

Abstract
Successful drug delivery and overcoming drug resistance are the primary clinical challenges for management and treatment of cancer. The ability to rapidly screen drugs and delivery systems within physiologically relevant environments is critically important; yet is currently limited due to lack of appropriate tumor models. To address this problem, we developed the Tumor-microenvironment-on-chip (T-MOC), a new microfluidic tumor model simulating the interstitial flow, plasma clearance, and transport of the drug within the tumor. We demonstrated T-MOC’s capabilities by assessing the delivery and efficacy of doxorubicin in small molecular form versus hyaluronic acid nanoparticle (NP) formulation in MCF-7 and MDA-MB-231, two cell lines representative of different molecular subtypes of breast cancer. Doxorubicin accumulated and penetrated similarly in both cell lines while the NP accumulated more in MDA-MB-231 than MCF-7 potentially due to binding of hyaluronic acid to CD44 expressed by MDA-MB-231. However, the penetration of the NP was less than the molecular drug due to its larger size. In addition, both cell lines cultured on the T-MOC showed increased resistance to the drug compared to 2D culture where MDA-MB-231 attained a drug-resistant tumor-initiating phenotype indicated by increased CD44 expression. When grown in immunocompromised mice, both cell lines exhibited cell-type-dependent resistance and phenotypic changes similar to T-MOC, confirming its predictive ability for in vivo drug response. This initial characterization of T-MOC indicates its transformative potential for in vitro testing of drug efficacy towards prediction of in vivo outcomes and investigation of drug resistance mechanisms for advancement of personalized medicine.
Keywords: Doxorubicin, breast cancer, cancer stem cell, drug transport, tumor-microenvironment-on-chip, chemoresistance
Graphical abstract
Introduction
Achieving effective delivery of therapeutics to the target tumor tissue and overcoming the drug resistance exhibited by cancer cells are the significant challenges of cancer management and treatment. The recent years have witnessed significant efforts in development of novel cancer nanomedicine aimed for improved delivery by fine-tuning their physiochemical properties such as size, shape, and surface functionalization [1, 2]. As a result, various drug delivery systems, including nanoparticle (NP) formulations, have been proposed and developed [3–6]. While these novel formulations have shown promising outcomes in preclinical studies; their clinical benefits have often found to be diminished due to multi-faceted mechanisms of drug transport and resistance present at the tumor microenvironment (TME).
TME poses structural and pathophysiological barriers such as dense extra cellular matrix (ECM) microstructure[7, 8], high cell packing density [9, 10], and an elevated interstitial fluid pressure (IFP) [11, 12]. These barriers hinder transport of drugs and drug delivery vehicles at various stages of their in vivo journey, including blood circulation, transvascular transport (i.e., extravasation), interstitial transport and cellular uptake [3, 5, 6]. In addition to the hostile and adverse conditions within the TME affecting drug delivery, many cancers may acquire multidrug resistance (MDR) by a group of membrane proteins that exclude cytotoxic molecules. These proteins belong to the ATP binding cassette superfamily of membrane transporters [13]. Other molecular mechanisms are also reported to cause the MDR by alteration of survival/apoptosis pathways, and increased DNA damage repair. For instance, eukaryotic translation initiation factor 3 subunit A (eIF3a) expression has been associated with poor clinical response to DNA-damaging drugs [14] and its reduced expression was shown to contribute to cisplatin and doxorubicin resistance possibly by inhibiting expression of DNA repair proteins and, thus, DNA repair activities [14]. Increased expression of 14–3-3σ protein has also been found in doxorubicin-selected and resistant cancer cells and was shown to contribute to doxorubicin resistance [2, 15, 16]. In addition to the over-expression of the MDR-associated proteins, cellular drug resistance also appears to be mediated by integrin signaling associated with adhesion of cancer cells to the ECM [17–19].
It is further complicated by the presence of tumor heterogeneity with different subtypes of cancer cells showing highly variable drug response [20]. For instance, multiple subtypes of breast cancer have been identified based on the expression of estrogen receptor, progesterone receptor, and human epidermal growth factor receptor-2 [20, 21]. Each subtype has a different prognosis and response to chemotherapy, and several subtypes including luminal B, basal, claudin-low, and HER2-enriched subtypes show variable or poor response to chemotherapy [22–25]. In order to develop personalized treatment strategies, thus, drug response of breast cancers should be evaluated as a compounding consequence of all these mechanisms.
Currently available tumor models show significant limitations in screening drugs for efficacy and chemoresistance in a way predictive of their in vivo performance. This is mainly due to the complex drug transport and resistance mechanisms above that are present in the host environment but are not typically realized in vitro cell culture settings. Although conventional in vitro cell cultures are valuable tools for cancer research, their two-dimensional (2D) culture do not adequately represent the conditions present in in vivo three-dimensional (3D) TME. Therefore, the outcome of traditional culture of cancer cells often fails to be indicative of in vivo or clinical outcomes. Furthermore, while 3D culturing systems have demonstrated a step forward in applicability from 2D [26], few 3D in vitro culture systems have been empirically shown to approximate the in vivo TME for accurate drug response, and many of these are unique to individual laboratories with limited availability. A growing number of studies reported that the physical, chemical and mechanical microenvironment of cancer cells significantly affects their behaviors [27–30]. Multiple breast cancer cell lines have the same morphology when cultured in 2D, but show different morphologies and organized structures when cultured in a 3D matrix [29, 30]. This difference depends on their subtypes [28]. Different drug sensitivity in 2D versus 3D culture is also reported [2]. Although small animal models can provide a more realistic tumor microenvironment, these only provide end results. It is not suitable to rapidly screen various chemotherapeutic agents with respect to heterogeneous subtypes of breast cancers. Thus, new tumor models are required to be capable of rapidly screening the efficacy of various drugs and drug delivery systems in the context of the in vivo tumor microenvironment where complex interactions occur among cells, ECM and interstitial fluid.
In order to address this critical gap, we developed a new 3D microfluidic in vitro tumor model, named tumor-microenvironment-on-chip (T-MOC), capable of simulating the complex transport at the in vivo TME and study cell-type-specific drug resistance of cancerous cells. The T-MOC is based on a microfluidic platform culturing cancer cells in 3D tissue architecture mimicking complex transport around tumors [31]. Specifically, the T-MOC is designed to mimic pharmacokinetics, extravasation, interstitial transport and cellular uptake of drugs and drug delivery systems. We previously utilized the T-MOC platform to investigate the transport of polymeric nanoparticles under different microenvironmental conditions by varying parameters such as cut-off pore size, interstitial fluid pressure, and tumor tissue microstructure [31]. In addition, we demonstrated the combination of T-MOC with preexisting in vitro cultures such as externally cultured multi-cellular spheroids that were transferred into the microfluidic device to simulate a dynamic in vivo microenvironment during drug testing [32]. We also investigated cell-type dependent drug uptake and efflux across the cellular membrane in the T-MOC platform [33].
In the present study, we used the T-MOC platform to characterize the transport and efficacy of doxorubicin (Dox) and dox-encapsulated hyaluronic acid nanoparticles (Dox-HANPs), in order to demonstrate its feasibility to rapidly characterize and predict drug transport, action and chemoresistance of breast cancers. Two human breast cancer cell lines (MDA-MB-231 and MCF-7) were introduced to the T-MOC platform, and were cultured in type I collagen matrices under an elevated IFP condition. These cell lines were selected to represent different molecular subtypes of breast cancers. MCF-7 is characterized as a member of luminal A subtype that expresses estrogen receptor and is typically responsive to chemotherapy and hormone therapy. On the other hand, MDA-MB-231 is a triple-negative breast cancer line identified by low expression of estrogen, progesterone and human epidermal growth factor 2 receptors as well as claudin-family tight junction proteins, exhibits markers associated with epithelial-mesenchymal transition and cancer stem cell and shows relatively poor response to chemotherapy [22–25]. Dox and Dox-HANPs were administered to T-MOCs in a way to simulate in vivo pharmacokinetics, specifically temporal clearance of drugs from plasma. During and after the administration, the transport and subsequent cellular response were characterized and compared with conventional 2D culture assay. The results were further validated against xenograft tumor models.
Materials and Methods
Cells and reagents
Human breast cancer cell lines (MCF-7 and MDA-MB-231) were cultured following standard practices for adherent cell culture. Culture medium (DMEM/F12, Invitrogen, NY) was supplemented by 2 mM L-glutamine, 100 μg/mL penicillin/streptomycin, 5% v/v fetal bovine serum for MCF-7 and 10% v/v fetal bovine serum for MDA-MB-231. Cells were routinely harvested at 80% confluence using 0.05% trypsin and 0.53 mM EDTA, used for experiments or sub-cultured accordingly. Cells were cultured up to 20 passages before being cryopreserved or discarded. Care was taken to conduct T-MOC and 2D culture experiments in parallel to control for potential phenotypic variations during the culture period. For experiments involving 2D culture, cells were plated in a 24 well plate at an initial density of 4000 cells/well and cultured up to 5 days. For 3D culture on T-MOC device, cells were seeded in a type-1 rat tail collagen (BD Bioscience) matrix. For this purpose, first a neutralized collagen solution with 6 mg/mL collagen concentration was prepared as described previously [34] and cells were suspended in the collagen solution at 2 × 107 cells/mL concentration. Next, approximately 20 μL of cell-laden collagen solution was loaded in the T-MOC interstitial channel by surface-tension bounded flow initiated by applying suction pressure at one of device ports. Devices loaded with cell-collagen mixture were placed in a CO2 incubator and maintained at 37 °C for an hour for polymerization of collagen matrix. Devices were perfused with culture media for 2 days before drug testing.
