

Abstract
Galectin-1 is an important immunoregulatory factor and can mediate the host–pathogen interaction via binding glycans on the surface of various viruses. We previously reported that avian respiratory viruses, including lentogenic Newcastle disease virus (NDV), can induce up-regulation of chicken galectin (CG)-1B in the primary target organ. In this study, we investigated whether CG-1B participated in the infectious process of NDV in chickens. We demonstrated that velogenic NDV induced up-regulation of CG-1B in target organs. We also found that CG-1B directly bound to NDV virions and inhibited their hemagglutination activity in vitro. We confirmed that CG-1B interacted with NDV hemagglutinin–neuraminidase (HN) glycoprotein, in which the specific G4 N-glycans significantly contributed to the interaction between CG-1B and HN glycoprotein. The presence of extracellular CG-1B, rather than the internalization process, inhibited adsorption of NDV. The interaction between intracellular CG-1B and NDV HN glycoproteins inhibited cell-surface expression of HN glycoprotein and reduced the titer of progeny virus in NDV-infected DF-1 cells. Significantly, the replication of parental and HN glycosylation mutant viruses in CG-1B knockdown and overexpression cells demonstrated that the replication of NDV was correlated with the expression of CG-1B in a specific glycan-dependent manner. Collectively, our results indicate that CG-1B has anti-NDV activity by binding to N-glycans on HN glycoprotein.
Keywords: galectin, glycoprotein, viral replication, virus, virus entry, Chicken galectin-1B, HN glycoprotein, Newcastle disease virus, adsorption, replication
Introduction
Newcastle disease virus (NDV)2 is a member of the genus Avulavirus in the family Paramyxoviridae (1, 2). It is an important pathogen of many species of birds, causing serious disease, and results in significant economic losses to the poultry industry (2). NDV contains a single-stranded, negative-sense, non-segmented RNA genome. The 15-kb genomic RNA contains six genes that encode nucleoprotein (NP), phosphoprotein (P), matrix, fusion (F), hemagglutinin–neuraminidase (HN), and polymerase (L). Infection by NDV requires two functional glycoproteins, HN and F, which are embedded in the envelope of NDV that surrounds the matrix protein and nucleocapsid core (3). The HN glycoprotein binds to sialic acid-containing receptors, and the F glycoprotein mediates membrane fusion between the viral and cellular membranes. The HN glycoprotein of NDV is a type II membrane protein, with N-terminal transmembrane domains followed by a stalk region and a C-terminal globular head domain (3, 4). NDV HN glycoprotein is a multifunctional protein. It is responsible for binding to sialic acid-containing cellular receptors, promoting the fusion activity of F protein, thereby allowing the virus to penetrate the cell surface, and removing the sialic acid from progeny virus particles to prevent viral self-agglutination via its neuraminidase (NA) activity (5).
N-Linked glycosylation is one of the most common forms of post-translational modification of protein. Viral envelope protein usually undergoes N-linked glycosylation by using this host cellular process, which ultimately affects the function of the viral glycoproteins in stability, antigenicity, and host cell invasion (6). It is believed that glycosylation in the envelope glycoproteins of Nipah virus, human immunodeficiency virus type-1 (HIV-1), West Nile virus, and influenza virus is associated with viral pathogenicity (7–11). NDV HN glycoprotein contains six potential N-linked glycosylation sites (residues 119, 341, 433, 481, 508, and 538), only four of which are utilized for N-linked glycosylation modification (12). One is located in the stalk region at residue 119 (G1), and the remaining three are located in the globular head at residues 341, 433, and 481 (G2, G3, and G4, respectively). Glycosylation mutation analysis demonstrates that G1 and G2 mainly mediate the biological activities of HN glycoprotein, whereas G3 and G4 are involved in folding and activity of HN glycoprotein (12). Loss of N-linked glycans of HN glycoprotein can alter virulence of NDV strains (5). During infection, N-glycans on viral glycoproteins can be recognized by various endogenous lectins that have important roles in the sensing and recognition of pathogens (6). It has also been shown that the direct interaction of endogenous lectins with viral glycoproteins is implicated in virus entry or clearance (6). For example, West Nile virus and Dengue virus can interact with cell-surface-bound lectin dendritic cell-specific ICAM-3 grabbing non-integrin (DC-SIGN) to facilitate entry of the virus into the target cells (13, 14), whereas host soluble mannose-binding lectin binds to Ebola and Marburg virus envelope glycoproteins and blocks the interaction between both viruses and DC-SIGN (15). Surfactant-associated protein (SP)-A and SP-D can interact with influenza virus to prevent associations of HA glycoprotein with its cell receptors (16).
