

Abstract
The Δ9-fatty acid desaturase introduces a double bond at the Δ9 position of the acyl moiety of acyl-CoA and regulates the cellular levels of unsaturated fatty acids. However, it is unclear how Δ9-desaturase expression is regulated in response to changes in the levels of fatty acid desaturation. In this study, we found that the degradation of DESAT1, the sole Δ9-desaturase in the Drosophila cell line S2, was significantly enhanced when the amounts of unsaturated acyl chains of membrane phospholipids were increased by supplementation with unsaturated fatty acids, such as oleic and linoleic acids. In contrast, inhibition of DESAT1 activity remarkably suppressed its degradation. Of note, removal of the DESAT1 N-terminal domain abolished the responsiveness of DESAT1 degradation to the level of fatty acid unsaturation. Further truncation and amino acid replacement analyses revealed that two sequential prolines, the second and third residues of DESAT1, were responsible for the unsaturated fatty acid–dependent degradation. Although degradation of mouse stearoyl-CoA desaturase 1 (SCD1) was unaffected by changes in fatty acid unsaturation, introduction of the N-terminal sequential proline residues into SCD1 conferred responsiveness to unsaturated fatty acid–dependent degradation. Furthermore, we also found that the Ca2+-dependent cysteine protease calpain is involved in the sequential proline–dependent degradation of DESAT1. In light of these findings, we designated the sequential prolines at the second and third positions of DESAT1 as a “di-proline motif,” which plays a crucial role in the regulation of Δ9-desaturase expression in response to changes in the level of cellular unsaturated fatty acids.
Keywords: Drosophila, fatty acid, lipid synthesis, phospholipid, protein degradation
Introduction
The number and position of double bonds and chain length in fatty acid moieties of phospholipids affect the physicochemical properties of membranes, thus affecting a broad range of cellular and physiological functions (1). Fatty acid desaturases are a family of enzymes that introduce a double bond into the acyl moiety of acyl-CoA and play a critical role in controlling membrane fluidity by modulating fatty acid composition of phospholipids (2). The Δ9-desaturase introduces a cis-double bond at the Δ9 position of acyl-CoAs and is a rate-limiting enzyme involved in the biosynthesis of monounsaturated fatty acids that are used to synthesize polyunsaturated fatty acids, phospholipids, triacylglycerol, cholesteryl esters, and wax esters (3). Therefore, the expression level of Δ9-desaturase should be strictly regulated to meet the demand for unsaturated fatty acids. Although the mechanisms underlying the regulation of fatty acid desaturase expression are reported for several desaturases as described below, it is unclear how cells recognize the changes in fatty acid desaturation of cellular lipids to regulate the expression of fatty acid desaturase.
Stearoyl-CoA desaturase 1 (SCD1), a mammalian Δ9-desaturase in the endoplasmic reticulum (ER),3 consists of four transmembrane helices with its N and C termini, as well as its catalytic sites, oriented toward the cytosolic side of the ER membrane (4, 5). The expression of SCD1 is mainly regulated by SREBP1, a master regulator of lipid biosynthesis at the transcriptional level (6), whereas the degradation of SCD1 is mediated by the ubiquitin–proteasome system irrespective of the cellular levels of unsaturated fatty acids that regulate Scd1 gene expression (7).
In Saccharomyces cerevisiae, the expression of Δ9-desaturase Ole1 is regulated at the levels of transcription and mRNA stability (8, 9). The transcriptional regulation of Ole1 is mediated through at least two elements, fatty acid-regulated sequence and the low-oxygen-response element, in the OLE1 promoter (10, 11). Furthermore, two ER-resident membrane proteins, Spt23 and Mga2, are activated in a 26S proteasome-dependent manner and positively regulate the expression of Ole1 (12). Repression of Ole1 by unsaturated fatty acids is mediated by the suppression of Spt23 processing and inhibition of the cleaved (activated) Mga2 fragment (12, 13). Recently, Mga2 processing was also shown to be regulated by the cellular levels of unsaturated fatty acids (14). Ole1 protein, a naturally short-lived protein, has been shown to be degraded by ubiquitin/proteasome-dependent ER-associated degradation with a half-life of <1 h, even in unsaturated fatty acid-depleted conditions (15).
Drosophila Δ9-desaturase, DESAT1, was identified by homology to vertebrate fatty acid desaturases (16), and subsequent genetic studies have shown unique features of DESAT1 in the control of sensory communications via pheromonal production as well as regulation of the double bond contents in the fatty acids of phospholipids (17–19). DESAT1 comprises 383 amino acids with four transmembrane helices and shows structural features similar to those of SCD1. Drosophila melanogaster as a model organism provides several advantages for studying the molecular mechanisms underlying the expression and physiological functions of Δ9-desaturase in multicellular organisms. First, in typical Drosophila cell lines such as S2 cells, DESAT1 is the sole fatty acid desaturase that introduces a double bond into acyl-CoAs because another fatty acid desaturase, DESAT2, is not expressed due to 16-bp deletion in the 5′ region of the Desat2 gene (20). Second, because Drosophila cannot synthesize sterols and only a trace amount of polyunsaturated fatty acids is detected in cellular membranes (21, 22), changes in DESAT1 activity are likely to directly affect the physicochemical properties of the membrane. Thus, Drosophila cells provide a useful model to study how cells recognize changes in membrane fluidity and regulate the expression of Δ9-desaturase. Although the mechanism of tissue-specific expression of DESAT1 was reported (23), the regulation of DESAT1 in response to changes in either the membrane fluidity or levels of fatty acid desaturation was not reported. In this study, we revealed a novel amino acid motif that regulates the degradation of DESAT1, which plays a dominant role in controlling the expression of DESAT1 in response to changes in the cellular levels of unsaturated fatty acids. Protein degradation pathways involved in DESAT1 degradation are also discussed.
Results
Effect of unsaturated fatty acids on the expression level of DESAT1
To explore the regulatory mechanisms of DESAT1 expression, we raised polyclonal antibodies against DESAT1 and examined the effect of exogenously added fatty acids on the expression of DESAT1. The amounts of DESAT1 protein were significantly reduced when S2 cells were incubated with 100 μm oleic acid (C18:1) or linoleic acid (C18:2) for 6 h, whereas no significant change was observed with stearic acid (C18:0) treatment (Fig. 1, A and B). Addition of a 16-carbon length monounsaturated fatty acid (palmitoleic acid; C16:1) also markedly reduced expression of the DESAT1 protein (supplemental Fig. 1, A and B). Incubation with C16:1, C18:1, or C18:2 for 6 h caused a significant increase in the proportion of fatty acid added in the acyl chain of phospholipids (supplemental Fig. 2A). Furthermore, C18:2 was effectively incorporated into phospholipids within 1 h (supplemental Fig. 2B). These results raised the possibility that unsaturated fatty acid contents in the phospholipids regulate the expression level of DESAT1.
Figure 1.
