

Abstract
Background
Intermittent hypoxia (IH) induced by obstructive sleep apnea is the key factor involved in cardiovascular fibrosis. Under persistent hypoxia condition, endothelial cells respond by endothelial‐to‐mesenchymal transition (EndMT), which is associated with cardiovascular fibrosis. Prolyl 4‐hydroxylase domain protein 3 (PHD3) is a cellular oxygen sensor and its expression increased in hypoxia. However, its role in obstructive sleep apnea–induced EndMT and cardiovascular fibrosis is still uncertain. We investigated the potential mechanism of obstructive sleep apnea–induced cardiac perivascular fibrosis and the role of PHD3 in it.
Methods and Results
In vivo, C56BL/6 mice were exposed to IH for 12 weeks. PHD3 expression was changed by lentivirus‐mediated short‐hairpin PHD3 and lentivirus carrying PHD3 cDNA. EndMT related protein levels, histological and functional parameters were detected after 12 weeks. In vitro, human umbilical vein endothelial cells were treated with IH/short‐hairpin PHD3/lentivirus carrying PHD3 cDNA to explore the mechanism of PHD3 in altered function of human umbilical vein endothelial cells. We found that chronic intermittent hypoxia increase PHD3 expression and EndMT. In vivo, IH accelerate cardiac dysfunction and aggravate collagen deposition via the process of EndMT. And, when PHD3 were overexpressed, cardiac dysfunction and collagen excessive deposition were improved. In vitro, IH induced EndMT, which endow human umbilical vein endothelial cells spindle morphology and an enhanced ability to migration and collagen secretion. PHD3 overexpression in cultured human umbilical vein endothelial cells ameliorated IH–induced EndMT through inactivating hypoxia‐inducible factor 1 alpha and small mothers against decapentaplegic 2 and 3.
Conclusions
Obstructive sleep apnea–induced cardiac perivascular fibrosis is associated with EndMT, and PHD3 overexpression might be beneficial in the prevention of it by inhibiting EndMT. PHD3 overexpression might have therapeutic potential in the treatment of the disease.
Keywords: cardiac perivascular fibrosis, endothelial‐to‐mesenchymal transition, intermittent hypoxia, obstructive sleep apnea, prolyl 4‐hydroxylase domain protein 3
Subject Categories: Basic Science Research, Fibrosis
Clinical Perspective
What Is New?
In our research, we explored an unrevealed pathogenic mechanism in obstructive sleep apnea–induced perivascular fibrosis, termed endothelial‐to‐mesenchymal transition, which can directly contribute to collagen deposition.
What Are the Clinical Implications?
We demonstrated that prolyl 4‐hydroxylase domain protein 3, a cellular low‐oxygen sensor, partially reversed the related protein expression of endothelial‐to‐mesenchymal transition, which may have therapeutic target in obstructive sleep apnea–induced perivascular fibrosis.
Introduction
Obstructive sleep apnea (OSA) is a common, yet an underdiagnosed, sleep‐related breathing disorder affecting predominantly middle‐aged men.1 It is a disordered breathing condition, characterized by intermittent airway obstruction during sleep, resulting in intermittent hypoxia, which is increasingly regarded as a risk factor for multiple organ systems. OSA is associated with various cardiovascular diseases, such as hypertension, congestive heart failure, atrial fibrillation, coronary artery disease, pulmonary hypertension, and metabolic syndrome.2, 3, 4 As we know, all these diseases could cause cardiac remodeling, including left ventricular hypertrophy, alterations in the coronary microcirculation, atrial electrical remodeling, and so on. The obvious modifications in cardiac remodeling are characterized of the disruption of normal myocardial structure through excessive deposition of extracellular matrix leading to myocardial fibrosis. Intermittent hypoxia (IH) is one of the most powerful incitants involved in OSA‐induced cardiac remodeling, which is considered as the main detrimental event leading to cardiovascular morbidity and mortality.5 However, the detailed mechanisms remain unclear. Given the increased risk of cardiac remodeling in OSA patients, a better understanding of the underlying mechanisms is necessary.
Interestingly, there is increasing evidence proving that a fraction of interstitial fibroblasts is derived from the endothelium, which is so‐called endothelial‐to‐mesenchymal transition (EndMT).6, 7 EndMT is a special form of epithelial‐mesenchymal transition that occurs during the embryonic development of the heart. Recent studies have indicated that EndMT plays a critical role in cardiac fibrosis,8, 9 which is predominantly characterized by perivascular and subendocardial fibrosis.8 EndMT is characterized by loss of cell‐cell junctions and a change in cell polarity, accompanied by downregulating endothelial cell (EC)‐specific markers, such as CD31 and vascular endothelial (VE)‐cadherin, and upregulating mesenchymal cell markers, such as α‐smooth muscle actin (α‐SMA), fibroblast‐specific protein‐1 (FSP‐1), and vimentin. Eventually, ECs acquire invasive and migratory phenotypes.10 EndMT could be induced by hypoxia, high glucose, transforming growth factor‐β, inflammation, and radiation.11 Studies have shown that transforming growth factor‐β2 stimulates EndMT through the small mothers against decapentaplegic (Smad), mitogen‐activated protein kinase, phosphoinositide 3‐kinase, and p38 mitogen‐activated protein kinase signaling pathways, and these pathways are essential for increasing expression of the cell‐adhesion–suppressing transcription factor, Snail,12 which may be a potential target molecule in cardiac fibrosis.13 However, no research reports that OSA could induce EndMT, and its mechanisms remain to be further elucidated.
Prolyl 4‐hydroxylase domain protein 3 (PHD3), located in the nuclear or cytoplasmic,14 belongs to 2‐oxoglutarate and iron‐dependent dioxygenases, which has been regarded as cellular low‐oxygen sensors.15 Under normoxia, PHD3 could hydroxylate the hypoxia‐inducible factor α (HIF‐α) through von Hippel‐Lindau protein, but under hypoxia, it can exert cell survival or proliferation‐supporting functions.15 Under hypoxia, PHD3 differs from other PHDs. First, PHD3 is abundantly expressed in the heart.16 Second, PHD3 mainly hydroxylates Pro564 of HIF‐1α and has little ability to hydroxylate Pro402.17 Third, PHD3 could retain its activity under prolonged milder hypoxia.18 Finally, compared with other PHDs, PHD3 expression is more significant under hypoxia.17 In most cells, PHD3 is mediated by HIF. In HIF‐deficient cells, PHD3 upregulation by hypoxia is hindered. However, nothing has been reported about the function of PHD3 in OSA‐induced cardiac remodeling and EndMT.
Based on these findings, we hypothesized that PHD3 may have a protective effect on IH‐induced cardiac remodeling by inhibiting EndMT. To uncover the mechanisms, we investigated its role in vitro in human umbilical vein endothelial cells (HUVECs) treating with IH and in vivo in an OSA mouse model.
Materials and Methods
The experiments conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health and Southeast University (Nanjing, China). The study protocol was approved by the Institutional Ethics Committee of Southeast University and Zhongda Hospital (Nanjing, China).
Study Design
C57BL/6J wild‐type male 6‐week‐old mice (21–23 g), purchased from Beijing Weitong Lihua Experimental Animal Technology (Beijing, China), were used for the animal experiments. Mice were housed at 22°C with a 12‐hour light/dark cycle. A total of 60 mice were randomly assigned to normoxia (n=10) or IH exposure (n=50) for 1 week. One week post‐IH exposure, IH mice were randomly divided into 5 groups: IH (n=10); IH+shRNA PHD3 NC (shNC; n=10; null lentivector at the dose of 108TU/mouse); IH+lentiviral PHD3 NC (LvNC; n=10; null lentivector at the dose of 108TU/mouse); IH+shRNA PHD3 (shPHD3; n=10; lentivector at the dose of 108TU/mouse); and IH+lentiviral PHD3 (LvPHD3; n=10; lentivector at the dose of 108TU/mouse). Lentivector was purchased from GenePharma (Shanghai, China). The target sequence for shPHD3 was 5′‐GCUGUAUCACCUGUAUCUATT‐3′, and the negative sequence was 5′‐UAGAUACAGGUGAUACAGCTT‐3′. At 3 months after IH induction, body weight and systolic blood pressure were measured.
Intermittent Hypoxia
For 3 months, mice in the IH, IH+shNC, IH+LvNC, IH+shPHD3, and IH+LvPHD3 received 60 hypoxic events per hour (20 seconds at 5% O2 followed by 40 seconds of room air), during 8 hours per day, corresponding to severe OSA. Control mice with normoxic breathing were placed in an identical device, but the hypoxic gas from the reservoir was replaced by room air.19, 20 The device, designed by PUHE Biotechnology Company LTD (Wuxi, China), was awarded a national patent (ZL 2014 1 0426971.2).
Blood Pressure and Cardiac Function Measurement
After 3 months, a noninvasive tail‐cuff system (Softron TMC‐203) was used to measure the systolic blood pressure. Blood pressure was reported as the mean value of 3 consecutive measurements. Cardiac diameter and function was measured under 2.0% isoflurane anesthesia by VIVID7 (GE, Little Chalfont, UK) with a 10‐MHz probe (MS400). Interventricular septum dimension, left ventricular end‐diastolic dimension, left ventricular end‐systolic dimension (LVIDs), left ventricular posterior wall (LVPWd), left ventricular ejection fraction (LVEF), and left ventricular shortening fraction (FS) were measured. Pulsed‐wave Doppler was used to measure the ratio of peak early diastolic ventricular filling velocity to peak atria filling velocity (E/A), and the ratio of diastolic mitral annulus velocities (E′/A′) were measured in tissue Doppler imaging.