In recent years, it has become clear that galectins (S-type lectins), which were initially thought to recognize endogenous host glycans and mediate diverse biological processes (17, 18), could also bind glycans on the surface of potentially pathogenic microorganisms to function as recognition and effective factors in innate immunity (7, 8, 19). Galectin-1 has been extensively reported as an important contributor to immune homeostasis and exhibits anti-inflammatory activity during infection (20–22). Galectin-1 specifically cross-links N-glycans on Nipah virus envelope glycoproteins, resulting in aberrant oligomerization and fusion inhibition (19). Galectin-1 also promotes an innate immune response to viral infection via stimulating production of proinflammatory cytokines (19). Galectin-1 binds directly to envelope glycoproteins of influenza virus and inhibits viral infection (8). However, galectin-1 facilitates Nipah virus and HIV-1 adsorption and infection through direct interaction with N-glycans on viral glycoproteins and host receptors (9, 23, 24).
So far, the contributions of host factors during the course of NDV infection remain largely unknown. Our previous proteomics analyses have shown that the abundance of chicken galectin (CG)-1B is up-regulated in the trachea of NDV-infected chickens, the primary target organ for this virus (25). In this study, we demonstrate for the first time that CG-1B directly bound to NDV and inhibited its hemagglutination (HA) activity in vitro. Furthermore, we show that CG-1B bound to NDV via interaction with HN glycoproteins, and specific G4 N-glycans significantly contributed to this interaction. The presence of CG-1B inhibited NDV infection by reducing adsorption and replication of NDV. Our results suggest that CG-1B plays important roles in antiviral defense against NDV infection.
Results
Expression of CG-1B mRNA is up-regulated in tissues of chicken infected with NDV
In our previous study, we found that the abundance of CG-1B in chicken tracheal tissue was increased after inoculation with lentogenic NDV La Sota (25). In this study, the viral loads in various tissue samples of chickens infected with velogenic NDV F48E9 were determined by real-time RT-PCR. As shown in Fig. 1A, viral RNA was detectable in trachea, cecal tonsil, proventriculus, harderian glands, liver, spleen, kidney, lung, and bursa of fabricius in F48E9-infected chickens at 24 and 48 h post-inoculation (hpi). The viral load was persistently elevated with time and significantly increased in trachea, harderian gland, kidney, lung, and bursa of fabricius (p < 0.05). No NDV RNA was detected in chickens from the control group.
Figure 1.

Viral load and expression of CG-1B mRNA were quantified using real-time RT-PCR. A, viral loads were determined in tissue samples of five NDV F48E9-infected and control birds. The average viral copy number per 1 μg of RNA of each tissue was calculated using a standard curve based on 10-fold dilution series of standard templates with known concentration. No NDV RNA was detected in chickens from the control group. We analyzed the change in viral load in each detected tissue from 24 to 48 hpi. B, transcript alteration of CG-1B gene in NDV F48E9-infected chickens. Comparison of mRNA levels of CG-1B gene at 24 and 48 hpi. Total RNA extracts were prepared from the tissue samples of F48E9-infected and control birds and measured by real-time RT-PCR. Data were normalized with expression of 18S rRNA gene, and mRNA expression of CG-1B gene was calculated relative to that of the control group (relative expression = 1). Change in mRNA level of CG-1B in each tissue at 24 hpi was compared with that of 48 hpi. Data represent means of five biological replicates per group. Error bars indicate S.D. of the mean. Data were compared using the Student's t test. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Transcriptional alteration analysis of CG-1B at 24 and 48 hpi was achieved using real-time RT-PCR. As shown in Fig. 1B, mRNA expression level of CG-1B in trachea, cecal tonsil, proventriculus, harderian gland, liver, spleen, kidney, lung, and bursa of fabricius in F48E9-infected chickens was significantly up-regulated in comparison with the birds in the control group at 24 hpi (p < 0.05). Notably, trends in CG-1B expression changes with time in detected tissues were not identical. The mRNA level of CG-1B increased persistently in trachea, cecal tonsils, and proventriculus with time (p < 0.05). The level of CG-1B in harderian gland was not significantly changed during 24–48 hpi. In contrast, the mRNA levels of CG-1B in liver, spleen, kidney, lung, and bursa of fabricius were dropped considerably at various degrees from 24 to 48 hpi. Our results indicated that CG-1B was up-regulated in target organs at early stages of velogenic NDV infection. Also, CG-1B produced by certain tissues may be insufficient to inhibit the F48E9 infection, which is a velogenic strain that can induce severe lesions and 100% mortality within 3–4 days post-infection (26). This further resulted in decreased expression of CG-1B with time.