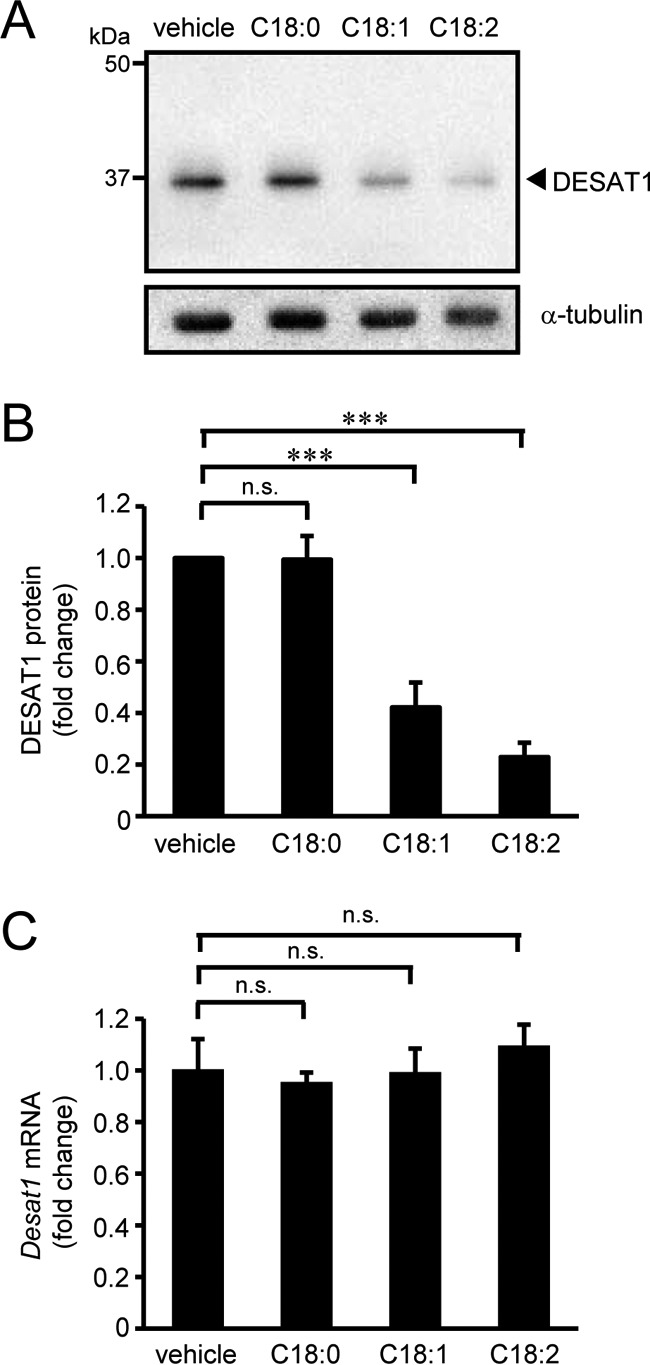
Effect of unsaturated fatty acids on DESAT1 expression. S2 cells were treated with indicated fatty acids (100 μm) for 6 h, and the amounts of DESAT1 and α-tubulin (loading control) proteins were detected with anti-DESAT1 antibody and anti-α-tubulin antibody, respectively (A and B). Band intensities were determined by ImageJ software, and levels of DESAT1 proteins are shown relative to the amount of DESAT1 protein in vehicle-treated cells (B). S2 cells were treated with indicated fatty acids (100 μm) for 6 h, and the expression levels of Desat1 mRNA were determined by real-time PCR (C). Mean ± S.D. (n = 3). ***, p < 0.001; n.s., not significant.
Given that the expression levels of mammalian SCD1 and yeast Ole1 are mainly controlled by transcriptional regulation, we examined the effect of exogenously added fatty acids on the expression level of Desat1 mRNA using real-time PCR. As shown in Fig. 1C and supplemental Fig. 1C, any of the fatty acids examined had no significant effect on the expression level of Desat1 mRNA.
We next examined the effect of SCD1 inhibitors on the expression of DESAT1. First, we evaluated whether the SCD1 inhibitors 23 and 37c (24, 25) inhibit the Δ9-desaturase activity of DESAT1 by measuring the conversion of a 17-carbon-long saturated fatty acid (C17:0) to the monounsaturated form (C17:1) in S2 cells. Production of C17:1 from C17:0 was decreased by the SCD1 inhibitors to <10% of vehicle-treated cells, demonstrating that the SCD1 inhibitors also inhibit the activity of DESAT1 (supplemental Fig. 3). We thus used these SCD1 inhibitors as DESAT1 inhibitors in the following experiments. Incubation of S2 cells with the DESAT1 inhibitors significantly decreased the proportion of monounsaturated fatty acids (C16:1 and C18:1) in the acyl chains of phospholipids and increased the content of saturated fatty acids (C16:0, C18:0, and C20:0) (Fig. 2, A and B). In contrast to the treatment with unsaturated fatty acids, DESAT1 inhibitors 23 and 37c increased the amount of DESAT1 protein by 2.7- and 2.9-fold, respectively (Fig. 2, C and D). To analyze the effect of the DESAT1 inhibitor on the degradation rate of DESAT1 protein, DESAT1 inhibitor-treated cells were incubated with cycloheximide for 2 h. Pre-treatment with DESAT1 inhibitor 37c suppressed the degradation of the DESAT1 protein (Fig. 2, E and F). Similar to unsaturated fatty acid treatment, DESAT1 inhibitors did not affect the amount of Desat1 mRNA (supplemental Fig. 4). These results indicate that the expression level of the DESAT1 protein is mainly regulated at the post-translational level where unsaturated fatty acid–dependent protein degradation is likely to play a dominant role.
Figure 2.

Effect of DESAT1 inhibitors on DESAT1 expression. S2 cells were treated with DESAT1 inhibitor 23 or 37c (1 μm) for 16 h, and fatty acid compositions of phospholipids were analyzed by gas chromatography (A and B). S2 cells were treated with DESAT1 inhibitor 23 or 37c (1 μm) for 16 h, and the amounts of DESAT1 and α-tubulin protein were detected with anti-DESAT1 antibody and anti-α-tubulin antibody, respectively (C and D). After treatment with DESAT1 inhibitor 37c (1 μm) for 16 h, S2 cells were incubated with cycloheximide (100 μg/ml) for 2 h, and the amounts of DESAT1 and α-tubulin protein were detected with specific antibodies (E and F). Band intensities were determined by ImageJ software, and levels of DESAT1 proteins are shown relative to the amount of DESAT1 protein in vehicle-treated cells (D) or cells not treated with cycloheximide and DESAT1 inhibitor (F). Mean ± S.D. (n = 3). *, p < 0.05; **, p < 0.01; ***, p < 0.001; n.s., not significant.
