpi plasma norepinephrine, P-Epi plasma epinephrine, P-Cortisol plasma cortisol, U-NorEpi urine norepinephrine-to-creatinine ratio, U-Epi urine epinephrine:creatinine ratio, U-Cortisol urine cortisol:creatinine ratio, P-TGF plasma transforming growth factor
Table 3.
Multiple linear regression models for neuroendocrine markers and plasma TGF-β in CFS patients with high fatigue score (≥ 20), CFS patients with low fatigue score (< 20), and healthy controls
| CFS patients, high fatigue score (n = 62)b | CFS patients, low fatigue score (n = 54)b | Healthy controls (n = 68) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Model 1 | |||||||||
| Dependent variable | |||||||||
| Plasma TGF-β1a | B (CI) | p value | R2 | B (CI) | p value | R2 | B (CI) | p value | R2 |
| Independent variables | |||||||||
| Plasma cortisol | 1.9 × 10−3 (0.18 to 3.5 × 10−3) | 0.031 | 0.17 | 0.26 × 10−3 (− 1.6 to 2.1 x 10−3) | 0.780 | 0.12 | − 0.19 × 10−3 (− 1.9 to 1.5 × 10−3) | 0.825 | 0.07 |
| Urine norepinephrine/creatinine ratioa | 0.99 (0.20 to 1.8) | 0.015 | 0.60 (0.01 to 1.2) | 0.048 | 0.62 (− 0.06 to 1.3) | 0.071 | |||
| Urine epinephrine/creatinine ratioa | − 0.65 (− 0.65 to − 0.06) | 0.031 | − 0.39 (− 0.77 to − 0.02) | 0.042 | 0.004 (− 0.47 to 0.48) | 0.987 | |||
| Model 2 | |||||||||
| Dependent variable | |||||||||
| Plasma TGF-β2 | B (CI) | p value | R2 | B (CI) | p value | R2 | B (CI) | p value | R2 |
| Independent variables | |||||||||
| Plasma cortisol | 0.64 (0.22 to 1.1) | 0.003 | 0.22 | 0.07 (− 0.49 to 0.62) | 0.814 | 0.12 | − 0.01 (− 0.49 to 0.46) | 0.950 | 0.05 |
| Urine norepinephrine/creatinine ratioa | 248 (50 to 447) | 0.015 | 133 (− 44 to 311) | 0.136 | 160 (− 28 to 349) | 0.094 | |||
| Urine epinephrine/creatinine ratioa | − 161 (− 308 to − 14) | 0.032 | − 132 (− 246 to − 18) | 0.024 | − 16 (− 148 to 115) | 0.806 | |||
| Model 3 | |||||||||
| Dependent variable | |||||||||
| Plasma TGF-β3a | B (CI) | p value | R2 | B (CI) | p value | R2 | B (CI) | p value | R2 |
| Independent variables | |||||||||
| Plasma cortisol | 2.0 × 10−3 (0.60 to 3.4 × 10−3) | 0.006 | 0.20 | 0.17 × 10−3 (− 1.6 to 1.9 × 10−3) | 0.851 | 0.11 | − 0.4 × 10−3 (− 1.8 to 0.99 × 10−3) | 0.565 | 0.07 |
| Urine norepinephrine/creatinine ratioa | 0.78 (0.12 to 1.4) | 0.022 | 0.40 (− 0.17 to 1.0) | 0.164 | 0.50 (− 0.06 to 1.1) | 0.076 | |||
| Urine epinephrine/creatinine ratioa | − 0.52 (− 1.0 to − 0.03) | 0.038 | − 0.41 (− 0.77 to − 0.05) | 0.027 | − 0.01 (− 0.40 to 0.38) | 0.964 | |||
P values in italics indicate statistical significance
B regression coefficient (unstandardized), CI confidence interval, R 2 explained variance of the dependent variable
aThe ln-transformed variable was used (better approximation to a normal distribution)
bFatigue score was missing from 4 CFS patients
Immune gene transcripts
Only a few differentially expressed immune gene transcripts were associated with TGF-β (Additional file 4: Table S3). However, there was a negative association, most evident for the TGF-β3 isoform, to the B cell annotated genes TNFRSF13C and CXCR5, which are both downregulated in CFS. Multiple linear regression analyses were consistent with a model in which TGF-β3 partly mediated the effect of plasma cortisol on the expression of these genes (Fig. 2).