Histology and Immunohistochemistry
Ten percent chloral hydrate (4 μL/g) was injected into the abdominal cavity to anesthetize mice, then mice hearts were taken out from the thoracic cavity. Mice hearts fixed in 4% paraformaldehyde were cut transversely at the papillary muscle level, embedded in paraffin, and cut into 4‐mm sections. Heart sections were stained with hematoxylin and eosin, Masson's trichrome, and picrosirius red to examine cardiac microvascular structure and extracellular matrix deposition, respectively. For immunohistochemistry, after deparaffinage, antigen retrieval, and blocking, sections were incubated with primary antibody against collagen I (1:1000; Abcam, Cambridge, UK), collagen III (1:1000; Abcam), CD31 (1:200; Abcam), and α‐SMA (1:200; Abcam) overnight at 4°C, then washed with PBS and incubated with secondary antibody (1:2000; Fuzhou Maixin Biotech Co, Ltd, Fuzhou, Fujian) at 37°C for 30 minutes. Cardiac microvascular intima‐media thickness (IMT) and lumen diameter were quantified by morphometric analysis of the hematoxylin and eosin–stained sections (20 measurements for each animal). The perivascular region fibrotic area was measured from all groups in every 6 randomly chosen views of each sample and analyzed by the Image‐Pro Plus 6.0 program.
Cell Culture
Human umbilical vein endothelial cells (HUVECs) were purchased from ScienCell Research Laboratories, Inc (Carlsbad, CA). EC medium supplemented with 5% FBS and 1% EC growth supplement was used. Cells were cultured in a humidified 5% CO2 incubator at 37°C and used between the third and sixth passages. Cells were treated with IH for 72 hours (air‐phase set point consisted of a 35‐minute hypoxic period, followed by 25 minutes of reoxygenation [21% O2 and 5% CO2]).21, 22 The medium was changed every 48 hours. Before IH treatment, HUVECs were infected with lentivirus at a multiplicity of infection of 10 for 36 hours. Cells were further cultured for 72 hours after confirming that more than 90% cells were positive for green fluorescent protein. For Smad2/3 inhibition, according to the manufacturer's instructions, cells were treated with smad2/3 inhibitor (12 nmol/L of A83‐01; Sigma‐Aldrich, St. Louis, MO).
Migration Assays
HUVECs' (exposed in IH or air; infected with lentivirus or not) migration ability was tested by transwell (Costar polycarbonate filters, 8‐μm pore size). HUVECs of all groups (0.5×105 cells/well) were added to the upper chamber without serum, and complete medium (5% FBS) was added to the lower chamber. Cells migrating into the lower chamber after 24 hours of incubation were manually counted.
Western Blot Analysis
Whole‐cell proteins were isolated from cell lysates and freshly dissected mice hearts. Western blot analysis was performed as described.23 PVDF membranes were incubated with primary antibodies against PHD3 (1:500; Novus Biologicals, Littleton, CO), HIF‐1α (1:1000; Cell Signaling Technology, Danvers, MA), CD31 (1:1000; Abcam), VE‐cadherin (1:1000; Cell Signaling Technology), α‐SMA (1:500; Abcam), FSP‐1 (1:250; Abcam), Snail (1:500; Cell Signaling Technology), Twist (1:500; Abcam), Collagen I (1:5000; Abcam), Collagen III (1:5000; Abcam), and β‐actin (1:1000; Abcam).
Immunofluorescence Microscopy
In brief, after fixation and blocking, cells were incubated with primary antibody against CD31 (1:200; Abcam) and α‐SMA (1:200; Abcam), CD31 (1:200; Abcam), and FSP‐1 (1:100; Abcam), HIF‐1α (1:200; Cell Signaling Technology), Smad2/3 (1:200; Cell Signaling Technology), and phosphorylated (p)‐Smad2 (ser465/467)/3 (ser423/425; 1:200; Cell Signaling Technology) at 4°C overnight, then washed with PBS and incubated with secondary antibody for 30 minutes at 37°C (Alexa Fluor 488 and 549 goat antirabbit; Alexa Fluor 549 and 488 goat antimouse 1:200). Nuclei were counterstained with 4′‐6‐diamidino‐2‐phenylindole (Beyotime Biotechnology, Shanghai, China). Specific fluorescence was acquired by fluorescence microscope (IX‐71; Olympus, Tokyo, Japan).
Statistical Analysis
Results are from 3 repeated experiments. Data are reported as mean±SD. Results were compared by 2‐tailed Student t test for 2 groups and 1‐way ANOVA followed by Tukey's t test (2‐tailed) for multiple groups. SPSS (v16.0; SPSS, Inc, Chicago, IL) was used for analysis. Differences were considered statistically significant at P<0.05.
Results
Body Weight and Systolic Blood Pressure
Body weight and blood pressure at baseline were similar in both groups. Interestingly, 3 months of IH did not show a significant difference on animal body weight (P>0.05; Figure S1A). Compared with control, systolic blood pressure elevated after 3 months of IH (112±6 versus 133±8 mm Hg; P<0.05; Figure S1B). Green fluorescent protein was used to evaluate transfection efficiency, which reached values of up to 90% (Figure S2B). Western blot revealed that lentiviral vector could change PHD3 expression successfully compared with the NC group in vivo and vitro (Figure S2A and S2C). But lentivirus treatment had no effect on body weight and blood pressure compared with the NC group (P>0.05; Figure S1B).
IH Induces Cardiac Dysfunction and Increases PHD3 Expression and Collagen Deposition
Echocardiography was performed to assess cardiac function at 3 months. At baseline, interventricular septum dimension, left ventricular diastolic diameter, LVEF, FS, E/A, and E′/A′ ratios did not differ among groups (data not shown). After 3 months, interventricular septum dimension and left ventricular diastolic diameter were of no change (P>0.05; Figure 1A, 1b1, and 1b2). However, compared with control, LVIDs and LVPWd were changed (LVIDs 2.8±0.2 versus 1.89±0.17; P<0.05; Figure 1A and 1b3; LVPWd 0.77±0.06 versus 0.89±0.02; P<0.05; Figure 1A and 1b4). Cardiac function features were lower in IH than control mice: LVEF (53.67±3.1% versus 75.67±4.7%; P<0.01; Figure 1A and 1b5), FS (25±2% versus 39.3±2.1%; P<0.01; Figure 1A and 1b6), E/A (0.72±0.11 versus 1.53±0.16; P<0.01; Figure 1A and 1b7), and E′/A′ (0.59±0.1 versus 1.6±0.1; P<0.01; Figure 1A and 1b8).
Figure 1.
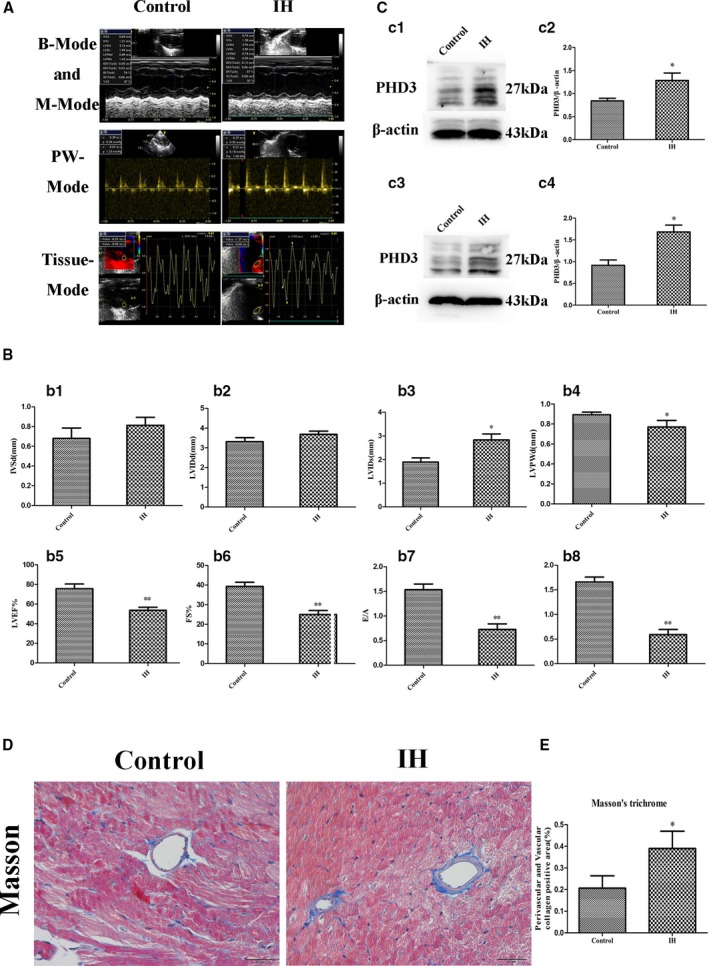
Change of cardiac function, PHD3 expression, and collagen deposition after IH treatment. A, Representative echocardiographic images of 2D echocardiograms, M‐mode echocardiograms, pulsed‐wave Doppler echocardiograms, and tissue Doppler echocardiograms in all groups. B, Quantitative analysis of echocardiographic measurements parameters. b1, Interventricular septum dimension (IVSD). b2, Left ventricular end‐diastolic dimension (LVIDd). b3, Left ventricular end‐systolic dimension (LVIDs). b4, Left ventricle posterior wall dimension (LVPWd). b5, Left ventricular ejection fraction (LVEF). b6, Fractional shortening (FS). b7, Early to late mitral flow (E/A). b8, Early to late ratio of diastolic mitral annulus velocities (E′/A′). Data are mean±SD; n=10 per group. *P<0.05 vs control; **P<0.01 vs control. C, Western blot analysis of PHD3 (c1) and quantitative analysis (c2) in vivo. Western blot analysis of PHD3 (c3) and quantitative analysis (c4) in vitro. *P<0.05 vs control. D, Representative images of the heart section with Masson's trichrome (blue staining) for each group (original magnification, ×400; bars=20 μm). E, Quantification of Masson's trichrome staining. Data are mean±SD; n=10 per group. *P<0.05 vs control. 2D indicates 2‐dimensional; IH, intermittent hypoxia; PHD3, prolyl 4‐hydroxylase domain protein 3.
