

Abstract
Background
Recent studies suggest that adult cardiac progenitor cells (CPCs) can produce new cardiac cells. Such cell formation requires an intricate coordination of progenitor cell proliferation and commitment, but the molecular cues responsible for this regulation in CPCs are ill defined.
Methods and Results
Extracellular matrix components are important instructors of cell fate. Using laminin and fibronectin, we induced two slightly distinct CPC phenotypes differing in proliferation rate and commitment status and analyzed the early transcriptomic response to CPC adhesion (<2 hours). Ninety‐four genes were differentially regulated on laminin versus fibronectin, consisting of mostly downregulated genes that were enriched for Yes‐associated protein (YAP) conserved signature and TEA domain family member 1 (TEAD1)‐related genes. This early gene regulation was preceded by the rapid cytosolic sequestration and degradation of YAP on laminin. Among the most strongly regulated genes was polo‐like kinase 2 (Plk2). Plk2 expression depended on YAP stability and was enhanced in CPCs transfected with a nuclear‐targeted mutant YAP. Phenotypically, the early downregulation of Plk2 on laminin was succeeded by lower cell proliferation, enhanced lineage gene expression (24 hours), and facilitated differentiation (3 weeks) compared with fibronectin. Finally, overexpression of Plk2 enhanced CPC proliferation and knockdown of Plk2 induced the expression of lineage genes.
Conclusions
Plk2 acts as coordinator of cell proliferation and early lineage commitment in CPCs. The rapid downregulation of Plk2 on YAP inactivation marks a switch towards enhanced commitment and facilitated differentiation. These findings link early gene regulation to cell fate and provide novel insights into how CPC proliferation and differentiation are orchestrated.
Keywords: cardiac progenitor cells, cell fate, extracellular matrix, polo‐like kinase 2, Yes‐associated protein
Subject Categories: Basic Science Research, Cell Signalling/Signal Transduction, Stem Cells
Clinical Perspective
What Is New?
Polo‐like kinase 2 (Plk2) acts as coordinator of proliferation and early lineage commitment of cardiac progenitor cells.
Downregulation of Plk2 slows proliferation and enhances lineage gene expression.
Downregulation of Plk2 follows loss of Yes‐associated protein activity and translates into facilitated endothelial differentiation.
What Are the Clinical Implications?
Molecules involved in the fate transition from a proliferating (ie, expanding) to a committed, ready to differentiate, cardiac progenitor cell could be therapeutically targeted to promote cardiac regeneration based on new cell formation.
Further studies are needed to examine whether the Yes‐associated protein/Plk2 axis qualifies as a potential target to therapeutically promote neovascularization.
Introduction
The adult mammalian heart has a limited capacity of cell replacement, which in case of injury, such as myocardial infarction, is insufficient to restore cardiac cellularity and function. The resulting heart failure is a major cause of morbidity and mortality, and an intense search for therapeutic strategies to promote cardiac regeneration is currently ongoing. Such an approach, however, requires a clear understanding of the molecular mechanisms orchestrating cardiac cell replacement. It is now well established that the adult heart harbors cardiac progenitor cells (CPCs), which are—in principal—capable of self‐renewal1, 2 and differentiation into cardiac lineages, including cardiomyocytes, endothelial cells, and vascular smooth muscle cells. A variety of markers and techniques for their identification and isolation have been reported.3 In preclinical studies, various populations, including c‐kit+ CPCs, cardiosphere‐derived cells, and cardiac side population (SP) CPCs, either endogenously or on in vitro expansion and transplantation, contribute to cardiac cell renewal after injury.4, 5, 6, 7 Clinical trials using c‐kit+ CPCs and cardiosphere‐derived cells to induce regeneration of the postinfarct myocardium in humans have been initiated.8, 9
Freshly isolated CPCs are low in numbers and require amplification before being transplanted into the injured heart for therapeutic application. Isolated CPCs can be readily expanded in vitro while keeping their multilineage differentiation potential. As shown for various CPC populations, including c‐kit+ and SP‐CPCs, successful differentiation on in vitro expansion requires additional steps and stimuli, including extracellular matrix (ECM), growth factors, and hormones.4, 10, 11 In addition, loss of multipotency associates with decreased cellular proliferation capacity.12 The identification of molecular cues that are responsible for the CPC fate transition from a proliferating (ie, expanding) to a committed, ready‐to‐differentiate phenotype, and that are also common to different CPC populations, will help identify therapeutic targets that could be exploited to promote cardiac regeneration based on new cell formation.
Cell fate decisions are characterized by dynamic shifts in the transcriptome that occur during specific phases of the cell cycle. Regulators of the cell cycle play an important role in the coordination of the cell cycle and gene expression, hence balancing proliferation and differentiation. An antagonistic relationship between cell cycle regulators and lineage gene expression was first described in skeletal muscle,13 but proliferation and differentiation decisions are complex and executed in a cell‐ and tissue‐dependent manner.12 How this coordination evolves in CPCs and what molecular cues regulate their fate transition are still poorly understood. The ECM is a major instructor of stem cell fate in terms of self‐renewal and lineage specification.14 It has recently been shown that proliferation of c‐kit+ CPCs is enhanced by fibronectin (FN)15 and that stiff matrix increases proliferation but diminishes differentiation of SP‐CPCs.16, 17, 18 Herein, we used a simplified model of ECM‐instructed CPC fate, using laminin (LN) and FN as substrates to produce two distinct phenotypes of c‐kit+ CPCs. These two phenotypes differed with respect to their proliferation rate, which was lower, and their commitment status, which was higher on LN compared with FN. To examine the early molecular cues of CPC lineage commitment, we investigated the early (<2 hours) transcriptomic response to CPC adhesion with particular emphasis on cell cycle regulatory genes. We identify the cell cycle regulator polo‐like kinase 2 (Plk2) as one of the most strongly downregulated genes on LN, and report a role for Plk2 as coordinator of proliferation and lineage commitment of c‐kit+ CPCs. To test whether Plk2 qualifies as a common fate cue of different populations of CPCs, this molecular regulation was validated in SP‐CPCs, a population that is distinct from c‐kit+ CPCs on their molecular signature.19
Methods
CPC Isolation and Culture
Rat CPCs (rCPCs) were a kind gift from Drs Piero Anversa and Annarosa Leri. They were established previously in the laboratory of Dr Anversa20 and used in a cell line‐like manner, such as different passages of cells were used for all experiments to support reproducibility. rCPCs were cultured in F12 medium (Thermo Fisher) containing 10% fetal bovine serum (FBS), 10 μg/L leukemia inhibitory factor (Millipore), 10 ng/mL basic fibroblast growth factor (PeproTech), 5 U/L erythropoietin and 0.2 mmol/L glutathione (both from Sigma) at 37°C with 5% CO2.
Isolation of Sca1+/CD31− SP‐CPCs from mice (mCPCs) was performed according to the Guide for the Care and Use of Laboratory Animals and with the approval of the Swiss Cantonal Authorities. Mice were euthanized by intraperitoneal injection of sodium pentobarbital (150–200 mg/kg body weight), followed by rapid excision of the heart. Cardiomyocyte‐depleted cardiac cell suspensions were prepared, as previously described,10 with some modifications. In brief, minced cardiac tissue from 4 mice was digested with 0.1% collagenase B (Roche) and 2.5 mmol/L CaCl2 at 37°C for 30 minutes, filtered, and washed with Hanks’ balanced salt solution buffer with 2% FBS. Cardiac cells were further treated with red blood cell lysis buffer (Biolegend). Erythrocyte‐depleted non‐myocytes were then resuspended (106 cells/mL) in DMEM supplemented with 10% FBS and 25 mmol/L HEPES (Thermo Fisher) and stained with bisbenzimide H33342 trihydrochloride (Hoechst) (Sigma #B2261; 5 μg/106 cells) at 37°C for 90 minutes in the dark. Cell surface antigen staining was performed at 4°C for 30 minutes using fluorescein isothiocyanate‐conjugated anti‐Sca‐1 antibody (BD Biosciences #557405, 1:200) and allophycocyanin‐conjugated anti‐CD31 antibody (BD Biosciences #551262, 1:200). 7‐Aminoactinomycin D (Thermo Fisher #A1310, 0.15 μg/106 cells) was added before fluorescence‐activated cell sorting to exclude dead cells. Sorting was performed using a BD Influx (BD Biosciences). The Hoechst dye was excited using UV 355 nm, fluorescence emission was collected with a 460/50‐nm band‐pass filter (Hoechst blue) and a 670/30‐nm band‐pass filter (Hoechst red). Verapamil (Sigma, 100 μmol/L) was used as negative control, as it suppresses extrusion of Hoechst. Sca1+/CD31− SP‐mCPCs were sorted and amplified using minimal essential medium α (Thermo Fisher) containing 20% FBS at 37°C with 5% CO2. SP‐mCPCs from passages 7 to 20 from 3 different isolations were used for all experiments, with each isolation containing cells from 4 mice. Experiments were performed on culture dishes coated with LN (Sigma #L2020, ready‐made in solution) and FN (Sigma #F4759, diluted in water).
Polymerase Chain Reaction Analysis for Cell Characterization
Whole hearts were cut into small pieces and homogenized with a polytron PT‐DA 07/2EC‐E107 homogenizer (Kinematica, Switzerland). Bone marrow was taken from rat femur. RNA was extracted from whole heart homogenates, bone marrow, mouse embryonic stem cells (used as positive control for stemness genes), and cultured rCPCs and SP‐mCPCs using TRI Reagent (Sigma). RNA was treated with a DNAse kit (Promega), according to the manufacturer's protocol. RNA (2 μg) was reverse transcribed using the High‐Capacity cDNA Reverse Transcription Kit (Applied Biosystems) according to the manufacturer's instructions. Polymerase chain reaction (PCR) was performed on a thermocycler (Biometra, Germany). Primer sequences (all from Microsynth) are described in Table S1. PCR was performed under the following conditions: 95°C for 2 minutes; X cycles of 95°C for 15 seconds; Y°C for 30 seconds; 72°C for 40 seconds; and a final elongation at 72°C for 10 minutes to amplify the genes (rat: Abcg2, Kit, Pou5f1, Abcb1a, Abcb1b, Nkx2.5, Gata4, Myh7b, Tnni3, Vwf, Pecam1, Gaphd, Tbx18, Wt1, Aldh1a2, and Tcf21); and 95°C for 2 minutes; X cycles of 94°C for 45 seconds; Y°C for 45 seconds; 72°C for 60 seconds; and a final elongation at 72°C for 10 minutes (rat: Nanog and Myh6; and all mouse genes). PCR products were then subjected to electrophoresis in a 2% agarose gel. X and Y are described in Table S1 number (#) of cycles and °C, respectively.
c‐Kit Staining
rCPCs were trypsinized and washed with PBS, incubated with 2 μg/106 cells of anti‐c‐Kit antibody (Santa Cruz #sc‐5535) or IgG isotype (#sc‐3888) for 1 hour and washed with PBS, and then stained with Alexa Fluor 568 conjugated anti‐rabbit antibody (1:100, Thermo Fisher) for 1 hour. Dead cells were removed by 7‐Aminoactinomycin D staining. Flow cytometry analysis was carried out with Fortessa (BD Biosciences).
