

Abstract
Despite clinical remission of epithelial ovarian cancer (EOC) after surgical resection and first-line chemotherapy, about 60% of patients will re-develop peritoneal metastasis and about 50% will relapse with chemoresistant disease. Clinical studies suggest that intra-peritoneal (i.p.) chemotherapy effectively treats residual EOC after cyto-reduction by gaining direct access into the peritoneal cavity, enabling elevated drug levels versus intravenous (i.v.) injection. However, chemoresistant disease is still problematic. To overcome resistance against microtubule stabilizing agents such as taxanes, epothilone B (EpoB) has merit, especially in combination with molecular targeted agents that inhibit heat shock protein 90 (Hsp90) and/or mammalian target of rapamycin (mTOR). In this paper, we report on the successful loading and solubilization of EpoB in a poly(D,L-lactic-co-glycolic acid)-block-poly(ethylene glycol)-block-poly(D,L-lactic-co-glycolic acid) (PLGA-b-PEG-b-PLGA) thermosensitive gel (g–E). Further, we report on successful co-loading of 17-AAG (Hsp90) and rapamycin (mTOR) (g-EAR). After i.p. injection in mice, g-EAR showed gelation in the peritoneum and sustained, local-regional release of EpoB, 17-AAG, and rapamycin. In a luciferase-expressing ES-2 (ES-2-luc) ovarian cancer xenograft model, single i.p. injections of g-E and g-EAR delayed bioluminescence from metastasizing ES-2-luc cells for 2 and 3 weeks, respectively, despite fast drug release for g-EAR in vivo versus in vitro. In summary, a PLGA-b-PEG-b-PLGA sol-gel has loading and release capacities for EpoB and its combinations with 17-AAG and rapamycin, enabling a platform for i.p. delivery, sustained multi-drug exposure, and potent antitumor efficacy in an ES-2-luc, ovarian cancer i.p. xenograft model.
Keywords: Drug combination, epothilone B, intraperitoneal injection, ovarian cancer, peritoneal carcinomatosis, thermogel
Graphical abstract

1. Introduction
Ovarian cancer has been called a “silent killer” due to its asymptomatic nature, and by the time of diagnosis, most patients have stage III-IV disease, extensive peritoneal metastatic lesions, and dismal outcomes [1]. While ovarian cancer patients initially respond well to chemotherapy, i.e. taxanes and platinates, ca. 50% relapse with chemoresistant disease [2]. To overcome drug resistance, two major tactics have been explored in pre-clinical and clinical studies: Intraperitoneal (i.p.) drug delivery and drug combinations involving molecularly-targeted agents.
In the former case, the i.p. route permits direct access to ovarian cancer in the form of numerous flat lesions on visceral or parietal peritoneum; higher intraperitoneal drug levels versus the intravenous (i.v.) route; and higher tumor exposure, especially with intraoperative hyperthermic i.p. chemotherapy (HIPEC) [3–5]. While HIPEC has demonstrated survival benefit in clinical trials, its use is somewhat controversial due to conflicting results in clinical trials and treatment-related morbidities, such as infection, abdominal pain, and hematologic toxicity. An alternative to HIPEC is i.p. chemotherapy polymeric depots that maintain therapeutic levels for several hours or perhaps to several days and reduce the likelihood of drug resistance [6]. In this tactic, i.p. depots must not cause peritoneal adhesions that limit drug distribution in the peritoneal cavity [7].
In the latter case, drug combinations involving molecularly-targeted agents aim to overcome de novo and acquired resistance towards chemotherapy, e.g. paclitaxel, in ovarian cancer. Pre-clinical and clinical studies have validated drug targets involved in angiogenesis, phosphatidylinositol-3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) signaling, and DNA repair, i.e. Poly(ADP-ribose) polymerase (PARP) inhibitors [8–10]. In most cases, molecularly-targeted agents rely on i.v. or oral routes of drug administration, and somewhat surprisingly, the i.p. route for molecularly-targeted agents has not received attention for the treatment of ovarian cancer, particularly in combination with i.p. chemotherapy.
We propose that a thermosensitive hydrogel can act as a platform controlled release technology for pre-clinical and clinical testing of chemotherapy and molecularly-targeted agents via the i.p. route (Figure 1). Poly(D,L-lactic-co-glycolic acid)-block-poly(ethylene glycol)-block-poly(D,L-lactic acid-co-glycolic acid) (PLGA-b-PEG-b-PLGA) is a hydrogel (Regel®) that undergoes a reversible, temperature-dependent sol-gel transition, enabling injection or placement as a sol and transformation into a gel for i.p. sustained release (Figure 1). Regel® has a high capacity for paclitaxel and has progressed into a phase 2b clinical trial for the local treatment of esophageal cancer [11]. Besides paclitaxel, Regel® has a high capacity for other poorly water-soluble anticancer agents, e.g. 17-AAG and rapamycin, enabling pre-clinical evaluation of microtubule-stabilizing agents and concurrent inhibition of mTOR and heat shock protein 90 (Hsp90) [12]. In this study, epothilone B (EpoB) has been chosen as an alternative to paclitaxel as a microtubule-stabilizing anticancer agent, owing to its higher potency than paclitaxel and lower susceptibility to drug efflux transporters, and we report on the pre-clinical characterization and evaluation of a Regel® containing EpoB, and its combination with 17-AAG, and rapamycin in an ES-2-luc i.p. ovarian tumor model.
Fig. 1.