CG-1B binds to NDV and inhibits viral HA activity
We used an ELISA-based assay to examine whether CG-1B bound directly to NDV. The standard ELISA curves were established based on the optical density (OD) values of 2-fold serially diluted NDVs. The linear range of the La Sota and F48E9 standard curves was 3–8 log2 hemagglutinating units (HAU) (Fig. 2A). Then, 6 log2 HAU of NDVs that were located within the linear range of the standard curve were applied to CG-1B-coated solid-phase plates. CG-1B bound 5.44 log2 HAU of lentogenic La Sota and 5.72 log2 HAU of velogenic F48E9 based on the linear standard curve (Fig. 2B). Binding of CG-1B and allantoic fluid, CG-1B and BSA, mock preparations and NDVs, and mock preparations and allantoic fluid was below the limit of the linear standard curves (Fig. 2B). Binding between NDVs and CG-1B was abolished by adding β-galactose rather than mannose, which specifically competed for the carbohydrate recognition domains (CRDs) of CG-1B and acted as an antagonist (Fig. 2B). These results indicate that CG-1B directly binds to NDV, and this interaction is dependent on carbohydrate recognition.
Figure 2.

Binding of CG-1B on the surface of NDV. A, standard curves of NDV La Sota and F48E9 were established based on OD values of 2-fold serially diluted NDVs. B, binding of CG-1B to different NDV strains. NDV F48E9 and La Sota were added to 96-well plates coated with CG-1B (2 μg). β-Galactose or mannose was used as an antagonist. Bound virus particles were detected by ELISA with chicken anti-NDV La Sota antiserum. Uninfected chick embryo allantoic fluid and BSA were used as negative controls of NDV. The number of virions bound to CG-1B was calculated based on the linear standard curves. Mock-purified preparation-coated wells served as the negative control of CG-1B. Each value represents the mean and S.D. Data were compared by using Student's t test. ***, p < 0.001.
It has been shown that chicken C-type lectin and human galectin-1, which is known as S-type lectin, inhibit HA activity of influenza virus (8, 27). Thus, we further examined whether chicken CG-1B inhibited HA activity of NDV using an HA assay. Because high concentrations of galectin-1 can induce HA per se via binding to erythrocytes, we estimated the bioactivity of CG-1B using an HA assay. CG-1B (8 μm) induced HA by binding to chicken peripheral red blood cells (PBRCs), whereas 4 μm CG-1B did not (Fig. 3A). To avoid interference of HA activity of CG-1B, 4 μm CG-1B was used to evaluate the effects of CG-1B on the HA activity of NDV. Consequently, we observed that 4 μm CG-1B inhibited the HA activity of 2 HAU of NDV (Fig. 3B). This agrees with previous studies that showed that lectins can inhibit HA activity of 2–4 HAU of influenza virus (8, 28). Inhibition of NDV HA induction by CG-1B was abolished by addition of β-galactose rather than mannose (Fig. 3B), indicating that hemagglutination inhibition (HI) activity of CG-1B on NDVs was specific and glycan-dependent. We demonstrated that CG-1B directly binds to NDV and inhibits HA activity of NDV, suggesting that CG-1B participates in antiviral defense of chickens against NDV.
Figure 3.

CG-1B inhibits HA activity of NDV. A, various doses of CG-1B (0.5–32 μm) were incubated with 1% chicken PBRCs, and HA was recorded after 30 min. B, NDV F48E9 and La Sota (0.5–128 HAU) were incubated with 4 μm CG-1B solely or in the presence of β-galactose or mannose for 1 h, followed by addition of 1% PBRCs. HA was recorded after 30 min. Data were compared by using Student's t test. ***, p < 0.001.
CG-1B binds to HN glycoprotein of NDV
Microscale affinity chromatography was used to determine whether CG-1B bound to the envelope glycoprotein of NDV. NDV F48E9 and La Sota allantoic fluid extracts were applied to a CG-1B affinity chromatography column, after removing unbound proteins, and bound glycoprotein was eluted with β-galactose. A 70-kDa NDV protein was eluted from the column by β-galactose, and Western blotting indicated that this protein could be specifically recognized by the antibody of NDV HN glycoprotein (Fig. 4A). To identify the viral proteins eluted from the column, the protein bands were manually excised from gels and subjected to LC-MS/MS identification. Database searches positively identified HN of NDV La Sota strain and Italian strain, which shares significant amino acid similarity with F48E9. Detailed information about the identified proteins is shown in Table 1. To confirm the direct interaction of CG-1B and HN glycoprotein, co-immunoprecipitation (Co-IP) assay using His and HN antibodies was performed. As shown in Fig. 4B, HN protein was co-precipitated with His-tagged CG-1B and vice versa. No HN protein was precipitated with anti-His antibody in the absence of CG-1B. Neither HN nor CG-1B was precipitated with the isotype control antibody. In summary, we identified HN glycoprotein of NDV as a viral protein that binds to CG-1B.