Fig. 2.
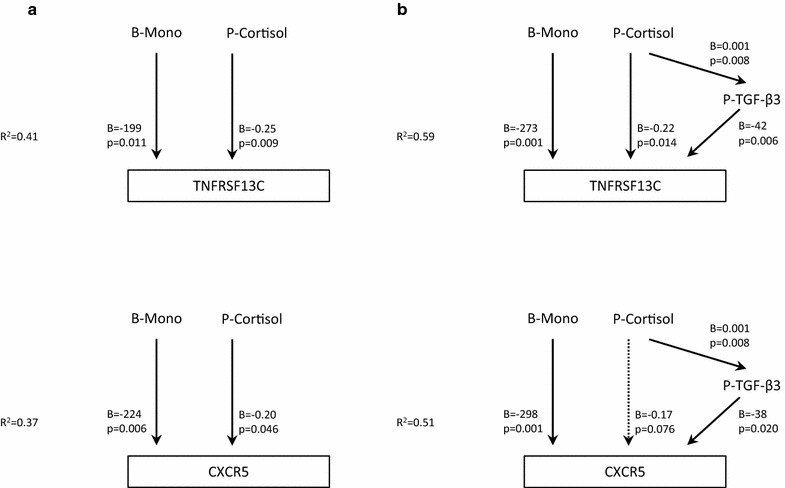
Multiple linear regression models in a subgroup of CFS patients (n = 29). a Previous established associations between blood monocyte count and plasma cortisol, and the expression in whole blood of the genes TNFRSF13C (tumor necrosis factor receptor superfamily member 13C) and CXCR5 (C-X-C motif chemokine receptor 5) (Nguyen et al. [13]). b The associations after introducing TGF-β3 as a mediating variable in the regression model. Significant associations are indicated with a continuous arrow, associations close to the level of significance are indicated with a dotted arrow. B-Mono blood monocyte count, P-Cortisol plasma cortisol, P-TGF plasma transforming growth factor
Discussion
There were three main finding of this study: (1) plasma levels of the three TGF-β isoforms did not differ in adolescent CFS patients as compared to healthy controls. (2) Within the CFS group, plasma levels of all TGF-β isoforms were associated with plasma levels of cortisol and urine levels of catecholamines. In addition, TGF-β3 might partly mediate the association between plasma cortisol and downregulation of some B cell annotated genes. (3) Plasma level of TGF-β was not directly related to CFS symptoms; however, the association of neuroendocrine mediators with TGF-β were related to fatigue score.
The lack of differences between CFS patients and healthy controls corroborate previous findings of normal plasma cytokine levels in CFS [17, 18]. Of note, subgrouping according to the Fukuda criteria or the Canada 2003-criteria did not reveal differential results. This is also in line with previous observations [17, 45]. While immunological alterations have been reported in many CFS studies, there seems to be no consistent pattern of plasma cytokine disturbances, calling for more specific investigations of immune mechanisms in different bodily compartments.
Previous studies have demonstrated a complex and possible bi-directional relationship between TGF-β levels and HPA axis activity [32, 46]. Similarly, TGF-β secretion has been associated with sympathetic nervous activity [47, 48] as well as plasma catecholamines [49]. Interestingly, in the present study, linear associations between TGF-β levels and neuroendocrine markers were found among CFS patients only. Moreover, within the CFS group, this picture was remarkably consistent across all TGF-β isoforms (Fig. 1), and related to the clinical symptom of fatigue (Table 3). Taken together, the results of the present study seem to suggest that underlying disease mechanisms of CFS are more related to altered immunological control than to altered levels of immune markers per se. Similar observations have been reported in a study of neuroendocrine regulatory systems [29], and are also in line with findings of a relationship between HPA axis dynamics and clinical symptoms [50, 51].
