

Abstract
Background
Telomere length heterogeneity has been detected in various cell types, including stem cells and cancer cells. Cell heterogeneity in pluripotent stem cells, such as embryonic stem cells (ESCs), is of particular interest; however, the implication and mechanisms underlying the heterogeneity remain to be understood. Single-cell analysis technology has recently been developed and effectively employed to investigate cell heterogeneity. Yet, methods that can simultaneously measure telomere length and analyze the global transcriptome in the same cell have not been available until now.
Results
We have established a robust method that can simultaneously measure telomere length coupled with RNA-sequencing analysis (scT&R-seq) in the same human ESC (hESC). Using this method, we show that telomere length varies with pluripotency state. Compared to those with long telomere, hESCs with short telomeres exhibit the lowest expressions of TERF1/TRF1, and ZFP42/REX1, PRDM14 and NANOG markers for pluripotency, suggesting that these hESCs are prone to exit from the pluripotent state. Interestingly, hESCs ubiquitously express NOP10 and DKC1, stabilizing components of telomerase complexes. Moreover, new candidate genes, such as MELK, MSH6, and UBQLN1, are highly expressed in the cluster of cells with long telomeres and higher expression of known pluripotency markers. Notably, short telomere hESCs exhibit higher oxidative phosphorylation primed for lineage differentiation, whereas long telomere hESCs show elevated glycolysis, another key feature for pluripotency.
Conclusions
Telomere length is a marker of the metabolic activity and pluripotency state of individual hESCs. Single cell analysis of telomeres and RNA-sequencing can be exploited to further understand the molecular mechanisms of telomere heterogeneity.
Electronic supplementary material
The online version of this article (doi:10.1186/s12915-017-0453-8) contains supplementary material, which is available to authorized users.
Keywords: Single cell analysis, Telomere length, Transcriptome, Human embryonic stem cell, Heterogeneity
Background
Telomeres are the highly repetitive ribonucleoprotein structures (TTAGGG)n that protect chromosome ends and maintain genomic instability [1, 2]. Short telomeres are associated with aging and tumorigenesis [3–5]. Yet, tumors, and particularly those positive for alternative lengthening of telomeres (ALT), are characterized by remarkable telomere length heterogeneity [6]. Notably, functional telomeres are required for embryo development and for the pluripotency and differentiation capacity of embryonic stem cells (ESCs) [7, 8]. Pluripotent stem cells, including ESCs, induced pluripotent stem cells (iPSCs), and nuclear transfer ESCs with sufficient telomere lengths, are able to give rise to offspring shown by germline chimera production or tetraploid embryo complementation tests, the most stringent and functional test of naïve pluripotency [7, 9–11]. While short telomeres impair stem cell differentiation [12], ESCs with hyper-long telomeres may delay aging as evidenced by the generation of healthier chimera mice that exhibit reduced cell senescence and DNA damage with age and better skin wound healing [13].
Telomeres are elongated during early expansion of human ESCs (hESCs) and then reach a relatively stable length [14]. Variations in telomere lengths are found in human iPSCs [15, 16]. Additionally, ESC cultures exhibit heterogeneous expression of transcription factors of naïve pluripotency such as NANOG [17, 18], REX1/ZFP42 [19, 20], STELLA/DPPA3 [17, 18], ESSRB [17, 19], KLF4 [19, 21], and TBX3 [17, 18]. Unlike mouse ESCs, functional tests of pluripotency of hESCs are limited due to ethics issues [22]. It remains elusive how telomere lengths and heterogeneity are implicated in pluripotency and differentiation of human ESCs/iPSCs.
Conventional methods for the measurement of telomere lengths include telomere restriction fragment (TRF) [23], Q-FISH (quantitative fluorescence in situ hybridization) [24], Flow-FISH (flow cytometry method using fluorescence in situ hybridization) [25], T-OLA (telomeric-oligonucleotide ligation assays) [26], qPCR (real-time quantitative polymerase chain reaction) [27], and STELA (single telomere length analysis) [28]. TRF measurement requires large amounts of DNA (micrograms) and consistently produces large telomere DNA fragment smears, which itself may indicate telomere length heterogeneity. Conventional qPCR does not need large amounts of DNA (nanogram), but still requires a cell population rather than a single cell. Q-FISH is normally used to estimate telomere lengths of metaphase chromosome spread prepared from a cell population.
Telomeres may be influenced by gene mutations shown by genomic sequencing analysis in a large cell population or complex tissues [29, 30]. The bulk cell population in the same environment also displays cell-to-cell variations in gene expression [31]. Single-cell genome sequencing (DNA-seq and RNA-seq) has rapidly developed into a powerful method to investigate genetic and transcriptome heterogeneity in a cell population [32–34]. By combining methods to amplify the telomere-specific sequences and mRNA/cDNA separately, we were able to simultaneously measure telomere length coupled with RNA-sequencing analysis in the same cell, and employed this method to investigate telomere length heterogeneity and pluripotency of hESCs.
Results
Simultaneous measurement of telomere length and gene expression in the same cell
To acquire the total mRNA and genomic DNA (gDNA) from the same cell, we used the biotinylated oligo-dT primer [35], targeting to mRNA containing a polyadenylated tail (poly (A)+) and applied a magnet to separate the gDNA from the same cell (Fig. 1a). We named this novel method as single cell telomere length and transcriptome sequencing (scT&R-seq; see Methods).
Fig. 1.
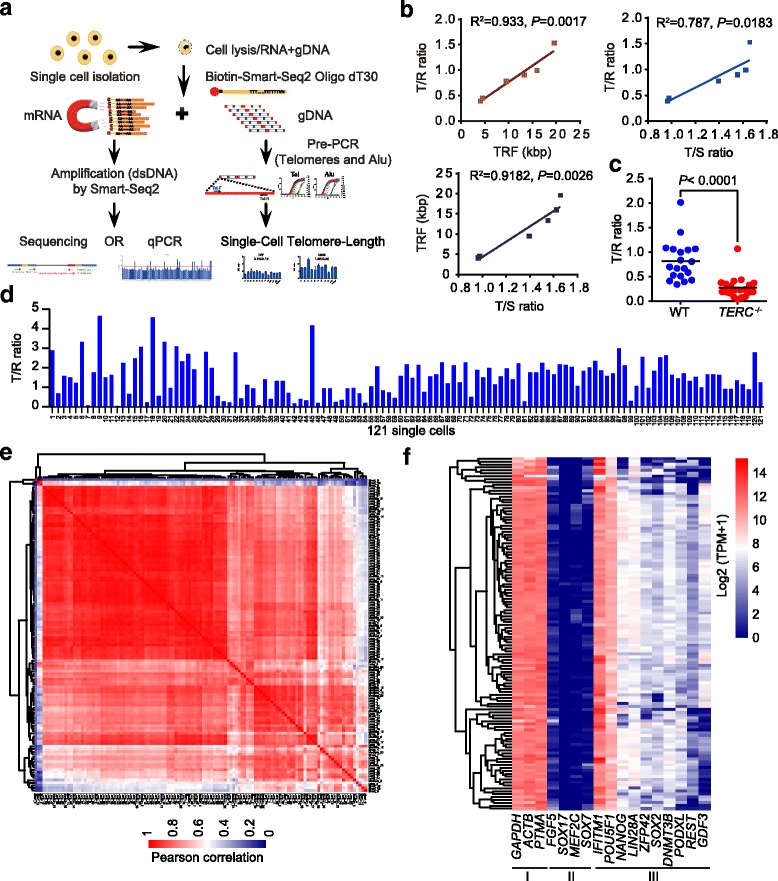
Simultaneous measurement of telomere length and transcriptome in the same cell. a Overview and design of single-cell telomere length measurement and RNA-sequencing analysis (scT&R-seq) in the same cell. b Average telomere length in single cells shown as T/R ratio by scT&R-seq is significantly correlated with that of other methods in various human cell lines. Each point represents one cell line, and different colors distinguish between methods. c Relative telomere length of TERC knockout and wild-type hESCs using scT&R-seq. d Relative telomere length distribution of all individual human embryonic stem cell (hESC, WA26). e Hierarchical clustering analysis of whole-gene expression similarity (measured by Pearson correlation coefficients) for all hESCs. f Heat-map showing expression of selected marker genes for housekeeping (I), differentiation (II), and pluripotency (III) in hESCs. Each row represents one cell
To validate the feasibility of the method, we analyzed telomere length and relative gene expression in single U2OS (osteosarcoma cells) and hESCs (WA26) (Additional file 1: Figure S1a–c). Following pre-amplification specific for telomeres (T) and multicopy repeat reference gene Alu (R) of gDNA in parallel, the Ct value for Tel and Alu in negative controls were much greater than those of single cells by qPCR (Additional file 1: Figure S1a). Following reverse transcription of mRNA and amplification of cDNA, the variation in the expression of GAPDH by qPCR, a housekeeping gene, was minimal. The pluripotency gene POU5F1/OCT4 was only highly expressed in WA26 but not in U2OS, as expected. No product was detected in the negative controls (Additional file 1: Figure S1c).
Because this new method involves physical separation of DNA from mRNA, we tested whether the separation step influenced the telomere length detection at single cell level from the same cell line (WA26). Twenty cells were randomly picked and subject to single cell telomere length analysis using single-cell telomere length measurement by qPCR, without a separation step [36], or scT&R-seq (Additional file 1: Figure S1d). Ct values of Tel (P = 0.1879) and Alu (P = 0.1044), as well as telomere length, did not differ (P = 0.6525) in single cells measured by the methods regardless of the separation technique (Additional file 1: Figure S1d). We further validated single-cell telomere length measurement by TRF (in kb) and conventional qPCR (T/S ratio) in hESCs (WA26, RuES2), U2OS, human colorectal cancer cells (HCT116), cervical cancer cells (HeLa S3), and human embryonic fibroblast (HEF) cells (Additional file 1: Figure S1e–h). The average relative telomere length shown as T/R ratio (Additional file 1: Figure S1e) of single cells measured by scT&R-seq was highly correlated with that measured by the two conventional methods applied to a cell population (Fig. 1b). Further, the T/S ratio also correlated well with TRF in all six cell lines tested (Fig. 1b). Variations in telomere length revealed by scT&R-seq were readily detected by TRF (Additional file 1: Figure S1h). Remarkably, telomere lengths differed among single cells within the same cell population. hESCs (WA26) were observed to normally express POU5F1/OCT4 by fluorescence microscopy and telomerase activity was observed by Telomeric Repeat Amplification Protocol (TRAP) assay (Additional file 1: Figure S1i, j).
To further validate the reliability of scT&R-seq, we measured telomere length by both Southern blot and scT&R-seq methods of telomerase gene-knockout hESCs after deletion of TERC by CRISPR/Cas9 and compared these with wild-type (WT) controls (Additional file 2: Figure S2a). Compared to WT hESCs, telomerase-deficient (TERC -/-) hESCs showed decreased telomerase activity by TRAP assay (Additional file 2: Figure S2b), and generally shorter telomeres measured by TRF (Additional file 2: Figure S2c), as expected. Telomere length measured at single cell level by scT&R-seq was notably shorter (P < 0.0001) in TERC -/- than in WT hESCs (Fig. 1c; Additional file 2: Figure S2d). While relative expression levels shown as Ct values of housekeeping gene GAPDH did not differ (P = 0.0726) between TERC -/- and WT hESCs (Additional file 2: Figure S2e), the expression level of NANOG was reduced at the single cell level in TERC -/- compared with that of WT hESCs (Additional file 2: Figure S2f), consistent with the bulk results (Additional file 2: Figure S2g). Further, the expression level of NANOG was correlated with telomere length, shown as T/R ratio (Additional file 2: Figure S2h).