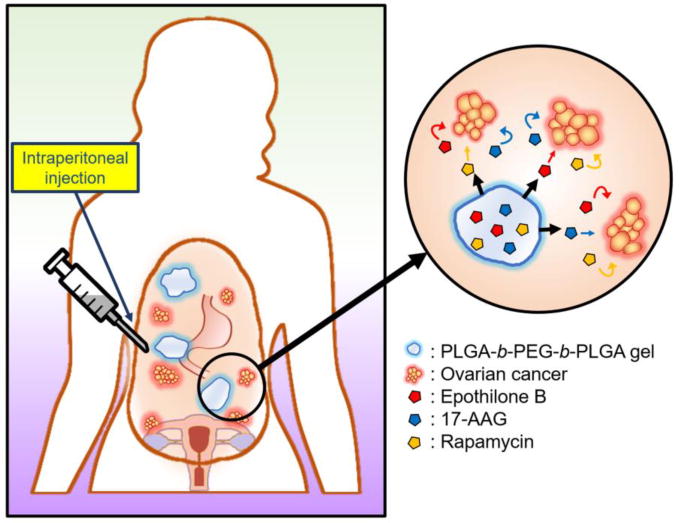
Schematic representation of i.p. injection of Regel® containing EpoB, 17-AAG, and rapamycin; i.p. gelation at body temperature; and i.p. multi-drug release.
2. Materials and methods
2.1. Materials
Phosphate-buffered saline (PBS), penicillin/streptomycin, trypsin-EDTA, McCoy’s 5a medium, and fetal bovine serum (FBS) were purchased from American Type Culture Collection (ATCC; Manassas, VA). Bovine serum albumin (BSA), Phosphoric acid and tert-butanol were purchased from Sigma-Aldrich Inc. (St. Louis, MO). Acetonitrile and methanol were obtained from Thermo Fisher Scientific Inc. (Waltham, MA). Cell Titer-Blue® reagent was purchased from Promega (Madison, WI). PLGA1500-b-PEG1000-b-PLGA1500 was purchased from Polyscitech. (West Lafayette, IN). EpoB, 17-AAG, and rapamycin were purchased from LC laboratories. (Woburn, MA). All other chemicals were analytical grade or better.
2.2. Cell culture
ES-2 human ovarian cancer cells, purchased from ATCC, were cultured in McCoy’s 5a medium supplemented with 1% penicillin/streptomycin and 10% FBS. To encode luciferase reporter gene, the cells were transfected with pGL4.51 luciferase expressing plasmid. The prepared luciferase-expressing ES-2 cells (ES-2-luc cells) were cultured in the same medium and maintained at 37 °C in a humidified 5% CO2 atmosphere.
2.3. Reverse-phase HPLC analysis
Quantification of EpoB, 17-AAG and rapamycin was accomplished by reverse-phase high performance liquid chromatography (Prominence HPLC system, Shimadzu, Japan) with a diode array detector (SPD-M20A, Shimadzu, Japan). The mobile phase was a mixture of acetonitrile-water-methanol-phosphoric acid (55:45:1:0.1, v/v/v/v). The flow rate of mobile phase was 1.0 mL/min. The analysis was performed on a Zorbax SB-C8 rapid resolution cartridge column (4.6 mm × 75 mm, 3.5 μm, Agilent, Santa Clara, CA), and temperature of the column was kept at 40 °C. The injection volume of each sample was 10 μL. EpoB, 17-AAG, or rapamycin was monitored at 211, 333, and 279 nm, respectively and retention time of each drug was 2.63, 3.61, and 10.5 min, respectively. Gossypol was used as an internal standard (IS).
2.4. Cell proliferation assay
Cell proliferation was measured using the Cell Titer-Blue® viability assay. Briefly, ES-2-luc cells were placed onto 96-well plates and incubated overnight at 37 °C at 5% CO2. The medium in each well was replaced with fresh medium containing varied drug concentrations. After 72 hours of incubation, the culture medium was removed and the medium containing 20% Cell Titer-Blue® reagent was added to each well. Cells were incubated for 1 hour, and the fluorescence intensity of the solution in the wells was measured with a Spectra M2 plate reader (Molecular Device, Sunnyvale, CA) at an excitation wavelength (λex) of 560 nm and an emission wavelength (λem) of 590 nm. The drug concentration that induces x% inhibition of cell growth (ICx) was calculated by curve fitting.
2.5. Combination index (CI) analysis
Combination index analysis was calculated according to Chou’s method to assess drug interaction between drugs [13]. CI was calculated from:
where A, B, and C are the concentrations of drug A, B, and C in combination that inhibit cell growth by x% (isoeffective compared with the individual drugs), and Ax, Bx, and Cx are the concentrations of drug A, B, and C, which induce a x% inhibition of cell growth. Values of CI>1, CI=1, and CI<1 indicate antagonism, additive, and synergism, respectively.
2.6. Clonogenic assay
The clonogenic assay was performed according to the protocols of previous literatures [14, 15]. ES-2-luc cells were dissociated to single cells and seeded in a 6-well plate with McCoy’s 5a medium containing 10% FBS (200 cells/well). Cells were incubated for 4 hours to allow the cells to adhere on the bottom of the plates and treated with drug(s). Cells were treated with single EpoB (0.4 nM), 17-AAG (2.6 nM), rapamycin (0.83 nM), and 2 or 3-drug combinations. After 2 weeks of incubation, the medium was removed, and the crystal violet was added to each well to stain colonies. Unstained dye was washed gently with clean water, and the number of colonies in each well were observed and quantified. The percentage of colony formation was calculated by the following equation:
2.7. Preparation of Regel® containing EpoB, 17-AAG, and rapamycin
EpoB gel (g-E), 17-AAG gel (g-A), rapamycin gel (g-R), EpoB/17-AAG gel (g-EA), 17-AAG/rapamycin gel (g-AR), EpoB/rapamycin gel (g-ER) and EpoB/17-AAG/rapamycin gel (g-EAR) were prepared by modified freeze-drying method [12]. Briefly, 150 mg of PLGA-b-PEG-b-PLGA polymer was incubated in 1.0 mL of pre-cooled PBS (10 mM, pH 7.4) with gentle agitation at 4 °C until the polymer was completely dissolved. For drug preparation, 0.4 mg of EpoB, 3.0 mg of 17-AAG, 1.5 mg of rapamycin, and their two or three drug combinations were added to 1.0 mL of tert-butanol in the scintillation glass vials and agitated in the shaking water bath (YB-521; American Scientific, Columbus, OH) at 60 °C until drugs were completely dissolved. The prepared polymer solution (1.0 mL) was added to the drug solution, and the whole mixture was vortexed vigorously. The vials were rapidly frozen at −70 °C and lyophilized overnight. The lyophilized cake was rehydrated with 1.0 mL of distilled water at 4 °C. The rehydrated solution was centrifuged at 13,000 rpm for 5 min and filtered with a 0.22 μm cellulose filter to remove unincorporated drug. After dissolution of the gel in acetonitrile, the level of the drug(s) in Regel® was quantified by reverse-phase HPLC analysis.