Colony‐Forming Assay
Cells (12.5 mCPCs/cm2; 25 rCPCs/cm2) were seeded on 35‐mm dishes and cultured in growth medium with 10% FBS for rCPCs and 2% FBS for mCPCs for 6, 9, and 14 days. The cells were then washed with PBS, fixed with 100% ice‐cold methanol for 10 minutes on ice, and stained with 0.5% crystal violet in water for 10 minutes at room temperature. The cells were then washed with water 5 to 6 times and dried at room temperature.
CardioStem Sphere Formation
For generation of CardioStem spheres,21 0.25 to 0.5×105 mCPCs were seeded on 6‐well dishes with culture medium (35% Iscove's modified Dulbecco's medium, 32.5% DMEM, 32.5% F12, and 3.5% bovine growth serum [HyClone]) with 100 nmol/L oxytocin (Sigma) for 3 days, then trypsinized and cultured on bacterial dishes (P100) for 2 to 3 days to induce CardioStem sphere formation. For rCPCs, 1.0×105 cells were seeded on 6‐well dishes with culture medium (F12 with 10% FCS) with 100 nmol/L oxytocin for 3 days, then trypsinized and cultured on bacterial dishes (P100) for 3 days.
Cardiomyogenic Differentiation
Neonatal rat ventricular myocytes were isolated from 1‐ to 3‐day‐old Sprague Dawley rats, as previously described,22 according to the Guide for the Care and Use of Laboratory Animals, and with the approval of the Swiss Cantonal Authorities. SP‐mCPCs were isolated from green fluorescence protein transgenic mice and cocultured with neonatal rat ventricular myocytes at a ratio of 1:10 in DMEM (containing 1 g/L d‐glucose, l‐glutamine, and pyruvate, 7% FBS and 25 mmol/L HEPES) for 3 weeks. For differentiation of rCPCs, CardioStem sphere‐derived cells were plated on culture dishes with cardiomyogenic medium (minimal essential medium α, 2% bovine growth serum, oxytocin, 50 μg/mL ascorbic acid, 1 μmol/L dexamethasone, and 10 mmol/L α‐glycerol phosphate) for 7 to 12 days.21 The medium was changed every 3 to 4 days.
Endothelial Differentiation
A total of 1.5×105 mCPCs were seeded on culture dishes precoated with LN or FN (10 μg/mL) and kept in culture medium7 (35% Iscove's modified Dulbecco's medium, 32.5% DMEM, 32.5% F12, 6.5 ng/mL epidermal growth factor, 13 ng/mL basic fibroblast growth factor, 1.3% B27, 2 mmol/L l‐glutamine, 0.2 mmol/L glutathione, 0.0005 U/mL thrombin [Diagnotec AG, Switzerland], and 0.65 ng/mL cardiothrophin1 [PeproTech]) containing 0.1% bovine growth serum for 24 to 48 hours, then cultured with endothelial cell growth medium (EGM)‐2 (Lonza) for 3 weeks. rCPCs were seeded 60% confluent with 0.1% FBS‐containing F12 medium on LN‐ or FN‐coated dishes (10 μg/mL) and kept for 24 hours, then cultured with EGM‐2 for 3 weeks. EGM‐2 was changed every 3 to 4 days.
Tube Formation Assay
Plates (96 wells) were coated with 100 μL of matrigel (Corning) per well, and 4×104 cells in 100 μL of EGM‐2 medium were seeded on the gel and incubated for 24 hours. Cells were cultured with EGM‐2 for 3 weeks before being used for tube formation assays. Imaging was performed with Olympus IX50. The number of meshes and nodes was counted as described by DeCicco‐Skinner et al.23
ECM‐Cell Interaction Experiments
rCPCs were cultured in 75‐mL flasks with F12 medium containing 10% FBS and the indicated supplements for 2 to 3 days. Cells were then trypsinized and seeded on culture dishes that were pre‐coated with 10 μg/mL of either LN or FN at a cell density of <60% confluency in F12 medium containing 0.1% FBS and supplements, and incubated for the times indicated. mCPCs were cultured in 75‐mL flasks with minimal essential medium α containing 20% FBS for 3 to 5 days, and the medium was changed every 2 to 3 days. Cells were trypsinized and seeded on LN or FN (10 μg/mL)‐coated dishes at <60% confluency in minimal essential medium α with 0.1% to 0.5% FBS, and incubated for the times indicated.
Cell Proliferation and Viability
rCPCs were seeded on LN‐ or FN‐coated dishes with F12 medium containing 0.1% FBS and indicated supplements, and cells were counted after 2 and 3 days. For assessment of viability, cells were stained with trypan blue after 2 and 3 days, and trypan blue‐positive (dead) and trypan blue‐negative (viable) cells were quantified. Viable cells are given in relation to the total cell number.
Quantitative Reverse Transcription‐PCR Analysis
Total RNA was isolated using TRI Reagent, and 2 μg of RNA were reverse transcribed using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems), according to the manufacturer's instructions. The amplification was performed using Power SYBR Green PCR Master Mix and the respective primers (all from Microsynth, Table S1), with an ABI PRISM 7500 sequence detection system (Applied Biosystems). mRNA levels were calculated using the comparative CT method with 18S rRNA as endogenous control. Data are presented as −ddCT values (corresponding to the log2‐fold induction).
3‐Dimensional Cell‐Based Constructs
rCPCs were loaded on 3‐dimensional collagen discs of 3‐mm thickness and 8‐mm diameter (Ultrafoam™) pre‐coated for ≈18 hours with 10 μg/mL of either LN or FN using a perfusion bioreactor with direct perfusion of the culture medium through the scaffold pores, as previously described.24 rCPCs were seeded at a density of 14.6×106 cells/cm3, corresponding to 2.2×106 cells per scaffold, with a bidirectional flow rate of 1 mL/min for 4 hours, and cultured at 0.1 mL/min for the following 24 hours.25 The culture medium was supplemented with 1% FBS.
Immunocytochemistry and Immunohistochemistry
To assess cardiomyogenic differentiation, SP‐mCPC cocultured with neonatal rat ventricular myocytes were fixed with 4% paraformaldehyde for 10 minutes, permeabilized with 0.1% Triton for 30 minutes, and washed with PBS. Then, they were stained with DAPI (1:1000), anti‐α‐actinin antibody (1:20, Sigma #A7811) and Alexa Fluor 568 conjugated anti‐mouse secondary antibody (1:800). rCPCs were fixed with 100% ice‐cold methanol on ice for 15 minutes and washed with PBS, then stained with DAPI (1:500), anti‐α‐actinin antibody (1:50) and Alexa Fluor 568 conjugated anti‐mouse secondary antibody (1:1000). For all other experiments, cells were incubated in 8‐well chamber slides for the times indicated, fixed with 4% paraformaldehyde in PBS for 15 minutes, and incubated with 100% methanol for 2 minutes and 0.1% Triton X in PBS for 20 minutes, and then blocked with 10% normal goat serum ready to use (Thermo Fisher). The slides were then incubated with anti‐YAP antibody (Cell Signaling #14074; 1:200), Alexa Fluor 488 conjugated anti‐rabbit secondary antibody (1:1000), anti‐vWF antibody (Abcam #ab6994; 1:400), Alexa Fluor 647 dye‐conjugated anti‐rabbit secondary antibody (1:1000), and Hoechst (0.01 mg/mL). Scaffolds were fixed with 1% paraformaldehyde, treated with 30% sucrose overnight, embedded within optimal cutting temperature compound, cut into 10‐μm‐thick sections in a cryostat (Microm International GmbH, Germany) and stained with anti‐LN antibody (Abcam #ab11575; 1:200) and Alexa Fluor 546 conjugated anti‐rabbit secondary antibody (1:1000). Imaging was performed with Olympus BX61 and BX63.
Immunoblotting
Whole cell lysates were prepared with radioimmunoprecipitation assay (RIPA) buffer (Cell Signaling) containing PhosSTOP and Complete Protease Inhibitor Cocktail (both Roche). Cell lysates were incubated on ice and then subjected to SDS‐PAGE. Blots were probed with anti‐YAP (Cell Signaling #4912, 1:2000), anti‐phospho‐YAP (Ser 127) (Cell Signaling #4911; 1:2000), anti‐GAPDH (Sigma #SAB1405848; 1:5000), anti‐ubiquitin (Sigma #U5379; 1:100), anti‐Plk2 (Sigma #SAB4500156; 1:5000) and anti‐FLAG (Sigma #F3165; 1:2000). Quantification was performed with Image J software.
RNA Sequence Analysis
Triplicate samples of each experimental condition were used for RNA sequence analysis. Cells were washed with PBS and collected with TRI Reagent. Total RNA was isolated using the Direct‐zol RNA MiniPrep kit (Zymo Research), according to the manufacturer's protocol. Library preparations and RNA sequencing were performed by the Quantitative Genomics Facility at the Department of Biosystems Science and Engineering of the Swiss Federal Institute of Technology Zurich (Basel, Switzerland), using the TruSeq Stranded Total RNA LT kit and sequencing single end reads of 51 bp on a HiSeq 2000 (Illumina). Obtained single‐end RNA‐sequencing reads (51 mers) were mapped to the rat genome assembly, version rn4, with SpliceMap26, 27 included in the R/Bioconductor package QuasR (version 1.8.4)28 using the following command: qAlign(samples.txt, BSgenome.Rnorvegicus.UCSC.rn4, splicedAlignment=TRUE). Using RefSeq mRNA coordinates from University of California, Santa Cruz (http://genome.ucsc.edu, downloaded in June 2014), and the qCount function, we quantified gene expression as the number of reads that started within any annotated exon of a gene. The differentially expressed genes were identified using the edgeR package (version 3.10.5).29 To remove the batch effect from the data, we included it into the fitted model. RNA sequencing data have been deposited at the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE77231.
Proteasome Inhibitor Assay
rCPCs were incubated for 2 hours with 10 μmol/L of MG132 (Sigma) or dimethyl sulfoxide. Cells were then trypsinized and seeded on LN‐ or FN‐coated dishes with F12 medium containing 0.1% FBS, leukemia inhibitory factor, basic fibroblast growth factor, erythropoietin, and glutathione, as indicated above (CPC isolation and culture).
Immunoprecipitation
Cytosolic lysates were prepared with CytoBuster (Novagen) containing PhosSTOP and Complete Protease Inhibitor Cocktail and incubated on ice for 50 minutes, during which they were vortexed every 10 minutes for 10 seconds, and centrifuged at 15 000g for 5 minutes at 4°C. Immunoprecipitation was performed with anti‐YAP antibody (Cell Signaling #14074, 5 μL/sample), anti‐rabbit IgG (Cell Signaling #2729), and Protein G Sepharose 4 fast flow (GE Healthcare). Immunoprecipitates were washed with CytoBuster and eluted at 95°C for 5 minutes. Samples were then separated by SDS‐PAGE.
RNA Interference
Cells were cultured overnight, transfected with 20 to 40 nmol/L siRNA (from Qiagen: Control: #SI03650318, rPlk2: #SI01962163) using DharmaFECT 1 (Dharmacon), and maintained for 2 days before experiments.
Plasmid Transfections
pCMV‐flag S127A YAP was a gift from Dr Kunliang Guan30 (Addgene plasmid #27370). pCMV‐flag S127A YAP, pCMV‐Myc‐flag‐rPlk2 plasmid (OriGene #RR203879) and pCMV‐Myc‐flag plasmid (OriGene #PS100001) were transfected using Lipofectamin LTX (Thermo Fisher), according to the manufacturer's protocol. G418 (500 μg/mL, Thermo Fisher) was used for selection of drug resistant clones.