On a more general level, the findings of the present study seem to comply with the “sustained arousal” model of CFS [30]. In this model, a maladaptive stress response is considered a central pathophysiological element, eliciting autonomic and neuroendocrine alterations which in turn are responsible for subtle immunological alterations. Thus, an association between neuroendocrine markers and immune markers is predicted from this model, in line with the reported findings. It should be noted, though, that the cross-sectional design of the present study does not allow inference of causality. Further research should aim at uncover underlying mechanisms; one promising field of study might be epigenetic alterations of the glucocorticoid receptor gene [52].
The findings of a possible relationship between TGF-β3 and B cell annotated gene transcripts should be interpreted with great caution due to the limited number of CFS patients undergoing RNA-seq (n = 29). However, the findings were rather specific for the TGF-β3 isoform, reducing the risk of a type 1-error. Besides, it is not biologically implausible that TGF-β3 might act as a mediator of a cortisol effect on B cell gene expression. This observation should be explored further in mechanistic studies of immune cell function, in particular since the Epstein–Barr virus (EBV) is known to be a common precipitating factor in CFS [1]. Infection of EBV in human is persistent and tightly associated with B cell differentiation pathways. EBV establishes its long-term latency in memory B cells where its ability of switching from latent to lytic form of infection is related to the differentiation state of the cell [53]. Interestingly, it has been shown that TGF-β can induce EBV reactivation in vitro [54], and that expression of some of EBV promoters is activated in B cells upon TGF-β binding to TGF receptors [55].
In general, however, our data indicate that all three isoforms of TFG-β behave similar and that the previous data published with TFG-β, using isoform 1, is transferable to the other two isoform. To the best of our knowledge, this is the first study comparing the three TGF-isotypes. Of even more importance is that the only cytokine from the meta-analysis of 38 articles and 77 cytokines [18] shown to be a possible mediator discriminating CFS patients from controls, namely the TGF-β, fail to be shown in the present study, leaving no systemic cytokines left for this purpose.
Limitations of the present study encompass the wide inclusion criteria which might have obscured results pertaining to a subgroup. However, adjusting for adherence to the Fukuda and Canada 2003 case definitions did not alter the cross-sectional comparison. Also, the fact that our inclusion criteria are not identical to common international standards might reduce generalizability. As this study only included adolescents, we do not know to what extent the results apply to adult CFS patients. The number of patients that underwent whole blood gene expression by RNA-seq was rather low, increasing the risk of type II-errors. Although blood samples for cortisol analyses were obtained at a standardized time point, we cannot rule out that sleep problems and disturbances of the diurnal rhythm in the CFS group might have confounded the results. The composite scores reflecting inflammatory symptoms and symptoms of postexertional malaise has not been formally validated, and it is possible that a relationship between TGF-β levels and symptoms might have been discovered using inventories with higher sensitivity. Furthermore, we have not assessed responses to exercise and other stimuli. Normal levels at rest do not rule out abnormal response patterns. Finally, the nature of a cross-sectional study does not allow any causal inferences to be made.
Conclusions
The main finding of this study is that plasma levels of the three TGF-β isoforms were equal in a large group of adolescent CFS patients compared to healthy controls. In addition, within the CFS group, plasma level of all TGF-β isoforms were associated with plasma levels of cortisol and urine levels of catecholamines, and these associations were related to fatigue score. Finally TGF-β3 might partly mediate an association between plasma cortisol and downregulation of some B cell annotated genes. These latter observations should be addressed in further studies aiming at unraveling the complex causal interplays between neuroendocrine signaling, subtle immune alterations and clinical symptoms in CFS.
Additional files
Additional file 1. Methods for post hoc-subgrouping of patients in the NorCAPITAL project according to the Canada 2003 and Fukuda case definitions.
Additional file 2: Table S1. Plasma levels of TGF-β1, TGF-β2, TGF-β3 in individual CFS patients and healthy controls (raw data).
