

Abstract
Hematopoietic stem cell transplant (HSCT) treats or cures a variety of hematological and inherited disorders. Unfortunately, patients that undergo HSCT are susceptible to infections by a wide array of opportunistic pathogens. Pseudomonas aeruginosa bacteria can have life-threatening effects in HSCT patients by causing lung pathology that has been linked to high levels of the potent pro-inflammatory cytokine, interleukin-1β (IL-1β). Using a murine bone marrow transplant (BMT) model, we show overexpression of prostaglandin E2 (PGE2) post-BMT signals via EP2 or EP4 to induce cyclic adenosine monophosphate (cAMP) which activates protein kinase A (PKA) or the exchange protein activated by cAMP (Epac) to induce cAMP response element binding (CREB)-dependent transcription of IL-1β leading to exacerbated lung injury in BMT mice. Induction of IL-1β by PGE2 is time and dose dependent. Interestingly, IL-1β processing post-P. aeruginosa infection occurs via the enzymatic activity of either caspase-1 or caspase-8. Furthermore, PGE2 can limit autophagy-mediated killing of P. aeruginosa in alveolar macrophages, yet, autophagy doesn't play a role in PGE2-mediated up-regulation of IL-1β. Reducing PGE2 levels with indomethacin improved bacterial clearance and reduced IL-1β-mediated acute lung injury (ALI) in P. aeruginosa-infected BMT mice.
Introduction
Hematopoietic stem cell transplant (HSCT) utilizes stem cells derived from bone marrow, umbilical cord blood, or peripheral blood to treat or cure a variety of hematological disorders1. This procedure has become a standard of care with more than 18,000 HSCT performed every year in the United States alone1. HSCT requires a conditioning regimen (e.g. total body irradiation and/or chemotherapy) to allow the patient's hematopoietic system to be replaced by donor cells. Depending on disease, the patient can undergo autologous (self-donation of stem cells) or allogeneic (stem cells from a Human Leukocyte Antigen (HLA)-matched donor) transplants. Unfortunately, patients that undergo HSCT become immunosuppressed and susceptible to infections by opportunistic pathogens2. Around 60% of HSCT patients develop pulmonary complications, many of them due to bacterial infections, which correlate with high mortality and morbidity1-3. Infections post-HSCT are independent of conditioning regimen and type of transplant as both autologous and allogenic patients are highly susceptible. Infections can take place in the pre-engraftment phase (before 30 days post-transplant) as well as after immune reconstitution (after 30 days post-transplant)3. Although infections can be caused by viral, bacterial, or fungal organisms, bacterial infections are increasingly problematic due to the rise in drug-resistant bacteria. Bacterial infections post-HSCT can occur in 51.3% of HSCT patients2. These infections, including infections by the Gram negative bacterial pathogen, Pseudomonas aeruginosa, cause life-threatening complications2.
P. aeruginosa is an opportunistic pathogen that normally infects immunocompromised individuals such as HSCT patients2, 4-7. It is a leading nosocomial pathogen, and it is the most frequently isolated Gram negative bacteria in the intensive care unit 8. This pathogen causes urinary tract infection, hospital-acquired pneumonia, and bacteremia in burn patients9. It is also the predominant cause of morbidity and mortality in Cystic Fibrosis patients. There has been a significant increase in research effort studying P. aeruginosa due to the difficulty of treating infected patients, as 26% of its isolates are resistant to antibiotics and disinfectants 8. Understanding how to modulate P. aeruginosa infections in an antibiotic-independent method is likely to have positive impacts on the outcome of infected patients. Although many antimicrobial pathways have been linked to P. aeruginosa clearance, induction of inflammasome activation and Interleukin 1β (IL-1β) secretion play pathogenic roles during P. aeruginosa infection5.
The inflammasome is a multi-protein complex expressed mainly in immune cells and activated by pathogenic stimuli. Its activation leads to secretion of two potent inflammatory cytokines, IL-1β and IL-18. Their secretion pathways are complex and partially unknown, but have been well established to require two signals. Signal one leads to up-regulation of immature pro-cytokines and is mediated by Toll-like receptor (TLR) stimulation by pathogen-associated molecular patterns (PAMPs). Gram negative bacteria like P. aeruginosa can trigger signal one by stimulation of TLR4 and TLR5 via lipopolysaccharide (LPS) and flagella, respectively10-12. Signal two can be triggered by a wide array of pathogenic stimuli that culminate in aggregation of inflammasome components (e.g. caspase-1 or NOD-like receptor proteins) and result in secretion of mature IL-1β and IL-18. P. aeruginosa can induce signal two via recognition of type III secretion proteins. Although a protective mechanism against many pathogens, overproduction of IL-1β has been associated with auto-inflammatory syndromes such as gout and periodic fever syndromes, such as Familial Mediterranean Fever and cryopyrin-associated periodic fever syndromes (CAPS)13. Therapies targeting IL-1β signaling have shown better outcomes in CAPS patients14. Moreover, asbestos and silica inhalation can cause IL-1β-dependent pulmonary fibrosis mediated by alveolar macrophage (AMs)15. In the lung, P. aeruginosa can induce IL-1β secretion by AMs5. Interestingly, depleting AMs prior to P. aeruginosa infection leads to significantly lower levels of IL-1β in the lung, improving survival5. Apart from IL-1β secretion, P. aeruginosa infection has also been shown to be regulated by prostaglandin E2 (PGE2)6, 7, 16.
PGE2 is a lipid mediator derived from arachidonic acid by the enzymatic activity of cyclooxygenase (COX) and PGE synthases that signals through 4 different G-protein-coupled plasma membrane receptors (GPCRs) of the E-prostanoid (EP) family termed EP1, EP2, EP3, and EP4. Each receptor activates different intracellular pathways. Stimulation of EP1 receptor increases intracellular calcium and activation of protein kinase C which stimulates the transcription factors, NFAT and NFκB 17. EP2 and EP4 receptors are stimulators of adenylyl cyclase (AC) and phosphoinositide 3-kinase (PI3K), respectively. AC mediates conversion of ATP to cyclic adenosine monophosphate (cAMP) leading to activation of protein kinase A (PKA) and the transcription factor CREB. The EP3 receptor is a regulator of the EP2-EP4 signaling pathway as its activation leads to inhibition of AC. P. aeruginosa infection increases levels of inducible COX-2 leading to high levels of PGE2 and inhibition of COX2, with subsequent diminished production of PGE2, can lead to a better outcome in P. aeruginosa-infected mice 7. Interestingly, HSCT patients possess higher levels of PGE2 in blood and bronchoalveolar lavage (BAL) when compared to healthy non-transplanted individuals18, 19.
PGE2 is dysregulated in HSCT patients18 and suppression of PGE2 confers protection against P. aeruginosa infection in murine HSCT models 6. However, other roles of PGE2 in the context of HSCT and pulmonary P. aeruginosa infection remain elusive. To study bacterial lung infections post-HSCT, we use a mouse model of syngeneic bone marrow transplant (BMT). Total body irradiation is used as a conditioning regimen6, 20-22. Experiments are performed in the post-engraftment period, 5 weeks post-BMT when lung leukocytes are composed of 82% donor cells and splenic leukocytes are 95% donor cells20. We previously reported BMT mice are deficient in phagocytosis and killing of P. aeruginosa6, 23, correlating with observations in humans2. In the present study, we compare the cytokine profile and lung tissue injury of control and BMT mice post P. aeruginosa infection. We also tested the direct relationship of PGE2 with P. aeruginosa-induced IL-1β and examined the effect that PGE2 stimulation had on autophagy, a main mechanism of P. aeruginosa clearance by AMs that has also been linked to inflammasome regulation24,25.
Results
Bone Marrow Transplant (BMT) mice are deficient in clearing P. aeruginosa infection and experience exacerbated lung tissue injury
BMT mice are deficient in clearing a P. aeruginosa PAO-1 infection compared to healthy control mice 24 hours post-infection (Fig.1A) confirming published data6. Deficiency in clearing bacteria is not due to low numbers of immune cells in the alveolar compartment as there are no differences in cell numbers or percentages of monocyte/macrophages and lymphocytes in the bronchoalveolar lavage (Supplemental Fig.1A, B) between groups. We also noted higher levels of albumin in the bronchoalveolar lavage fluid (BALF) of BMT mice compared to control mice (Fig. 1b) suggesting more severe pulmonary injury (Fig.1c). Acute lung injury (ALI) is a leading cause of death in the intensive care unit, characterized by accumulation of leukocytes, protein leakage, and epithelial injury. ALI has been linked to high levels of IL-1β in the lung 26. Thus, we measured levels of several pro-inflammatory cytokines. We found higher levels of IL-1β, but not other pro-inflammatory cytokines (IL-6, IL-12, TNF-α; data not shown) in BALF from infected BMT mice compared to control mice (Fig.2A). We tested levels of PGE2 in BALF, and detected higher levels of PGE2 in BMT mice infected with PA01 compared to infected control mice. Moreover, we noticed mice expressing higher levels of PGE2 have direct and significant correlations between levels of IL-1β and tissue injury, but not with other cytokines such as TNF-α (Fig.2C-F). Previous research has suggested a role for PGE2 in IL-1β induction27, 28. Thus, exacerbated levels of IL-1β post-P. aeruginosa infection might mediate lung tissue injury and be dependent on PGE2. Additionally, P. aeruginosa-mediated IL-1β release has been shown to be dependent on alveolar macrophages (AMs) 5, 26. Thus we directed our attention to AMs in BMT mice.