2.8. In vitro drug release
Regel® formulations (0.5 mL) were placed on the bottom of scintillation glass vials and incubated in ambient temperature until gel formation. PBS containing 2% BSA (5.0 mL) was added to the vials, and the vials were placed in a water bath at 37 °C with gentle stirring. At each sampling time point, the buffer in the vials was collected carefully and pre-warmed fresh PBS containing 2% BSA was added. The sampling time points were 0, 0.5, 3, 6, 10, 24, 48, 96, 120, 144, 168, 196, and 240 hours. The collected sample was mixed with chloroform, and the mixture was vortexed and centrifuged at 5,000 rpm for 10 min. The organic phase was collected, transferred into a vial, and evaporated using SpeedVac Concentrator (SVC-100H; Savant instruments, Mumbai, India). The residue in vials was reconstituted with acetonitrile, and the level of drug(s) was determined by reverse-phase HPLC analysis.
2.9. In vivo gel observation and drug release
The care and treatment of experimental mice were performed according to the institutional guidelines of University of Wisconsin-Madison. Eighteen athymic nude mice, purchased from Harlan Laboratories (Madison, WI), were randomly divided into 6 groups. All the mice were i.p. injected with EpoB/17-AAG/rapamycin-loaded PEG-b-PLA micelle (m-EAR, 2.0, 15.0, and 7.5 mg/kg for EpoB, 17-AAG, and rapamycin, respectively) and pre-cooled g-EAR (2.0, 15.0, and 7.5 mg/kg). The volume of gel for each rat was approximately 1.0 mL. To observe the distribution of gel in the peritoneal cavity, the mice in each group were sacrificed and subjected to abdominal incision at 2, 5, 9, 24, 48, and 96 hours after injection. The collectable g-EAR gel was harvested from the peritoneal cavity. The samples were dissolved in acetonitrile and centrifuged at 15,000 rpm for 10 min. The level of drug(s) in Regel® was quantified by reverse-phase HPLC analysis.
2.10. In vivo antitumor efficacy
Briefly, 1×106 ES-2-luc cancer cells resuspended in sterile PBS were injected i.p. into each mouse. After 3 days, mice were divided into 3 groups (4 mice in each group). Each group of mice was i.p. injected with various formulations. After 0, 3, 7, 14, 21, and 28 days, D-luciferin (Caliper Life Science, Hopkinton, MA,) at 100.0 mg/kg was injected i.p. into each mouse. After 15 min, whole body bioluminescence image was obtained using Xenogen IVIS® 200 Series. The total photon counts in the region of interest (ROIs) of each bioluminescence image was quantified using Live imaging® software. Total body weight changes of the groups were also monitored periodically. The animals were euthanized by CO2 in the euthanasia device when mice reached a moribund condition, according to the ethical guideline of University of Wisconsin-Madison.
3. Results and discussion
Regel® containing EpoB, 17-AAG, or rapamycin (g-E, g-A, and g-R); 2-drugs (g-EA, g-AR, and g-ER); and 3-drugs (g-EAR) were prepared as a freeze-dried solid cake and successfully reconstituted as a solution at 4 to 15 °C (Table 1). At 10 °C, PLGA-b-PEG-b-PLGA at ca. 150 mg/mL exists as a flower-like polymeric micelle with a z-average diameter at ca. 140 nm, a shell of PEG loops, and a core of PLGA that presumably is the location of the solubilized drug(s) [16]. As g-E, the water solubility of EpoB increased 15-fold from 25 μg/mL to 0.371 mg/mL at 93% loading efficiency (Table 1). The levels of 17-AAG and rapamycin in g-A and g-R were 2.85 and 1.44 mg/mL, respectively. Confirming our earlier report, 2 or 3 different anticancer agents can be combined in Regel®, attaining drug levels that are close to values for single drug loading [12]. For g-EAR, levels were 0.36, 2.5, and 1.2 mg/mL for EpoB, 17-AAG, and rapamycin, respectively (Table 1). The sol-gel transition occurs as a result of dehydration and hydrophobic interaction of PLGA-b-PEG-b-PLGA at elevated temperature, and importantly, the sol-gel temperature of Regel® was mostly unaffected by drug loading even in the case of g-EAR; in this case transformation into a gel occurred at ca. 16 °C (Figure S1). In summary, EpoB was loaded and solubilized by Regel® as a single drug and drug combinations with 17-AAG and rapamycin, permitting sufficient drug dose(s) for the pre-clinical efficacy studies on i.p. g-E and i.p. g-EAR in a murine model of ovarian cancer.
Table 1.
