

Abstract

Orotidine 5′-monophosphate decarboxylase (OMPDC) catalyzes the decarboxylation of 5-fluoroorotate (FO) with kcat/Km = 1.4 × 10–7 M–1 s–1. Combining this and related kinetic parameters shows that the 31 kcal/mol stabilization of the transition state for decarboxylation of OMP provided by OMPDC represents the sum of 11.8 and 10.6 kcal/mol stabilization by the substrate phosphodianion and the ribosyl ring, respectively, and an 8.6 kcal/mol stabilization from the orotate ring. The transition state for OMPDC-catalyzed decarboxylation of FO is stabilized by 5.2, 7.2, and 9.0 kcal/mol, respectively, by 1.0 M phosphite dianion, d-glycerol 3-phosphate and d-erythritol 4-phosphate. The stabilization is due to the utilization of binding interactions of the substrate fragments to drive an enzyme conformational change, which locks the orotate ring of the whole substrate, or the substrate pieces in a caged complex. We propose that enzyme-activation is a possible, and perhaps probable, consequence of any substrate-induced enzyme conformational change.
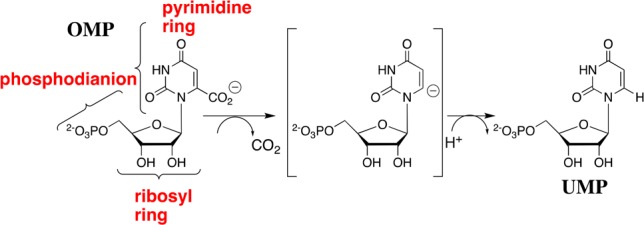
The underlying cause for enzymatic catalysis is stabilization of the transition state by interactions with the protein catalyst.1 Orotidine 5′-monophosphate decarboxylase (OMPDC) affects a 31 kcal/mol stabilization of the transition state for decarboxylation of orotidine 5′-monophosphate (OMP) to give uridine 5′-monophosphate (UMP),2 by a stepwise mechanism through a UMP carbanion intermediate (Scheme 1).3 This transition state stabilization has been modeled in computational studies.4 What has not been fully modeled is the extraordinary specificity of OMPDC in binding the decarboxylation transition state with a higher affinity (31 kcal/mol) compared with the substrate OMP (8 kcal/mol).2,5
Scheme 1. Substrate Fragments of OMP that Provide the Binding Energy Utilized in the Stabilization of the UMP Carbanion Intermediate of OMPDC-Catalyzed Decarboxylation.
Binding interactions between OMPDC and the phosphodianion of OMP provide 12 of the 31 kcal/mol transition state stabilization.6 These interactions do not only anchor OMP to the protein, because eliminating the anchoring connection results in only a 4 kcal/mol decrease, to 8 kcal/mol, in the stabilization of the transition state for decarboxylation of the truncated substrate piece 1-(β-d-erythrofuranosyl)orotate (EO) from interactions with 1.0 M phosphite dianion (Figure 1A).6,7 This activation is due to the utilization of binding energy from interactions between OMPDC and activator (Act, Figure 1) to drive a complex conformational change from inactive open OMPDC (EO) to the active closed caged complex (EC),8 where EC is stabilized relative to EO by interactions between dianions and the side chains of Q215, Y217, and R235.9
Figure 1.
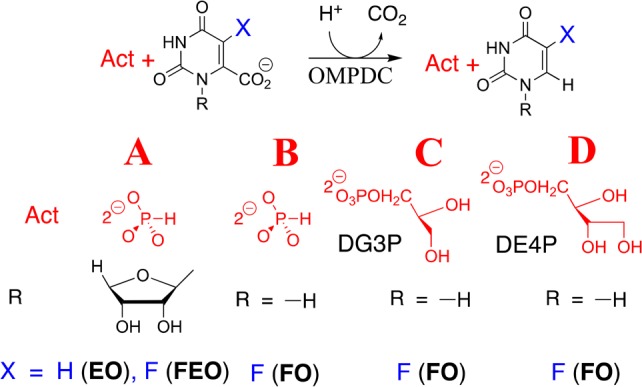
Activation of OMPDC-catalyzed decarboxylation reactions. (A) Activation of decarboxylation of EO and FEO by HPO32–.6,10 (B) Activation of decarboxylation of FO by HPO32–. (C, D) Activation of decarboxylation of FO by d-glycerol 3-phosphate (DG3P) and d-erythritol 4-phosphate (DE4P), respectively.