Degradation of exogenously expressed DESAT1 protein
To evaluate the molecular mechanisms underlying the post-translational regulation of DESAT1 expression, we expressed FLAG-tagged DESAT1 in S2 cells under the control of the Act5C promoter. The degradation rate of the C-terminally FLAG-tagged DESAT1 (DESAT1-FLAGC) protein was comparable with that of endogenous DESAT1 protein (Fig. 3, A and B). The effects of C18:1 (Fig. 3, C and D) and DESAT1 inhibitors (Fig. 3, E and F) on the expression levels of DESAT1-FLAGC protein were also comparable with those observed with the endogenous DESAT1 protein (Figs. 1 and 2).
Figure 3.

Effect of a C-terminal FLAG tag on DESAT1 expression. S2 cells expressing DESAT1-FLAGC were treated with cycloheximide (100 μg/ml) for 0, 2, and 4 h, and the amounts of DESAT1 and α-tubulin protein were detected with anti-DESAT1 antibody and anti-α-tubulin antibody, respectively (A and B). S2 cells expressing DESAT1-FLAGC were treated with C18:1 (100 μm) for 6 h (C and D) or DESAT1 inhibitor 23 or 37c (1 μm) for 16 h (E and F), and the amounts of DESAT1 and α-tubulin protein were detected with specific antibodies. Band intensities were determined by ImageJ software, and levels of DESAT1 proteins are shown relative to the amount of DESAT1 protein in cells not treated with cycloheximide (B) or vehicle-treated cells (D and F). Filled arrowhead, DESAT1-FLAGC; open arrowhead, endogenous DESAT1. Mean ± S.D. (n = 3). **, p < 0.01; ***, p < 0.001.
In contrast, N-terminally FLAG-tagged DESAT1 (FLAGN-DESAT1) protein was remarkably stable compared with endogenous DESAT1 protein (Fig. 4, A and B). Furthermore, the amount of FLAGN-DESAT1 protein was not affected by addition of either C18:1 (Fig. 4, C and D) or DESAT1 inhibitors (Fig. 4, E and F). These results suggest that the N-terminal domain of DESAT1 protein is involved in the fatty acid–dependent degradation of DESAT1 protein.
Figure 4.
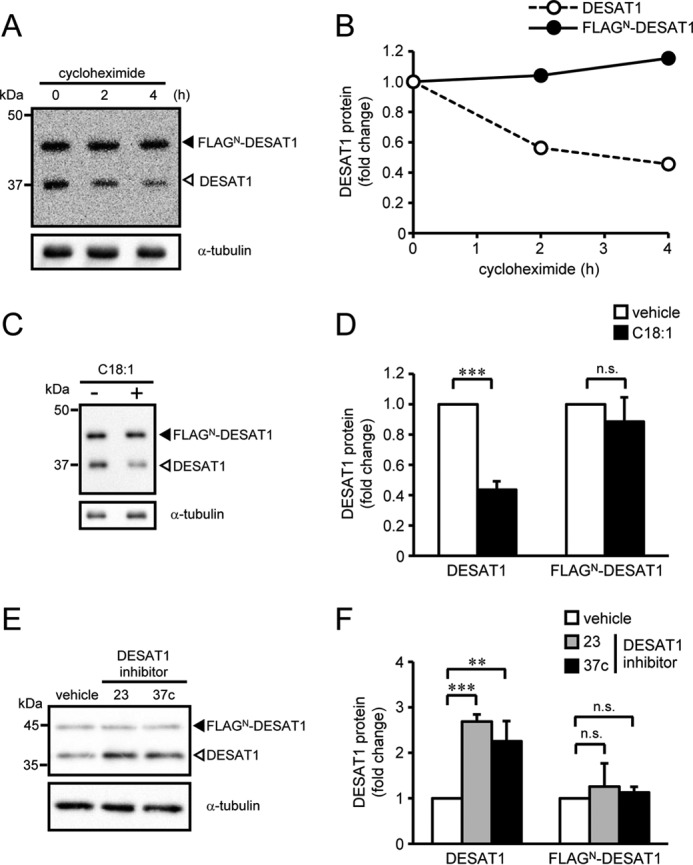
Effect of an N-terminal FLAG tag on DESAT1 expression. S2 cells expressing FLAGN-DESAT1 were treated with cycloheximide (100 μg/ml) for 0, 2, and 4 h, and the amounts of DESAT1 and α-tubulin protein were detected with anti-DESAT1 antibody and anti-α-tubulin antibody, respectively (A and B). S2 cells expressing FLAGN-DESAT1 were treated with C18:1 (100 μm) for 6 h (C and D) or DESAT1 inhibitor 23 or 37c (1 μm) for 16 h (E and F), and the amounts of DESAT1 and α-tubulin protein were detected with specific antibodies. Band intensities were determined by ImageJ software, and levels of DESAT1 proteins are shown relative to the amount of DESAT1 protein in cells not treated with cycloheximide (B) or vehicle-treated cells (D and F). Filled arrowhead, FLAGN-DESAT1; open arrowhead, endogenous DESAT1. Mean ± S.D. (n = 3). **, p < 0.01; ***, p < 0.001; n.s., not significant.
Identification of residues responsible for the degradation of DESAT1
To identify the residues of DESAT1 protein that determine the sensitivity against its protein degradation, we expressed N-terminal truncation mutants in S2 cells and analyzed the effect of C18:1 and DESAT1 inhibitor on the expression levels of the mutant proteins. Based on the crystal structures of SCD1 (4, 5), it is predicted that the first transmembrane helix of DESAT1 starts at around residue 70. The expression levels of mutant proteins lacking residues 2–11, 2–21, 2–41, or 2–60 of DESAT1 were not affected by treatment with C18:1 (supplemental Fig. 5). Furthermore, truncation of residues 2–6 of DESAT1 decreased the degradation rate of the mutant protein (Fig. 5, A and B) and abolished the effect of C18:1 and DESAT1 inhibitor on the expression levels of the mutant protein (Fig. 5, C–F). These results suggest that residues 2–6 contain residues required for the unsaturated fatty acid-induced degradation of DESAT1 protein.
Figure 5.

Effect of deletion of residues 2–6 on DESAT1 expression. S2 cells expressing Δ2–6 DESAT1-FLAGC were treated with cycloheximide (100 μg/ml) for 0, 2, and 4 h, and the amounts of DESAT1 and α-tubulin protein were detected with anti-DESAT1 antibody and anti-α-tubulin antibody, respectively (A and B). S2 cells expressing Δ2–6 DESAT1-FLAGC were treated with C18:1 (100 μm) for 6 h (C and D) or DESAT1 inhibitor 37c (1 μm) for 16 h (E and F), and the amounts of DESAT1 and α-tubulin protein were detected with specific antibodies. Band intensities were determined by ImageJ software, and levels of DESAT1 proteins are shown relative to the amount of DESAT1 protein in cells not treated with cycloheximide (B) or vehicle-treated cells (D and F). Filled arrowhead, Δ2–6 DESAT1-FLAGC; open arrowhead, endogenous DESAT1. Mean ± S.D. (n = 3). ***, p < 0.001; n.s., not significant.