Drug loading of Regel® (n=3).
| Type Formulation | g-E | 1-in-1 g-A | g-R | g-EA | 2-in-1 g-AR | g-ER | 3-in-1 g-EAR |
|---|---|---|---|---|---|---|---|
| Polymer (mg/mL) | 150 | 150 | 150 | 150 | 150 | 150 | 150 |
| Initial drug(s) added (mg/mL) | 0.4 | 3 | 1.5 | 0.4/3 | 3/1.5 | 0.4/1.5 | 0.4/3/1.5 |
| Drug loading efficiency (%) | 92.7±4.8 | 95.0±3.6 | 95.8±5.4 | 91.0±7.0/89.6±8.2 | 98.0±11.9/98.5±11.6 | 94.1±11.2/96.0±8.6 | 90.0±8.3/82.8±8.4/81.9±7.9 |
| Total drug level (mg/mL) | 0.371±0.019 | 2.85±0.11 | 1.44±0.08 | 0.364±0.028/2.69±0.25 | 2.94±0.36/1.48±0.17 | 0.376±0.045/1.44±0.13 | 0.360±0.033/2.48±0.25/1.23±0.12 |
In vitro drug release for g-E, g-A, g-R, g-EA, g-AR, g-ER, and g-EAR was evaluated in a gel state at 37 °C in the presence of PBS, pH=7.4 containing 2% BSA (Figure 2). In vitro release of EpoB was the fastest, mostly completed within 60 hours with a half-life (t1/2) = 4 hours. In vitro release of 17-AAG followed at a lower rate over 240 hours with a t1/2 = 50 hours. Lastly, in vitro release of rapamycin was slow from Regel®, with ca. 30% drug release at 240 hours, probably reflecting its hydrophobic nature versus EpoB and 17-AAG. For g-EAR, in vitro release of EpoB was noticeably slower than g-E: t1/2 = 7 versus 4 hours, respectively (Figure 2). Given that EpoB loading levels were similar for g-EAR and g-E, it is hypothesized that drug interaction inside Regel® contributes to a reduced rate of EpoB release from g-EAR. However, there was no difference for 17-AAG between g-EAR and g-A and only a slight difference for rapamycin between g-EAR and g-R, pointing to the need for further analysis of drug release to fully gain a mechanistic underpinning for g-EAR. In summary, in vitro release of EpoB from Regel® is relatively fast compared to 17-AAG and rapamycin; both showing sustained release over 240 hours in vitro. Nonetheless, g-EAR fulfills a central requirement for synergistic activity: EpoB release before 17-AAG and rapamycin, favoring action at the G2-M phase of cancer cell growth. Lastly, there are a range of strategies to control the release rate of the drugs from hydrogels by modifying (i) drug hydrogel interactions and (ii) gel network engineering [17]. Otherwise, chemical modification of the drugs could enable slow release of payloads from hydrogel, given that the release rate of hydrophobic oligo lactic acid-conjugated paclitaxel prodrug in PEG-b-PLA micelle was slower than that of paclitaxel micelle [18]. Such a modification of each drug release rate may alter anticancer efficacy of the formulation.
Fig. 2.
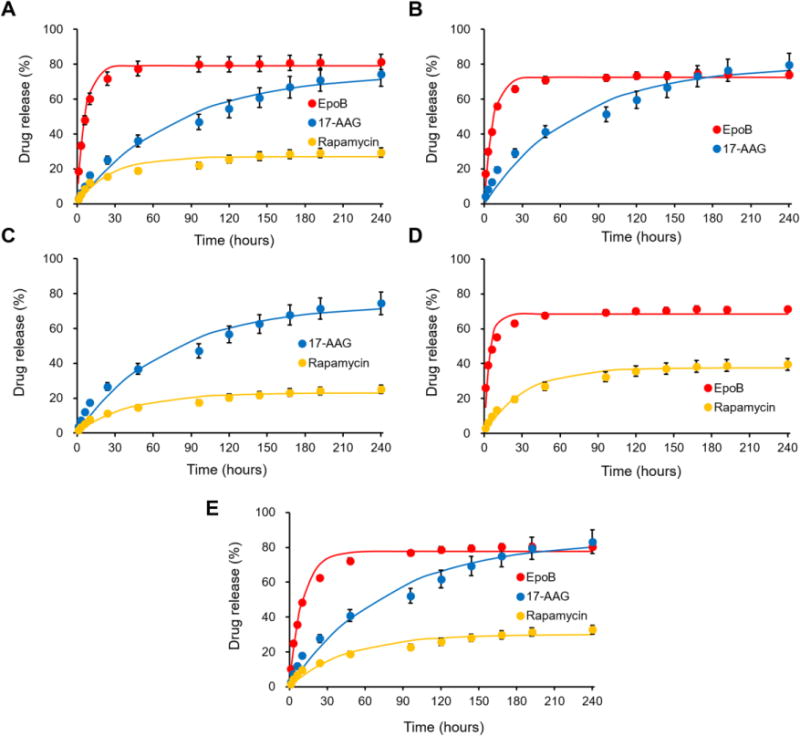
In vitro drug release profiles of various Regel® formulations containing a single drug (A: g-E, g-A, and g-R), 2-drug combinations (B: g-EA, C: g-AR, and D: g-ER) and 3-drug combination (E: g-EAR) (n=3).
EpoB was more potent than paclitaxel for ES-2-luc cells with IC50 values of 0.427 and 3.10 nM, respectively. EpoB has been shown to be more potent than paclitaxel against several human cancer cell lines. For example, Bollag et al. showed greater cytotoxic activity of EpoB over paclitaxel against P-glycoprotein-expressing multi-drug resistant KBV-1 cells [19]. Individually, 17-AAG and rapamycin were less cytotoxic than EpoB with IC50 values of 98.9 and 27.4 nM, respectively. The relatively high value for 17-AAG was somewhat surprising given that the common B-RAF p.V600E mutation in present in ES-2-luc cells and that B-RAF p.V600E mutants rely on Hsp90 for stability [20, 21]. However, EpoB and 17-AAG (0.4:3.0) was potent, having an IC50 value 180-fold lower than 17-AAG. Inhibition of mTOR by rapamycin did not effectively inhibit growth of ES-2-luc cells even at high levels. Based on CI analysis, 2- and 3-drug combinations of EpoB, 17-AAG, and rapamycin were mainly additive or synergistic, i.e. CI < 1 or CI = 1 across of a range of fraction affected of ES-2-luc cells (35–65%) (Table 2). At a 0.4:3.0:1.5 ratio, the IC50 and IC65 values of the 3-drug combination of EpoB, 17-AAG, and rapamycin were 3.00 and 10.9 nM, respectively.