Figure 2A shows a representation of the open form of OMPDC determined for unliganded OMPDC from yeast (EO, Scheme 2), with a hypothetical 6-aza uridine 5′-monophosphate (azaUMP) ligand inserted at the position determined for the OMPDC·azaUMP complex (Figure 2B).11 Many ligands induce a large conformational change in OMPDC that is driven by the development of strong protein–ligand interactions.12 The interactions between the protein and the phosphodianion, or the ribosyl hydroxyls, which develop at the complex to the tight-binding inhibitor azaUMP [or to substrate analogs],12 are illustrated in Figure 2B for the closed form of OMPDC (EC).11 Our model (Scheme 2) requires that the binding energy from all interactions that drive the conformational change from EO to EC activate OMPDC for catalysis, as has been demonstrated for protein–dianion interactions,6,10,13 because these interactions are only fully expressed at the decarboxylation transition state. This prediction is confirmed here by the observation that binding interactions between OMPDC and either phosphite dianion (Figure 1B) or sugar hydroxyls (Figure 1C and 1D) activate OMPDC for catalysis of decarboxylation of the definitive truncated substrate, 5-fluoroorotate (FO).
Figure 2.
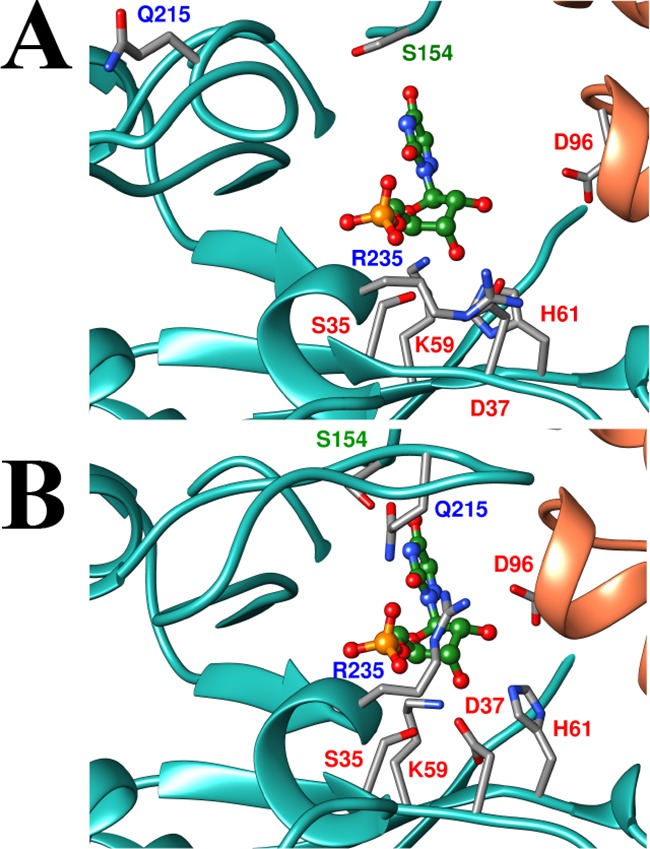
Representations of the open EO (PDB entry 3GDK) and the closed, liganded, (B, 3GDL) forms of yeast OMPDC.11 The azaUMP ligand is placed at structure A at the position determined for structure B. The ligand is stabilized by interactions from the side chains of R235 and Q215 with the phosphodianion, and of the D96, H61, D37 and K59 with the ribosyl hydroxyls.
Scheme 2. The Role of an Activator-Driven Conformational Change in Catalysis by OMPDC.