As shown in Fig. 6A, the N-terminal 6-amino acid sequence of DESAT1 is 1MPPNAQ6. To identify the critical residues responsible for the degradation of DESAT1, we constructed a series of alanine substitution mutants (Fig. 6A). Similar to wild-type DESAT1, the amount of DESAT1 mutant proteins with substitutions of alanine for Asn-4 and Gln-6 (DESAT1 (N4A/Q6A)-FLAGC) was altered by C18:1 and DESAT1 inhibitor treatments (Fig. 6, B and C, and supplemental Fig. 6, A and B). However, single mutations as well as a double mutation at Pro-2 and Pro-3 of DESAT1 (Fig. 6, B and C, supplemental Fig. 6, C–H) remarkably abolished the effect of C18:1 and DESAT1 inhibitor on the expression levels of the mutant proteins. Moreover, the mutations at Pro-2 and Pro-3 of DESAT1 decreased the degradation rate of the mutant protein (Fig. 6, D and E). These results clearly demonstrate that the N-terminal sequential proline motif (Pro-2 and Pro-3) is involved in the DESAT1 degradation pathway, and we named these residues the “di-proline motif.”
Figure 6.

Role of N-terminal residues of DESAT1 in the regulation of degradation of DESAT1. A, N-terminal amino acid sequences of wild-type and mutants of DESAT1. S2 cells expressing DESAT1 (N4A/Q6A)-FLAGC, DESAT1 (P2A/P3A)-FLAGC, DESAT1 (P2A)-FLAGC, and DEAT1 (P3A)-FLAGC were treated with C18:1 (100 μm) for 6 h (B) or DESAT1 inhibitor 37c (1 μm) for 16 h (C), and the amounts of DESAT1 and α-tubulin protein were detected with anti-DESAT1 antibody and anti-α-tubulin antibody, respectively. S2 cells expressing DESAT1 (P2A/P3A)-FLAGC were treated with cycloheximide (100 μg/ml) for 0, 2, and 4 h, and the amounts of DESAT1 and α-tubulin protein were detected with specific antibodies (D and E). Band intensities were determined by ImageJ software, and levels of DESAT1 proteins are shown relative to the amount of DESAT1 protein in vehicle-treated cells (B and C) or cells not treated with cycloheximide (E). The values for DESAT1-FLAGC (Fig. 3, D and F) are shown for comparison (B and C). Filled arrowhead, DESAT1 (P2A/P3A)-FLAGC; open arrowhead, endogenous DESAT1. Mean ± S.D. (n = 3). *, p < 0.05; **, p < 0.01; ***, p < 0.001; n.s., not significant.
Role of the di-proline motif in regulation of Δ9-desaturase degradation
By comparing the N-terminal sequences of Δ9-desaturases from several species, we found that most Δ9-desaturases have a single proline residue at N-terminal position 2, except for D. melanogaster DESAT1 with the di-proline motif (Fig. 7A). To evaluate the importance of the di-proline motif in the degradation of Δ9-desaturase, we produced SCD1-DESAT1 chimeric proteins and analyzed the effect of a di-proline motif on the stability of mouse SCD1 that has a proline residue at position 2 but not at position 3 (Fig. 7B). The amount of wild-type mouse SCD1 protein expressed in S2 cells was not altered by treatment with C18:1 or DESAT1 inhibitor (Fig. 7, C and D, and supplemental Fig. 7, A and B). In contrast, the expression level of the SCD1 chimeric protein that carries residues 1–74 of DESAT1 responded to the treatments with C18:1 and DESAT1 inhibitor (Fig. 7, C and D, and supplemental Fig. 7, C and D). Alanine substitution of the di-proline motif abolished the responsiveness of the chimera protein to C18:1 and DESAT1 inhibitor treatments (Fig. 7, C and D, and supplemental Fig. 7, E and F). Furthermore, introduction of proline at the third residue of mouse SCD1 conferred responsiveness to C18:1-sensitive and DESAT1 inhibitor-sensitive degradation (Fig. 7, C and D, and supplemental Fig. 7, G and H). Taken together, these results suggest that the di-proline motif in the N-terminal region is sufficient for the unsaturated fatty acid–dependent degradation of Δ9-desaturase.
Figure 7.

Role of the di-proline motif in degradation of Δ9-desaturase. N-terminal amino acid sequences of Δ9-desaturases were compared (D. melanogaster DESAT1 (NP_652731), Mus musculus SCD1 (NP_033153), M. musculus SCD3 (NP_077770), Homo sapiens SCD1 (NP_005054), Danio rerio stearoyl-CoA desaturase (AAO25582), and S. cerevisiae Ole1 (CAA96757)) (A). Schematic illustration of DESAT1, mouse SCD1, and constructed mutants is shown (B). S2 cells expressing mouse SCD1-FLAGC, chimera-FLAGC, chimera (AA)-FLAGC, and SCD1 (di-Pro)-FLAGC were treated with C18:1 (100 μm) for 6 h (C) or DESAT1 inhibitor 37c (1 μm) for 16 h (D), and the amounts of endogenous DESAT1, FLAG-tagged exogenous Δ9-desaturases, and α-tubulin protein were detected with anti-DESAT1 antibody, anti-FLAG antibody, and anti-α-tubulin antibody, respectively. Band intensities were determined by ImageJ software, and levels of SCD1 proteins are shown relative to the amount of SCD1 protein in vehicle-treated cells (C and D). Mean ± S.D. (n = 3). *, p < 0.05; ***, p < 0.001; n.s., not significant.
Identification of the protease involved in DESAT1 degradation via the di-proline motif
Next, we screened for the protease involved in DESAT1 protein degradation. Treatment with the proteasome inhibitor MG132 and the lysosome inhibitor chloroquine did not affect the expression level of the DESAT1 protein (Fig. 8A and supplemental Fig. 8A). Because treatment with MG132 significantly increased the amount of ubiquitinated proteins in S2 cells (supplemental Fig. 9), it is likely that the proteasome is not involved in the degradation of DESAT1. Because anti-DESAT1 antibody recognizes the C-terminal domain of DESAT1, there is a possibility that the C-terminal truncated DESAT1 is degraded by the proteasome system. To analyze the amount of DESAT1 protein without using the anti-C-terminal antibody, we introduced a FLAG tag in the cytosolic domain between transmembrane helices 2 and 3 of DESAT1 (DESAT1-FLAGInternal). Consistent with the results obtained with DESAT1-FLAGC, the amount of DESAT1-FLAGInternal protein was decreased by the treatment with C18:1 (supplemental Fig. 10). Furthermore, the amount of DESAT1-FLAGInternal was not altered by the MG132 treatment. These results suggest that the proteasome system is not involved in the regulation of DESAT1 degradation in response to changes in the amount of cellular unsaturated fatty acids. In contrast, treatment with the calpain inhibitor calpeptin caused a 2.1-fold increase in the expression of DESAT1 protein (Fig. 8A and supplemental Fig. 8A), and ionomycin treatment decreased the DESAT1 expression level (supplemental Fig. 11), suggesting the involvement of calpain, a calcium-dependent cysteine protease, in the degradation of the DESAT1 protein. Given that the D. melanogaster genome encodes four calpain genes (26), we suppressed the expression of each calpain by dsRNA in S2 cells. Among four calpains, suppression of calpain A or calpain B expression inhibited C18:1-induced degradation of the DESAT1 protein (Fig. 8B and supplemental Fig. 8B). The amount of exogenously expressed DESAT1-FLAGC was increased by calpeptin treatment, whereas the expression level of DESAT1 (P2A/P3A)-FLAGC protein was not affected, implying that the di-proline motif is required for calpain-mediated degradation of DESAT1 (Fig. 8C, supplemental Fig. 8, C and D).