Table 2.
In vitro inhibitory concentration values that inhibits cell growth by x% (ICx) and combination index (CIx) analysis of various drug combination on ES-2-luc cells (n=3).
| Drug combination (weight ratio) | Paclitaxel | EpoB | 17-AAG | Rapamycin | EpoB/17 AAG (0.4:3) | 17-AAG/Rapamycin (3:1.5) Q | EpoB/Rapamycin (0.4:1.5) | EpoB/17-AAG/Rapamycin (0.4:3:1.5) |
|---|---|---|---|---|---|---|---|---|
| IC35 (nM) | 0.475±0.062 | 0.164±0.028 | 5.19±0.47 | 1.08±0.14 | 0.0806±0.0121 | 1.63±0.20 | 0.218±0.024 | 0.825±0.165 |
| IC50 (nM) | 3.10±0.53 | 0.427±0.047 | 98.9±12.9 | 27.4±1.4 | 0.538±0.048 | 30.5±4.0 | 1.39±0.21 | 3.00±0.69 |
| IC65 (nM) | 20.2±3.0 | 1.11±0.22 | 1880±43 0 | 698±91 | 3.59±1.04 | 570±108 | 8.82±0.79 | 10.9±2.2 |
| CI35 | – | – | – | – | 0.0790 | 0.605 | 0.569 | 0.798 |
| CI50 | – | – | – | – | 0.173 | 0.503 | 1.09 | 0.777 |
| CI65 | – | – | – | – | 0.434 | 0.427 | 2.61 | 1.03 |
In the longer-term clonogenic survival assay, EpoB and 17-AAG slightly inhibited colony formation of ES-2-luc cells (25% and 30% inhibition, respectively), whereas rapamycin inhibited 61% of colony formation compared to PBS (control), indicating that long-term exposure of rapamycin has marked effects on clonogenicity of ES-2-luc cells (Figure 3). Similarly, the 2-drug combination of 17-AAG and rapamycin was surprisingly effective in inhibiting colony formation of ES-2-luc cells, indicating that long-term exposure is required for these 2 signal transduction inhibitors. Lastly, the 3-drug combination was the most effective inhibiting colony formation of ES-2-luc cells with ca. 7% remaining colonies compared to the PBS (control). In summary, the 3-drug combination of EpoB, 17-AAG, and rapamycin has potent in vitro anticancer activity in cell short-term proliferation and long-term clonogenic survival assays against ES-2-luc cells.
Fig. 3.
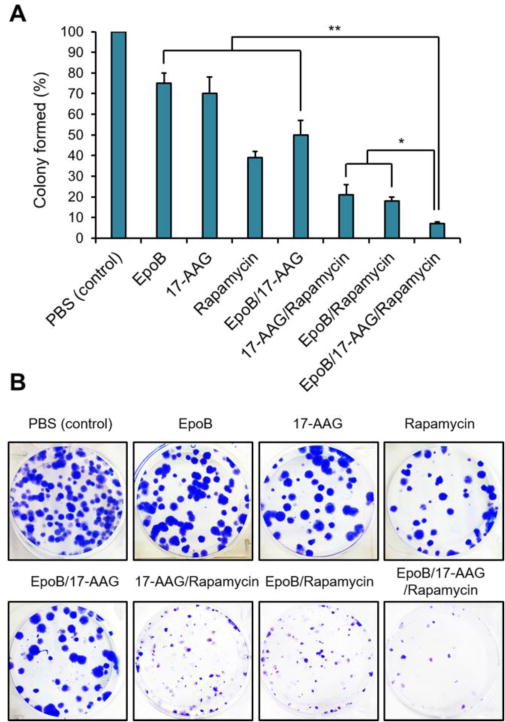
(A) In vitro clonogenic assay for ES-2-luc cells. EpoB, 17-AAG, and rapamycin at 0.4, 2.6, and 0.83 nM (same as weight ratio of 0.4:3.0:1.5), respectively (B) Representative photographs of colonies in each treated well.
A sol of Regel® containing EpoB, 17-AAG, or rapamycin was safely injected i.p. in athymic nude mice and transformed into a purple gel, distributed into spaces between internal organs (Figure 4A). Due to gravity, diaphragmatic pressure, and organ mobility, g-EAR appeared to split into smaller pieces in the peritoneal cavity over time. Over severaltime points (hours and days), athymic nude mice were sacrificed and g-EAR was collected to measure drug release in vivo (Figure 4B). Consistent with in vitro results, the release rate of EpoB from g-EAR was fastest, followed by 17-AAG and rapamycin. At 5 hours post i.p. injection of g-EAR, EpoB was not detected in g-EAR, whereas 25 and 37% of 17-AAG and rapamycin, respectively were detected in g-EAR. After 9 hours, less than 10% of the drugs remained in the collectable g-EAR, clearly showing that in vivo drug release is faster than in vitro drug release of g-EAR. Thus, fragmentation of g-EAR in the i.p. cavity due to mechanical forces and perhaps flow of peritoneal fluid, 100–250 μL [22], contributed to faster drug release in vivo, particularly for 17-AAG and rapamycin. It is known that the drug and/or protein release from both ABA and BAB copolymers occurs by two principal mechanisms: (i) drug diffusion from the hydrogel during the initial release phase; and (ii) release of drug by the erosion of the hydrogel matrix during the later phase [17]. It has been proposed that PLGA degrades primarily through hydrolytic degradation but it has also been suggested that enzymatic degradation may play a role in the process [23]. However, due to a lack of uniformity in in vivo tests, there is difficulty in comparing and demonstrating the choice of proposed enzymes and their contribution in the degradation process [24, 25]. Lastly, this study was done with normal mice without ascites, i.e. accumulation of fluid in the peritoneal cavity due to ovarian cancer, which will likely enhance drug release from g-EAR [26]. In an identical setting of experiment, m-EAR rapidly disappeared within 2 hours post i.p. injection (Fig. 4C)
Fig. 4.