3‐[4,5‐Dimethylthiazol‐2‐yl]‐2,5 Diphenyl Tetrazolium Bromide Assay
The 3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5 diphenyl tetrazolium bromide (MTT) assay was performed using the cell proliferation kit I (Roche), according to the manufacturer's protocol.
Statistical Analyses
Unless otherwise indicated, data are presented as mean±SEM. Statistical analyses were performed with GraphPad Prism version 6 software (GraphPad) on nonnormalized (ie, raw) data for all data sets with n≥4 independent experiments using non‐parametric testing, as indicated. Expression differences of the quantitative PCR data were tested for significance based on dCT values. P<0.05 was considered statistically significant.
Results
LN Slows CPC Proliferation and Induces the Expression of Cardiac Lineage‐Specific Genes
The ECM is an important regulator of cell fate decisions,31, 32 and biomechanical properties of the ECM have previously been linked to CPC fate.17, 18 However, the contribution of individual ECM proteins to CPC fate is less understood. We, therefore, sought to test how LN and FN affect proliferation and lineage commitment of CPCs. c‐kit+ rCPCs and Sca1+/CD31− SP‐mCPCs were plated on plastic dishes that were either noncoated or precoated with LN or FN (10 μg/mL). In‐laboratory characterization of c‐kit+ rCPCs and Sca1+/CD31− SP‐mCPCs, showing gene and surface marker expression, clonogenicity, cardio‐stem‐sphere formation, and endothelial and cardiomyogenic differentiation, is depicted in Figure S1. Both cell types have previously been shown to exhibit cardiomyogenic and vasculogenic differentiation potential in vitro and in vivo,4, 5, 7, 10, 20, 33 although meaningful in vivo cardiomyogenic differentiation of c‐kit+ CPCs is currently highly disputed.34, 35 Cells were plated at low density (<60% confluency) to avoid cell‐cell contact and under low serum conditions (≤0.5% FBS) to prevent activation of growth factor–dependent pathways, which both could act as potential confounders of adhesion‐dependent CPC regulation. To assess substrate‐dependent proliferation of CPCs, identical numbers of rCPCs were plated on LN‐ or FN‐coated dishes, and cells were counted at days 2 and 3. We observed a continuous increase in cell numbers over time on FN, which was markedly blunted on LN (Figure 1A). The lower cell numbers on LN were not because of cell death, as cell viability was comparable between LN and FN and virtually stable throughout the 3‐day follow‐up (Figure 1B). We further sought to test whether the lower cell proliferation on LN may be associated with initiation of lineage specification. rCPCs were plated on LN and FN for 24 hours. Gene expression of the cardiac transcription factor GATA binding protein 4 (Gata4), the cardiomyogenic markers troponin I and β‐myosin heavy chain, and the endothelial markers vWF and CD31 (Pecam1) was measured. While there was a nonsignificant trend towards higher expression of Gata4, troponin I, and β‐myosin heavy chain in CPCs on LN than on FN, gene expression of both vWF and CD31 was significantly upregulated on LN as compared with FN (Figure 1C and Figure S2A), indicative of enhanced lineage commitment with a higher propensity towards the endothelial than cardiomyogenic lineage at low serum conditions and in the absence of facilitating factors. Similar experiments with SP‐mCPCs yielded comparable results (Figure 1D and Figure S2B).
Figure 1.
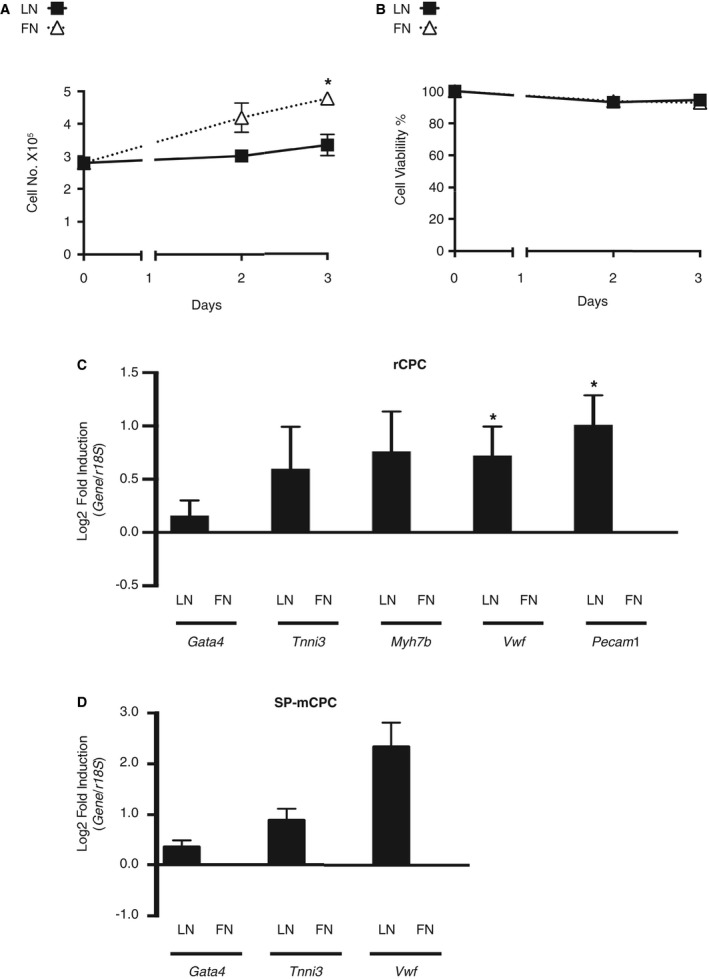
Phenotypic differences of cardiac progenitor cells (CPCs) on laminin (LN) vs fibronectin (FN). A and B, Absence of meaningful CPC proliferation on LN. A, Rat CPC (rCPC) growth curve on LN and FN with 0.1% fetal bovine serum (FBS) (n=4 different passages). *P<0.05 day 3 FN vs LN by Kruskal‐Wallis test, followed by Dunn's test. B, Corresponding cell viability (n=4). C and D, LN enhances lineage commitment with higher propensity towards the endothelial lineage. C, Gene expression by quantitative reverse transcription–polymerase chain reaction (qRT‐PCR) (n=6 different passages). rCPCs were plated on LN or FN with 0.1% FBS for 24 hours. *P<0.05 dCT FN vs LN for Vwf and Pecam1 (Wilcoxon signed rank test). D, Side population mouse CPCs (SP‐mCPCs) were plated on LN‐ and FN‐coated dishes with 0.5% FBS for 16 hours. Gene expression was assessed by qRT‐PCR (n=4 different passages from 3 different isolations; P=0.0625 for all 3 genes).
To test whether LN and FN regulate cell fate in a similar manner in a more physiological culture system, 3‐dimensional rCPC cultures were created using scaffolds precoated with LN (Figure S3A and S3B) or FN in a perfusion‐based bioreactor. While under perfusion on both LN and FN, the expression of endothelial lineage markers increased compared with nonadhering rCPCs in suspension, rCPCs cultured on LN‐coated scaffolds showed higher gene expression of vWF and CD31 (Figure S3C). Taken together and similar to pluripotent stem cells,36, 37 LN and FN appear to play an active role in CPC fate decision.
LN Facilitates Endothelial Differentiation of CPCs
Under growth conditions, rCPCs maintained their shape (Figure S4A), but grew confluent after 3 weeks in culture, exhibiting a cobblestone pattern similar to epicardial progenitor cells when cultured on LN or FN and kept under low serum within the first 24 hours (Figure 2A). However, although rCPCs expressed T‐box 18 (Tbx18) at a similar level as found in the neonatal heart, they did not express other epicardial markers, such as Wilms tumor protein, aldehyde dehydrogenase 1 family member a2, and transcription factor 21, neither at baseline (Figure S4B) nor after culturing (data not shown). To test whether the observed differences in the expression levels of lineage genes, which were more pronounced for the endothelial lineage, translate into differences in differentiation potential, cells were plated at low density and under low serum conditions on LN or FN for 24 hours, followed by 3 weeks of culturing in endothelial differentiation medium. Although rCPCs did not express vWF protein when cultured in growth medium (Figure S4C), rCPCs on both LN and FN adopted an endothelial cell phenotype after 3 weeks in differentiation medium, with more pronounced expression of vWF protein in rCPCs on LN than on FN (Figure 2A and 2B). Similarly, rCPCs that were primed and differentiated on LN had a higher capacity of tube formation than rCPCs on FN (Figure 2C and 2D). These findings suggest a sustained effect of LN facilitating endothelial differentiation of CPCs.
Figure 2.
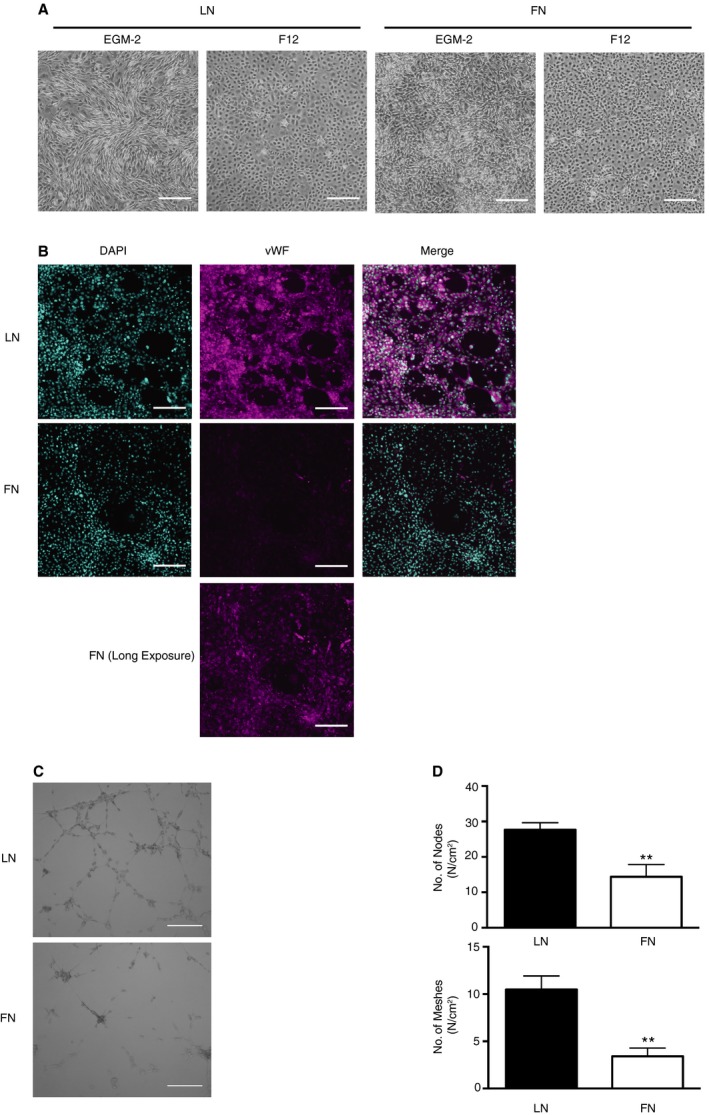
Laminin (LN) facilitates endothelial differentiation of cardiac progenitor cells (CPCs). A, Bright‐field (BF) images of rat CPCs (rCPCs) seeded on LN‐ or fibronectin (FN)‐coated dishes with 0.1% fetal bovine serum (FBS)–containing F12 medium for 24 hours, and then changed to either 2% FBS‐containing F12 or endothelial differentiation medium (endothelial cell growth medium [EGM]‐2) for 3 weeks (magnification ×2; bar=100 μm). B, More robust von Willebrand factor (vWF) protein expression on LN. rCPCs were seeded on LN‐ or FN‐coated dishes with 0.1% FBS‐containing F12 medium for 24 hours, then cultured for 3 weeks in 2% FBS‐containing EGM‐2 medium. Representative images from n=3 different passages (magenta: vWF; cyan: DAPI; magnification ×10; bar=100 μm). C and D, Enhanced tube formation capacity of CPCs differentiated on LN. Endothelial differentiation was induced in rCPCs, as per B; cells were then plated on matrigel for 24 hours, and numbers of nodes and meshes were counted (C and D). **P<0.01 by Mann‐Whitney test (n=6 experiments from 2 different passages) (BF, magnification ×2; bar=100 μm).