Single-cell analysis of telomere length and global transcriptome in hESCs by scT&R-seq
To investigate the potential relationship in heterogeneity of transcriptome and telomere length, 121 single hESCs were randomly selected for single cell analysis using scT&R-seq. Both telomere length and transcriptome displayed high heterogeneity (Fig. 1d, e, Additional file 3: Table S1). Telomere length of single cells ranged from 0.04 to 4.64 (Fig. 1d). The correlation coefficient of the global gene expression profile varied from 0.1 to 1.0 (Fig. 1e), revealing the heterogeneity of hESCs at global transcriptome levels. Housekeeping (class I) and pluripotency-related transcripts (class III) at single cell level were highly expressed in hESCs, and differentiation-related genes (class II, e.g., SOX7, MEF2C, SOX17, FGF5) were maintained at relatively lower expression levels (Fig. 1f).
All single cell RNA-seq libraries showed remarkably uniform gene body coverage profiles from the 5′ to 3′ end of each gene (Additional file 4: Figure S3a). Most of the expressed genes were detected even with down-sampling of 50% of the reads (Additional file 4: Figure S3a). The sequencing depth of all individual libraries was enough for a subsequent reliable analysis based on previous reports [37]. All single cell RNA-seq data were used for downstream analyses. The RNA-seq libraries with an average of 3.3 million single cell RNA-seq reads per cell were obtained and the average ratio mapped was 69.65% (Additional file 4: Figure S3b, c). Most of the detected genes had transcripts per million values (TPM) of over 10, and 26,321 genes were identified when all single cell libraries were combined (Additional file 4: Figure S3d, Additional file 5: Table S2). Of these, the top 40 genes expressed in single cells included housekeeping genes, ribosomal protein genes, and pluripotency genes (e.g., POU5F1/OCT4 and IFITM1). In addition, TMSB4X, FTL, GSTP1, H2AFZ, SLIRP, and UBB had highly expressed abundance in hESCs (Additional file 4: Figure S3e). A strong correlation was found between single-cell RNA-seq and qPCR for TERF1 and POU5F1/OCT4 (Additional file 6: Figure S4), further validating the single cell RNA-seq data. Cell cycle stage may have influenced the heterogeneity of gene expression [38]. We employed the prediction method (single-cell latent variable model, scLVM) to classify cells into cell cycle phases based on gene expression data. Only 11.6% of variation was attributable to the cell cycle phases, and cell cycle stage had minimal effects on the clustering results. No obvious differences in telomere length were found at different cell cycle stages (Additional file 4: Figure S3f).
Telomere length heterogeneity linked to specific gene expression profile
Telomere length (T/R ratio) distribution in hESCs varied, with a mean value of 1.52. Therefore, we used cutoffs for the three telomere length groups based on the T/R ratio (single cell telomere length) (see Methods). hESCs and telomere ALT positive cells (U2OS) showed longer telomere lengths (long group), and differentiated HEF cells had shorter telomeres (short group) (Fig. 2a, Additional file 3: Table S1). Eighty-one cells showed a T/R ratio centered from 1.00 to 4.64 (long group), and fewer hESCs (25 and 15, respectively) exhibited medium or short telomeres (inset figures in Fig. 2a).
Fig. 2.
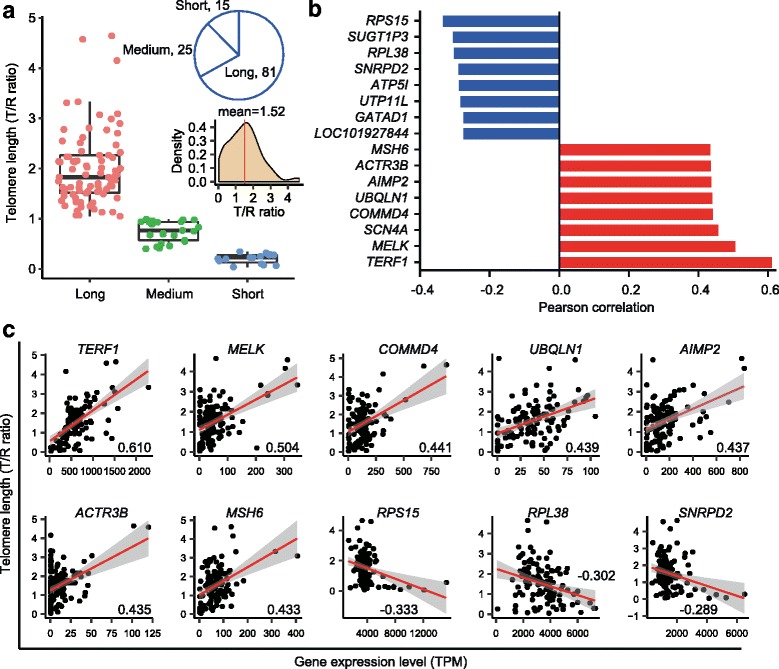
Telomere length heterogeneity and candidate genes potentially involved in telomere length maintenance of human embryonic stem cells (hESCs). a hESCs were clustered into three telomere length groups based on T/R ratio by single-cell analysis. We defined the cells with T/R ratio over 1.0 as the “long” telomere group, those with T/R ratio 0.4–1.0 as the “medium” group, and those with T/R ratio under 0.4 as the “short” group. Inset pie figure shows cell number in each telomere length group. Inset density figure indicates distribution of telomere length of hESCs. b Top eight genes with expression patterns remarkably correlated with telomere length (T/R ratio). Red and blue bars show positive and negative correlation with each other. c Scatter plots showing correlation between gene expression level and T/R ratio for selected genes in all samples. The line represents fitted values and shaded band shows 95% confident regions. Correlation value of each gene is shown on the plot
By searching for the relationship of telomere lengths and gene expression levels, we identified the candidate genes that might be directly or indirectly involved in telomere length regulation according to the top positive and negative correlation coefficient (Fig. 2b, Additional file 7: Table S3, Additional file 8: Table S4). Expression of TERF1, a key component factor of telomere shelterin complex important for telomere maintenance [39], was highly and positively correlated with telomere length (Fig. 2c). Expression levels of DNA mismatch repair protein MSH6 were also highly associated with hESC telomere lengths. Others, including MELK, SCN4A, COMMD4, UBQLN1, AIMP2, and ACTR3B, also showed a positive correlation with telomere length (Fig. 2c). In contrast, RPS15, SUGT1P3, RPL38, SNRPD2, ATP5I, UTP11L, GATAD1, and LOC101927844 were negatively correlated with telomere length (Fig. 2c).
To explore the molecular identities of individual cells by combination of telomere length and gene expression, we performed global principal component analysis (PCA) projection of all single cells. Long or short telomere hESCs could be mostly separated by clustering of PC1 and PC2 (Fig. 3a, Additional file 9: Table S5). To exclude the possibility that the cluster might be caused by confounder and artifact of low mapping ratios, we employed the scater package (single-cell analysis toolkit for expression in R) to compute the cumulative proportion of reads [40]. Cells from these three telomere length groups displayed the same cumulative proportion of reads (Additional file 10: Figure S5a). The mapping ratio of raw reads showed no significant correlation (P = 0.3884) with single cell telomere length (Additional file 10: Figure S5b), indicating that the result of PCA was not an artifact.
Fig. 3.
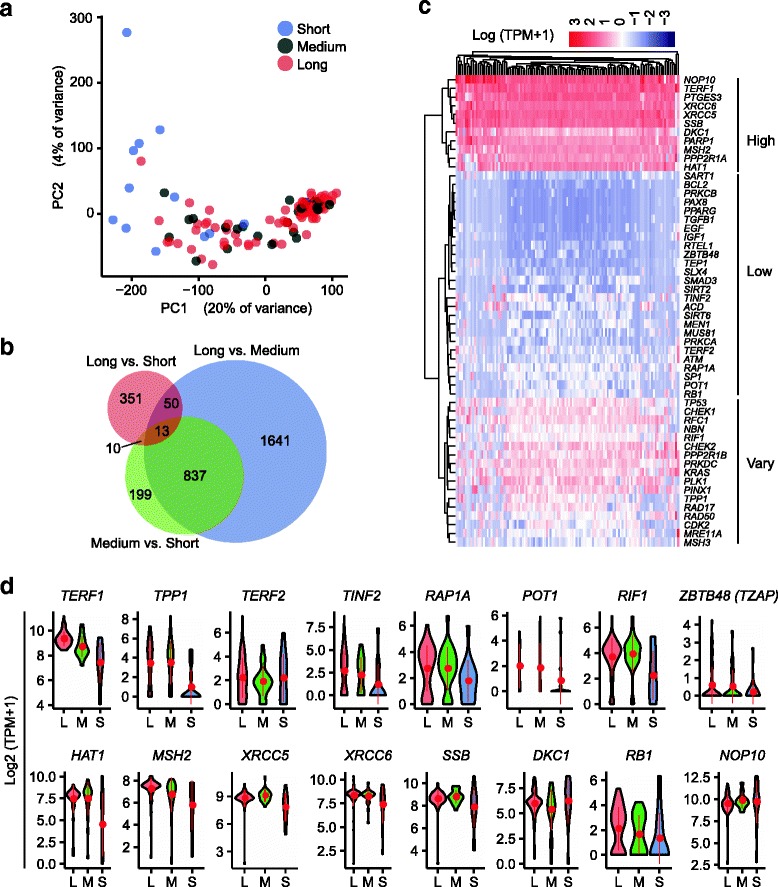
Classification of telomere lengths in association with global transcriptome of human embryonic stem cell (hESCs). a Classification of all single cells based on whole transcriptome profile using principal component analysis (PCA, scatter plot of component 1 and 2), with cells colored according to telomere length group. b Venn diagram depicting the overlap among three telomere groups. c Heatmap representing expression levels of 54 selected genes related to telomere length maintenance from GESA gene sets (http://software.broadinstitute.org/gsea). “High” means gene expression at high levels; “Low” means low expression levels; “Vary” indicates gene expression levels in a wide range. d Violin plots showing relative expression levels of genes associated with shelterin complex in each telomere length group (L long, M medium, S short). Y-axis indicates Log2 (TPM + 1)
Analysis of differentially expressed genes between long, medium, or short telomere groups showed that 13 genes were commonly shared among these three separate comparisons (long vs. short, long vs. medium, medium vs. short) (Fig. 3b, Additional file 11: Table S7). The 351 genes were differentially expressed in long telomere compared with short telomere cells, and 50 genes were shared in long versus short and medium versus short telomere cells (Fig. 3b). We performed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses for the differentially expressed genes in long versus short telomere cells. The highest enrichment was linked to neuroactive ligand-receptor interaction, extracellular matrix-receptor interaction of KEGG pathway, and the metabolic biological process in long telomere cells, relative to short telomere cells (Additional file 12: Figure S6).