Fig 1. BMT Mice are Deficient in Clearing Pseudomonas aeruginosa Infection and Experience Exacerbated Lung Tissue Injury.

(a) Colony Forming Units (CFUs) were counted 24 hours after infecting non-transplanted control and BMT mice (C57BL/6J→C57BL/6J) with 5×105 CFUs of Pseudomonas aeruginosa (PA01). (n=5 control; n=5 BMT). (b) Albumin measurements from bronchoalveolar lavage fluid (BALF) from PA01 infected control and BMT mice. (n=7 control; n=6 BMT) (c) Hematoxylin and Eosin stain (H&E) of lungs from saline or PA01 infected control and BMT mice; images taken at 40× magnification (representative of n=3 control; n=3 BMT). Statistics are student T test between comparative groups. *P<0.05. Data is representative of at least two independent experiments.
Fig 2. Pseudomonas aeruginosa Infection Induces Higher Levels of IL-1β and PGE2 post-BMT Correlating with Increased Lung Injury.
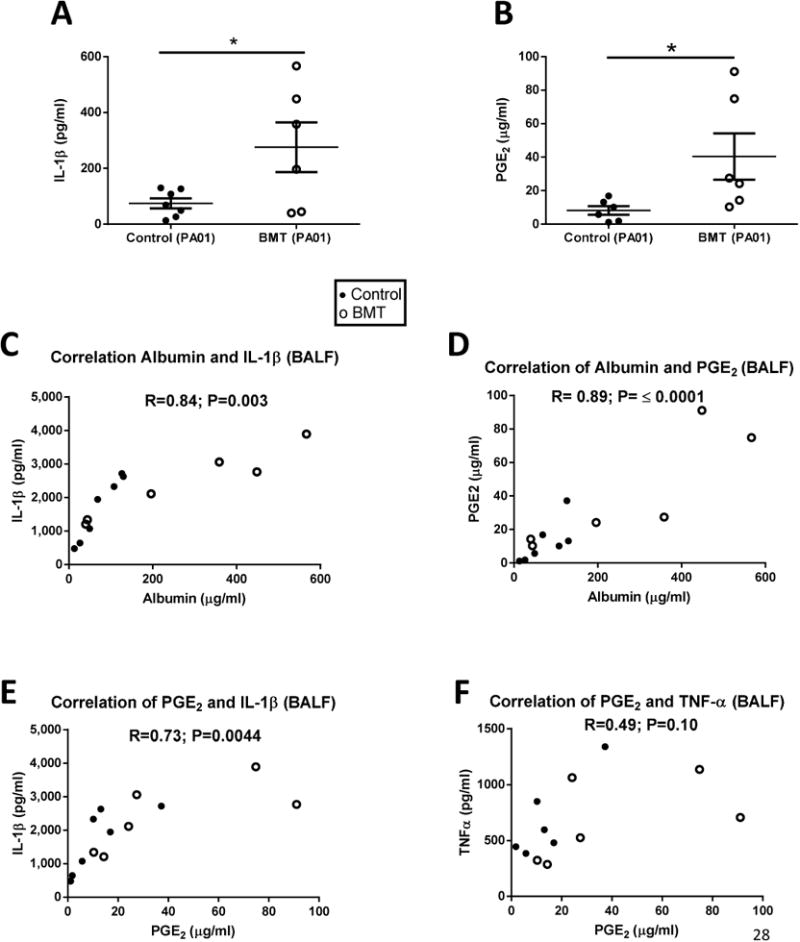
(a) IL-1β and (b) Prostaglandin E2 (PGE2) measurements from the BALF of control and BMT mice 24 hours after infection with PA01; measurements done by ELISA. (n=7 control; n=6 BMT); (c) Correlation between albumin and IL-1β in BALF 24h post-PAO1; (d) Correlation between albumin and PGE2 in BALF 24h post-PAO1; (e) Correlation between PGE2 and IL-1β in BALF 24h post-PAO1 ; (f) Correlation between PGE2 and TNF-α in BALF 24h post-PAO1. In all correlations, closed symbols represent control mice whereas open circles are BMT mice. R= Pearson correlation coefficient; *P<0.05.
Pro-inflammatory AMs in BMT mice account for higher levels of IL-1β in response to P.aeruginosa infection
To determine sources of IL-1β and PGE2 in the lung, we compared mRNA levels from AMs obtained by BALF to lung interstitium. Transcripts for the rate limiting enzymes in PGE2 synthesis, cyclooxygenase (COX) 1 and 2, were higher in AMs from BMT mice compared to control (Fig.3A). This was unique to AMs, but not interstitial samples. In addition, AMs from BMT mice have higher levels of IL-1β transcripts compared to control cells (Fig.3A). Consistent with our past studies, overnight culture of AMs from BMT mice secreted higher levels of PGE2 compared to cells from control mice (Fig.3B). To assess whether PGE2 can have an effect on IL-1β secretion, we pre-treated AMs with or without PGE2 prior to PA01 infection, in vitro. We detected higher levels of IL-1β in supernatants of AMs pretreated with PGE2 and infected with PAO1 compared to AMs infected with PA01 alone (Fig.3C). Moreover, to determine whether higher bacterial burden in BMT mice was responsible for higher levels of IL-1β, we induced acute lung injury with lipopolysaccharide instillation in control and BMT mice. We detected higher IL-1β, but not IL-6 or TNF-α in BALF from BMT mice compared to control mice post-LPS instillation (Fig.3 D-F).
Fig 3. Alveolar Macrophages in BMT Mice Account for Higher IL-1β Release post-P.aeruginosa in Response to PGE2.
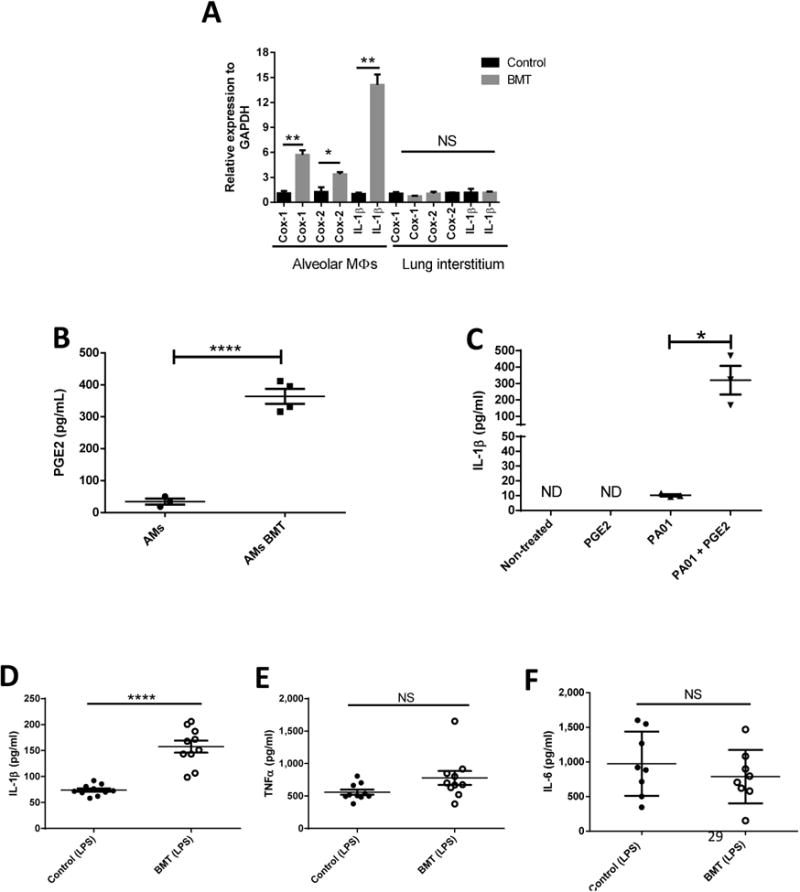
(a) RTqPCR measurement of relative gene expression of Cox-1, Cox-2, and IL-1β from AMs and interstitial lung from uninfected control and BMT mice normalized to GAPDH (n=3 control; n=3 BMT/group). (b) PGE2 measurements by ELISA from overnight culture of untreated AMs from control and BMT mice (n=4 Control; n= 4 BMT). (c) IL-1β measurements by ELISA from AMs infected or not in vitro with PA01 (MOI:10), treated or not with 100nM of PGE2 (n=3). (d) IL-1β, (e) TNF-α and (f) IL-6 measurements by ELISA from BALF of LPS (50ug)-treated control and BMT mice (n=10 control, n=10 BMT). Statistics are student T test between comparative groups. *P<0.05 ,**P<0.01, ***P<0.001, ****P<0.0001. Data is representative of at least two independent experiments.
PGE2 induces higher levels of IL-1β upon pathogenic stimuli
To assess whether PGE2 could induce IL-1β in macrophages other than AMs, we prepared bone marrow derived macrophages (BMDMs) from healthy mice and pretreated them with PGE2 prior to PA01 infection, in vitro. Similar to AMs, we detected higher IL-1β, but not IL-10, in supernatants of PGE2- stimulated cells compared to non-treated cells post-PA01 (Fig.4A-B). In addition, to detect whether PGE2-mediated higher levels of IL-1β with different stimuli; we pretreated BMDMs with and without PGE2 and stimulated cells with LPS, and/or heat-killed PA01. In all cases, PGE2 pretreatment increased levels of IL-1β compared to non-treated cells (Fig.4C). A canonical method to achieve IL-1β secretion is to activate the NLRP3 inflammasome by stimulation of the pannexin-1 channel and the purinergic P2X7 by LPS priming and adenosine triphosphate (ATP) treatment25, 28-30. We also detected higher IL-1β in BMDMs pretreated with PGE2 and treated with LPS and ATP when compared to treatment with LPS and ATP alone (Fig.4D). Furthermore, we noted higher levels of IL-1β in BMDMs pre-treated with PGE2 and infected with methicillin-resistant Staphylococcus aureus (MRSA) or Streptococcus pneumoniae compared to infected BMDMs not treated with PGE2(Supplemental Fig.2B). MRSA and S.pneumoniae are also main pathogens that infect immunosuppressed individuals6, 19, 31, 32.
