

Abstract
Influenza viruses are negative strand RNA viruses that replicate in the nucleus of the cell. The viral nucleoprotein (NP) is the major component of the viral ribonucleoprotein. In this paper we show that the NP of influenza B has a long N-terminal tail of 70 residues with intrinsic flexibility. This tail contains the Nuclear Location Signal (NLS). The nuclear trafficking of the viral components mobilizes cellular import factors at different stages, making these host-pathogen interactions promising targets for new therapeutics. NP is imported into the nucleus by the importin-α/β pathway, through a direct interaction with importin-α isoforms. Here we provide a combined nuclear magnetic resonance and small-angle X-ray scattering (NMR/SAXS) analysis to describe the dynamics of the interaction between influenza B NP and the human importin-α. The NP of influenza B does not have a single NLS nor a bipartite NLS but our results suggest that the tail harbors several adjacent NLS sequences, located between residues 30 and 71.
Introduction
Both transcription and replication of influenza virus take place in the nucleus of the infected cell. To ensure these two mechanisms, a number of viral proteins have to be transported from the cytoplasm to the nucleus1. These are in particular the three subunits of the RNA-dependent RNA-polymerase of the virus and the nucleoprotein (NP), which is the major protein of the ribonucleoproteins (RNPs) that binds the viral RNA2 and has been shown to be essential for viral proliferation.
Transport of large proteins (>40 kDa) into the nucleus is generally mediated by the presence of a nuclear localization signal (NLS). NLSs are often made by short motifs with basic amino acids3–6, which mediate the interaction with proteins of the karyopherin family, in particular different importin-α variants7,8. By specifically recognizing NLSs of cargo proteins, importin-alphas act as an adaptor protein for the nuclear transport, through a complex with an importin-β receptor9. Once inside the nucleus, the heterotrimeric importin-β:importin-α:cargo complex interacts with Ran:GTP, resulting in dissociation of the cargo from its carrier concomitant to the hydrolysis of GTP8,10,11.
Two putative NLSs have been described in influenza A NP (A/NP): NLS1 is a non-classical (ncNLS) motif in the first 14 amino acids of the unfolded N-terminal region, while NLS2 consists of residues 198–216 located at the surface in the middle of the protein12–18. Recently, two crystal structures of the NLSs of influenza virus NP bound to importin-α have been published: the structure of NLS1 (residues 3–14) bound to the minor NLS-binding pocket of importin-α and the structure of NLS2 (residues 213–216) interacting with the major pocket of importin-α19,20. Both NLSs bind to importin-α with high dissociation constants (Kds between 2 and 5 µM for NLS1 and 70 µM for NLS2). Wu and coworkers suggest that importin-α can bind to both sites, either on the same monomer of NP or on two different protomers inside the NP-trimer, so that the synergy of the two sites is strong enough for the transport of NP into the nucleus20. Phosphorylation sites have been identified in the NLS1 of A/NP (S/T3, S9 and Y10) suggesting that phosphorylation at these sites prevent the interaction of NLS1 with importin-α21,22.
Except in their respective oligomeric state, the overall structures of A/NP and B/NP are highly similar15,23,24. The main difference between the two proteins lies in their N-terminal extremities: A/NP has an N-terminal tail of about 20 residues with NLS1 whereas B/NP has a 70-residue N-terminal tail (Fig. 1). Both were present in the protein used for the crystallogenesis but were not observed in the X-ray structures. We used several algorithms25 to predict the probability of B/NPTAIL to contain secondary structures. Figure 1 gives the D-score (disordered score) of B/NP showing that the first 70 residues are most likely disordered without any stable secondary structures. However, the location of the NLS in B/NP cannot be clearly identified. Stevens and Barclay have shown that deletions of the N-terminus up to residue 69 do not impair the nuclear accumulation of B/NP26 whereas others suggested that residues 44-KRTR-47 is the putative NLS27–29.
Figure 1.

Computational analysis of B/NP. The sequences of the N-termini of B/NP (strain B/Memphis/13/03) and A/NP (strain A/WSN/1933) have been aligned using Clustal W71. On the sequence alignment, the putative NLSs motifs of the two proteins are highlighted in yellow. The black triangles show the phosphorylation sites of each protein72. D-score is an algorithm to find structured and disordered regions in proteins25. The prediction is based on 22 predictor web servers. The value 1.0 means that the protein is fully ordered and 0.0 means the peptide is fully disordered.
With this paper we present biochemical and biophysical data that demonstrate that the 70 N-terminal amino acids of B/NP are intrinsically disordered. We show by size exclusion chromatography, NMR and SAXS, that this tail binds to human importin-α7, with a K d value for this interaction estimated by isothermal titration calorimetry. The precise interaction site of the NP tail (NPTAIL) in complex with importin-α was mapped using NMR spectroscopy, revealing the extent of the interacting region.
Results
The N-terminus of the B/NP is disordered
We aimed to characterise the structural propensities of the first 70 N-terminal residues (B/NPTAIL) using NMR spectroscopy and we therefore obtained the complete backbone resonance assignment of B/NPTAIL. We calculated secondary structure propensities (SSPs) based on experimental Cα and Cβ chemical shifts, which indicate that no strong propensities for either α-helical or extended structure exist (Fig. 2a). We then developed a multi-conformational model of B/NPTAIL using Flexible-Meccano30 and the genetic algorithm ASTEROIDS31 that allowed us to select sub-ensembles of 200 conformers on the basis of the experimental 13C, 15N and 1H chemical shifts (Fig. 2b and Supplementary Figure 1). The obtained conformational ensembles describe B/NPTAIL as a protein behaving much like a statistical coil, with the exception of a region starting around residue 59 that had a slightly increased propensity to form right handed α-helices as compared to random coil. The backbone dihedral angles describing B/NPTAIL were then used to calculate a model of full length B/NP using the crystal structure of the folded domain23 and Flexible-Meccano30 to add the intrinsically disordered tail (Fig. 2c). The experimental SAXS curve of full length B/NP was in reasonable agreement (chi2 below 1) with this conformational ensemble (Fig. 2d).
Figure 2.