Design and fabrication of T-MOC
The T-MOC has a 3D structure formed by stacking two layers of microchannels with a porous membrane sandwiched between the layers as shown in Figure 1A. The top layer has a 300 μm wide channel that is 100 μm in thickness, which simulates the capillary of the tumor vasculature. Culture medium flows along the capillary channel at a physiologically relevant velocity and pressure. The porous membrane has nominal pore size of 400 nm, which is at the lower end of the range of vascular pore sizes i.e 0.4 – 2.0 μm observed in fenestrated and leaky tumor vasculatures [35]. The bottom layer is 100 μm in thickness and includes an interstitial channel with a 1 mm by 2.7 mm central chamber for tumor cells and ECM as well as two 300 μm wide side channels simulating passive drainage to lymphatics (Figure 1B). The lymphatic channels are separated from the interstitial channel by equally spaced posts approximately 50 μm apart from each other. The hydrostatic pressure in each compartment is maintained by connection of elevated culture medium reservoirs to individual channel inlet and outlets where the pressure levels are adjusted by changing the height of the reservoirs with respect to the base of the device. During the drug treatment, 20 mmHg was applied at capillary and interstitial channels while 5 mmHg was applied at the lymphatic channels, mimicking the fluid pressure levels observed in breast tumors [12]. We mainly considered steady state flow and passive drainage within capillary and lymphatic channels that were driven by hydrostatic pressure gradients. Pulsatile vascular and lymph flows generated by smooth and skeletal muscle contractions were not considered. The T-MOC was fabricated via a two-step photolithography technique. Briefly, SU-8 photoresist was spin-coated on silicon dioxide (SiO2) wafers, then baked. The wafer was then exposed to UV light through masks with channel configurations of top and bottom layers of the T-MOC. After the post-baking procedure, the wafers were immersed into SU-8 developer solution and rinsed with isopropyl alcohol. Then, a mixture of polydimethylsiloxane (PDMS) and a curing agent were poured on the resulting silicon wafer molds, degased and baked. After polymerization, the PDMS layers were peeled off from the mold, cleaned, and punched through to generate the inlet and outlet ports. A polycarbonate membrane (Whatman Cyclopore, GE Healthcare Life Sciences, PA) was treated with 3-aminopropyltriethoxysilane (APTES) solution, dried, and bonded to the top and then the bottom PDMS layer using oxygen plasma treatment. The devices were perfused with distilled water for several hours to remove excess APTES, dried on a hot plate, and sterilized using ozone prior to use in experiments.
Figure 1. Design and fabrication of T-MOC to simulate the drug transport at the TME.
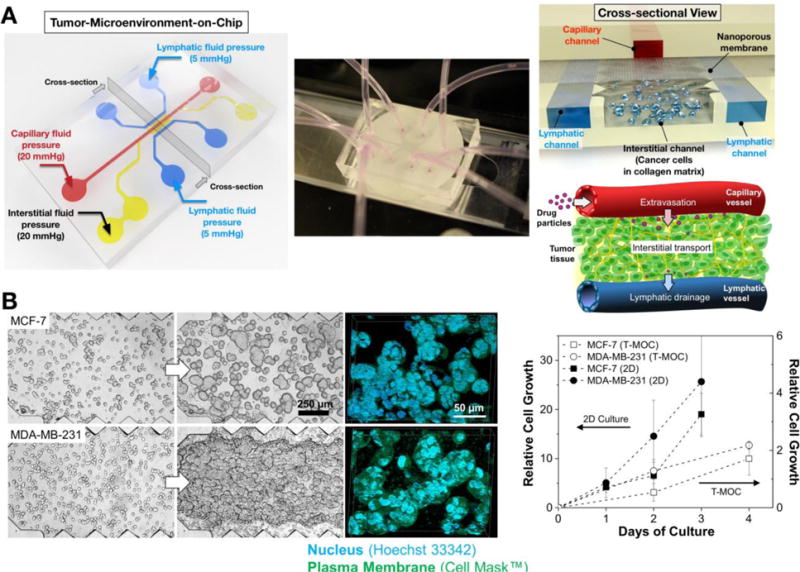
(A) Schematic of the fabricated T-MOC platform and its operating pressure conditions. Detailed 3D configuration of the device is illustrated in cross-sectional view – top layer with capillary channel, nanoporous membrane, and bottom layer with interstitial and lymphatic channels. This design is to mimic a pair of capillary-lymphatic vessels with tumor tissues. (B) 3D morphology of breast cancer cells grown on the T-MOC: MCF-7 and MDA-MB-231. Comparison of growth rate of MCF-7 and MDA-MB-231 under 2D culture and 3D T-MOC culture configurations.
Quantitative time-lapse microscopy of drug transport and action
Doxorubicin was considered as a model drug and used as either free molecular drug (Dox) or encapsulated in 250 nm hyaluronic acid nanoparticles (Dox-HANP). For free molecular drug, doxorubicin-HCI was purchased in powder (Sigma Aldrich) and prepared into 2 mM concentrated stock in DMSO. Dox-HANP was synthesized as described previously [9]. Briefly, Dox-loaded HANPs were prepared by a simple dialysis method. HA-NPs were dispersed in distilled water. The Dox in DMSO was slowly added into the HANP solution under sonication. The resulting mixture was dialyzed for one day against an excess amount of distilled water to remove unloaded drugs and organic solvent. Dox-HANPs were then resuspended in physiological buffer at 200 μM Dox-equivalent concentration. Both solutions were diluted to 2μM in supplemented culture medium and used immediately in experiments. Drug solution was perfused through the capillary channel for a prescribed duration depending whether slow or rapid plasma clearance is simulated. In the meantime, fluorescence images (Ex: 547 nm, Em: 572 nm) were acquired every 10 minutes using an inverted microscope (Olympus IX71, Japan). At the end of the treatment time, drug containing culture medium was replaced by drug free culture medium and devices were incubated for an additional 24 hours before viability assessment. Drug tests in 2D culture were carried out in 24 well plates where cells were exposed to drug containing culture medium for prescribed amount of time, replaced with drug free medium and further cultured up to 3 days before viability assessment.
Calculation of drug transport metrics
Intrinsic fluorescence of doxorubicin enabled real time observation of drug transport within T-MOC including quantification of drug concentration, as well as calculation of various metrics that describe its overall accumulation and spatial distribution at a given time. First, time-lapse images were registered and corrected for non-specific background fluorescence. The image pixels were then segmented into non-overlapping foreground and background regions using Trainable Weka Segmentation plugin in ImageJ [36, 37]. Foreground pixels, denoted by Rcell, contained cells, while background pixels, denoted by RECM, contained ECM only. In addition, fluorescence intensity was converted into concentration based on calibration curves determined from experiments conducted under similar imaging conditions.
We used MATLAB® to calculate the following metrics based on the image segmentation and concentration measured as a function of pixel coordinates and time, C(xi, yj, t). First of all, drug cell and ECM accumulation profiles, denoted by Ak(t), represent the mean temporal variation of concentration within respective regions and were calculated by averaging the concentration at each time point as follows:
| (0) |
where i and j are the pixel indices across x- and y-directions, i.e. the directions parallel and perpendicular to the capillary channel, respectively. t represents time.
Drug penetration profile, P(y, t)represents the spatiotemporal distribution of concentration across the interstitial space in the direction perpendicular to the capillary channel. It was calculated as follows:
| (0) |
Finally, an area under the curve (AUC) metric was calculated by integration of drug accumulation profile over the treatment duration:
| (0) |
In particular, we considered AUCcell as a measure of total drug exposure since it accounts for both the duration and concentration at which the cells were exposed to the drug.
Post-treatment viability assay
After treatments with Dox and Dox-HANP, the cells were further cultured in drug-free medium for 24 hours. Then, the cell viability on T-MOC and 2D culture were assessed by a membrane integrity assay based on Hoechst 33342 that stains all cell nuclei, and propidium idodide that stains cell nuclei with compromised membrane. Fluorescence images were acquired, segmented and analyzed for nuclear area coverage by ImageJ. Live cell nuclear area was calculated by subtracting the area stained by propidium iodide from the area stained by Hoechst 33342. Survival fraction as an indicator of cell viability was calculated by normalizing the live cell nuclear area of the drug treatment with respect to that of control without the drug treatment.
