

Abstract
Background
Congenital heart defects are the most common birth defects worldwide. Although defective Notch signaling is the major cause of mouse embryonic death from cardiovascular defects, how Notch signaling is regulated during embryonic vasculogenesis and heart development is poorly understood.
Methods and Results
Regulator of G protein signaling 6 (RGS6)−/−/Ca2+/calmodulin‐dependent protein kinase II (CaMKII)VV double mutant mice were developed by crossing RGS6−/− mice with mice expressing an oxidation‐resistant CaMKIIδ (CaMKIIVV), and the resulting embryonic defects/lethality were investigated using E7.5 to E15.5 embryos. While loss of either RGS6 or oxidized CaMKIIδ does not alter embryogenesis, their combined loss causes defective Notch signaling, severe cardiovascular defects, and embryonic lethality (≈E10.5–11.5). Embryos lacking RGS6 and expressing oxidation‐resistant CaMKIIδ exhibit reduced myocardial wall thickness, abnormal trabeculation, and arterial specification defects. Double mutants show vascular remodeling defects, including reduced neurovascularization, delayed neural tube maturation, and small dorsal aortae. These striking cardiovascular defects were accompanied by placental and yolk sac defects in angiogenesis, hematopoiesis, and vascular remodeling similar to what is seen with defective Notch1 signaling. Double mutant hearts, embryos, and yolk sacs exhibit profound downregulation of Notch1, Jagged 1, and Notch downstream target genes Hey1, Hey2, and Hey1L as well as impaired Notch1 signaling in embryos/hearts.
Conclusions
RGS6 and oxidized CaMKIIδ together function as novel critical upstream modulators of Notch signaling required for normal cardiovascular development and embryo survival. Their combined need indicates that they function in parallel pathways needed for Notch1 signaling in yolk sac, placenta and embryos. Thus, dysregulated embryonic RGS6 expression and oxidative activation of CaMKII may potentially contribute to congenital heart defects.
Keywords: cardiovascular development, embryonic lethality, Notch signaling, oxidized Ca2+/calmodulin‐dependent protein kinase II, Regulator of G protein signaling 6
Subject Categories: Congenital Heart Disease, Animal Models of Human Disease
Clinical Perspective
What Is New?
Defective Notch signaling during human and mouse development is known to cause congenital heart defects. However, it remains unclear how Notch signaling is regulated during embryonic vasculogenesis and heart development.
Here, Regulator of G protein signaling 6 and oxidized CaMKIIδ are found to function in parallel pathways as novel critical upstream modulators of Notch signaling and cardiovascular development. In evidence of this, their combined but not individual loss in mouse causes defective Notch signaling, severe cardiovascular defects, and embryonic lethality.
What Are the Clinical Implications?
The findings presented in this article suggest that altered embryonic expression of Regulator of G protein signaling 6 and oxidation of CaMKII may lead to the development of human congenital heart defects, the most common birth defects worldwide.
Further understanding of the possible underlying genetic causes of congenital heart defects will allow us to more accurately identify individuals whose children will experience these disorders and will lay the groundwork for future research into gene therapies that could be used to treat these disorders.
Congenital heart defects represent the most common developmental problem in newborns, occurring in 3% of live births.1, 2 The embryonic circulatory system includes vascular, cardiac, and hematopoietic components, which are derived from the mesoderm during gastrulation.3 Accurate vasculogenic, arteriogenic, and angiogenic mechanisms, all relying on precise control of gene expression, are needed for proper embryonic cardiovascular system development.4 Vasculogenesis, one of the earliest key events during embryogenesis, is a process whereby vascular endothelial progenitor cells (angioblasts) differentiate and coalesce to form the primary vascular plexus. This process is followed by a series of events leading to vascular remodeling and maturation including angiogenesis (sprouting, branching, splitting, and differential growth of vessels in primary plexus), arterial/venous specification, and recruitment of mural cells to vascular walls.5, 6
Numerous signaling molecules and pathways, including vascular endothelial growth factor (VEGF), transforming growth factor‐β, angiopoietin/Tie receptor, ephrin/Eph receptor, and Notch signaling, have been implicated in various stages of vascular development and endothelial function.7, 8, 9, 10, 11 The Notch signaling pathway, in particular, plays a major role in vertebrate development, functioning as a direct paracrine signaling system involved in cell fate determination, specification, proliferation, differentiation, and apoptosis.12 The Notch family consists of 4 Notch receptors (Notch 1–4) and 5 Notch ligands, Jagged (Jag) 1, Jag2, Delta‐like (Dll) 1, Dll3, and Dll4. Ligand‐receptor interaction between neighboring cells triggers γ‐secretase complex‐mediated proteolytic cleavage of the Notch receptor releasing the Notch intracellular domain (NICD) into the cytoplasm. The Notch intracellular domain then translocates into the nucleus and associates with the DNA‐binding protein RBPJK/CBF1/Su(H) and the co‐activator of the Mastermind‐like family, leading to transcription of downstream target genes, including hairy/enhancer of split‐related family with BHLH transcription factor (Hes) or with YRPW motif (Hey) family members.13, 14 In mice, global and endothelial‐specific gain or loss of function mutations in genes encoding the Notch receptor (Notch1, Notch3, Notch4, and Notch1/4)15, 16, 17, 18, 19 or its ligands (Jag1 and Dll1)20, 21, 22, 23 leads to death from vascular and cardiac defects at gestational days E9.5 to 11.5 or after birth. Likewise, mice with genetic deletions in Notch downstream pathway genes die at E10.5 as a result of defective vasculature.24, 25, 26 Importantly, defective Notch signaling in humans is associated with both the degenerative vascular disease cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL: missense mutation in Notch 3 gene) and Allagile syndrome (mutation in Jag1 or Notch 2).27, 28
Regulator of G protein signaling 6 (RGS6) is a member of the RGS protein family that function as GTPase‐activating proteins for specific Gα subunits. By promoting the inactivation of Gα subunits, RGS proteins control the intensity and duration of signaling through G protein–coupled receptors. RGS6 is a GTPase‐activating protein for Gαi/o subunits and regulates Gαi/o‐coupled receptors in the brain.29, 30, 31 Previous studies from our laboratory have shown that RGS6 is robustly expressed in the heart where it controls not only M2R signaling through its canonical G protein actions,32 but also mediates doxorubicin and alcohol‐induced myocardial cell apoptosis/cardiomyopathy by functioning as an upstream activator of NADPH oxidase (Nox)‐dependent reactive oxygen species (ROS) generation.33, 34 ROS act as secondary messengers or signaling molecules to mediate cardiomyocyte apoptosis. We hypothesized that this cardiomyocyte apoptosis could be facilitated by the ROS‐mediated oxidation of Ca2+/calmodulin‐dependent protein kinase II δ (ox‐CaMKII) on 2 key regulatory domain methionines (M281/282), as we demonstrated that ox‐CaMKII promotes myocardial injury and death.35 The present study was initiated to test the role of this RGS6‐Nox‐ROS‐ox‐CaMKII signaling axis in heart injury by determining whether mice with individual or combined loss of RGS6 and ox‐CaMKII phenocopied each other in terms of protection from ROS‐induced cardiomyocyte apoptosis. In this process, we discovered the critical role of RGS6 and ox‐CaMKII as upstream co‐modulators of Notch signaling and embryonic development.
Here, we report severe cardiovascular defects leading to early embryonic lethality in mice lacking RGS6 and expressing an oxidation‐resistant form of CaMKIIδ (RGS6−/−/CaMKIIVV) by knock‐in replacement of regulatory domain methionines with valines. These mice allowed us to directly interrogate the relationship between RGS6, ROS, and ox‐CaMKII in embryonic development. Mice lacking either RGS6 or ox‐CaMKII did not exhibit embryonic lethality. However, RGS6−/−/CaMKIIVV mice display extensive defects in cardiac, hematopoietic, and vascular remodeling that impacts the embryo, yolk sac, and placenta. Our findings demonstrate a critical combined need for RGS6 and ox‐CaMKII regulation of Notch signaling as well as downstream arterial/venous specification (vascular development) and cardiac development.
Methods
An expanded Methods section is available in Data S1.
Mice
RGS6 knockout (RGS6−/−) mice were backcrossed onto a C57BL6 background for 12 generations.32 Knock‐in mice expressing an oxidation‐resistant form of CaMKIIδ were generated on a C57BL6 background by replacing methionine 281 and 282 with valines.36 Wild‐type, heterozygous, and mutant CaMKIIδ mice are referred to as CaMKIIMM, CaMKIIMV, and CaMKIIVV, respectively. CaMKIIVV mice were crossed with RGS6−/− mice to generate RGS6−/−/CaMKIIMV mice. RGS6−/−/CaMKIIVV embryos were generated by intercrossing these RGS6−/−/CaMKIIMV mice. Mice/embryos were genotyped using polymerase chain reaction as previously described.32, 36 Primers and representative genotyping results are shown in Table S1 and Figure S1, respectively. Embryos from the same litter were used for experiments whenever possible to minimize litter effects. All animal procedures were approved and experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals.
Histology
Embryos, yolk sacs, and placenta were fixed with 4% paraformaldehyde at 4°C overnight. Tissues were dehydrated and embedded in paraffin wax for sectioning as described.37 Paraffin‐embedded slices (10 μm) of embryos, yolk sacs, and placentas were prepared in the Central Microscopy Research Facility of the University of Iowa. Hematoxylin and eosin staining was also performed in the Central Microscopy Research Facility.
Myocardium Thickness and Extent of Trabeculation
Tissue sections (stained with hematoxylin and eosin) were examined by a boarded veterinary pathologist. For morphometry, post‐examination masking to groups was applied.38 Myocardial wall area (not including trabeculae) was enumerated and this value was divided by the circumference of the heart in each section to prevent sectioning artifacts. These were then normalized to the control mean to produce relative myocardial diameter values. Trabeculation was assessed through quantification of the trabecular area (not including the myocardial wall) and these values were divided by the circumference and normalized to the mean values of the CON as described above.
Quantitative Polymerase Chain Reaction
Total RNA isolation, cDNA synthesis, and quantitative polymerase chain reaction was performed as previously described39 and details are provided in Data S1.
Western Blot Analysis
Western blotting was performed as previously described34 and details are provided in Data S1.
MEFs Isolation and Culture
Mouse embryonic fibroblasts (MEFs) were isolated from E10.5 RGS6+/+/CaMKIIMM, RGS6−/−/CaMKIIMM, and RGS6−/−/CaMKIIVV embryos according to standard protocols.40 MEFs were grown in DMEM supplemented with 10% FBS and penicillin/streptomycin (10 U/mL). At passage 2, MEFs of these genotypes were seeded in 96‐well plates (104 cells per well) and allowed to grow for 2, 6, and 8 days. At different days, MTT was added and absorbance at 550 nm was determined as previously described.41 Intracellular ROS generation was estimated using the cell‐permeable oxidation‐sensitive probe CM‐H2DCFDA (Sigma) as previously described.41
Statistical Analysis
Unless otherwise stated, data were analyzed using Student t test. Results were considered significantly different at P<0.05. Values are expressed as means±SE.