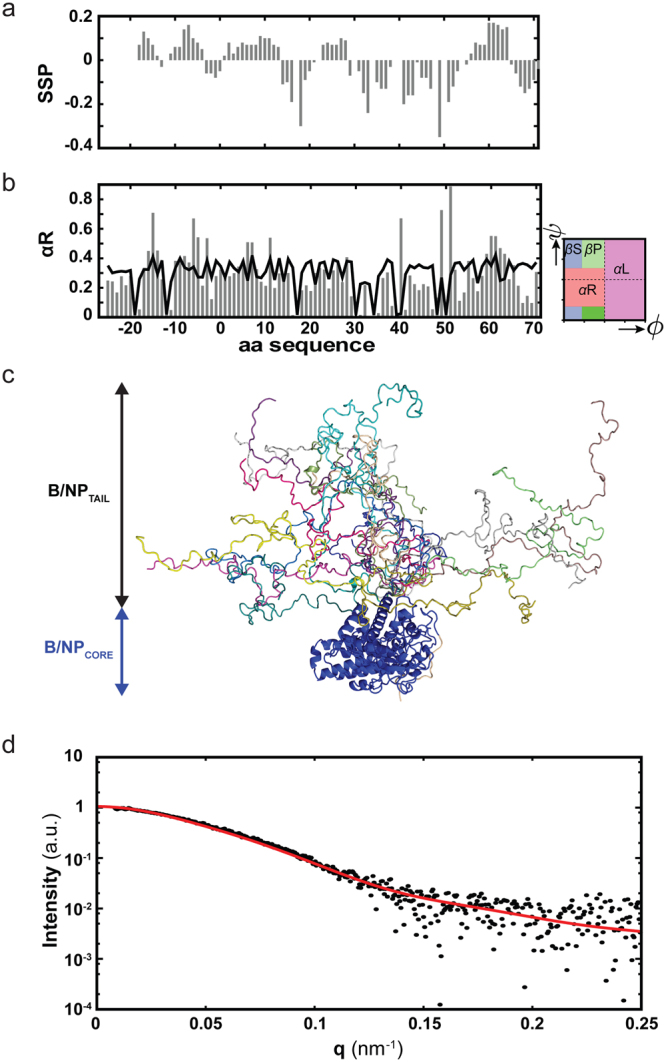
Conformational propensities of B/NPTAIL. (a) Secondary structure propensities (SSPs) calculated from Cα chemical shifts as obtained from the assignment of the N-terminally tagged protein. Residues are numbered with 1 starting at the methionine of B/NPTAIL. SSP scales from −1 to 1. SSP > 0: α-helical propensity; SSP < 0: propensity for extended structure. (b) Ramachandran plot showing regions of β-sheet (βS), poly-proline (βP), right (αR) and left handed (αL) helical conformations (right) and corresponding αR propensities (grey bars) as derived from a set of 200 conformers describing the structural propensities of B/NPTAIL in a combination of Flexible-Meccano30 and ASTEROIDS31 using chemical shifts. Supplementary Figure 1 details the data obtained for αL, αR, βP and βS. The black trace reflects the propensities of a statistical coil ensemble without selection based on experimental data. (c) Conformational ensemble (10 of 1000 conformers are shown) of full length B/NP calculated from the crystal structure of B/NPCORE (PDB 3TJ023) and Flexible-Meccano using the backbone dihedral angles as obtained for B/NPTAIL to describe the N-terminal intrinsically disordered region. The 10 different conformations of B/NPTAIL are shown in different colours with the core of B/NP in blue. (d) Averaged SAXS curves calculated from the ensemble as described in (c) (red line) overlaid with the experimental SAXS curve of B/NP full length (black dots).
Binding of B/NP and importin-α7 analysed by size-exclusion chromatography
We have previously shown that, under the same experimental conditions as used here (i.e. 20 mM Tris-HCl pH 7.5; 150 mM NaCl), B/NP is mainly monomeric32. B/NP (residues 1 to 561) and B/NPCORE (residues 71 to 561) were injected separately or in complex with importin-α7, on a size exclusion chromatography column (SuperdexTM increase 200 10/300 GL column). B/NP and importin-α7 alone are both eluted as a single peak, with a respective elution volume of 13.8 mL and 13.7 mL (Fig. 3a and Supplementary Figure 2). When the two proteins are mixed before injection, the elution profile presents a single peak at 12 mL and the SDS-PAGE confirms the presence of the two proteins in the peak. For B/NPCORE and importin-α7 (Fig. 3b and Supplementary Figure 2), both samples are eluted as a single peak, respectively at 15 mL and 13.7 mL. By a SEC-MALLS-RI experiment, we confirmed the molecular mass of B/NPCORE (Supplementary Figure 3). When B/NPCORE and importin-α7 are mixed before injection, we observed an elution of the two proteins in two separate peaks and no shift in the elution volume is observed. We can conclude that without the N-terminal tail on NP, the two proteins do not interact under the experimental conditions. For B/NPTAIL and importin-α7 (Fig. 3c and Supplementary Figure 2), the samples were injected on a SuperdexTM 75 10/300 GL column with an excess of B/NPTAIL. The peak eluted at 12.5 mL corresponds to the B/NPTAIL and the SDS-PAGE gel confirms the presence of the protein contained in that elution peak (band at 15 kDa). The elution peak at 10.5 mL corresponds to importin-α7 alone and the peak at 10.1 mL corresponds to the complex between B/NPTAIL and importin-α7. These results confirm that importin-α7 binds the full-length influenza B nucleoprotein and its N-terminal tail but not B/NPCORE.
Figure 3.

Interaction of B/NP with importin-α7. The binding experiments with importin-α7 were performed by size elution chromatography, respectively using a SuperdexTM 200 increase 10/300GL column for (a) B/NP and (b) B/NPCORE and a SuperdexTM 75 10/300GL column for (c) B/NPTAIL. On the left are the elution profiles of the binding tests and on the right are the corresponding coomassie blue colored SDS-PAGE gels (Tris-Glycine, 4–20% polyacrylamide) for the mixture between the two partners. The ratio used for these experiments are 1.2:1 for B/NP:importin-α7 and B/NPCORE:importin-α7 and 2:1 B/NPTAIL:importin-α7. The SDS-PAGE gels for the controls are shown in Supplementary Figure 2.