Western blot analysis for MDR proteins
MCF7 and MDA-MB-231 cells cultured in 2D and in T-MOC were harvested and lysed in TNN-SDS buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 50 mM NaF, 1 mM sodium orthovanadate, 1 mM DTT, 2 mM PMSF, 0.5% NP-40, and 0.1% SDS) at 4°C for 30 min followed by centrifugation (10,000 g for 10 min at 4°C) to remove cell debris. The supernatants were collected and equal amount of proteins from each preparation were subjected to separation on SDS-PAGE and Western blot analysis probed using eIF3a and 14–3-3σ specific antibodies as previously described [14].
Validation with in vivo animal tumor models
Cell surface analysis was performed to compare the phenotypic changes induced by culturing on the T-MOC platform against that of xenografted tumors. Briefly, after 3 day culture, tumor cells embedded in collagen gels were retrieved from the TMOC platform. Then, the gels were treated with 0.5 mg/ml collagenase (Liberase TM, Roche, Indianapolis, IN) and 50 μg DNase I (Sigma, St. Loius, MO) for 15 min at 37°C with shaking to isolate tumor cells. Collagenase was neutralized with cold culture medium containing 10% calf serum and single cells were isolated. Cells were stained with fluorescently labeled anti-CD24 and anti-CD44 (Biolegend, San Diego CA) antibodies for 30 min at 4°C, washed twice and fixed prior to analysis using flow cytometry (LSR II cytometer, Beckton Dickinson, San Jose, CA). For xenografted tumors, 5×106 cells in log phase growth were implanted on the flank of 6 week old female NSG mice. When tumors reached 200 mm3 doxorubicin or vehicle was administered intravenously at 2 mg/kg two times a week for 3 weeks. Mice were euthanized and tumors excised and minced for digestion with 0.5 mg/ml and 50 μg DNase I and collagenase for 60 min at 37°C with shaking. Collagenase was neutralized with cold culture medium containing 10% calf serum and single cells were isolated. Cells were stained as above.
Statistical Analysis
Experiments were repeated 3 to 6 times for each treatment group. Experimental data are reported as mean ± standard deviation unless otherwise is noted. Differences in treatment means were tested by one-way analysis of variance and pairwise comparisons were performed by Student’s t-test. Differences were considered statistically significant when p-value was less than 0.05.
Results
Design and fabrication of T-MOC to simulate drug transport at the TME
The structure of the T-MOC platform and its operating pressure conditions are presented in Figure 1A. T-MOC is a microfluidic platform with three types of channels - capillary, interstitial and lymphatic channels (noted with red, yellow and blue colors in Figure 1A). These channels are independently pressurized as illustrated to mimic the elevated interstitial fluid pressure at the TME, which has been reported to be in a range between 10 and 140 mmHg with an average of 20 mmHg for breast cancer [11]. These channels are configured in a 3D structure by stacking two PDMS layers of microchannels with a porous membrane sandwiched between the layers (cross-sectional view in Figure 1A). The capillary channel on the top layer mimics a capillary vessel of tumor microvasculature, where drug-suspended medium flows at a physiologically relevant velocity on the order of 1 mm/s [38]. The capillary wall is mimicked by a porous membrane with a pore size of 400 nm, which is within a range of tumor vasculature pore size [39, 40]. The interstitial channel and two lymphatic channels are patterned on the bottom layer. In the interstitial channel, cancer cells grow within a 3D collagen matrix, and the interstitial fluid flows through the matrix and exerts elevated IFP. Since the pressure of the lymphatic channels is lower than that of the interstitial channel, the drug-suspended medium flow through the interstitial channel and eventually drain to these lymphatic channels.
This configuration is relevant to simulate the drug transport processes through tumor tissue between a capillary and a lymphatic vessel. After systemic administration, drugs and NPs are thought to reach the tumor via blood-borne circulation along capillary, extravasation across the capillary wall, interstitial transport through tumor interstitium and cellular uptake by tumor cells. The excess drugs may be further drained to lymphatic vessels. The features of the T-MOC platform are analogous to these key conditions that arise in vivo tumors.
Cellular morphology and growth of both breast cancer cells (MCF-7 and MDA-MB-231) on the T-MOC platform are shown in Figure 1B. Both cell lines proliferate and form 3D tissue-like structures of tightly packed cell aggregates as confirmed by confocal microscopy. The growth rate of MDA-MB-231 was higher than that of MCF-7, but both cell lines proliferated slower in the T-MOC platform than in the 2D culture.
Transport of doxorubicin and doxorubicin-encapsulated hyaluronic acid nanoparticles
One of the primary aims in developing NP formulations for existing molecular drugs is to achieve improved tumor delivery by prolonged blood circulation. Doxorubicin in free form has a considerably limited plasma half-life of approximately 5 minutes [41] which could be prolonged beyond 10 hours by its encapsulation in drug carriers such as polymeric micelles [42]. Here we consider Dox-HANP, a doxorubicin-HA conjugate whose plain [43, 44] and PEGylated [45–47] variants have shown tumor targeting potential due to slow plasma clearance and specific binding of HA with CD44 receptor over-expressed in certain tumor types [44]. As illustrated in Figure 2A, Dox-HANPs were synthesized by loading free Dox to hyaluranic acid-5β-cholanic acid (HA-CA) conjugate to form NPs [5, 9, 48].
Figure 2. Transport of doxorubicin and its nanoparticle formulation in T-MOC.
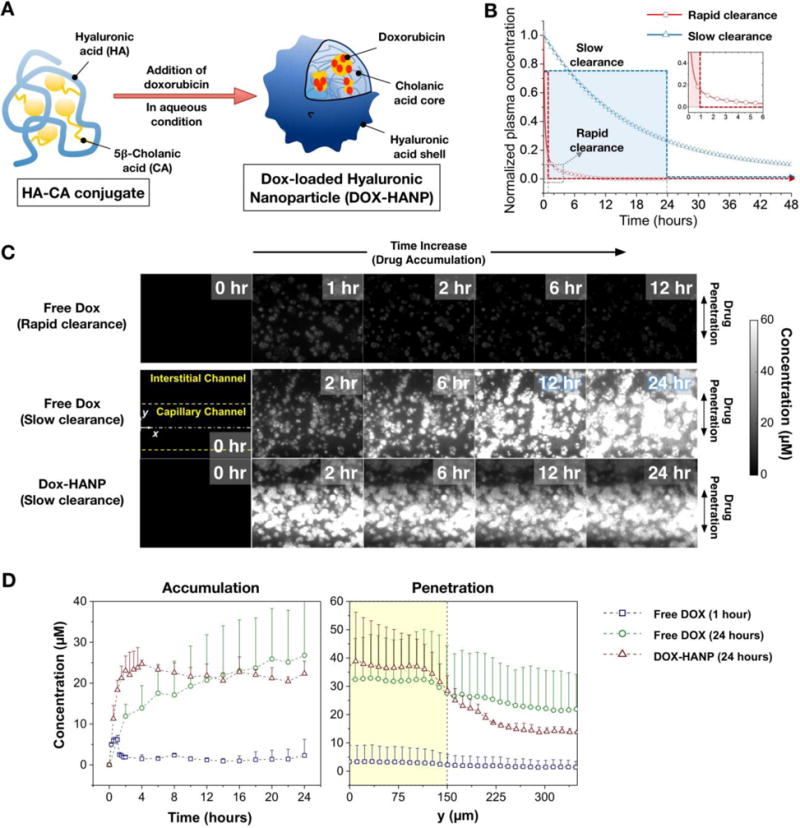
(A) Synthesis of hyaluronic acid based NP delivery system. (B) Pharmacokinetics scenarios for free Dox and Dox-HANP considering slow and rapid plasma clearance. (C) Time-lapse fluorescence micrograph of free Dox and Dox-HANP on the T-MOC. The fluorescence intensity is proportional to the doxorubicin concentration.
In this study, we make a qualitative distinction between the plasma pharmacokinetics of free Dox and Dox-HANP by considering them in generic terms as short and long circulating drugs, realized through rapid clearance (red line) and slow clearance (blue line) treatments, respectively, as illustrated in Figure 2B. In order to approximate plasma clearance in the device in the absence of in vivo clearance mechanisms by mononuclear phagocytic system [49], we employed a one-step on/off perfusion scheme where the drug was perfused through the capillary channel at fixed concentration for a prescribed duration of time, after which it was immediately replaced with drug free culture medium and perfusion was continued, if applicable, for a total of 24 hours. The Dox concentration was 2 μM for both Free Dox and Dox-HANP. The drug was perfused for 1 hour to model rapid clearance, and for 24 hours to model slow clearance. This technique offers a simple-to-implement first approximation to exponentially decaying plasma concentration profiles. A similar perfusion scheme was previously used to investigate the clearance of free Dox and Doxil NP formulation in a microfluidic device [50].
Time-lapse fluorescence microscopy images of Dox and Dox-HANP transport on the T-MOC platform of MCF-7 cells are presented in Figure 2C (Movie S1, Movie S2, and Movie S3 in Supporting Information). In all experimental groups, T-MOC can visualize the accumulation, penetration, and cellular uptake of doxorubicin. These images indicate spatiotemporal changes of doxorubicin concentration by use of pre-calibrated doxorubicin concentration against fluorescence intensity. As for the rapid clearance pharmacokinetics profile of free Dox, fluorescence is weakly observed at 1 hour, which indicates drug accumulation at the interstitial channel. In the following hours, the fluorescence gradually disappears implying the accumulated drug is cleared from the interstitial channel. As for the slow clearance profile, the free Dox slowly accumulates as noted by the increase in fluorescence intensity. During the accumulation, the drug is diffused and distributed throughout the interstitial channel (i.e., penetration). Interestingly, Dox-HANP accumulates much faster than free Dox with the same PK profile. However, the penetration is significantly limited where the fluorescence is mostly observed just underneath the capillary channel.