Fig 4. PGE2 Increases IL-1β in Bone Marrow Derived Macrophages (BMDMs) Upon Pathogenic Stimulation.

(a-b) IL-1β and IL-10 measurements from supernatants of PA01 infected (MOI:10) BMDMs treated or not with 100nM PGE2. (c) IL-1β measurements from supernatant of BMDMs treated with lipopolysaccharide (LPS) derived from P. aeruginosa (500ng/ml), heat killed PA01 (MOI:10), and (c) ATP (1mM) with LPS with or without 100nM of PGE2. Supernatant taken 2 hours post-stimulation. One-way ANOVA with Bonferroni's post-test. *P<0.05 ,**P<0.01, ***P<0.001, ****P<0.0001. Data is representative of three independent experiments.
PGE2-mediated increase in IL-1β is dependent on activation of transcription factor CREB via increased levels of cAMP dependent on EP2 and EP4 signaling
We next probed PGE2 signaling pathways. We stimulated PGE2 receptors using pharmacologic agonists for the EP2, EP3, and EP4 receptors17. A selective EP1 agonist is not available. We found pre-stimulating BMDMs with EP2 and EP4 agonists lead to higher levels of IL-1β post-P.aeruginosa infection (Fig.5A). EP3 stimulation did not lead to higher IL-1β (data not shown). EP2 and EP4 receptors activate adenylate cyclase (AC) and increase cyclic adenosine monophosphate (cAMP) from cytosolic ATP. We pretreated BMDMs with the AC stimulator forskolin prior to PA01 infection and observed higher levels of IL-1β in forskolin-pretreated BMDMs (Fig.5B). Moreover, as increased levels of cAMP lead to the activation of protein kinase A (PKA) and Epac (exchange protein directly activated by cAMP), we used agonists for the activation of these two proteins 33. We found higher levels of IL-1β in supernatant of BMDMs pre-stimulated with PKA and Epac agonists prior to PA01 infection (Fig.5C). PKA and Epac activation lead to activation of the transcription factor CREB, thus, we used a CREB inhibitor during PGE2 stimulation prior to PA01 infection. We decreased the levels of PGE2 mediated-IL-1β using the CREB inhibitor (Fig.5D). Thus, PGE2 increases IL-1β by activation of the transcription factor CREB via stimulation of EP2 and/or EP4 receptors.
Fig 5. PGE2 Mediated Increase in IL-1β is Dependent on Activation of Transcription Factor CREB by Increasing Levels of cAMP Dependent on EP2 and EP4 Signaling.
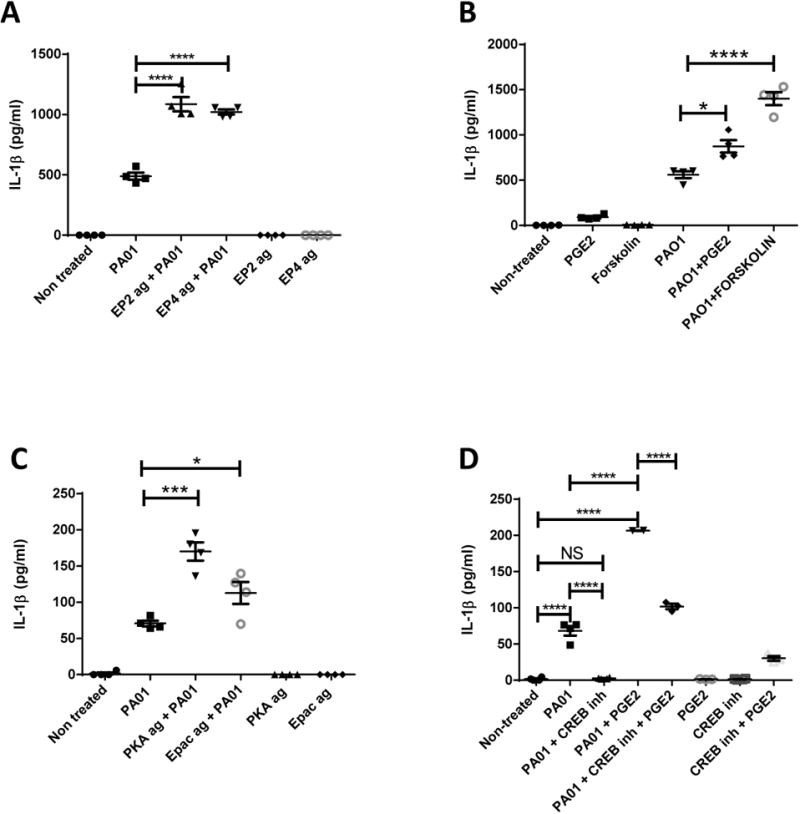
IL-1β measurements from supernatant of BMDMs treated or not with PAO1 (MOI:10) with or without (a) EP2 agonist (1μM, Butaprost), EP4 agonist (500nM, ONO-AE1-329) (b) forskolin (25μM), PGE2 (100nM), (c) PKA agonist (50μM, 6-BNZ-cAMP), Epac agonist (50μM, 8-pcpt-2′-OM-cAMP). (d) IL-1β protein measurement from supernatants of BMDMs treated or not with 100nM of PGE2, CREB inhibitor (100μM, Naphthol AS-E phosphate) and infected or not with PA01 (MOI:10). In all cases, supernatant taken 2 hours after infection. One-way ANOVA with Bonferroni's post-test. *P<0.05 ,**P<0.01, ***P<0.001, ****P<0.0001. Data is representative of two independent experiments.
PGE2-dependent IL-1β release post-P. aeruginosa infection can be mediated by canonical or non-cannonical inflammasomes and is independent of autophagy inhibition
Mycobacterium tuberculosis and Candida albicans have been shown to induce IL-1β by a non-canonical inflammasome pathway dependent on caspase-8 in macrophages34, 35. Although a role of caspase-8-mediated IL-1β processing post-P.aeruginosa infection hasn't been proposed, research has shown that P.aeruginosa-infected caspase-1 deficient mice can secrete normal levels of IL-1β compared to control mice36. Thus, other proteases might play a role in processing pro-IL-1β post- P.aeruginosa infection. We stimulated macrophages with or without caspase-1 and caspase-8 inhibitors. Interestingly, in both cases, IL-1β secretion was inhibited with or without the effects of PGE2 (Fig.6A-B). Stimulating macrophages with LPS and ATP in the presence or absence of caspase-1 and/or caspase-8 inhibitors confirmed the specificity of these inhibitors (Supplemental Fig. 3). AMs can clear P.aeruginosa infection by autophagy and IL-1β release can be negatively regulated by autophagy24, 25. Autophagy can be induced by serum starvation; thus, we tested effects of PGE2 on autophagy-enhanced clearance of P. aeruginosa carried out under conditions of serum deprivation. Our results indicate PGE2 can inhibit autophagy-dependent clearance in AMs (Fig.7A). To detect whether there is a direct effect of PGE2 on autophagy, we treated BMDMs with different concentrations of PGE2 and detected levels of the autophagy-related proteins, LC3 and P62. We detected downregulation of LC3 and accumulation of P62 when we treated AMs with PGE2. These changes are characteristic of autophagy inhibition (Fig.7B and Supplemental Fig. 4). In addition, we induced autophagy by serum starvation with or without PGE2 stimulation and determined that PGE2 inhibits levels of the autophagy-related protein, ATG5, by western blot and RTqPCR (Fig.7C). Although we found PGE2 inhibits autophagy, autophagy-deficient BMDMs were still able to upregulate IL-1β release when PGE2 was present, suggesting PGE2 inhibition of autophagy is not required for processing and upregulation of IL-1β (Fig.7D).
Fig 6. PAO1 can use cannonical or non-cannonical inflammasomes to make IL-1β.
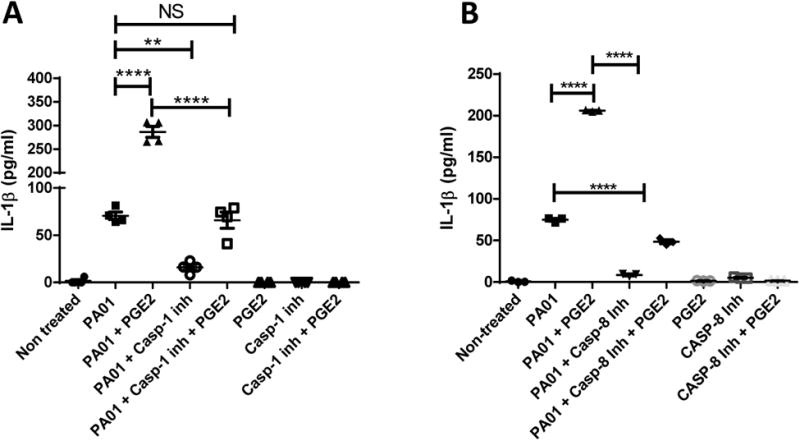
IL-1β measurements from supernatant of BMDMs treated or not with (a) Caspase 8 inhibitor (10nM) or Caspase 1 inhibitor (10nM) with or without PGE2 (100nM). Supernatant taken 2 hours after infection. One-way ANOVA with Bonferroni's post-test. *P<0.05 ,**P<0.01, ***P<0.001, ****P<0.0001. Data is representative of two independent experiments.