Analysis of the importin-α binding site on B/NPTAIL by NMR
In order to precisely map the residues of B/NPTAIL that are involved in the interaction with importin-α, we titrated 15N labelled B/NPTAIL with unlabelled importin-α7 and measured 1H-15N HSQC spectra at each titration step. Peak intensities were extracted and compared to those of the unbound B/NPTAIL (Fig. 4a and b). Interestingly, the interaction between B/NPTAIL and importin-α involves all residues of the disordered tail between amino acids 30 and 71 (Fig. 4c). This may be relevant to the recent observation that the cytoplasmic fraction of B/NP is directly proportional to truncations made on its N-terminal tail28.
Figure 4.

Mapping of B/NPTAIL:importin-α7 interaction by NMR. (a) 1H-15N HSQC spectra of B/NPTAIL alone (red) and in the presence of 77% (dark blue) and 211% importin-α7 (light blue) respectively. The assignment is displayed as one letter code. (b) Zoom into the 1H-15N HSQC spectra from (a), demonstrating localized chemical shift changes. (c) Intensity ratio of bound as compared to unbound B/NPTAIL. B/NPTAIL:importin-α7 ratios were 1:0.77, 1:1.41, and 1:2.11 (dark to light blue). Intensities were normalized to 1 at the maximum intensity ratio. (d) 15N transverse relaxation rates (R2) were measured in the absence (grey bars) and presence (dark blue line) of 77% importin-α at a 1H frequency of 950 MHz. All measurements were done at 25 °C.
We then analyzed B/NPTAIL for the presence of other putative NLS motifs that could explain this interaction using the eukaryotic linear motif database (ELM)33. A classical NLS is predicted, starting from residue 41. The analysis of the sequence by a prediction server that is specialized for the prediction of composite motifs, cNLS Mapper34 predicts indeed a bipartite NLS between residues 41 and 68. Interestingly the region of residues 41 to 68 also contains the region with increased helical propensity – a frequent observation in case small recognition motifs are encoded in an otherwise disordered chain. Residues 30–39, however, are additionally involved in the interaction and show characteristics of two conformational states in slow exchange on the NMR chemical shift time scale, visualized by peaks splitting in the course of the titration. While the ncNLS of influenza A has been shown to bind to the minor groove of importin-α119,20, bipartite NLS motifs are thought to bind both the major and the minor binding site35. The extended NLS region in B/NP may explain the convolved dynamic features observed in the interaction, switching between slow exchange from residues 30–39 and fast or intermediate exchange in the region of residues 41–50 (Fig. 4d).
SAXS analysis
We used in-line SEC-SAXS analysis for the characterization in solution of B/NP, importin-α7 and the complexes B/NP:importin-α7 and B/NPTAIL:importin-α7. Each dataset has been processed individually and provides coherent statistics (Table 1). By using the volume of correlation determination methods36, the molecular weights (MR) were estimated (Table 1) in accordance with the respective calculated masses from the sequences. The hydrodynamic radius (Rg) and Dmax values of each sample are listed in Table 1 and the Guinier plots are shown in Supplementary Figure 4. The Guinier plot estimates two essential SAXS parameters, the size (through the Rg) and the mass (through the extrapolated intensity at zero scattering angle (I(0))) of the analyzed sample. An absence of linearity in the Guinier plot reflects the presence of aggregates and/or attractive/repulsive interactions between the scattering particles. We have combined the processing of the dataset in order to further localize the B/NP NLS binding site on the importin-α7. The program MONSA builds ab initio shapes using the scattering curves for the complexes together with those of their individual components37,38. While the shapes obtained from a protein containing an intrinsically disordered tail will not be expected to provide realistic shapes (compare Fig. 2c with Fig. 5), we still expect this analysis to provide qualitative information on the orientation of the two proteins when bound to each other. For the B/NP:importin-α7 complex (Fig. 5a and b), the resulting shapes seem to be made by two parts: a globular green domain with an extension and a more flat entity (purple). We can postulate that the extension of the globular green domain corresponds to B/NPTAIL which we have shown to be flexible in solution, and the globular part is then B/NPCORE. In this hypothesis, the flat entity would be the importin-α7. This is a reasonable description of the ab initio shapes as the NLS sequence is located within NPTAIL that would be, in that case, the main contact between the two partners of the complex. For the shape corresponding to the importin-α7, the contact seems to occur with only one third of the shape, meaning that only a part of the protein is involved in the interaction.
Table 1.
SAXS data collection and scattering-derived parameters.
| B/NP | importin-α7 | B/NP:importin-α7 | B/NPTAIL:importin-α7 | |
|---|---|---|---|---|
| Data collection parameters | ||||
| Instrument | ESRF - BM29 | |||
| Beam size at sample (µm) | 700 × 700 | |||
| Wavelength (Å) | 0.9919 | |||
| q range (Å−1) | 0.25–50 | |||
| Detector | Pilatus 1 M | |||
| Detector distance | 2.867 | |||
| Exposure (s per image) | 1 | |||
| Column | SuperdexTM increase 200 5/150 GL | |||
| Flow rate (mL.min−1) | 0.5 | |||
| Injected sample concentration (mg.mL−1) | 10 | |||
| Injected volume (µL) | 50 | |||
| Temperature (K) | 293 | |||
| Structural parameters | ||||
| Rg (Å) [from P(r)] | 33.5 | 38.3 | 47.5 | 35.8 |
| Rg (Å) [from Guinier]* | 34.0 | 37.8 | 47.2 | 34.4 |
| Dmax (Å) | 119 | 133.5 | 163.6 | 120.3 |
| Porod volume estimate (Å3) | 109 290 | 81 420 | 246 570 | 83 120 |
| Molecular mass MR (kDa) from Rambo | 63 | 54 | 151 | 57 |
| Calculated MR (kDa) from sequence | 61.6 | 57.0 | 118.6 | 64.5 |
| Software employed | ||||
| Primary Data reduction | Primus | |||
| Data processing | Primus | |||
| Ab initio analysis | MONSA | |||
| 3D graphics representation | PyMOL | |||
*The Guinier plots are shown on Supplementary Figure 4.