Comparison of Dox transport in T-MOC between different treatment conditions and the two cell lines is presented in Figure 3. Accumulation and penetration profiles are largely similar for MCF-7 and MDA-MB-231 with the exception of Dox-HANP case (Figure 3A and 3B, respectively). Free Dox with the rapid clearance PK shows a peak of approximately 5 μM between 1 ~ 2 hours, but is eventually cleared from the interstitial channel. For the slow PKs, Dox-HANP quickly accumulates up to 25 to 30 μM within first 4 hour duration. Free Dox accumulates at a slower rate and reach the similar concentrations at 12 ~ 14 hours for MCF-7 case but remains well below the accumulation of Dox-HANP in the MDA-MB-231 case. Drug distribution at 24 hour is also shown as a measure of drug penetration. The amount of drug within the interstitial channel at this time point is almost non-existent in the case of Free Dox with rapid clearance PK, indicated by uniform distribution with levels below 5 μM. In the case of Free Dox with the slow clearance PK, the doxorubicin concentration ranges from 20 to 30 μM across the interstitial channel with slightly elevated drug concentration underneath the capillary and nearly uniform distribution throughout the interstitial channel. For the Dox-HANP, although the peak concentration is slightly higher at 40 to 50 μM, the drug concentration rapidly decreases as the distance from the capillary channel increases. This is thought to be largely due to the size difference between free Dox and Dox-HANP, which affects extravasation and interstitial transport of drugs. This suggests that T-MOC can mimic key biophysical barriers of drug transport at the tumor microenvironment.
Figure 3. Doxorubicin accumulation, penetration and dosage to MCF-7 and MDA-MB-231 in T-MOC.
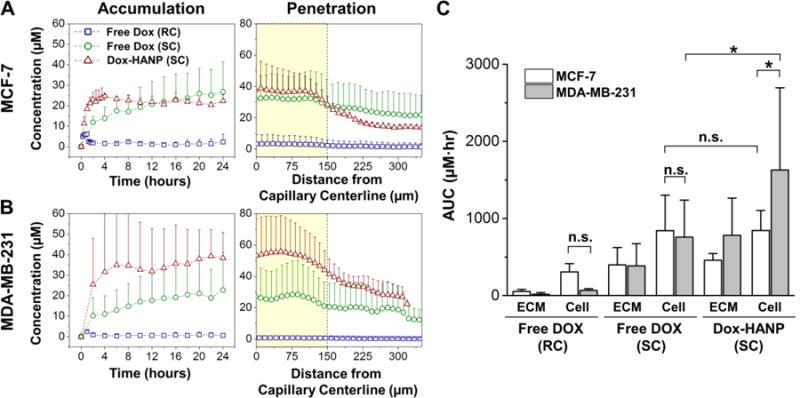
Spatiotemporal ECM accumulation and penetration profiles of free Dox and Dox-HANP on the T-MOC for (A) MCF-7 and (B) MDA-MB-231. (C) Area under the curve (AUC) based on cell and ECM accumulation profiles, representing cumulative drug exposure of the two cell lines during different treatments. RC: Rapid Clearance, SC: Slow Clearance, * p-value < 0.05. n.s: not significant.
Area under the curve (AUC) was calculated by integration of drug accumulation profiles in Figure 3A and Figure 3B as a measure of total drug exposure. The comparison of MCF-7 and MDA-MB-231 drug exposures is presented in Figure 3C. In Free Dox treatments, the AUC values for the two cell lines were similar (p-value > 0.05). However, exposure of MDA-MB-231 to Dox-HANP was significantly higher than that of MCF-7 (p-value < 0.05) as well as exposure of MDA-MB-231 to free Dox (p-value < 0.05). The increased accumulation of Dox-HANP in MDA-MB-231 with respect that in MCF-7 suggest cell-type-specific binding HANP on cancer cells [44]. In our previous study, Dox-HANP showed faster tumor accumulation than free anticancer agents. It is thought that slow drainage due to its larger size is a potential cause. In addition, structural and electrostatic properties of collagen matrix, which formed fibril structure with positive charged secondary amines make negative charged HANP-DOX preferentially bound to the collagen. Moreover, HANP could be bound to CD44 receptor which was overexpressed on the tumor cell surface, resulting in improving accumulation profile in tumor tissue or tumor cells [45]. However, the present results also suggest that Dox-HANP diffusion is limited to the adjacent region near the capillary channel, which does not guarantee increased cellular drug uptake throughout the tumors.
Post-treatment analysis of drug response
Time-lapse fluorescence microscopy images of cellular drug uptake and response are presented in Figure 4A. Prior to the perfusion of doxorubicine, both cellular aggregates are highly motile with random rotational motion as observed in previous studies [51–53]. As both cells uptake the drugs during perfusion, drug accumulated around the cellular aggregates and the motions of both cells slowed down. MCF-7 cells seem to uptake the drug faster than MDA-MB-231 based on brighter fluorescence around the aggregates noted with arrows. Interestingly, MCF-7 aggregates gradually enlarged suggesting their disintegration over the course of the treatment. The disintegration was well-correlated with cell death as confirmed by viability assay. In the meantime, MDA-MB-231 aggregates remained tightly packed even after the drug was accumulated. The molecular mechanisms of the spheroid disintegration in this study are not yet clear. In particular, relative importance of cell-cell and cell-matrix adhesions in DOX-induced disintegration of MCF-7 spheroids needs to be further investigated. We consider loss of E-cadherin mediated cell-cell adhesion as a possible mechanism as elevated levels of E-cadherin are reported for MCF-7 spheroids compared to their 2D culture and inhibition of E-cadherin results in disintegration of MCF-7 spheroids [54]. On the other hand, the role of cell-matrix adhesion proteins such as integrin-β1 in integrity of MCF-7 spheroids is limited with integrin-β1 being necessary for the formation of MDA-MB-231 spheroids [54]. Overall, these results imply that the action of doxorubicin can induce cell death via cell-type-specific response mechanisms [55]. Further research is warranted to identify these mechanisms on the two cell types studied.
Figure 4. Post-treatment analysis of drug response of breast cancer cells on T-MOC platform.
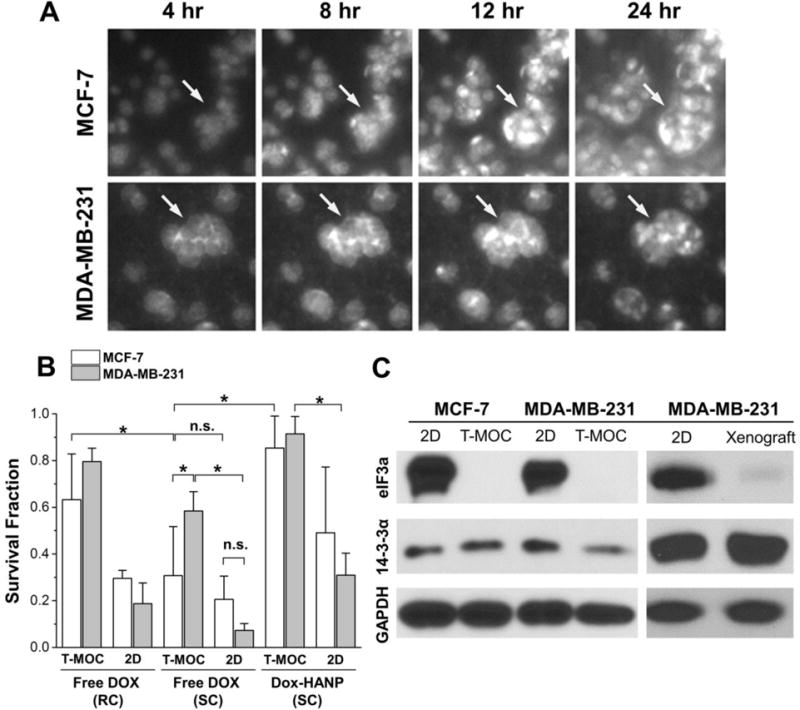
(A) Time-lapse fluorescence micrographs of cellular drug uptake and cell-type specific response. (B) Post-treatment survival fraction of breast cancer cells cultured on T-MOC and 2D conditions. Results reported in terms of mean ± standard error. Both cell lines became significantly more resistant to Dox on T-MOC, compared to 2D culture (p-value < 0.05). While the two cell lines showed similar levels of resistance to Dox in 2D culture, MDA-MB-231 became significantly more resistant than MCF-7 on T-MOC (p-value < 0.05). (C) Western-blot of doxorubicin resistance-associated proteins of breast cancer cells cultured on T-MOC and 2D conditions as well as in vivo tumor xenograft model.