By hierarchical clustering [41] of telomere maintenance genes from GSEA (Gene Set Enrichment Analysis) (Additional file 13: Table S6), we observed that NOP10, DKC1, TERF1, PTGES3, XRCC5, XRCC6, and SSB were expressed at high levels in almost all hESCs (Fig. 3c); NOP10 and DKC1 are two components known as stabilizers of the telomerase complex [42]. Analysis of six shelterin complex proteins TERF1, TERF2, TINF2, TPP1, RAP1A, and POT1 [39] in different telomere length groups revealed that TERF1 was consistently highly expressed in long telomere hESCs and less expressed in short telomere cells (Fig. 3d). TPP1 and RAP1A were also expressed at higher levels in long and medium telomere hESCs than in short telomere cells (Fig. 3d). TZAP (also named ZBTB48) was recently identified as a new telomere-associated gene involved in telomere length control [43]; hESCs generally expressed TZAP at a low level, and rarely at a high expression level (Fig. 3d). Of other genes potentially expressed for telomere maintenance, HAT1, MSH2, XRCC5, XRCC6, and SSB were expressed at lower levels in short telomere hESCs (Fig. 3d).
Telomere length associated with pluripotency
To investigate whether telomere lengths correlated with expression levels of pluripotent genes, we defined a pluripotency score of hESCs by average expression of 299 pluripotent genes from PluriNet [44]. We validated the pluripotency score (named PluriNet score) using the single cell data obtained from human pluripotent stem cells and differentiated cells reported by Chu et al. [45]. As expected, the PluriNet score was higher in undifferentiated hESCs than in differentiated cells from the same precursor cell (Additional file 14: Figure S7a). We used this score to define the pluripotency of hESCs with various telomere lengths. Short telomere hESCs displayed a lower PluriNet score compared with hESCs in the long and medium telomere groups (Fig. 4a, Additional file 14: Figure S7b). Pearson’s correlation analysis of telomere length (Log(T/R ratio)) and Log(PluriNet score) showed a positive association of the two factors with a coefficient of approximately 0.48 (Fig. 4b, Additional file 3: Table S1). We divided hESCs into three groups, Group 1 representing high pluripotency with long telomere length, Group 2 representing high pluripotency with short telomere length, and Group 3 representing low pluripotency with short telomere length (Fig. 4b).
Fig. 4.
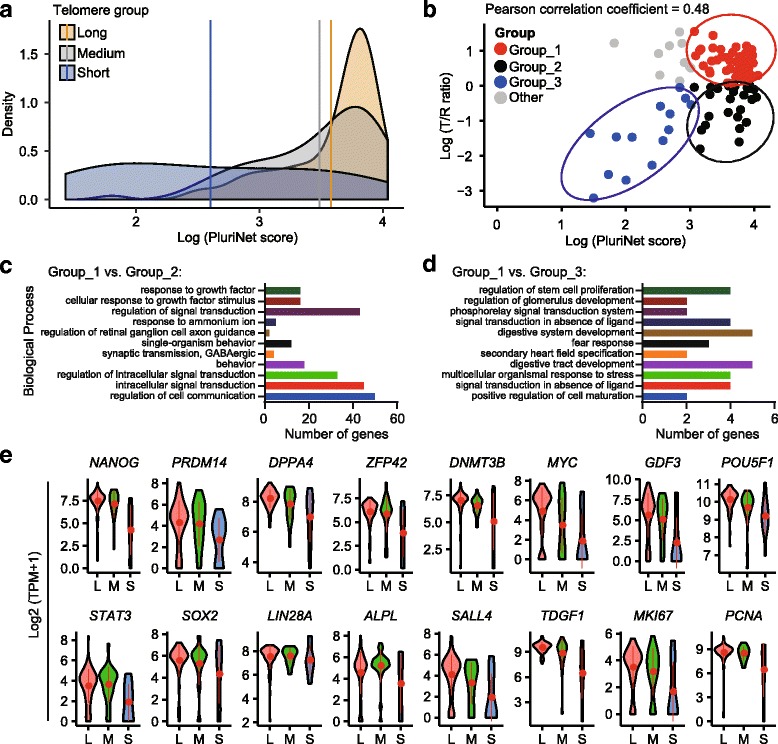
scT&R-seq analysis of heterogeneity in pluripotency gene expression of hESCs. a Distribution of PluriNet score. Gene set for PluriNet score was generated from study in GEO (Chu et al. [45]). b Correlation between the averaged PluriNet score and telomere length (T/R ratio). hESCs were divided into three groups by Log (PluriNet score) and Log (T/R ratio), Group_1 (Log (T/R ratio) > 0; Log (PluriNet score) > 3.0), Group_2 (Log (T/R ratio) ≥ 0; Log (PluriNet score) ≥ 3.0), and Group_3 (Log (T/R ratio) < 0; Log (PluriNet score) < 3.0). c Functional annotation of genes showing higher expression in Group_1 compared to Group_2. d Functional annotation of genes with higher expression in Group_1 compared to Group_3. e Violin plots detailing single cell telomere length distribution and expression of pluripotency genes in each telomere length (L long, M medium, S short) group. Y-axis indicates Log2 (TPM + 1)
GO biological process enrichment indicates that the genes highly expressed in Group 1 compared with Group 2 involved regulation of cell communication, cellular response to growth factor stimulus, and response to growth factor (Fig. 4c, Additional file 15: Table S8). Genes highly expressed in Group 1 compared with Group 3 involved regulation of stem cell proliferation (Fig. 4d).
Further analysis of 16 selected genes related to pluripotency showed that genes for pluripotency, NANOG, PRDM14, and ZFP42/REX1 [17, 18], were expressed at lower levels in hESCs with short telomeres (Fig. 4e). DNMT3B, MYC, and GDF3 were expressed at higher levels in general in long and medium telomere hESCs than in short telomere hESCs (Fig. 4e), consistent with their potential roles in maintaining the pluripotent state of human ESCs [17, 46, 47]. However, the relative expression level of each gene still varied among hESCs with long and medium telomere hESCs, and whether this relates to a naïve or primed state remains unclear. POU5F1/OCT4, SOX2, and LIN28A did not differ in their expression levels regardless of telomere lengths (Fig. 4e). LIN28A is expressed at markedly reduced levels in naïve versus primed states [48]. In contrast, cells with short telomeres expressed the genes primed for differentiation at higher levels, e.g., FGF5 (Additional file 16: Figure S8). Thus, telomere length can mark the pluripotency state of hESCs.
Distinct metabolic pathway in association with telomere length heterogeneity of hESCs
We performed KEGG pathway enrichment analysis of the differentially expressed genes of Group 1 compared with Group 3 (Fig. 5a, Additional file 17: Table S9). Specifically, genes involved in the oxidative phosphorylation metabolic pathway were more enriched in hESCs with short telomeres than in those of long telomeres (Fig. 5b). Genes associated with oxidative phosphorylation included ATP5I, NDUFA13, NDUFA3, UQCRQ, NDUFB7, and ATP5J2 (Fig. 5b). In contrast, genes related to glycolysis metabolism ENO3, SLC16A1, PFKM, and LDHA [49] were up-regulated in hESCs with long telomeres, compared with those of short telomeres (Fig. 5b).
Fig. 5.
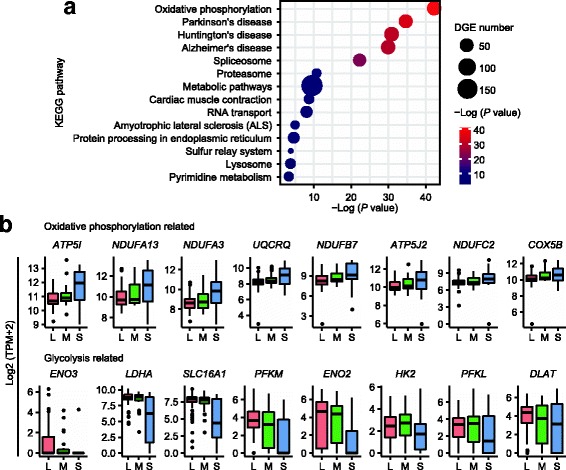
Short telomere human embryonic stem cells (hESCs) display increased oxidative phosphorylation and decreased glycolysis. a Top 14 KEGG pathway annotations of significantly up-regulated genes in short telomere group compared with long telomere group. The terms are sorted by their enrichment P value and the shape indicates the number of differentially expressed genes. b Boxplots illustrating distribution in the expression of genes associated with oxidative phosphorylation and glycolysis in the various telomere length (L long, M medium, S short) groups. Gene list of oxidative phosphorylation from KEGG and genes related to glycolysis were obtained from Gu et al. [49]
Pluripotent ESCs are metabolically characterized with high glycolysis activity, whereas primed ESCs and differentiated cells have an increased and even higher oxidative phosphorylation activity, respectively [49]. Our data support the idea that hESCs with short telomeres may tend to rely on the oxidative phosphorylation pathway in metabolism, further suggesting the reduced pluripotency or tendency to differentiate.
Validation of NANOG and TERF1 expression related to telomere length heterogeneity and pluripotency of hESCs
Immunofluorescence-FISH (IF-FISH) was employed to reveal NANOG expression and telomere length fluorescence signals in the same cell (Fig. 6a). The expression level of NANOG correlated well with telomere length (P < 0.01) (Fig. 6b). We also observed that expression levels of NANOG were positively correlated with telomere length in TERC -/- and WT hESCs at the single cell level using scT&R-seq (Additional file 2: Figure S2h). However, OCT4/POU5F1 expression showed much less heterogeneity in hESCs (Fig. 6c) and its expression level by the relative fluorescence intensity was minimally correlated with telomere length (Fig. 6d), further supporting RNA-seq data (Fig. 4e).
Fig. 6.