Results
RGS6 and ox‐CaMKII are Expressed in Embryos During Early Developmental Stages
We first analyzed the level and distribution of RGS6 and ox‐CaMKII in wild‐type (RGS6+/+/CaMKIIMM) embryos during embryonic development by western blot and immunostaining. RGS6 protein expression was detected in embryos at E7.5 by western blot and was present through E15.5, while ox‐CaMKII and p‐CaMKII were detectable only from E9.5 to E15.5 (Figure 1A, Figure S2). Both RGS6 and ox‐CaMKII are expressed at E10.5 in the developing endothelium of the dorsal aorta in RGS6+/+/CaMKIIMM embryos, as both proteins were co‐localized with platelet endothelial cell adhesion molecule 1 (PECAM1; endothelial marker) (Figure 1C and 1E). Furthermore, RGS6 and ox‐CaMKII are expressed in heart (endocardium and myocardium) of E10.5 RGS6+/+/CaMKIIMM embryos as both proteins were present in PECAM1‐positive and ‐negative cells (Figure 1D and 1F). These findings clearly demonstrate that RGS6 and ox‐CaMKII are expressed at similar developmental stages in the heart and developing vasculature.
Figure 1.
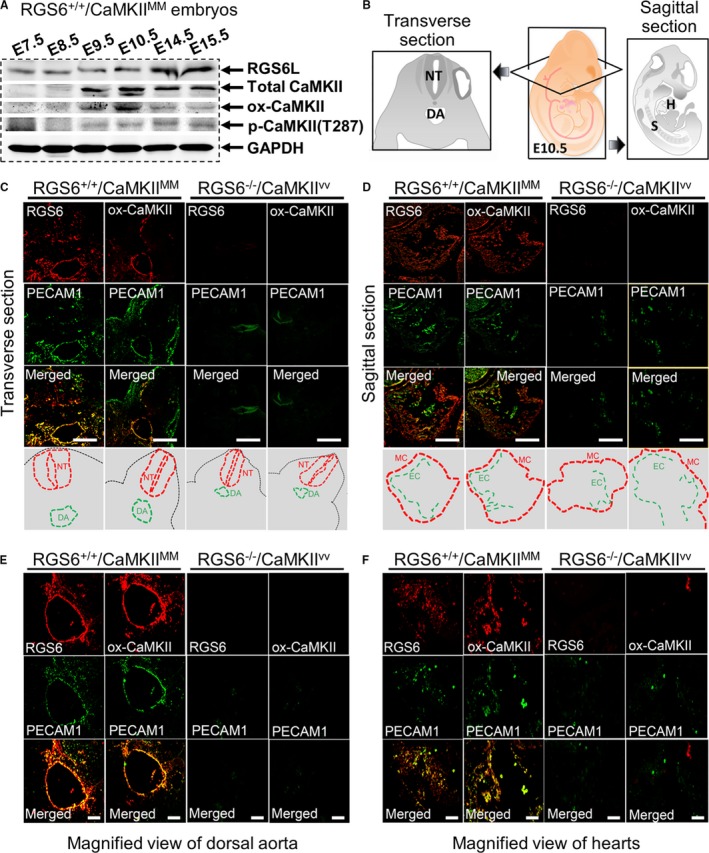
Expression of Regulator of G protein signaling 6 (RGS6) and oxidized Ca2+/calmodulin‐dependent protein kinase II (ox‐CaMKII) in mouse embryos. A, Western blot analysis of RGS6, ox‐CaMKII, phosphorylated CaMKII (p‐CaMKII), and total Ca2+/calmodulin‐dependent protein kinase II (CaMKII) expression in RGS6+/+/CaMKIIMM embryos at different gestation periods. B, Schematic illustration of transverse and sagittal sections of embryos used for immunostaining. C, Immunohistochemical analyses of RGS6 and ox‐CaMKII (red) expression in the dorsal aorta of embryos (transverse section) at E10.5 and co‐localization with the endothelial marker platelet endothelial cell adhesion molecule 1 (PECAM1) (green, upper). The schematic view showing the location of the neural tube (NT) and dorsal aorta (DA), in the respective pictures, are indicated by dotted red lines and green lines, respectively (lower). D, Immunohistochemical analysis of RGS6 and ox‐CaMKII (red) expression in the hearts of embryos (sagittal section) at E10.5 and co‐localization with PECAM1 (green, upper). The schematic view showing the location of the myocardium (MC) and endocardium (EC), for the respective pictures, are indicated by dotted red lines and green lines, respectively (lower). E, Co‐localization of RGS6 and ox‐CaMKII (red) with PECAM1 (green) in the DA region at higher magnification from embryo sections at E10.5. F, Co‐localization of RGS6 and ox‐CaMKII (red) with PECAM1 (green) immunostaining of endothelial cells in heart at higher magnification from E10.5 embryo sections. Decreased numbers of positive PECAM1‐stained endothelial cells were found in RGS6−/−/CaMKIIVV embryos compared with RGS6+/+/CaMKIIMM embryos. Scale bars, 50 μm (C and D) and 20 μm (E and F). H indicates heart; S, somites.
Combined Loss of RGS6 and ox‐CaMKII Leads to Embryonic Lethality Between E10.5 and E11.5
Both RGS6−/− and CaMKIIVV mice are born according to expected Mendelian ratios, have no apparent phenotypic defects, and are fertile.32, 36 However, when we crossed RGS6−/−/CaMKIIMV mice to generate RGS6−/−/CaMKIIVV mice we observed a striking deviation (≈1:2:0) from expected inheritance ratios (1:2:1) with no RGS6−/−/CaMKIIVV offspring being identified at weaning/genotyping age (P21) (Table). To determine the age of embryonic lethality, we performed a genotypic analysis of yolk sacs from staged embryos derived from crossing of RGS6−/−/CaMKIIMV mice. Embryos of all 3 CaMKII genotypes (on RGS6−/− background) had normal inheritance ratios from E7.5 to E9.5 (Table) and were phenotypically indistinguishable. However, at E10.5, a marked difference in phenotype between the genotypes was seen as double mutant embryos (RGS6−/−/CaMKIIVV) now appeared smaller when compared with RGS6−/−/CaMKIIMM (Figure 2A) or RGS6−/−/CaMKIIMV embryos (not shown). No RGS6−/−/CaMKIIVV embryos survived past E10.5 and were only visible as resorbed embryos from E11.5 to E15.5 (Table). Therefore, the combined loss of RGS6 and ox‐CaMKII results in lethal embryonic abnormalities that manifest by E10.5.
Table 1.
Genotypes of Offspring Produced by Crossing RGS6−/−/CaMKIIMV Mice
| Age | RGS6−/−/CaMKIIMM | RGS6−/−/CaMKIIMV | RGS6−/−/CaMKIIVV | Total |
|---|---|---|---|---|
| P21 | 51 | 136 | 0 | 187 |
| E15.5 | 4 | 13 | 7a | 24 |
| E12.5 | 10 | 15 | 7a | 32 |
| E11.5 | 7 | 12 | 5a | 24 |
| E10.5 | 49 | 110 | 45 | 204 |
| E9.5 | 8 | 14 | 11 | 33 |
| E8.5 | 2 | 4 | 11 | 17 |
| E7.5 | 2 | 5 | 2 | 9 |
CaMKII indicates Ca2+/calmodulin‐dependent protein kinase II; E, embryonic; P, postnatal; RGS6, Regulator of G protein signaling 6.
Embryos were partially resorbed at the time of isolation.
Figure 2.
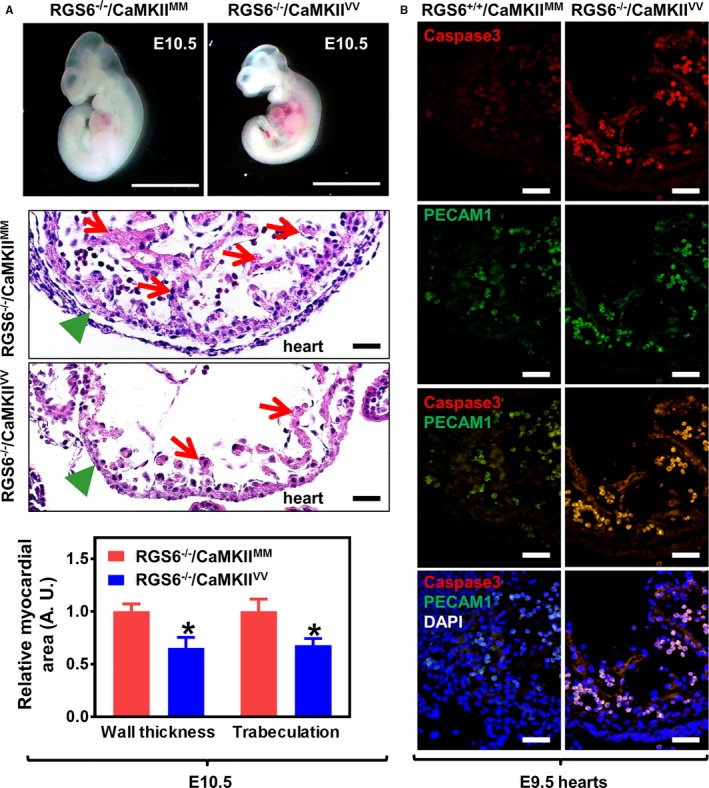
Cardiovascular abnormalities in Regulator of G protein signaling 6 (RGS6)−/−/Ca2+/calmodulin‐dependent protein kinase II (CaMKII)VV embryos. A, Representative images of freshly dissected live E10.5 embryos (upper panel, scale bar=200 μm) and hematoxylin and eosin (H&E) staining of heart sections taken from E10.5 embryos (middle panel, scale bar=20 μm). Green arrowheads indicate normal (RGS6−/−/CaMKIIMM) and thinned (RGS6−/−/CaMKIIVV) myocardium and red arrows indicate regions of normal trabeculation in RGS6−/−/CaMKIIMM hearts and its loss in RGS6−/−/CaMKIIVV hearts of H&E‐stained sections (middle panel). Both myocardial thickness and the level of trabeculation were significantly reduced in RGS6−/−/CaMKIIVV embryos when compared with RGS6−/−/CaMKIIMM embryos, analyzed by Mann–Whitney test (lower panel). N=7 to 9 embryos, *P<0.05. B, Increased caspase 3 (red) and platelet endothelial cell adhesion molecule 1 (PECAM1) (green)–stained double‐positive cells (yellow) in the heart ventricle of RGS6−/−/CaMKIIVV embryos at E9.5 compared with RGS6−/−/CaMKIIMM. Scale bar=20 μm. DAPI indicates 4′,6‐diamidino‐2‐phenylindole.