To ascertain whether PHD3 plays a role in myocardial fibrosis and dysfunction associated with IH, we measured the expression of PHD3 in vivo and vitro. We found that IH increased PHD3 expression in vivo (0.84±0.05 versus 1.28±0.16; P<0.05; Figure 1c1 and 1c2) and vitro (0.91±0.12 versus 1.68±0.16; P<0.05; Figure 1c3 and 1c4) compared with control. Masson's trichome revealed greater extracellular matrix in the perivascular and vascular of the IH than control. Quantitative analysis of those staining showed a 1.45‐fold increase in collagen deposition in IH group as compared with control mice (0.39±0.07 versus 0.2±0.05; P<0.05; Figure 1D and 1E).
PHD3 Overexpression Improve IH‐Induced Cardiac Dysfunction
To further explore PHD3's role in cardiac dysfunction, we then investigated wild‐type/PHD3 gene silencing/PHD3 gene overexpression in IH mice. Compared with the vehicle, PHD3 overexpression (LvPHD3) improve LVIDs (2.19±0.08 versus 2.73±0.15; P<0.05; Figure 2A and 2b3), LVPWd (0.88±0.05 versus 0.72±0.02; P<0.05; Figure 2A and 2b4), LVEF (64.67±3% versus 56±4%; P<0.05; Figure 2A and 2b5), FS (30.67±2.1% versus 24.67±1.5%; P<0.05; Figure 2A and 1b6), E/A (1.2±0.17 versus 0.8±0.04; P<0.05; Figure 2A and 2b7), and E′/A′ (1.22±0.07 versus 0.75±0.05; P<0.01; Figure 2A and 2b8), but improved LVIDs, LVPWd, LVEF, FS, E/A, and E′/A′ were not observed in the PHD3 interference (shPHD3) group (P>0.05; Figure 2A, 2b3, and 2b8).
Figure 2.
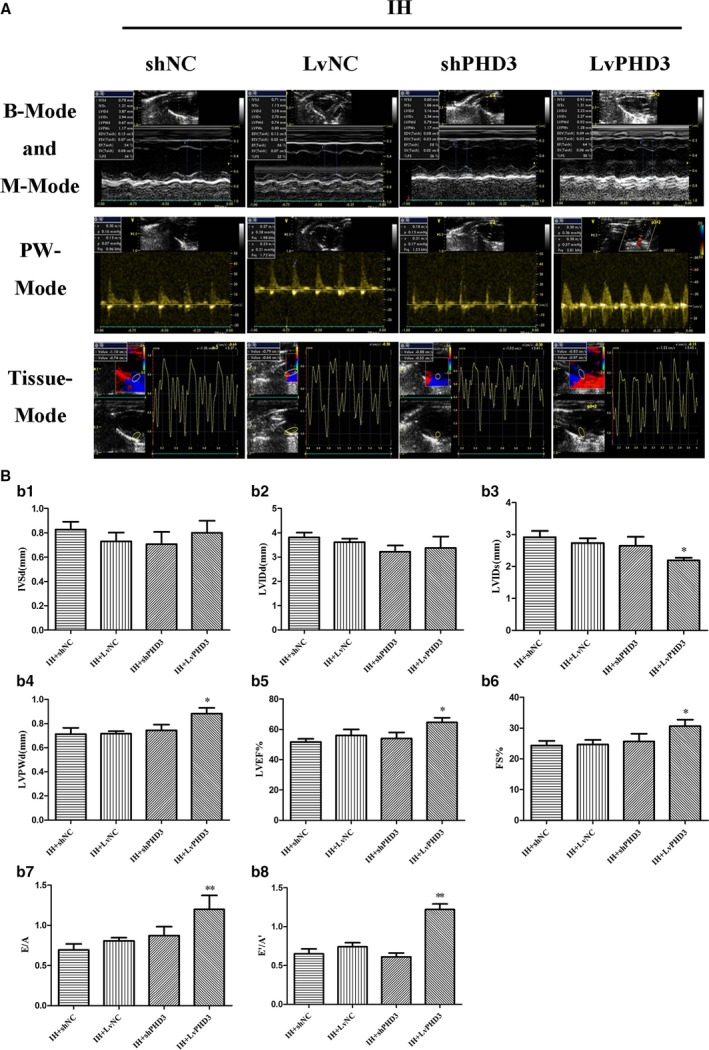
PHD3 overexpression improves IH‐induced cardiac dysfunction. A, Representative echocardiographic images of 2D echocardiograms, M‐mode echocardiograms, pulsed‐wave Doppler echocardiograms, and tissue Doppler echocardiograms in all groups. B, Quantitative analysis of echocardiographic measurements parameters. b1, Interventricular septum dimension (IVSD). b2, Left ventricular end‐diastolic dimension (LVIDd). b3, Left ventricular end‐systolic dimension (LVIDs). b4, Left ventricle posterior wall dimension (LVPWd). b5, Left ventricular ejection fraction (LVEF). b6, Fractional shortening (FS). b7, Early to late mitral flow (E/A). b8, Early to late ratio of diastolic mitral annulus velocities (E′/A′). Data are mean±SD; n=10 per group. *P<0.05 vs IH+LvNC; **P<0.01 vs IH+LvNC. 2D indicates 2‐dimensional; IH, intermittent hypoxia; LvPHD3, lentivirus carrying PHD3 cDNA; PHD3, prolyl 4‐hydroxylase domain protein 3; shPHD3, short‐hairpin PHD3.
PHD3 Overexpression Improves IH‐Induced Cardiac Perivascular Fibrosis
Compared with the control, the cardiac microvascular IMT/lumen diameter was increased by IH exposure from hematoxylin‐eosin staining (0.23±0.03 versus 0.14±0.02; P<0.05; Figure 3A and 3b1). However, when compared with the vehicle, decreased IMT/lumen diameter were observed in LvPHD3 group (0.15±0.04 versus 0.23±0.03; P<0.05; Figure 3A and 3b1), not in shPHD3 (P>0.05; Figure 3A and 3b1).
Figure 3.
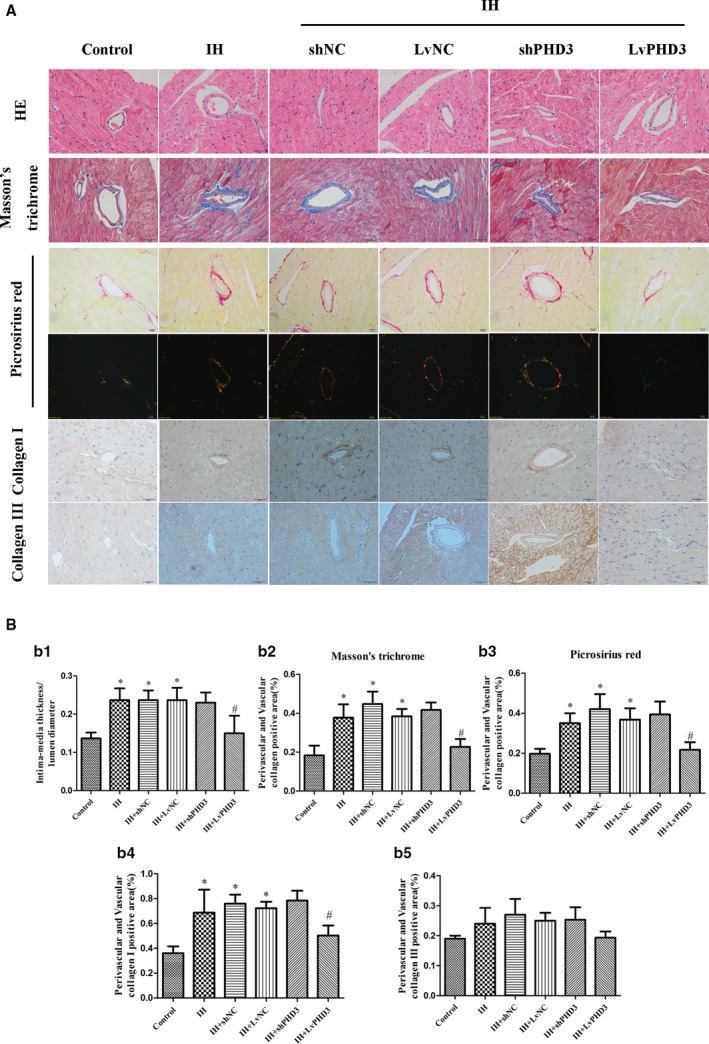
PHD3 overexpression improves IH‐induced cardiac perivascular fibrosis. A, Representative images of the heart section with HE staining, Masson's trichrome (blue staining), Picrosirius Red staining (red, yellow, and green staining) and immunohistochemical staining of collagen I collagen III (yellow staining) for each group (original magnification, ×400; bars=20 μm). B, Quantification of intima‐media thickness/lumen diameter (b1), Masson's trichrome staining (b2), Picrosirius Red staining (b3), collagen I (b4), and collagen III (b5). Data are mean±SD; n=10 per group. *P<0.05 vs control; # P<0.05 vs IH+LvNC. HE indicates hematoxylin‐eosin; IH, intermittent hypoxia; LvPHD3, lentivirus carrying PHD3 cDNA; PHD3, prolyl 4‐hydroxylase domain protein 3; shPHD3, short‐hairpin PHD3.
Masson's trichome and picrosirius red staining of heart sections revealed greater extracellular matrix in the perivascular and vascular of the IH than control. Quantitative analysis of those staining showed a 2.05‐ and 1.84‐fold increase in collagen deposition in the IH group as compared with control mice (0.37±0.07 versus 0.18±0.05; P<0.05; 0.35±0.05 versus 0.19±0.03; P<0.05; Figure 3A, 3b2, and 3b3). LvPHD3 treatment reduced collagen deposition as compared with vehicle treatment (0.22±0.04 versus 0.38±0.04; P<0.05; 0.21±0.04 versus 0.36±0.06; P<0.05; Figure 3A, 3b2, and 3b3). However, shPHD3 treatment did not change collagen deposition as compared with vehicle treatment (P>0.05; Figure 3A, 3b2, and 3b3).