Fig 7. Inhibition of Autophagy Impacts Bacterial Killing but Not IL-1β Release.
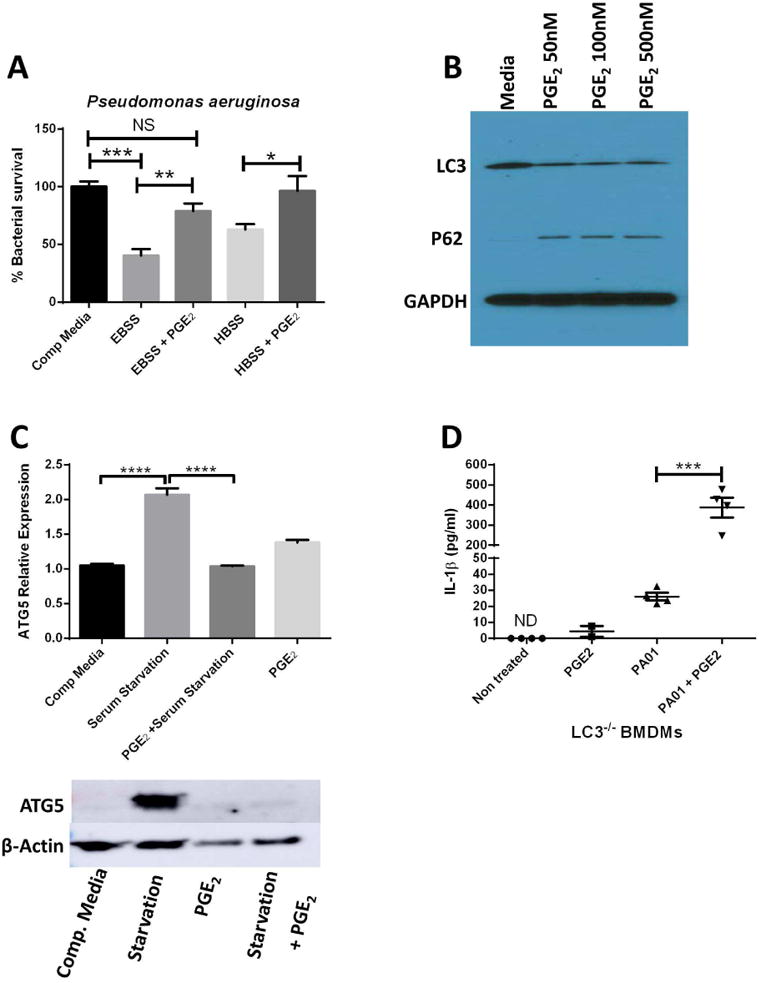
(a) AMs were cultured in complete media or were nutrient starved in Earle's Balanced Salt Solution (EBSS) (#1) or Hanks' Balanced Salt solution (HBSS) (#2) in the presence or absence of PGE2 (100nM) for 2 hours. (b) AMs were collected from control mice and stimulated for 1 hour in the presence of serum-free media alone with or without a dose response of PGE2 added (50-500 nM) for 1h. Cell lysates were then analyzed for levels of LC3 and p62 by Western blot compared to GAPDH. Blot shown is representative of 3 experiments. (c) AMs were subjected to culture in complete media or were serum-starved for 1 h in the presence or absence of 100nM PGE2 before RNA was prepared and analyzed for expression of the autophagy gene ATG5 relative to β-actin by qRT-PCR (n=4); (bottom) cell lysates were also taken from samples assessed for levels of ATG5 and β-actin by Western blot.(d) BMDMs from autophagy-deficient LC3-/- mice were stimulated with 100nM PGE2, PAO1 (MOI:10) or the combination for 2 hours before supernatants were collected and measured for IL-1β by ELISA (n=4); Data are representative of two independent experiments and statistics were measured by one-way ANOVA with Bonferroni post-hoc test. *P<0.05 ,**P<0.01, ***P<0.001, ****P<0.0001.
PGE2 elevated the levels of IL-1β transcripts by EP2 and EP4 stimulation in human and mouse cells
Considering that neither autophagy inhibition nor inflammasome activation explain why PGE2 boosted IL-1β release; we next studied the transcriptional effects of PGE2 on IL-1β. We treated BMDMs with PGE2 in a dose and time dependent manner and detected significant increases in IL-1β transcripts as soon as 2 hours post-PGE2 stimulation (Fig.8A-B). Furthermore, we confirmed elevated levels of IL-1β transcripts by PGE2 stimulation are dependent on increasing levels of cAMP by stimulation of EP2 and/or EP4 receptors but not by EP3 stimulation (Fig.8C). Moreover, we confirmed our findings in human peripheral monocytes and human AMs (Fig.9).
Fig 8. PGE2 Elevates the Levels of IL-1β Transcripts by EP2 and EP4 Stimulation.
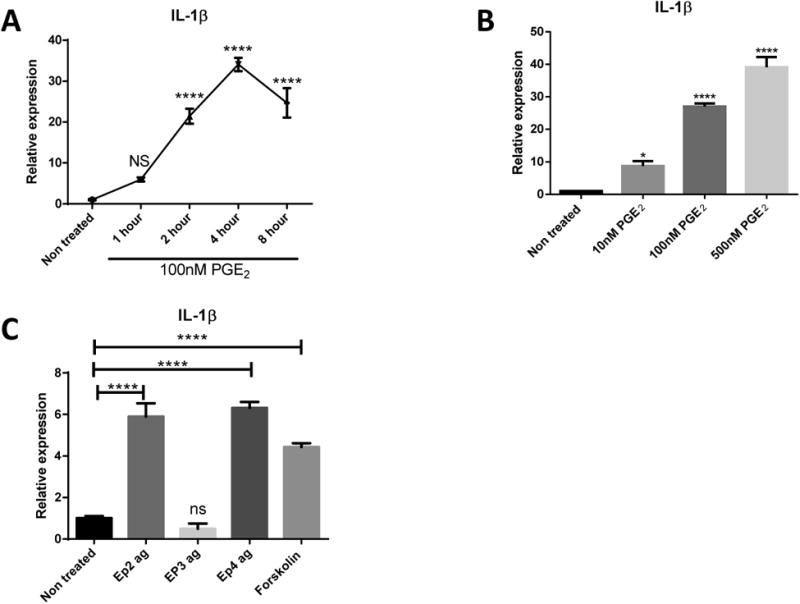
(a) BMDMs were treated or not with 100nM PGE2 for 1, 2, 4, and 8 hours before RNA was prepared and analyzed for expression of the IL-1β gene. (b) BMDMs were treated or not with 10nM, 100nM, and 500nM of PGE2 during 4 hours before RNA was prepared and analyzed for expression of IL-1β. (c) BMDMs were treated with EP2 agonist (1μM, Butaprost), EP3 agonist (10nM, Sulprostone), EP4 agonist (500nM, ONO-AE1-329), and forskolin (25μM) before RNA was prepared and analyzed. All RNA data was normalized to expression levels of GAPDH. Data are representative of two independent experiments and statistics were measured by one-way ANOVA with Bonferroni post-hoc test. *P<0.05 ,**P<0.01, ***P<0.001, ****P<0.0001.
Fig 9. Human AMs and Peripheral Monocytes Upregulate IL-1β under PGE2 stimulation.
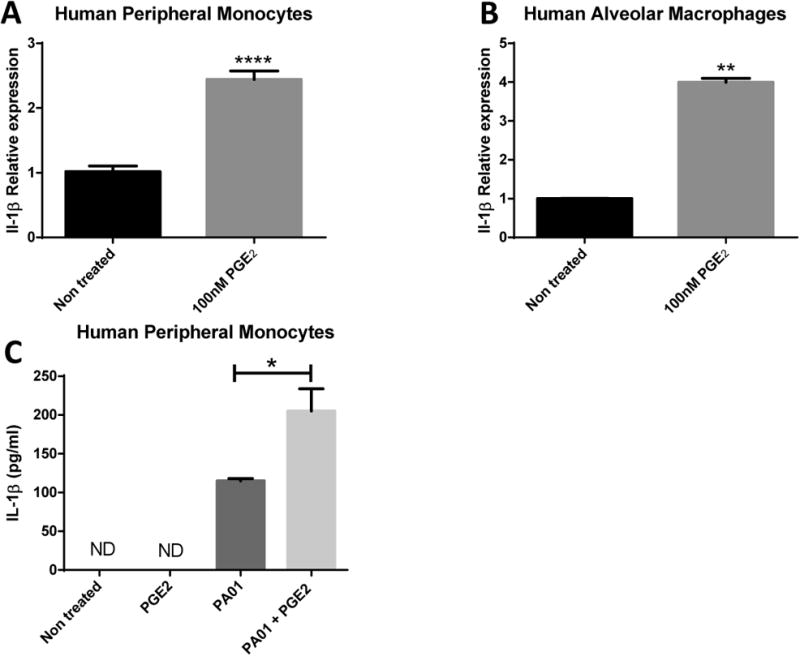
IL-1β relative expression from 100nM PGE2 stimulated (a) Human peripheral monocytes and (b) human AMs normalized to GAPDH. (c) IL-1β protein measurements from supernatant of PGE2-treated or not peripheral macrophages during or not PA01 infection. Statistics are student T test between comparative groups. *P<0.05 ,**P<0.01, ***P<0.001, ****P<0.0001.