Figure 4.

CG-1B binds to HN glycoprotein of NDV. A, viral extracts of La Sota and F48E9 were applied to the CG-1B affinity chromatography column; bound viral proteins were eluted with 0.1 m β-galactose in PBS. After SDS-PAGE separation, viral proteins of La Sota (lane 1) and F48E9 (lane 2) eluted from CG-1B column and control column La Sota (lane 3) and F48E9 (lane 4) were detected by Western blotting using HN–glycoprotein-specific antibody. B, Co-IP assay. CG-1B was incubated with NDV viral lysates, which were immunoprecipitated with anti-His or anti-HN monoclonal antibody. The Co-IP assay was also performed with anti-His antibody in the absence of CG-1B, and normal mouse IgG2 was used as the isotype control antibody of His and HN monoclonal antibody, both of which belong to the IgG2 subclass. The precipitated proteins were detected by Western blotting using anti-NDV HN and anti-His monoclonal antibodies, respectively.
Table 1.
Identification of NDV viral protein interacted with CG-1B by LC-MS/MS
| No. | Accession no.a | Ion scoresb | Mass | Matched peptidesc | Sequence coverage | Protein name |
|---|---|---|---|---|---|---|
| % | ||||||
| 1 | P35743 | 3762 | 63,854 | AVSQVALENDEREAK | 65 | HN OS = NDV (strain chicken/United States/LaSota/46) GN = HN PE = 3 SV = 1 |
| ISRAEEKITSTLGSNQDVVDRIYK | ||||||
| ELIVDDASDVTSFYPSAFQEHLNFIPAPTTGSGCTRIPSFDMSATHYCYTHNVILSGCRDHSHSYQYLALGVLR | ||||||
| VFFSTLRSINLDDTQNRKSCSVSATPLGCDMLCSKVTETEEEDYNSAVPTR | ||||||
| LGFDGQYHEKDLDVTTLFGDWVANYPGVGGGSFIDSRVWFSVYGGLKPNSPSDTVQEGKYVIYKR | ||||||
| SSYKPGR | ||||||
| RIQQAILSIKVSTSLGEDPVLTVPPNTVTLMGAEGRILTVGTSHFLYQR | ||||||
| TATLHSPYTFNAFTRPGSIPCQASAR | ||||||
| GVFGTMLDGVQARLNPASAVFDSTSR | ||||||
| AAYTTSTCFK | ||||||
| TYCLSIAEISNTLFGEFRIVPLLVEILKDDGVR | ||||||
| 2 | P12556 | 533 | 63,307 | ELIVDDASDVTSFYPSAFQEHLNFIPAPTTGSGCTR | 28 | HN OS = NDV (strain Italian/45) GN = HN PE = 3 SV = 1 |
| VFFSTLRSINLDDNQNR | ||||||
| LGFDGQYHEK | ||||||
| VWFPVYGGLKPNSPSDTAQEGR | ||||||
| SSYKPGR | ||||||
| RVQQAILSIK | ||||||
| GVFGTMLDDKQARLNPVSAVFDNISR | ||||||
| TYCLSIAEISNTLFGEFRIVPLLVEILKEDGV |
a Accession numbers are according to Uniprot database.
b Ion scores were from LC-MS/MS identification. Proteins with significant ion score of >21 (p < 0.05) were considered successfully identified.
c Peptides identified by LC-MS/MS matched the successfully identified protein.
Generation and characterization of HN mutant NDVs
A reverse genetics system was used to generate mutant NDVs containing HN N-linked glycosylation site mutations. All recovered mutant viruses were subjected to RT-PCR, and the HN genes were sequenced to confirm the presence of the introduced mutations (Fig. 5A). Viral proteins extracted from parental and HN mutant viruses containing equivalent HAU were subjected to SDS-PAGE, and the levels of HN and NP proteins were analyzed by Western blotting. The expression levels of HN mutants were comparable with the parental HN (Fig. 5B). Our results also indicated that all HN mutants migrated faster than the parental HN. The HN proteins with single mutation at sites 341 and 433 (G2 and G3) showed similar migration rates and migrated faster than HN protein with a single mutation at site 119 (G1). HN protein with two mutations at sites 119 and 341 (G12) migrated faster than these single mutant HN proteins. HN protein with a single mutation at site 481 (G4) migrated faster than the other single mutant HN proteins (Fig. 5B).