IH enhanced the expression of the fibrotic markers collagen I as compared with the control (0.68±0.18 versus 0.36±0.06; P<0.05; Figure 3A and 3b4), whereas it is LvPHD3 not shPHD3 transfection which significantly reduced the levels as compared with vehicle treatment (0.5±0.08 versus 0.72±0.05; P<0.05; Figure 3A and 3b4). However, IH had no effect on collagen III (P>0.05; Figure 3A and 3b5). Interesting, compared with control, IH did not increase collagen deposition in myocardium (P<0.05; Figure S3).
IH Induces EndMT
EndMT has been shown to contribute to the progression of cardiac fibrosis. To determine whether IH induced EndMT, we performed in vitro and in vivo experiments.
In vivo, CD31, a specificity marker of ECs, was stained by immunohistochemistry. The location of CD31 was restricted to the intima endothelium in the control group. CD31 is a specificity marker of ECs and is expressed only in the intima and not in the vascular medial smooth muscle. However, we observed diffuse staining, which was not specifically localized in intima (endothelium), but in both intima (endothelium) and media (smooth muscle), after IH treatment (Figure 4A). Then we used immunofluorescence‐colocalized staining to prove the existence of EndMT in vitro. Immunofluorescence demonstrated that IH treatment significantly increased the levels of mesenchymal marker α‐SMA and decreased the levels of endothelial marker CD31 as compared with control (Figure 4C).
Figure 4.
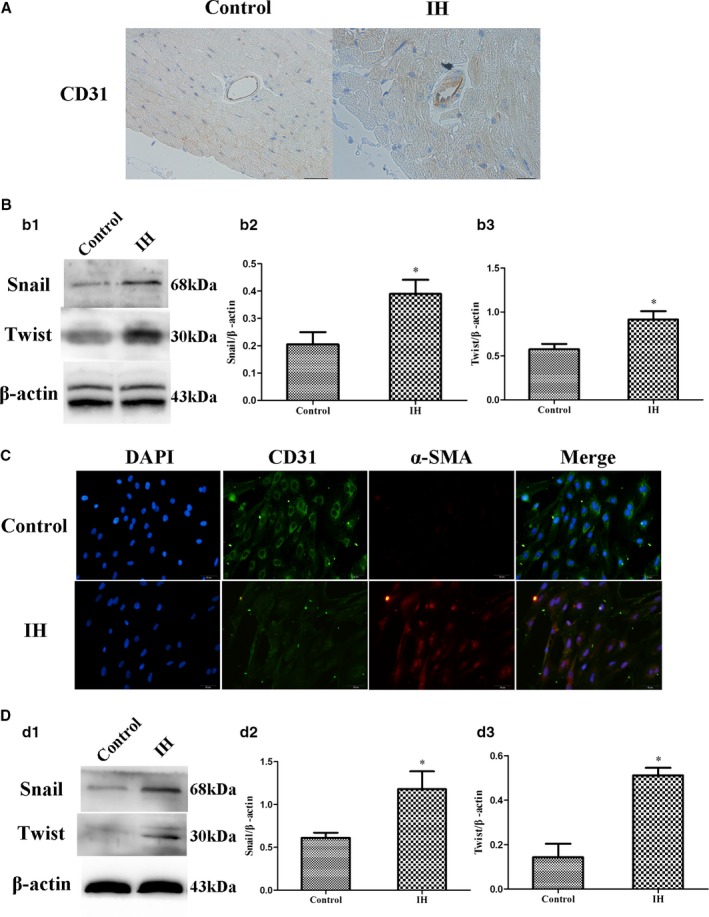
IH induces EndMT in vivo and vitro. A, Representative images of immunohistochemical staining of CD31 (yellow staining) in the perivascular and vascular region. Presence of CD31 is detected in EC, subendothelial cells, and media (original magnification, ×400; bars=20 μm). B, Western blot analysis and quantification of snail (b1 and b2) and twist (b1 and b3) in vivo. C, Representative images of double immunofluorescence staining with antibodies to CD31 (green) and α‐SMA (Red). Nuclei were counterstained with DAPI (blue; original magnification, ×400; bars=20 μm). D, Western blot analysis and quantification of snail (d1 and d2) and twist (d1 and d3) in vitro. Data are mean±SD; n=10 per group. *P<0.05 vs control. DAPI indicates 4',6‐diamidino‐2‐phenylindole; EC, endothelial cells; EndMT, endothelial‐to‐mesenchymal transition; IH, intermittent hypoxia; α‐SMA, alpha‐smooth muscle actin.
Western blot revealed that IH increase snail and twist expression in vivo and vitro, indicators which result in EndMT,24 as compared with control (0.38±0.05 versus 0.2±0.04, 0.91±0.09 versus 0.58±0.06, 0.18±0.2 versus 0.61±0.06, and 0.51±0.03 versus 0.14±0.06; P<0.05; Figure 4B and 4D). It proved that IH induces EndMT from another aspect.
PHD3 Overexpression Inhibits IH‐Induced EndMT
To further explore the mechanism through which PHD3 overexpression improve IH induced cardiovascular fibrosis, we performed the next experiments. In vivo, CD31 was stained by immunohistochemistry. We observed a diffused staining in intima and media after IH treatment; however, LvPHD3 transfection in IH mice improved it as compared with vehicle treatment (Figure 5A). The immunohistochemistry data were confirmed by western blot from another aspect. Compared with vehicle treatment, LvPHD3 could improve Snail and Twist expression (0.06±0.02 versus 0.14±0.04 and 0.57±0.05 versus 1±0.22; P<0.05; Figure 5b1, 5b3, and 5b4), but shPHD3 could not decrease their expression (P>0.05; Figure 5b1, 5b3, and 5b4). For further verification, we stained α‐SMA, which is a specificity marker of vascular smooth cells. In the control group, the α‐SMA location was stained only in the media as observed in the immunohistochemistry results. However, after IH treatment, the presence of α‐SMA was detected in ECs (red arrow). In other words, some ECs expressed α‐SMA as indicated by immunohistochemistry (Figure S4). LvPHD3 transfection in IH mice can improve this expression as compared with vehicle treatment (Figure S4). In vitro, HUVECs were treated with IH with or without shPHD3/LvPHD3. Western blot analysis demonstrated that IH treatment significantly increased the levels of mesenchymal markers α‐SMA and FSP‐1 (0.22±0.04 versus 0.07±0.02 and 0.57±0.07 versus 0.08±0.01; P<0.01; Figure 5c1, 5c5, and 5c6) and caused a reduction in the levels of endothelial markers CD31 and VE‐Cadherin (0.53±0.07 versus 1.12±0.1 and 0.15±0.02 versus 0.45±0.05; P<0.01; Figure 5c1, 5c3, and 5c4) as compared with controls. However, the features above were alleviated when PHD3 was overexpressed: α‐SMA (0.1±0.03 versus 0.24±0.02; P<0.01; Figure 5c1 and 5c5); FSP‐1 (0.23±0.06 versus 0.52±0.07; P<0.01; Figure 5c1 and 5c6); CD31 (0.79±0.08 versus 0.5±0.09; P<0.05; Figure 5c1 and 5c3); and VE‐Cadherin (0.29±0.02 versus 0.19±0.03; P<0.05; Figure 5c1 and 5c4). Similar to the results in vivo, compared with vehicle treatment, LvPHD3 could improve Snail and Twist expression (0.35±0.04 versus 0.62±0.04 and 0.43±0.1 versus 0.85±0.09; P<0.05; Figure 5c2, 5c7, and 5c8), but shPHD3 could not decrease their expression (P>0.05; Figure 5c2, 5c7, and 5c8).
Figure 5.
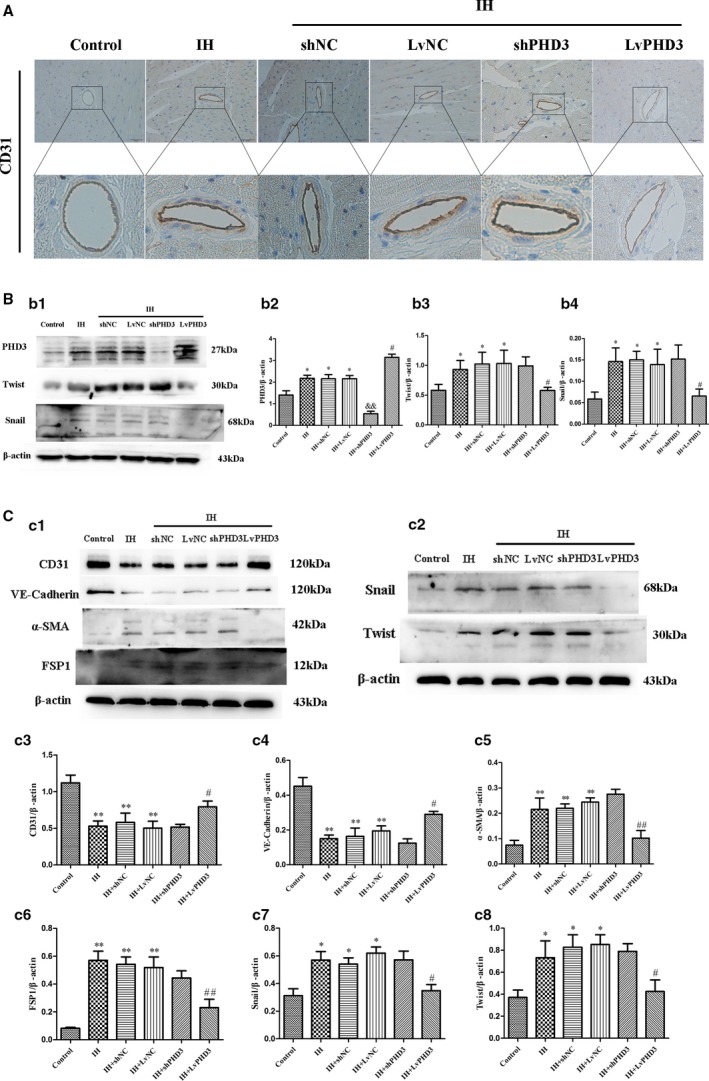
PHD3 overexpression improve IH‐induced EndMT. A, Representative images of immunohistochemical staining of CD31 (yellow staining) in the perivascular and vascular region (original magnification, ×400; bars=20 μm). B, Western blot analysis and quantification of snail (b1 and b2) and twist (b1 and b3) in vivo. C, Western blot analysis and quantification of CD31 (c1 and c3), VE‐Cadherin (c1 and c4), α‐SMA (c1 and c5), FSP1 (c1 and c6), snail (c2 and c7), and twist (c2 and c8) in vivo. Data are mean±SD; *P<0.05 vs control; **P<0.01 vs control; # P<0.05 vs IH+LvNC; ## P<0.01 vs IH+LvNC; && P<0.01 vs IH+shNC. EndMT indicates endothelial‐to‐mesenchymal transition; FSP1, fibroblast‐specific protein‐1; IH, intermittent hypoxia; LvPHD3, lentivirus carrying PHD3 cDNA; PHD3, prolyl 4‐hydroxylase domain protein 3; shPHD3, short‐hairpin PHD3; α‐SMA, alpha‐smooth muscle actin; VE, vascular endothelial.