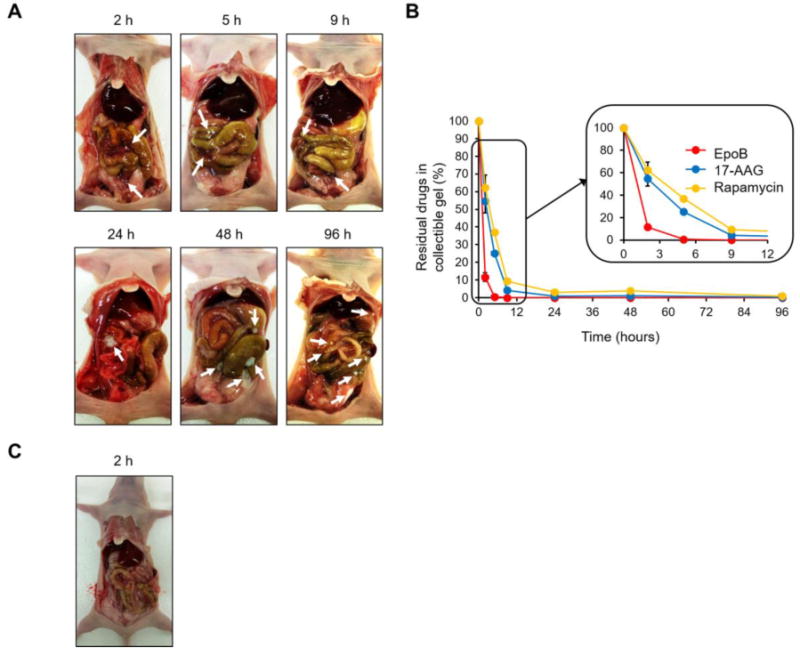
In vivo drug release (B) and appearance (A) of collected g-EAR after i.p. injection (n=5). (C) Detection of m-EAR in the peritoneum. The white arrows indicate gel form of g-EAR distributed into spaces between internal organs.
In an ES-2-luc ovarian cancer xenograft model, mice dosed with a single i.p. injection of g-EAR at 2.0, 15.0, and 7.5 mg/kg for EpoB, 17-AAG, and rapamycin, respectively, showed only a slight increase in bioluminescence signal from ES-2-luc cells that was apparent at day 21 and 28 (Figure 5A and 5B). By contrast, bioluminescence from ES-2-luc cells in the PBS control increased by ca. 5000-fold, over the first 14 days, accompanied by increased girth of the mice to due to the accumulation of ascites. On day 28, bioluminescence signal from g-E mice was 2.3-fold higher than g-EAR. In terms of survival, the median survival times of g-EAR, g-E, and PBS control mice were 58, 33, and 22 days, respectively, consistent with the longitudinal bioluminescence imaging study (Fig. 5B).
Fig. 5.
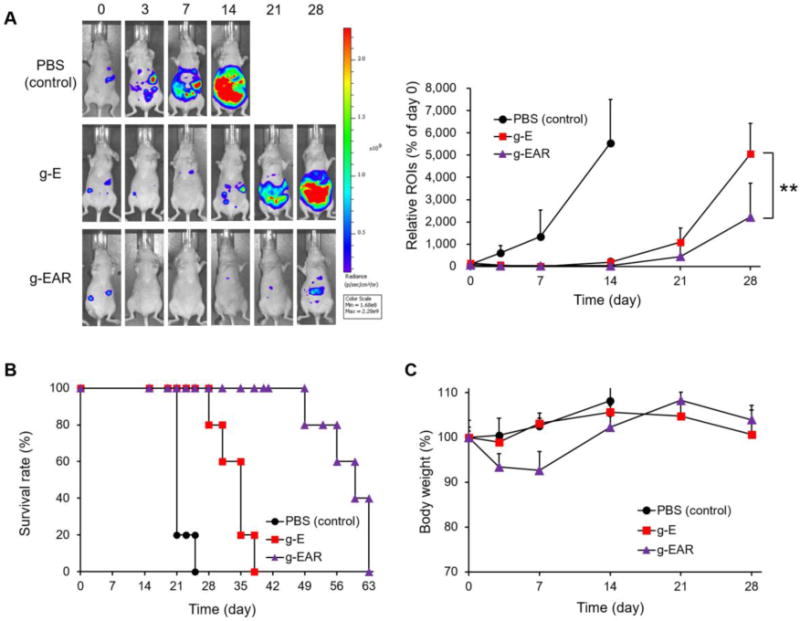
Antitumor efficacy of i.p. g-E and i.p. g-EAR in an ES-2-luc ovarian cancer xenograft model (A). Survival rate (B) and body weight change (C) were monitored periodically.