NANOG and OCT4/POU5F1 expression levels in association with telomere length and pluripotency of human embryonic stem cell (hESCs). a Telomere fluorescence signal (green, TelC-FITC, F1009, Panagene) and NANOG expression (red) in hESCs revealed by IF-FISH. DAPI-stained nuclei. b Linear regression analysis of telomere length and NANOG protein expression at single cell level. c Telomere fluorescence signal (red, TelC-Cy3, F1002, Panagene) and OCT4/POU5F1 expression (green) in hESCs revealed by IF-FISH. DAPI-stained nuclei. d Linear regression analysis of telomere length and OCT4/POU5F1 protein expression at single cell level
TERF1 was highly expressed in hESCs with longer telomeres. To understand the role of TERF1 in telomere length regulation and pluripotency of hESCs, we performed TERF1 knock-out experiments in hESCs by CRISPR/Cas9. Homozygous TERF1 -/- cell lines were not achievable by repeated experiments, suggesting that complete knockout of TERF1 in hESCs might be lethal. Heterozygous TERF1 +/- indicated deletion of 399 bp covering the whole exon1 of TERF1 (Fig. 7a). Compared to WT hESCs, TERF1 +/- hESCs showed smaller clones, reduced level of TERF1 and down-regulation of pluripotency genes, including POU5F1/OCT4, NANOG, SOX2, KLF4, and REX1/ZFP42, and of telomerase genes TERT and TERC (Fig. 7b–d), suggesting that TERF1 is important for pluripotency maintenance of hESCs. In addition, TERF1 +/- hESCs displayed an altered cell cycle and elongated G2/M phase (Fig. 7e), and decreased telomerase activity with increasing passages (Fig. 7f). Telomeres were slightly shortened in TERF1 +/- hESCs, compared with WT hESCs (Fig. 7g, h). It is unclear whether telomeres altered more in TERF1 -/- hESCs, as the complete TERF1 -/- knockout cell line was not achieved. It also will be necessary to assess whether TERF1 functions differently in human ESCs in comparison with other somatic cell types. Terf1 deficiency could increase telomere fragility and cancer in mice [50]. We showed that γH2AX foci were increased in telomeres in TERF1 +/- cells (Fig. 7i–k), supporting the notion that telomeres of TERF1 defective cells are prone to breakage [50]. Additionally, we analyzed the influence of TERF1 deficiency on telomeres at single cell level using scT&R-seq. As a result, telomeres were shorter in TERF1 +/- cells compared with WT hESCs (Fig. 7l), largely in agreement with TRF telomere distribution data (Fig. 7g, h). Moreover, expression levels of TERF1 and NANOG were reduced in TERF1 +/- hESCs (Fig. 7m), consistent with the bulk results (Fig. 7c). Expression levels of NANOG and TERF1 were also positively correlated with telomere lengths at the single cell level measured by scT&R-seq (Fig. 7n). Other causes, aside from the reduced telomerase and shortened telomeres, might also drive gene expression changes in these cells.
Fig. 7.
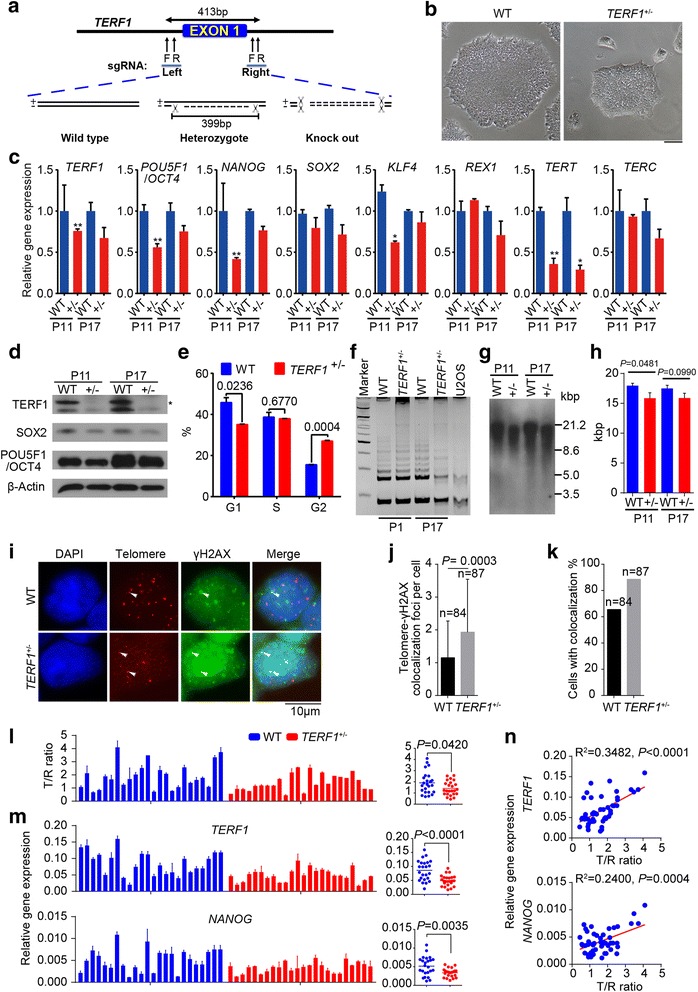
TERF1 expression levels associated with telomere length and pluripotency of human embryonic stem cell (hESCs). a Scheme for hTERF1 knockout by CRISPR/Cas9. b Morphology of TERF1 +/- and wild-type (WT) hESCs (WA26) at passage 11. Scale bar = 100 μm. c Expression levels of pluripotency and telomerase genes in TERF1 +/- and WT hESCs by qPCR analysis. *P < 0.05, **P < 0.01, compared with WT controls. The data values of each gene are provided in Additional file 18: Table S10. d Western blot analysis of TERF1, SOX2, and OCT4/POU5F1 protein levels in TERF1 +/- and WT hESCs. β-actin served as a loading control. e Cell cycle analysis of TERF1 +/- and WT hESCs by flow cytometry. Data represent mean ± SD of three independent experiments. f Telomerase activity by TRAP assay of TERF1 +/- and WT hESCs at different passages. U2OS served as a negative control. g Telomere length measurement by southern blot analysis of TERF1 +/- and WT hESCs. h Quantification of southern blot results. Data represent mean ± SD of two independent experiments. i Immunofluorescence detection of γH2AX and telomere FISH of TERF1 +/- and WT hESCs. j, k Number of telomere-γH2AX colocalization foci per cell (j) and percentage of cells with the colocalization (k). l Relative telomere length of individual TERF1 +/- and WT hESCs by scT&R-seq. Right panel shows the mean telomere length by scT&R-seq. m Gene expression measured in single cells of TERF1 +/- and WT hESCs by scT&R-seq. Right panel shows the mean level of gene expression. n Linear regression analysis of telomere length and gene expression (TERF1 and NANOG) at single cell level
Discussion
Using simultaneous measurement of both transcriptome and telomere length in the same cell, we observed that telomere length heterogeneity links to pluripotency and, interestingly, to metabolism signaling representing a pluripotent state in human ESCs.
High expression levels of NOP10 and DKC1 may suggest their potential role in telomere maintenance of hESCs, presumably through the activities of the telomerase enzyme complex with the shelterin complex (TERF1, TRF2, TINF2, POT1, TPP1, and RAP1) [51]. ESCs with short telomeres have lower pluripotency, and pluripotent cells show more gene copy number variation than differentiation cells [52]. In addition to the genes already known for telomere length maintenance [53], our data reveals 16 candidate genes potentially associated with telomere lengths in hESCs (Additional file 7: Table S3). For instance, MELK and MSH6 were expressed at higher levels in long telomere hESCs relative to those of short telomere cells. MELK is expressed in the mouse egg and preimplantation embryo [54] and may regulate proliferation of cancer stem cells [55]. MSH6 is required for repair of mismatched DNA bases to avoid mutation and repress homologous recombination [56]. MSH6 also is involved in NHEJ by association with Ku70/80 in DSB repair [57], and Ku70/80 interacts with both the protein and RNA components of human telomerase involved in telomere maintenance [58]. Further experiments are required to assess whether these genes are involved in telomere length regulation of hESCs.
Simultaneous measurement of telomere and transcriptome in the same cell reveals that long telomeres are linked to high expression of pluripotency marker genes of hESCs. This is of particular significance given that developmental pluripotency of hESCs could not be functionally tested by the methods that can be applied to mouse ESCs, including germline chimera tests or tetraploid embryo complementation. hESC pluripotent marker genes, such as ZFP42 (REX1), PRDM14, and NANOG [59], are expressed at lower levels in short telomere ESCs than those of long telomere hESCs. Moreover, IF-FISH confirms higher NANOG expression level in long telomere hESCs than in short telomere cells, consistent with the notion that telomere lengths could be used to mark the pluripotency state of human ESCs. In addition, CRISPR/Cas9-mediated genome editing of TERF1 suggests that TERF1 plays an important role in hESCs, as TERF1 -/- cells could be lethal, and pluripotency-related genes are down-regulated in TERF1 +/- hESCs.
Excitingly, telomere length is implicated in metabolism signaling. Long telomere hESCs display higher glycolysis metabolic signaling than do short telomere cells. Primed and naïve hESCs exhibit a different metabolic state [17, 49, 60], and naïve hESCs show increased glycolysis compared to primed counterparts [49]. We find that genes for glycolysis metabolism, including ENO3, SLC16A1, LDHA, and PFKM, are expressed at higher levels in hESCs with long rather than short telomeres, and these are reportedly highly expressed in naïve hESCs relative to primed hESCs [49]. In contrast, genes for oxidative phosphorylation are highly expressed in short telomere cells. These data indicate that hESCs with long telomeres might maintain pluripotency by high glycolytic metabolism, whereas short telomeres may signal a tendency to differentiation by increased oxidative phosphorylation [49, 60]. Together, telomere lengths can indicate the pluripotency state of hESCs.
Conclusions
Our analysis focused on heterogeneity in telomere length and pluripotency of hESCs. It has been recognized that tumors are more heterogeneous in their gene expression and telomere lengths than those of normal cells. We anticipate that the method of simultaneous analysis of telomere length and global transcription in the same cell will be useful in the further understanding of the molecular signaling underlying tumor heterogeneity and in the discovery of new targets for cancer treatment.
Methods
Cell culture
WA26 and RuES2 hESCs were routinely maintained in the undifferentiated state in E8 medium (A1517001, Life technologies) on Matrigel-coated (356230, BD Bioscience) tissue culture plates with daily medium change and passaged every 3 days using 0.5 mM EDTA in PBS at a 1:20 ratio with Rocki (sc-281642A, Santa Cruz). The colon carcinoma cell line (HCT116) was cultured at 37 °C in 5% CO2 in RPMI1640 (11875085, Life technologies) plus 10% FBS. Osteosarcoma cells (U2OS, ALT positive cell line), cervix adenocarcinoma cells (HeLa), and HEF cells were cultured in high glucose DMEM plus 10% FBS with 1% penicillin and streptomycin.
Single-cell isolation and lysis
hESCs were separated following digestion with 0.5 mM EDTA in PBS and other cell types were separated following treatment with 0.25% Trypsin-EDTA. Single cells were resuspended in PBS with 0.1% BSA (A3311-10g, SIGMA), picked up in 1 μL 0.1% BSA using a micropipette with an epT.I.P.S. pipette tip (0030000838, Eppendorf) under a dissecting microscope, and transferred to the bottom of a 200-μL PCR tube (8-strip, nuclease-free, thin-walled PCR tubes with caps, PCR-0208-C, Axygen) consisting of 4 μL lysis buffer containing 3.45 μL of Buffer RLT plus (1053393, Qiagen), 0.5 μL Biotin-oligo-dT primer (5′-biotin-TEG-AAGCAGTGGTATCAACGCAGAGTACT30VN-3′), and 0.05 μL of Recombinant RNase inhibitor (2313A, Clontech).
Separation of the genome and transcriptome in the same cell
Separation of gDNA and mRNA was performed as described previously [35]. Briefly, samples were incubated at 72 °C for 3 min (in our preliminary experiments, we found that this hybridization step could increase the efficiency of mRNA capture and bring better gene body coverage profile from 5′ to 3′), followed by hybridization of the Biotin-oligo-dT primer to poly(A)+ tail mRNA. Dynabeads in 4 μL (65001, Life technologies) were added to capture poly(A)+ tail mRNA, collected by a magnet, and the gDNA supernatant was transferred to a fresh PCR tube. Single-cell cDNA was synthesized in the tube containing mRNA, based on Smart-seq2 protocol [61]. The gDNA was purified by 8 μL Agencourt AMPure beads (A63881, Beckman Coulter). After washing with 80% (vol/vol) ethanol, the gDNA beads were freshly used or stored at −80 °C.