PHD3 Overexpression Improved EndMT‐Induced Morphologic Alteration, Enhanced Migration, and Collagen Secretion
HUVECs migration and collagen secretion plays an important role in myocardial fibrosis. To further evaluate the effect of PHD3 on HUVECs migration and collagen secretion, we used the transwell migration assay. Fluorescence microscopy revealed that control HUVECs showed a typical rounded or cobblestone shape (Figure 6A). HUVECs had a shape change from a cobblestone‐like to spindle‐shaped feature with IH induction (Figure 6A). Interestingly, they had an enhanced ability of migration compared with the control (1493±100.7 versus 866±117.2; P<0.05; Figure 6A and 6B). Compared with vehicle treatment, LvPHD3 could improve the enhanced migration (1133±251.7 versus 1577±186.1; P<0.05; Figure 6A and 6B) and HUVECs' spindle‐shaped feature (Figure 6A), but shPHD3 group has no ability to improve them (P>0.05; Figure 6A and 6B).
Figure 6.
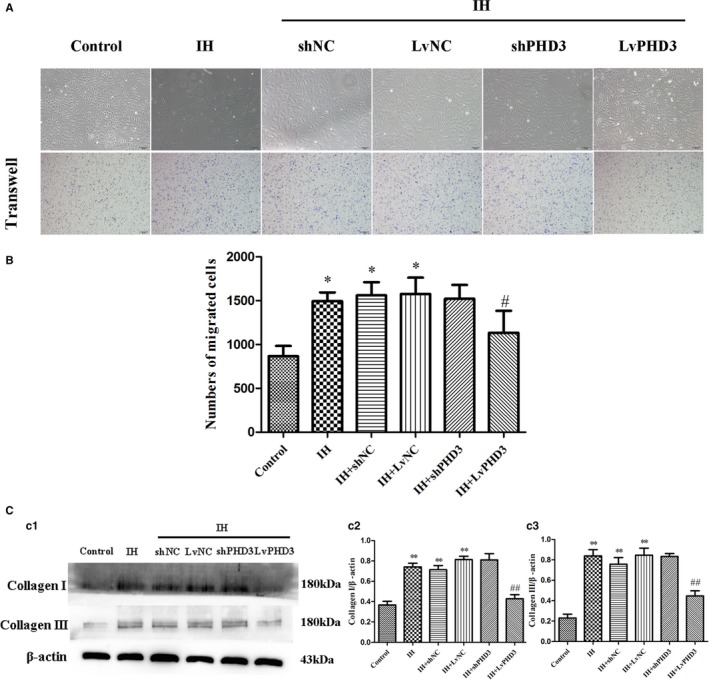
PHD3 overexpression improve EndMT‐induced morphologic alteration, enhanced migration, and collagen secretion. A, Morphological changes in HUVECs were observed by microscopy. Representative images of crystal violet staining (purple; original magnification, ×100; bars=50 μm). B, Quantification of crystal violet staining. C, Western blot analysis and quantification of Collagen I (c1 and c2) and Collagen III (c1 and c3). *P<0.05 vs control; **P<0.01 vs control; # P<0.05 vs IH+LvNC; ## P<0.01 vs IH+LvNC. EndMT indicates endothelial‐to‐mesenchymal transition; HUVECs, human umbilical vein endothelial cells; IH, intermittent hypoxia; PHD3, prolyl 4‐hydroxylase domain protein 3.
When HUVECs experience EndMT induced by IH, we found that the expression of collagen I and collagen III increased (0.74±0.04 versus 0.37±0.03 and 0.84±0.06 versus 0.23±0.04; P<0.01; Figure 6C). However, their expression decreased after PHD3 overexpression compared with the LvNC group (0.43±0.04 versus 0.81±0.03 and 0.45±0.05 versus 0.84±0.07; P<0.01; Figure 6c1, 6c2, and 6c3). Compared with vehicle treatment, collagen I and collagen III expression did not decrease in the shPHD3 group (P>0.05; Figure 6C).
PHD3 Overexpression Mitigates IH‐Induced HIF‐1α and smad2/3 Activation in Endothelial Cells
To clarify the mechanism of PHD3 function in EndMT further, we examined the potential signal transduction pathways involved in IH‐induced cardiac fibrosis. Western blot revealed that HIF‐1α expression increased after IH treatment compared with control (0.65±0.04 versus 0.36±0.03; P<0.05; Figure 7a1 and 7a2), but PHD3 overexpression could improve the increased expression as compared with vehicle treatment (0.29±0.09 versus 0.62±0.08; P<0.05; Figure 7a1 and 7a2). Immunofluorescence revealed that IH induced relocation of HIF‐1α (Figure 7B), Smad2/3 (Figure 8A), and p‐Smad2 (ser465/467)/3 (ser423/425; Figure 8A), from cytoplasm to nucleus. This means that they are activated by IH. Compared with vehicle treatment, LvPHD3 could keep most HIF‐1α (Figure 7B), Smad2/3 (Figure 8A), and p‐Smad2 (ser465/467)/3 (ser423/425; Figure 8A) staying in cytoplasm, but shPHD3 did not inactivate HIF‐1α (Figure 7B), Smad2/3 (Figure 8A), and p‐Smad2 (ser465/467)/3 (ser423/425; Figure 8A) as compared with vehicle treatment. So, PHD3 may improve EndMT by inactivating HIF‐1α and Smad2/3. To further clarify, we used A83‐01 to inhibit phosphorylation of smad2/3. Immunofluorescence showed that A83‐01 was effective at inhibiting p‐Smad2 (ser465/467)/3 (ser423/425; Figure 8B). From the above results, IH activated the Smad2/3 signaling pathway and induced EndMT in HUVECs as compared with the control group. While compared with the IH group, A83‐01 decreased α‐SMA (0.32±0.03 versus 0.51±0.09; P<0.05; Figure 8c1 and 8c4), snail (0.67±0.06 versus 0.94±0.06; P<0.01; Figure 8c2 and 8c5), and twist expression (0.18±0.04 versus 0.54±0.04; P<0.01; Figure 8c2 and 8c6) and increase CD31 expression (0.95±0.06 versus 0.67±0.07; P<0.05; Figure 8c1 and 8c3), which further proved that the smads pathway participated in EndMT.
Figure 7.
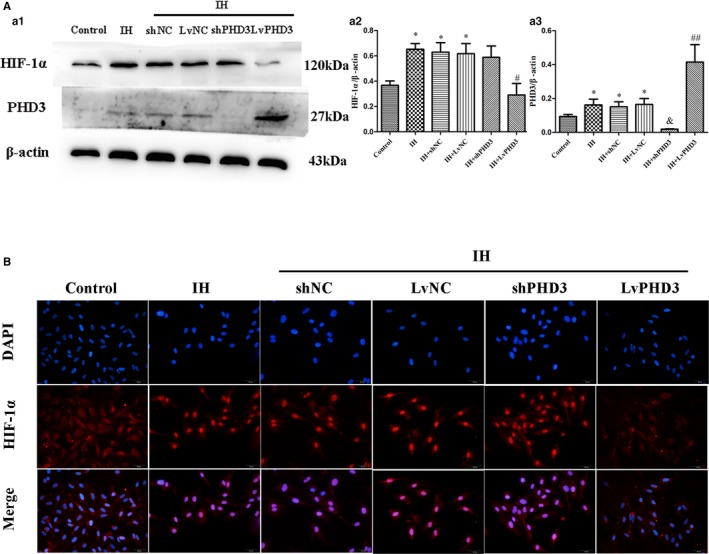
PHD3 overexpression mitigates IH‐induced HIF‐1α activation in endothelial cells. A, Western blot analysis and quantification of HIF‐1α (a1 and a2), PHD3 (a1 and a3). B, Representative images of immunofluorescence staining with antibodies to HIF‐1α (red). Nuclei were counterstained with DAPI (blue; original magnification, ×400; bars=20 μm). *P<0.05 vs control; # P<0.05 vs IH+LvNC; ## P<0.01 vs IH+LvNC; & P<0.05 vs IH+shNC. DAPI indicates 4',6‐diamidino‐2‐phenylindole; HIF‐1, hypoxia‐inducible factor 1 alpha; IH, intermittent hypoxia; LvPHD3, lentivirus carrying PHD3 cDNA; PHD3, prolyl 4‐hydroxylase domain protein 3; shPHD3, short‐hairpin PHD3.
Figure 8.