To verify enhanced antitumor efficacy of g-EAR in detail, separate antitumor efficacy experiment with various formulations was additionally performed. As seen in Fig. 6A and 6B, bioluminescence from ES-2-luc cells in the PBS control increased by ca. 2000-fold, over the first 21 days. On day 21, bioluminescence signals from g-E (2.0 mg/kg), g-EA (2.0 and 15.0 mg/kg), and g-ER (2.0 and 7.5 mg/kg) mice were significantly (p value<0.05) higher than g-EAR (2.0, 15.0, and 7.5 mg/kg) consistent with the higher anticancer activity with the 3-drug combination, validating mTOR and Hsp90 as targets in ovarian cancer. The single i.p. injection of m-EAR (2.0, 15.0, and 7.5 mg/kg) was less effective than g-EAR (2.0, 15.0, and 7.5 mg/kg), presumably due to the rapid elimination of drugs, which was shown in Fig. 4C. The bioluminescence intensity of g-EAR group in different drug ratio (2.0, 7.5, and 3.75 mg/kg) seemed to be slightly higher than that of g-EAR (2.0, 15.0, and 7.5 mg/kg), however, no significant difference was observed between them. Lastly, a single i.p. injection of micelles and various gels caused < 10% loss in body weight compared to PBS control group, suggesting the low toxicity of g-EAR while gaining potent antitumor efficacy (Figure 5C and 6C).
Fig. 6.
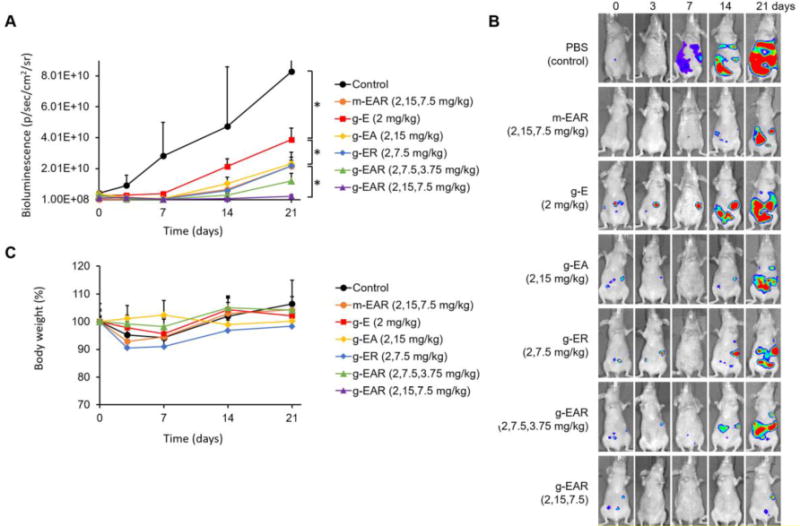
Antitumor efficacy of single i.p. injection of m-EAR or various gels in an ES-2-luc ovarian cancer xenograft model (A and B). Body weight change (C) were monitored periodically (n=3).
4. Conclusions
For ovarian cancer patients, the goal of i.p. chemotherapy is to treat residual disease after cytoreductive surgery. As an alternative to HIPEC, Regel® may serve as a controlled release technology for pre-clinical and clinical testing of chemotherapy and molecularly-targeted agents via the i.p. route, coupling concurrent drug exposure and controlled release. Besides taxanes like paclitaxel, Regel® also has a capacity for other poorly water-soluble anticancer agents, e.g. EpoB, 17-AAG, and rapamycin, singly and as synergistic drug combinations. Regel® as a sol containing EpoB, 17-AAG, or rapamycin was safely injected i.p. and transformed into a purple gel (17-AAG) in athymic nude mice. Release of EpoB, 17-AAG, and rapamycin from Regel® after i.p. injection was faster in vivo than in vitro, presumably due to loss of Regel® integrity in the peritoneal cavity, resulting in release over ca. 12 hours in the rank order of EpoB>17-AAG>rapamycin. In an ES-2-luc ovarian tumor model, a single i.p. injection of g-EAR had greater efficacy than g-E against ES-2-luc metastasis based on whole-body bioluminescence imaging and survival. While EpoB may have higher potency than taxanes such as paclitaxel, further research with Regel®, e.g. disposition kinetics, toxicology, are needed, perhaps in other tumor xenograft models to make meaningful conclusions. Given that Regel® plus paclitaxel has been safely evaluated in humans in a phase 2b clinical trial for esophageal cancer, drug combinations including microtubule stabilizing agents, Hsp90 inhibitor, and mTOR inhibitor can be safely tested using i.p. Regel®, aiming for clinical trials for ovarian cancer patients.
Supplementary Material
Acknowledgments
This work was supported in part by the National Institutes of Health [R01 AI101157].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The author(s) confirmed that this article content has no conflict of interest.