Library construction and sequencing
The libraries were prepared by using TruePrep DNA Library Prep Kit V2 for Illumina® (TD503-02, Vazyme Biotech) according to the instruction manual. Samples were barcoded during library preparation and multiplex sequenced, with a 50-bp single-end sequencing strategy on a HiSeq 2500 (Illumina) in fast mode.
Single-cell RNA-seq data analysis
Reads were mapped to hg19 from iGenome (https://support.illumina.com/sequencing/sequencing_software/igenome.html) by TopHat (v2.1.0) [62] with default parameters. Read counts of each gene annotated in RefGene were calculated by HTSeq with default parameters. Raw counts were normalized by library size via ‘estimateSizeFactorsForMatrix’ function from R package ‘DESeq’. High variable genes were identified based on the method developed by Brennecke et al. [63]. The squared coefficient of variation (CV2) and average count of each gene was calculated and fitted to a curve with the parameterization of CV2 = a1/μ + α0.
The gene counts were loaded into the R package scater and standard quality control metrics were calculated [40]. The package provides a convenient, flexible workflow to process raw sequencing reads into a high-quality expression dataset ready for downstream analysis.
PCA was conducted using log2 transformed TPM values, the PCA_int() function in R and the first two principal components of variance. The PluriNet score of single cells was calculated by averaging expression levels of PluriNet gene set [44]. Differential expression and function enrichment of genes between the putative groups were conducted using the R package SCDE [64].
Analysis of cell cycle phase in association with gene expression profile
We examined the effect of the cell cycle on our clustering results by scLVM [38] and using cell cycle-related genes, including a previously defined core cell cycle gene set of cell cycle phase [65].
Single-cell telomere length assay
Single-cell telomere length was measured using single-cell telomere length measurement by qPCR assay, as previously described [36]. A multiplex pre-amplification (pre-PCR) step that can amplify telomere repeats (T) and Alu reference gene (R) were simultaneously employed with telomere primers (forward primer: CGGTTTGTTTGGGTTTGGGTTTGGGTTTGGGTTTGGGTT, reverse primer: GGCTTGCCTTACCCTTACCCTTACCCTTACCCTTACCCT), and Alu primers (forward primer: GACCATCCCGGCTAAAACG, reverse primer: CGGGTTCACGCCATTCTC). The reactions were set up by 25-μL aliquota of a master mix with single-cell gDNA beads. Each reaction was set up with 2.5 μL of 10X iTaq buffer, 1.5 mM MgCl2, 0.625U iTaq DNA polymerase (170-8870, Bio-Rad), 0.5 μL of 10 mM dNTP mix (170-8874, Bio-Rad), 1 μL each of telomere forward and reverse primers (10 μM), and 1 μL each of Alu forward and reverse primers (10 μM). Thermal cycler reaction conditions were set at 95 °C for 3 min followed by 18 cycles of 95 °C for 30 s, 60 °C annealing for 30 s, and extension at 72 °C for 30 s. PCR products were purified using DNA clean and concentrator-5 kit (D4004, Zymo Research) and eluted in 64 μL of double distilled water. iQ™ SYBR® Green-based real-time PCR (170-8882, Bio-Rad) was performed using the same primer sets. Relative telomere length (T/R ratio) was calculated by comparing the values of telomere (T) and reference gene Alu (R) in individual cells by the 2-ΔΔCt method when the standard curves of telomere and Alu showed similar high amplification efficiency. We defined the cells with T/R ratio over 1.0 as the “long” telomere group, T/R ratio ranges from 0.4 to 1.0 as the “medium” group, and T/R ratio under 0.4 as the “short” group under our experimental conditions.
Telomere measurement by quantitative real-time PCR (T/S ratio)
Genomic DNA was extracted using DNeasy Blood & Tissue kit (69504, Qiagen) and the ratio of 260 to 280 was between 1.8 and 2.1. Average telomere length was measured using qPCR assay, as previously described [27, 36]. Each 20 μL reaction was performed as follows: 35 ng gDNA, 1 × SYBR Green master mix (QPK-201, TOYOBO), 250 nM telomere forward primer and 250 nM telomere reverse primer, or 36B4 primers (forward primer: CAGCAAGTGGGAAGGTGTAATCC, reverse primer: CCCATTCTATCATCAACGGGTACAA). The telomere signal (T) was normalized to the signal from single copy gene (S) human 36B4 to generate a T/S ratio indicative of relative telomere length according to a standard curve. Three repeat reactions were performed for each sample.
Telomere terminal restriction fragment (TRF) by Southern blot analysis
TRF analysis was performed as described using TeloTAGGG Telomere Length Assay Kit (12209136001, Roche). DNA (1.5 μg) was digested using Hinf I and Rsa I restriction enzymes. Digested DNA underwent electrophoresis through a 0.8% agarose gel (111860, Biowest) for 4 h at 6 V/cm in 1× TAE buffer. Gels were denatured, neutralized, and transferred to positively charged nylon membranes (RPN2020B, GE Healthcare). The membranes were hybridized in DIG Easy Hyb containing telomere probe at 42 °C overnight. Mean TRF length was quantitatively measured according to the kit instructions.
Single-cell qPCR
Single-cell mRNA was directly reverse transcribed to cDNA by Smart-seq2, and the product diluted at 0.25 ng/μL. Real-time qPCR reaction was performed using FastStart Universal SYBR Green Master (4913914001, Roche). All genes were confirmed for their specificity by dissociation curves and amplification curves, and GAPDH served as housekeeping gene for normalization of gene expression. Primers used for qPCR experiments are listed in Additional file 19: Table S11.
Immunofluorescence microscopy and quantification
Cells were washed twice with PBS, then fixed in freshly prepared 3.7% paraformaldehyde for 15 min at 4 °C, permeabilized with 0.1% Triton X-100 in blocking solution (3% goat serum plus 0.5% BSA in PBS) for 40 min at room temperature, washed three times (each for 15 min), and left in blocking solution for 1 h. Cells were incubated overnight at 4 °C with primary antibodies against OCT4/POU5F1 (Cat# sc-9081, Lot# G0607, RRID: AB_2167703, Santa Cruz), NANOG (Cat# sc-293121, Lot# F2716, RRID:AB_2665475, Santa Cruz), and γH2AX (Cat# ab11175, RRID:AB_297814) and incubated for 1 h with fluorescence-labeled secondary antibodies. Samples were washed and counterstained with 0.5 μg/mL DAPI in Vectashield mounting medium. Fluorescence was detected and imaged using a Zeiss inverted fluorescence microscope. IF-FISH was performed as described [66]. Integrated fluorescence intensity was estimated using ImageJ software, and the threshold was defined using non-specific background staining fluorescence.
Knockout of TERF1 or TERC by CRISPR/Cas9
pSpCas9(BB)-2A-Puro (PX459, Addgene plasmid #48139) and pSpCas9(BB)-2A-GFP (PX458, Addgene plasmid #48138) were a gift from Feng Zhang. Guide RNAs were designed using the online design tool available at http://crispr.genome-engineering.org/. PX458/PX459 was digested with BbsI and then gel purified. Two pairs of oligos including targeting sequences were annealed, guide RNAs of TERF1 were cloned into BbsI-digested PX459, and guide RNAs of TERC were cloned into BbsI-digested PX458. Primers used for CRISPR/Cas9 experiments are listed in Additional file 19: Table S11.
Western blot
Western blot was performed as described previously [66] and the antibodies used were TRF1/TERF1 (Cat# ab10579, Lot# GR253870-4, RRID:AB_2201461, Abcam), OCT4/POU5F1 (Cat# sc-9081, Lot# G0607, RRID: AB_2167703, Santa Cruz), SOX2 (Cat# AB5603, Lot# 2762353, RRID: AB_2286686, Millipore), and β-actin (Cat# sc-1616R, Lot# A3009, RRID: AB_630836, Santa Cruz). The protein bands were detected by Enhanced ECL AmershamTM prime Western blotting detection reagent (RPN2232, GE Healthcare).
Telomerase activity assay
Telomerase activity was determined using TeloChaser Telomerase assay kit (TLK-101, TOYOBO). Approximately 2.5 × 104 cells from each sample were lysed, and heated at 70 °C for 10 min to serve as negative controls. PCR products of cell lysates were separated on non-denaturing TBE-based 12% polyacrylamide gel electrophoresis and visualized by ethidium bromide staining.
Gene expression by quantitative real-time PCR
Total RNA was isolated from cells using RNeasy mini kit (74104, Qiagen). RNA (2 μg) was subjected to cDNA synthesis using M-MLV Reverse Transcriptase (28025021, Invitrogen). Real-time quantitative PCR reactions were set up in duplicate with FastStart Universal SYBR Green Master (4913914001, Roche) and run on the iCycler iQ5 2.0 Standard Edition Optical System (Bio-Rad). Each sample was repeated three times and analyzed using GAPDH as the internal control. Primers used for qPCR experiments are listed in Additional file 19: Table S11.
Cell cycle analysis
Cells were fixed in freshly prepared 70% ethanol at 4 °C overnight, then centrifuged at 1000 g for 5 min and stained with propidium iodide at 37 °C for 30 min in a water bath. FACS analysis was used to determine cell cycle phases.
Statistics and reproducibility
Correlation between telomere length and expression level of genes was analyzed using Pearson’s correlation. Significance in different groups was analyzed by Student’s t test (two-paired groups) and ANOVA (multiple groups). Significant differences were defined as *P < 0.05 and **P < 0.01. The results were shown as mean ± SD.