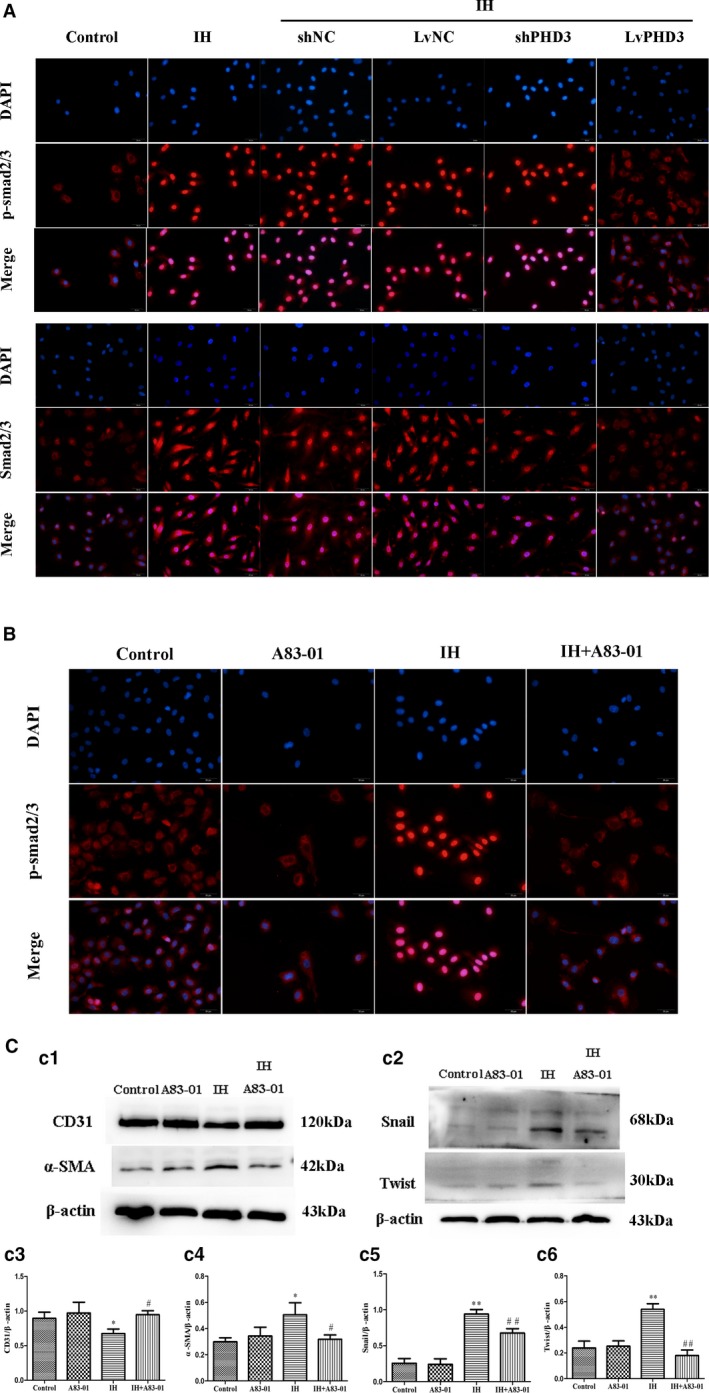
PHD3 overexpression mitigates IH‐induced smad2/3 activation in endothelial cells. A, Representative images of immunofluorescence staining with antibodies to p‐smad2/3 (red) and smad2/3 (red). Nuclei were counterstained with DAPI (blue; original magnification, ×400; bars=20 μm). B, Representative images of immunofluorescence analysis of p‐smad2/3 (red) localization in HUVECs with or without IH or A83‐01 treatment. Nuclei were counterstained with DAPI (blue; original magnification, ×400; bars=20 μm). C, Western blot analysis and quantitative of CD31 (c1 and c3), α‐SMA (c1 and c4), Snail (c2 and c5), and Twist (c2 and c6). *P<0.05 vs control; **P<0.01 vs control; # P<0.05 vs IH+LvNC; ## P<0.01 vs IH+LvNC. DAPI indicates 4',6‐diamidino‐2‐phenylindole; HIF‐1, hypoxia‐inducible factor 1 alpha; HUVECs, human umbilical vein endothelial cells; IH, intermittent hypoxia; LvPHD3, lentivirus carrying PHD3 cDNA; PHD3, prolyl 4‐hydroxylase domain protein 3; p‐ smad2/3, phosphorylated small mothers against decapentaplegic 2 and 3; shPHD3, short‐hairpin PHD3; α‐SMA, alpha‐smooth muscle actin.
Discussion
The present study demonstrates that PHD3 overexpression after a period of chronic IH reverts the cardiovascular remodeling, which is induced by this injurious challenge that characterizes OSA. In the field of experimental sleep apnea, considerable researches have focused on analyzing the effects of IH, but few studies have figured out a novel target spot for preventing cardiovascular remodeling in patients with OSA. Our novel experimental setting strongly suggests that PHD3 overexpression had a protective role in cardiac dysfunction and IH‐induced cardiac fibrosis by suppressing EndMT. This is the first report demonstrating that PHD3 overexpression alleviates IH‐induced EndMT and myocardial dysfunction.
OSA is a cardiovascular disease factor, but it is IH that plays the most important role in the progression of cardiovascular diseases.25 Although some studies have demonstrated beneficial effects of IH in animal models with OSA,26 the opposing effects induced by IH depend mainly on the experimental time; long‐term exposure (4–8 weeks) often cause detrimental effects.27 In our research, we assessed several morphological cardiovascular changes resulting from IH for 3 months, a common experimental paradigm to mimic severe OSA. Hypoxia can induce injury of cardiac myocytes; hence, we did not exclude the possibility that this condition directly affects cardiac myocytes. However, after long‐term exposure (3 months), IH induced cardiac dysfunction and fibrosis in our study. Immunohistochemical staining indicates that collagen was only deposited in perivascular and vascular media. This type of collagen deposition can reduce the elasticity of microvessels, increase the vascular wall thickness, and induce microvessel occlusion, which can cause a relatively insufficient oxygen supply. For myocardium, these changes can aggravate hypoxia injury and change IH into chronic continuous hypoxia. PHD3, which is deemed as a cellular oxygen sensor, can hydroxylate the HIF‐1α and HIF‐2α subunits at the conserved proline residues (Pro402 and Pro564 in HIF‐1α).28 Jaakkola and Rantanen believe that, under restricted oxygen, PHD3 is not an activator of cell death, but, in contrast, is a promoter of cell survival and proliferation.15 Interestingly, cardiac dysfunction and fibrosis improved when PHD3 was overexpressed. Furthermore, Broberg et al considered that the fibrosis index was correlated with the ventricular ejection fraction, but not with age and oxygen saturation, in cyanotic patients whose characteristics are similar to those with acquired heart failure.29 Thus, we concluded that PHD3 improves cardiac function by remodeling inhibition. Whether or not PHD3 can also increase the myocardial ability of antihypoxia or improve oxygen supply will be further explored.
Vascular remodeling is a response to long‐standing changes in hemodynamic conditions. IH is an independent factor that promotes vascular remodeling.30 IMT remodeling is an early stage in plaque formation and is associated with increased cardiovascular risk. Consistent with other researches, our finding confirmed that cardiac microvascular IMT increased with IH exposure in mice. Importantly, our experimental data showed that IMT returns to normal after PHD3 overexpression. Thus, it is speculated that PHD3 overexpression may be of therapeutic potential in ameliorating this process. IH‐induced perivascular fibrosis was observed in the left and right ventricle, whereas interstitial fibrosis was not obviously increased, which was in agreement with previous studies.20, 31 Perivascular fibrosis substantially impairs coronary blood flow seriously, leading to heart failure eventually.32 Consistent with previous studies, we did not observe left ventricular hypertrophy after IH treatment either.20 However, our results showed that cardiac systolic and diastolic function both decreased. Several studies demonstrated that cardiac hypertrophy could be induced by IH.31 The large disparity may reflect differences in species or strain, which could explain our negative result. Surprisingly, immunohistochemistry showed that there was a significant increase in expression of collagen I in the perivascular regions compared with the control under IH, but no significant difference was observed in collagen III expression compared with the control in vivo. Interestingly, both collagen I and III expression increased after IH treatment in vitro. Thus, we conjectured that collagen deposition was in a progress of dynamic equilibrium, and IH could increase not only its production, but also its degradation, which finally contributed to the equilibrium state of collagen III. Besides, we also speculate that IH may affect collagen I only in vivo. The present study revealed that PHD3 overexpression reduced perivascular deposition of collagen I in the IH treatment group. Consistent with this, echocardiography revealed that PHD3 overexpression also improved IH‐induced cardiac dysfunction.
To elucidate the mechanism through which PHD3 overexpression reduces perivascular fibrosis and improves cardiac function, we designed the experiment in vivo. Increasing evidences have shown that a large proportion of this collagen is derived from the endothelium through a process called EndMT.33 Hypoxia is an important initiator of EndMT.34 As we know, heart fibrosis resulting from EndMT is predominantly characterized by perivascular and subendocardial fibrosis.8 In the control group, we observed a single thin layer of ECs expressing CD31 adjacent to microvascular media by immunohistochemistry staining. After IH treatment, we observed a luminal layer of cells expressing diffuse endothelial markers (CD31). However, the diffuse staining disappears after PHD3 overexpression treatment. From western blot, snail and twist, which were deemed as the EndMT master regulators,35, 36 exerted higher expression than the control, but its expression decreased after PHD3 overexpression treatment. Therefore, we confirm that EndMT is involve in IH‐induced cardiac fibrosis and that PHD3 may improve it by attenuating EndMT.