Figure 8.
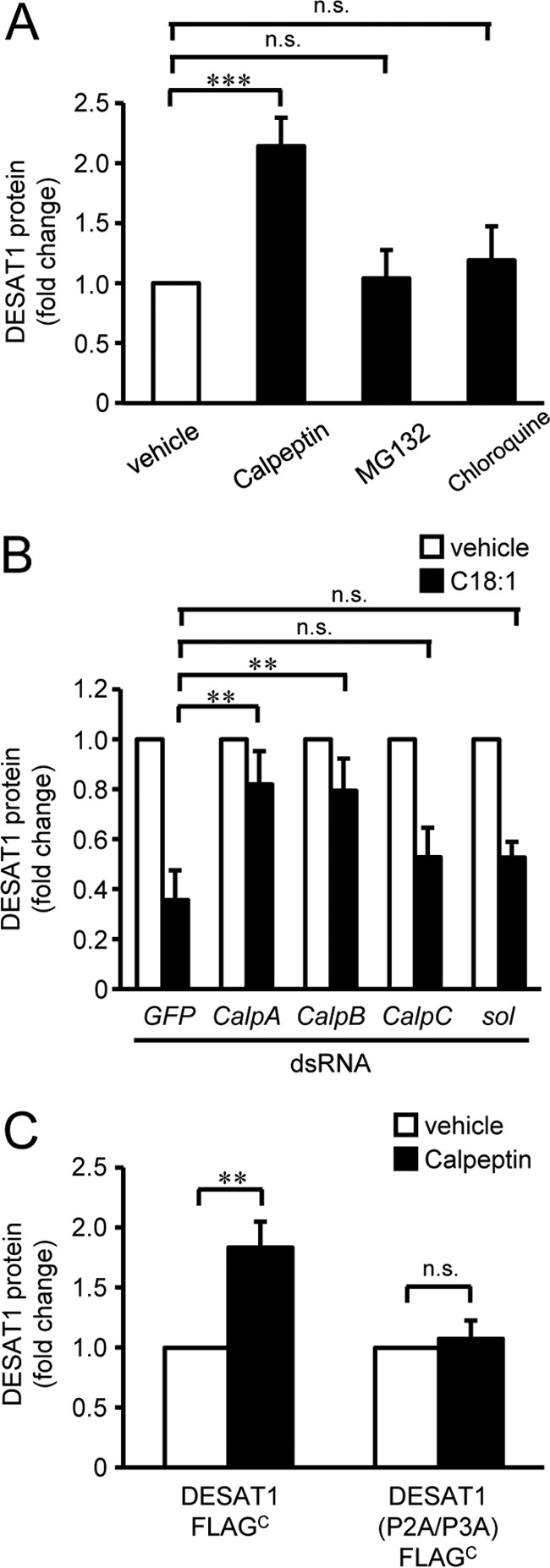
Identification of protease involved in the degradation of DESAT1. S2 cells were treated with calpeptin (50 μm), MG132 (50 μg/ml), or chloroquine (50 μm) for 6 h (A). S2 cells were treated with dsRNA against GFP, CalpA, CalpB, CalpC, or sol for 72 h and then exposed to C18:1 (100 μm) for 6 h (B). S2 cells expressing DESAT1-FLAGC and DESAT1 (P2A/P3A)-FLAGC were treated with calpeptin (50 μm) for 6 h (C). The amounts of endogenous DESAT1, FLAG-tagged exogenously expressed DESAT1, and α-tubulin protein were detected with anti-DESAT1 antibody, anti-FLAG antibody, and anti-α-tubulin antibody, respectively (A–C). Band intensities were determined by ImageJ software, and levels of DESAT1 proteins are shown relative to the amount of DESAT1 protein in vehicle-treated cells (A–C). Mean ± S.D. (n = 3). **, p < 0.01; ***, p < 0.001; n.s., not significant.
Discussion
Although DESAT1, a Δ9-desaturase in D. melanogaster, plays a role in various crucial physiological functions, such as pheromonal communication, regulation of lipid metabolism, and control of membrane fluidity, little is known about how the expression of DESAT1 is regulated at the cellular level. In this study, we identified a unique N-terminal di-proline motif that plays a crucial role in unsaturated fatty acid–dependent degradation of DESAT1.
DESAT1 protein was rapidly degraded in S2 cells with a half-life of ∼2 h (Figs. 3–6), which is comparable with those observed for mammalian and yeast Δ9-desaturases, SCD1 and Ole1, respectively (7, 15). The degradation of DESAT1 was significantly enhanced by addition of unsaturated fatty acids such as oleic (C18:1) and linoleic (C18:2) acids (Figs. 1 and 3), in which exogenously added fatty acids were rapidly incorporated into the acyl chains of phospholipids (supplemental Fig. 2). In contrast, the addition of DESAT1 inhibitors significantly suppressed the degradation of DESAT1 (Figs. 2 and 3). The treatment with DESAT1 inhibitors significantly decreased the contents of unsaturated fatty acyl chains of phospholipids (Fig. 2, A and B). These results suggest that changes in the composition of fatty acyl chains of membrane phospholipids are responsible for regulating the stability of the DESAT1 protein.
It has been reported that N-terminal sequences determine the stability of proteins (27). For example, in the N-end rule pathway, N-terminal degradation signal (N-degron)-containing proteins are recognized by N-recognins and degraded by the ubiquitin–proteasome system (28). Both mammalian SCD1 and yeast Ole1 have been shown to be degraded by the ubiquitin–proteasome system, and deletion of the N-terminal residues of SCD1 was also reported to suppress its degradation (7, 15, 29). In this study, we identified that the two sequential proline residues in the N-terminal sequence 1MPPNAQ6 are responsible for unsaturated fatty acid–dependent degradation of DESAT1 (Fig. 6). This observation was further confirmed by studies using SCD1-DESAT1 chimeric proteins where the introduction of Pro-2 to Pro-3 residues into the N terminus of SCD1 significantly enhanced unsaturated fatty acid–dependent degradation of SCD1 in S2 cells (Fig. 7). From these findings, we designated the sequential prolines at the second and third positions of DESAT1 as a di-proline motif, which plays a crucial role in the regulation of Δ9-desaturase expression in response to changes in the level of cellular unsaturated fatty acids.