Figure 5.

Confirmation of the loss of N-linked glycans in HN mutant viruses. A, schematic diagram of the positions of the N-linked glycans in HN mutant viruses. RNA was extracted from the recovered mutant viruses; the fragments containing the mutant sites were amplified by RT-PCR using primers LaFull-F and LaFull-R and sequenced to confirm the presence of the introduced mutations in the HN mutant viruses. B, viral proteins extracted from parental and HN mutant viruses containing equivalent HAU were subjected to SDS-PAGE; the expression of HN and NP protein was detected by corresponding antibodies. The IRDye800DX-conjugated secondary antibodies were used, and the membranes were scanned using a LI-COR infrared imaging system with Odyssey software (Li-Cor Biosciences, Lincoln, NE). The densitometric intensity was determined using ImageJ; the level of HN protein was normalized by the expression of NP protein. The intensity ratios of La Sota-G1, La Sota-G2, La Sota-G3, La Sota-G4, and La Sota-G12 were normalized to the intensity of La Sota.
Growth and virulence of HN mutant NDVs
The multicycle growth kinetics of La Sota, La Sota-G1, -G2, -G3, -G4, and -G12 in specific pathogen-free (SPF) embryonated chicken eggs were determined to compare the replication efficiencies of parental and HN mutant viruses in vitro. La Sota, La Sota-G1, -G2, -G3, -G4, and -G12 had similar replication kinetics. Significantly, all mutant viruses showed delayed growth characteristics before 72 hpi. The titers of all mutant viruses at 24 hpi were significantly lower than the parental virus (p < 0.05), in which the titer of La Sota-G1 was significantly higher than that of the other mutant viruses (p < 0.05). At 48 hpi, the virus titers of La Sota-G1 and parental virus were comparable and significantly higher than those of other mutant viruses, in which La Sota-G4 and -G12 had the lowest virus titers (p < 0.05). All viruses reached maximum titers at 72 hpi, and the maximum titers of La Sota-G1, -G2, -G3, and -G4 were comparable with those of parental La Sota, except that La Sota-G12 had a lower virus titer than the parental and other HN mutant viruses (Fig. 6A).
Figure 6.

Growth kinetics of HN mutant viruses in vitro and in vivo. A, growth kinetics of HN mutant viruses in embryonated eggs. The parental La Sota and rescued La Sota-G1, La Sota-G2, La Sota-G3, La Sota-G4, and La Sota-G12 (0.5 HAU) were inoculated into the allantoic cavities of 9-day-old embryonated SPF eggs, the allantoic fluid of five eggs receiving each virus was harvested at 1–5 dpi, and the viral titers were determined by HA assay. Viral titers of HN mutant viruses at each time point were compared with that of parental virus. B, growth kinetics of HN mutant viruses in 1-day-old chicks. Chicks were inoculated with La Sota, La Sota-G1, -G2, -G3, -G4, or -G12 by oculonasal application at 1 day of age with a dose of log106 EID50/100 μl per chick. Five birds from each group were killed humanely at 1, 3, 6, and 12 dpi, and fresh tissue samples from trachea, lung, and brain were quickly harvested. Viral load in tissues was analyzed using real-time RT-PCR as described in Fig. 1A. No viral RNA was detected in brains from any infected chickens. Viral loads in trachea and lung from HN mutant virus-infected chickens at each time point were compared with those in the trachea and lung from parental virus-infected chickens. C, sera were collected at 1, 3, 6, and 12 dpi, and NDV HI antibody titers were determined by HI assay. HI antibody titers induced by HN mutant viruses at each time point were compared with that of parental virus. Each value represents the mean and S.D. Data were compared by using Student's t test. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
The replication efficiency of La Sota, La Sota-G1, -G2, -G3, -G4, and -G12 in vivo was further determined in 1-day-old chickens. NDV RNA was detected in trachea and lung tissues at 1 dpi from all infected groups, and viral copy number persistently increased and peaked at 6 dpi, whereas no viral RNA was detected at 12 dpi (Fig. 6B). The results in brain tissues from all groups were negative. The viral loads in the trachea from all mutant virus-infected chickens were significantly lower than those of parental La Sota-infected chickens at 1 and 3 dpi (p < 0.05) (Fig. 6B). The viral loads in the trachea from La Sota-G1-, -G2-, and G3-infected groups were comparable with those of the La-Sota-infected group at 6 dpi. The viral loads in the trachea from La Sota-G4- and -G12-infected groups were significantly lower than those of the other groups at 1, 3, and 6 dpi (p < 0.05). The viral loads in the trachea from La Sota-G4-infected groups were lowest among all groups at 1 and 3 dpi. Similar results were observed in lungs (Fig. 6B). Serum HI antibody was detected from 6 dpi and increased with time in all groups. HI antibody titers in La Sota-G4- and G12-infected groups were significantly lower than those in other groups at 6 and 12 dpi, which may be attributed to the low replication levels of these two viruses in vivo (Fig. 6C). What is noteworthy is that although the numerical values of virus titers in vitro and viral loads in vivo of mutant viruses at indicated time points were statistically lower than that of parental virus, there were no considerable differences in replication level among the parental and mutant viruses.