To further certify our observations in vivo, we designed experiments in vitro. Like our study in vivo, HUVECs induced by IH decreased the level of the endothelial markers (CD31, VE‐cadherin) and increased expression of the mesenchymal cell markers (α‐SMA, FSP‐1). These effects were markedly reversed with PHD3 overexpression in accord with our observation in vivo. Hypoxia is a principal inducer of angiogenesis. In this regard, sprouting ECs, which were called tip cells, share similarities with ECs undergoing EndMT: Both tip cells and EndMT cells display spindle, not cobblestone, morphology because of the increased expression of mesenchymal cytoskeletal constituents.37 However, the principal differences are the decreased expression of endothelial markers (CD31, VE‐cadherin), which only appear in EndMT, but are not observed in tip cells. Another difference is an increased secretion of collagen I and III, which is only observed in ECs undergoing EndMT, but not in tip cells.38 Based on our results, we discovered that endothelial markers decreased and collagen I and III expression increased after IH treatment in HUVECs. Furthermore, IH increased the EndMT master regulators, snail and twist, expression. So, we confirmed that IH truly results in EndMT and endows HUVECs with an enhanced ability to secret collagen I and III, which may lead to cardiac fibrosis. However, these effects were markedly reversed with PHD3 overexpression. We further explored the molecular mechanism of PHD3 in vitro. Hypoxia inhibits prolyl hydroxylases, such as PHDs, which act to hydroxylate HIF‐1α under normoxic conditions. However, PHD3 best retains its activity under prolonged hypoxia and its expression is more marked as compared with other PHDs.18 HIF‐1α is able to promote the expression of TWIST39 and SNAIL.24 In the present study, we found that IH not only increased HIF‐1α expression, but also activated it, resulting in nuclear translocation. After PHD3 overexpression, the effect induced by IH is reversed. Thus, PHD3‐HIF‐1α‐snail/twist pathway is an essential mechanism that may contribute to IH‐induced EndMT, which was associated with cardiac remodeling. PHD3 may improve IH‐induced cardiac remodeling through other mechanisms. The transforming growth factor‐β/Smads pathway and their downstream receptors are considered as the major regulators of EndMT, because they are the pivotal cause of fibrosis.40 We found that PHD3 overexpression could inhibit IH‐induced Smad2/3 and p‐smad2/3 activity, which present as stopping IH‐induced nuclear translocation, and attenuate IH‐induced EndMT in HUVECs. Then, we used A83‐01 to inhibit p‐smad2/3; we found that IH‐induced EndMT was improved. These results showed that the smads pathway might be another mechanism that contributes to IH‐induced EndMT.
There are several limitations in our research. (1) OSA results in IH, hypercapnia, sleep arousal, and changes in intrathoracic pressure. All the negative factors contribute to cardiovascular remodeling together. We only focused on IH stress. It is a limitation because the mice model of IH does not represent the total disorder. Jordan considered that OSA is linked to cardiovascular disorders, such as myocardial infarction and congestive heart failure, which further increase the morbidity and mortality in OSA population.41 However, IH is the most powerful incitant involved in OSA‐induced cardiac remodeling, which is the main detrimental event leading to cardiovascular diseases.5 Our model can effectively mimic IH induced by severe OSA. Complications, such as cardiac remodeling and heart failure, were reported in our research. Furthermore, the model has been used to explore OSA and its complications, such as cardiac remodeling, by international and domestic academicians.20, 42, 43 (2) We did not consider the exact contribution of mesenchymal cells by endothelial cells to cardiac fibrosis, which is also a limitation in our study. (3) In vivo, we believed that collagen was derived from coronary artery ECs. We used human HUVECs to explore the mechanism in vitro. Mouse coronary microvessel ECs in vitro are a reasonable option. We finally selected human cells because the former is difficult to extract and cannot be purchased from companies, such as ScienceCell. Furthermore, the use of human cells in in vitro research is accepted in the cardiovascular fundamental field.44, 45 In addition, PHD3 is an ortholog found in both humans and mice. Hence, we firmly believe that human cells are involved in these mechanisms in vitro. (4) We used lentiviral to interfere or overexpress PHD3, and its effect is not better than knockout or transgenic technology. However, the lentiviral basically achieved the anticipated effect.
In conclusion, we illustrate that PHD3 could protect against cardiac perivascular fibrosis and improve myocardial function in an OSA mice model by inhibiting EndMT. The mechanism may be involved with the translocation of HIF‐1α smad2/3 p‐smad2/3 to the nucleus. Although the exact underlying mechanisms are not fully understood, given the cardioprotective effects of PHD3 overexpression, PHD3 may be a potential therapeutic target for OSA‐induced heart diseases.
Sources of Funding
This work was supported by the National Science Foundation of China (Nos. 81300159 and 81300227), Jiangsu University's Innovation Project Foundation of China (No. KYLX16_0296), and Nanjing Technological Development Foundation of China (No. 201605074).
Disclosures
None.
Supporting information
Figure S1. Basic characteristic of mice. A, Weight of mice. B, Systolic blood pressure. Data are mean±SD; n=10 per group. *P<0.05 vs control.
Figure S2. Change of PHD3 expression in vivo and vitro by lentivirus transfection. A, Western blot analysis of PHD3 protein level relative to that of β‐actin (a1) and quantitative analysis (a2) in vivo. B, Representative photograph of GFP‐labeled scramble transfection efficiency in HUVECs by fluorescence microscopy; the transfection efficiency was evaluated at more than 90% (original magnification, ×100; bar=50 μm). C, Western blot analysis of PHD3 protein level relative to that of β‐actin (c1) and quantitative analysis (c2) in vitro. Data are mean±SD; n=10 per group. *P<0.05 vs shNC or # P<0.05 vs LvNC.
Figure S3. Masson and collagen I III staining in myocardium region. Representative images of the heart section with Masson's trichrome (blue staining), and immunohistochemical staining of collagen I collagen III (yellow staining) for each group (original magnification, ×400; bars=20 μm).
Figure S4. PHD3 overexpression improved IH‐induced EndMT. Representative images of immunohistochemical staining of α‐SMA (yellow staining) in the perivascular and vascular regions. The presence of α‐SMA is detected in endothelial cells (red arrows; original magnification, ×400; bars=20 μm).
Acknowledgment
The IH device and IH experimental process was performed under the supervision of PUHE Biotechnology Company LTD
(J Am Heart Assoc. 2017;6:e006680 DOI: 10.1161/JAHA.117.006680.)
This work was presented as a poster at the European Society of Cardiology Congress, August 26–30, 2017, in Barcelona, Spain.
References
- 1. Phillipson EA. Sleep apnea—a major public health problem. N Engl J Med. 1993;328:1271–1273. [DOI] [PubMed] [Google Scholar]
- 2. Gopalakrishnan P, Tak T. Obstructive sleep apnea and cardiovascular disease. Cardiol Rev. 2011;19:279–290. [DOI] [PubMed] [Google Scholar]
- 3. Fava C, Montagnana M, Favaloro EJ, Guidi GC, Lippi G. Obstructive sleep apnea syndrome and cardiovascular diseases. Semin Thromb Hemost. 2011;37:280–297. [DOI] [PubMed] [Google Scholar]
- 4. Javaheri S, Barbe F, Campos‐Rodriguez F, Dempsey JA, Khayat R, Javaheri S, Malhotra A, Martinez‐Garcia MA, Mehra R, Pack AI, Polotsky VY, Redline S, Somers VK. Sleep apnea: types, mechanisms, and clinical cardiovascular consequences. J Am Coll Cardiol. 2017;69:841–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baguet JP, Barone‐Rochette G, Tamisier R, Levy P, Pepin JL. Mechanisms of cardiac dysfunction in obstructive sleep apnea. Nat Rev Cardiol. 2012;9:679–688. [DOI] [PubMed] [Google Scholar]
- 6. LeBleu VS, Taduri G, O'Connell J, Teng Y, Cooke VG, Woda C, Sugimoto H, Kalluri R. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. 2013;19:1047–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma‐associated fibroblasts. Cancer Res. 2007;67:10123–10128. [DOI] [PubMed] [Google Scholar]
- 8. Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. Endothelial‐to‐mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. [DOI] [PubMed] [Google Scholar]
- 9. Zhang Y, Wu X, Li Y, Zhang H, Li Z, Zhang Y, Zhang L, Ju J, Liu X, Chen X, Glybochko PV, Nikolenko V, Kopylov P, Xu C, Yang B. Endothelial to mesenchymal transition contributes to arsenic‐trioxide‐induced cardiac fibrosis. Sci Rep. 2016;6:33787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ranchoux B, Antigny F, Rucker‐Martin C, Hautefort A, Pechoux C, Bogaard HJ, Dorfmuller P, Remy S, Lecerf F, Plante S, Chat S, Fadel E, Houssaini A, Anegon I, Adnot S, Simonneau G, Humbert M, Cohen‐Kaminsky S, Perros F. Endothelial‐to‐mesenchymal transition in pulmonary hypertension. Circulation. 2015;131:1006–1018. [DOI] [PubMed] [Google Scholar]
- 11. Yu W, Liu Z, An S, Zhao J, Xiao L, Gou Y, Lin Y, Wang J. The endothelial‐mesenchymal transition (EndMT) and tissue regeneration. Curr Stem Cell Res Ther. 2014;9:196–204. [DOI] [PubMed] [Google Scholar]