The slow decarboxylation of FO to form 5-fluorouracil (FU) catalyzed by yeast OMPDC was monitored by HPLC analyses, as described in the Supporting Information (SI). Observed first-order rate constants kobs = v/[E] for the decarboxylation of FO (5 or 10 mM) catalyzed by 0.7 mM OMPDC at 25 °C were determined from the initial reaction velocity v during the first 0.01% reaction, over a two-week reaction time, during which OMPDC maintained full activity. The second-order rate constant (kcat/Km)o = (1.4 ± 0.1) × 10–7 M–1 s–1 for OMPDC-catalyzed decarboxylation of FO (Table 1) was determined as the slope of the linear plot of kobs = v[E] against [FO] (Figure 3A). These apparent first-order rate constants from HPLC analyses are reproducible to better than ±10%. The 5-F substituent provides strong stabilization of the UMP carbanion intermediate of OMPDC-catalyzed reactions.3a,10,13b,14 The value of (kcat/Km)o = 3 × 10–10 M–1 s–1 for OMPDC-catalyzed decarboxylation of orotate (Table 1) was determined from (kcat/Km)o = 1.4 × 10–7 M–1 s–1 for decarboxylation of FO and a 500-fold effect of the 5-F substituent. This is a rough average of the 5-F effect on mutant OMPDC-catalyzed decarboxylation of OMP, when chemistry is strongly rate determining for both OMPDC-catalyzed reactions.14
Table 1. Contribution of the IBE from Substrate Fragments to the 31 kcal/mol IBE of OMP2 for OMPDCa.
Figure 3.
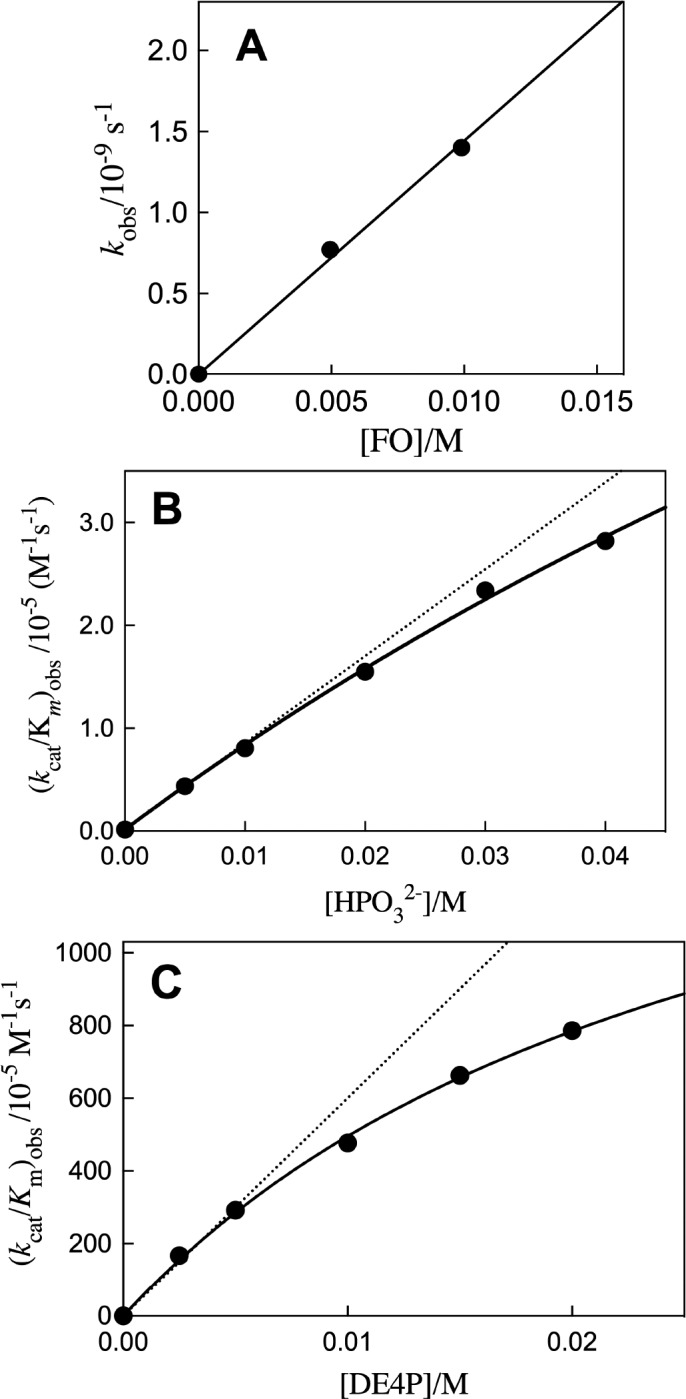
Plots of kinetic data for OMPDC-catalyzed decarboxylation of FO. (A) The dependence of kobs = v[E] on [FO]. (B) The dependence of (kcat/Km)obs for OMPDC-catalyzed decarboxylation of FO on [HPO32–]. (C) The dependence of (kcat/Km)obs for OMPDC-catalyzed decarboxylation of FO on the concentration of DE4P.