The N-terminal domains of DESAT1 and mouse SCD1 have low sequence homology. Furthermore, the amounts of mutant proteins lacking residues 7–33 or 34–59 of DESAT1 were altered by C18:1 (supplemental Fig. 12). Therefore, it is likely that both residues 7–33 and 34–59 of DESAT1 do not have an essential sequence for DESAT1 protein degradation. However, truncation of residues 7–59 of DESAT1 abolished the unsaturated fatty acid–dependent degradation of DESAT1 (supplemental Fig. 12). Thus, these results suggest that the appropriate distance between the di-proline motif and the first transmembrane helix is required for the degradation of DESAT1 protein.
We demonstrated that treatment with the calpain inhibitor calpeptin or depletion of calpain A and calpain B expression significantly suppressed the degradation of DESAT1, but the expression level of DESAT1 (P2A/P3A)-FLAGC was not increased by calpeptin treatment, suggesting that calpains are responsible for di-proline motif-mediated degradation of DESAT1 (Fig. 8). The D. melanogaster genome encodes four calpain genes (26). Calpain A and calpain B are typical calpains and contain motifs for Ca2+ and lipid binding (26). In contrast, calpain C does not have proteolytic activity due to mutations in all three of its active-site residues (30). SOL is an atypical calpain and the first gene of the calpain superfamily in Drosophila to be identified (31). Although the roles of calpains in lipid metabolism have not been clarified in Drosophila, the proteolytic activity of calpain A is shown to be enhanced by phosphatidylinositol 4,5-bisphosphate, phosphatidylinositol 4-monophosphate, phosphatidylinositol, and phosphatidic acid (32). Recently, it was also reported that SOL mediates temperature reset of the circadian clock (33), anticipating the participation of calpain in the temperature-sensitive regulation of membrane fluidity. Therefore, it is plausible that calpains are involved in the regulation of DESAT1 expression that modulates phospholipid composition and lipid membrane fluidity.
In Drosophila, tissue-specific expression of the Desat1 gene is precisely controlled by distinct putative regulatory regions targeting either pheromone biosynthetic cells, neurons involved in pheromone perception, or non-neuronal cells (23). The expression of Desat1 gene in pheromone biosynthetic cells called oenocytes is under the control of circadian clock genes, which also affect pheromone production and mating behavior of Drosophila (34). It is conceivable that changes in fatty acid composition, which may result from dietary lipids or seasonal change in environmental temperature (35), affect the expression of DESAT1, thus modifying pheromone production and controlling the fluidity of cellular membranes. Elucidation of the mechanism underlying recognition of the di-proline motif will facilitate our understanding about how cells recognize change in membrane fluidity and coordinate the various physiological functions of Drosophila.
Experimental procedures
Materials and cell culture
Drosophila S2 cells (36) were maintained in Schneider's Drosophila medium supplemented with 10% fetal bovine serum (FBS), 50 units/ml penicillin, and 50 μg/ml streptomycin at 25 °C. BSA-fatty acid complex (molar ration of 1:9) was prepared by incubating fatty acid with a 0.9% NaCl solution containing BSA (111 mg/ml). Cells were incubated with BSA-fatty acid complex (100 μm fatty acid) in serum-free Schneider's Drosophila medium for 6 h. SCD1 inhibitors 23 (DS40410855) and 37c (DS18220913) were obtained from Daiichi Sankyo Co. Ltd. (24, 25). A rabbit anti-DESAT1 polyclonal antibody was generated against the C-terminal 18 amino acid residues (DQPKEEIEDAVITHKKSE) of DESAT1. Anti-α-tubulin antibody (PM054), anti-FLAG antibody (M2), and anti-ubiquitin antibody (SPC-119) were purchased from MBL, Sigma, and StressMarq Biosciences, respectively. DESAT1 coding sequence was isolated from a cDNA library of strain Canton-S. cDNA encoding mouse SCD1 was purchased from ORIGENE. Expression vectors were established by inserting the coding sequences of DESAT1 and mouse SCD1 into EcoRI and XhoI sites of pAc5.1/V5-His A plasmid (Invitrogen). Mutations were introduced by site-directed mutagenesis using PrimeSTAR Max DNA polymerase (TaKaRa) and specific primers (supplemental Table 1). Chimeras and mutants of DESAT1 and mouse SCD1 were prepared using an In-Fusion HD cloning kit (TaKaRa) and specific primers (supplemental Table 2). FLAG epitope tag (DYKDDDDDK) was inserted into the N or C terminus of the gene of interest. Internal FLAG tag was introduced between Gly-199 and Val-200 of DESAT1 using specific primers (supplemental Table 2). S2 cells were transfected with an expression vector with pCoBlast plasmid using TransFectin lipid reagent (Bio-Rad), and stable transformants were selected by 20 μg/ml blasticidin.
Measurement of the fatty acid composition of phospholipids by gas chromatography
Total lipids were extracted by Bligh and Dyer procedures (37). Phospholipids were separated by thin-layer chromatography (TLC) using hexane/diethyl ether/acetic acid (60:40:1, v/v/v) and incubated with methanolic HCl at 100 °C for 3 h. Fatty acid methyl esters were extracted and analyzed with a Shimadzu GC-14A with a flame ionization detector and an Omegawax Capillary GC column (Supelco). The temperature of the injector and the flame ionization detector were held at 200 and 280 °C, respectively. The column temperature was programmed as follows: held at 180 °C for 5 min; ramped to 220 °C at 3 °C/min; held for 7 min; ramped to 240 °C at 3 °C/min; and held for 10 min. The peak areas of methyl esters of C14:0, C16:0, C16:1, C18:0, C18:1, C18:2, C20:0, C20:4, and C22:0 were determined.
Measurement of Δ9-desaturase activity
Cells were incubated with BSA-margaric acid complex (20 μm margaric acid) in serum-free Schneider's Drosophila medium for 6 h. Conversion of margaric acid (17:0) to heptadecenoic acid (C17:1) was evaluated by gas chromatography as described above. The Δ9-desaturase activity was defined as the ratio of the amount of C17:1 to the total amount of C17 fatty acid species.
Immunoblotting
Cells were washed with PBS and lysed in lysis buffer (10 mm Tris-HCl, pH 7.4, 1% Triton X-100, 0.1% SDS, 1% sodium deoxycholate) containing 1% protease inhibitors (Nacalai Tesque). The lysates were centrifuged at 14,000 × g for 10 min at 4 °C. 40 μl of supernatant was mixed with 10 μl of sampling buffer 1 (50% sucrose, 50 mm Tris-HCl, pH 8.0, 1% SDS, 5 mm EDTA, 0.4% bromphenol blue), incubated for 10 min at 50 °C, and then mixed with 50 μl of sampling buffer 2 (10% sucrose, 10 mm Tris-HCl, pH 8.0, 0.2% SDS, 1 mm EDTA, 0.08% bromphenol blue, 60% urea). Samples were electrophoresed on SDS-polyacrylamide gel and blotted on PVDF membrane (Wako) using Trans-Blot SD Semi-Dry Electrophoretic Transfer Cell (Bio-Rad). Immunoblotting analysis was performed by using anti-DESAT1 antibody, anti-FLAG antibody (M2), anti-ubiquitin antibody, and anti-α-tubulin antibody as a loading control. Bound antibodies were detected with horseradish peroxidase-conjugated anti-rabbit IgG antibody and anti-mouse IgG antibody using Super Signal West Pico (Thermo Fisher Scientific) and Ez-Capture MG (Atto). Band intensity was determined by ImageJ software.