RGS6−/−/CaMKIIVV Embryos Exhibit Cardiac Developmental Defects
The cardiovascular system is the first organ system to develop and is required for functional blood circulation and embryonic survival after E9.5.42 Given the expression of RGS6 and ox‐CaMKII in hearts and developing vasculature of RGS6+/+/CaMKIIMM embryos and the death of RGS6−/−/CaMKIIVV embryos after E10.5, we examined the hearts of these double mutant embryos before their death. Figure 2A shows that E10.5 RGS6−/−/CaMKIIVV embryos had thin ventricular walls and loss of trabeculation. Furthermore, BMP2 and Tbx2, genes critical for patterning of atrioventricular myocardium, were downregulated in double mutant embryonic hearts (Figure S3). Histological examination of these double mutant embryos 1 day earlier at E9.5 revealed a substantial increase in caspase 3‐positive apoptotic endothelial (PECAM1+) cells compared with hearts of RGS6−/−/CaMKIIMM embryos (Figure 2B). Consistent with endothelial cell apoptosis at E9.5, we found few PECAM1‐stained cells in the heart of E10.5 RGS6−/−/CaMKIIVV embryos (Figure 1D and 1F). These findings demonstrate a combined need for RGS6 and ox‐CaMKII in cardiac embryogenesis.
Vascular Remodeling and Hematopoietic Defects in RGS6−/−/CaMKIIVV Placentas, Yolk Sacs, and Embryos
We undertook studies to determine the cause of the apparent cardiovascular failure in RGS6−/−/CaMKIIVV embryos. First, we analyzed extraembryonic structures and yolk sacs at E10.5 for any defects that might lead to developmental arrest at this stage. The placenta, which is composed of the giant cell, spongiotrophoblast, and labyrinthine regions (Figure 3A), is essential for the exchange of gases and nutrients between embryo and mother during gestation.43 Figure 3B shows that while RGS6−/−/CaMKIIVV placentas exhibited a normal morphology of trophoblast giant cells and spongiotrophoblasts, the labyrinth region had a marked loss of maternal (anucleated) and fetal (nucleated) red blood cells (RBCs) in the vasculature compared with RGS6−/−/CaMKIIMM placentas. To determine whether embryonic blood vessel development was defective, we performed immunostaining for fetal liver kinase 1, a marker of endothelial‐containing embryonic blood vessels. Fetal liver kinase 1 immunostaining confirmed a lack of viable embryonic blood vessels in the labyrinthine region of RGS6−/−/CaMKIIVV placentas (Figure 3C), suggesting that combined loss of RGS6 and ox‐CaMKII results in defective placental angiogenesis. Consistent with these findings, we found that both RGS6 and ox‐CaMKII are expressed in the labyrinthine region of RGS6+/+/CaMKIIMM placenta (Figure S4).
Figure 3.
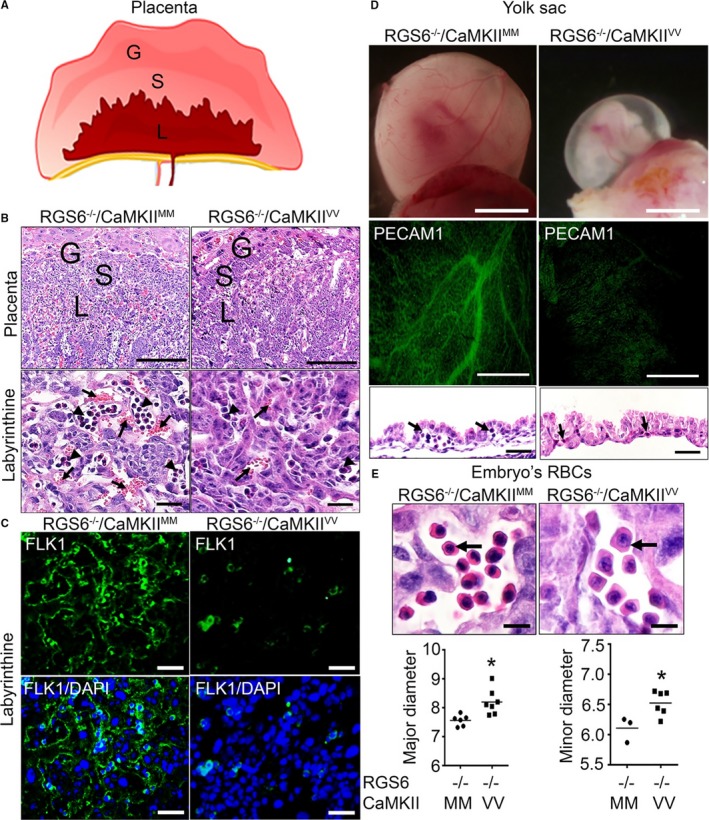
Vascular remodeling and hematopoietic defects in Regulator of G protein signaling 6 (RGS6)−/−/Ca2+/calmodulin‐dependent protein kinase II (CaMKII)VV placentas, yolk sacs, and embryos. A, Schematic representation of different layers of placenta in mice; giant (G), spongiotrophoblast (S), and labyrinthine (L) layers. B, Hematoxylin and eosin (H&E) staining of RGS6−/−/CaMKIIMM and RGS6−/−/CaMKIIVV placenta illustrating G, S, and L layers. Higher magnification images of the labyrinthine layer reveals reduced maternal and fetal blood–filled vasculature in RGS6−/−/CaMKIIVV placentas. Arrows indicate maternal blood (anucleated) and arrowheads indicate fetal blood (nucleated). Scale bars=200 μm (placenta) and 5 μm (L). C, Fetal liver kinase 1 immunostaining of the placental L region revealed a reduction in embryonic vessels in RGS6−/−/CaMKIIVV placentas compared with RGS6−/−/CaMKIIMM placentas. Scale bars=50 μm. D, Representative images of embryos within their yolk sacs show what appears to be an absence of blood‐filled large vitelline vessels, which we later confirmed using whole‐mount platelet endothelial cell adhesion molecule 1 (PECAM1) immunostaining. H&E staining of yolk sacs section reveals fewer blood/hematopoietic cells in vessels (black arrows) in RGS6−/−/CaMKIIVV yolk sacs at E10.5 compared with RGS6−/−/CaMKIIMM yolk sacs, suggesting altered vascular remodeling and hematopoiesis from blood islands. Scale bars=200 μm (whole‐mount and PECAM1) and 20 μm (yolk sac), and (E) larger red blood cell (RBC) nuclei and cell sizes were observed in RGS6−/−/CaMKIIVV embryo sections. Scale bar=20 μm. Both the major and minor diameter of RBCs were significantly larger in RGS6−/−/CaMKIIVV embryos compared with RGS6−/−/CaMKIIMM embryos analyzed by Mann–Whitney statistical methods. N=3, *P<0.05. DAPI indicates 4′,6‐diamidino‐2‐phenylindole.
Vascular and hematopoietic lineages are derived from hemangioblasts in yolk sac that lead to the formation of both blood cells and vessels.6 Yolk sacs of RGS6−/−/CaMKIIVV embryos lacked larger branching vitelline vessels and blood circulation in comparison with RGS6−/−/CaMKIIMM yolk sacs (Figure 3D, upper), a finding confirmed by immunostaining for the early endothelial marker PECAM1 (Figure 3D, middle). In addition, findings from hematoxylin and eosin staining revealed that RGS6−/−/CaMKIIVV yolk sacs were thicker overall with more collapsed vessels and fewer RBCs in blood island pools than RGS6−/−/CaMKIIMM yolk sacs (Figure 3D, lower). To further characterize the apparent hematopoietic defect in yolk sacs, we performed hematopoietic colony formation assays using semisolid methylcellulose containing medium. We observed a marked reduction in the number of erythroid colonies (BFU‐E colonies) derived from RGS6−/−/CaMKIIVV yolk sacs as compared with RGS6−/−/CaMKIIMM yolk sacs (Figure S5A). We then analyzed the expression level of genes required for hematopoiesis in yolk sacs derived from mice of these genotypes and found a significant decrease in the expression of GATA‐binding protein 1, β‐globin, and β‐major in RGS6−/−/CaMKIIVV yolk sacs compared with RGS6−/−/CaMKIIMM yolk sacs (Figure S5B). The expression of c‐kit (KIT proto‐oncogene receptor tyrosine kinase), LIM domain‐only 2, stem cell leukemia protein, and runt‐related transcription factor 1 did not differ in the yolk sacs from mice of these two genotypes (Figure S5B). These findings indicate that RGS6−/−/CaMKIIVV yolk sacs exhibit a failure in both remodeling and hematopoiesis of the primary vascular plexus.
RGS6−/−/CaMKIIVV E10.5 embryos also exhibited both hematopoietic and vascular defects similar to those observed in placenta and yolk sac. RBCs in RGS6−/−/CaMKIIVV embryos were larger with less eosinophilic cytoplasm and had larger nuclei than RBCs of RGS6−/−/CaMKIIMM embryos (Figure 3E). Morphometric analysis of the RBC diameter in both the major and minor planes showed significant increases in RBC size in RGS6−/−/CaMKIIVV embryos (Figure 3E). Further, results from whole‐mount PECAM1 immunostaining revealed a reduction in the number of large blood vessels in the fetal brain of RGS6−/−/CaMKIIVV when compared with RGS6−/−/CaMKIIMM embryos, although this reduction was not obvious in other regions, such as in the intersomitic vessels of the tail (Figure 4A). Histological sections of E10.5 RGS6−/−/CaMKIIVV embryos revealed that neural tubes appeared smaller with the presence of apoptotic cells (Figure 4C). Further, the dorsal aortae of RGS6−/−/CaMKIIVV, the first functional intra‐embryonic blood vessels,44 were smaller and had little endothelial PECAM1 immunoreactivity (Figures 1E and 4D). Thus, the combined loss of both RGS6 and ox‐CaMKII results in extensive hematopoietic and vascular remodeling defects in both extraembryonic tissues as well as embryos themselves.
Figure 4.