Figure 5.

Modelling of the B/NP:importin-α7 interaction by SAXS. (a) Observed scattering (dots) profiles for B/NP alone, B/NP:Imp-α7 and human Imp-α7 alone (from left to right) with MONSA-generated fits (solid red lines). (b) Low-resolution structure of the B/NP:Imp-α7 complex with B/NP in green and importin-α7 in pink. (c) Observed scattering (dots) profiles for B/NPTAIL:Imp-α7 and Imp-α7 alone (from left to right) with MONSA-generated fits (solid red lines). (d) 90°-rotation view of the B/NPTAIL:Imp-α7 low-resolution structure with B/NPTAIL shown as a blue grid density and human importin-α7 in pink. The right part corresponds to the docking of the X-Ray structure of A/NPTAIL:Imp-α1(mouse) complex (PDB id: 4ZDU;19) in the MONSA envelop obtained for B/NPTAIL:Imp-α7.
In order to get a closer view for the localization of the NLS major binding site on the importin-α7, we have generated ab initio shapes with MONSA, corresponding to B/NPTAIL bound to the importin-α7 (Fig. 5c and d). An extra density corresponding to B/NPTAIL can be observed in one third of the shape of the importin-α7 (blue ball on Fig. 5d). This observation is coherent with the shapes obtained for the B/NP:importin-α7 complex. The X-ray model of the complex between the mouse importin-α1 and the NLS1 of A/NP (PDB id: 4ZDU) has been docked in our ab initio SAXS envelope (Fig. 5d on the right, red part). The superimposition shows that the NLS of A/NP fits in the additional density of our shape.
Determination of the affinity constant between B/NPTAIL and importin-α with ITC
We have determined the binding affinity of B/NPTAIL to importin-α7 using ITC (isothermal calorimetry). The experiment was done in triplicates. One of the titration curves is shown in Fig. 6 and all the data are compiled in a table on the same figure. B/NPTAIL binds human importin-α7 with a K d of 844 nM, similar to the affinity measured between A/NP and the mouse importin-α1 (K d between 2 and 5 µM)19,20. We tried to repeat the experiment with the full-length B/NP, but because of its tendency to adopt tetramers under these conditions, a meaningful interpretation of the ITC data could not be obtained.
Figure 6.

ITC determination of the binding thermodynamics of B/NPTAIL to importin-α7. Titration of 150 µM of importin-α7 into a solution of 15 mM of B/NPTAIL. The experiments were performed in 20 mM Tris-HCl pH 7.5; 150 mM NaCl at 25 °C in triplicate.
Discussion
This paper shows that the long N-terminal region of influenza B nucleoprotein is disordered. Figure 2c shows that the B/NPTAIL samples a large space. The long N-terminal tail of influenza B NP can therefore change how the B/RNPs (RNPs from influenza B virus) pack together in comparison to what has been shown for influenza A39. CryoEM of paramyxoviruses like Sendai or measles viruses40,41, which have very long NTAILS 42,43 shows that the nucleocapsids take a large volume inside the viral particle, unlike the nucleocapsids from rhabdovirus particles, because the nucleocapsids of rabies and VSV have no NTAILS 44,45. In the influenza A viral particle the RNPs form the oval shape of the virus with a tight enwrapping of the envelope46,47 whereas the particles from Influenza virus B seem to be more loose and spherical48–50.
Like the shorter N-terminal end of influenza A, the B/NPTAIL carries the NLS of the protein. Whereas the non-classical NLS1 of A/NP is rather short with about 11 residues, the NLS of B/NP show three overlapping NLS sequences from residue 30 up to the end of the tail at residue 71. This result could explain why stepwise shortening of the tail gradually decreases the propensity of NP to go into the nucleus implicating a longer stretch in the interaction with importin-α rather than just a 4 residue linear motif between 44 and 4728. The complex between importin-α7 and B/NP was studied with SAXS. The analysis shows the volumes of the two proteins. Although the SAXS envelope of a disordered chain cannot give a single structure for B/NTAIL the complex shows that importin-α binds two thirds of the tail, concordant with the results from NMR, and no other parts of B/NP.
For the complex between the B/NTAIL and importin-α we used importin-α7. For efficient virus replication in vivo, avian influenza viruses depend on importin-α3, whereas mammalian viruses depend on importin-α751. However, all importin-alphas are very similar in their sequence and their structures and a specific importin-α can bind a very large range of proteins. The affinity between B/NTAIL and importin-α is rather low (K d around 900 nM) as measured with ITC and close to the affinity between the A/NTAIL and mouse importin-α1 (1.7 µM). This affinity is much lower than the affinity between the influenza polymerase protein PB2 and importin-α which has a K d value of around 5 nM52. Maybe one of the reasons is that the infected cell makes a lot of NP and only very little PB2, at least in earlier hours of the infection53,54.
Despite its intrinsic flexibility, the disordered N-terminal tail of NP may present a viable target for inhibition of viral infection, due to its essential role in nuclear import. Although the development of inhibitory molecules for intrinsically disordered targets represents a significant challenge55,56, recent advances have demonstrated that these flexible domains can be targeted57–60 and biophysical descriptions of the behavior of linear motifs such as the NLS, and their complex interaction modes, will no doubt aid in the conception of rational peptide or small-molecule based strategies.