The drug responses of MCF-7 and MDA-MB-231 were studied and characterized as post-treatment survival fraction as shown in Figure 4B. In 2D cell culture, both cell lines are generally sensitive to doxorubicin for both 1 and 24-hour 2 μM free DOX treatments. The survival fraction reduces to approximately at or below 20%. Interestingly, triple-negative breast cancer cells, MDA-MB-231 cells, which are known to be more aggressive and drug-resistant in vivo, seem to be comparable or more sensitive than MCF-7 cells in 2D culture (p-value < 0.05). As the cells were cultured in the T-MOC platform, both cell lines became substantially resistant to doxorubicin, compared to when they are cultured in 2D culture, so that the survival fraction increases (p-value < 0.05). In addition, MDA-MB-231 cells become more resistant than MCF-7 cells. The same trend is noted for DOX-HANP treatment. However, the survival fraction in the T-MOC platform is higher than that of free DOX treatment. This is thought to be due to less penetration of the NPs than free DOX. This result suggests that the advantage of NP-based drug delivery could be prolonged circulation and increased dosage due to lower toxicity rather than increased targeting efficacy.
The increased drug resistance in the T-MOC platform is thought to be a compounding result of -i) biophysical drug resistance mechanisms associated with drug transport, which includes pharmacokinetics, accumulation, penetration and cellular uptake of drugs as illustrated in Figure 2; and ii) 3D induced cellular drug resistance mechanisms, which are linked to intracellular signaling via integrin and other receptor engagement [17]. The T-MOC platform can mimic these key drug resistance mechanisms, which are absent in 2D culture configurations.
It has been reported that down-regulation of eIF3a expression [14] or over-expression of 14–3-3σ [14] contribute to doxorubicin resistance. The mechanism is thought that eIF3a down-regulation increases synthesis of double strand DNA repair proteins. eIF3a is also a proto-oncogene and its over-expression drives tumorigenesis. Thus, we compared the expression of these genes in MCF7 and MDA-MB-231 cells in both 2D and T-MOC cultures as well as tumor xenograft model. As shown in Figure 4C, the expression of eIF3a was completely suppressed in the T-MOC condition while that of 14–3-3σ had no significant change between the two culturing conditions with only a slight downregulation in the case of MDA-MB-231. eIF3a expression in MDA-MB-231 was also decreased in xenograft tumor model compared to 2D cultures, consistent with T-MOC. These findings suggest that the reduced eIF3a expression due to culture on the T-MOC platform mimicks the in vivo animal model, may have contributed to doxorubicin resistance, and was not replicated in 2D culture.
Validation of the drug response from the T-MOC platform against animal models
The results of flow cytometry analysis for CD44 and CD24 expressions are presented in Figure 4. It has been reported that cells with a surface phenotype of CD44+CD24lo/− are tumorigenic and intrinsically drug-resistant [15, 23, 56–58]. Although the results in Figures 2 and 4 demonstrate that the T-MOC platform can delineate several key drug resistance mechanisms including pharmacokinetic clearance, interstitial transport barriers, and doxorubicin resistance proteins, these results cannot fully explain the significant increase of chemoresistance of MDA-MB-231 cells as compared to MCF-7. This cell-type-specific resistance suggests other drug resistance mechanisms are still present in the T-MOC platform.
Results from the flow cytometry analysis indicate that MDA-MB-231 cells show a substantial increase of a cell population with CD44+CD24lo/− expression from the T-MOC only (Figure 5A). On the other hand, MCF-7 cells show low expressions of a discernable CD44+CD24lo/− population regardless of whether they are cultured on 2D or T-MOC. Since tumor-initiating cells, having a phenotype of CD44+CD24lo/−, are intrinsically drug-resistant [15, 23, 56–58]. This is a vital observation consistent with MDA-MB-231 becoming more chemoresistant on the T-MOC as compared to MCF-7. This also matches with the flow cytometry analysis of xenograft tumors of these two cell lines (Figure 5A).
Figure 5. Comparison of cell phenotypes cultured in T-MOC and xenograft.
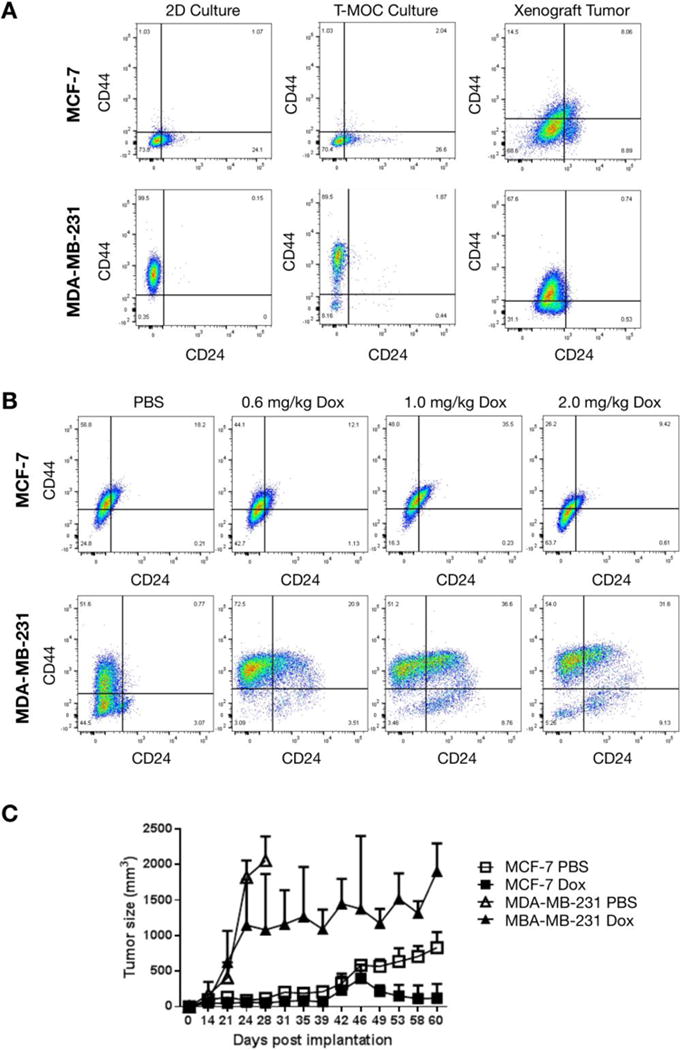
(A) Flow cytometry of CD44 and CD24 expression of cells cultured in different conditions. (B) Cellular phenotype change of xenografted MCF-7 and MDA-MB-231 cells after doxorubicin treatments. (C) Tumor progression during 2.0mg/kg doxorubicin treatments.
We further characterized the doxorubicin treatments of both cell lines to demonstrate the drug response on the T-MOC platform matches what is observed for in vivo xenografts. Similarly, while the sub-populations of MCF-7 tumors do not vary significantly, those of MDA-MB-231 tumors show significantly increased CD44+CD24lo/− population compared to control (Figure 5B). This enrichment of CD44+CD24lo/− population implies that MDA-MB-231 tumors become more chemoresistant after drug treatment and may be more tumorigenic. This would be consistent with the changes of tumor size during the treatment as shown in Figure 5C. For untreated control groups, tumors of MDA-MB-231 grew significantly faster than those of MCF-7 so that their size reached to approximately 2000 mm3 within a month. As treated with 2.0 mg/kg of doxorubicin twice a week, the size of tumor shrank while the tumors of MDA-MB-231 still slowly enlarged throughout the treatment durations. At the same dosage, the MCF-7 tumors remain small. This cell-type-specific drug response agrees well with the response from the T-MOC platform.
The T-MOC platform, thus, is able to reproduce a predictive surface phenotype and behavior of drug response as observed in vivo with both of the breast cancer cell lines. This result provides evidence that T-MOC can be a viable predictor of in vivo drug response and has the potential to provide a surrogate platform for initial drug development/validation that minimizes the need for animal studies.
Discussion
Using breast cancer cell lines, we have shown that the present T-MOC platform can delineate three key drug resistance mechanisms of tumors: (i) biophysical transport barriers at the TME, (ii) doxorubicin-resistance associated proteins, and (iii) cancer stem cell phenotypes. Moreover, T-MOC can provide detailed quantitative information to evaluate drug response cell-type specifically. The T-MOC is configured to mimic leaky vasculature, elevated IFP, dense ECM and high cell packing density [3–6]. Although it is not directly TME-associated transport process, pharmacokinetics, i.e. drug clearance from the plasma, has significant impacts on the delivery efficacy. The present T-MOC platform can simulate various circulation scenarios as shown in Figure 2B.