Decreasing levels of PGE2 reduces P. aeruginosa-mediated lung tissue injury
Increased levels of IL-1β post-P.aeruginosa infection aggravate lung tissue injury4, 5. Therapeutic strategies using caspase-1 inhibitors reduced severity of IL-1β pulmonary injury 26. However, other research has shown that inhibiting IL-1β signaling has no impact on bacterial burden or immune cell recruitment, with minimal effects on lung injury36. As we have shown that PGE2 elevation in BMT mice aggravates lung injury, we tested the effect that PGE2 inhibition had on lung injury outcomes. We instilled P. aeruginosa to control and BMT mice with or without administration of the COX inhibitor, indomethacin. We noted reduced IL-1β in the BALF as well as lower levels of protein leakage, suggesting inhibition of PGE2 can decrease levels of ALI post-P.aeruginosa infection (Fig.10).
Fig 10. Decreasing Levels of PGE2 by Indomethacin Treatment Leads to Decreased IL-1β in the Lung after P. aeruginosa Infection in BMT mice.
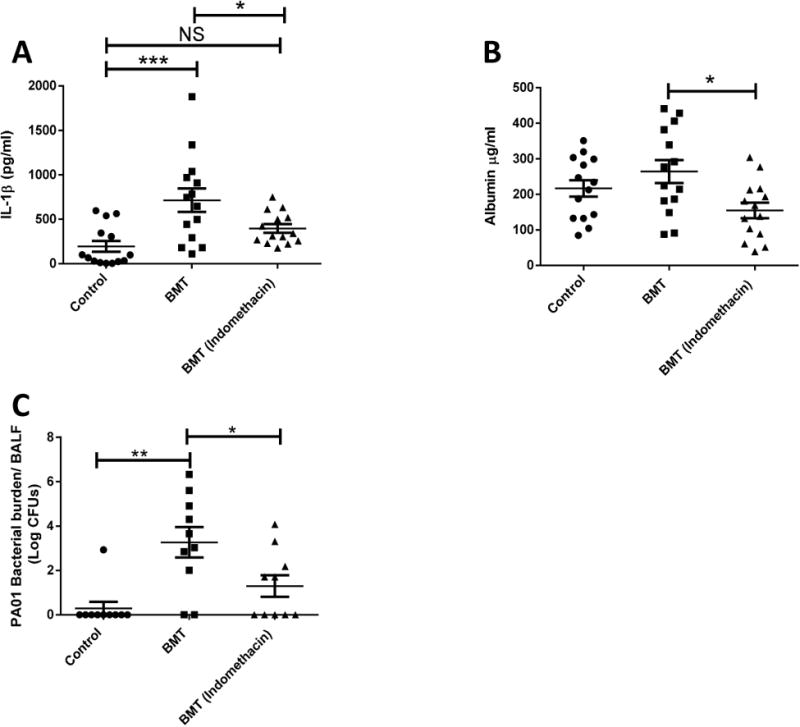
(a) IL-1β and (b) albumin measurements from the BALF of control, and BMT mice treated or not with Indomethacin (1.2mg/kg) for 24 hours after PA01 infection; measurements done by ELISA. (n=14 control; n=14 BMT; n=14 BMT treated with Indomethacin); (C) PA01 CFU measurement in BAL from infected mice, 24 hours. (n=10 control; n=10 BMT; n=10 BMT treated with Indomethacin). Data is representative of at least two independent experiments. Statistics were measured by one-way ANOVA with Bonferroni post-hoc test. *P<0.05 ,**P<0.01, ***P<0.001, ****P<0.0001; non-significant (ns).
Discussion
In February 2017, the World Health Organization (WHO) published a report containing a list of 12 bacterial pathogens for which new therapeutic strategies are urgently needed (http://www.who.int/medicines/publications/global-priority-list-antibiotic-resistant-bacteria/en/). Pseudomonas aeruginosa was considered to be of critical importance. P. aeruginosa is an opportunistic pathogen which causes minimal pathogenicity in healthy individuals, but major mortality and morbidity in HSCT patients 2, 6, 19, 31, 37, 38. Susceptibility to this pathogen has been reported to occur even after immune reconstitution following HSCT2, 6, 31. HSCT patients have high levels of PGE2 in the blood and BALF when compared to healthy individuals18-20. We have previously reported that elevated PGE2 is causally related to the impaired ability of AMs from BMT mice to phagocytize and kill bacteria 20, 23, 39.
The purpose of the current study was to determine whether PGE2 production post-BMT was responsible for enhanced lung injury post-infection as well, and if so, by what mechanism. We now know that inflammasome-dependent IL-1β secretion is induced by P. aeruginosa, and better health outcomes have been reported by inhibiting IL-1β signaling4, 5, 12, 26, 36. In addition, recent articles have linked PGE2 signaling with IL-1β regulation27, 28, 40. However, reports have shown contradictory effects of PGE2 on IL-1β release27, 28. Thus, in this study we focused on the PGE2-mediated effects on IL-1β response to P. aeruginosa infection by AMs from BMT mice in our quest to determine new therapeutic strategies.
A dose of 2 × 106 CFU P. aeruginosa instilled into un-transplanted mice causes a moderate infection 41. Yet, BMT mice have difficulty clearing this bacterial dose and 100% of them die within 48 hours23. Interestingly, the susceptibility to P. aeruginosa in BMT mice is seen even after immune reconstitution6, 23, 42. Here, we show evidence that BMT mice have severe vascular and epithelial leakage indicating a more severe ALI post-P.aeruginosa infection. ALI can be caused by exacerbated levels of pro-inflammatory cytokines. Thus, we searched for upregulated levels of different pro-inflammatory cytokines (IL-12, IL-6, TNF-α, and IL-1β) and found that only IL-1β was significantly higher in BMT mice when compared to un-transplanted mice post-infection. In addition, we noticed a direct correlation between IL-1β, PGE2, and albumin levels in the BALF, but not a correlation with other pro-inflammatory cytokines such as TNF-α.
Because we observed exacerbated levels of IL-1β post-P.aeruginosa infection in BMT mice, we analyzed the capability of AMs from BMT mice to secrete IL-1β. Interestingly, AMs from BMT mice have higher levels of IL-1β transcripts compared to control mice, correlating with higher levels of cyclooxygenase 1 and 2 transcripts. Furthermore, stimulating AMs with PGE2 prior to infection with P. aeruginosa also leads to higher levels of IL-1β compared to untreated AMs. These findings show that higher levels of PGE2 in AMs from BMT mice might influence the higher levels of IL-1β post-P.aeruginosa.
As higher amounts of bacterial burden in BMT mice might influence the levels of IL-1β, we adopted a bacterial-free model of ALI dependent on lipopolysaccharide instillation 43. We were able to get exacerbated levels of IL-1β, but not IL-6 and TNF-α, in BMT mice compared to un-transplanted mice. In addition, in an in-vitro model where we controlled the stimulants, we detected that PGE2 could increase secretion of IL-1β in bone marrow derived macrophages by a wide array of pathogenic stimuli such as LPS, live and heat killed P. aeruginosa, MRSA infection, and Streptococcus pneumoniae infection. All stimuli induce increased IL-1β after previous exposure to PGE2 in macrophages. Thus, PGE2 signaling acts as signal 1 for IL-1β secretion. Interestingly, dual stimulation of macrophages with PGE2 and another signal-1 stimulant, LPS, can lead to increased IL-1β secretion. This phenomenon is likely explained by the moderate activation of caspase-1 (signal 2) in macrophages by LPS stimulation as previously noted 44 .
Recent reports have shown an opposite effect of PGE2 regulation on IL-1β27 than our results. We compared their PGE2 stimulation method, based on 5 minutes of stimulation by PGE2, side by side with our stimulation method, based on 4 hours pre-stimulation with PGE2. As expected, we were able to obtain exacerbated levels of IL-1β post-4 hours of PGE2 stimulation whereas 5 minutes of stimulation had no effect on IL-1β regulation (Supplemental Fig. 5). Thus, these comparative results suggest that the kinetics of PGE2 exposure is important. However, in the setting of HCST, it is important to remember that PGE2 levels are chronically elevated18, 19, and thus, in this setting PGE2 is promoting IL-1β.
PGE2 signaling is mediated by 4 members of the G-coupled protein receptor family termed EP1-EP417. We determined that stimulating the EP2 and EP4 receptor in macrophages prior to P. aeruginosa infection leads to higher levels of IL-1β when compared to non-stimulated macrophages. Furthermore, as EP2/EP4 receptors share the ability to activate PKA and Epac, we activated PKA or Epac with the use of intracellular agonists prior to P. aeruginosa infection and detected higher levels of IL-1β when compared to non-stimulated macrophages. We were able to abolish IL-1β secretion in P. aeruginosa-infected mice by stimulating macrophages with an inhibitor for the CREB transcription factor. These data indicate that the PGE2-mediated increase in IL-1β is due to CREB transcription factor activation by EP2/EP4-mediated stimulation of PKA and/or Epac. Although we present evidence that CREB is mediating PGE2-dependent elevation of IL-1β, we do not discard the possibility that other transcription factors such as NF-κB 28 may also play a role. Furthermore, while both EP2 and EP4 can mediate the transcriptional effect of PGE2 in macrophages from control mice, in the setting of BMT, we have previously shown that levels of EP2 are upregulated, while EP4 is slightly downregulated on alveolar macrophages and an EP2 antagonist, AH6809 was able to mimic effects of indomethacin on alveolar macrophage phagocytosis39.