Additional file 3: Table S2. Correlations (Pearson’s r) between the three isoforms of TGF-β and immune markers, neuroendocrine markers and clinical markers in CFS patients and healthy controls.
Additional file 4: Table S3. Correlations (Pearson’s r) between the three isoforms of TGF-β and immune annotated single gene transcriptional counts in a subgroup of CFS patients (n = 29).
Authors’ contributions
Conceived and designed the study: VBW, TEM. Collected and analyzed the data: CBN, JAL. Interpreted the results and wrote the paper: VBW, CBN, JAL, TEM. All authors read and approved the final manuscript.
Acknowledgements
We thank Kari Gjersum for secretary assistance; Annette Winger, Dag Sulheim and Even Fagermoen for practical assistance.
Competing interests
The authors declare that that they have no competing interests.
Availability of data and materials
The dataset supporting the conclusions of this article is included within the article and its additional files.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The study was approved by the Norwegian National Committee for Ethics in Medical Research and the Norwegian Medicines Agency. Written informed consent was obtained from all participants and from parents/next-of-kin if required.
Funding
This study was funded by: The University of Oslo, The Research Council of Norway, The Southern and Eastern Norway Regional Health Authority, The Northern Norway Regional Health Authority. The funding has covered payroll and operating expenses for the involved researchers. No one of the funding sources has otherwise been involved in the study. The study has not received funding from the pharmaceutical industry or other commercial sources.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- CFS
chronic fatigue syndrome
- TGF
transforming growth factor
- NorCAPITAL
The Norwegian Study of Chronic Fatigue Syndrome in Adolescents: Pathophysiology and Intervention Trial
- HPA axis
hypothalamus–pituitary–adrenal axis
- IL
interleukin
- TFN
tumor necrosis factor
- CRP
C-reactive protein
- RNA-Seq
RNA sequencing
- DEG
differentially expressed genes
- HPLC
high-performance liquid chromatography
- HRV
heart rate variability
- LF
low frequency
- HF
high frequency
- CFQ
Chalder Fatigue Questionnaire
- MFQ
Moods and Feelings Questionnaire
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12967-017-1350-1) contains supplementary material, which is available to authorized users.
Contributor Information
Vegard Bruun Wyller, Email: brwylle@online.no.
Chinh Bkrong Nguyen, Email: c.b.t.t.nguyen@medisin.uio.no.
Judith Anita Ludviksen, Email: Judith.anita.ludviksen@nordlandssykehuset.no.
Tom Eirik Mollnes, Email: t.e.mollnes@medisin.uio.no.
References
- 1.Institute of Medicine . Beyond myalgic encephalomyelitis/chronic fatigue syndrome: redefining an illness. Washington, DC: The National Academies Press; 2015. [PubMed] [Google Scholar]
- 2.Royal College of Paediatrics and Child Health . Evidence based guidelines for the management of CFS/ME (chronic fatigue syndrome/myalgic encephalopathy) in children and young adults. London: Royal College of Paediatrics and Child Health; 2004. [Google Scholar]
- 3.Crawley EM, Emond AM, Sterne JA. Unidentified Chronic Fatigue Syndrome/myalgic encephalomyelitis (CFS/ME) is a major cause of school absence: surveillance outcomes from school-based clinics. BMJ Open. 2011;1:e000252. doi: 10.1136/bmjopen-2011-000252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nijhof SL, Rutten JM, Uiterwaal CS, Bleijenberg G, Kimpen JL, Putte EM. The role of hypocortisolism in chronic fatigue syndrome. Psychoneuroendocrinology. 2014;42:199–206. doi: 10.1016/j.psyneuen.2014.01.017. [DOI] [PubMed] [Google Scholar]