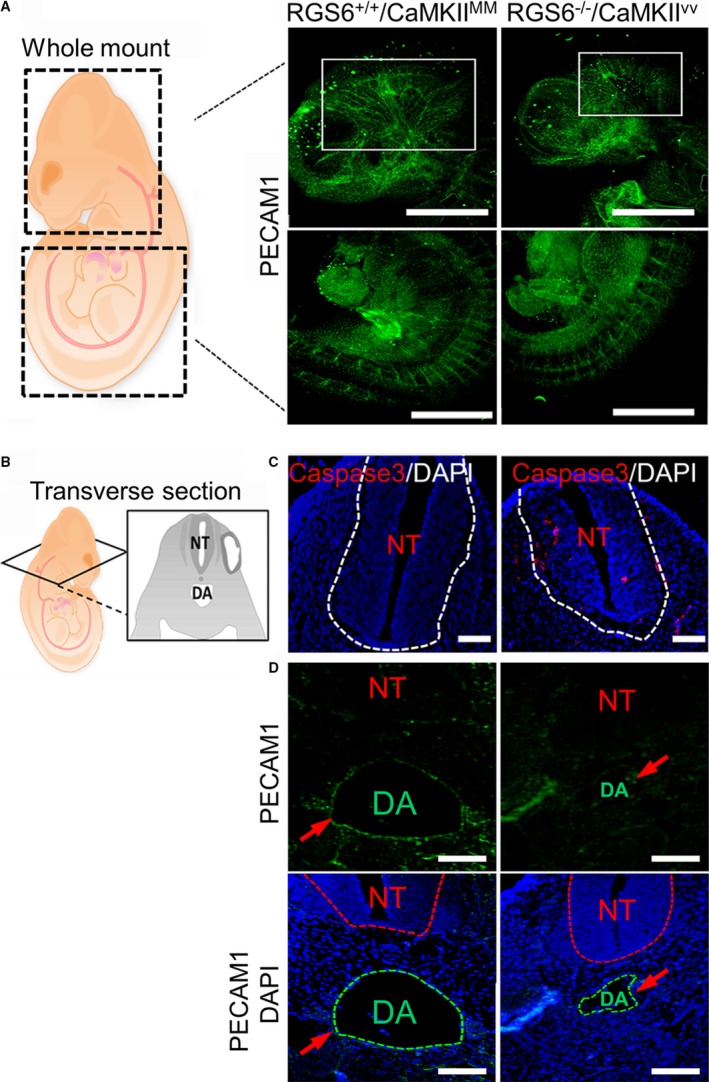
Vascular remodeling defects in Regulator of G protein signaling 6 (RGS6)−/−/Ca2+/calmodulin‐dependent protein kinase II (CaMKII)VV embryos. A, Schematic view of embryos represents the head and tail region used for whole‐mount platelet endothelial cell adhesion molecule 1 (PECAM1) immunostaining. Results from whole‐mount PECAM1 immunostaining of embryos revealed a lack of large blood vessels within the fetal brain region (indicated by white rectangles) of RGS6−/−/CaMKIIVV compared with RGS6−/−/CaMKIIMM embryos, although no defects on intersomitic vessels in the tail region of embryos were observed. Scale bars=200 μm. B, Schematic view of transverse section of embryos used for immunostaining (C and D). RGS6−/−/CaMKIIVV embryos show a developmentally delayed neural tube (smaller in size) (C), which may be caused by increased cell death, as represented by the caspase 3–positive cells. Scale bars=20 μm. D, Embryo sections showed a reduction in the size of the dorsal aorta and a thin endothelial layer around the dorsal aorta as indicated by red arrows in RGS6−/−/CaMKIIVV relative to RGS6−/−/CaMKIIMM embryos. The dotted lines in 4′,6‐diamidino‐2‐phenylindole (DAPI) staining indicate different regions as shown by abbreviation; NT (neural tube), and DA (dorsal aorta). Scale bars=20 μm.
Defects of Arterial Differentiation and Maturation in RGS6−/−/CaMKIIVV Embryos
The vascular and cardiac defects we observed in RGS6−/−/CaMKIIVV embryos are highly reminiscent of those seen with global and endothelial‐specific deletion of Notch1, Notch1/Notch4, Jag1, and Tmem100 as well as double knockout (DKO) of Hey1/Hey2 and Epsin1/Epsin2.16, 19, 20, 21, 25, 45, 46 Jag1, Notch1, Hey1, Hey2, and Tmem100 genes are essential in establishing the arterial identity of vascular endothelial cells.25, 45, 47, 48 To determine the cause of the vascular defects in RGS6−/−/CaMKIIVV embryos and yolk sacs we analyzed via quantitative polymerase chain reaction the expression of arterial and venous endothelial markers in these tissues. Figure 5A shows that expression of arterial endothelial markers Ephrin B2 (EfnB2), gap junction protein alpha 4, and gap junction protein alpha 5 were significantly decreased in E10.5 RGS6−/−/CaMKIIVV yolk sacs and embryos when compared with RGS6−/−/CaMKIIMM embryos and yolk sacs. In contrast, combined loss of RGS6 and ox‐CaMKII had no effects on expression of Fms‐related tyrosine kinase 1 (also known as VEGFR1), fetal liver kinase 1 (VEGFR2), vascular endothelial marker VE‐cadherin, and venous endothelial marker EPH receptor B4 (EphB4) in either embryos or yolk sacs. Further, immunohistochemical analysis demonstrated a remarkable loss of Sm22 (smooth muscle precursor marker) expression around the dorsal aorta of RGS6−/−/CaMKIIVV embryos (Figure 5B), suggesting defects in recruitment of Sm22 for proper maturation of aortic vessel.49 Thus, the vascular defects we observed in RGS6−/−/CaMKIIVV embryos are associated with defective arterial differentiation and maturation.
Figure 5.
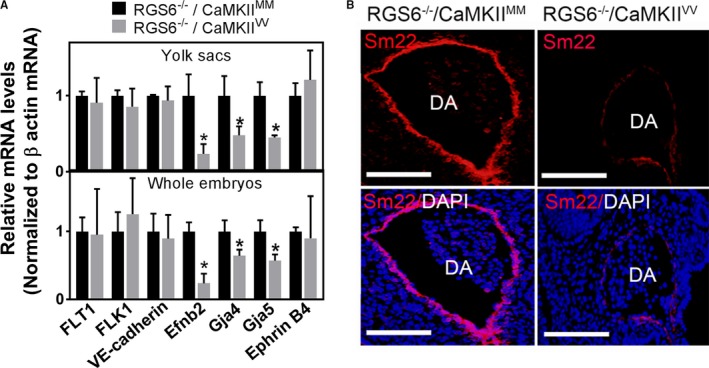
Arterial differentiation and maturation defects in Regulator of G protein signaling 6 (RGS6)−/−/Ca2+/calmodulin‐dependent protein kinase II (CaMKII)VV embryos. A, mRNA expression levels of endothelial precursors (fms related tyrosine kinase 1 [FLT1] and fetal liver kinase 1 [FLK1]), the vascular endothelial marker (VE‐cadherin), arterial markers (Ephrin B2, gap junction protein alpha 4, and gap junction protein alpha 5), and the venous marker (EphrinB4) were analyzed by quantitative polymerase chain reaction in E10.5 embryos and their yolk sacs. N=3, *P<0.001. B, Sm22 immunostaining of the dorsal aorta revealed a lack of smooth muscle recruitment in RGS6−/−/CaMKIIVV embryos compared with RGS6−/−/CaMKIIMM embryos. Scale bars=20 μm.
Impaired Notch Signaling in RGS6−/−/CaMKIIVV Embryos
Given the role of Notch‐mediated juxtacrine signaling in arterial fate determination50 and the defects in arterial differentiation we observed in RGS6−/−/CaMKIIVV embryos and yolk sacs, we investigated whether there is a combined need for RGS6 and ox‐CaMKII for normal Notch signaling. First, we performed quantitative polymerase chain reaction for all Notch family members as well as Notch ligands and Notch transcriptional targets. We found significant decreases in the expression of Notch1, Jag1, Hey1, Hey2, and Hey1L, as well as upregulation of Dll4 in RGS6−/−/CaMKIIVV versus RGS6−/−/CaMKIIMM embryos, yolk sacs, and hearts (Figure 6A). In contrast, no differences were observed in other Notch family members or Jag2 among these genotypes. Given the deficits in Notch1 expression and downstream Notch targets in embryonic tissues of RGS6−/−/CaMKIIVV mice, we measured expression of the Notch1 intracellular domain (N1ICD), the initiator and mediator of Notch1 signaling, in E10.5 embryos by western blot. Figure 6B shows a marked loss of N1ICD expression in RGS6−/−/CaMKIIVV versus RGS6−/−/CaMKIIMM embryos. Consistent with these findings, immunohistochemical staining of E10.5 embryos for N1ICD showed a severe loss of N1ICD expression in the heart endothelium of RGS6−/−/CaMKIIVV embryos compared with controls (Figure 6C). These findings reveal a remarkable loss in expression of Notch1 gene expression and signaling in embryos caused by a combined loss of RGS6 and ox‐CaMKII.
Figure 6.
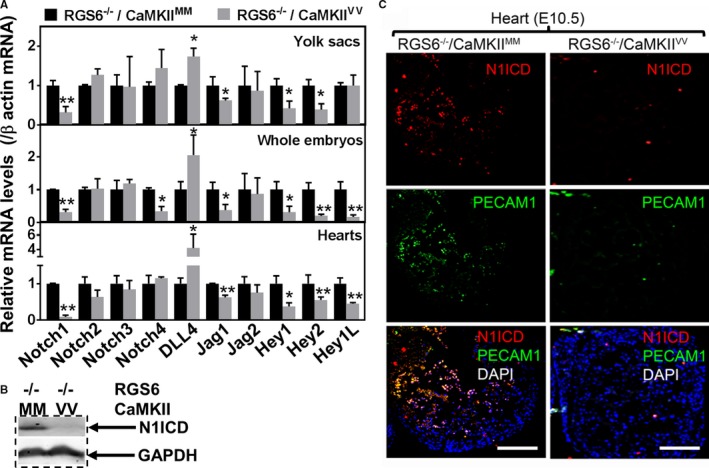
Impaired Notch signaling in Regulator of G protein signaling 6 (RGS6)−/−/Ca2+/calmodulin‐dependent protein kinase II (CaMKII)VV embryos. A, quantitative polymerase chain reaction indicated that expression of the Notch receptor (Notch1), Notch ligand (Jagged [Jag] 1), and the Notch downstream target genes Hey1 and Hey2 were decreased in RGS6−/−/CaMKIIVV yolk sacs, embryos, and hearts relative to RGS6−/−/CaMKIIMM embryos. In contrast, Hey1L was only decreased in embryos and heart in double mutants N=3, *P<0.05, **P<0.01 RGS6−/−/CaMKIIVV vs RGS6−/−/CaMKIIMM. B, Western blot analysis indicated that Notch1 intracellular domain (N1ICD) expression was reduced in RGS6−/−/CaMKIIVV embryos compared with RGS6−/−/CaMKIIMM embryos. C, Immunohistochemistry using N1ICD antibody showed decreased expression levels of N1ICD levels in the heart endothelial layer of RGS6−/−/CaMKIIVV embryos compared with RGS6−/−/CaMKIIMM embryos as Notch intracellular domain (red) co‐localizes with platelet endothelial cell adhesion molecule 1 (PECAM1) (green). Scale bars=50 μm. DAPI indicates 4′,6‐diamidino‐2‐phenylindole.