A comparison of the second-order rate constants kcat/Km for OMPDC-catalyzed decarboxylation of orotate, EO, and OMP (Table 1) shows that the ribosyl and phosphodianion fragments contribute 10.6 and 11.8 kcal/mol intrinsic binding energies, respectively, to stabilization of the transition state for OMPDC-catalyzed decarboxylation of OMP: the sum is 22.4 kcal/mol of the total 31 kcal/mol total intrinsic binding energy of OMP, leaving 8.6 kcal/mol for stabilization of the transition state by interactions with orotate.2 This provides a graphic and readily generalizable illustration of how enormous enzymatic rate accelerations may be obtained through recruitment of several modest, additive, binding energies of substrate fragments.
Figure 3B,C shows the effect of increasing [HPO32–] and [DE4P], respectively, on (kcat/Km)obs for OMPDC-catalyzed decarboxylation of FO (5 mM). These data were fit to eq 1 (Scheme 3A) to give the values for (kcat/Km)Act and Kd reported in Table 2. Table 2 reports values for (kcat/Km)Act/Kd obtained as the slope of the linear correlations shown as dashed lines in Figure 3B,C. Data for the effect of increasing [D,LG3P] (Figure S1A) and [LG3P] (Figure S1B) on (kcat/Km)obs for OMPDC-catalyzed decarboxylation of FO were evaluated to give the kinetic parameters in Table 2. The value of (kcat/Km)Act/Kd for OMPDC-catalyzed decarboxylation of DG3P was calculated from these kinetic parameters using eq 2, where (kAct)XG3P = [(kcat/Km)Act/Kd)]XG3P (X = D, L, or D, L). The intrinsic fragment binding energies (IBEs), determined using eq 3 (Scheme 3B), are reported in Table 2.
Scheme 3. (a) Kinetic Scheme Used for the Derivation of eq 1. (b) Kinetic Scheme Used for the Derivation of eq 3.

Table 2. Kinetic Parameters for Unactivated and Activated OMPDC-Catalyzed Decarboxylation of FOa.
| Activator | (kcat/Km)Act M–1 s–1b | Kd (M)c | [(kcat/Km)Act]/Kd M–2 s–1d | IBE (kcal/mol)e |
|---|---|---|---|---|
| HPif | (1.6 ± 0.4) × 10–4 | 0.18 ± 0.05 | (8.4 ± 0.4) × 10–4 | 5.2 |
| D,LG3Pg | (7.0 ± 0.7) × 10–4 | 0.05 ± 0.01 | (1.3 ± 0.1) × 10–2 | 6.8 |
| LG3Pg,h | (9.9 ± 0.2) × 10–4 | 5.2 | ||
| DG3Pi | (2.5 ± 0.01) × 10–2 | 7.2 | ||
| DE4Pj | (1.9 ± 0.2) × 10–2 | 0.030 ± 0.003 | (6.0 ± 0.2) × 10–1 | 9.0 |
| DR5Pg | ≈3 × 10–5 | 3.1 |
At 25 °C, pH 7.0 and I = 0.15 (NaCl). The quoted errors are the standard deviations from the least-squares fits of the data.
Second-order rate constant for breakdown of E·Act·FO to form FU.
Dissociation constant for the activator.
Determined as described in the text.
See SI.
No detectable saturation of OMPDC.