- 12. Medici D, Potenta S, Kalluri R. Transforming growth factor‐beta2 promotes snail‐mediated endothelial‐mesenchymal transition through convergence of smad‐dependent and smad‐independent signalling. Biochem J. 2011;437:515–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee SW, Won JY, Kim WJ, Lee J, Kim KH, Youn SW, Kim JY, Lee EJ, Kim YJ, Kim KW, Kim HS. Snail as a potential target molecule in cardiac fibrosis: paracrine action of endothelial cells on fibroblasts through snail and CTGF axis. Mol Ther. 2013;21:1767–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Metzen E, Berchner‐Pfannschmidt U, Stengel P, Marxsen JH, Stolze I, Klinger M, Huang WQ, Wotzlaw C, Hellwig‐Burgel T, Jelkmann W, Acker H, Fandrey J. Intracellular localisation of human HIF‐1 alpha hydroxylases: implications for oxygen sensing. J Cell Sci. 2003;116:1319–1326. [DOI] [PubMed] [Google Scholar]
- 15. Jaakkola PM, Rantanen K. The regulation, localization, and functions of oxygen‐sensing prolyl hydroxylase PHD3. Biol Chem. 2013;394:449–457. [DOI] [PubMed] [Google Scholar]
- 16. Lieb ME, Menzies K, Moschella MC, Ni R, Taubman MB. Mammalian EGLN genes have distinct patterns of mRNA expression and regulation. Biochem Cell Biol. 2002;80:421–426. [DOI] [PubMed] [Google Scholar]
- 17. Appelhoff RJ, Tian YM, Raval RR, Turley H, Harris AL, Pugh CW, Ratcliffe PJ, Gleadle JM. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia‐inducible factor. J Biol Chem. 2004;279:38458–38465. [DOI] [PubMed] [Google Scholar]
- 18. Ginouves A, Ilc K, Macias N, Pouyssegur J, Berra E. PHDs overactivation during chronic hypoxia “desensitizes” HIFalpha and protects cells from necrosis. Proc Natl Acad Sci USA. 2008;105:4745–4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Torres M, Laguna‐Barraza R, Dalmases M, Calle A, Pericuesta E, Montserrat JM, Navajas D, Gutierrez‐Adan A, Farre R. Male fertility is reduced by chronic intermittent hypoxia mimicking sleep apnea in mice. Sleep. 2014;37:1757–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Castro‐Grattoni AL, Alvarez‐Buve R, Torres M, Farre R, Montserrat JM, Dalmases M, Almendros I, Barbe F, Sanchez‐de‐la‐Torre M. Intermittent hypoxia‐induced cardiovascular remodeling is reversed by normoxia in a mouse model of sleep apnea. Chest. 2016;149:1400–1408. [DOI] [PubMed] [Google Scholar]
- 21. Dyugovskaya L, Polyakov A, Lavie P, Lavie L. Delayed neutrophil apoptosis in patients with sleep apnea. Am J Respir Crit Care Med. 2008;177:544–554. [DOI] [PubMed] [Google Scholar]
- 22. Dyugovskaya L, Polyakov A, Ginsberg D, Lavie P, Lavie L. Molecular pathways of spontaneous and TNF‐{alpha}‐mediated neutrophil apoptosis under intermittent hypoxia. Am J Respir Cell Mol Biol. 2011;45:154–162. [DOI] [PubMed] [Google Scholar]
- 23. Yan F, Zhang GH, Feng M, Zhang W, Zhang JN, Dong WQ, Zhang C, Zhang Y, Chen L, Zhang MX. Glucagon‐like peptide 1 protects against hyperglycemic‐induced endothelial‐to‐mesenchymal transition and improves myocardial dysfunction by suppressing poly(ADP‐ribose) polymerase 1 activity. Mol Med. 2015;21:15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu X, Tan X, Tampe B, Sanchez E, Zeisberg M, Zeisberg EM. Snail is a direct target of hypoxia‐inducible factor 1alpha (HIF1alpha) in hypoxia‐induced endothelial to mesenchymal transition of human coronary endothelial cells. J Biol Chem. 2015;290:16653–16664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fletcher EC. Invited review: physiological consequences of intermittent hypoxia: systemic blood pressure. J Appl Physiol (1985). 2001;90:1600–1605. [DOI] [PubMed] [Google Scholar]
- 26. Almendros I, Wang Y, Gozal D. The polymorphic and contradictory aspects of intermittent hypoxia. Am J Physiol Lung Cell Mol Physiol. 2014;307:L129–L140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Campen MJ, Shimoda LA, O'Donnell CP. Acute and chronic cardiovascular effects of intermittent hypoxia in C57BL/6J mice. J Appl Physiol (1985). 2005;99:2028–2035. [DOI] [PubMed] [Google Scholar]
- 28. Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ. C. elegans EGL‐9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. [DOI] [PubMed] [Google Scholar]
- 29. Broberg CS, Chugh SS, Conklin C, Sahn DJ, Jerosch‐Herold M. Quantification of diffuse myocardial fibrosis and its association with myocardial dysfunction in congenital heart disease. Circ Cardiovasc Imaging. 2010;3:727–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gileles‐Hillel A, Almendros I, Khalyfa A, Zhang SX, Wang Y, Gozal D. Early intermittent hypoxia induces proatherogenic changes in aortic wall macrophages in a murine model of obstructive sleep apnea. Am J Respir Crit Care Med. 2014;190:958–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ramirez TA, Jourdan‐Le Saux C, Joy A, Zhang J, Dai Q, Mifflin S, Lindsey ML. Chronic and intermittent hypoxia differentially regulate left ventricular inflammatory and extracellular matrix responses. Hypertens Res. 2012;35:811–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dai Z, Aoki T, Fukumoto Y, Shimokawa H. Coronary perivascular fibrosis is associated with impairment of coronary blood flow in patients with non‐ischemic heart failure. J Cardiol. 2012;60:416–421. [DOI] [PubMed] [Google Scholar]
- 33. Goumans MJ, van Zonneveld AJ, ten Dijke P. Transforming growth factor beta‐induced endothelial‐to‐mesenchymal transition: a switch to cardiac fibrosis? Trends Cardiovasc Med. 2008;18:293–298. [DOI] [PubMed] [Google Scholar]
- 34. Choi SH, Hong ZY, Nam JK, Lee HJ, Jang J, Yoo RJ, Lee YJ, Lee CY, Kim KH, Park S, Ji YH, Lee YS, Cho J, Lee YJ. A hypoxia‐induced vascular endothelial‐to‐mesenchymal transition in development of radiation‐induced pulmonary fibrosis. Clin Cancer Res. 2015;21:3716–3726. [DOI] [PubMed] [Google Scholar]
- 35. Kokudo T, Suzuki Y, Yoshimatsu Y, Yamazaki T, Watabe T, Miyazono K. Snail is required for TGFbeta‐induced endothelial‐mesenchymal transition of embryonic stem cell‐derived endothelial cells. J Cell Sci. 2008;121:3317–3324. [DOI] [PubMed] [Google Scholar]
- 36. Kovacic JC, Mercader N, Torres M, Boehm M, Fuster V. Epithelial‐to‐mesenchymal and endothelial‐to‐mesenchymal transition: from cardiovascular development to disease. Circulation. 2012;125:1795–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dave JM, Bayless KJ. Vimentin as an integral regulator of cell adhesion and endothelial sprouting. Microcirculation. 2014;21:333–344. [DOI] [PubMed] [Google Scholar]
- 38. Davis GE, Senger DR. Endothelial extracellular matrix: biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ Res. 2005;97:1093–1107. [DOI] [PubMed] [Google Scholar]
- 39. Yang MH, Wu MZ, Chiou SH, Chen PM, Chang SY, Liu CJ, Teng SC, Wu KJ. Direct regulation of TWIST by HIF‐1alpha promotes metastasis. Nat Cell Biol. 2008;10:295–305. [DOI] [PubMed] [Google Scholar]
- 40. Hein S, Arnon E, Kostin S, Schonburg M, Elsasser A, Polyakova V, Bauer EP, Klovekorn WP, Schaper J. Progression from compensated hypertrophy to failure in the pressure‐overloaded human heart: structural deterioration and compensatory mechanisms. Circulation. 2003;107:984–991. [DOI] [PubMed] [Google Scholar]
- 41. Jordan AS, McSharry DG, Malhotra A. Adult obstructive sleep apnoea. Lancet. 2014;383:736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Polotsky VY, Rubin AE, Balbir A, Dean T, Smith PL, Schwartz AR, O'Donnell CP. Intermittent hypoxia causes REM sleep deficits and decreases EEG delta power in NREM sleep in the C57BL/6J mouse. Sleep Med. 2006;7:7–16. [DOI] [PubMed] [Google Scholar]
- 43. McGuire M, MacDermott M, Bradford A. The effects of chronic episodic hypercapnic hypoxia on rat upper airway muscle contractile properties and fiber‐type distribution. Chest. 2002;122:1400–1406. [DOI] [PubMed] [Google Scholar]
- 44. Chen XY, Lv RJ, Zhang W, Yan YG, Li P, Dong WQ, Liu X, Liang ES, Tian HL, Lu QH, Zhang MX. Inhibition of myocyte‐specific enhancer factor 2A improved diabetic cardiac fibrosis partially by regulating endothelial‐to‐mesenchymal transition. Oncotarget. 2016;7:31053–31066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yao J, Guihard PJ, Blazquez‐Medela AM, Guo Y, Moon JH, Jumabay M, Bostrom KI, Yao Y. Serine protease activation essential for endothelial‐mesenchymal transition in vascular calcification. Circ Res. 2015;117:758–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Basic characteristic of mice. A, Weight of mice. B, Systolic blood pressure. Data are mean±SD; n=10 per group. *P<0.05 vs control.
Figure S2. Change of PHD3 expression in vivo and vitro by lentivirus transfection. A, Western blot analysis of PHD3 protein level relative to that of β‐actin (a1) and quantitative analysis (a2) in vivo. B, Representative photograph of GFP‐labeled scramble transfection efficiency in HUVECs by fluorescence microscopy; the transfection efficiency was evaluated at more than 90% (original magnification, ×100; bar=50 μm). C, Western blot analysis of PHD3 protein level relative to that of β‐actin (c1) and quantitative analysis (c2) in vitro. Data are mean±SD; n=10 per group. *P<0.05 vs shNC or # P<0.05 vs LvNC.
Figure S3. Masson and collagen I III staining in myocardium region. Representative images of the heart section with Masson's trichrome (blue staining), and immunohistochemical staining of collagen I collagen III (yellow staining) for each group (original magnification, ×400; bars=20 μm).
Figure S4. PHD3 overexpression improved IH‐induced EndMT. Representative images of immunohistochemical staining of α‐SMA (yellow staining) in the perivascular and vascular regions. The presence of α‐SMA is detected in endothelial cells (red arrows; original magnification, ×400; bars=20 μm).