- 5.Kennedy G, Underwood C, Belch JJ. Physical and functional impact of chronic fatigue syndrome/myalgic encephalomyelitis in childhood. Pediatrics. 2010;125:e1324–e1330. doi: 10.1542/peds.2009-2644. [DOI] [PubMed] [Google Scholar]
- 6.Missen A, Hollingwort W, Eaton N, Crawley E. The financial and psychological impacts on mothers of children with chronic fatigue syndrome (CFS/ME) Child Care Health Dev. 2012;38:505–512. doi: 10.1111/j.1365-2214.2011.01298.x. [DOI] [PubMed] [Google Scholar]
- 7.Bansal AS, Bradley AS, Bishop KN, Kiani-Alikhan S, Ford B. Chronic fatigue syndrome, the immune system and viral infection. Brain Behav Immun. 2012;26:24–31. doi: 10.1016/j.bbi.2011.06.016. [DOI] [PubMed] [Google Scholar]
- 8.Klimas NG, Broderick G, Fletcher MA. Biomarkers for chronic fatigue. Brain Behav Immun. 2012;26:1202–1210. doi: 10.1016/j.bbi.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raison CL, Lin JM, Reeves WC. Association of peripheral inflammatory markers with chronic fatigue in a population-based sample. Brain Behav Immun. 2009;23:327. doi: 10.1016/j.bbi.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 10.Bradley AS, Ford B, Bansal AS. Altered functional B cell subset populations in patients with chronic fatigue syndrome compared to healthy controls. Clin Exp Immunol. 2013;172:73. doi: 10.1111/cei.12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brenu EW, Huth TK, Hardcastle SL, Fuller K, Kaur M, Johnston S, Ramos SB, Staines DR, Marshall-Gradisnik SM. Role of adaptive and innate immune cells in chronic fatigue syndrome/myalgic encephalomyelitis. Int Immunol. 2014;26:233–242. doi: 10.1093/intimm/dxt068. [DOI] [PubMed] [Google Scholar]
- 12.Mensah F, Bansal A, Berkovitz S, Sharma A, Reddy V, Leandro MJ, Cambridge G. Extended B cell phenotype in patients with myalgic encephalomyelitis/chronic fatigue syndrome: a cross-sectional study. Clin Exp Immunol. 2016;184:237–247. doi: 10.1111/cei.12749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nguyen CB, Alsøe L, Lindvall JM, Sulheim D, Fagermoen E, Winger A, Kaarbø M, Nilsen H, Wyller VB. Whole blood gene expression in adolescent chronic fatigue syndrome: an exploratory cross-sectional study suggesting altered B cell differentiation and survival. J Transl Med. 2017;15:102. doi: 10.1186/s12967-017-1201-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maes M, Twisk FN, Ringel K. Inflammatory and cell-mediated immune biomarkers in myalgic encephalomyelitis/chronic fatigue syndrome and depression: inflammatory markers are higher in myalgic encephalomyelitis/chronic fatigue syndrome than in depression. Psychother Psychosom. 2012;81:286–295. doi: 10.1159/000336803. [DOI] [PubMed] [Google Scholar]
- 15.Fletcher MA, Zeng XR, Barnes Z, Levis S, Klimas NG. Plasmas cytokines in women with chronic fatigue syndrome. J Transl Med. 2009;7:96. doi: 10.1186/1479-5876-7-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vollmer-Conna U, Cameron B, Pavlonic DH, Singletary K, Davenport T, Vernon S, Reeves WC, Hickie I, Wakenfield D, Lloyd AR. Postinfective fatigue syndrome is not associated with altered cytokine production. Clin Infect Dis. 2007;45:732–735. doi: 10.1086/520990. [DOI] [PubMed] [Google Scholar]
- 17.Wyller VB, Vitelli V, Sulheim D, Fagermoen E, Ueland T, Mollnes TE. Plasma cytokine expression in adolescent chronic fatigue syndrome. Brain Behav Immun. 2015;46:80–86. doi: 10.1016/j.bbi.2014.12.025. [DOI] [PubMed] [Google Scholar]