Real-time PCR
Cellular RNA was extracted using TRIzol reagent (Ambion), and cDNA was prepared by using ReverTra Ace qPCR RT Master Mix (Toyobo). Expression levels of Desat1 and Rpl32 (as a reference gene) were analyzed by StepOnePlus real-time PCR system using TaqMan Fast Advanced Master Mix and TaqMan Gene Expression Assays (Dm02147300_m1 and Dm02151827_g1) (Applied Biosystems).
Suppression of calpain gene expression by dsRNA
Templates with a T7 promoter flanking both sides were amplified by a traditional PCR method from cDNA encoding calpain A, calpain B, calpain C, Sol, and GFP using specific primers (supplemental Table 3) (33). The templates were used for in vitro RNA transcription with an Ambion MEGAscript T7 kit according to the manufacturer's instructions. The quality of dsRNAs was confirmed using agarose gel electrophoresis. Cells were incubated with 15 μg/ml dsRNA for 3 days.
Statistical analysis
Values are presented as means ± S.D. The statistical significance of differences between the mean values was analyzed using the non-paired t test. Multiple comparisons were performed using Tukey's test or Dunnett's test following analysis of variance. A p value of <0.05 was considered statistically significant.
Author contributions
K. N. and M. U. designed the project. A. M., K. N., and M. U. wrote the manuscript. A. M. performed most of the experiments. N. J. and Y. H. provided support in the design and performance of experiments. All authors analyzed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Kenichi Takeuchi for production of the polyclonal antibody against DESAT1. We thank Dr. Kenichi Takeuchi, Dr. Utako Kato, Ryuta Ishii (Kyoto University), Atsumi Yamaguchi, Mizuho Kaneda, and Masako Aizu (Tokyo Metropolitan Institute of Medical Science) for technical assistance and helpful discussions and Dr. Yoshikazu Uto (Daiichi Sankyo Co., Ltd.) for providing SCD1 inhibitors.
This work was supported by Grants-in-aid for Scientific Research 15H05930 (to M. U.), 15K21744 (to M. U.), 15K14476 (to M. U.), 17H03805 (to M. U.), 15K07389 (to K. N.), and 25221203 (to K. N.) from Japan Society for the Promotion of Science (JSPS) and Ministry of Education, Culture, Sports, Science and Technology (MEXT). The authors declare that they have no conflicts of interest with the contents of this article.
This article was selected as one of our Editors' Picks.
This article contains supplemental Tables S1–S3 and Figs. S1–S12.
- ER
- endoplasmic reticulum
- C14:0
- myristic acid
- C16:0
- palmitic acid
- C16:1
- palmitoleic acid
- C17:0
- margaric acid
- C17:1
- heptadecenoic acid
- C18:0
- stearic acid
- C18:1
- oleic acid
- C18:2
- linoleic acid
- C20:0
- arachidic acid
- C20:4
- arachidonic acid
- C22:0
- behenic acid.
References
- 1. Holthuis J. C., and Menon A. K. (2014) Lipid landscapes and pipelines in membrane homeostasis. Nature 510, 48–57 [DOI] [PubMed] [Google Scholar]
- 2. Los D. A., and Murata N. (1998) Structure and expression of fatty acid desaturases. Biochim. Biophys. Acta 1394, 3–15 [DOI] [PubMed] [Google Scholar]
- 3. Ntambi J. M., and Miyazaki M. (2004) Regulation of stearoyl-CoA desaturases and role in metabolism. Prog. Lipid Res. 43, 91–104 [DOI] [PubMed] [Google Scholar]
- 4. Wang H., Klein M. G., Zou H., Lane W., Snell G., Levin I., Li K., and Sang B. C. (2015) Crystal structure of human stearoyl-coenzyme A desaturase in complex with substrate. Nat. Struct. Mol. Biol. 22, 581–585 [DOI] [PubMed] [Google Scholar]
- 5. Bai Y., McCoy J. G., Levin E. J., Sobrado P., Rajashankar K. R., Fox B. G., and Zhou M. (2015) X-ray structure of a mammalian stearoyl-CoA desaturase. Nature 524, 252–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mauvoisin D., and Mounier C. (2011) Hormonal and nutritional regulation of SCD1 gene expression. Biochimie 93, 78–86 [DOI] [PubMed] [Google Scholar]
- 7. Kato H., Sakaki K., and Mihara K. (2006) Ubiquitin-proteasome-dependent degradation of mammalian ER stearoyl-CoA desaturase. J. Cell Sci. 119, 2342–2353 [DOI] [PubMed] [Google Scholar]
- 8. McDonough V. M., Stukey J. E., and Martin C. E. (1992) Specificity of unsaturated fatty acid-regulated expression of the Saccharomyces cerevisiae OLE1 gene. J. Biol. Chem. 267, 5931–5936 [PubMed] [Google Scholar]
- 9. Gonzalez C. I., and Martin C. E. (1996) Fatty acid-responsive control of mRNA stability. Unsaturated fatty acid-induced degradation of the Saccharomyces OLE1 transcript. J. Biol. Chem. 271, 25801–25809 [DOI] [PubMed] [Google Scholar]
- 10. Choi J. Y., Stukey J., Hwang S. Y., and Martin C. E. (1996) Regulatory elements that control transcription activation and unsaturated fatty acid-mediated repression of the Saccharomyces cerevisiae OLE1 gene. J. Biol. Chem. 271, 3581–3589 [DOI] [PubMed] [Google Scholar]
- 11. Vasconcelles M. J., Jiang Y., McDaid K., Gilooly L., Wretzel S., Porter D. L., Martin C. E., and Goldberg M. A. (2001) Identification and characterization of a low oxygen response element involved in the hypoxic induction of a family of Saccharomyces cerevisiae genes. Implications for the conservation of oxygen sensing in eukaryotes. J. Biol. Chem. 276, 14374–14384 [DOI] [PubMed] [Google Scholar]
- 12. Hoppe T., Matuschewski K., Rape M., Schlenker S., Ulrich H. D., and Jentsch S. (2000) Activation of a membrane-bound transcription factor by regulated ubiquitin/proteasome-dependent processing. Cell 102, 577–586 [DOI] [PubMed] [Google Scholar]
- 13. Chellappa R., Kandasamy P., Oh C. S., Jiang Y., Vemula M., and Martin C. E. (2001) The membrane proteins, Spt23p and Mga2p, play distinct roles in the activation of Saccharomyces cerevisiae OLE1 gene expression. Fatty acid-mediated regulation of Mga2p activity is independent of its proteolytic processing into a soluble transcription activator. J. Biol. Chem. 276, 43548–43556 [DOI] [PubMed] [Google Scholar]