The virulence of HN mutant viruses was evaluated by mean death time (MDT) in 9-day-old SPF embryonated chicken eggs. The MDTs for parental and HN mutant viruses were 100 h (La Sota), 112 h (La Sota-G1), 116 h (La Sota-G2), 110 h (La Sota-G3), 144 h (La Sota-G4), and 118 h (La Sota-G12), which indicated that loss of glycosylation from the HN protein leads to decreased virulence of La Sota. La Sota-G4, which has lost glycosylation at residue 481, has greatly decreased virulence.
CG-1B binds to specific N-glycans on HN glycoprotein
The ability of CG-1B to bind to NDV and NDV-HN mutants was tested by the above-mentioned ELISA. The levels of HN and HN mutants in parental and HN mutant viruses were first quantified by ELISA based on the linear standard curve of HN protein. Then, La Sota, La Sota-G1, -G2, -G3, -G4, and -G12 containing equivalent HN proteins (Fig. 7A) were applied to ELISA plates coated with CG-1B. Based on the linear range of the La Sota standard curve, insufficient La Sota-G4 was detected, indicating that NDV HN G4 N-glycans contribute significantly to CG-1B binding to NDV HN glycoprotein (Fig. 7B). However, the other HN N-glycans may also contribute to CG-1B binding to HN glycoprotein because CG-1B also bound less La Sota-G1 (0.60), -G2 (0.58), -G3 (0.64), and -G12 (0.61) than La Sota. Similar to parental La Sota, the binding of CG-1B to La Sota-G1, -G2, -G3, and -G12 was abolished in the presence of β-galactose rather than mannose, suggesting that the binding of these HN mutant viruses to CG-1B was still glycan-specific (Fig. 7B). The Co-IP assay showed that although CG-1B precipitated less HN G1 (0.63), G2 (0.64), G3 (0.65), and G12 (0.63), significantly less HN G4 (0.07) was precipitated when compared with parental HN. Also, in comparison with parental HN, less CG-1B was precipitated by HN G1 (0.62), G2 (0.61), G3 (0.68), and G12 (0.64), and significantly less CG-1B was precipitated by HN G4 (0.09) (Fig. 7C). These results indicated that G4 N-glycans are critical for the interaction between CG-1B and HN protein.
Figure 7.
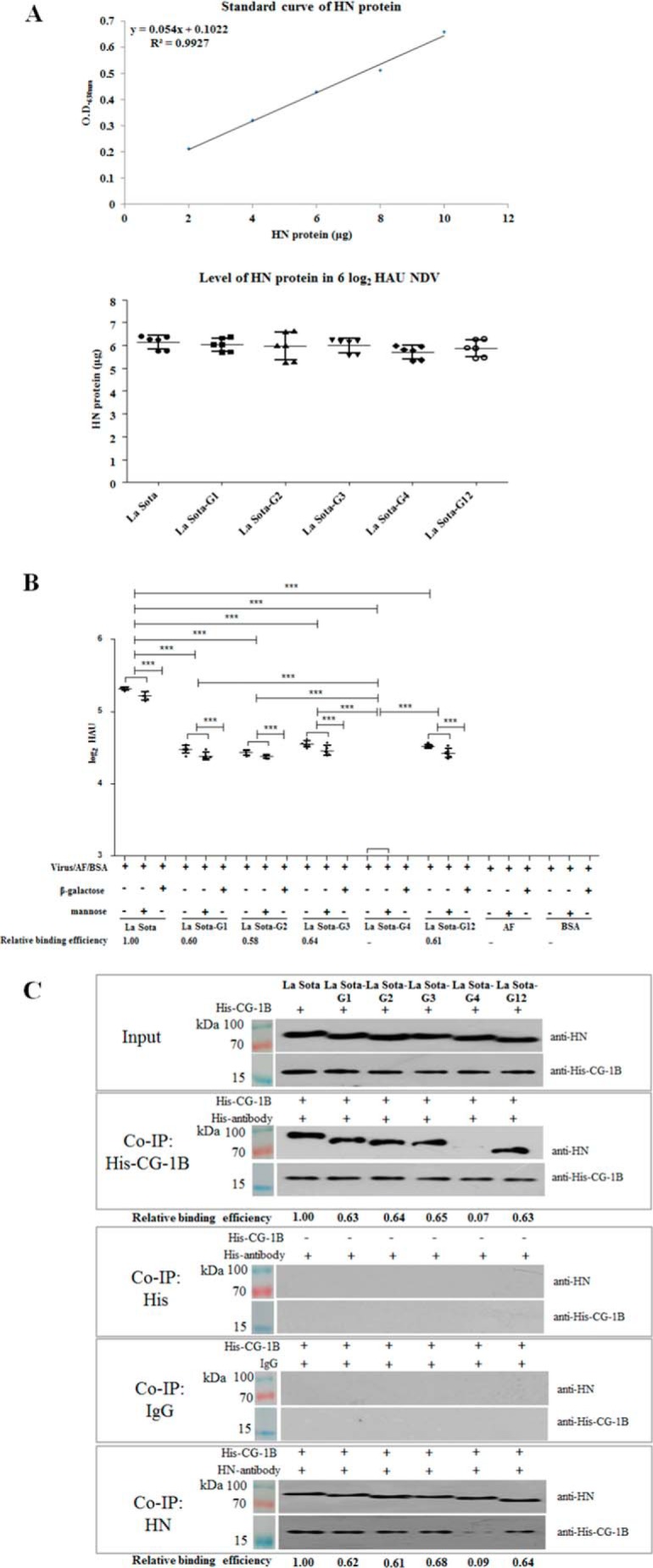
CG-1B binds to specific N-glycans on NDV HN glycoprotein. A, quantification of HN protein levels in parental and HN mutant viruses. NDV HN proteins with known concentration were used to coat ELISA plates and detected by anti-HN monoclonal antibody. Standard curve of HN protein was established based on OD values. 2-Fold serially diluted La Sota, La Sota-G1, -G2, -G3, -G4, and -G12 were used to coat ELISA plates; HN protein was detected by anti-HN monoclonal antibody. The amounts of HN protein in NDVs were calculated based on the linear standard curves. Each value represents the mean and S.D. The results of quantification of 6 log2 HAU of parental and HN mutant viruses are presented. B, La Sota and La Sota-G1, -G2, -G3, -G4, and -G12 containing equivalent HN proteins were applied