The compounding effects of all these pathophysiological barriers are extremely difficult, or often impossible, to study using currently available in vitro tumor models as well as in vivo small animal models. In particular, translation of tumor penetration, accumulation and tumor growth metrics typically measured in in vivo studies remain to be difficult to translate into relevant metric in in vitro models due to differences in length and time scales between the models as well as limited spatial and temporal resolution of transport characterization in in vivo models. The present T-MOC can provide a solution to bridge these two model systems. Particularly, cancer cell survival rate was significantly higher in T-MOC compared to the control assay involving static culture of cells in monolayer. In addition, simulating limited plasma lifetime of doxorubicin on the device lead to even higher cell survival rates due to rapid clearance of drug from interstitial space and diminished overall exposure of cells to the drug. These different treatment outcomes on the two model systems signify the importance of 3D culture environment and transport barriers limiting drug exposure in determining the apparent drug resistance measured in different assays. While the control assay in this study involved 2D culture, it will also be interesting to test and compare the drug treatment outcomes evaluated on T-MOC with other mainstream in vitro models such as multicellular spheroids that also emulate the 3D tumor microenvironment but lack the dynamic culture settings and specific tumor tissue architecture realized in our model system.
In this study, we limited ourselves to use of a porous membrane with a pore size of 400 nm, which is at the lower end of typical pore size observed in tumor vasculature [35]. It is important to note that a porous membrane alone cannot fully recapitulate the passive and active transvascular transport mechanisms mediated by endothelial cell layer and basement membrane. Culture of microvascular endothelial cells on the nanoporous membrane to improve the barrier function of the vascular wall is possible and was previously investigated in T-MOC platform. We refer the reader to our previous study with T-MOC on transport across a combined cell and membrane layer [31].
We used type I collagen as the matrix of the engineered tumor tissue in T-MOC. Collagen is present in mammary carcinoma tissues at amounts 3 to 10 times higher than other ECM proteins such as HA and GAG [59, 60]. Markers of mammary carcinoma such as increased tissue density and stiffness are primarily due to overexpressed collagen in tumor ECM. The resulting increase in tissue tension strengthens the linkage between cytoskeleton and integrins and promotes cell matrix adhesions, contributes to EMT and cancer invasion [61]. These indicate that collagen is a compositionally dominant and functionally significant component of tumor ECM making it suitable for use in in vitro assays seeking to mimic physiological functionalities of tumor ECM. T-MOC platform is adoptable for use with other ECM components such as HA, proteoglycans and fibronectin, which also play important roles in regulating interstitial fluid pressure and cell-matrix adhesion. Their adoption in biomimetic in vitro assays in addition to collagen grants further study [62, 63]. Furthermore, due to gas permeability of PDMS, T-MOC is compatible with environmental chambers that control O2 and CO2 levels. Therefore, it is possible to use T-MOC to investigate cell culture and drug response under hypoxia conditions.
The first difference regarding drug resistance-associated proteins is eIF3a, which is a translation initiation factor and has been suggested to play important regulatory role in synthesis of subset of proteins [14, 64, 65]. eIF3a has also been shown to play a major role in cisplatin and doxorubicin sensitivity possibly by suppressing synthesis of DNA repair proteins [14]. It is tempting to speculate that suppression of eIF3a expression in T-MOC results in up-regulation of synthesis of DNA repair proteins that can repair damages induced by doxorubicin, resulting in doxorubicin resistance. The finding that culturing cancer cells in T-MOC lead to suppression of eIF3a expression is also very intriguing. It may reveal a novel mechanism to understand the intrinsic drug resistance of solid tumors, which may have reduced eIF3a expression. Further studies are clearly needed to investigate these possibilities. Secondly, notable cell phenotypic difference could be distinguished. Although no phenotypic difference was observed in MCF-7, significant increase of CD44+CD24lo/− cell phenotype was noted in MDA-MB-231. This observation concurs with many of MDA-MB-231 characteristics including higher tumorigenicity, chemoresistance and metastatic potential than MCF-7.
It is desirable to measure in vivo drug uptake profiles to further validate T-MOC, yet is technically challenging to measure based on Dox fluorescence. In the meantime, a primary outcome of this study is that the TMOC-generated phenotype accurately predicts the resulting drug-induced phenotype in vivo (i.e. CD24/44 expressions in Figure 5B). Because the results were strikingly similar, we conclude that the TMOC accurately predicts the in vivo drug effect on cancer cells and expect that the drug uptake profiles would be similar. Regardless, the conclusions from this experiment still stand and would not be altered by any result obtained which assessed in vivo drug uptake profiles.
The present results provide detailed information to explain the transport characteristics of NPs around the TME. As shown in Figure 2C, if both free DOX and DOX encapsulated HANP are cleared from the plasma at the same rate (i.e., 24 hr perfusion), the DOX accumulation was not significantly improved by NP formulation for MCF-7, but notable increase for MDA-MB-231. However, penetration through the tumor tissue was significantly hampered for both cell types due to larger particle size of the NP formation compared to free drugs. However, since the key advantages of NP formulation are prolonged circulation and improved solubility of hydrophobic drugs, the comparison between free DOX and DOX-HANP should be made considering the difference in their pharmacokinetic profiles. This implies that higher NP accumulation at tumor sites may be a result of prolonged circulation rather than enhanced permeation. This also suggests that, when targeted tumor is well vascularized such as in large primary tumors or xenografts, NP mediated drug delivery may be a good drug delivery strategy. However, if the tumor is poorly vascularized, the NP formulation is not as effective as anticipated.
While T-MOC can provide a reliable test bed for drug and NP delivery, it is carefully used considering potential absorption of molecules to PDMS, which the T-MOC is made of. Having a network polymer structure, PDMS is permeable to different materials to varying degrees [66]. While gas-permeable nature of PDMS enables free exchange of oxygen and carbon dioxide and is useful in cell culture applications, adsorption of molecular reagents within PDMS in often undesirable in drug screening applications where adsorption can confound the amount of drug present in the channel intended by the tested treatment. However, we do not expect adsorption of doxorubicin in PDMS to be a significant factor in our experiments. It is reported that PDMS is particularly vulnerable to adsorption by hydrophobic molecules where adsorption becomes significant for molecules with partition coefficients greater than 2.47 [5]. Doxorubicin, having a partition coefficient of 1.27 [67], is relatively hydrophilic and is expected to be only minimally adsorbed within PDMS (>10%). There are no known studies on potential PDMS adsorption of DOX-HANP. Surface modification of PDMS with HA often requires chemical conjugation [68], indicating that HA and PDMS alone may not form a stable adsorption layer. Furthermore, HANP is hydrophilic with negative charge and is expected to only weakly interact with PDMS that is also negatively changed but hydrophobic.
In summary, we demonstrated the feasibility of the present T-MOC platform for rapid characterization of drug and NP delivery. The present T-MOC platform allows systematic and mechanistic information related to multiple chemoresistance mechanisms and their compounding effects. Moreover, cell-type specific drug response and resistance can be rapidly detected with mechanistic explanation of corresponding drug resistance. These capabilities of T-MOC can ultimately be used to examine the properties of cancer cells obtained from individual patients for personalized medicine. Although the T-MOC platform still requires further research to replicate many other aspects of TME including hypoxia, complex ECM protein compositions, and various stromal cells, it can be used for fast and accurate assessment of tumor cell properties.
Supplementary Material
Movie S1 - MCF-7 Free Dox (RC).avi: Time-lapse recording of doxorubicin fluorescence during Dox (RC) treatment of MCF-7.
Movie S2 - MCF-7 Free Dox (SC).avi: Time-lapse recording of doxorubicin fluorescence during Dox (SC) treatment of MCF-7.
Movie S3 - MCF-7 Dox-HANP (SC).avi: Time-lapse recording of doxorubicin fluorescence during Dox-HANP (SC) treatment of MCF-7.
Movie S4 - MCF-7 and MDA-MB-231 Drug Responses.avi: Time-lapse recording of close-up on MCF-7 and MDA-MB-231 multicellular aggregates showing comparison of their responses to the Free Dox (SC).
Acknowledgments
This work was partially supported by NIH HHSN261201400021C, a CTR Award from Indiana CTSI funded in part by UL1 TR000006 from NIH, a Grant from Walther Cancer Foundation, and the Digital Human Project from Purdue University. The authors also acknowledged the support from the Purdue University Center for Cancer Research, NIH grant P30 CA023168.