- 14. Covino R., Ballweg S., Stordeur C., Michaelis J. B., Puth K., Wernig F., Bahrami A., Ernst A. M., Hummer G., and Ernst R. (2016) A eukaryotic sensor for membrane lipid saturation. Mol. Cell 63, 49–59 [DOI] [PubMed] [Google Scholar]
- 15. Braun S., Matuschewski K., Rape M., Thoms S., and Jentsch S. (2002) Role of the ubiquitin-selective CDC48(UFD1/NPL4)chaperone (segregase) in ERAD of OLE1 and other substrates. EMBO J. 21, 615–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wicker-Thomas C., Henriet C., and Dallerac R. (1997) Partial characterization of a fatty acid desaturase gene in Drosophila melanogaster. Insect Biochem. Mol. Biol. 27, 963–972 [DOI] [PubMed] [Google Scholar]
- 17. Labeur C., Dallerac R., and Wicker-Thomas C. (2002) Involvement of desat1 gene in the control of Drosophila melanogaster pheromone biosynthesis. Genetica 114, 269–274 [DOI] [PubMed] [Google Scholar]
- 18. Ueyama M., Chertemps T., Labeur C., and Wicker-Thomas C. (2005) Mutations in the desat1 gene reduces the production of courtship stimulatory pheromones through a marked effect on fatty acids in Drosophila melanogaster. Insect Biochem. Mol. Biol. 35, 911–920 [DOI] [PubMed] [Google Scholar]
- 19. Dallerac R., Labeur C., Jallon J. M., Knipple D. C., Roelofs W. L., and Wicker-Thomas C. (2000) A delta 9 desaturase gene with a different substrate specificity is responsible for the cuticular diene hydrocarbon polymorphism in Drosophila melanogaster. Proc. Natl. Acad. Sci. U.S.A. 97, 9449–9454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Takahashi A., Tsaur S. C., Coyne J. A., and Wu C. I. (2001) The nucleotide changes governing cuticular hydrocarbon variation and their evolution in Drosophila melanogaster. Proc. Natl. Acad. Sci. U.S.A. 98, 3920–3925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Clark A. J., and Block K. (1959) The absence of sterol synthesis in insects. J. Biol. Chem. 234, 2578–2582 [PubMed] [Google Scholar]
- 22. Shen L. R., Lai C. Q., Feng X., Parnell L. D., Wan J. B., Wang J. D., Li D., Ordovas J. M., and Kang J. X. (2010) Drosophila lacks C20 and C22 PUFAs. J. Lipid Res. 51, 2985–2992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bousquet F., Nojima T., Houot B., Chauvel I., Chaudy S., Dupas S., Yamamoto D., and Ferveur J. F. (2012) Expression of a desaturase gene, desat1, in neural and nonneural tissues separately affects perception and emission of sex pheromones in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 109, 249–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Uto Y., Ogata T., Harada J., Kiyotsuka Y., Ueno Y., Miyazawa Y., Kurata H., Deguchi T., Watanabe N., Takagi T., Wakimoto S., Okuyama R., Abe M., Kurikawa N., Kawamura S., et al. (2009) Novel and potent inhibitors of stearoyl-CoA desaturase-1. Part I: discovery of 3-(2-hydroxyethoxy)-4-methoxy-N-[5-(3-trifluoromethylbenzyl)thiazol-2-yl]benzamide. Bioorg. Med. Chem. Lett. 19, 4151–4158 [DOI] [PubMed] [Google Scholar]
- 25. Uto Y., Ogata T., Kiyotsuka Y., Miyazawa Y., Ueno Y., Kurata H., Deguchi T., Yamada M., Watanabe N., Takagi T., Wakimoto S., Okuyama R., Konishi M., Kurikawa N., Kono K., and Osumi J. (2009) Novel and potent inhibitors of stearoyl-CoA desaturase-1. Part II: Identification of 4-ethylamino-3-(2-hydroxyethoxy)-N-[5-(3-trifluoromethylbenzyl)thiazol-2-yl]benzamide and its biological evaluation. Bioorg. Med. Chem. Lett. 19, 4159–4166 [DOI] [PubMed] [Google Scholar]
- 26. Friedrich P., Tompa P., and Farkas A. (2004) The calpain-system of Drosophila melanogaster: coming of age. Bioessays 26, 1088–1096 [DOI] [PubMed] [Google Scholar]
- 27. Bachmair A., Finley D., and Varshavsky A. (1986) In vivo half-life of a protein is a function of its amino-terminal residue. Science 234, 179–186 [DOI] [PubMed] [Google Scholar]
- 28. Tasaki T., Sriram S. M., Park K. S., and Kwon Y. T. (2012) The N-end rule pathway. Annu. Rev. Biochem. 81, 261–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mziaut H., Korza G., and Ozols J. (2000) The N terminus of microsomal delta 9 stearoyl-CoA desaturase contains the sequence determinant for its rapid degradation. Proc. Natl. Acad. Sci. U.S.A. 97, 8883–8888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Spadoni C., Farkas A., Sinka R., Tompa P., and Friedrich P. (2003) Molecular cloning and RNA expression of a novel Drosophila calpain, calpain C. Biochem. Biophys. Res. Commun. 303, 343–349 [DOI] [PubMed] [Google Scholar]
- 31. Delaney S. J., Hayward D. C., Barleben F., Fischbach K. F., and Miklos G. L. (1991) Molecular cloning and analysis of small optic lobes, a structural brain gene of Drosophila melanogaster. Proc. Natl. Acad. Sci. U.S.A. 88, 7214–7218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jékely G., and Friedrich P. (1999) Characterization of two recombinant Drosophila calpains. CALPA and a novel homolog, CALPB. J. Biol. Chem. 274, 23893–23900 [DOI] [PubMed] [Google Scholar]
- 33. Tataroglu O., Zhao X., Busza A., Ling J., O'Neill J. S., and Emery P. (2015) Calcium and SOL protease mediate temperature resetting of circadian clocks. Cell 163, 1214–1224 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34. Krupp J. J., Kent C., Billeter J. C., Azanchi R., So A. K., Schonfeld J. A., Smith B. P., Lucas C., and Levine J. D. (2008) Social experience modifies pheromone expression and mating behavior in male Drosophila melanogaster. Curr. Biol. 18, 1373–1383 [DOI] [PubMed] [Google Scholar]
- 35. Tattersall G. J., Sinclair B. J., Withers P. C., Fields P. A., Seebacher F., Cooper C. E., and Maloney S. K. (2012) Coping with thermal challenges: physiological adaptations to environmental temperatures. Compr. Physiol. 2, 2151–2202 [DOI] [PubMed] [Google Scholar]
- 36. Schneider I. (1972) Cell lines derived from late embryonic stages of Drosophila melanogaster. J. Embryol. Exp. Morphol. 27, 353–365 [PubMed] [Google Scholar]
- 37. Bligh E. G., and Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.