Researchers have established autophagy as a main mechanism of P. aeruginosa clearance 24 as well as IL-1β regulation10, 25. We tested the effects of PGE2 in the regulation of autophagy and determined that PGE2 can inhibit autophagy-induced clearance of P. aeruginosa. Therefore, these data suggested that PGE2-mediated IL-1β increase might be due to autophagy inhibition. However, we detected high levels of IL-1β protein in autophagy-deficient macrophages after PGE2 administration and infection. Thus, the mechanism of PGE2-mediated increase in IL-1β production cannot be attributed to impaired clearance of inflammasome components as a result of defective autophagy 25. While we previously demonstrated that PGE2 was associated with impaired autophagy in neutrophils19, this is the first description of its ability to limit autophagy in macrophages. Thus, PGE2 may be a common negative regulator of autophagic flux in other cell types as well. Future work will be needed to understand what impact this negative regulation may have on processes such as epithelial repair.
In macrophages, IL-1β secretion can be mediated by the cysteine proteases, caspase-1 and caspase-835. Thus, we tested the role of these two caspases during P. aeruginosa infection. We determined that we could abolish the secretion of IL-1β prior to P. aeruginosa infection with the use of inhibitors to caspase-1 or caspase-8. To our knowledge, these are the first data to link IL-1β secretion post-P. aeruginosa to caspase-8; this pathway has been previously established for Mycobacterium tuberculosis and fungal infection 34.These results suggested that P. aeruginosa infection can induce the activation of IL-1β release using either caspase-1 and/or caspase-8. If true, this suggests that P. aeruginosa may be able to stimulate non-cannonical inflammasome activation, possibly via cross-reactivity caused by recognition of P. aeruginosa by dectin receptors 45 which are known to be linked to caspase 8 activation34 .
Our data suggest that PGE2 can influence a pro-inflammatory environment by exacerbating levels of IL-1β, in macrophages. We detected that PGE2 can mediate a massive increase in IL-1β transcripts. We detected increased IL-1β transcripts in macrophages within an hour post-stimulation with PGE2. Increases in IL-1β transcripts were dependent on stimulation of EP2 and EP4 receptors but not by EP3. While we did not have access to an EP1-selective agonist, we wouldn't anticipate this receptor to regulate cAMP levels17. Additionally, we were able to detect an increase in IL-1β transcripts following activation of cAMP with forskolin. Moreover, PGE2 stimulation increased transcription of IL-1β in human alveolar macrophages and human monocyte-derived macrophages. Altogether, we conclude that PGE2 can strongly prime macrophages for IL-1β, but not other cytokines, and upon pathogenic stimulation will lead to exacerbated levels of IL-1β causing IL-1β-mediated injury. Thus, we expected to decrease the IL-1β-mediated lung injury post-BMT with the use of COX inhibitors. When we tested the effects of PGE2 inhibition in BMT mice, we were able decrease bacterial load as previously reported7, but also reduce protein leakage and IL-1β in the lung. When our results are taken together, we identified new mechanisms by which P. aeruginosa causes life threatening effects in HSCT patients. These findings can help in the development of new therapeutic strategies that can improve outcome of HSCT patients with pulmonary complications due to P. aeruginosa, and possibly other pathogens. We speculate that COX inhibitors or possibly Anakinra may offer therapeutic benefit at limiting lung injury caused by bacterial infection post-HSCT.
Methods
Mice
Male C57BL/6J mice were purchased from Jackson Laboratories and used at 6-8 weeks of age. Experiments were approved by the University of Michigan Institutional Animal Care and Use Committee.
Reagents
PGE2 (1-1000nM; Cayman Chemicals, Ann Arbor, MI); Forskolin (25μM; Cayman Chemical, Ann Arbor, MI); Pseudomonas aeruginosa LPS (100ng/ml; Sigma-Aldrich, St. Louis, MO); ATP (1μM; Sigma-Aldrich, St. Louis, MO); Caspase-1 inhibitor (10nM Ac-YVAD-CHO; Enzo Life Sciences, Farmingdale, NY); Caspase-8 inhibitor (10nM Ac-IETD-CHO; BD Biosciences, San Jose, CA); CREB inhibitor (100μM Naphthol AS-E phosphate; Sigma-Aldrich, St. Louis, MO); EP2 agonist (1μM Butaprost; Cayman Chemical, Ann Arbor, MI), EP3 agonist (10nM Sulprostone; Cayman Chemical, Ann Arbor, MI) EP4 agonist (500nM ONO-AE1-329; Sigma-Aldrich, St. Louis, MO); Protein Kinase A (PKA) agonist (50μM 6-BNZ-cAMP; Biolog, Hayward, CA); Epac agonist (50μM 8-pcpt-2′-OM-cAMP; Biolog, Hayward, CA).
Cells
AMs were harvested by performing bronchoalveolar lavage (BAL) on C57BL/6J mice using 20mls of supplemented Dulbecco's Modified Eagle Medium (DMEM) (89% DMEM, 10% fetal bovine serum, 1% pen-strep, and 5mM EDTA) as described in22. To prepare RNA from interstitial cells, lungs were homogenized in TRIzol (Thermo Fisher Scientific; Waltham, MA) after BAL using a tissue homogenizer (OMNI International; cat# Th115). BMDMs were obtained by flushing bone marrow cells from the femur and tibia of C57BL/6J mice as explained in 46 . Briefly, bone marrow cells were incubated at 37°C in 5% CO2 for 7 days in BMDM differentiation media (59% Iscove's Modified Dulbecco's Medium (IMDM), 30% L-929 cell supernatant, 10% fetal calf serum (FCS), and 1% Pen-Strep). Human AMs were collected by ex-vivo BAL of human lungs that were not used for transplant. Human monocyte-derived macrophages were harvested by gradient centrifugation with the use of Ficoll-Paque Plus (GE healthcare) from peripheral blood of healthy donors followed by differentiation with human recombinant colony stimulation factor-1 (50ng/ml; R&D System, Minneapolis, MN) for 7 days.
Bacteria
Pseudomonas aeruginosa (PA01) and Staphylococcus aureus (US300) were grown in tryptic soy broth and nutrient broth, respectively, and incubated with gentle agitation overnight at 37°C. Streptococcus pneumoniae (serotype 3, 6303) was grown in Todd Hewitt Broth with 0.5% yeast extract and incubated overnight at 37°C and 5% CO2. CFU were determined by absorbance relative to known standard curves.
Bone Marrow Transplantation
Healthy 6-8 week old C57BL/6J male mice were lethally irradiated with a split dose of 13 grey (13-Gy) with the use of an X-rad 320 irradiator. Irradiated mice were infused with 5×106 bone marrow cells from a genetically identical donor (C57BL/6J). Mice were housed for 5 weeks after infusion to achieve full reconstitution of their immune system. The percentage of donor-derived cells was ∼95 ± 1% in the spleen and 82 ± 2% in the lung at this time point, as assessed by transplanting CD45.1+ bone marrow into C57BL/6J CD45.2+ mice as showed in 20.
Model of Infection
Pseudomonas aeruginosa infection was done by intratracheal inoculation of 5×105 CFU of PA01 in 50μl of saline solution. The control group received saline solution as placebo. Harvest was done 24 hours after infection; mice were first euthanized with CO2 asphyxiation and BAL was performed with 1ml of phosphate-buffered saline (PBS) containing 5mM EDTA, followed by lung perfusion and harvest. CFU measurements were obtained by serial dilution and plating on nutrient agar plates. LPS was given by intratracheal inoculation of 50μg P. aeruginosa-derived LPS in 40μl of saline solution.
Tetrazolium Dye Reduction Assay of Bacterial Killing
AMs from C57BL/6 mice or BMDMs were placed into duplicate 96-well plates: one experimental plate and one control plate. Cells from both plates were infected with IgG-opsonized P. aeruginosa (multiplicity of infection 50:1) for 30 min at 37°C. The cells on the experimental plate were washed and then incubated with or without treatments at 37°C for 120 min, whereas the cells on the control plate were washed and then lysed with 0.5% saponin in TSB (Sigma-Aldrich, St. Louis, MO) and placed at 4°C. After 2 hours, the cells from the experimental plate were lysed with 0.5% saponin in TSB. Both plates were then incubated in a shaker at 37°C for 4h. Thiazolyl blue Tetrazolium Bromide (MTT) assay was performed as recommended by the company. Briefly, a total of 5 mg/ml MTT (Sigma) was added to each plate and incubated for 30 min. Solubilization solution was added to dissolve formazan salts, and the absorbance was read at 595 nm (A595). Results were expressed as percentage of survival of ingested bacteria normalized to the percentage of control, where the A595 experiment values were divided by the average of the A595 control values. Survival of ingested bacteria = (A595 experimental plate/A595 control plate) × 100%.
ELISA, Protein Measurement, Immunofluorescence and Immunoblotting
Cytokine measurement was done with the use of R&D duoset ELISA kits for IL-1β, IL-10, and IL-6. Albumin measurements were done with the Bethyl laboratory mouse albumin ELISA kit. Protein quantification was achieved with the use of Pierce BCA assay kit from Thermo Fisher. Immunofluorescence images were taken with an Olympus 500 confocal microscope.