Nomenclature
- AUC
Area under the curve
- Dox
Doxorubicin
- ECM
Extracellular matrix
- eIF3a
Eukaryotic translation initiation factor 3 subunit A
- GAG
Glycosaminoglycan
- HA
Hyaluronic acid
- HER2
Human epidermal growth factor receptor 2
- IFP
Interstitial fluid pressure
- MDR
Multidrug resistance
- NP
Nanoparticle
- PDMS
Polydimethlysiloxane
- T-MOC
Tumor-microenvironment-on-chip
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Albanese A, Tang PS, Chan WC. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annual Review of Biomedical Engineering. 2012;14:1–16. doi: 10.1146/annurev-bioeng-071811-150124. [DOI] [PubMed] [Google Scholar]
- 2.Duan XP, Li YP. Physicochemical Characteristics of Nanoparticles Affect Circulation, Biodistribution, Cellular Internalization, and Trafficking. Small. 2013;9:1521–1532. doi: 10.1002/smll.201201390. [DOI] [PubMed] [Google Scholar]
- 3.Bae YH, Park K. Targeted drug delivery to tumors: Myths, reality and possibility. Journal of Controlled Release. 2011;153:198–205. doi: 10.1016/j.jconrel.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kanapathipillai M, Brock A, Ingber DE. Nanoparticle targeting of anti-cancer drugs that alter intracellular signaling or influence the tumor microenvironment. Advanced Drug Delivery Reviews. 2014;79–80:107–118. doi: 10.1016/j.addr.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 5.Choi KY, Jeon EJ, Yoon HY, Lee BS, Na JH, Min KH, Kim SY, Myung SJ, Lee S, Chen X, Kwon IC, Choi K, Jeong SY, Kim K, Park JH. Theranostic nanoparticles based on PEGylated hyaluronic acid for the diagnosis, therapy and monitoring of colon cancer. Biomaterials. 2012;33:6186–6193. doi: 10.1016/j.biomaterials.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ozcelikkale A, Ghosh S, Han B. Multifaceted transport characteristics of nanomedicine: Needs for characterization in dynamic environment. Molecular Pharmaceutics. 2013;10:2111–2126. doi: 10.1021/mp3005947. [DOI] [PubMed] [Google Scholar]
- 7.Holback H, Yeo Y. Intratumoral Drug Delivery with Nanoparticulate Carriers. Pharmaceutical Research. 2011;28:1819–1830. doi: 10.1007/s11095-010-0360-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Research. 2000;60:2497–2503. [PubMed] [Google Scholar]
- 9.Choi KY, Yoon HY, Kim JH, Bae SM, Park RW, Kang YM, Kim IS, Kwon IC, Choi K, Jeong SY, Kim K, Park JH. Smart Nanocarrier Based on PEGylated Hyaluronic Acid for Cancer Therapy. ACS Nano. 2011;5:8591–8599. doi: 10.1021/nn202070n. [DOI] [PubMed] [Google Scholar]
- 10.Lu D, Wientjes MG, Lu Z, Au JLS. Tumor Priming Enhances Delivery and Efficacy of Nanomedicines. Journal of Pharmacology and Experimental Therapeutics. 2007;322:80–88. doi: 10.1124/jpet.107.121632. [DOI] [PubMed] [Google Scholar]
- 11.Heldin CH, Rubin K, Pietras K, Ostman A. High interstitial fluid pressure - an obstacle in cancer therapy. Nature Review Cancer. 2004;4:806–813. doi: 10.1038/nrc1456. [DOI] [PubMed] [Google Scholar]
- 12.Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, Jain RK. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91:1071–1121. doi: 10.1152/physrev.00038.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allikments R, Schrim LM, Hutchinson A, Romano-Spica V, Dean M. Characterization of the human ABC superfamily: isolation and mapping of 21 new genes using the expressed sequence tags database. Hum Mol Genet. 1966;5:1649–1655. doi: 10.1093/hmg/5.10.1649. [DOI] [PubMed] [Google Scholar]
- 14.Liu R-Y, Dong Z, Liu J, Yin J-Y, Zhou L, Wu X, Yang Y, Mo W, Huang W, Khoo SK, et al. Role of eIF3a in regulating cisplatin sensitivity and in translational control of nucleotide excision repair of nasopharyngeal carcinoma. Oncogene. 2011;30:4814–4823. doi: 10.1038/onc.2011.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC, Wong H, Rosen J, Chang JC. Intrinsic Resistance of Tumorigenic Breast Cancer Cells to Chemotherapy. Journal of the National Cancer Institute. 2008;100:672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- 16.Hermeking H. The 14-3-3 cancer connection. Nat Rev Cancer. 2003;3:931–943. doi: 10.1038/nrc1230. [DOI] [PubMed] [Google Scholar]
- 17.Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS. Cell adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood. 1999;93:1658–1667. [PMC free article] [PubMed] [Google Scholar]
- 18.Jean C, Gravelle P, Fournie JJ, Laurent G. Influence of stress on extracellular matrix and integrin biology. Oncogene. 2011;30:2697–2706. doi: 10.1038/onc.2011.27. [DOI] [PubMed] [Google Scholar]
- 19.Shain KH, Dalton WS. Cell adhesion is a key determinant in de Novo multidrug resistance (MDR): New targets for the prevention of acquired MDR. Molecular Cancer Therapeutics. 2001;1:69–78. [PubMed] [Google Scholar]
- 20.Perou CM, Jeffrey SS, Rijn Mvd, Rees CA, Eisen MB, Ross DT, Pergamenschikov A, Williams CF, Zhu SX, Lee JCF, Lashkari D, Shalon D, Brown PO, Botstein D. Distinctive gene expression patterns in human mammary epithelial cells and breast cancers. Proceedings of the National Academy of Sciences. 1999;96:9212–9217. doi: 10.1073/pnas.96.16.9212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge Ø, Pergamenschikov A, Williams C, Zhu SX, Lønning PE, Børresen-Dale AL, Brown PO, Botstein D. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 22.Holliday DL, Speirs V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011;13:215. doi: 10.1186/bcr2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prat A, Karginova O, Parker JS, Fan C, He X, Bixby L, Harrell JC, Roman E, Adamo B, Troester M, Perou CM. Characterization of cell lines derived from breast cancers and normal mammary tissues for the study of the intrinsic molecular subtypes. Breast Cancer Research and Treatment. 2013;142:237–255. doi: 10.1007/s10549-013-2743-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sørlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, Rijn Mvd, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Lønning PE, Børresen-Dale A-L. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proceedings of the National Academy of Sciences. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gogineni K, DeMichele A. Current approaches to the management of Her2-negative metastatic breast cancer. Breast Cancer Research. 2012;14:205. doi: 10.1186/bcr3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Justice BA, Badr NA, Felder RA. 3D cell culture opens new dimensions in cell-based assays. Drug Discovery Today. 2009;14:102–107. doi: 10.1016/j.drudis.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 27.Albini A, Sporn M. The tumor microenvironment as a target for chemoprevention. Nat Rev Cancer. 2007;7:139–147. doi: 10.1038/nrc2067. [DOI] [PubMed] [Google Scholar]
- 28.Kenny PA, Lee GY, Myers CA, Neve RM, Semeiks JR, Spellman PT, Lorenz K, Lee EH, Barcellos-Hoff MH, Petersen OW, Gray JW, Bissell MJ. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol Oncol. 2007;1:84–96. doi: 10.1016/j.molonc.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weigelt B, Lo AT, Park CC, Gray JW, Bissell MJ. HER2 signaling pathway activiation and response of breast cancer cells to HER2-targeting agents is dependent strongly on the 3D microenvironment. Breast Cancer Res Treat. 2010;122:35–43. doi: 10.1007/s10549-009-0502-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krause S, Maffini MV, Soto A, Sonnenschein C. The microenvironment determines the breast cancer cells’ phenotype: organization of MCF7 cells in 3D cultures. BMC Cancer. 2010;10:263. doi: 10.1186/1471-2407-10-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kwak B, Ozcelikkale A, Shin CS, Park K, Han B. Simulation of complex transport of nanoparticles around a tumor using tumor-microenvironment-on-chip. Journal of Controlled Release. 2014;194:157–167. doi: 10.1016/j.jconrel.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shin CS, Kwak B, Han B, Park K. Development of an in Vitro 3D Tumor Model to Study Therapeutic Efficiency of an Anticancer Drug. Molecular Pharmaceutics. 2013;10:2167–2175. doi: 10.1021/mp300595a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shin K, Klosterhoff BS, Han B. Characterization of Cell-Type-Specific Drug Transport and Resistance of Breast Cancers Using Tumor-Microenvironment-on-Chip. Molecular Pharmaceutics. 2016;13:2214–2223. doi: 10.1021/acs.molpharmaceut.6b00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teo KY, Dutton JC, Han B. Spatiotemporal measurement of freezing-induced deformation of engineered tissues. J Biomech Eng. 2010;132:31003. doi: 10.1115/1.4000875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuan F. Transvascular drug delivery in solid tumors. Semin Radiat Oncol. 1998;8:164–175. doi: 10.1016/s1053-4296(98)80042-8. [DOI] [PubMed] [Google Scholar]