- 18.Blundell S, Ray KK, Buckland M, White PD. Chronic fatigue syndrome and circulating cytokines: a systematic review. Brain Behav Immun. 2015;50:186–195. doi: 10.1016/j.bbi.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 19.Kelly A, Houston SA, Sherwood E, Casulli J, Travis MA. Regulation of innate and adaptive immunity by TGFβ. Adv Immunol. 2017;134:137–233. doi: 10.1016/bs.ai.2017.01.001. [DOI] [PubMed] [Google Scholar]
- 20.Sanjabi S, Oh SA, Li MO. Regulation of the immune response by TGF-β: From conception to autoimmunity and infection. Cold Spring Harb Perspect Biol. 2017;9:a022236. doi: 10.1101/cshperspect.a022236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brenu EW, van Driel ML, Staines DR, Ashton KJ, Ramos SB, Keane J, Klimas NG, Marshall-Gradisnik SM. Immunological abnormalities as potential biomarkers in Chronic fatigue syndrome/myalgic encephalomyelitis. J Transl Med. 2011;9:81. doi: 10.1186/1479-5876-9-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Curriu M, Carillo J, Massanella M, Riqau J, Aleqe J, Puig J, Garcia-Quintana AM, Castro-Marrerro J, Negredo E, Clotet B, Cabrera C, Blanco J. Screening NK-, B- and T-cell phenotype and function in patients suffering from Chronic fatigue syndrome. J Transl Med. 2013;11:68. doi: 10.1186/1479-5876-11-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martínez-Martínez LA, Mora T, Varqas A, Fuentes-Iniestra M, Martínez-Lavin M. Sympathetic nervous system dysfunction in fibromialgia, chronic fatigue syndrome, irritable bowel syndrome, and interstitial cystitis: a review of case–control studies. J Clin Rhematol. 2014;20:146–150. doi: 10.1097/RHU.0000000000000089. [DOI] [PubMed] [Google Scholar]
- 24.Wyller VB, Barbieri R, Saul P. Blood pressure variability and closed-loop baroreflex assessment in adolescent chronic fatigue syndrome during supine rest and orthostatic stress. Eur J Appl Physiol. 2011;111:497–502. doi: 10.1007/s00421-010-1670-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wyller VB, Barbieri R, Thaulow E, Saul JP. Enhanced vagal withdrawal during mild orthostatic stress in adolescents with chronic fatigue. Ann Noninvasive Electrocardiol. 2008;13:67–73. doi: 10.1111/j.1542-474X.2007.00202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wyller VB, Due R, Saul JP, Amlie JP, Thaulow E. Usefulness of an abnormal cardiovascular response during low-grade head-up tilt-test for discriminating adolescents with chronic fatigue from healthy controls. Am J Cardiol. 2007;99:997–1001. doi: 10.1016/j.amjcard.2006.10.067. [DOI] [PubMed] [Google Scholar]
- 27.Papadopoulos AS, Cleare AJ. Hypothalamic–pituitary–adrenal axis dysfunction in chronic fatigue syndrome. Nat Rev Endocrinol. 2011;27:22–32. doi: 10.1038/nrendo.2011.153. [DOI] [PubMed] [Google Scholar]
- 28.Segal TY, Hindmarsh PC, Viner RM. Disturbed adrenal function in adolescents with chronic fatigue syndrome. J Pediatr Endocrinol Metab. 2005;18:295–301. doi: 10.1515/JPEM.2005.18.3.295. [DOI] [PubMed] [Google Scholar]
- 29.Wyller VB, Vitelli V, Sulheim D, Fagermoen E, Winger A, Godang K, Bollerslev J. Alterered neuroendocrine control and association to clinical symptoms in adolescent chronic fatigue syndrome: a cross-sectional study. J Transl Med. 2016;14:121. doi: 10.1186/s12967-016-0873-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wyller VB, Malterud K, Eriksen HR. Can sustained arousal explain chronic fatigue syndrome? Behav Brain Funct. 2009;5:10. doi: 10.1186/1744-9081-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yanagawa Y, Hiraide S, Lizuka K. Isoform-specific regulation of transforming growth factor-β mRNA expression in macrophages in response to adrenoceptor stimulation. Microbiol Immunol. 2016;60:56–63. doi: 10.1111/1348-0421.12344. [DOI] [PubMed] [Google Scholar]