YAP Is Differentially Regulated in CPCs on LN Versus FN
General fluctuations in the transcriptome contribute to lineage choice.38 The transcriptional coactivator YAP can act as an intracellular checkpoint in the integration of (bio)mechanical signals by transducing ECM signals into the nucleus. YAP is regulated by the Hippo or noncanonical pathways and responds to cell adhesion and biomechanical properties of the substrate.39, 40 In addition, YAP is a known regulator of cell proliferation in many cell types, including organ‐specific progenitor cells,41, 42, 43, 44 and its expression is decreased during embryonic stem cell differentiation.45 We, therefore, hypothesized that YAP may be regulated on early CPC‐ECM protein interaction. Indeed, CPCs plated on LN showed rapid degradation of YAP protein within 2 hours (Figure 3A through 3F), with increasing concentrations of LN coating leading to more pronounced degradation (Figure 3G and 3H). In contrast, YAP protein abundance was virtually maintained in CPCs plated on FN, whereas CPCs on noncoated dishes showed intermediate to LN‐like YAP protein regulation.
Figure 3.
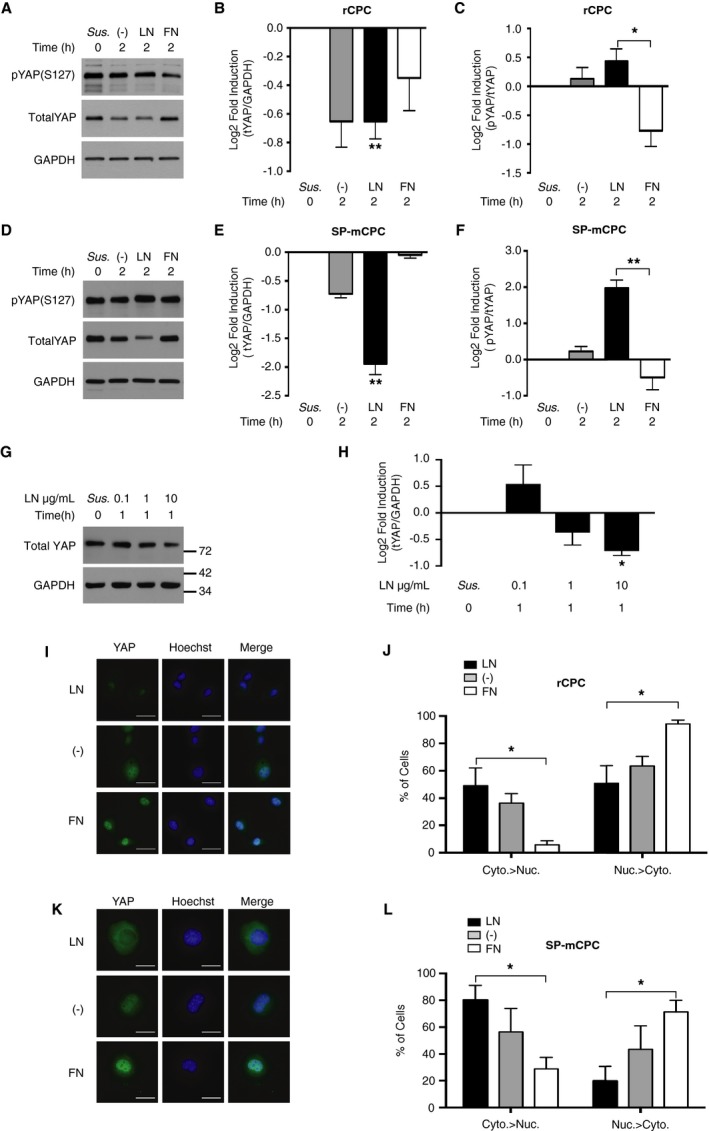
Laminin (LN), but not fibronectin (FN), decreases Yes‐associated protein (YAP) abundance and reduces its nuclear availability. A through F, YAP phosphorylation and protein abundance depending on the substrate. Rat cardiac progenitor cells (rCPCs) (A through C; n=5 different passages) and side population mouse CPCs (SP‐mCPCs) (D through F; n=4 different passages from at least 2 different isolations) were plated on LN‐ or FN‐coated dishes with 0.1% fetal bovine serum for 2 hours, and total and phosphorylated (serine 127) YAP (tYAP and pYAP, respectively) was detected by immunoblotting. Friedman test, followed by Dunn's test, on nonnormalized data for rCPCs: **P<0.01 for tYAP/GAPDH LN vs CPCs in suspension (Sus.), and *P<0.05 for pYAP/tYAP LN vs FN; for SP‐mCPCs: **P<0.01 for tYAP/GAPDH LN vs Sus., and for pYAP/tYAP LN vs FN. G and H, Dose‐response to different concentrations of LN coating. rCPCs were plated for 1 hour on dishes precoated with the indicated concentrations of LN, and YAP protein was assessed by immunoblotting (n=5). Friedman test, followed by Dunn's test, on nonnormalized data: *P<0.05 LN 10 vs Sus. I through L, Intracellular distribution of YAP. rCPCs (G and H; n=3) and SP‐mCPCs (I and J; n=4) were plated for 1 hour (SP‐mCPCs) or 2 hours (rCPCs), as above, and YAP was detected by immunocytochemistry. Quantitative data of the percentage of cells showing preferential cytosolic (Cyto.) or nuclear (Nuc.) localization of YAP are given.*P<0.05 for the distribution of cytosolic YAP and nuclear YAP LN vs FN for rCPCs and SP‐mCPCs, Kruskal‐Wallis test, followed by Dunn's test (bar=20 μm). −, noncoated.
YAP shuttles between the cytosol and the nucleus, where it exerts its activity as a transcriptional coactivator. Phosphorylation of YAP at serine 127 leads to the cytosolic sequestration of YAP, where it can be proteasomally degraded.46 Paralleling the degree of YAP protein loss, the ratio of serine 127–phosphorylated YAP/total YAP was significantly increased in CPCs plated on LN as compared with FN (Figure 3A through 3F). Furthermore, the percentage of cells exhibiting predominantly cytosolic localization of YAP, as assessed by immunocytochemistry, was significantly higher on LN than on FN, in which case most cells showed predominant nuclear YAP localization (Figure 3I through 3L). This was the case for both rCPCs and mCPCs and suggests that serine 127 phosphorylation‐mediated cytosolic sequestration precedes YAP degradation in CPCs on LN. Similar as for YAP protein expression, CPCs on noncoated dishes exhibited an intermediate to LN‐like response in terms of YAP phosphorylation and intracellular distribution.
YAP Inactivation on LN Is Associated With Early Differences in Gene Expression
YAP is a transcriptional coactivator, and its nuclear localization is required for YAP activity and YAP‐dependent gene expression. We, therefore, sought to identify potential YAP target genes that could mediate the adhesion‐dependent CPC phenotype in our model. rCPCs were either kept nonadhering in suspension or plated on LN, FN, or noncoated dishes for 2 hours, and early response gene expression was assessed by RNA sequencing (n=3). Gene expression was remarkably changed through cell adhesion to all 3 substrates (LN, FN, and noncoated dishes) as compared with CPCs in suspension. In total, 4984 genes were significantly (adjusted P<0.05) regulated on LN, 2395 on FN, and 3261 on noncoated dishes when compared with suspension.
Because nuclear YAP was depleted on LN and enriched on FN, we further compared gene regulation on LN versus FN. Ninety‐four genes were significantly different between LN and FN (adjusted P<0.05; Table S2). Among the top‐ranked gene sets enriched were YAP conserved signature (CORDENONSI_YAP_CONSERVED_SIGNATURE; Figure S5A) and genes related to the YAP‐interacting transcription factor TEAD1 (WGGAATGY_V$TEF1_Q6; Figure S5B), whereby the vast majority of the genes were downregulated on LN versus FN for both sets. To identify potential target genes directly downregulated in response to YAP inactivation, genes that were at least 1 log2‐fold downregulated on LN versus FN (17 genes; Table), but showed no significant regulation on FN versus suspension, were further examined (9 genes, marked with an asterisk). The 3 top‐ranked genes, which all showed >1.5 log2‐fold downregulation on LN versus FN, were cysteine‐rich angiogenic inducer 61 (Cyr61), connective tissue growth factor (Ctgf), and Plk2. To confirm the adhesion‐dependent regulation of these 3 candidate genes, we performed single gene expression analyses by quantitative reverse transcription–PCR for Cyr61, Ctgf, and Plk2 in suspended rCPCs and after plating on LN and FN. Consistent with the results from RNA sequencing, single gene expression analyses showed downregulation of all 3 genes on LN (Figure 4A through 4C), and this was also true in mCPCs (Figure 4D through 4F). Plk2 is serum inducible.47 To avoid growth factor and/or mitogenic stimulation, we performed our experiments under low serum conditions, but we observed similar regulation of YAP and Plk2 when 10% FBS was used. However, these results did not reach statistical significance (Figure 4G through 4I).
Table 1.
Genes With ≥Log2‐Fold Upregulation on FN Versus LN
| FN vs LN | FN vs Sus. | LN vs Sus. | NC vs Sus. | Candidate genes | ||||
|---|---|---|---|---|---|---|---|---|
| Symbol | EntrezID | Log2 FC | Adj.P.Val | |||||
| 1 | Cyr61 | 83476 | 3.4 | 2.5E‐40 | −3.3 | −2.8 | a | |
| 2 | Ctgf | 64032 | 2.0 | 2.7E‐30 | −2.9 | −3.0 | a | |
| 3 | Plk2 | 83722 | 1.7 | 4.2E‐28 | −1.6 | −1.7 | a | |
| 4 | Nlrp3 | 287362 | 1.7 | 1.4E‐14 | −1.1 | −2.8 | −2.8 | |
| 5 | Rn28s | 100861535 | 1.5 | 1.1E‐03 | ||||
| 6 | Rn45s | 24723 | 1.5 | 2.4E‐06 | ||||
| 7 | Amotl2 | 65157 | 1.4 | 1.3E‐27 | ||||
| 8 | Ajuba | 85265 | 1.4 | 3.5E‐29 | −1.5 | −1.5 | a | |
| 9 | Nuak2 | 289419 | 1.3 | 3.8E‐20 | −1.3 | −1.5 | a | |
| 10 | Vof16 | 259227 | 1.3 | 9.2E‐10 | −2.3 | −2.0 | a | |
| 11 | Ankrd1 | 27064 | 1.2 | 2.4E‐06 | −1.8 | −3.0 | −3.3 | |
| 12 | Gadd45b | 299626 | 1.2 | 2.2E‐13 | ||||
| 13 | Gata3 | 85471 | 1.2 | 9.0E‐11 | −1.4 | −1.7 | a | |
| 14 | Rnd1 | 362993 | 1.1 | 1.3E‐10 | −1.1 | a | ||
| 15 | Hbegf | 25433 | 1.1 | 1.1E‐03 | −1.9 | −3.0 | −2.7 | |
| 16 | Hmgcs1 | 29637 | 1.1 | 1.7E‐06 | ||||
| 17 | Epha2 | 366492 | 1.0 | 1.1E‐06 | −1.8 | −1.6 | a | |
Results from RNA sequence analyses (n=3). Adj.P.Val, adjusted P value; EX=10X; FC, fold change; FN, fibronectin; LN, laminin; NC, noncoated dishes; Sus., rat cardiac progenitor cells in suspension.