Histology
Hematoxylin and Eosin (H&E) stain was done after perfusion of the lung with PBS and tissue fixation with 10% formalin. Tissues were left in 10% formalin overnight before replacing fluid with 70% ethanol. Lung samples were processed and stained by McClinchey Histology Services (Stockbridge, MI).
Quantitative Real Time-PCR
mRNA was isolated using TRIzol according to manufacturer's instructions. Relative gene expression measurements were achieved with the use of a Step-one plus real time PCR system from Applied Biosystem. Gene specific primers and probes were designed with the GenScript Real-time PCR primer design software.
Statistical analysis
Graphpad Prism version 6 software was used to analyze experimental results. When groups of 2 were compared, student's t-test was used to determine statistical significance. Groups of 3 or more were compared using one way-ANOVA with Bonferroni multiple mean comparisons. Fisher exact test was used to compare clearance of bacteria and albumin levels between infected mice.
Supplementary Material
Supplemental Fig 1. Leukocytes in Bronchoalveolar Lavage Post-BMT (a) Absolute number of leukocytes in bronchoalveolar lavage fluid (BALF), quantified with a hemocytometer, (n=3). (b) Percentage of leukocytes from BALF, taken via differential staining, (n=4). Statistics done by student T test. Not significant (NS).
Supplemental Fig 2. PGE2 Enhances IL-1β Release Under Different Pathogenic Stimuli. (a) IL-1β measurements of supernatant from BMDMs treated or not with PGE2 (100nM) and infected or not with PA01 (MOI:10) for 30 minutes after which supernatant was replaced with complete media containing 1% gentamycin. (b) IL-1β measurements of supernatant after methicillin-resistant Staphylococcus aureus (MRSA) (MOI:10) or Streptococcus pneumoniae (MOI:10) infection with or without PGE2 (100nM). Samples were taken two hours after infection. Statistics used one-way ANOVA with Bonferroni's post-test. *P<0.05, **P<0.01, ***P<0.001. Data is representative of three independent experiments.
Supplemental Fig 3. Specificity of caspase-1 and caspase-8 inhibitors. IL-1β measurements by ELISA of supernatant samples from cultured BMDMs treated or not for 4 hours with LPS (100ng/ml) with or without caspase-8 inhibitor (10nM) or caspase-1 inhibitor (10nM) prior 45 minutes stimulation with/without ATP (1mM). One-way ANOVA with Bonferroni's post-test. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Supplemental Fig 4. PGE2 Limits Autophagosome Formation in Response to Serum Starvation. BMDMs were serum-starved for 1 h in the presence or absence of 100nM PGE2. Cells were fixed and stained with FITC-labeled LC3 or DAPI to visualize autophagosomes and nuclei and immunofluorescence images were captured singly or were merged. Panels shown are representative of 2 different experiments.
Supplemental Fig 5. PGE2 Enhances IL-1β Release after 4h, but not 5′ Stimulation. IL-1β measurements by ELISA of supernatant samples from BMDMs treated for 3.5 hours or 5 minutes with LPS (100ng/ml) and/or PGE2 (500nM) followed by 30 minutes of ATP (5mM). Statistics used one-way ANOVA with Bonferroni's post-test. *P<0.05, **P<0.01, ***P<0.001.
Acknowledgments
Supported by NIH grants: AI117229, HL127805 and HL119682 (BBM). GM-C was supported by T32AI007413 and a University of Michigan Rackham Merit fellowship. We thank Dr. Nick Lukacs (University of Michigan, Ann Arbor) for provision of LC3b-/- mice.
Footnotes
Authorship Contributions: GM-C and BBM designed the experiments and wrote the manuscript. Experiments were performed by GM-C, QMT, ABP and CAW. All authors approved the final version of the manuscript.
References
- 1.Gratwohl A, Baldomero H, Aljurf M, Pasquini MC, Bouzas LF, Yoshimi A, et al. Hematopoietic stem cell transplantation: a global perspective. JAMA. 2010;303(16):1617–1624. doi: 10.1001/jama.2010.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ullah K, Raza S, Ahmed P, Chaudhry QU, Satti TM, Ahmed S, et al. Post-transplant infections: single center experience from the developing world. Int J Infect Dis. 2008;12(2):203–214. doi: 10.1016/j.ijid.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 3.Danby R, Rocha V. Improving engraftment and immune reconstitution in umbilical cord blood transplantation. Front Immunol. 2014;5:68. doi: 10.3389/fimmu.2014.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wonnenberg B, Bischoff M, Beisswenger C, Dinh T, Bals R, Singh B, et al. The role of IL-1beta in Pseudomonas aeruginosa in lung infection. Cell Tissue Res. 2016;364(2):225–229. doi: 10.1007/s00441-016-2387-9. [DOI] [PubMed] [Google Scholar]
- 5.Cohen TS, Prince AS. Activation of inflammasome signaling mediates pathology of acute P. aeruginosa pneumonia J Clin Invest. 2013;123(4):1630–1637. doi: 10.1172/JCI66142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Domingo-Gonzalez R, Katz S, Serezani CH, Moore TA, Levine AM, Moore BB. Prostaglandin E2-induced changes in alveolar macrophage scavenger receptor profiles differentially alter phagocytosis of Pseudomonas aeruginosa and Staphylococcus aureus post-bone marrow transplant. Journal of immunology (Baltimore, Md : 1950) 2013;190(11):5809–5817. doi: 10.4049/jimmunol.1203274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sadikot RT, Zeng H, Azim AC, Joo M, Dey SK, Breyer RM, et al. Bacterial clearance of Pseudomonas aeruginosa is enhanced by the inhibition of COX-2. Eur J Immunol. 2007;37(4):1001–1009. doi: 10.1002/eji.200636636. [DOI] [PubMed] [Google Scholar]
- 8.Aloush V, Navon-Venezia S, Seigman-Igra Y, Cabili S, Carmeli Y. Multidrug-resistant Pseudomonas aeruginosa: risk factors and clinical impact. Antimicrob Agents Chemother. 2006;50(1):43–48. doi: 10.1128/AAC.50.1.43-48.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lavoie EG, Wangdi T, Kazmierczak BI. Innate immune responses to Pseudomonas aeruginosa infection. Microbes and infection. 2011;13(14-15):1133–1145. doi: 10.1016/j.micinf.2011.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jabir MS, Ritchie ND, Li D, Bayes HK, Tourlomousis P, Puleston D, et al. Caspase-1 cleavage of the TLR adaptor TRIF inhibits autophagy and beta-interferon production during Pseudomonas aeruginosa infection. Cell Host Microbe. 2014;15(2):214–227. doi: 10.1016/j.chom.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 11.Nakamura S, Iwanaga N, Seki M, Fukudome K, Oshima K, Miyazaki T, et al. Toll-Like Receptor 4 Agonistic Antibody Promotes Host Defense against Chronic Pseudomonas aeruginosa Lung Infection in Mice. Infection and immunity. 2016;84(7):1986–1993. doi: 10.1128/IAI.01384-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tolle L, Yu FS, Kovach MA, Ballinger MN, Newstead MW, Zeng X, et al. Redundant and cooperative interactions between TLR5 and NLRC4 in protective lung mucosal immunity against Pseudomonas aeruginosa. Journal of innate immunity. 2015;7(2):177–186. doi: 10.1159/000367790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim YK, Shin JS, Nahm MH. NOD-Like Receptors in Infection, Immunity, and Diseases. Yonsei Med J. 2016;57(1):5–14. doi: 10.3349/ymj.2016.57.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Imagawa T, Nishikomori R, Takada H, Takeshita S, Patel N, Kim D, et al. Safety and efficacy of canakinumab in Japanese patients with phenotypes of cryopyrin-associated periodic syndrome as established in the first open-label, phase-3 pivotal study (24-week results) Clin Exp Rheumatol. 2013;31(2):302–309. [PubMed] [Google Scholar]
- 15.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320(5876):674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dennis EA, Norris PC. Eicosanoid storm in infection and inflammation. Nat Rev Immunol. 2015;15(8):511–523. doi: 10.1038/nri3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Callaghan G, Houston A. Prostaglandin E2 and the EP receptors in malignancy: possible therapeutic targets? Br J Pharmacol. 2015;172(22):5239–5250. doi: 10.1111/bph.13331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cayeux SJ, Beverley PC, Schulz R, Dorken B. Elevated plasma prostaglandin E2 levels found in 14 patients undergoing autologous bone marrow or stem cell transplantation. Bone Marrow Transplant. 1993;12(6):603–608. [PubMed] [Google Scholar]