- 32.Smith EL, Batuman OA, Trost RC, Coplan JD, Rosenblum LA. Transforming growth factor-beta 1 and cortisol in differentially reared primates. Brain Behav Immun. 2002;16:140–149. doi: 10.1006/brbi.2001.0629. [DOI] [PubMed] [Google Scholar]
- 33.Carruthers BM, Jain AK, De Meirleir KL, Peterson DL, Klimas NG, Lerner AM, et al. Myalgic encephalomyelitis/chronic fatigue syndrome: clinical working case definition, diagnostic and treatment protocols. J Chron Fatigue Syndr. 2003;11:7–11. doi: 10.1300/J092v11n01_02. [DOI] [Google Scholar]
- 34.Sulheim D, Fargermoen E, Winger A, Andersen AM, Godang K, Müller F, Rowe PC, Saul JP, Skovlund E, Øie MG, Wyller VB. Disease mechanisms and clonidine treatment in adolescent chronic fatigue syndrome: a combined cross-sectional and randomized controlled trial. JAMA Pediatr. 2014;168:351–360. doi: 10.1001/jamapediatrics.2013.4647. [DOI] [PubMed] [Google Scholar]
- 35.Fukuda K, Straus SE, Hickie I, Sharpe MC, Dobbins JG, Komaroff A. The chronic fatigue syndrome: a comprehensive approach to its definition and study. Ann Int Med. 1994;121:953–959. doi: 10.7326/0003-4819-121-12-199412150-00009. [DOI] [PubMed] [Google Scholar]
- 36.Fagermoen E, Sulheim D, Winger A, Andersen AM, Gjerstad J, Godang K, Rowe PC, Saul JP, Skovlund E, Wyller VB. Effects of low-dose clonidine on cardiovascular and autonomic variables in adolescents with chronic fatigue syndrome: a randomized controlled trial. BMC Pediatr. 2015;15:117. doi: 10.1186/s12887-015-0428-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liao Y, Smyth GK, Shi W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013;41:e108. doi: 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anders S, McCarthy DJ, Chen Y, Okoniewsky M, Smyth GK, Huber W, Robinson MD. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat Protoc. 2013;8:1765–1786. doi: 10.1038/nprot.2013.099. [DOI] [PubMed] [Google Scholar]
- 40.HumanMine Database, University of Cambridge, UK. https://www.humanmine.org. Accessed 23 Feb 2017.
- 41.Chalder T, Berelowitz G, Pawlikowska T, et al. Development of a fatigue scale. J Psychosom Res. 1993;37:147–153. doi: 10.1016/0022-3999(93)90081-P. [DOI] [PubMed] [Google Scholar]
- 42.Daviss WB, Birmaher B, Melhem NA, Axelson DA, Michaels SM, Brent DA. Criterion validity of the mood and feelings questionnaire for depressive episodes in clinic and non-clinic subjects. J Child Psychol Psychiatry. 2006;47:927–934. doi: 10.1111/j.1469-7610.2006.01646.x. [DOI] [PubMed] [Google Scholar]
- 43.Kendall PC, Finch A, Jr, Auerback SM, Hooke JF, Mikulka PJ. The state-trait anxiety inventory: a systematic evaluation. J Consult Clin Psychol. 1976;44:406–412. doi: 10.1037/0022-006X.44.3.406. [DOI] [PubMed] [Google Scholar]
- 44.Grant PM, Ryan CG, Tigbe WW, Granat MH. The validation of a novel activity monitor in the measurement of posture and motion during everyday activities. Br J Sports Med. 2006;40:992–997. doi: 10.1136/bjsm.2006.030262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Asprusten TT, Fagermoen E, Sulheim D, Skovlund E, Sørensen Ø, Mollnes TE, Wyller VB. Study finding challenge the content validity of the Canadian Consensus Criteria for adolescent chronic fatigue syndrome. Acta Paediatr. 2015;104:498–503. doi: 10.1111/apa.12950. [DOI] [PubMed] [Google Scholar]