Parallel Pathways: RGS6‐Dependent ROS Does Not Mediate CaMKII Oxidation
The finding that combined, but not individual, loss of RGS6 and ox‐CaMKII leads to embryonic lethality and vascular defects in embryos, yolk sacs, and placenta indicates that RGS6 and ox‐CaMKII function in parallel versus in linear pathways. On the other hand, we previously showed that RGS6 is a critical upstream modulator of ROS generation33 and is needed for doxorubicin‐induced CaMKII oxidation in isolated cardiac myocytes (J. Huang PhD and R. Fisher PhD, unpublished data, 2012). Therefore, we assessed the relationship between RGS6, ROS, and CaMKII oxidation by staining for ox‐CaMKII and ROS (dihydroethidium) in embryos of different genotypes. While we observed that ox‐CaMKII expression was unaffected by RGS6 loss (Figure 7A), a reduction in ROS expression was observed in the head region and heart of RGS6−/−/CaMKIIMM embryos compared with RGS6+/+/CaMKIIMM embryos (Figure 7B and Figure S6). As expected, we observed a loss of ox‐CaMKII expression in RGS6−/−/CaMKIIVV compared with RGS6−/−/CaMKIIMM embryos but saw no further alterations in ROS expression when comparing embryos of these two genotypes (Figure 7A and 7B). Thus, ROS sources not dependent on RGS6, which is abundant in RGS6−/− embryos, mediates oxidative activation of CaMKII, consistent with RGS6 and CaMKII functioning in parallel pathways to promote normal embryogenesis.
Figure 7.
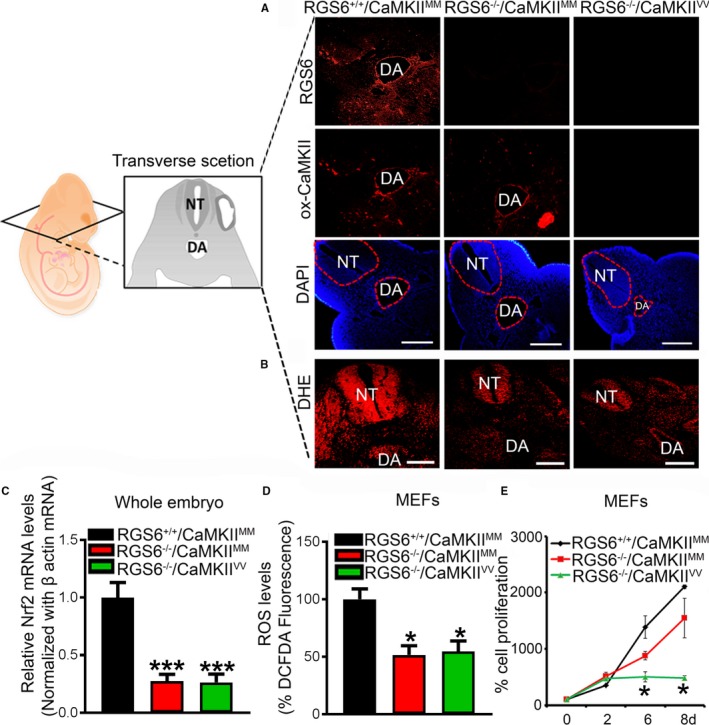
Regulator of G protein signaling 6 (RGS6), oxidized Ca2+/calmodulin‐dependent protein kinase II (ox‐CaMKII), and reactive oxygen species (ROS) expression in embryos of different genotypes. A, Detection of RGS6 (red) and ox‐CaMKII (red) levels in wild‐type (RGS6+/+/CaMKIIMM), RGS6−/− (RGS6−/−/Ca2+/calmodulin‐dependent protein kinase II [CaMKII]MM), and RGS6−/−/CaMKIIVV double mutant embryos by immunostaining. Scale bars=50 μm. B, Effects of combined loss of RGS6 and ox‐CaMKII on superoxide (red) generation in the head region of embryos as shown by dihydroethidium (DHE) staining. The transverse sectioning plane of embryos is represented in the schematic diagram. Scale bars=50 μm. C, Relative Nrf2 mRNA expression levels were analyzed by quantitative polymerase chain reaction in RGS6+/+/CaMKIIMM, RGS6−/−/CaMKIIMM, and RGS6−/−/CaMKIIVV embryos. D, ROS levels in mouse embryonic fibroblasts (MEFs) using 2′,7′‐dichlorodihydrofluorescein diacetate fluorescence. E, Cell proliferation of MEFs at different days using MTT assay. N=3. *P<0.05, ***P<0.01 RGS6−/−/CaMKIIMM or RGS6−/− CaMKIIVV vs RGS6+/+/CaMKIIMM. DA indicates dorsal aorta; DAPI, 4′,6‐diamidino‐2‐phenylindole; NT, neural tube.
RGS6 functions canonically to negatively regulate Gαi/o proteins, although its noncanonical functions include promoting ROS generation.33, 34, 40 A possible link between RGS6‐derived ROS and Notch signaling is the finding that ROS flux modulates Nrf2 activity, which activates the Notch pathway to maintain airway basal cell homeostasis.51 Figure 7C shows that embryos lacking RGS6 (RGS6−/−/CaMKIIMM and RGS6−/−/CaMKIIVV) showed downregulation of Nrf2 compared with RGS6+/+/CaMKIIMM embryos (Figure 7C), raising the possibility that RGS6 may work through a noncanonical ROS‐Nrf2 pathway to modulate Notch signaling.
To corroborate our in vivo findings, we isolated MEFs to study the influence of RGS6 and ox‐CaMKII on ROS and cell death. We found a similar reduction of ROS in both RGS6−/−/CaMKIIMM and RGS6−/−/CaMKIIVV MEFs in comparison to ROS levels in RGS6+/+/CaMKIIMM MEFs (Figure 7D). This analysis also revealed that RGS6−/−/CaMKIIVV MEFs proliferated more slowly compared with RGS6+/+/CaMKIIMM or RGS6−/−/CaMKIIMM MEFs (Figure 7E). These findings indicate that the reduced cell proliferation of double mutant MEFs is not the result of loss of RGS6‐mediate ROS production but instead could be the result of defective Notch signaling in these cells.
Discussion
This study establishes a critical need for RGS6 and ox‐CaMKII in normal cardiac development, vascular remodeling with arterial specification, and hematopoiesis in developing embryos, yolk sacs, and placenta. While loss of either RGS6 or ox‐CaMKII does not alter embryonic development, their combined loss leads to lethality between E10.5 and 11.5 as a result of severe cardiovascular defects associated with defective Notch1 signaling. While RGS6 and ox‐CaMKII each have been implicated in heart disease, including heart injury and failure, no studies have examined their role individually or together in cardiovascular development. Here, we show that RGS6 and ox‐CaMKII are expressed in the early vasculature as well as heart and function in parallel pathways to ensure normal cardiovascular development.
We provide the first evidence of a requirement for RGS6 and ox‐CaMKII in the regulation of Notch signaling. This was first suggested by the nature and timing of the cardiovascular defects observed in RGS6−/−/CaMKIIVV embryos. Indeed, Notch signaling is required for the beginning of organogenesis at E9.5 to 10.5 at which time embryonic lethality is frequently attributed to cardiovascular and placental defects. Multiple aspects of cardiovascular development including atrioventricular canal formation, endocardial cushion formation, ventricular maturation, and trabeculation52 are controlled by Notch signaling. Normal heart development begins between E7.5 and 9.0, with the endothelial tube undergoing looping to start heart chamber formation by the process of septation at E9.5, followed by maturation and trabeculation of the ventricles between E9.5 and 10.5. We found severe cardiac defects in E9.5 to 10.5 RGS6−/−/CaMKIIVV embryos associated with a reduction in myocardium thickness and abnormal trabeculation caused by increased endothelial cell apoptosis (Figure 2). These same cardiac defects have also been observed in Hey1/Hey2 DKO, Epsin1/Epsin2 DKO, Jag1 KO mice, and mice with an endothelial‐specific deletion of Notch1,16, 20, 25, 46 although these defects were not accompanied by endothelial cell apoptosis.
The striking cardiovascular defects in RGS6−/−/CaMKIIVV embryos were accompanied by placental and yolk sac defects also similar to those caused by defective Notch signaling. Placental development begins at E3.5 and chorioallantoic fusion takes place at E8.5. Thereafter, the trophoblast compartment with its associated fetal blood vessel forms the labyrinthine layer of placenta where exchange of material between maternal and fetal blood system occurs.43 Notch signaling molecules are present in the placenta and are required for trophoblast functions, vasculogenesis, and angiogenesis.53 Similarly, our data demonstrate defective angiogenesis in RGS6−/−/CaMKIIVV placentas as there is a lack of embryo‐derived blood vessels in the labyrinth region as revealed by fetal liver kinase 1 staining (Figure 3C), similar to those seen in Hey1/Hey2 DKO.25 Likewise, the defective vascular remodeling we observed in RGS6−/−/CaMKIIVV yolk sacs (Figure 3D) was also reported in Notch1 KO, Notch1/4 DKO, Hey1/Hey2 DKO mice, as well as in mice in which the N1ICD is overexpressed,16, 19, 25, 54 while the hematopoietic defects we observed have previously been studied in detail only in N1ICD overexpressing mice.54
Combined loss of RGS6 and ox‐CaMKII also provoked vascular remodeling defects in embryos. These embryos exhibited a lack of major blood vessels in brain, developmentally delayed neural tube maturation likely caused by increased apoptosis, and a reduction in the size of the dorsal aorta (Figure 4), as has been reported in Notch1 KO mice.19 Likewise, Notch1 is essential for generating hematopoietic stems cells.55 Interestingly, we also observed hematopoietic defects in histological sections of E10.5 RGS6−/−/CaMKIIVV embryos (Figure 3E). However, since we did see blood cells in double mutant embryos, these findings also demonstrate that blood flow was established between the yolk sac and embryo despite the presence of yolk sac vascular degeneration (Figure 3D, middle) and hematopoiesis defects (Figure 3D, lower; Figure S5A) as RBCs in these embryos must originate from the yolk sac, the primary site of hematopoiesis until E11.5 when embryonic erythropoiesis begins.56 Yolk sac–associated defects in double mutant mice were also associated with downregulation of genes essential for erythroblast maturation (GATA‐a, β‐globin, and β‐major; Figure S5B), similar to what has been reported in N1ICD embryos.54 This finding, together with the lack of differences in heart rates in RGS6−/−/CaMKIIMM and RGS6−/−/CaMKIIVV embryos (Figure S7), suggests that the vascular defects reported here may not result from cardiac insufficiency. On the other hand, combined loss of RGS6 and ox‐CaMKII provoked cardiac defects, which, accompanied by the observed vascular remodeling and hematopoietic defects, might be expected to compromise cardiovascular function in embryos. The vascular defects we observed in double mutant mouse embryos were associated with disrupted artery/vein specification as evidenced by decreased expression levels of arterial markers Efnb2, gap junction protein alpha 4, and gap junction protein alpha 5, as well as lack of smooth muscle recruitment around the dorsal aorta, which is required for proper arterial maturation (Figure 5). Arterial/venous differentiation is a critical step for vascular development and function.5, 57 Efnb2 is expressed in arteries while EPH receptor B4 is expressed in veins.58, 59 Their expression is an indicator of arterial/venous specification and their imbalance leads to defective vascular development.44, 60 Consistent with the downregulation of Efnb2 expression and the impaired Notch1 signaling we observed in RGS6−/−/CaMKIIVV embryos, the Notch signaling pathway genes Notch1, Jag1, Hey1, and Hey2, which reportedly confer arterial fate by increasing Efnb2 and decreasing EPH receptor B4 expression,60 were also downregulated (Figure 6A).