Identified candidate genes (significantly downregulated on LN vs Sus. in the absence of regulation on FN vs Sus.).
Figure 4.
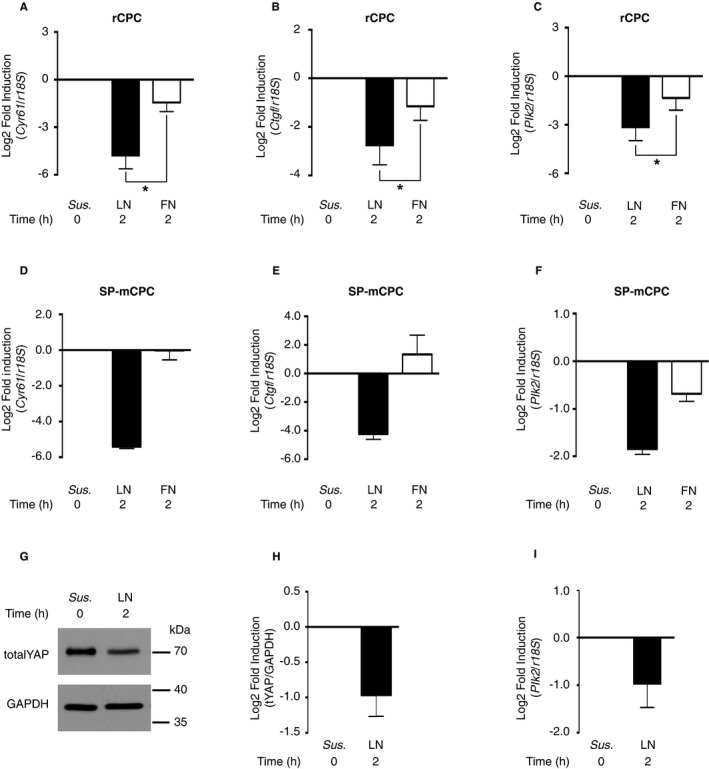
Polo‐like kinase 2 (Plk2) is downregulated on laminin (LN). A through C, Confirmatory gene expression analyses. Rat cardiac progenitor cells (rCPCs) were plated as per Figure 2, and gene expression of the known Yes‐associated protein (YAP)‐1 target genes Cyr61 and Ctgf and of the candidate gene Plk2 was analyzed by quantitative reverse transcription–polymerase chain reaction (qRT‐PCR) (n=5 different passages). *P<0.05 dCT fibronectin (FN) vs LN for Cyr61, Ctgf, and Plk2 (Wilcoxon signed rank test). D through F, Side population mouse CPCs (SP‐mCPCs) were plated on LN‐ and FN‐coated dishes with 0.5% fetal bovine serum (FBS) for 2 hours (n=3 different passages from at least 2 different isolations). G through I, LN‐induced downregulation of the serum‐inducible kinase Plk2 also occurs in the presence of serum. rCPCs were plated on LN‐coated dishes for 2 hours in the presence of 10% FBS. G and H, Protein expression of YAP by immunoblotting (n=3). I, Gene expression of Plk2 by qRT‐PCR (n=5; P=0.0938). Sus, suspension.
Cyr61 and Ctgf have previously been identified as YAP target genes,44, 48 but little is known about the regulation and role of Plk2. We, therefore, tested whether stabilization of YAP protein allows for mRNA levels of the known YAP target genes Cyr61 and Ctgf, and of the newly identified candidate gene Plk2, to be maintained on LN. On cytosolic retention, YAP can be ubiquitinated and proteasomally degraded. Consistently, YAP ubiquitination was present in rCPCs plated on LN for 30 minutes (Figure 5A), and under treatment with MG132, a cell‐permeable proteasome inhibitor, YAP protein abundance on LN was preserved (Figure 5B). Interestingly, although mRNA levels of Cyr61 were not rescued in the presence of MG132 (Figure 5C), the LN‐induced decrease of Ctgf and Plk2 mRNA was attenuated (Figure 5D and 5E), suggesting that Plk2 mRNA levels depend on YAP stability in our model. To corroborate YAP dependency of Plk2 regulation, rCPCs were transfected with mutant YAP, in which serine 127 is replaced by alanine (Flag‐hYAPS127A), preventing the serine 127 phosphorylation‐induced cytosolic sequestration of YAP. Transfection with this construct significantly upregulated Plk2 mRNA, supporting that Plk2 may represent a to date unrecognized downstream effector of YAP (Figure 5F and 5G).
Figure 5.
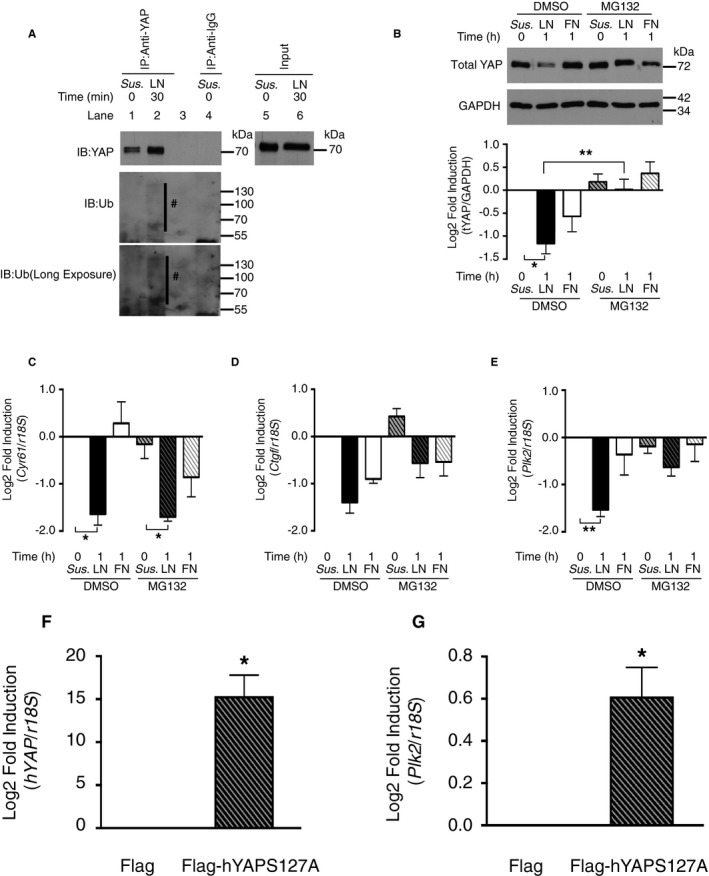
Polo‐like kinase 2 (Plk2) regulation depends on Yes‐associated protein (YAP) stability. A, Laminin (LN) induces YAP ubiquitination. Rat cardiac progenitor cells (rCPCs) were plated as described for 30 minutes. #: ubiquitinated (Ub) YAP. B, MG132 stabilizes YAP on LN. rCPCs were treated with MG132 (10 μmol/L) for 2 hours before trypsinization and plating on the respective substrates for 1 hour. n=5–7 different passages. *P<0.05 for dimethyl sulfoxide (DMSO)/LN vs DMSO/rCPCs in suspension (Sus.) and **P<0.01 for DMSO/LN vs MG132/LN (Kruskal‐Wallis test, followed by Dunn's test). C through E, Stabilization of YAP rescues connective tissue growth factor (Ctgf) and Plk2 mRNA expression on LN. Gene expression by quantitative reverse transcription–polymerase chain reaction (qRT‐PCR) (n=4) after plating of MG132‐treated rCPCs for 1 hour. For cysteine‐rich angiogenic inducer 61 (Cyr61): *P<0.05 dCT DMSO/LN vs DMSO/Sus. and MG132/LN vs MG132/Sus.; for Ctgf: P=0.1657 dCT DMSO/LN vs. DMSO/Sus. and MG132/LN vs. MG132/Sus.; and for Plk2: **P<0.01 dCT DMSO/LN vs DMSO/Sus. (Friedman test, followed by Dunn's test). F, hYAP serine 127A transfection efficiency in rCPCs. *P<0.05 dCT Flag‐hYAPSer127A vs Flag (Wilcoxon signed rank test, n=6). G,rPlk2 expression in hYAPSer127A‐overexpressing cells. Cells were transfected with Flag or Flag‐hYAPS127A plasmid and maintained for 2 days before RNA isolation. Gene expression was analyzed by qRT‐PCR (n=6). *P<0.05 dCT Flag‐hYAPSer127A vs Flag. FN, fibronectin; IB, Immunoblotting.
Loss and Gain of Function of Plk2 Affect CPC Proliferation and Lineage Commitment
While YAP has been implicated in cell proliferation and differentiation in various cell types, the role of Plk2 in the regulation of cell fate is less clear. Plk2 mRNA and protein are upregulated during the G1 and early S phase of the cell cycle, and their activation facilitates cell cycle progression, thereby regulating cell proliferation.49 To assess whether Plk2 qualifies as mediator of the LN‐induced CPC phenotype, we first examined the effects of Plk2 knockdown by siRNA on rCPC proliferation and expression of cardiac lineage markers. Although knockdown of Plk2 was only moderate (Figure 6A, 6B, and 6D), Plk2 siRNA blunted CPC proliferation on FN (Figure 6C). In addition, Plk2 siRNA induced the expression of cardiomyogenic and endothelial lineage markers (Figure 6E) in the absence of permissive factors, such as differentiation medium. These findings show that downregulation of Plk2 gene expression is sufficient to mimic the CPC phenotype observed on LN and suggest a so far unknown role for Plk2 in the regulation of CPC fate. Next, we sought to test whether overexpression of Plk2 may revert the decreased cell proliferation on LN and diminish CPC lineage commitment. mCPCs were transfected with Plk2 plasmid, and cell proliferation was assessed on LN after 24 hours by MTT assay. Consistent with our hypothesis, MTT reduction was higher in Flag‐Plk2–expressing mCPCs as compared with control (Figure 6F and 6G). Furthermore, expression of endothelial and—to a lesser degree—cardiomyogenic lineage markers was diminished in Flag‐Plk2–expressing rCPCs (Figure 6H and 6I). The observed differences in lineage gene expression are strongly suggestive of an inverse relationship between Plk2 and lineage gene expression in both directions (ie, not only when Plk2 is downregulated), but our results did not reach statistical significance.
Figure 6.