Methods
Expression and Purification
The cloning of the human importin-α7 (KPNA6; Uniprot O60684) DNA coding sequence was described in52. The full length B/NP (B/NP1-560 called B/NP), its N-terminus (B/NP1-70 called B/NPTAIL) and its core (B/NP71-560 called B/NPCORE), were cloned in the pETM11 vector (EMBL) to express N-terminal His-tag TEV protease-cleavable constructs. Escherichia coli BL21 (DE3) cells transformed with the corresponding plasmids were induced 12 hours by adding 0.3 mM isopropyl-β-D-thiogalactopyranoside (IPTG) at 18 °C and collected by centrifugation. For B/NP constructs, pellets were resuspended and sonicated in lysis buffer composed of 50 mM Tris-HCl pH 7.5, 300 mM NaCl, 1 M NDSB201 (Sigma), 5 mM β-mercaptoethanol (β-ME) and cOmplete EDTA-free protease inhibitor cocktail (Roche). For importin-α7, pellets were resuspended and sonicated in lysis buffer composed of 50 mM Tris-HCl pH 8, 500 mM NaCl, 1 mM β-ME and cOmplete EDTA-free protease inhibitor cocktail. Purifications were performed at room temperature. Proteins were purified by Ni2+ affinity chromatography (Ni-NTA, Qiagen). For the B/NP and the B/NPCORE, this step was followed by a heparin column (GE-Healthcare). Heparin elution fractions or nickel elution fractions were dialyzed overnight at 4 °C with TEV (1/100) against 20 mM Tris-HCl pH 7.5 at 150 mM, 5 mM β-ME and 20 mM imidazol, followed by Ni2+ affinity chromatography to remove the His-tag and the TEV protease. The proteins were then purified by size-exclusion chromatography using a SuperdexTM 200 increase 10/300 GL column (GE healthcare). For ITC and NMR, the His-tag of B/NPTAIL was removed and purified by size-exclusion chromatography (SuperdexTM 75 10/300 GL column; GE Healthcare). The purities of the samples were confirmed by SDS-PAGE. Proteins were concentrated by centrifugation (Amicon concentrators with cutoff of 3 and/or 10 kDa). Protein concentrations were determined using the extinction molar coefficient at 280 nm ε = 46 785 M−1.cm−1 for importin-α7, ε = 24 995 M−1.cm−1 for B/NP and B/NPCORE and ε = 2980 M−1.cm−1 for B/NPTAIL. For B/NPTAIL without the his-tag, the concentration was determined using a bicinchoninic acid protein assay (BCA protein assay kit, Pierce).
NMR experiments
Samples for NMR spectroscopy were produced in M9 minimal medium containing MEM vitamins (Gibco). For producing 13C, 15N proteins the medium was supplemented with 1.0 g.L−1 of 15NH4Cl (Cambridge Isotope Laboratories, INC; USA) and 2.0 g.L−1 of D-glucose U-13C6 (euriso-top; France). Purification protocol was the same as above and all NMR experiments were performed in 20 mM Bis-Tris buffer pH 6.5, 50 mM NaCl.
Spectral assignment of 13C, 15N B/NPTAIL was obtained from a set of BEST-type triple resonance spectra: HNCO, intra-residue HN(CA)CO, HN(CO)CA, intra-residue HNCA, HN(COCA)CB, and intra-residue HN(CA)CB61. All assignment spectra were recorded at a 1H frequency of 700 MHz and at 5 °C. The spectra were processed with NMRPipe62 and automatic assignment was done with the program MARS63 and manually verified. Secondary chemical shifts were calculated using the random coil values from refDB64.
A multi-conformational model of B/NPTAIL was calculated based on the chemical shifts as obtained from the assignment using a combination of Flexible-Meccano and the genetic algorithm ASTEROIDS. Five times 200 conformers were selected from a statistical coil ensemble of 10,000 conformers31. A new ensemble of 8,500 conformers was generated based on the φ and ψ angles of the selected conformers, and supplemented with 1500 structures from the initial ensemble. This ensemble was subjected to another round of ASTEROIDS selection and this iteration was repeated four times30 until convergence was achieved with respect to the experimental chemical shifts.
For titration of 15N labelled B/NPTAIL with importin-α, 1H-15N HSQC spectra of 71 μM B/NPTAIL were recorded at increasing concentrations of importin-α (55, 100, 150 μM) at 25 °C and a 1H frequency of 850 MHz. 15N R2 (CPMG) were recorded at a 1H frequency of 950 MHz in the absence and presence of 55 μM importin-α at 25 °C using delay times of 4, 8, 20, 40, and 68 ms to measure the magnetization decay65. The time point at 8 ms was repeated for error estimation. Assignment experiments were performed with the purification tag present, whereas all interaction experiments with importin-α were performed with the purification tag cleaved off B/NPTAIL.
Interaction assays by size exclusion chromatography
All size exclusion chromatography (SEC) experiments were performed in 20 mM Tris-HCl pH 7.5, 150 mM NaCl and 5 mM β-ME using a SuperdexTM 200 increase 10/300GL column for B/NP and B/NPCORE and a SuperdexTM 75 10/300GL column for B/NPTAIL. 300 μL of each sample (30 μM B/NP; 30 μM B/NPCORE; 60 μM B/NPTAIL; 25 μM importin-α7; 30 μM B/NP + 25 μM importin-α7; 30 μM B/NPCORE + 25 μM importin-α7; 50 μM B/NPTAIL + 25 μM importin-α7) were incubated 1 hour at room temperature before injection.
SAXS analysis
All datasets were collected on BM29 (ESRF). For in-line SEC-SAXS, the experimental setup consists of High Pressure Liquid Chromatography (HPLC) system connected to an analytical SuperdexTM increase 200 5/150 GL column (GE Healthcare) followed down-stream by SAXS sample capillary. SAXS measurements were performed every second with a Pilatus 1 M detector (distance of 2.87 m) allowing a q range of 0.03 to 4.5 nm with a wavelength of 0.01 nm.
Experimental curves were subtracted and analyzed using Primus (ATSAS programs suite)66. To verify the molecular mass, the Rambo and Tainer method was used36,67. Rg predictions using Guinier extrapolation were plotted against the elution volume to select the most monodisperse part of the protein elution peak. SAXS datasets within this zone were scaled and averaged to produce one unique I(q) curve. Distance distribution functions p(r) were calculated using the program GNOM68. The ab-initio models were generated by MONSA using when available, the individual data sets in order to fit them simultaneously37. Homologue PDB structure comparison was assessed using Crysol69. Homologue structure fitting within the DAMAVER envelope was performed with PyMOL70 and curve representations using Graphpad (Prism). SAXS curves with B/NP alone contained the poly-His tag, which is cleaved off for all samples probing interaction with importin-α.
A description of the conformational ensemble of full length B/NP was obtained using the crystal structure of the folded domain (PDB 3TJ023) and Flexible-Meccano30 to add the intrinsically disordered tail as a conformational ensemble. The backbone dihedral angle distribution identified by a Flexible-Meccano/ASTEROIDS31 combination to describe B/NPTAIL (see above) was assumed to be valid also within the full length protein construct. 200 conformers were generated and SAXS curves for each of the conformers were calculated using CRYSOL69 and averaged to obtain the expected SAXS curve for the full length B/NP ensemble.