In agreement with the striking similarity between disrupted embryonic phenotypes caused by impaired Notch1 signaling or combined loss of RGS6 and ox‐CaMKII, we provide direct evidence that RGS6 and ox‐CaMKII function in parallel pathways as critical upstream modulators of Notch1 signaling. Indeed, we found downregulation of Notch1, Jag1, and Notch downstream target genes Hey1, Hey2, and Hey1L in RGS6−/−/CaMKIIVV embryos and yolk sacs (Figure 6A), favoring the arterial specification defects we observed. Moreover, RGS6 and ox‐CaMKII are expressed in the developing endothelium of the heart (Figure 1) where they cooperate to modulate expression of these same Notch signaling components as well as Notch1 signaling, as measured by N1ICD expression in heart (Figure 6B and 6C). The cardiac defects in RGS6−/−/CaMKIIVV embryos are likely the result of impaired Notch1 signaling pathway gene expression and signaling. Defects in trabeculation are observed in mice with global or endothelial deletion of Notch1 as well as in RBP Jk−/− and Hey1−/−/Hey 2−/− mice.61 In addition, endothelial or endocardial deletion of Dll4 and Notch 1, but not Jag1, severely disrupts trabeculation.62 Our results suggest that trabeculation defects in double mutant mice (Figure 2A) are likely caused by loss of Notch1 expression and signaling rather than alterations in Dll4 expression, whose upregulation may be a response to Notch1 deficiency. Indeed, the trabeculation defects we observe in double mutant mice are comparable to those in mice with endocardial deletion of Notch1.16 The decrease in myocardium thickness and trabeculation and increased endothelial cell apoptosis we observed in hearts of RGS6−/−/CaMKIIVV embryos likely reflect the failure of Notch‐dependent endothelial cell interactions needed for myocardial development.
Our findings suggest that RGS6 and ox‐CaMKII function in parallel pathways to modulate Notch signaling, as embryos with individual loss of RGS6 or ox‐CaMKII developed properly and had no defects in Notch signaling. Consistent with this idea, we found that while RGS6 loss resulted in a reduction of ROS and Nrf2 levels in embryos it had no effects on ox‐CaMKII levels (Figure 7), suggesting that CaMKII is oxidized by ROS produced by sources independent of RGS6. RGS6‐dependent sources of ROS include Nox,34 which is intriguing given that Nox2 deficiency, like loss of RGS6 and ox‐CaMKII, promotes arterial specification defects caused by defective Notch1 signaling.63 Together, these findings suggest that RGS6‐mediated ROS generation may regulate Notch pathways underlying cardiovascular development. One of these pathways may include Nrf2, which, like RGS6, is dispensable for embryonic development.64 The precise nature of the pathway by which ox‐CaMKII cooperates with RGS6 to ensure proper Notch signaling is not yet clear. Among numerous possible mechanisms is the potential role of ox‐CaMKII in trafficking/recycling of Notch ligands, as observed in zebrafish inner ear,65 or in phosphorylation‐mediated degradation of a Notch co‐repressor (silencing mediator of retinoic acid and thyroid hormone receptor),66 the only known links between activated (phosphorylated) CaMKII and Notch signaling. Importantly, the knock‐in MM to VV mutation eliminates ROS‐mediated activation, but does not interfere with CaMKII autophosphorylation.67
We illustrate a possible mechanism by which RGS6 and ox‐CaMKII may cooperate to promote Notch signaling within the developing embryo, which is required for proper vasculature and heart development (Figure 8). We depict RGS6 and ox‐CaMKII working via parallel pathways within a “signaling or receiving cell” to modulate Notch signaling, a depiction that is supported by our data in dorsal aorta and heart showing that both RGS6 and ox‐CaMKII are expressed in endothelial cells (Figure 1). On the basis of these findings, Figure 8 demonstrates that these parallel pathways, whether present in the same cell or adjacent cells, do work in heart and yolk sacs to ensure proper Notch signaling and embryonic development.
Figure 8.
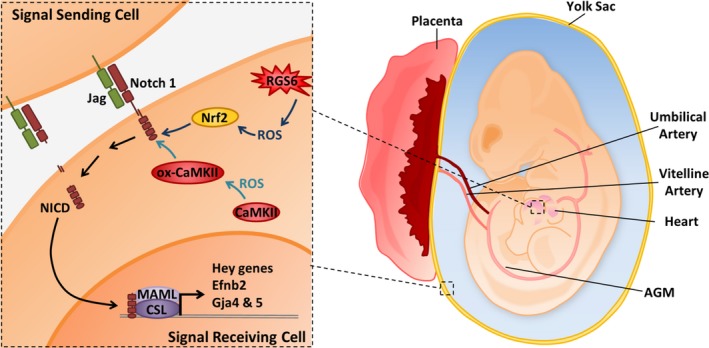
Schematic illustration of Regulator of G protein signaling 6 (RGS6)– and/or Ca2+/calmodulin‐dependent protein kinase II (CaMKII)–dependent activation of Notch signaling in embryonic heart and vasculature. In this article we provide evidence that RGS6 and oxidated CaMKII (ox‐CaMKII) function in parallel pathways as critical upstream modulators of Notch signaling. RGS6‐mediated reactive oxygen species (ROS) generation modulates Nrf2 levels while RGS6‐independent ROS oxidizes CaMKII to control Notch signaling for proper cardiovascular development. We hypothesize that together, RGS6 and ox‐CaMKII in signal‐receiving cells (receptor‐ expressing cells), which promote Notch1 receptor and ligand (Jagged [Jag]) interaction and culminate in the proteolytic cleavage of Notch and the release of the Notch intracellular domain (NICD) into the cytoplasm, NICD translocation into the nucleus, and NICD‐mediated gene transcription upon association with other transcriptional regulators CBF1/Su(H)/Lag‐1, CSL, and mastermind‐like (MAML). However, it is possible that RGS6 and ox‐CaMKII are also present in signal‐sending cells (ligand‐expressing cells) and work in a similar manner. In addition, the parallel pathways utilized by RGS6 and ox‐CaMKII (including their respective ROS; RGS6‐dependent ROS [moderate‐high level] and non‐RGS6‐ROS [basal‐low level]) are denoted using different shades of blue to emphasize their separate and independent functions. AGM indicates aorta‐gonad‐mesonephros (contains the dorsal aorta); Efnb2, Ephrin B2.
Conclusions
We have shown that RGS6 and ox‐CaMKII cooperatively ensure normal Notch signaling, which is crucial for proper cardiovascular development and embryo survival. Our findings suggest that alterations in the embryonic expression of RGS6 and ox‐CaMKII may potentially contribute to congenital heart defects, which represent the most common birth defects in humans.
Sources of Funding
This work was supported by National Institutes of Health CA161882 and HL014388, American Heart Association 14GRNT20460208 (Fisher) and 11SDG7580008 (Yang), and Iowa Cardiovascular Interdisciplinary Research Fellowship (NIH HL007121).
Disclosures
None.
Supporting information
Data S1. Supplemental Methods.
Table S1. Primers Used for qPCR and Genotyping
Figure S1. Regulator of G protein signaling 6 (RGS6) and Ca2+/calmodulin‐dependent protein kinase II (CaMKII) genotyping of mice and embryos. Representative gel images used within the analysis of mouse RGS6 and CaMKII. Key bands used in the determination of RGS6 and CaMKII genotype are indicated to the right of each respective gel. A 100 bp DNA ladder was used in this analysis. We have identified the 200 and 500 bp bands within the ladder at the left of each gel image. Primers required for this analysis can be found in Table S1.
Figure S2. Summary data of expression of regulator of G protein signaling 6 (RGS6), oxidized Ca2+/calmodulin‐dependent protein kinase II (ox‐CaMKII), phosphorylated CaMKII (p‐CaMKII), and total Ca2+/calmodulin‐dependent protein kinase II (CaMKII) in mouse embryos at different gestation periods. Western blot analysis of RGS6, ox‐CaMKII, p‐CaMKII, and total CaMKII expression levels in mouse embryos at different gestation stages was performed and quantified using ImageQuant 5.2 program (N=3). Representative blot can be seen in Figure 1A.
Figure S3. Expression of BMP2 and Tbx2 in E10.5 hearts. Analysis of BMP2 and Tbx2 expression, quantified via quantitative polymerase chain reaction, revealed that Tbx2, but not BMP2, is significantly downregulated in regulator of G protein signaling 6 (RGS6)−/−/Ca2+/calmodulin‐dependent protein kinase II (CaMKII)VV hearts relative to RGS6−/−/CaMKIIMM hearts. *P<0.05, **P<0.01.
Figure S4. Regulator of G protein signaling 6 (RGS6) and oxidized Ca2+/calmodulin‐dependent protein kinase II (ox‐CaMKII) expression at E10.5 in RGS6+/+/Ca2+/calmodulin‐dependent protein kinase II (CaMKII)MM placentas. RGS6 and ox‐CaMKII (red) were expressed in the labyrinthine region of placentas as detected by immunohistochemistry. Scale bars=20 μm.
Figure S5. Hematopoietic defects in regulator of G protein signaling 6 (RGS6)−/−/Ca2+/calmodulin‐dependent protein kinase II (CaMKII)VV yolk sacs. A, In vitro differentiation analysis of yolk sac hematopoietic cells derived from RGS6−/−/Ca2+/calmodulin‐dependent protein kinase II (CaMKII)MM and RGS6−/−/CaMKIIVV yolk sacs at E10.5 using methylcellulose colony assay. The number of erythroid burst‐forming units (BFU‐E) colonies (>20 cells) produced from RGS6−/−/CaMKIIMM and RGS6−/−/CaMKIIVV yolk sacs were quantified after 7 days in culture, as shown in the representative images. This analysis revealed that the combined loss of RGS6 and ox‐CaMKII reduces the ability of hematopoetic cells to form colonies. Scale bars=50 μm. N=3. *P<0.01. B, Quantitative polymerase chain reaction analysis of hematopoietic gene expression in E10.5 yolk sac revealed a significant reduction in the expression of the GATA‐binding protein 1, β globin, and β‐major genes in RGS6−/−/CaMKIIVV yolk sacs relative to RGS6−/−/CaMKIIMM yolk sacs. N=3. *P<0.01.