- 46.Coplan JD, Gopinath S, Abdallah CG, Marqolis J, Chen W, Scharf BA, Rosenblum LA, Batuman OA, Smith EL. Effects of acute confinement stress-induced hypothalamic-pituitary-adrenal axis activation and concomitant peripheral and central transforming growth factor-β1 measures in nonhuman primates. Chronic stress (Thousand Oaks). 2017 (Epub ahead of print). [DOI] [PMC free article] [PubMed]
- 47.Ebbinghaus M, Gajda M, Boettger MK, Schaible HG, Bräuer R. The anti-inflammatory effects of sympathectomy in murine antigen-induced arthritis are associated with a reduction of Th1 and Th17 responses. Ann Rheum Dis. 2012;71:253–261. doi: 10.1136/ard.2011.150318. [DOI] [PubMed] [Google Scholar]
- 48.Hoch H, Hering L, Crowley S, Zhang J, Yang G, Rump LC, Vonend O, Steqbauer J. 9A.05: sympathetic nervous system drives renal inflammation by alpha(2a)-adrenoceptors. J Hypertens. 2015;33(Suppl 1):e188. [Google Scholar]
- 49.Rassler B, Marx G, Schierle K, Zimmer HG. Catecholamines can induce pulmonary remodeling in rats. Cell Physiol Biochem. 2012;30:1134–1147. doi: 10.1159/000343304. [DOI] [PubMed] [Google Scholar]
- 50.Hall DL, Lattei EG, Antoni MH, Fletcher MA, Czaja S, Perdomo D, Klimas NG. Stress management skills, cortisol awakening response, and post-exertional malaise in chronic fatigue syndrome. Psychoneuroendocrinology. 2014;49:26–31. doi: 10.1016/j.psyneuen.2014.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nijhof SL, Maijer K, Bleijenberg G, Uiterwaal CS, Kimpen JL, van der Putte EM. Adolscent chronic fatigue syndrome: prevalence, incidence, and morbidity. Pediatrics. 2011;127:e1169–e1175. doi: 10.1542/peds.2010-1147. [DOI] [PubMed] [Google Scholar]
- 52.Vangeel E, Van den Eede F, Hompes T, Izzi B, Del Favero J, Moorkens G, Lambrechts D, Freson K, Claes S. Chronic fatigue syndrome and DNA hypomethylation of the glucocorticoid receptor gene promotor 1F region: association with HPA axis hypofunction and childhood trauma. Psychosom Med. 2015;77:853–862. doi: 10.1097/PSY.0000000000000224. [DOI] [PubMed] [Google Scholar]
- 53.Thorley-Lawson DA. EBV Persistence—introducing the virus. Curr Top Microbiol Immunol. 2015;390:151–209. doi: 10.1007/978-3-319-22822-8_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kenney SC, Mertz J. Regulation of the latent-lytic switch in Epstein–Barr virus. Semin Cancer Biol. 2014;26:60–68. doi: 10.1016/j.semcancer.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Iempridee T, Das S, Xu I, Mertz JE. Transforming growth factor beta-induced reactivation of Epstein–Barr virus involves multiple Smad-binding elements cooperatively activating expression of the latent-lytic switch BZLF1 gene. J Virol. 2011;85:7836–7848. doi: 10.1128/JVI.01197-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Methods for post hoc-subgrouping of patients in the NorCAPITAL project according to the Canada 2003 and Fukuda case definitions.
Additional file 2: Table S1. Plasma levels of TGF-β1, TGF-β2, TGF-β3 in individual CFS patients and healthy controls (raw data).
Additional file 3: Table S2. Correlations (Pearson’s r) between the three isoforms of TGF-β and immune markers, neuroendocrine markers and clinical markers in CFS patients and healthy controls.
Additional file 4: Table S3. Correlations (Pearson’s r) between the three isoforms of TGF-β and immune annotated single gene transcriptional counts in a subgroup of CFS patients (n = 29).
Data Availability Statement
The dataset supporting the conclusions of this article is included within the article and its additional files.