Isothermal titration calorimetry studies
B/NPTAIL and importin-α7 were dialyzed in the same buffer (20 mM Tris-HCl pH 7.5; 150 mM NaCl) before the titration. The ITC titration experiments were done at 20 °C using a MicroCal ITC200 (GE Healthcare) with 16 × 2.4 µL injections of 150 µM importin-α7 into a 20 µM B/NPTAIL solution. Integration of the titration curves was performed using the ORIGIN software (OriginLab, Northampton, United Kingdom) to extract thermodynamic parameters, stoichiometry N, equilibrium association constant Ka and the binding enthalpy ΔH. The Gibbs free energy of binding ΔG was calculated from the relation ΔG = −RT ln(Ka) and the binding entropy ΔS was deduced from the equation (ΔG = ΔH − TΔS). All titrations fit the single-binding site mechanism with 1:1 stoichiometry and binding parameters were calculated as the average of three independent experiments ± SD.
Electronic supplementary material
Acknowledgements
We are highly grateful to Caroline Mas and Laura Tengo for their help and Nicola Salvi for providing the 15N R2 pulse sequence. We thank Darren J Hart for the plasmid to express the human importin-α7. We thank Guy Schoehn, Bernard Delmas and Anny Slama-Schwok for discussion. AL and AD were funded through the Labex GRAL (ANR-10-LABX-49-01). SM acknowledges the EMBO longterm fellowship (ALTF 468-2014) and EC (EMBOCOFUND2012, GA-2012-600394) through Marie Curie Action. This work was supported by the French agency for Research through the ANR RNAP-IAV (ANR-14-CE09-0017). This work used the beamlines of the and European Synchrotron Radiation Facility (ESRF) and the platforms of the Grenoble Instruct-ERIC Center (ISBG; UMS 3518 CNRS-CEA-UGA-EMBL) with support from FRISBI (ANR-10-INSB-05-02) and GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology (PSB).
Author Contributions
T.C., R.W.H.R. and M.B. conceived the experiments. A.L., S.M., A.D., J.M.B. and T.C. performed the experiments. A.L., S.M., A.D., M.R.J., M.B., J.M.B., R.W.H.R. and T.C. analysed the data. A.L., S.M., M.B., J.M.B., R.W.H.R. and T.C. wrote the paper.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Alice Labaronne and Sigrid Milles contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-017-17458-z.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Boulo S, Akarsu H, Ruigrok RW, Baudin F. Nuclear traffic of influenza virus proteins and ribonucleoprotein complexes. Virus research. 2007;124:12–21. doi: 10.1016/j.virusres.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 2.Te Velthuis AJ, Fodor E. Influenza virus RNA polymerase: insights into the mechanisms of viral RNA synthesis. Nature reviews. Microbiology. 2016;14:479–493. doi: 10.1038/nrmicro.2016.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalderon D, Richardson WD, Markham AF, Smith AE. Sequence requirements for nuclear location of simian virus 40 large-T antigen. Nature. 1984;311:33–38. doi: 10.1038/311033a0. [DOI] [PubMed] [Google Scholar]
- 4.Kalderon D, Roberts BL, Richardson WD, Smith AE. A short amino acid sequence able to specify nuclear location. Cell. 1984;39:499–509. doi: 10.1016/0092-8674(84)90457-4. [DOI] [PubMed] [Google Scholar]
- 5.Kosugi S, et al. Six classes of nuclear localization signals specific to different binding grooves of importin alpha. The Journal of biological chemistry. 2009;284:478–485. doi: 10.1074/jbc.M807017200. [DOI] [PubMed] [Google Scholar]
- 6.Robbins J, Dilworth SM, Laskey RA, Dingwall C. Two interdependent basic domains in nucleoplasmin nuclear targeting sequence: identification of a class of bipartite nuclear targeting sequence. Cell. 1991;64:615–623. doi: 10.1016/0092-8674(91)90245-T. [DOI] [PubMed] [Google Scholar]
- 7.Gorlich D, Mattaj IW. Nucleocytoplasmic transport. Science. 1996;271:1513–1518. doi: 10.1126/science.271.5255.1513. [DOI] [PubMed] [Google Scholar]
- 8.Pemberton LF, Paschal BM. Mechanisms of receptor-mediated nuclear import and nuclear export. Traffic. 2005;6:187–198. doi: 10.1111/j.1600-0854.2005.00270.x. [DOI] [PubMed] [Google Scholar]
- 9.Mattaj IW, Englmeier L. Nucleocytoplasmic transport: the soluble phase. Annual review of biochemistry. 1998;67:265–306. doi: 10.1146/annurev.biochem.67.1.265. [DOI] [PubMed] [Google Scholar]
- 10.Kuersten S, Ohno M, Mattaj IW. Nucleocytoplasmic transport: Ran, beta and beyond. Trends in cell biology. 2001;11:497–503. doi: 10.1016/S0962-8924(01)02144-4. [DOI] [PubMed] [Google Scholar]
- 11.Lott K, Cingolani G. The importin beta binding domain as a master regulator of nucleocytoplasmic transport. Biochimica et biophysica acta. 2011;1813:1578–1592. doi: 10.1016/j.bbamcr.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boulo S, et al. Human importin alpha and RNA do not compete for binding to influenza A virus nucleoprotein. Virology. 2011;409:84–90. doi: 10.1016/j.virol.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 13.Cros JF, Garcia-Sastre A, Palese P. An unconventional NLS is critical for the nuclear import of the influenza A virus nucleoprotein and ribonucleoprotein. Traffic. 2005;6:205–213. doi: 10.1111/j.1600-0854.2005.00263.x. [DOI] [PubMed] [Google Scholar]
- 14.Neumann G, Castrucci MR, Kawaoka Y. Nuclear import and export of influenza virus nucleoprotein. Journal of virology. 1997;71:9690–9700. doi: 10.1128/jvi.71.12.9690-9700.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ng AK, et al. Structure of the influenza virus A H5N1 nucleoprotein: implications for RNA binding, oligomerization, and vaccine design. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2008;22:3638–3647. doi: 10.1096/fj.08-112110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ozawa M, et al. Contributions of two nuclear localization signals of influenza A virus nucleoprotein to viral replication. Journal of virology. 2007;81:30–41. doi: 10.1128/JVI.01434-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang P, Palese P, O’Neill RE. The NPI-1/NPI-3 (karyopherin alpha) binding site on the influenza a virus nucleoprotein NP is a nonconventional nuclear localization signal. Journal of virology. 1997;71:1850–1856. doi: 10.1128/jvi.71.3.1850-1856.