to 96-well plates coated with CG-1B (2 μg); β-galactose or mannose was used as an antagonist. Bound virus particles were detected by ELISA with chicken anti-NDV La Sota antiserum. Uninfected chick embryo allantoic fluid and BSA acted as negative virus controls. The number of virions bound to CG-1B were calculated based on the linear standard curves. The bars represent means and S.D. Data were compared using Student's t test. ***, p < 0.001. C, interaction of HN mutants with CG-1B. CG-1B was incubated with viral lysates of parental and HN mutant viruses, and the lysates were immunoprecipitated with anti-His and anti-HN monoclonal antibody. The Co-IP assay was also performed with anti-His antibody in the absence of CG-1B, and normal mouse IgG2 was used as the isotype control antibody. The precipitated proteins were detected by Western blotting using anti-NDV HN and anti-His monoclonal antibodies, respectively.
CG-1B inhibits NDV adsorption rather than internalization
Although galectin-1 lacks a typical secretion signal peptide, it can present in the cytosol and nucleus and be secreted into the extracellular space by an alternative pathway (17). Therefore, we evaluated whether extracellular CG-1B interfered with the process of NDV infection in vitro. Entry of NDV into target cells requires two steps as follows: HN glycoprotein attachment to sialic acid-containing receptors on the cell surface (adsorption), and then F glycoprotein mediates membrane fusion between the viral and cellular membranes (internalization). Thus, to examine whether CG-1B specifically affected the adsorption and/or internalization process of NDV, DF-1 cells were incubated with NDV in the presence or absence of CG-1B for 1 h at 4 °C to allow viral attachment but not virus-mediated cell fusion. Viral RNA was quantified by real-time RT-PCR. NDV genomic RNA copies in velogenic F48E9-infected and lentogenic La Sota-infected DF-1 cells were significantly reduced when CG-1B was present during adsorption, indicating that CG-1B during the 4 °C inoculation step significantly inhibited NDV attachment to target cells (Fig. 8A). To determine whether the effects of CG-1B were mediated by binding to NDVs or the target cells during adsorption, NDVs or DF-1 cells were pre-incubated with CG-1B, respectively. Pre-treatment of NDVs with CG-1B prior to exposure to the DF-1 cells significantly inhibited NDV adsorption (Fig. 8B), whereas pre-treatment of DF-1 cells with CG-1B prior to virus inoculation had no significant effect on the adsorption of NDV (Fig. 8C). These results indicated that the effect of CG-1B on NDV adsorption was mediated by specific interaction of CG-1B with NDV HN protein. This inhibitory effect of CG-1B on NDV adsorption was reversed by addition of β-galactose (Fig. 8, A and B), suggesting that the role of CG-1B in the course of NDV adsorption is specific and glycan-dependent.