Phosphite dianion (1.0 M) provides similar 5.010 and 5.2 (Table 2) kcal/mol stabilization, respectively, of the transition states for OMPDC-catalyzed decarboxylation of truncated substrates 1-(β-d-erythrofuranosyl)-5-fluoroorotate (FEO) and FO. The small apparent effect of the ribosyl group of FEO on activation for decarboxylation shows that the effect of the dianion driven protein conformational change on the reactivity of the fluoroorotate ring is transmitted across both the ribosyl group and the vacant protein core. DG3P and DE4P provide 7.2 and 9.0 kcal/mol transition state stabilization, respectively, which corresponds to ca. 2 kcal/mol transition state stabilization/sugar hydroxyl. OMPDC shows specificity for activation by DG3P, because the IBE determined for LG3P is similar to that for HPO32– alone. The small activation of OMPDC-catalyzed decarboxylation of FO for a reaction in the presence of 40 mM d-ribose 5′-phosphate (DR5P, SI) is consistent with a ligand IBE of only 3.1 kcal/mol (Table 2). This shows that a tight and precise fit of the ligand is required for strong transition state binding.10
| 1 |
| 2 |
| 3 |
These results provide strong support for the conclusion that each of the many OMPDC–substrate interactions, which stabilize the closed enzyme EC relative to the open enzyme EO, contribute to activation of OMPDC for catalysis of decarboxylation of orotate and fluoroorotate rings. We conclude that these protein–ligand interactions act in concert to construct a tight, catalytically active form of OMPDC from the floppy open enzyme. These results provide a dramatic example of the effect of such preorganization of protein structure on enzyme activity.15
X-ray crystallographic analyses and other protocols for the evaluation of the role of conformational changes in enzyme catalysis, failed to suggest the activating nature of phosphodianion driven conformational changes in catalysis by triosephosphate isomerase, and other enzymes.7b,16 Our report of the, likewise, unrecognized activating role of protein sugar–hydroxyl interactions in catalysis by OMPDC provides compelling motivation for a re-evaluation of the role of substrate-driven conformational changes in enzyme catalysis;1b,8a,8b,17 and, for experiments to test the hypothesis that many, or most, such protein conformational changes activate enzymes for catalysis of the reaction of their bound substrates.
Experiments to probe the activation of OMPDC by substrate induced conformational changes have outpaced other mechanistic studies on this enzyme. Consequently, there is now strong evidence that the binding interactions of the nonreacting substrate parts are used to construct a protein cage that shows a high reactivity toward substrate decarboxylation. The results of our earlier work have emphasized the role the protein plays in strongly stabilizing the UMP carbanion intermediate relative to the carbon acid substrate.3 This suggests that an unusually strong stabilization of this carbanion by interaction with the cationic side chain of K93.4c,16a This single interaction seems unlikely to enable the entire rate acceleration, so that there remains much to be learned about the origin of the high-reactivity of the caged OMPDC–substrate complex
Acknowledgments
This work was supported by grants GM116921 and GM39754 from the National Institutes of Health.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b08897.
References for experimental procedures used to purify yeast OMPDC, and a description of the procedures for the assay of OMPDC-catalyzed decarboxylation of FO; kinetic data for the activation of OMPDC by d-ribose 5′-phosphate; effect on increasing [LG3P] and [D,LG3P], respectively on (kcat/Km)obs for OMPDC-catalyzed decarboxylation of FO (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Pauling L. Nature 1948, 161, 707–709. 10.1038/161707a0. [DOI] [PubMed] [Google Scholar]; b Amyes T. L.; Richard J. P. Biochemistry 2013, 52, 2021–2035. 10.1021/bi301491r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radzicka A.; Wolfenden R. Science 1995, 267, 90–93. 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- a Tsang W.-Y.; Wood B. M.; Wong F. M.; Wu W.; Gerlt J. A.; Amyes T. L.; Richard J. P. J. Am. Chem. Soc. 2012, 134, 14580–14594. 10.1021/ja3058474. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Amyes T. L.; Wood B. M.; Chan K.; Gerlt J. A.; Richard J. P. J. Am. Chem. Soc. 2008, 130, 1574–1575. 10.1021/ja710384t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Vardi-Kilshtain A.; Doron D.; Major D. T. Biochemistry 2013, 52, 4382–4390. 10.1021/bi400190v. [DOI] [PubMed] [Google Scholar]; b Wu N.; Mo Y.; Gao J.; Pai E. F. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 2017–2022. 10.1073/pnas.050417797. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Warshel A.; Strajbl M.; Villa J.; Florian J. Biochemistry 2000, 39, 14728–14738. 10.1021/bi000987h. [DOI] [PubMed] [Google Scholar]; d Gao J. Curr. Opin. Struct. Biol. 2003, 13, 184–192. 10.1016/S0959-440X(03)00041-1. [DOI] [PubMed] [Google Scholar]
- Porter D. J. T.; Short S. A. Biochemistry 2000, 39, 11788–11800. 10.1021/bi001199v. [DOI] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P.; Tait J. J. J. Am. Chem. Soc. 2005, 127, 15708–15709. 10.1021/ja055493s. [DOI] [PubMed] [Google Scholar]
- a Spong K.; Amyes T. L.; Richard J. P. J. Am. Chem. Soc. 2013, 135, 18343–18346. 10.1021/ja4107513. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Reyes A. C.; Zhai X.; Morgan K. T.; Reinhardt C. J.; Amyes T. L.; Richard J. P. J. Am. Chem. Soc. 2015, 137, 1372–1382. 10.1021/ja5123842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Amyes T. L.; Malabanan M. M.; Zhai X.; Reyes A. C.; Richard J. P. Protein Eng., Des. Sel. 2017, 30, 159–168. 10.1093/protein/gzw064. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Richard J. P.; Amyes T. L.; Goryanova B.; Zhai X. Curr. Opin. Chem. Biol. 2014, 21, 1–10. 10.1016/j.cbpa.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Go M. K.; Amyes T. L.; Richard J. P. Biochemistry 2009, 48, 5769–5778. 10.1021/bi900636c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Amyes T. L.; Ming S. A.; Goldman L. M.; Wood B. M.; Desai B. J.; Gerlt J. A.; Richard J. P. Biochemistry 2012, 51, 4630–4632. 10.1021/bi300585e. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Goldman L. M.; Amyes T. L.; Goryanova B.; Gerlt J. A.; Richard J. P. J. Am. Chem. Soc. 2014, 136, 10156–10165. 10.1021/ja505037v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryanova B.; Spong K.; Amyes T. L.; Richard J. P. Biochemistry 2013, 52, 537–546. 10.1021/bi301650d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K. K.; Wood B. M.; Fedorov A. A.; Fedorov E. V.; Imker H. J.; Amyes T. L.; Richard J. P.; Almo S. C.; Gerlt J. A. Biochemistry 2009, 48, 5518–5531. 10.1021/bi900623r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Fujihashi M.; Wei L.; Kotra L. P.; Pai E. F. J. Mol. Biol. 2009, 387, 1199–1210. 10.1016/j.jmb.2009.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wittmann J. G.; Heinrich D.; Gasow K.; Frey A.; Diederichsen U.; Rudolph M. G. Structure 2008, 16, 82–92. 10.1016/j.str.2007.10.020. [DOI] [PubMed] [Google Scholar]
- a Goryanova B.; Goldman L. M.; Amyes T. L.; Gerlt J. A.; Richard J. P. Biochemistry 2013, 52, 7500–7511. 10.1021/bi401117y. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Goryanova B.; Amyes T. L.; Gerlt J. A.; Richard J. P. J. Am. Chem. Soc. 2011, 133, 6545–6548. 10.1021/ja201734z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryanova B.; Goldman L. M.; Ming S.; Amyes T. L.; Gerlt J. A.; Richard J. P. Biochemistry 2015, 54, 4555–4564. 10.1021/acs.biochem.5b00591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshel A. J. Biol. Chem. 1998, 273, 27035–27038. 10.1074/jbc.273.42.27035. [DOI] [PubMed] [Google Scholar]
- a Miller B. G.; Hassell A. M.; Wolfenden R.; Milburn M. V.; Short S. A. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 2011–2016. 10.1073/pnas.030409797. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ou X.; Ji C.; Han X.; Zhao X.; Li X.; Mao Y.; Wong L.-L.; Bartlam M.; Rao Z. J. Mol. Biol. 2006, 357, 858–869. 10.1016/j.jmb.2005.12.074. [DOI] [PubMed] [Google Scholar]; c Davenport R. C.; Bash P. A.; Seaton B. A.; Karplus M.; Petsko G. A.; Ringe D. Biochemistry 1991, 30, 5821–6. 10.1021/bi00238a002. [DOI] [PubMed] [Google Scholar]; d Kholodar S. A.; Allen C. L.; Gulick A. M.; Murkin A. S. J. Am. Chem. Soc. 2015, 137, 2748–2756. 10.1021/ja512911f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabanan M. M.; Amyes T. L.; Richard J. P. Curr. Opin. Struct. Biol. 2010, 20, 702–710. 10.1016/j.sbi.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.