Figure S6. Oxidized Ca2+/calmodulin‐dependent protein kinase II (ox‐CaMKII) and reactive oxygen species (ROS) expression in hearts of embryos of different genotypes. A, Detection of ox‐CaMKII (green) levels in wild‐type (regulator of G protein signaling 6 [RGS6]+/+/Ca2+/calmodulin‐dependent protein kinase II [CaMKII]MM), RGS6−/− (RGS6−/−/CaMKIIMM), and RGS6−/−/CaMKIIVV double mutant embryo hearts (arrows) by immunostaining reveals that RGS6 loss does not alter ox‐CaMKII expression. Note: embryos were not in the same sectioning planes. Scale bars=200 μm. B, Effects of combined loss of RGS6 and ox‐CaMKII on superoxide (red) generation in hearts of embryos as shown by dihydroethidium staining. Scale bars=50 μm.
Figure S7. Heart rates in regulator of G protein signaling 6 (RGS6)−/−/Ca2+/calmodulin‐dependent protein kinase II (CaMKII)MM and RGS6−/−/CaMKIIVV live embryos. The heart rate of live E10.5 embryos was manually counted by viewing the embryo through a dissection microscope. This analysis revealed no significant difference in the heart rate of RGS6−/−/CaMKIIVV double mutant embryos relative to RGS6−/−/CaMKIIMM embryos. N=3.
Acknowledgments
We thank Dr Bibi Ayesha (University of Iowa) for providing advice and support in mouse developmental biology and clinical relevance in humans. We also acknowledge the University of Iowa Central Microscopy Facility for the sectioning of embryos and hematoxylin and eosin staining.
(J Am Heart Assoc. 2017;6:e007038 DOI: 10.1161/JAHA.117.007038.)29079565
This article was handled independently by Hossein Ardehali, MD, PhD, as a guest editor.
References
- 1. Hoyert DL, Mathews TJ, Menacker F, Strobino DM, Guyer B. Annual summary of vital statistics: 2004. Pediatrics. 2006;117:168–183. [DOI] [PubMed] [Google Scholar]
- 2. Bruneau BG. The developmental genetics of congenital heart disease. Nature. 2008;451:943–948. [DOI] [PubMed] [Google Scholar]
- 3. Niessen K, Karsan A. Notch signaling in cardiac development. Circ Res. 2008;102:1169–1181. [DOI] [PubMed] [Google Scholar]
- 4. Brand T. Heart development: molecular insights into cardiac specification and early morphogenesis. Dev Biol. 2003;258:1–19. [DOI] [PubMed] [Google Scholar]
- 5. Marcelo KL, Goldie LC, Hirschi KK. Regulation of endothelial cell differentiation and specification. Circ Res. 2013;112:1272–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Risau W, Flamme I. Vasculogenesis. Annu Rev Cell Dev Biol. 1995;11:73–91. [DOI] [PubMed] [Google Scholar]
- 7. Coultas L, Chawengsaksophak K, Rossant J. Endothelial cells and VEGF in vascular development. Nature. 2005;438:937–945. [DOI] [PubMed] [Google Scholar]
- 8. Goumans MJ, Liu Z, ten Dijke P. TGF‐beta signaling in vascular biology and dysfunction. Cell Res. 2009;19:116–127. [DOI] [PubMed] [Google Scholar]
- 9. Thurston G, Daly C. The complex role of angiopoietin‐2 in the angiopoietin‐tie signaling pathway. Cold Spring Harb Perspect Med. 2012;2:a006550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hamada K, Oike Y, Ito Y, Maekawa H, Miyata K, Shimomura T, Suda T. Distinct roles of ephrin‐B2 forward and EphB4 reverse signaling in endothelial cells. Arterioscler Thromb Vasc Biol. 2003;23:190–197. [DOI] [PubMed] [Google Scholar]
- 11. Hofmann JJ, Iruela‐Arispe ML. Notch signaling in blood vessels: who is talking to whom about what? Circ Res. 2007;100:1556–1568. [DOI] [PubMed] [Google Scholar]
- 12. Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. de la Pompa JL, Epstein JA. Coordinating tissue interactions: Notch signaling in cardiac development and disease. Dev Cell. 2012;22:244–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678–689. [DOI] [PubMed] [Google Scholar]
- 15. Conlon RA, Reaume AG, Rossant J. Notch1 is required for the coordinate segmentation of somites. Development. 1995;121:1533–1545. [DOI] [PubMed] [Google Scholar]
- 16. Limbourg FP, Takeshita K, Radtke F, Bronson RT, Chin MT, Liao JK. Essential role of endothelial Notch1 in angiogenesis. Circulation. 2005;111:1826–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Domenga V, Fardoux P, Lacombe P, Monet M, Maciazek J, Krebs LT, Klonjkowski B, Berrou E, Mericskay M, Li Z, Tournier‐Lasserve E, Gridley T, Joutel A. Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes Dev. 2004;18:2730–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carlson TR, Yan Y, Wu X, Lam MT, Tang GL, Beverly LJ, Messina LM, Capobianco AJ, Werb Z, Wang R. Endothelial expression of constitutively active Notch4 elicits reversible arteriovenous malformations in adult mice. Proc Natl Acad Sci USA. 2005;102:9884–9889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Krebs LT, Xue Y, Norton CR, Shutter JR, Maguire M, Sundberg JP, Gallahan D, Closson V, Kitajewski J, Callahan R, Smith GH, Stark KL, Gridley T. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000;14:1343–1352. [PMC free article] [PubMed] [Google Scholar]
- 20. Xue Y, Gao X, Lindsell CE, Norton CR, Chang B, Hicks C, Gendron‐Maguire M, Rand EB, Weinmaster G, Gridley T. Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum Mol Genet. 1999;8:723–730. [DOI] [PubMed] [Google Scholar]
- 21. High FA, Lu MM, Pear WS, Loomes KM, Kaestner KH, Epstein JA. Endothelial expression of the Notch ligand Jagged1 is required for vascular smooth muscle development. Proc Natl Acad Sci USA. 2008;105:1955–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hofmann JJ, Briot A, Enciso J, Zovein AC, Ren S, Zhang ZW, Radtke F, Simons M, Wang Y, Iruela‐Arispe ML. Endothelial deletion of murine Jag1 leads to valve calcification and congenital heart defects associated with Alagille syndrome. Development. 2012;139:4449–4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sorensen I, Adams RH, Gossler A. DLL1‐mediated Notch activation regulates endothelial identity in mouse fetal arteries. Blood. 2009;113:5680–5688. [DOI] [PubMed] [Google Scholar]
- 24. Oka C, Nakano T, Wakeham A, de la Pompa JL, Mori C, Sakai T, Okazaki S, Kawaichi M, Shiota K, Mak TW, Honjo T. Disruption of the mouse RBP‐J kappa gene results in early embryonic death. Development. 1995;121:3291–3301. [DOI] [PubMed] [Google Scholar]
- 25. Fischer A, Schumacher N, Maier M, Sendtner M, Gessler M. The Notch target genes Hey1 and Hey2 are required for embryonic vascular development. Genes Dev. 2004;18:901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Alabi RO, Glomski K, Haxaire C, Weskamp G, Monette S, Blobel CP. ADAM10‐dependent signaling through Notch1 and Notch4 controls development of organ‐specific vascular beds. Circ Res. 2016;119:519–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, Alamowitch S, Domenga V, Cecillion M, Marechal E, Maciazek J, Vayssiere C, Cruaud C, Cabanis EA, Ruchoux MM, Weissenbach J, Bach JF, Bousser MG, Tournier‐Lasserve E. Notch3 mutations in CADASIL, a hereditary adult‐onset condition causing stroke and dementia. Nature. 1996;383:707–710. [DOI] [PubMed] [Google Scholar]
- 28. McDaniell R, Warthen DM, Sanchez‐Lara PA, Pai A, Krantz ID, Piccoli DA, Spinner NB. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am J Hum Genet. 2006;79:169–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Maity B, Stewart A, Yang J, Loo L, Sheff D, Shepherd AJ, Mohapatra DP, Fisher RA. Regulator of G protein signaling 6 (RGS6) protein ensures coordination of motor movement by modulating GABAB receptor signaling. J Biol Chem. 2012;287:4972–4981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stewart A, Maity B, Wunsch AM, Meng F, Wu Q, Wemmie JA, Fisher RA. Regulator of G‐protein signaling 6 (RGS6) promotes anxiety and depression by attenuating serotonin‐mediated activation of the 5‐HT(1A) receptor‐adenylyl cyclase axis. FASEB J. 2014;28:1735–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Garzon J, Lopez‐Fando A, Sanchez‐Blazquez P. The R7 subfamily of RGS proteins assists tachyphylaxis and acute tolerance at mu‐opioid receptors. Neuropsychopharmacology. 2003;28:1983–1990. [DOI] [PubMed] [Google Scholar]
- 32. Yang J, Huang J, Maity B, Gao Z, Lorca RA, Gudmundsson H, Li J, Stewart A, Swaminathan PD, Ibeawuchi SR, Shepherd A, Chen CK, Kutschke W, Mohler PJ, Mohapatra DP, Anderson ME, Fisher RA. RGS6, a modulator of parasympathetic activation in heart. Circ Res. 2010;107:1345–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang J, Maity B, Huang J, Gao Z, Stewart A, Weiss RM, Anderson ME, Fisher RA. G‐protein inactivator RGS6 mediates myocardial cell apoptosis and cardiomyopathy caused by doxorubicin. Cancer Res. 2013;73:1662–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stewart A, Maity B, Anderegg SP, Allamargot C, Yang J, Fisher RA. Regulator of G protein signaling 6 is a critical mediator of both reward‐related behavioral and pathological responses to alcohol. Proc Natl Acad Sci USA. 2015;112:E786–E795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Anderson ME. Oxidant stress promotes disease by activating CaMKII. J Mol Cell Cardiol. 2015;89:160–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Luo M, Guan X, Luczak ED, Lang D, Kutschke W, Gao Z, Yang J, Glynn P, Sossalla S, Swaminathan PD, Weiss RM, Yang B, Rokita AG, Maier LS, Efimov IR, Hund TJ, Anderson ME. Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J Clin Invest. 2013;123:1262–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xie J, Wu T, Xu K, Huang IK, Cleaver O, Huang CL. Endothelial‐specific expression of WNK1 kinase is essential for angiogenesis and heart development in mice. Am J Pathol. 2009;175:1315–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gibson‐Corley KN, Olivier AK, Meyerholz DK. Principles for valid histopathologic scoring in research. Vet Pathol. 2013;50:1007–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bushdid PB, Osinska H, Waclaw RR, Molkentin JD, Yutzey KE. NFATc3 and NFATc4 are required for cardiac development and mitochondrial function. Circ Res. 2003;92:1305–1313. [DOI] [PubMed] [Google Scholar]