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu WW, Sun YH, Pante N. Nuclear import of influenza A viral ribonucleoprotein complexes is mediated by two nuclear localization sequences on viral nucleoprotein. Virology journal. 2007;4:49. doi: 10.1186/1743-422X-4-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakada R, Hirano H, Matsuura Y. Structure of importin-alpha bound to a non-classical nuclear localization signal of the influenza A virus nucleoprotein. Scientific reports. 2015;5:15055. doi: 10.1038/srep15055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu W, et al. Synergy of two low-affinity NLSs determines the high avidity of influenza A virus nucleoprotein NP for human importin alpha isoforms. Scientific reports. 2017;7:11381. doi: 10.1038/s41598-017-11018-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hutchinson EC, et al. Mapping the phosphoproteome of influenza A and B viruses by mass spectrometry. PLoS pathogens. 2012;8:e1002993. doi: 10.1371/journal.ppat.1002993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng W, et al. Phosphorylation controls the nuclear-cytoplasmic shuttling of influenza A virus nucleoprotein. Journal of virology. 2015;89:5822–5834. doi: 10.1128/JVI.00015-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ng AK, et al. Structural basis for RNA binding and homo-oligomer formation by influenza B virus nucleoprotein. Journal of virology. 2012;86:6758–6767. doi: 10.1128/JVI.00073-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ye Q, Krug RM, Tao YJ. The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature. 2006;444:1078–1082. doi: 10.1038/nature05379. [DOI] [PubMed] [Google Scholar]
- 25.Gerard FC, et al. Modular organization of rabies virus phosphoprotein. Journal of molecular biology. 2009;388:978–996. doi: 10.1016/j.jmb.2009.03.061. [DOI] [PubMed] [Google Scholar]
- 26.Stevens MP, Barclay WS. The N-terminal extension of the influenza B virus nucleoprotein is not required for nuclear accumulation or the expression and replication of a model RNA. Journal of virology. 1998;72:5307–5312. doi: 10.1128/jvi.72.6.5307-5312.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu M, et al. The Functional Study of the N-Terminal Region of Influenza B Virus Nucleoprotein. PloS one. 2015;10:e0137802. doi: 10.1371/journal.pone.0137802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sherry L, Smith M, Davidson S, Jackson D. The N terminus of the influenza B virus nucleoprotein is essential for virus viability, nuclear localization, and optimal transcription and replication of the viral genome. Journal of virology. 2014;88:12326–12338. doi: 10.1128/JVI.01542-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wanitchang A, Narkpuk J, Jongkaewwattana A. Nuclear import of influenza B virus nucleoprotein: involvement of an N-terminal nuclear localization signal and a cleavage-protection motif. Virology. 2013;443:59–68. doi: 10.1016/j.virol.2013.04.025. [DOI] [PubMed] [Google Scholar]
- 30.Ozenne V, et al. Flexible-meccano: a tool for the generation of explicit ensemble descriptions of intrinsically disordered proteins and their associated experimental observables. Bioinformatics. 2012;28:1463–1470. doi: 10.1093/bioinformatics/bts172. [DOI] [PubMed] [Google Scholar]
- 31.Jensen MR, Salmon L, Nodet G, Blackledge M. Defining conformational ensembles of intrinsically disordered and partially folded proteins directly from chemical shifts. Journal of the American Chemical Society. 2010;132:1270–1272. doi: 10.1021/ja909973n. [DOI] [PubMed] [Google Scholar]
- 32.Labaronne, A. et al. Binding of RNA by the Nucleoproteins of Influenza Viruses A and B. Viruses8, 10.3390/v8090247 (2016). [DOI] [PMC free article] [PubMed]
- 33.Dinkel H, et al. ELM 2016–data update and new functionality of the eukaryotic linear motif resource. Nucleic acids research. 2016;44:D294–300. doi: 10.1093/nar/gkv1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kosugi S, Hasebe M, Tomita M, Yanagawa H. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:10171–10176. doi: 10.1073/pnas.0900604106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pang X, Zhou HX. Design rules for selective binding of nuclear localization signals to minor site of importin alpha. PloS one. 2014;9:e91025. doi: 10.1371/journal.pone.0091025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rambo RP, Tainer JA. Accurate assessment of mass, models and resolution by small-angle scattering. Nature. 2013;496:477–481. doi: 10.1038/nature12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Svergun DI. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophysical journal. 1999;76:2879–2886. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Svergun DI, Nierhaus KH. A map of protein-rRNA distribution in the 70 S Escherichia coli ribosome. The Journal of biological chemistry. 2000;275:14432–14439. doi: 10.1074/jbc.275.19.14432. [DOI] [PubMed] [Google Scholar]
- 39.Fournier E, et al. A supramolecular assembly formed by influenza A virus genomic RNA segments. Nucleic acids research. 2012;40:2197–2209. doi: 10.1093/nar/gkr985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liljeroos L, Huiskonen JT, Ora A, Susi P, Butcher SJ. Electron cryotomography of measles virus reveals how matrix protein coats the ribonucleocapsid within intact virions. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:18085–18090. doi: 10.1073/pnas.1105770108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Loney C, Mottet-Osman G, Roux L, Bhella D. Paramyxovirus ultrastructure and genome packaging: cryo-electron tomography of sendai virus. Journal of virology. 2009;83:8191–8197. doi: 10.1128/JVI.00693-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jensen MR, et al. Intrinsic disorder in measles virus nucleocapsids. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:9839–9844. doi: 10.1073/pnas.1103270108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Longhi S, et al. The C-terminal domain of the measles virus nucleoprotein is intrinsically disordered and folds upon binding to the C-terminal moiety of the phosphoprotein. The Journal of biological chemistry. 2003;278:18638–18648. doi: 10.1074/jbc.M300518200. [DOI] [PubMed] [Google Scholar]