- 36.Arganda-Carreras I, Kaynig V, Rueden C, Eliceiri KW, Schindelin J, Cardona A, Sebastian Seung H. Trainable Weka Segmentation: a machine learning tool for microscopy pixel classification. Bioinformatics. 2017;33:2424–2426. doi: 10.1093/bioinformatics/btx180. [DOI] [PubMed] [Google Scholar]
- 37.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koutsiaris AG, Tachmitzi SV, Batis N. Wall shear stress quantification in the human conjunctival pre-capillary arterioles in vivo. Microvascular Research. 2013;85:34–39. doi: 10.1016/j.mvr.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 39.Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP, Jain RK. Vascular Permeability in a Human Tumor Xenograft: Molecular Size Dependence and Cutoff Size. Cancer Research. 1995;55:3752–3756. [PubMed] [Google Scholar]
- 40.Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, Torchilin VP, Jain RK. Regulation of Transport Pathways in Tumor Vessels: Role of Tumor Type and Microenvironment. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:4607–4612. doi: 10.1073/pnas.95.8.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robert J, Illiadis A, Hoerni B, Cano JP, Durand M, Lagarde C. PHARMACOKINETICS OF ADRIAMYCIN IN PATIENTS WITH BREAST-CANCER - CORRELATION BETWEEN PHARMACOKINETIC PARAMETERS AND CLINICAL SHORT-TERM RESPONSE. European Journal of Cancer & Clinical Oncology. 1982;18:739–745. doi: 10.1016/0277-5379(82)90072-4. [DOI] [PubMed] [Google Scholar]
- 42.Lee SY, Kim S, Tyler JY, Park K, Cheng JX. Blood-stable, tumor-adaptable disulfide bonded mPEG-(Cys)4-PDLLA micelles for chemotherapy. Biomaterials. 2013;34:552–561. doi: 10.1016/j.biomaterials.2012.09.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Choi KY, Min KH, Na JH, Choi K, Kim K, Park JH, Kwon IC, Jeong SY. Self-assembled hyaluronic acid nanoparticles as a potential drug carrier for cancer therapy: synthesis, characterization, and in vivo biodistribution. Journal of Materials Chemistry. 2009;19:4102–4107. [Google Scholar]
- 44.Choi KY, Chung H, Min KH, Yoon HY, Kim K, Park JH, Kwon IC, Jeong SY. Self-assembled hyaluronic acid nanoparticles for active tumor targeting. Biomaterials. 2010;31:106–114. doi: 10.1016/j.biomaterials.2009.09.030. [DOI] [PubMed] [Google Scholar]
- 45.Choi KY, Yoon HY, Kim JH, Bae SM, Park RW, Kang YM, Kim IS, Kwon IC, Choi K, Jeong SY, Kim K, Park JH. Smart Nanocarrier Based on PEGylated Hyaluronic Acid for Cancer Therapy. ACS Nano. 2011;5:8591–8599. doi: 10.1021/nn202070n. [DOI] [PubMed] [Google Scholar]
- 46.Choi KY, Jeon EJ, Yoon HY, Lee BS, Na JH, Min KH, Kim SY, Myung SJ, Lee S, Chen X, Kwon IC, Choi K, Jeong SY, Kim K, Park JH. Theranostic nanoparticles based on PEGylated hyaluronic acid for the diagnosis, therapy and monitoring of colon cancer. Biomaterials. 2012;33:6186–6193. doi: 10.1016/j.biomaterials.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Han HS, Lee J, Kim HR, Chae SY, Kim M, Saravanakumar G, Yoon HY, You DG, Ko H, Kim K, Kwon IC, Park JC, Park JH. Robust PEGylated hyaluronic acid nanoparticles as the carrier of doxorubicin: Mineralization and its effect on tumor targetability in vivo. Journal of Controlled Release. 2013;168:105–114. doi: 10.1016/j.jconrel.2013.02.022. [DOI] [PubMed] [Google Scholar]
- 48.Choi KY, Chung H, Min KH, Yoon HY, Kim K, Park JH, Kwon IC, Jeong SY. Self-assembled hyaluronic acid nanoparticles for active tumor targeting. Biomaterials. 2010;31:106–114. doi: 10.1016/j.biomaterials.2009.09.030. [DOI] [PubMed] [Google Scholar]
- 49.Bertrand N, Leroux JC. The journey of a drug-carrier in the body: An anatomo-physiological perspective. Journal of Controlled Release. 2012;161:152–163. doi: 10.1016/j.jconrel.2011.09.098. [DOI] [PubMed] [Google Scholar]
- 50.Toley BJ, Lovatt ZGT, Harrington JL, Forbes NS. Microfluidic technique to measure intratumoral transport and calculate drug efficacy shows that binding is essential for doxorubicin and release hampers Doxil. INTEGRATIVE BIOLOGY. 2013;5:1184–1196. doi: 10.1039/c3ib40021b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tanner K, Mori H, Mroue R, Bruni-Cardoso A, Bissell MJ. Coherent angular motion in the establishment of multicellular architecture of glandular tissues. PNAS. 2012;109:1973–1978. doi: 10.1073/pnas.1119578109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shin CS, Kwak B, Han B, Park K. Development of an in vitro 3D tumor model to study therapeutic efficiency of an anticancer drug. Molecular Pharmaceutics. 2013;10:2167–2175. doi: 10.1021/mp300595a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shin K, Klosterhoff BS, Han B. Characterization of cell-type specific drug transport and resistance of breast cancers using tumor-microenvironment-on-chip. Molecular Pharmaceutics. 2016 doi: 10.1021/acs.molpharmaceut.6b00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ivascu A, Kubbies M. Diversity of cell-mediated adhesions in breast cancer spheroids. International Journal of Oncology. 2007;31:1403–1413. [PubMed] [Google Scholar]
- 55.Thorn CF, Oshiro C, Marsh S, Hernandez-Boussard T, McLeod H, Klein TE, Altman RB. Doxorubicin pathways: pharmacodynamics and adverse effects. Pharmacogenet Genomics. 2011;21:440–446. doi: 10.1097/FPC.0b013e32833ffb56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, Rimm DL, Wong H, Rodriguez A, Herschkowitz JI, Fan C, Zhang X, He X, Pavlick A, Gutierrez MC, Renshaw L, Larionov AA, Faratian D, Hilsenbeck SG, Perou CM, Lewis MT, Rosen JM, Chang JC. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:13820–13825. doi: 10.1073/pnas.0905718106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, Speed T, Spellman PT, DeVries S, Lapuk A, Wang NJ, Kuo WL, Stilwell JL, Pinkel D, Albertson DG, Waldman FM, McCormick F, Dickson RB, Johnson MD, Lippman M, Ethier S, Gazdar A, Gray JW. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Prat A, Perou CM. Deconstructing the molecular portraits of breast cancer. Molecular Oncology. 2011;5:5–23. doi: 10.1016/j.molonc.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ozcelikkale A, Moon H-R, Linnes M, Han B. In vitro microfluidic models of tumor microenvironment to screen transport of drugs and nanoparticles. Wiley Interdisciplinary Reviews: Nanomedicine and Nanobiotechnology. 2017:e1460. doi: 10.1002/wnan.1460. n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Beachley VZ, Wolf MT, Sadtler K, Manda SS, Jacobs H, Blatchley MR, Bader JS, Pandey A, Pardoll D, Elisseeff JH. Tissue matrix arrays for high-throughput screening and systems analysis of cell function. Nature Methods. 2015;12:1197–1204. doi: 10.1038/nmeth.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Insua-Rodríguez J, Oskarsson T. The extracellular matrix in breast cancer. Advanced Drug Delivery Reviews. 2016;97:41–55. doi: 10.1016/j.addr.2015.12.017. [DOI] [PubMed] [Google Scholar]
- 62.Provenzano PP, Hingorani SR. Hyaluronan, fluid pressure, and stromal resistance in pancreas cancer. British Journal of Cancer. 2013;108:1–8. doi: 10.1038/bjc.2012.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Theocharis AD, Skandalis SS, Gialeli C, Karamanos NK. Extracellular matrix structure. Advanced Drug Delivery Reviews. 2016;97:4–27. doi: 10.1016/j.addr.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 64.Dong Z, Liu LH, Han B, Pincheira R, Zhang JT. Role of eIF3 p170 in controlling synthesis of ribonucleotide reductase M2 and cell growth. Oncogene. 2004;23:3790–3801. doi: 10.1038/sj.onc.1207465. [DOI] [PubMed] [Google Scholar]
- 65.Dong Z, Zhang JT. Initiation factor eIF3 and regulation of mRNA translation, cell growth, and cancer. Crit Rev Oncol Hematol. 2006;59:169–180. doi: 10.1016/j.critrevonc.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 66.Toepke MW, Beebe DJ. PDMS absorption of small molecules and consequences in microfluidic applications. Lab Chip. 2006;6:1484–1486. doi: 10.1039/b612140c. [DOI] [PubMed] [Google Scholar]
- 67.Malfanti A, Miletto I, Bottinelli E, Zonari D, Blandino G, Berlier G, Arpicco S. Delivery of Gemcitabine Prodrugs Employing Mesoporous Silica Nanoparticles. Molecules. 2016;21:16. doi: 10.3390/molecules21040522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yue Z, Liu X, Molino PJ, Wallace GG. Bio-functionalisation of polydimethylsiloxane with hyaluronic acid and hyaluronic acid – Collagen conjugate for neural interfacing. Biomaterials. 2011;32:4714–4724. doi: 10.1016/j.biomaterials.2011.03.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1 - MCF-7 Free Dox (RC).avi: Time-lapse recording of doxorubicin fluorescence during Dox (RC) treatment of MCF-7.
Movie S2 - MCF-7 Free Dox (SC).avi: Time-lapse recording of doxorubicin fluorescence during Dox (SC) treatment of MCF-7.
Movie S3 - MCF-7 Dox-HANP (SC).avi: Time-lapse recording of doxorubicin fluorescence during Dox-HANP (SC) treatment of MCF-7.
Movie S4 - MCF-7 and MDA-MB-231 Drug Responses.avi: Time-lapse recording of close-up on MCF-7 and MDA-MB-231 multicellular aggregates showing comparison of their responses to the Free Dox (SC).