Additional files
Measurement of relative telomere length in single cells by scT&R-seq of various human cell lines and validation by other established methods. To validate the feasibility of this method, we analyzed the telomere length and gene expression of six single cultured osteosarcoma cells (U2OS, ALT pathway), and six single human ESCs (WA26, telomerase activity-positive). (a) Mean Ct value of single human cell after preamplification by telomere and Alu primers in WA26 and U2OS. Both Ct value of Tel and Alu of negative controls are much greater than those of single cells. (b) Standard curves used for calculation of relative telomere length by scT&R-seq. Standard curves of telomeres and Alu using human embryonic fibroblasts (HEF) show remarkable linear correlation from DNA concentrations 0.2 ng to 125 ng, indicating a high amplification efficiency of telomeres and Alu. (c) Mean Ct value of GAPDH from the same cell by scT&R-seq and value of GAPDH does not differ in these cells. OCT4/POU5F1 is highly expressed in hESC but not in U2OS, so WA26 has a low Ct value for POU5F1. (d) Comparison of telomere length by the method without separation step and scT&R-seq method. (e) Relative telomere length of individual cells from human ESC WA26 and RuES2, human cancer cells U2OS, HCT116 and HeLa, and HEF. (f) Telomere length measurement by scT&R-seq. (g) Telomere length measurement by qPCR (T/S ratio). (h) Measurement of telomere length by southern blot of terminal restriction fragments (TRF). (i) Immunofluorescence images of OCT4/POU5F1 protein expression level in WA26 and HEF cells. Scale bar = 50 μm. (j) TRAP assay of telomerase activity in hESC (WA26) and U2OS. IC, internal control; HI, heat-inactivated; NT, not treated. All values indicate mean ± SD from three independent experiments. (PDF 888 kb)
Validation of telomere length and gene expression by scT&R-seq of TERC knockout and WT hESCs. (a) Morphology of TERC -/- and WT hESCs. Scale bar = 200 μm. Two pairs of guide RNAs were cloned into pSpCas9(BB)-2A-GFP (PX458, Addgene plasmid # 48138, a gift from Feng Zhang) for construction of the TERC knockout cells. Primers used for CRISPR/Cas9 experiments are listed in Additional file 19: Table S11. (b) Telomerase activity by TRAP assay of TERC -/- and WT hESCs. (c) Telomere length by TRF of hESCs after TERC knockout. (d) Relative telomere length of single cells from TERC -/- and WT hESCs using scT&R-seq. (e) Comparison of Ct values for housekeeping gene (GAPDH) in TERC -/- and WT hESCs. (f) Single cell qPCR analysis of expression of NANOG in the same cell from TERC -/- and WT hESCs using scT&R-seq. (g) Relative expression levels by qPCR of NANOG in bulk TERC -/- and WT hESCs. (h) Linear regression analysis of telomere length and NANOG expression at single cell level. (PDF 590 kb)
Single cell telomere length T/R ratio and PluriNet score. Telomere length of single hESCs by scT&R-seq and PluriNet score of single cell by average of the expression level of genes from PluriNet gene set. (XLSX 16 kb)
Quality control of single-cell RNA-seq analysis. (a) Cumulative gene diversity and gene-body coverage profile across lengths of all genes, and the reads coverage along the position from 5′ to 3′. (b, c) Total mapped reads and mapped ratio of single cell RNA-seq data. The reads were filtered and mapped to hg19 (ftp://igenome:G3nom3s4u@ussd-ftp.illumina.com/Homo_sapiens/UCSC/hg19/Homo_sapiens_UCSC_hg19.tar.gz) by TopHat (v2.1.0) with default parameters. Read counts for each gene were calculated for each replicate using HTSeq with default parameters. (d) Total number of genes detected with different transcripts per million value (TPM). (e) Heat-map shows top 40 genes that are highly expressed in all hESCs. The genes are ranked based on average of TPM value of all cells. (f) Plot of gene expression profile related to cell-cycle phases of hESCs, and distribution of telomere length at various cell cycle phases. (PDF 1307 kb)
Normalized counts (TPM) for all 121 single cells. All detected genes’ (>10 counts in ≥ 5 single cells) raw counts were normalized by library size via ‘estimateSizeFactorsForMatrix’ function from R package DESeq. (XLSX 15470 kb)
Validation of single-cell RNA-seq data by quantitative real-time PCR. (a, c) Single cell gene expression profile revealed by single cell RNA-seq. (b, d) Relative single cell gene expression level by single cell qPCR. (e, f) Correlation between RNA-seq and qPCR evaluated by Pearson’s correlation coefficient. (PDF 416 kb)
Top genes in the expression levels showing positive or negative association with telomere length of hESCs. (XLSX 9 kb)
Telomere length in association with expression level of all detected genes. (XLSX 588 kb)
Results of principal component analysis (PCA). All PCAs presented in this manuscript were conducted using log2 (TPM + 1) values of all detected genes using the PCA_int() function. (XLS 178 kb)
The mapping ratio of single cell libraries is not correlated with telomere length (T/R ratio). (a) Cumulative gene diversity plot showing the proportion of the library size accounted for by the expressed features across different telomere groups. (b) Linear regression analysis of telomere length (T/R ratio) and mapping ratio at single cell level. (PDF 223 kb)
Differential gene expression in various groups based on T/R ratio and PluriNet score. (XLSX 307 kb)
Functional annotation analysis of the differentially expressed genes from long, medium and short telomere groups. Functional annotation was performed by the assignment of Gene Ontology (GO) terms, and differentially expressed genes were annotated based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) using the R package SCDE. (PDF 1040 kb)
Complete list of gene sets used in this study, including selected housekeeping, pluripotency and differentiation genes, and genes for PluriNet and telomere length maintenance from GSEA. (XLSX 14 kb)
PluriNet score analysis of pluripotency state of human ESCs and differentiated cells. (a) Verification of PluriNet score for characterization of the pluripotency level. We validated the pluripotency score (named PluriNet score) using the single cell data of human pluripotent stem cells and differentiation cells from Thomson’s group [45]. PluriNet score displayed higher in undifferentiated hESCs than did differentiated cells from the same precursor cell. (b) Heat-map shows expression of PluriNet genes in all hESCs. Gene list of PluriNet was obtained from the GSEA website (Additional file 14: Table S6). The vertical color scale is shown as log(TPM + 1). (PDF 4032 kb)
GO Biological Process enrichment for up-regulated genes based on T/R ratio and PluriNet score group by SCDE. (XLSX 51 kb)
Heat-map showing expression of marker genes for human naïve ESCs. Of the 12 selected genes related to pluripotency, genes for naïve pluripotency, NANOG, KLF2, KLF4, TBX3, DPPA3 (STELLA), DPPA5, PRDM14, ESRRB, and REX1 are expressed at high levels in human naïve ESCs, whereas OTX2, ZIC2, ZIC3, and FGF5 decreased in human naïve ESCs. The vertical color scale is shown as log(TPM + 1). (PDF 1132 kb)
Enriched KEGG pathways for down-regulated genes in long vs. short telomere groups. (XLS 4 kb)
Quantification of gene expression by RT-qPCR. Gene expression levels were calculated by 2-ΔΔCt method, GAPDH served as an internal control. (XLSX 10 kb)
Primer sequences. Primers for knock-out of hTERF1 and hTERC by CRISPR/Cas9, and primers for qPCR. (XLSX 10 kb)
Acknowledgments
We thank Fang Wang for technical help with developing scT&R-seq, Fang Wang and David Keefe for providing WA26 and RuES2 hESCs, Feng Qi for HCT116 cell line, and Xinghua Pan and Sherman Weissman for advice.
Funding
This work was supported by funding from the China Ministry of Science and Technology Program of International S&T Cooperation (no. 2014DFA30450) and PCSIRT (no. IRT13023).
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its additional files. The scRNA-seq raw data have been deposited on GEO under accession number GSE98644.
Abbreviations
- ALT
alternative lengthening of telomeres
- ESC
embryonic stem cell
- GO
Gene ontology
- HEF
human embryonic fibroblast cell
- IF-FISH
immunofluorescence-fluorescence in situ hybridization
- iPSC
induced pluripotent stem cell
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- PCA
principal component analysis
- Q-FISH
quantitative fluorescence in situ hybridization
- qPCR
real-time quantitative polymerase chain reaction
- scT&R-seq
single cell telomere length and transcriptome sequencing
- TPM
transcripts per million values
- TRAP
telomeric repeat amplification protocol
- TRF
telomere terminal restriction fragment
- WT
wild-type
Authors’ contributions
LL conceived of the study. HW and LL designed the experiments. KSZ, YFL, YDF, HYW, and SG assisted in experimental design. HW performed the experiments and analyzed the data. KSZ performed data analysis. YDF, SG, PG, MZ, ZFW, YS, and TC helped the experiments or data analysis. SGL contributed the data analysis tool and supervised data analysis. HW, SGL and LL wrote the manuscript. HW and KSZ contributed equally to the study. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12915-017-0453-8) contains supplementary material, which is available to authorized users.
Contributor Information
Hua Wang, Email: moodswh@gmail.com.
Kunshan Zhang, Email: mart555@163.com.
Yifei Liu, Email: yifei.liu@yale.edu.
Yudong Fu, Email: 1203781972@qq.com.
Shan Gao, Email: gao_shan@mail.nankai.edu.cn.
Peng Gong, Email: perylgong@163.com.
Haiying Wang, Email: 1066525604@qq.com.
Zhongcheng Zhou, Email: loyal.zhongcheng@163.com.
Ming Zeng, Email: zengming700@163.com.
Zhenfeng Wu, Email: wu_zhenfeng@mail.nankai.edu.cn.
Yu Sun, Email: 531784197@qq.com.
Tong Chen, Email: ct@ehbio.com.
Siguang Li, Email: siguangli@163.com.
Lin Liu, Email: liulin@nankai.edu.cn, Email: liutelom@yahoo.com.