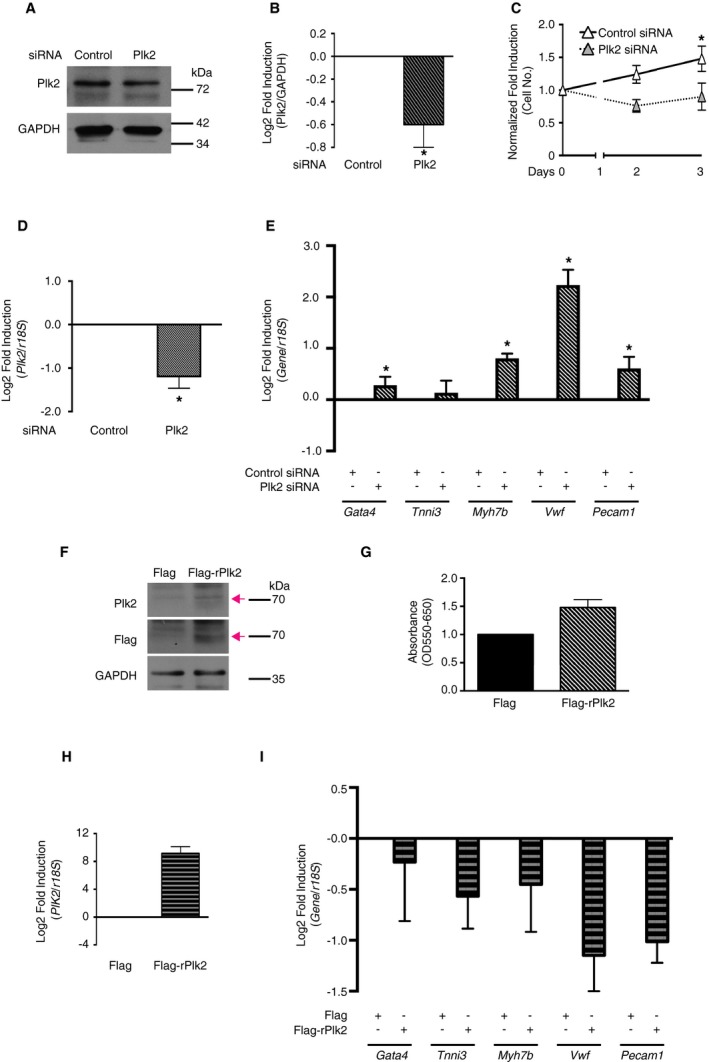
Transcriptional regulation of polo‐like kinase 2 (Plk2) affects cardiac progenitor cell (CPC) proliferation and lineage gene expression. A through E, Knockdown of Plk2 inhibits CPC proliferation on fibronectin (FN) and mimics the CPC response to laminin (LN). A and B, Efficiency of Plk2 knockdown (immunoblotting; n=5 different passages, *P<0.05 control siRNA (siCtr) vs Plk2 siRNA (siPlk2), Wilcoxon signed rank test). C, Increase in cell number on FN with Plk2 siRNA (n=5). Rat CPCs (rCPCs) were transfected with siRNA and maintained for 2 days, then plated on FN with 0.1% fetal bovine serum (FBS). *P<0.05 day 3 siCtr vs siPlk2 by Kruskal‐Wallis test, followed by Dunn's test. D and E, Plk2 and lineage marker gene expression by quantitative reverse transcription–polymerase chain reaction (qRT‐PCR) after plating for 24 hours (n=5). For siCtr vs siPlk2: *P<0.05 dCT Plk2, Gata4, Myh7b, Vwf, and Pecam1 (Wilcoxon signed rank test). F through I, Overexpression of Plk2 promotes CPC proliferation on LN and mimics the CPC response to FN. F, Efficiency of Plk2 overexpression (immunoblotting). G, The 3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5 diphenyl tetrazolium bromide assay on LN (n=3 different passages from at least 2 different isolations). Side population mouse CPCs‐were transfected with Flag or Flag‐rPlk2, selected, and amplified, then plated on LN with 0.5% FBS for 24 hours. H and I, Plk2 and lineage marker gene expression by qRT‐PCR (n=4). rCPCs were transfected with Flag or Flag‐rPlk2 and maintained for 2 days before RNA isolation (P=0.0625 for Vwf and Pecam1, and >0.1 for all other genes).
Discussion
This report is unique in that it focuses on the early molecular events in the process of CPC lineage commitment and differentiation. Using a simplified model of matrix protein‐instructed CPC fate, we identify Plk2 among a set of genes that are regulated early (ie, within 2 hours of CPC adhesion) and describe a role for Plk2 in the coordination of proliferation and early lineage commitment of CPCs as a key finding of this study. We also show that this early transcriptomic response is prompted by the rapid intracellular trafficking of the mechanosensor YAP and is associated with the facilitated endothelial differentiation of CPCs on LN (Figure 7).
Figure 7.
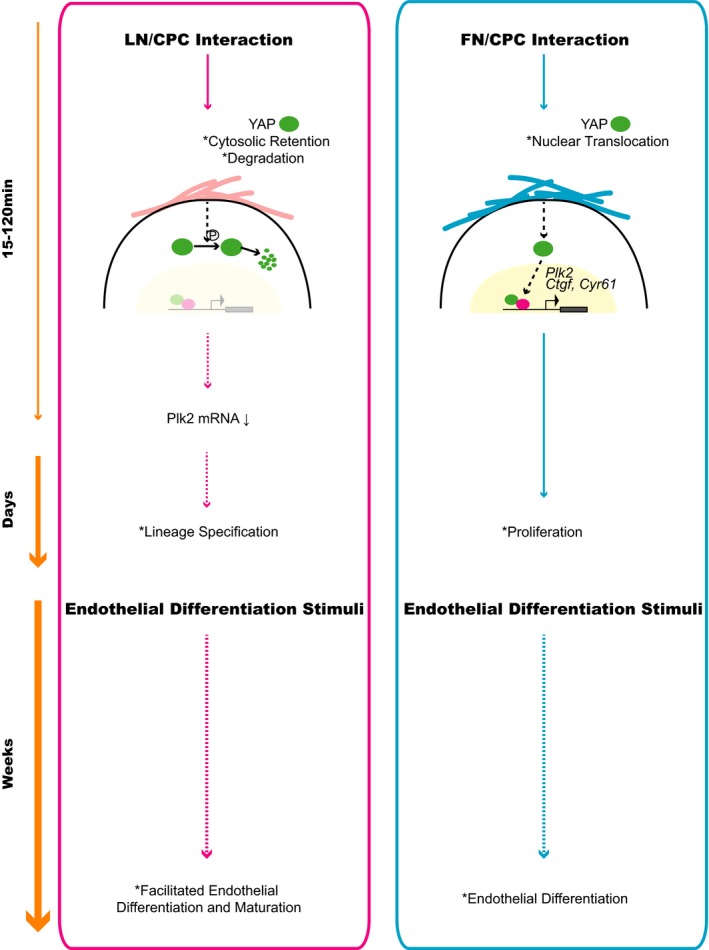
Proposed model of the molecular mechanisms facilitating endothelial differentiation of cardiac progenitor cells (CPCs). FN, fibronectin; LN, laminin; YAP, Yes‐associated protein.
Plk2 is a serine‐threonine kinase that acts as a critical regulator of cell cycle progression.50, 51, 52 In fact, the rapid decrease in Plk2 mRNA expression on LN‐CPC contact preceded, and was causatively linked to, the reduced CPC proliferation, which was absent when Plk2 mRNA expression was preserved, such as in CPCs on FN or in Flag‐Plk2–transfected CPCs on LN. Slowing of cell proliferation is associated with prolongation of the gap phases, the most critical phases for lineage programming and initiation of differentiation.53 Considering the spontaneous differentiation that can be observed in pluripotent stem cells when the G1 phase is prolonged54, 55 and the prolongation of the G1 phase because of delayed entry into the S phase that occurs in Plk2‐/‐ embryonic fibroblasts,56 decreased expression of Plk2 may mark the timing of early lineage programming. Consistent with this hypothesis, we found that Plk2 expression inversely regulated the expression of cardiac lineage genes. Not only was the decrease in Plk2 mRNA in CPCs on LN followed by the enhanced expression of lineage genes, but knockdown of Plk2 was sufficient to mimic the LN‐induced CPC phenotype as it decreased CPC proliferation on FN and increased lineage gene expression. Its forced expression accelerated CPC proliferation on LN and suppressed lineage gene expression. Our data, therefore, propose an as yet unknown function of Plk2 as a fate cue and molecular link between cell cycle and early lineage commitment in CPCs.
Plk2 was among a set of genes that were regulated early on LN‐CPC contact, and this early transcriptomic response was preceded by the phosphorylation, cytosolic retention, and degradation of YAP, which result in YAP inactivation. Inactivation of YAP occurs during embryonic stem cell differentiation,45 whereas activated YAP regulates epidermal stem cell proliferation,57 which are both in accordance with our observations in CPCs. We also found that the genes regulated early on LN compared with FN were enriched for YAP conserved signature genes and TEAD1‐associated genes, which were mostly downregulated and support decreased YAP activity on LN. In addition to the canonical YAP target genes Cyr61 and Ctgf, Plk2 was among the 3 genes showing the most pronounced regulation on LN. Although Plk2 has not yet been identified as a YAP target gene, upregulation of Plk2 was previously observed in YAP‐overexpressing MCF‐10A cells.58 We found Plk2 upregulated in CPCs transfected with S127A‐mutant YAP, positioning Plk2, in principal, downstream of YAP. Furthermore, Plk2 mRNA was consistently rescued by YAP protein stabilization. Expression of both Cyr61 and Ctgf depends on TEAD transcription factors,44, 48 but the mechanisms regulating Plk2 expression are poorly understood.59 Plk2 is a target of microRNAs, and YAP was recently found to suppress microRNA processing when present in the nucleus.60 One could speculate that YAP cytosolic trafficking may unleash microRNA activity, hence leading to Plk2 mRNA degradation.
The regulation of YAP localization within CPCs has recently been linked to the stiffness of the substrate, whereby a stiff substrate favors nuclear localization and a soft substrate favors cytosolic retention.61 YAP nuclear localization correlates with increased cell spreading area, as previously reported in other cell types,39 and shows within 3 to 5 hours of CPC contact with the substrate. For both CPC types studied, we found predominantly nuclear localization of YAP on FN versus predominantly cytosolic localization on LN. However, in our model and in contrast to the reported findings,61 differences in YAP intracellular localization and abundance were established within 15 to 120 minutes on plating (data not shown), which suggests a mechanism of signal transduction that is distinct from previously described pathways of mechanosensing.39, 40 In fact, integrin–focal adhesion kinase–Src has recently been deemed responsible for YAP nuclear localization on FN through suppression of canonical Hippo signaling in MCF‐10A cells.62
CPCs respond to the biomechanical properties of their environment. Mechanical cues guide the endothelial differentiation of cardiosphere‐derived CPCs,18 and the stiffening of the ECM after cardiac injury enhances proliferation, but impairs differentiation, of SP‐CPCs.17 We observed a higher proliferation rate of CPCs on FN, but increased expression of cardiovascular lineage genes on LN, which was more pronounced for the endothelial lineage and translated into facilitated endothelial differentiation. These findings are consistent with a previous report on the role of FN in CPC expansion15 and support that specific ECM proteins act as CPC fate cues. We found that CPCs that were primed under low serum conditions on LN for 24 hours and then exposed to endothelial differentiation conditions reached a higher degree of maturity after 3 weeks in culture than cells on FN. This showed in the more robust expression of vWF protein and a higher capacity of tube formation. Whether this mechanism is unique for endothelial differentiation or may apply to cardiomyogenic differentiation as well remains to be established. The true nature of c‐kit+ cells in the heart is currently under intense debate,34, 35 with evidence suggesting a limited propensity towards a cardiomyogenic lineage in vivo.5, 63, 64 Still, cardiomyogenic genes were increased in CPCs on LN versus FN, as well as in Plk2‐deficient rCPCs, indicating a potential role of the identified pathway in commitment towards both endothelial and cardiomyogenic lineages, depending on cell type and context. However, it has to be emphasized that successful differentiation of CPCs requires additional corroborative stimuli, and numerous cofactors, including cell‐cell interactions, soluble factors, oxygen tension, and others, may interfere with this process. In particular, cardiomyogenic differentiation into functional cardiomyocytes with intact excitation‐contraction coupling and calcium transients relies on CPC‐cardiomyocyte contact, which may compete with matrix‐dependent YAP regulation, and includes features such as gap junction formation.10 But evidence is accumulating that cardiomyogenesis, endogenously or on cell transplantation, may be an unlikely event in the injured heart. Instead, using an advanced fate‐tracking method, both c‐kit+ and SP‐CPCs (ATP‐binding cassette subfamily G member 2‐expressing cells) have recently been shown to mostly produce endothelial cells.5, 65 Although our data demonstrate a role for Plk2 in CPC lineage commitment and endothelial differentiation, this study does not address the in vivo implications of this regulation, which is the focus of our ongoing investigation. Such work is crucial in view of a potential therapeutic exploitation of the described pathway in the future (eg, to promote neovascularization by inducing endothelial differentiation of CPCs through Plk2 modulation).