- 40. Huang J, Yang J, Maity B, Mayuzumi D, Fisher RA. Regulator of G protein signaling 6 mediates doxorubicin‐induced ATM and p53 activation by a reactive oxygen species‐dependent mechanism. Cancer Res. 2011;71:6310–6319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Maity B, Yang J, Huang J, Askeland RW, Bera S, Fisher RA. Regulator of G protein signaling 6 (RGS6) induces apoptosis via a mitochondrial‐dependent pathway not involving its GTPase‐activating protein activity. J Biol Chem. 2011;286:1409–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Moorman A, Webb S, Brown NA, Lamers W, Anderson RH. Development of the heart: (1) formation of the cardiac chambers and arterial trunks. Heart. 2003;89:806–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rossant J, Cross JC. Placental development: lessons from mouse mutants. Nat Rev Genet. 2001;2:538–548. [DOI] [PubMed] [Google Scholar]
- 44. Sato Y. Dorsal aorta formation: separate origins, lateral‐to‐medial migration, and remodeling. Dev Growth Differ. 2013;55:113–129. [DOI] [PubMed] [Google Scholar]
- 45. Somekawa S, Imagawa K, Hayashi H, Sakabe M, Ioka T, Sato GE, Inada K, Iwamoto T, Mori T, Uemura S, Nakagawa O, Saito Y. Tmem100, an ALK1 receptor signaling‐dependent gene essential for arterial endothelium differentiation and vascular morphogenesis. Proc Natl Acad Sci USA. 2012;109:12064–12069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen H, Ko G, Zatti A, Di Giacomo G, Liu L, Raiteri E, Perucco E, Collesi C, Min W, Zeiss C, De Camilli P, Cremona O. Embryonic arrest at midgestation and disruption of Notch signaling produced by the absence of both epsin 1 and epsin 2 in mice. Proc Natl Acad Sci USA. 2009;106:13838–13843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Villa N, Walker L, Lindsell CE, Gasson J, Iruela‐Arispe ML, Weinmaster G. Vascular expression of Notch pathway receptors and ligands is restricted to arterial vessels. Mech Dev. 2001;108:161–164. [DOI] [PubMed] [Google Scholar]
- 48. Fischer A, Gessler M. Hey genes in cardiovascular development. Trends Cardiovasc Med. 2003;13:221–226. [DOI] [PubMed] [Google Scholar]
- 49. Fouillade C, Monet‐Lepretre M, Baron‐Menguy C, Joutel A. Notch signalling in smooth muscle cells during development and disease. Cardiovasc Res. 2012;95:138–146. [DOI] [PubMed] [Google Scholar]
- 50. Quillien A, Moore JC, Shin M, Siekmann AF, Smith T, Pan L, Moens CB, Parsons MJ, Lawson ND. Distinct Notch signaling outputs pattern the developing arterial system. Development. 2014;141:1544–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Paul MK, Bisht B, Darmawan DO, Chiou R, Ha VL, Wallace WD, Chon AT, Hegab AE, Grogan T, Elashoff DA, Alva‐Ornelas JA, Gomperts BN. Dynamic changes in intracellular ROS levels regulate airway basal stem cell homeostasis through Nrf2‐dependent Notch signaling. Cell Stem Cell. 2014;15:199–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. High FA, Epstein JA. The multifaceted role of Notch in cardiac development and disease. Nat Rev Genet. 2008;9:49–61. [DOI] [PubMed] [Google Scholar]
- 53. Zhao WX, Lin JH. Notch signaling pathway and human placenta. Int J Med Sci. 2012;9:447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Venkatesh DA, Park KS, Harrington A, Miceli‐Libby L, Yoon JK, Liaw L. Cardiovascular and hematopoietic defects associated with Notch1 activation in embryonic Tie2‐expressing populations. Circ Res. 2008;103:423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kumano K, Chiba S, Kunisato A, Sata M, Saito T, Nakagami‐Yamaguchi E, Yamaguchi T, Masuda S, Shimizu K, Takahashi T, Ogawa S, Hamada Y, Hirai H. Notch1 but not Notch2 is essential for generating hematopoietic stem cells from endothelial cells. Immunity. 2003;18:699–711. [DOI] [PubMed] [Google Scholar]
- 56. Baron MH, Isern J, Fraser ST. The embryonic origins of erythropoiesis in mammals. Blood. 2012;119:4828–4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kume T. Specification of arterial, venous, and lymphatic endothelial cells during embryonic development. Histol Histopathol. 2010;25:637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang HU, Chen ZF, Anderson DJ. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin‐B2 and its receptor Eph‐B4. Cell. 1998;93:741–753. [DOI] [PubMed] [Google Scholar]
- 59. Adams RH, Wilkinson GA, Weiss C, Diella F, Gale NW, Deutsch U, Risau W, Klein R. Roles of ephrinB ligands and EphB receptors in cardiovascular development: demarcation of arterial/venous domains, vascular morphogenesis, and sprouting angiogenesis. Genes Dev. 1999;13:295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kim YH, Hu H, Guevara‐Gallardo S, Lam MT, Fong SY, Wang RA. Artery and vein size is balanced by Notch and ephrin B2/EphB4 during angiogenesis. Development. 2008;135:3755–3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Luxan G, D'Amato G, MacGrogan D, de la Pompa JL. Endocardial Notch signaling in cardiac development and disease. Circ Res. 2016;118:e1–e18. [DOI] [PubMed] [Google Scholar]
- 62. D'Amato G, Luxan G, del Monte‐Nieto G, Martinez‐Poveda B, Torroja C, Walter W, Bochter MS, Benedito R, Cole S, Martinez F, Hadjantonakis AK, Uemura A, Jimenez‐Borreguero LJ, de la Pompa JL. Sequential Notch activation regulates ventricular chamber development. Nat Cell Biol. 2016;18:7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kang X, Wei X, Wang X, Jiang L, Niu C, Zhang J, Chen S, Meng D. Nox2 contributes to the arterial endothelial specification of mouse induced pluripotent stem cells by upregulating Notch signaling. Sci Rep. 2016;6:33737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci USA. 1996;93:13943–13948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rothschild SC, Lahvic J, Francescatto L, McLeod JJ, Burgess SM, Tombes RM. CaMK‐II activation is essential for zebrafish inner ear development and acts through Delta‐Notch signaling. Dev Biol. 2013;381:179–188. [DOI] [PubMed] [Google Scholar]
- 66. Ann EJ, Kim HY, Seo MS, Mo JS, Kim MY, Yoon JH, Ahn JS, Park HS. Wnt5a controls Notch1 signaling through CaMKII‐mediated degradation of the SMRT corepressor protein. J Biol Chem. 2012;287:36814–36829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O'Donnell SE, Aykin‐Burns N, Zimmerman MC, Zimmerman K, Ham AJ, Weiss RM, Spitz DR, Shea MA, Colbran RJ, Mohler PJ, Anderson ME. A dynamic pathway for calcium‐independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental Methods.
Table S1. Primers Used for qPCR and Genotyping
Figure S1. Regulator of G protein signaling 6 (RGS6) and Ca2+/calmodulin‐dependent protein kinase II (CaMKII) genotyping of mice and embryos. Representative gel images used within the analysis of mouse RGS6 and CaMKII. Key bands used in the determination of RGS6 and CaMKII genotype are indicated to the right of each respective gel. A 100 bp DNA ladder was used in this analysis. We have identified the 200 and 500 bp bands within the ladder at the left of each gel image. Primers required for this analysis can be found in Table S1.
Figure S2. Summary data of expression of regulator of G protein signaling 6 (RGS6), oxidized Ca2+/calmodulin‐dependent protein kinase II (ox‐CaMKII), phosphorylated CaMKII (p‐CaMKII), and total Ca2+/calmodulin‐dependent protein kinase II (CaMKII) in mouse embryos at different gestation periods. Western blot analysis of RGS6, ox‐CaMKII, p‐CaMKII, and total CaMKII expression levels in mouse embryos at different gestation stages was performed and quantified using ImageQuant 5.2 program (N=3). Representative blot can be seen in Figure 1A.
Figure S3. Expression of BMP2 and Tbx2 in E10.5 hearts. Analysis of BMP2 and Tbx2 expression, quantified via quantitative polymerase chain reaction, revealed that Tbx2, but not BMP2, is significantly downregulated in regulator of G protein signaling 6 (RGS6)−/−/Ca2+/calmodulin‐dependent protein kinase II (CaMKII)VV hearts relative to RGS6−/−/CaMKIIMM hearts. *P<0.05, **P<0.01.
Figure S4. Regulator of G protein signaling 6 (RGS6) and oxidized Ca2+/calmodulin‐dependent protein kinase II (ox‐CaMKII) expression at E10.5 in RGS6+/+/Ca2+/calmodulin‐dependent protein kinase II (CaMKII)MM placentas. RGS6 and ox‐CaMKII (red) were expressed in the labyrinthine region of placentas as detected by immunohistochemistry. Scale bars=20 μm.
Figure S5. Hematopoietic defects in regulator of G protein signaling 6 (RGS6)−/−/Ca2+/calmodulin‐dependent protein kinase II (CaMKII)VV yolk sacs. A, In vitro differentiation analysis of yolk sac hematopoietic cells derived from RGS6−/−/Ca2+/calmodulin‐dependent protein kinase II (CaMKII)MM and RGS6−/−/CaMKIIVV yolk sacs at E10.5 using methylcellulose colony assay. The number of erythroid burst‐forming units (BFU‐E) colonies (>20 cells) produced from RGS6−/−/CaMKIIMM and RGS6−/−/CaMKIIVV yolk sacs were quantified after 7 days in culture, as shown in the representative images. This analysis revealed that the combined loss of RGS6 and ox‐CaMKII reduces the ability of hematopoetic cells to form colonies. Scale bars=50 μm. N=3. *P<0.01. B, Quantitative polymerase chain reaction analysis of hematopoietic gene expression in E10.5 yolk sac revealed a significant reduction in the expression of the GATA‐binding protein 1, β globin, and β‐major genes in RGS6−/−/CaMKIIVV yolk sacs relative to RGS6−/−/CaMKIIMM yolk sacs. N=3. *P<0.01.
Figure S6. Oxidized Ca2+/calmodulin‐dependent protein kinase II (ox‐CaMKII) and reactive oxygen species (ROS) expression in hearts of embryos of different genotypes. A, Detection of ox‐CaMKII (green) levels in wild‐type (regulator of G protein signaling 6 [RGS6]+/+/Ca2+/calmodulin‐dependent protein kinase II [CaMKII]MM), RGS6−/− (RGS6−/−/CaMKIIMM), and RGS6−/−/CaMKIIVV double mutant embryo hearts (arrows) by immunostaining reveals that RGS6 loss does not alter ox‐CaMKII expression. Note: embryos were not in the same sectioning planes. Scale bars=200 μm. B, Effects of combined loss of RGS6 and ox‐CaMKII on superoxide (red) generation in hearts of embryos as shown by dihydroethidium staining. Scale bars=50 μm.
Figure S7. Heart rates in regulator of G protein signaling 6 (RGS6)−/−/Ca2+/calmodulin‐dependent protein kinase II (CaMKII)MM and RGS6−/−/CaMKIIVV live embryos. The heart rate of live E10.5 embryos was manually counted by viewing the embryo through a dissection microscope. This analysis revealed no significant difference in the heart rate of RGS6−/−/CaMKIIVV double mutant embryos relative to RGS6−/−/CaMKIIMM embryos. N=3.