References
- 1.Menon U, Griffin M, Gentry-Maharaj A. Ovarian cancer screening–current status, future directions. Gynecol Oncol. 2014;132:490–495. doi: 10.1016/j.ygyno.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.du Bois A, Quinn M, Thigpen T, Vermorken J, Avall-Lundqvist E, Bookman M, Bowtell D, Brady M, Casado A, Cervantes A, Eisenhauer E, Friedlaender M, Fujiwara K, Grenman S, Guastalla JP, Harper P, Hogberg T, Kaye S, Kitchener H, Kristensen G, Mannel R, Meier W, Miller B, Neijt JP, Oza A, Ozols R, Parmar M, Pecorelli S, Pfisterer J, Poveda A, Provencher D, Pujade-Lauraine E, Randall M, Rochon J, Rustin G, Sagae S, Stehman F, Stuart G, Trimble E, Vasey P, Vergote I, Verheijen R, Wagner U. 2004 consensus statements on the management of ovarian cancer: final document of the 3rd International Gynecologic Cancer Intergroup Ovarian Cancer Consensus Conference (GCIG OCCC 2004) Ann Oncol. 2005;16(Suppl 8):viii7–viii12. doi: 10.1093/annonc/mdi961. [DOI] [PubMed] [Google Scholar]
- 3.Hasovits C, Clarke S. Pharmacokinetics and pharmacodynamics of intraperitoneal cancer chemotherapeutics. Clin Pharmacokinet. 2012;51:203–224. doi: 10.2165/11598890-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.Chambers SK, Chow HH, Janicek MF, Cragun JM, Hatch KD, Cui H, Laughren C, Clouser MC, Cohen JL, Wright HM, Abu Shahin N, Alberts DS. Phase I trial of intraperitoneal pemetrexed, cisplatin, and paclitaxel in optimally debulked ovarian cancer. Clin Cancer Res. 2012;18:2668–2678. doi: 10.1158/1078-0432.CCR-12-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Helm CW. The role of hyperthermic intraperitoneal chemotherapy (HIPEC) in ovarian cancer. Oncologist. 2009;14:683–694. doi: 10.1634/theoncologist.2008-0275. [DOI] [PubMed] [Google Scholar]
- 6.Vassileva V, Grant J, De Souza R, Allen C, Piquette-Miller M. Novel biocompatible intraperitoneal drug delivery system increases tolerability and therapeutic efficacy of paclitaxel in a human ovarian cancer xenograft model. Cancer Chemother Pharmacol. 2007;60:907–914. doi: 10.1007/s00280-007-0449-0. [DOI] [PubMed] [Google Scholar]
- 7.Bajaj G, Yeo Y. Drug delivery systems for intraperitoneal therapy. Pharm Res. 2010;27:735–738. doi: 10.1007/s11095-009-0031-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Papa A, Caruso D, Strudel M, Tomao S, Tomao F. Update on Poly-ADP-ribose polymerase inhibition for ovarian cancer treatment. J Transl Med. 2016;14:267. doi: 10.1186/s12967-016-1027-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu H, Xiao F, Serebriiskii IG, O’Brien SW, Maglaty MA, Astsaturov I, Litwin S, Martin LP, Proia DA, Golemis EA, Connolly DC. Network analysis identifies an HSP90-central hub susceptible in ovarian cancer. Clin Cancer Res. 2013;19:5053–5067. doi: 10.1158/1078-0432.CCR-13-1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andorfer P, Heuwieser A, Heinzel A, Lukas A, Mayer B, Perco P. Vascular endothelial growth factor A as predictive marker for mTOR inhibition in relapsing high-grade serous ovarian cancer. BMC Syst Biol. 2016;10:33. doi: 10.1186/s12918-016-0278-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho H, Gao J, Kwon GS. PEG-b-PLA micelles and PLGA-b-PEG-b-PLGA sol-gels for drug delivery. J Control Release. 2016;240:191–201. doi: 10.1016/j.jconrel.2015.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cho H, Kwon GS. Thermosensitive poly-(d,l-lactide-co-glycolide)-block-poly(ethylene glycol)-block-poly-(d,l-lactide-co-glycolide) hydrogels for multi-drug delivery. J Drug Target. 2014;22:669–677. doi: 10.3109/1061186X.2014.931406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 14.Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1:2315–2319. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- 15.Han NK, Shin DH, Kim JS, Weon KY, Jang CY, Kim JS. Hyaluronan-conjugated liposomes encapsulating gemcitabine for breast cancer stem cells. Int J Nanomedicine. 2016;11:1413–1425. doi: 10.2147/IJN.S95850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McKenzie M, Betts D, Suh A, Bui K, Tang R, Liang K, Achilefu S, Kwon GS, Cho H. Proof-of-Concept of Polymeric Sol-Gels in Multi-Drug Delivery and Intraoperative Image-Guided Surgery for Peritoneal Ovarian Cancer. Pharm Res. 2016;33:2298–2306. doi: 10.1007/s11095-016-1968-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoare TR, Kohane DS. Hydrogels in drug delivery: Progress and challenges. Polymer. 2008;49:1993–2007. [Google Scholar]
- 18.Tam YT, Gao J, Kwon GS. Oligo(lactic acid)n-Paclitaxel Prodrugs for Poly(ethylene glycol)-block-poly(lactic acid) Micelles: Loading, Release, and Backbiting Conversion for Anticancer Activity. J Am Chem Soc. 2016;138:8674–8677. doi: 10.1021/jacs.6b03995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bollag DM, McQueney PA, Zhu J, Hensens O, Koupal L, Liesch J, Goetz M, Lazarides E, Woods CM. Epothilones, a new class of microtubule-stabilizing agents with a taxol-like mechanism of action. Cancer Res. 1995;55:2325–2333. [PubMed] [Google Scholar]
- 20.Estep AL, Palmer C, McCormick F, Rauen KA. Mutation analysis of BRAF, MEK1 and MEK2 in 15 ovarian cancer cell lines: implications for therapy. PLoS One. 2007;2:e1279. doi: 10.1371/journal.pone.0001279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Acquaviva J, Smith DL, Jimenez JP, Zhang C, Sequeira M, He S, Sang J, Bates RC, Proia DA. Overcoming acquired BRAF inhibitor resistance in melanoma via targeted inhibition of Hsp90 with ganetespib. Mol Cancer Ther. 2014;13:353–363. doi: 10.1158/1535-7163.MCT-13-0481. [DOI] [PubMed] [Google Scholar]
- 22.Hartveit F, Thunold S. Peritoneal fluid volume and the oestrus cycle in mice. Nature. 1966;210:1123–1125. doi: 10.1038/2101123a0. [DOI] [PubMed] [Google Scholar]
- 23.Makadia HK, Siegel SJ. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers (Basel) 2011;3:1377–1397. doi: 10.3390/polym3031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cai Q, Shi G, Bei J, Wang S. Enzymatic degradation behavior and mechanism of poly(lactide-co-glycolide) foams by trypsin. Biomaterials. 2003;24:629–638. doi: 10.1016/s0142-9612(02)00377-0. [DOI] [PubMed] [Google Scholar]
- 25.Li S, Girard A. Enzymatic degradation of PLA stereocopolymers with predominant D-lactyl contents. Polymer Degradation and Stability. 2001;71:61–67. [Google Scholar]
- 26.Ahmed N, Stenvers KL. Getting to know ovarian cancer ascites: opportunities for targeted therapy-based translational research. Front Oncol. 2013;3:256. doi: 10.3389/fonc.2013.00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.