References
- 1.Hackett JA, Feldser DM, Greider CW. Telomere dysfunction increases mutation rate and genomic instability. Cell. 2001;106(3):275–86. doi: 10.1016/S0092-8674(01)00457-3. [DOI] [PubMed] [Google Scholar]
- 2.O’Sullivan JN, Bronner MP, Brentnall TA, Finley JC, Shen WT, Emerson S, Emond MJ, Gollahon KA, Moskovitz AH, Crispin DA, et al. Chromosomal instability in ulcerative colitis is related to telomere shortening. Nat Genet. 2002;32(2):280–4. doi: 10.1038/ng989. [DOI] [PubMed] [Google Scholar]
- 3.Rudolph KL, Millard M, Bosenberg MW, DePinho RA. Telomere dysfunction and evolution of intestinal carcinoma in mice and humans. Nat Genet. 2001;28(2):155–9. doi: 10.1038/88871. [DOI] [PubMed] [Google Scholar]
- 4.Shay JW, Reddel RR, Wright WE. Cancer and telomeres-an ALTernative to telomerase. Science. 2012;336(6087):1388–90. doi: 10.1126/science.1222394. [DOI] [PubMed] [Google Scholar]
- 5.Gunes C, Rudolph KL. The role of telomeres in stem cells and cancer. Cell. 2013;152(3):390–3. doi: 10.1016/j.cell.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 6.Bryan TM, Englezou A, Dalla-Pozza L, Dunham MA, Reddel RR. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat Med. 1997;3(11):1271–4. doi: 10.1038/nm1197-1271. [DOI] [PubMed] [Google Scholar]
- 7.Huang J, Wang F, Okuka M, Liu N, Ji G, Ye X, Zuo B, Li M, Liang P, Ge WW, et al. Association of telomere length with authentic pluripotency of ES/iPS cells. Cell Res. 2011;21(5):779–92. doi: 10.1038/cr.2011.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marion RM, Strati K, Li H, Tejera A, Schoeftner S, Ortega S, Serrano M, Blasco MA. Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell. 2009;4(2):141–54. doi: 10.1016/j.stem.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 9.Wang F, Yin Y, Ye XY, Liu K, Zhu HY, Wang LL, Chiourea M, Okuka M, Ji GZ, Dan JM, et al. Molecular insights into the heterogeneity of telomere reprogramming in induced pluripotent stem cells. Cell Res. 2012;22(4):757–68. doi: 10.1038/cr.2011.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Le RR, Kou ZH, Jiang YH, Li M, Huang B, Liu WQ, Li H, Kou XC, He WZ, Rudolph KL, et al. Enhanced telomere rejuvenation in pluripotent cells reprogrammed via nuclear transfer relative to induced pluripotent stem cells. Cell Stem Cell. 2014;14(1):27–39. doi: 10.1016/j.stem.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 11.Sung LY, Chang WF, Zhang Q, Liu CC, Liou JY, Chang CC, Ou-Yang H, Guo R, Fu H, Cheng WT, et al. Telomere elongation and naive pluripotent stem cells achieved from telomerase haplo-insufficient cells by somatic cell nuclear transfer. Cell Rep. 2014;9(5):1603–9. doi: 10.1016/j.celrep.2014.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pucci F, Gardano L, Harrington L. Short telomeres in ESCs lead to unstable differentiation. Cell Stem Cell. 2013;12(4):479–86. doi: 10.1016/j.stem.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Varela E, Munoz-Lorente MA, Tejera AM, Ortega S, Blasco MA. Generation of mice with longer and better preserved telomeres in the absence of genetic manipulations. Nat Commun. 2016;7:11739. doi: 10.1038/ncomms11739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeng SC, Liu LJ, Sun Y, Xie PY, Hu L, Yuan D, Chen DH, Ouyang Q, Lin G, Lu GX. Telomerase-mediated telomere elongation from human blastocysts to embryonic stem cells. J Cell Sci. 2014;127(4):752–62. doi: 10.1242/jcs.131433. [DOI] [PubMed] [Google Scholar]
- 15.Agarwal S, Loh YH, McLoughlin EM, Huang JJ, Park IH, Miller JD, Huo HG, Okuka M, dos Reis RM, Loewer S, et al. Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients. Nature. 2010;464(7286):292–6. doi: 10.1038/nature08792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Batista LF, Pech MF, Zhong FL, Nguyen HN, Xie KT, Zaug AJ, Crary SM, Choi J, Sebastiano V, Cherry A, et al. Telomere shortening and loss of self-renewal in dyskeratosis congenita induced pluripotent stem cells. Nature. 2011;474(7351):399–402. doi: 10.1038/nature10084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takashima Y, Guo G, Loos R, Nichols J, Ficz G, Krueger F, Oxley D, Santos F, Clarke J, Mansfield W, et al. Resetting transcription factor control circuitry toward ground-state pluripotency in human. Cell. 2014;158(6):1254–69. doi: 10.1016/j.cell.2014.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pastor WA, Chen D, Liu W, Kim R, Sahakyan A, Lukianchikov A, Plath K, Jacobsen SE, Clark AT. Naive human pluripotent cells feature a methylation landscape devoid of blastocyst or germline memory. Cell Stem Cell. 2016;18(3):323–9. doi: 10.1016/j.stem.2016.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weinberger L, Ayyash M, Novershtern N, Hanna JH. Dynamic stem cell states: naive to primed pluripotency in rodents and humans. Nat Rev Mol Cell Biol. 2016;17(3):155–69. doi: 10.1038/nrm.2015.28. [DOI] [PubMed] [Google Scholar]
- 20.Theunissen TW, Powell BE, Wang H, Mitalipova M, Faddah DA, Reddy J, Fan ZP, Maetzel D, Ganz K, Shi L, et al. Systematic identification of culture conditions for induction and maintenance of naive human pluripotency. Cell Stem Cell. 2014;15(4):524–6. doi: 10.1016/j.stem.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sahakyan A, Kim R, Chronis C, Sabri S, Bonora G, Theunissen TW, Kuoy E, Langerman J, Clark AT, Jaenisch R, et al. Human naive pluripotent stem cells model X chromosome dampening and X inactivation. Cell Stem Cell. 2017;20(1):87–101. doi: 10.1016/j.stem.2016.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boroviak T, Nichols J. Primate embryogenesis predicts the hallmarks of human naïve pluripotency. Development. 2017;144(2):175–86. doi: 10.1242/dev.145177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345(6274):458–60. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 24.Zijlmans JM, Martens UM, Poon SS, Raap AK, Tanke HJ, Ward RK, Lansdorp PM. Telomeres in the mouse have large inter-chromosomal variations in the number of T2AG3 repeats. Proc Natl Acad Sci U S A. 1997;94(14):7423–8. doi: 10.1073/pnas.94.14.7423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rufer N, Dragowska W, Thornbury G, Roosnek E, Lansdorp PM. Telomere length dynamics in human lymphocyte subpopulations measured by flow cytometry. Nat Biotechnol. 1998;16(8):743–7. doi: 10.1038/nbt0898-743. [DOI] [PubMed] [Google Scholar]
- 26.Cimino-Reale G, Pascale E, Battiloro E, Starace G, Verna R, D’Ambrosio E. The length of telomeric G-rich strand 3′-overhang measured by oligonucleotide ligation assay. Nucleic Acids Res. 2001;29(7):E35. doi: 10.1093/nar/29.7.e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cawthon RM. Telomere measurement by quantitative PCR. Nucleic Acids Res. 2002;30(10):e47. doi: 10.1093/nar/30.10.e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baird DM, Rowson J, Wynford-Thomas D, Kipling D. Extensive allelic variation and ultrashort telomeres in senescent human cells. Nat Genet. 2003;33(2):203–7. doi: 10.1038/ng1084. [DOI] [PubMed] [Google Scholar]
- 29.Stuart BD, Choi J, Zaidi S, Xing C, Holohan B, Chen R, Choi M, Dharwadkar P, Torres F, Girod CE, et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat Genet. 2015;47(5):512–7. doi: 10.1038/ng.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Codd V, Nelson CP, Albrecht E, Mangino M, Deelen J, Buxton JL, Hottenga JJ, Fischer K, Esko T, Surakka I, et al. Identification of seven loci affecting mean telomere length and their association with disease. Nat Genet. 2013;45(4):422–7. doi: 10.1038/ng.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shapiro E, Biezuner T, Linnarsson S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat Rev Genet. 2013;14(9):618–30. doi: 10.1038/nrg3542. [DOI] [PubMed] [Google Scholar]
- 32.Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, Wang X, Bodeau J, Tuch BB, Siddiqui A, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6(5):377–82. doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- 33.Reuter JA, Spacek DV, Pai RK, Snyder MP. Simul-seq: combined DNA and RNA sequencing for whole-genome and transcriptome profiling. Nat Methods. 2016;13(11):953–8. doi: 10.1038/nmeth.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wen L, Tang F. Single-cell sequencing in stem cell biology. Genome Biol. 2016;17:71. doi: 10.1186/s13059-016-0941-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Macaulay IC, Haerty W, Kumar P, Li YI, Hu TX, Teng MJ, Goolam M, Saurat N, Coupland P, Shirley LM, et al. G&T-seq: parallel sequencing of single-cell genomes and transcriptomes. Nat Methods. 2015;12(6):519–22. doi: 10.1038/nmeth.3370. [DOI] [PubMed] [Google Scholar]
- 36.Wang F, Pan XH, Kalmbach K, Seth-Smith ML, Ye XY, Antumes DMF, Yin Y, Liu L, Keefe DL, Weissman SM. Robust measurement of telomere length in single cells. Proc Natl Acad Sci U S A. 2013;110(21):E1906–12. doi: 10.1073/pnas.1306639110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li L, Dong J, Yan L, Yong J, Liu X, Hu Y, Fan X, Wu X, Guo H, Wang X, et al. Single-cell RNA-Seq analysis maps development of human germline cells and gonadal niche interactions. Cell Stem Cell. 2017;20(6):858–73. doi: 10.1016/j.stem.2017.03.007. [DOI] [PubMed] [Google Scholar]
- 38.Buettner F, Natarajan KN, Casale FP, Proserpio V, Scialdone A, Theis FJ, Teichmann SA, Marioni JC, Stegle O. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat Biotechnol. 2015;33(2):155–60. doi: 10.1038/nbt.3102. [DOI] [PubMed] [Google Scholar]
- 39.Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–34. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- 40.McCarthy DJ, Campbell KR, Lun ATL, Wills QF. Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics. 2017;33(8):1179–86. doi: 10.1093/bioinformatics/btw777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ji HK, Liu XS. Analyzing ‘omics data using hierarchical models. Nat Biotechnol. 2010;28(4):337–40. doi: 10.1038/nbt.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamaguchi H, Calado RT, Ly H, Kajigaya S, Baerlocher GM, Chanock SJ, Lansdorp PM, Young NS. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med. 2005;352(14):1413–24. doi: 10.1056/NEJMoa042980. [DOI] [PubMed] [Google Scholar]
- 43.Li JS, Miralles Fuste J, Simavorian T, Bartocci C, Tsai J, Karlseder J, Lazzerini Denchi E. TZAP: a telomere-associated protein involved in telomere length control. Science. 2017;355(6325):638–41. doi: 10.1126/science.aah6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muller FJ, Laurent LC, Kostka D, Ulitsky I, Williams R, Lu C, Park IH, Rao MS, Shamir R, Schwartz PH, et al. Regulatory networks define phenotypic classes of human stem cell lines. Nature. 2008;455(7211):401–5. doi: 10.1038/nature07213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chu LF, Leng N, Zhang J, Hou Z, Mamott D, Vereide DT, Choi J, Kendziorski C, Stewart R, Thomson JA. Single-cell RNA-seq reveals novel regulators of human embryonic stem cell differentiation to definitive endoderm. Genome Biol. 2016;17(1):173. doi: 10.1186/s13059-016-1033-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adewumi O, Aflatoonian B, Ahrlund-Richter L, Amit M, Andrews PW, Beighton G, Bello PA, Benvenisty N, Berry LS, Bevan S, et al. Characterization of human embryonic stem cell lines by the International Stem Cell Initiative. Nat Biotechnol. 2007;25(7):803–16. doi: 10.1038/nbt1318. [DOI] [PubMed] [Google Scholar]
- 47.Felgentreff K, Du L, Weinacht KG, Dobbs K, Bartish M, Giliani S, Schlaeger T, DeVine A, Schambach A, Woodbine LJ, et al. Differential role of nonhomologous end joining factors in the generation, DNA damage response, and myeloid differentiation of human induced pluripotent stem cells. Proc Natl Acad Sci U S A. 2014;111(24):8889–94. doi: 10.1073/pnas.1323649111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang J, Ratanasirintrawoot S, Chandrasekaran S, Wu Z, Ficarro SB, Yu C, Ross CA, Cacchiarelli D, Xia Q, Seligson M, et al. LIN28 regulates stem cell metabolism and conversion to primed pluripotency. Cell Stem Cell. 2016;19(1):66–80. doi: 10.1016/j.stem.2016.05.009. [DOI] [PubMed] [Google Scholar]