In conclusion, we identify Plk2 as a coordinator of cell proliferation and early lineage commitment of CPCs, whose early regulation is associated with the facilitated endothelial differentiation of CPCs on LN downstream of YAP. These findings link early gene regulation to cell fate and provide novel insights into how CPC proliferation and differentiation are orchestrated. While we do not believe that Plk2 is the only responsible gene involved in the coordination of cell cycle and early lineage commitment in CPCs, an improved understanding of the molecular mechanisms of early lineage programming, which includes the identification of novel fate cues, as shown in this study, will ultimately lead to improved strategies for therapeutic regeneration.
Author Contributions
Conceptualization, Mochizuki, Pfister, Kuster; Methodology, Mochizuki, Marsano, Pfister, Kuster; Formal Analysis, Mochizuki, Ivanek, Kuster; Investigation, Mochizuki, Lorenz, Della Verde, Gaudiello; Resources, Ivanek; Writing—Original Draft, Mochizuki, Kuster; Writing—Review and Editing, all; Supervision, Kuster; Funding Acquisition, Mochizuki, Kuster.
Sources of Funding
This work was supported by a grant from the Swiss National Science Foundation (grant number 310030_144208 to Kuster), the Foundation for Cardiovascular Research, Basel, and the Medical Division of the Margarete and Walter Lichtenstein Foundation, University of Basel (all to Kuster) as well as a grant‐in‐aid from the University of Basel (to Mochizuki).
Disclosures
None.
Supporting information
Table S1. List of Primers, Cycle Numbers (No. of Cycles), and Temperatures (°C)
Table S2. Complete List of Genes Regulated on FN Versus LN
Figure S1. Characterization of mouse and rat cardiac progenitor cell (CPCs).
Figure S2. Nonnormalized data of gene expression as per Figure 1.
Figure S3. Lineage marker expression in cardiac progenitor cells (CPCs) in 3‐dimensional culture on laminin (LN)‐ and fibronectin (FN)‐coated scaffolds using a perfusion bioreactor.
Figure S4. A, Morphological features of rat cardiac progenitor cells (rCPCs) under growth conditions. rCPCs are an inhomogeneous cell population encompassing cells with different shapes. B, Gene expression of epicardial markers in rCPCs. Tbx18 is the only marker expressed in rCPCs. C, rCPCs do not express von Willebrand factor (vWF) protein in culture medium (F12). Human microvascular endothelial cells (HMECs) were used as positive control (bar=25 µm). Aldh1a2, aldehyde dehydrogenase 1 family member a2; rHeart, RNA from neonatal rat heart serving as positive control; Tcf21, transcription factor 21; Wt1, Wilms tumor protein 1.
Figure S5. Heat maps of gene expression from enriched gene sets as identified by Gene Set Enrichment Analysis (GSEA) in rat cardiac progenitor cells (CPCs) on laminin (LN) and fibronectin (FN).
Acknowledgments
We thank Drs Annarosa Leri and Piero Anversa from the Brigham and Women's Hospital and Harvard Medical School (Boston, MA) for the generous gift of rat cardiac progenitor cells; the staff from the Flow Cytometry Facility from the Department of Biomedicine (DBM), University and University Hospital Basel, for cell sorting; Dr Christian Beisel, Katja Eschbach, and colleagues from the Quantitative Genomics Facility at the Department of Biosystems Science and Engineering of the Swiss Federal Institute of Technology Zurich in Basel for RNA libraries and sequencing; Dr Takafumi Shimizu for support with flow cytometry analyses; Daria Monogiou Belik for neonatal rat heart RNA extraction; Dr Florian Geier (DBM) for statistical advice; and Dr Primo Schär (DBM) for helpful discussions.
(J Am Heart Assoc. 2017;6:e005920 DOI: 10.1161/JAHA.117.005920.)
References
- 1. Sereti KI, Oikonomopoulos A, Unno K, Cao X, Qiu Y, Liao R. ATP‐binding cassette G‐subfamily transporter 2 regulates cell cycle progression and asymmetric division in mouse cardiac side population progenitor cells. Circ Res. 2013;112:27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shenje LT, Andersen P, Uosaki H, Fernandez L, Rainer PP, Cho GS, Lee DI, Zhong W, Harvey RP, Kass DA, Kwon C. Precardiac deletion of numb and numblike reveals renewal of cardiac progenitors. Elife. 2014;3:e02164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Martin‐Puig S, Wang Z, Chien KR. Lives of a heart cell: tracing the origins of cardiac progenitors. Cell Stem Cell. 2008;2:320–331. [DOI] [PubMed] [Google Scholar]
- 4. Ellison GM, Vicinanza C, Smith AJ, Aquila I, Leone A, Waring CD, Henning BJ, Stirparo GG, Papait R, Scarfo M, Agosti V, Viglietto G, Condorelli G, Indolfi C, Ottolenghi S, Torella D, Nadal‐Ginard B. Adult c‐kit(pos) cardiac stem cells are necessary and sufficient for functional cardiac regeneration and repair. Cell. 2013;154:827–842. [DOI] [PubMed] [Google Scholar]
- 5. van Berlo JH, Kanisicak O, Maillet M, Vagnozzi RJ, Karch J, Lin SC, Middleton RC, Marban E, Molkentin JD. C‐kit+ cells minimally contribute cardiomyocytes to the heart. Nature. 2014;509:337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Malliaras K, Zhang Y, Seinfeld J, Galang G, Tseliou E, Cheng K, Sun B, Aminzadeh M, Marban E. Cardiomyocyte proliferation and progenitor cell recruitment underlie therapeutic regeneration after myocardial infarction in the adult mouse heart. EMBO Mol Med. 2013;5:191–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Noseda M, Harada M, McSweeney S, Leja T, Belian E, Stuckey DJ, Abreu Paiva MS, Habib J, Macaulay I, de Smith AJ, Al‐Beidh F, Sampson R, Lumbers RT, Rao P, Harding SE, Blakemore AI, Eirik Jacobsen S, Barahona M, Schneider MD. PDGFRalpha demarcates the cardiogenic clonogenic Sca1(+) stem/progenitor cell in adult murine myocardium. Nat Commun. 2015;6:6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bolli R, Chugh AR, D'Amario D, Loughran JH, Stoddard MF, Ikram S, Beache GM, Wagner SG, Leri A, Hosoda T, Sanada F, Elmore JB, Goichberg P, Cappetta D, Solankhi NK, Fahsah I, Rokosh DG, Slaughter MS, Kajstura J, Anversa P. Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): initial results of a randomised phase 1 trial. Lancet. 2011;378:1847–1857. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9. Makkar RR, Smith RR, Cheng K, Malliaras K, Thomson LE, Berman D, Czer LS, Marban L, Mendizabal A, Johnston PV, Russell SD, Schuleri KH, Lardo AC, Gerstenblith G, Marban E. Intracoronary cardiosphere‐derived cells for heart regeneration after myocardial infarction (CADUCEUS): a prospective, randomised phase 1 trial. Lancet. 2012;379:895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pfister O, Mouquet F, Jain M, Summer R, Helmes M, Fine A, Colucci WS, Liao R. CD31− but not CD31+ cardiac side population cells exhibit functional cardiomyogenic differentiation. Circ Res. 2005;97:52–61. [DOI] [PubMed] [Google Scholar]
- 11. Oyama T, Nagai T, Wada H, Naito AT, Matsuura K, Iwanaga K, Takahashi T, Goto M, Mikami Y, Yasuda N, Akazawa H, Uezumi A, Takeda S, Komuro I. Cardiac side population cells have a potential to migrate and differentiate into cardiomyocytes in vitro and in vivo. J Cell Biol. 2007;176:329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ruijtenberg S, van den Heuvel S. Coordinating cell proliferation and differentiation: antagonism between cell cycle regulators and cell type‐specific gene expression. Cell Cycle. 2016;15:196–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Skapek SX, Rhee J, Spicer DB, Lassar AB. Inhibition of myogenic differentiation in proliferating myoblasts by cyclin D1‐dependent kinase. Science. 1995;267:1022–1024. [DOI] [PubMed] [Google Scholar]
- 14. Discher DE, Mooney DJ, Zandstra PW. Growth factors, matrices, and forces combine and control stem cells. Science. 2009;324:1673–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Konstandin MH, Toko H, Gastelum GM, Quijada P, De La Torre A, Quintana M, Collins B, Din S, Avitabile D, Volkers M, Gude N, Fassler R, Sussman MA. Fibronectin is essential for reparative cardiac progenitor cell response after myocardial infarction. Circ Res. 2013;113:115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ausoni S, Sartore S. From fish to amphibians to mammals: in search of novel strategies to optimize cardiac regeneration. J Cell Biol. 2009;184:357–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Qiu Y, Bayomy AF, Gomez MV, Bauer M, Du P, Yang Y, Zhang X, Liao R. A role for matrix stiffness in the regulation of cardiac side population cell function. Am J Physiol Heart Circ Physiol. 2015;308:H990–H997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kshitiz, Hubbi ME, Ahn EH, Downey J, Afzal J, Kim DH, Rey S, Chang C, Kundu A, Semenza GL, Abraham RM, Levchenko A. Matrix rigidity controls endothelial differentiation and morphogenesis of cardiac precursors. Sci Signal. 2012;5:ra41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dey D, Han L, Bauer M, Sanada F, Oikonomopoulos A, Hosoda T, Unno K, De Almeida P, Leri A, Wu JC. Dissecting the molecular relationship among various cardiogenic progenitor cells. Circ Res. 2013;112:1253–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, Leri A, Kajstura J, Nadal‐Ginard B, Anversa P. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114:763–776. [DOI] [PubMed] [Google Scholar]
- 21. Smith AJ, Lewis FC, Aquila I, Waring CD, Nocera A, Agosti V, Nadal‐Ginard B, Torella D, Ellison GM. Isolation and characterization of resident endogenous c‐Kit+ cardiac stem cells from the adult mouse and rat heart. Nat Protoc. 2014;9:1662–1681. [DOI] [PubMed] [Google Scholar]
- 22. Hauselmann SP, Rosc‐Schluter BI, Lorenz V, Plaisance I, Brink M, Pfister O, Kuster GM. Beta1‐integrin is up‐regulated via Rac1‐dependent reactive oxygen species as part of the hypertrophic cardiomyocyte response. Free Radic Biol Med. 2011;51:609–618. [DOI] [PubMed] [Google Scholar]
- 23. DeCicco‐Skinner KL, Henry GH, Cataisson C, Tabib T, Gwilliam JC, Watson NJ, Bullwinkle EM, Falkenburg L, O'Neill RC, Morin A, Wiest JS. Endothelial cell tube formation assay for the in vitro study of angiogenesis. J Vis Exp. 2014;91:e51312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wendt D, Marsano A, Jakob M, Heberer M, Martin I. Oscillating perfusion of cell suspensions through three‐dimensional scaffolds enhances cell seeding efficiency and uniformity. Biotechnol Bioeng. 2003;84:205–214. [DOI] [PubMed] [Google Scholar]