- 19.Domingo-Gonzalez R, Martinez-Colon GJ, Smith AJ, Smith CK, Ballinger MN, Xia M, et al. Inhibition of Neutrophil Extracellular Trap Formation after Stem Cell Transplant by Prostaglandin E2. Am J Respir Crit Care Med. 2016;193(2):186–197. doi: 10.1164/rccm.201501-0161OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hubbard LL, Ballinger MN, Wilke CA, Moore BB. Comparison of conditioning regimens for alveolar macrophage reconstitution and innate immune function post bone marrow transplant. Exp Lung Res. 2008;34(5):263–275. doi: 10.1080/01902140802022518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hubbard LL, Ballinger MN, Thomas PE, Wilke CA, Standiford TJ, Kobayashi KS, et al. A Role for IL-1 Receptor-Associated Kinase-M in Prostaglandin E2-Induced Immunosuppression Post-Bone Marrow Transplantation. Journal of immunology (Baltimore, Md : 1950) 2010;184(11):6299–6308. doi: 10.4049/jimmunol.0902828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Domingo-Gonzalez R, Wilke CA, Huang SK, Laouar Y, Brown JP, Freeman CM, et al. Transforming growth factor-beta induces microRNA-29b to promote murine alveolar macrophage dysfunction after bone marrow transplantation. Am J Physiol Lung Cell Mol Physiol. 2015;308(1):L86–95. doi: 10.1152/ajplung.00283.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ojielo CI, Cooke K, Mancuso P, Standiford TJ, Olkiewicz KM, Clouthier S, et al. Defective phagocytosis and clearance of Pseudomonas aeruginosa in the lung following bone marrow transplantation. Journal of immunology (Baltimore, Md : 1950) 2003;171(8):4416–4424. doi: 10.4049/jimmunol.171.8.4416. [DOI] [PubMed] [Google Scholar]
- 24.Yuan K, Huang C, Fox J, Laturnus D, Carlson E, Zhang B, et al. Autophagy plays an essential role in the clearance of Pseudomonas aeruginosa by alveolar macrophages. J Cell Sci. 2012;125(Pt 2):507–515. doi: 10.1242/jcs.094573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13(3):255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu DD, Pan PH, Liu B, Su XL, Zhang LM, Tan HY, et al. Inhibition of Alveolar Macrophage Pyroptosis Reduces Lipopolysaccharide-induced Acute Lung Injury in Mice. Chin Med J (Engl) 2015;128(19):2638–2645. doi: 10.4103/0366-6999.166039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mortimer L, Moreau F, MacDonald JA, Chadee K. NLRP3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease CAPS mutations. Nat Immunol. 2016;17(10):1176–1186. doi: 10.1038/ni.3538. [DOI] [PubMed] [Google Scholar]
- 28.Zoccal KF, Sorgi CA, Hori JI, Paula-Silva FW, Arantes EC, Serezani CH, et al. Opposing roles of LTB4 and PGE2 in regulating the inflammasome-dependent scorpion venom-induced mortality. Nat Commun. 2016;7:10760. doi: 10.1038/ncomms10760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38(6):1142–1153. doi: 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang D, He Y, Munoz-Planillo R, Liu Q, Nunez G. Caspase-11 Requires the Pannexin-1 Channel and the Purinergic P2X7 Pore to Mediate Pyroptosis and Endotoxic Shock. Immunity. 2015;43(5):923–932. doi: 10.1016/j.immuni.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Domingo-Gonzalez R, Moore BB. Innate Immunity Post-Hematopoietic Stem Cell Transplantation: Focus on Epigenetics. Adv Neuroimmune Biol. 2014;5(3):189–197. doi: 10.3233/NIB-140079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ballinger MN, McMillan TR, Moore BB. Eicosanoid regulation of pulmonary innate immunity post-hematopoietic stem cell transplantation. Arch Immunol Ther Exp (Warsz) 2007;55(1):1–12. doi: 10.1007/s00005-007-0001-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng X, Ji Z, Tsalkova T, Mei F. Epac and PKA: a tale of two intracellular cAMP receptors. Acta Biochim Biophys Sin (Shanghai) 2008;40(7):651–662. doi: 10.1111/j.1745-7270.2008.00438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gringhuis SI, Kaptein TM, Wevers BA, Theelen B, van der Vlist M, Boekhout T, et al. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1beta via a noncanonical caspase-8 inflammasome. Nat Immunol. 2012;13(3):246–254. doi: 10.1038/ni.2222. [DOI] [PubMed] [Google Scholar]
- 35.Afonina IS, Muller C, Martin SJ, Beyaert R. Proteolytic Processing of Interleukin-1 Family Cytokines: Variations on a Common Theme. Immunity. 2015;42(6):991–1004. doi: 10.1016/j.immuni.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 36.Al Moussawi K, Kazmierczak BI. Distinct contributions of interleukin-1alpha (IL-1alpha) and IL-1beta to innate immune recognition of Pseudomonas aeruginosa in the lung. Infection and immunity. 2014;82(10):4204–4211. doi: 10.1128/IAI.02218-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lossos IS, Breuer R, Or R, Strauss N, Elishoov H, Naparstek E, et al. Bacterial pneumonia in recipients of bone marrow transplantation. A five-year prospective study Transplantation. 1995;60(7):672–678. doi: 10.1097/00007890-199510150-00010. [DOI] [PubMed] [Google Scholar]
- 38.Whittle AT, Davis M, Shovlin CL, Ganly PS, Haslett C, Greening AP. Alveolar macrophage activity and the pulmonary complications of haematopoietic stem cell transplantation. Thorax. 2001;56(12):941–946. doi: 10.1136/thorax.56.12.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ballinger MN, Aronoff DM, McMillan TR, Cooke KR, Okiewicz K, Toews GB, et al. Critical Role of Prostaglandin E2 Overproduction in Impaired Pulmonary Host Response Following Bone Marrow Transplantation. J Immunol. 2006;177:5499–5508. doi: 10.4049/jimmunol.177.8.5499. [DOI] [PubMed] [Google Scholar]
- 40.Sokolowska M, Chen LY, Liu Y, Martinez-Anton A, Qi HY, Logun C, et al. Prostaglandin E2 Inhibits NLRP3 Inflammasome Activation through EP4 Receptor and Intracellular Cyclic AMP in Human Macrophages. Journal of immunology (Baltimore, Md : 1950) 2015;194(11):5472–5487. doi: 10.4049/jimmunol.1401343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Li X, Carpinteiro A, Goettel JA, Soddemann M, Gulbins E. Kinase suppressor of Ras-1 protects against pulmonary Pseudomonas aeruginosa infections. Nature medicine. 2011;17(3):341–346. doi: 10.1038/nm.2296. [DOI] [PubMed] [Google Scholar]
- 42.Hubbard LL, Wilke CA, White ES, Moore BB. PTEN limits alveolar macrophage function against Pseudomonas aeruginosa after bone marrow transplantation. American journal of respiratory cell and molecular biology. 2011;45(5):1050–1058. doi: 10.1165/rcmb.2011-0079OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bosmann M, Grailer JJ, Russkamp NF, Ruemmler R, Zetoune FS, Sarma JV, et al. CD11c+ alveolar macrophages are a source of IL-23 during lipopolysaccharide-induced acute lung injury. Shock. 2013;39(5):447–452. doi: 10.1097/SHK.0b013e31828f9c92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schumann RR, Belka C, Reuter D, Lamping N, Kirschning CJ, Weber JR, et al. Lipopolysaccharide activates caspase-1 (interleukin-1-converting enzyme) in cultured monocytic and endothelial cells. Blood. 1998;91(2):577–584. [PubMed] [Google Scholar]
- 45.Mennink-Kersten MA, Ruegebrink D, Verweij PE. Pseudomonas aeruginosa as a cause of 1,3-beta-D-glucan assay reactivity. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2008;46(12):1930–1931. doi: 10.1086/588563. [DOI] [PubMed] [Google Scholar]
- 46.Trouplin V, Boucherit N, Gorvel L, Conti F, Mottola G, Ghigo E. Bone marrow-derived macrophage production. Journal of visualized experiments : JoVE. 2013;(81):e50966. doi: 10.3791/50966. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig 1. Leukocytes in Bronchoalveolar Lavage Post-BMT (a) Absolute number of leukocytes in bronchoalveolar lavage fluid (BALF), quantified with a hemocytometer, (n=3). (b) Percentage of leukocytes from BALF, taken via differential staining, (n=4). Statistics done by student T test. Not significant (NS).
Supplemental Fig 2. PGE2 Enhances IL-1β Release Under Different Pathogenic Stimuli. (a) IL-1β measurements of supernatant from BMDMs treated or not with PGE2 (100nM) and infected or not with PA01 (MOI:10) for 30 minutes after which supernatant was replaced with complete media containing 1% gentamycin. (b) IL-1β measurements of supernatant after methicillin-resistant Staphylococcus aureus (MRSA) (MOI:10) or Streptococcus pneumoniae (MOI:10) infection with or without PGE2 (100nM). Samples were taken two hours after infection. Statistics used one-way ANOVA with Bonferroni's post-test. *P<0.05, **P<0.01, ***P<0.001. Data is representative of three independent experiments.
Supplemental Fig 3. Specificity of caspase-1 and caspase-8 inhibitors. IL-1β measurements by ELISA of supernatant samples from cultured BMDMs treated or not for 4 hours with LPS (100ng/ml) with or without caspase-8 inhibitor (10nM) or caspase-1 inhibitor (10nM) prior 45 minutes stimulation with/without ATP (1mM). One-way ANOVA with Bonferroni's post-test. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Supplemental Fig 4. PGE2 Limits Autophagosome Formation in Response to Serum Starvation. BMDMs were serum-starved for 1 h in the presence or absence of 100nM PGE2. Cells were fixed and stained with FITC-labeled LC3 or DAPI to visualize autophagosomes and nuclei and immunofluorescence images were captured singly or were merged. Panels shown are representative of 2 different experiments.
Supplemental Fig 5. PGE2 Enhances IL-1β Release after 4h, but not 5′ Stimulation. IL-1β measurements by ELISA of supernatant samples from BMDMs treated for 3.5 hours or 5 minutes with LPS (100ng/ml) and/or PGE2 (500nM) followed by 30 minutes of ATP (5mM). Statistics used one-way ANOVA with Bonferroni's post-test. *P<0.05, **P<0.01, ***P<0.001.