- 44.Desfosses A, et al. Self-organization of the vesicular stomatitis virus nucleocapsid into a bullet shape. Nature communications. 2013;4:1429. doi: 10.1038/ncomms2435. [DOI] [PubMed] [Google Scholar]
- 45.Ge P, et al. Cryo-EM model of the bullet-shaped vesicular stomatitis virus. Science. 2010;327:689–693. doi: 10.1126/science.1181766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harris A, et al. Influenza virus pleiomorphy characterized by cryoelectron tomography. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:19123–19127. doi: 10.1073/pnas.0607614103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Noda T, et al. Architecture of ribonucleoprotein complexes in influenza A virus particles. Nature. 2006;439:490–492. doi: 10.1038/nature04378. [DOI] [PubMed] [Google Scholar]
- 48.Booy FP, Ruigrok RW, van Bruggen EF. Electron microscopy of influenza virus. A comparison of negatively stained and ice-embedded particles. Journal of molecular biology. 1985;184:667–676. doi: 10.1016/0022-2836(85)90312-2. [DOI] [PubMed] [Google Scholar]
- 49.Cusack S, Ruigrok RW, Krygsman PC, Mellema JE. Structure and composition of influenza virus. A small-angle neutron scattering study. Journal of molecular biology. 1985;186:565–582. doi: 10.1016/0022-2836(85)90131-7. [DOI] [PubMed] [Google Scholar]
- 50.Katz G, et al. Morphology of influenza B/Lee/40 determined by cryo-electron microscopy. PloS one. 2014;9:e88288. doi: 10.1371/journal.pone.0088288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gabriel G, et al. Differential use of importin-alpha isoforms governs cell tropism and host adaptation of influenza virus. Nature communications. 2011;2:156. doi: 10.1038/ncomms1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boivin S, Hart DJ. Interaction of the influenza A virus polymerase PB2 C-terminal region with importin alpha isoforms provides insights into host adaptation and polymerase assembly. The Journal of biological chemistry. 2011;286:10439–10448. doi: 10.1074/jbc.M110.182964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hay AJ, Lomniczi B, Bellamy AR, Skehel JJ. Transcription of the influenza virus genome. Virology. 1977;83:337–355. doi: 10.1016/0042-6822(77)90179-9. [DOI] [PubMed] [Google Scholar]
- 54.Shapiro GI, Gurney T, Jr, Krug RM. Influenza virus gene expression: control mechanisms at early and late times of infection and nuclear-cytoplasmic transport of virus-specific RNAs. Journal of virology. 1987;61:764–773. doi: 10.1128/jvi.61.3.764-773.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheng Y, et al. Rational drug design via intrinsically disordered protein. Trends in biotechnology. 2006;24:435–442. doi: 10.1016/j.tibtech.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 56.Uversky VN. Intrinsically disordered proteins and novel strategies for drug discovery. Expert opinion on drug discovery. 2012;7:475–488. doi: 10.1517/17460441.2012.686489. [DOI] [PubMed] [Google Scholar]
- 57.Iconaru LI, et al. Discovery of Small Molecules that Inhibit the Disordered Protein, p27(Kip1) Scientific reports. 2015;5:15686. doi: 10.1038/srep15686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Joshi P, et al. A Fragment-Based Method of Creating Small-Molecule Libraries to Target the Aggregation of Intrinsically DisorderedProteins. ACS combinatorial science. 2016;18:144–153. doi: 10.1021/acscombsci.5b00129. [DOI] [PubMed] [Google Scholar]
- 59.Krishnan N, et al. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nature chemical biology. 2014;10:558–566. doi: 10.1038/nchembio.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu C, et al. Structure-based Inhibitor Design for the Intrinsically Disordered Protein c-Myc. Scientific reports. 2016;6:22298. doi: 10.1038/srep22298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lescop E, Schanda P, Brutscher B. A set of BEST triple-resonance experiments for time-optimized protein resonance assignment. Journal of magnetic resonance. 2007;187:163–169. doi: 10.1016/j.jmr.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 62.Delaglio F, et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. Journal of biomolecular NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 63.Jung YS, Zweckstetter M. Mars–robust automatic backbone assignment of proteins. Journal of biomolecular NMR. 2004;30:11–23. doi: 10.1023/B:JNMR.0000042954.99056.ad. [DOI] [PubMed] [Google Scholar]
- 64.Zhang H, Neal S, Wishart DS. RefDB: a database of uniformly referenced protein chemical shifts. Journal of biomolecular NMR. 2003;25:173–195. doi: 10.1023/A:1022836027055. [DOI] [PubMed] [Google Scholar]
- 65.Kay LE, Torchia DA, Bax A. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry. 1989;28:8972–8979. doi: 10.1021/bi00449a003. [DOI] [PubMed] [Google Scholar]
- 66.Petoukhov MV, Svergun DI. Analysis of X-ray and neutron scattering from biomacromolecular solutions. Current opinion in structural biology. 2007;17:562–571. doi: 10.1016/j.sbi.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 67.Rambo RP, Tainer JA. Super-resolution in solution X-ray scattering and its applications to structural systems biology. Annual review of biophysics. 2013;42:415–441. doi: 10.1146/annurev-biophys-083012-130301. [DOI] [PubMed] [Google Scholar]
- 68.Franke D, Svergun DI. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. Journal of applied crystallography. 2009;42:342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Svergun DI, Barberato C, Koch MHJ. CRYSOL – a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. Journal of applied crystallography. 1995;28:768–773. doi: 10.1107/S0021889895007047. [DOI] [Google Scholar]
- 70.Schrodinger, L. L. C. The PyMOL Molecular Graphics System, Version 1.8 (2015).
- 71.Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 72.Hutchinson EC, Fodor E. Nuclear import of the influenza A virus transcriptional machinery. Vaccine. 2012;30:7353–7358. doi: 10.1016/j.vaccine.2012.04.085. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.