- 25. Cerino G, Gaudiello E, Grussenmeyer T, Melly L, Massai D, Banfi A, Martin I, Eckstein F, Grapow M, Marsano A. Three dimensional multi‐cellular muscle‐like tissue engineering in perfusion‐based bioreactors. Biotechnol Bioeng. 2016;113:226–236. [DOI] [PubMed] [Google Scholar]
- 26. Au KF, Jiang H, Lin L, Xing Y, Wong WH. Detection of splice junctions from paired‐end RNA‐seq data by SpliceMap. Nucleic Acids Res. 2010;38:4570–4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory‐efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gaidatzis D, Lerch A, Hahne F, Stadler MB. QuasR: quantification and annotation of short reads in R. Bioinformatics. 2015;31:1130–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, Zheng P, Ye K, Chinnaiyan A, Halder G, Lai ZC, Guan KL. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. [DOI] [PubMed] [Google Scholar]
- 32. Huebsch N, Arany PR, Mao AS, Shvartsman D, Ali OA, Bencherif SA, Rivera‐Feliciano J, Mooney DJ. Harnessing traction‐mediated manipulation of the cell/matrix interface to control stem‐cell fate. Nat Mater. 2010;9:518–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liang SX, Tan TY, Gaudry L, Chong B. Differentiation and migration of Sca1+/CD31− cardiac side population cells in a murine myocardial ischemic model. Int J Cardiol. 2010;138:40–49. [DOI] [PubMed] [Google Scholar]
- 34. Hatzistergos KE, Hare JM. Murine models demonstrate distinct vasculogenic and cardiomyogenic cKit+ lineages in the heart. Circ Res. 2016;118:382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. van Berlo JH, Molkentin JD. Most of the dust has settled: cKit+ progenitor cells are an irrelevant source of cardiac myocytes in vivo. Circ Res. 2016;118:17–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rowland TJ, Blaschke AJ, Buchholz DE, Hikita ST, Johnson LV, Clegg DO. Differentiation of human pluripotent stem cells to retinal pigmented epithelium in defined conditions using purified extracellular matrix proteins. J Tissue Eng Regen Med. 2013;7:642–653. [DOI] [PubMed] [Google Scholar]
- 37. Horejs CM, Serio A, Purvis A, Gormley AJ, Bertazzo S, Poliniewicz A, Wang AJ, DiMaggio P, Hohenester E, Stevens MM. Biologically‐active laminin‐111 fragment that modulates the epithelial‐to‐mesenchymal transition in embryonic stem cells. Proc Natl Acad Sci USA. 2014;111:5908–5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chang HH, Hemberg M, Barahona M, Ingber DE, Huang S. Transcriptome‐wide noise controls lineage choice in mammalian progenitor cells. Nature. 2008;453:544–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–183. [DOI] [PubMed] [Google Scholar]
- 40. Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, Dupont S, Piccolo S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin‐processing factors. Cell. 2013;154:1047–1059. [DOI] [PubMed] [Google Scholar]
- 41. Camargo FD, Gokhale S, Johnnidis JB, Fu D, Bell GW, Jaenisch R, Brummelkamp TR. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol. 2007;17:2054–2060. [DOI] [PubMed] [Google Scholar]
- 42. Cao X, Pfaff SL, Gage FH. YAP regulates neural progenitor cell number via the TEA domain transcription factor. Genes Dev. 2008;22:3320–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reddy BV, Rauskolb C, Irvine KD. Influence of fat‐Hippo and notch signaling on the proliferation and differentiation of Drosophila optic neuroepithelia. Development. 2010;137:2397–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang H, Pasolli HA, Fuchs E. Yes‐associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc Natl Acad Sci USA. 2011;108:2270–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lian I, Kim J, Okazawa H, Zhao J, Zhao B, Yu J, Chinnaiyan A, Israel MA, Goldstein LS, Abujarour R, Ding S, Guan KL. The role of YAP transcription coactivator in regulating stem cell self‐renewal and differentiation. Genes Dev. 2010;24:1106–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhao B, Tumaneng K, Guan KL. The Hippo pathway in organ size control, tissue regeneration and stem cell self‐renewal. Nat Cell Biol. 2011;13:877–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Winkles JA, Alberts GF. Differential regulation of polo‐like kinase 1, 2, 3, and 4 gene expression in mammalian cells and tissues. Oncogene. 2005;24:260–266. [DOI] [PubMed] [Google Scholar]
- 48. Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang CY, Chinnaiyan AM, Lai ZC, Guan KL. TEAD mediates YAP‐dependent gene induction and growth control. Genes Dev. 2008;22:1962–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. van de Weerdt BC, Medema RH. Polo‐like kinases: a team in control of the division. Cell Cycle. 2006;5:853–864. [DOI] [PubMed] [Google Scholar]
- 50. Barr FA, Sillje HH, Nigg EA. Polo‐like kinases and the orchestration of cell division. Nat Rev Mol Cell Biol. 2004;5:429–440. [DOI] [PubMed] [Google Scholar]
- 51. Ma S, Liu MA, Yuan YL, Erikson RL. The serum‐inducible protein kinase Snk is a G1 phase polo‐like kinase that is inhibited by the calcium‐ and integrin‐binding protein CIB. Mol Cancer Res. 2003;1:376–384. [PubMed] [Google Scholar]
- 52. Warnke S, Kemmler S, Hames RS, Tsai HL, Hoffmann‐Rohrer U, Fry AM, Hoffmann I. Polo‐like kinase‐2 is required for centriole duplication in mammalian cells. Curr Biol. 2004;14:1200–1207. [DOI] [PubMed] [Google Scholar]
- 53. Soufi A, Dalton S. Cycling through developmental decisions: how cell cycle dynamics control pluripotency, differentiation and reprogramming. Development. 2016;143:4301–4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Neganova I, Zhang X, Atkinson S, Lako M. Expression and functional analysis of G1 to S regulatory components reveals an important role for CDK2 in cell cycle regulation in human embryonic stem cells. Oncogene. 2009;28:20–30. [DOI] [PubMed] [Google Scholar]
- 55. Ruiz S, Panopoulos AD, Herrerias A, Bissig KD, Lutz M, Berggren WT, Verma IM, Izpisua Belmonte JC. A high proliferation rate is required for cell reprogramming and maintenance of human embryonic stem cell identity. Curr Biol. 2011;21:45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ma S, Charron J, Erikson RL. Role of Plk2 (Snk) in mouse development and cell proliferation. Mol Cell Biol. 2003;23:6936–6943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schlegelmilch K, Mohseni M, Kirak O, Pruszak J, Rodriguez JR, Zhou D, Kreger BT, Vasioukhin V, Avruch J, Brummelkamp TR, Camargo FD. Yap1 acts downstream of alpha‐catenin to control epidermal proliferation. Cell. 2011;144:782–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hao Y, Chun A, Cheung K, Rashidi B, Yang X. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J Biol Chem. 2008;283:5496–5509. [DOI] [PubMed] [Google Scholar]
- 59. Zitouni S, Nabais C, Jana SC, Guerrero A, Bettencourt‐Dias M. Polo‐like kinases: structural variations lead to multiple functions. Nat Rev Mol Cell Biol. 2014;15:433–452. [DOI] [PubMed] [Google Scholar]
- 60. Mori M, Triboulet R, Mohseni M, Schlegelmilch K, Shrestha K, Camargo FD, Gregory RI. Hippo signaling regulates microprocessor and links cell‐density‐dependent mirna biogenesis to cancer. Cell. 2014;156:893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mosqueira D, Pagliari S, Uto K, Ebara M, Romanazzo S, Escobedo‐Lucea C, Nakanishi J, Taniguchi A, Franzese O, Di Nardo P, Goumans MJ, Traversa E, Pinto‐do OP, Aoyagi T, Forte G. Hippo pathway effectors control cardiac progenitor cell fate by acting as dynamic sensors of substrate mechanics and nanostructure. ACS Nano. 2014;8:2033–2047. [DOI] [PubMed] [Google Scholar]
- 62. Kim NG, Gumbiner BM. Adhesion to fibronectin regulates Hippo signaling via the FAK‐Src‐PI3K pathway. J Cell Biol. 2015;210:503–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hatzistergos KE, Takeuchi LM, Saur D, Seidler B, Dymecki SM, Mai JJ, White IA, Balkan W, Kanashiro‐Takeuchi RM, Schally AV, Hare JM. cKit+ cardiac progenitors of neural crest origin. Proc Natl Acad Sci USA. 2015;112:13051–13056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sultana N, Zhang L, Yan J, Chen J, Cai W, Razzaque S, Jeong D, Sheng W, Bu L, Xu M, Huang GY, Hajjar RJ, Zhou B, Moon A, Cai CL. Resident c‐kit(+) cells in the heart are not cardiac stem cells. Nat Commun. 2015;6:8701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Doyle MJ, Maher TJ, Li Q, Garry MG, Sorrentino BP, Martin CM. Abcg2‐labeled cells contribute to different cell populations in the embryonic and adult heart. Stem Cells Dev. 2016;25:277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of Primers, Cycle Numbers (No. of Cycles), and Temperatures (°C)
Table S2. Complete List of Genes Regulated on FN Versus LN
Figure S1. Characterization of mouse and rat cardiac progenitor cell (CPCs).
Figure S2. Nonnormalized data of gene expression as per Figure 1.
Figure S3. Lineage marker expression in cardiac progenitor cells (CPCs) in 3‐dimensional culture on laminin (LN)‐ and fibronectin (FN)‐coated scaffolds using a perfusion bioreactor.
Figure S4. A, Morphological features of rat cardiac progenitor cells (rCPCs) under growth conditions. rCPCs are an inhomogeneous cell population encompassing cells with different shapes. B, Gene expression of epicardial markers in rCPCs. Tbx18 is the only marker expressed in rCPCs. C, rCPCs do not express von Willebrand factor (vWF) protein in culture medium (F12). Human microvascular endothelial cells (HMECs) were used as positive control (bar=25 µm). Aldh1a2, aldehyde dehydrogenase 1 family member a2; rHeart, RNA from neonatal rat heart serving as positive control; Tcf21, transcription factor 21; Wt1, Wilms tumor protein 1.
Figure S5. Heat maps of gene expression from enriched gene sets as identified by Gene Set Enrichment Analysis (GSEA) in rat cardiac progenitor cells (CPCs) on laminin (LN) and fibronectin (FN).