- 49.Gu W, Gaeta X, Sahakyan A, Chan AB, Hong CS, Kim R, Braas D, Plath K, Lowry WE, Christofk HR. Glycolytic metabolism plays a functional role in regulating human pluripotent stem cell state. Cell Stem Cell. 2016;19(4):476–90. doi: 10.1016/j.stem.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martinez P, Thanasoula M, Munoz P, Liao C, Tejera A, McNees C, Flores JM, Fernandez-Capetillo O, Tarsounas M, Blasco MA. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev. 2009;23(17):2060–75. doi: 10.1101/gad.543509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Savage SA. Connecting complex disorders through biology. Nat Genet. 2012;44(3):238–40. doi: 10.1038/ng.2206. [DOI] [PubMed] [Google Scholar]
- 52.Laurent LC, Ulitsky I, Slavin I, Tran H, Schork A, Morey R, Lynch C, Harness JV, Lee S, Barrero MJ, et al. Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell. 2011;8(1):106–18. doi: 10.1016/j.stem.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet. 2012;13(10):693–704. doi: 10.1038/nrg3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heyer BS, Warsowe J, Solter D, Knowles BB, Ackerman SL. New member of the Snf1/AMPK kinase family, Melk, is expressed in the mouse egg and preimplantation embryo. Mol Reprod Dev. 1997;47(2):148–56. doi: 10.1002/(SICI)1098-2795(199706)47:2<148::AID-MRD4>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 55.Nakano I, Masterman-Smith M, Saigusa K, Paucar AA, Horvath S, Shoemaker L, Watanabe M, Negro A, Bajpai R, Howes A, et al. Maternal embryonic leucine zipper kinase is a key regulator of the proliferation of malignant brain tumors, including brain tumor stem cells. J Neurosci Res. 2008;86(1):48–60. doi: 10.1002/jnr.21471. [DOI] [PubMed] [Google Scholar]
- 56.Rizki A, Lundblad V. Defects in mismatch repair promote telomerase-independent proliferation. Nature. 2001;411(6838):713–6. doi: 10.1038/35079641. [DOI] [PubMed] [Google Scholar]
- 57.Shahi A, Lee JH, Kang Y, Lee SH, Hyun JW, Chang IY, Jun JY, You HJ. Mismatch-repair protein MSH6 is associated with Ku70 and regulates DNA double-strand break repair. Nucleic Acids Res. 2011;39(6):2130–43. doi: 10.1093/nar/gkq1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Doksani Y, Wu JY, de Lange T, Zhuang X. Super-resolution fluorescence imaging of telomeres reveals TRF2-dependent T-loop formation. Cell. 2013;155(2):345–56. doi: 10.1016/j.cell.2013.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Los AA, Ferrari F, Xi R, Fujiwara Y, Benvenisty N, Deng H, Hochedlinger K, Jaenisch R, Lee S, Leitch HG, et al. Hallmarks of pluripotency. Nature. 2015;525(7570):469–78. doi: 10.1038/nature15515. [DOI] [PubMed] [Google Scholar]
- 60.Sperber H, Mathieu J, Wang Y, Ferreccio A, Hesson J, Xu Z, Fischer KA, Devi A, Detraux D, Gu H, et al. The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat Cell Biol. 2015;17(12):1523–35. doi: 10.1038/ncb3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Picelli S, Faridani OR, Bjorklund AK, Winberg G, Sagasser S, Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc. 2014;9(1):171–81. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- 62.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brennecke P, Anders S, Kim JK, Kolodziejczyk AA, Zhang X, Proserpio V, Baying B, Benes V, Teichmann SA, Marioni JC, et al. Accounting for technical noise in single-cell RNA-seq experiments. Nat Methods. 2013;10(11):1093–5. doi: 10.1038/nmeth.2645. [DOI] [PubMed] [Google Scholar]
- 64.Kharchenko PV, Silberstein L, Scadden DT. Bayesian approach to single-cell differential expression analysis. Nat Methods. 2014;11(7):740–2. doi: 10.1038/nmeth.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161(5):1202–14. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dan J, Liu Y, Liu N, Chiourea M, Okuka M, Wu T, Ye X, Mou C, Wang L, Wang L, et al. Rif1 maintains telomere length homeostasis of ESCs by mediating heterochromatin silencing. Dev Cell. 2014;29(1):7–19. doi: 10.1016/j.devcel.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Measurement of relative telomere length in single cells by scT&R-seq of various human cell lines and validation by other established methods. To validate the feasibility of this method, we analyzed the telomere length and gene expression of six single cultured osteosarcoma cells (U2OS, ALT pathway), and six single human ESCs (WA26, telomerase activity-positive). (a) Mean Ct value of single human cell after preamplification by telomere and Alu primers in WA26 and U2OS. Both Ct value of Tel and Alu of negative controls are much greater than those of single cells. (b) Standard curves used for calculation of relative telomere length by scT&R-seq. Standard curves of telomeres and Alu using human embryonic fibroblasts (HEF) show remarkable linear correlation from DNA concentrations 0.2 ng to 125 ng, indicating a high amplification efficiency of telomeres and Alu. (c) Mean Ct value of GAPDH from the same cell by scT&R-seq and value of GAPDH does not differ in these cells. OCT4/POU5F1 is highly expressed in hESC but not in U2OS, so WA26 has a low Ct value for POU5F1. (d) Comparison of telomere length by the method without separation step and scT&R-seq method. (e) Relative telomere length of individual cells from human ESC WA26 and RuES2, human cancer cells U2OS, HCT116 and HeLa, and HEF. (f) Telomere length measurement by scT&R-seq. (g) Telomere length measurement by qPCR (T/S ratio). (h) Measurement of telomere length by southern blot of terminal restriction fragments (TRF). (i) Immunofluorescence images of OCT4/POU5F1 protein expression level in WA26 and HEF cells. Scale bar = 50 μm. (j) TRAP assay of telomerase activity in hESC (WA26) and U2OS. IC, internal control; HI, heat-inactivated; NT, not treated. All values indicate mean ± SD from three independent experiments. (PDF 888 kb)
Validation of telomere length and gene expression by scT&R-seq of TERC knockout and WT hESCs. (a) Morphology of TERC -/- and WT hESCs. Scale bar = 200 μm. Two pairs of guide RNAs were cloned into pSpCas9(BB)-2A-GFP (PX458, Addgene plasmid # 48138, a gift from Feng Zhang) for construction of the TERC knockout cells. Primers used for CRISPR/Cas9 experiments are listed in Additional file 19: Table S11. (b) Telomerase activity by TRAP assay of TERC -/- and WT hESCs. (c) Telomere length by TRF of hESCs after TERC knockout. (d) Relative telomere length of single cells from TERC -/- and WT hESCs using scT&R-seq. (e) Comparison of Ct values for housekeeping gene (GAPDH) in TERC -/- and WT hESCs. (f) Single cell qPCR analysis of expression of NANOG in the same cell from TERC -/- and WT hESCs using scT&R-seq. (g) Relative expression levels by qPCR of NANOG in bulk TERC -/- and WT hESCs. (h) Linear regression analysis of telomere length and NANOG expression at single cell level. (PDF 590 kb)
Single cell telomere length T/R ratio and PluriNet score. Telomere length of single hESCs by scT&R-seq and PluriNet score of single cell by average of the expression level of genes from PluriNet gene set. (XLSX 16 kb)
Quality control of single-cell RNA-seq analysis. (a) Cumulative gene diversity and gene-body coverage profile across lengths of all genes, and the reads coverage along the position from 5′ to 3′. (b, c) Total mapped reads and mapped ratio of single cell RNA-seq data. The reads were filtered and mapped to hg19 (ftp://igenome:G3nom3s4u@ussd-ftp.illumina.com/Homo_sapiens/UCSC/hg19/Homo_sapiens_UCSC_hg19.tar.gz) by TopHat (v2.1.0) with default parameters. Read counts for each gene were calculated for each replicate using HTSeq with default parameters. (d) Total number of genes detected with different transcripts per million value (TPM). (e) Heat-map shows top 40 genes that are highly expressed in all hESCs. The genes are ranked based on average of TPM value of all cells. (f) Plot of gene expression profile related to cell-cycle phases of hESCs, and distribution of telomere length at various cell cycle phases. (PDF 1307 kb)
Normalized counts (TPM) for all 121 single cells. All detected genes’ (>10 counts in ≥ 5 single cells) raw counts were normalized by library size via ‘estimateSizeFactorsForMatrix’ function from R package DESeq. (XLSX 15470 kb)
Validation of single-cell RNA-seq data by quantitative real-time PCR. (a, c) Single cell gene expression profile revealed by single cell RNA-seq. (b, d) Relative single cell gene expression level by single cell qPCR. (e, f) Correlation between RNA-seq and qPCR evaluated by Pearson’s correlation coefficient. (PDF 416 kb)
Top genes in the expression levels showing positive or negative association with telomere length of hESCs. (XLSX 9 kb)
Telomere length in association with expression level of all detected genes. (XLSX 588 kb)
Results of principal component analysis (PCA). All PCAs presented in this manuscript were conducted using log2 (TPM + 1) values of all detected genes using the PCA_int() function. (XLS 178 kb)
The mapping ratio of single cell libraries is not correlated with telomere length (T/R ratio). (a) Cumulative gene diversity plot showing the proportion of the library size accounted for by the expressed features across different telomere groups. (b) Linear regression analysis of telomere length (T/R ratio) and mapping ratio at single cell level. (PDF 223 kb)
Differential gene expression in various groups based on T/R ratio and PluriNet score. (XLSX 307 kb)
Functional annotation analysis of the differentially expressed genes from long, medium and short telomere groups. Functional annotation was performed by the assignment of Gene Ontology (GO) terms, and differentially expressed genes were annotated based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) using the R package SCDE. (PDF 1040 kb)
Complete list of gene sets used in this study, including selected housekeeping, pluripotency and differentiation genes, and genes for PluriNet and telomere length maintenance from GSEA. (XLSX 14 kb)
PluriNet score analysis of pluripotency state of human ESCs and differentiated cells. (a) Verification of PluriNet score for characterization of the pluripotency level. We validated the pluripotency score (named PluriNet score) using the single cell data of human pluripotent stem cells and differentiation cells from Thomson’s group [45]. PluriNet score displayed higher in undifferentiated hESCs than did differentiated cells from the same precursor cell. (b) Heat-map shows expression of PluriNet genes in all hESCs. Gene list of PluriNet was obtained from the GSEA website (Additional file 14: Table S6). The vertical color scale is shown as log(TPM + 1). (PDF 4032 kb)
GO Biological Process enrichment for up-regulated genes based on T/R ratio and PluriNet score group by SCDE. (XLSX 51 kb)
Heat-map showing expression of marker genes for human naïve ESCs. Of the 12 selected genes related to pluripotency, genes for naïve pluripotency, NANOG, KLF2, KLF4, TBX3, DPPA3 (STELLA), DPPA5, PRDM14, ESRRB, and REX1 are expressed at high levels in human naïve ESCs, whereas OTX2, ZIC2, ZIC3, and FGF5 decreased in human naïve ESCs. The vertical color scale is shown as log(TPM + 1). (PDF 1132 kb)
Enriched KEGG pathways for down-regulated genes in long vs. short telomere groups. (XLS 4 kb)
Quantification of gene expression by RT-qPCR. Gene expression levels were calculated by 2-ΔΔCt method, GAPDH served as an internal control. (XLSX 10 kb)
Primer sequences. Primers for knock-out of hTERF1 and hTERC by CRISPR/Cas9, and primers for qPCR. (XLSX 10 kb)
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its additional files. The scRNA-seq raw data have been deposited on GEO under accession number GSE98644.