Figure 8.

Reduction of NDV adsorption and replication in DF-1 cells by CG-1B. A–C, adsorption assay. A, DF-1 cells were infected with NDV F48E9 or La Sota at m.o.i. 1 in the presence or absence of 7 μm CG-1B during 1 h at 4 °C. B, F48E9 and La Sota were pre-incubated with 7 μm CG-1B for 1 h at 37 °C before inoculation into DF-1 cells at 4 °C for 1 h. C, DF-1 cells were pre-incubated with 7 μm CG-1B for 1 h at 37 °C. Cells were washed three times with PBS and incubated with F48E9 or La Sota for 1 h at 4 °C. D, internalization assays. DF-1 cells were inoculated with NDV F48E9 and La Sota at m.o.i. 1 at 4 °C for 1 h. Cells were washed and incubated at 37 °C for 1 h supplemented with 7 μm CG-1B or medium. Un-internalized viruses were inactivated with citrate buffer. E, panel a, effect of CG-1B on the adsorption of HN mutants. DF-1 cells were inoculated with La Sota, La Sota-G1, -G2, -G3, -G4, and -G12 in the presence or absence of 7 μm CG-1B during 1 h at 4 °C. Panel b, effect of CG-1B on the internalization of HN mutants. DF-1 cells were inoculated with La Sota, La Sota-G1, -G2, -G3, -G4, and -G12 at 4 °C for 1 h. Then, cells were washed and incubated at 37 °C for 1 h supplemented with 7 μm CG-1B or medium. Un-internalized viruses were inactivated with citrate buffer. In all cases, β-galactose was used as an antagonist. Viral RNA was quantified by real-time RT-PCR. Data were normalized with expression of 18S rRNA gene, and the copy number of viral RNA was calculated relative to that of the La Sota group (×100%). F and G, CG-1B reduced replication of NDV in DF-1 cells. F, panel a, expression of FLAG-tagged CG-1B protein in DF-1 cells was identified through indirect immunofluorescence and Western blotting using anti-FLAG antibody. Panel b, quantification of NDV produced from pCAGGS-FLAG-CG-1B-transfected, pCAGGS-FLAG-transfected, and untransfected DF-1 cells after infection with F48E9 and La Sota. G, quantification of NDV produced from F48E9- and La Sota-infected DF-1 cells treated with CG-1B after adsorption. Viral titer in the culture supernatants of F48E9-infected cells was determined at 16 and 24 hpi by the Reed-Muench method in DF-1 cells, whereas viral titer in the culture supernatants of La-Sota-infected cells was determined at 24 and 48 hpi by HA assay. Virus titer assay was performed three times for each condition and compared using Student's t test (*, p < 0.05; **, p < 0.01; ***, p < 0.001). The bars represent means and S.D.
Alternatively, NDV was adsorbed onto DF-1 cells in the absence of CG-1B and then incubated with or without CG-1B for 1 h at 37 °C to permit fusion. In contrast, the presence or absence of CG-1B during internalization did not result in significant differences in the NDV genomic RNA copy numbers in F48E9- and La Sota-infected DF-1 cells (Fig. 8D), indicating that CG-1B added after inoculation at 4 °C and present during the 37 °C fusion step did not inhibit viral entry. These results suggest that the interaction between CG-1B and HN glycoprotein results in inhibition of adsorption rather than internalization of NDV during infection.
To investigate whether CG-1B had similar effects on the entry process of NDV HN mutants, the adsorption and internalization assays of La Sota, La Sota-G1, -G2, -G3, -G4, and -G12 was performed in the presence of CG-1B and β-galactose. The results of adsorption assay demonstrated that there were no significant differences in the NDV genomic RNA copy numbers in parental and HN mutant virus-incubated DF-1 cells, indicating that loss of N-glycans in HN protein does not affect the adsorption ability of NDV (Fig. 8E, panel a). Significantly, the adsorption of La Sota-G1, -G2, -G3, and -G12 was significantly inhibited by CG-1B, and this inhibitory effect was reversed by addition of β-galactose (Fig. 8E, panel a). There was no significant effect of CG-1B on adsorption of La Sota-G4, which lacked the N-glycans that are critical for efficient CG-1B binding. Our results indicated that CG-1B also specifically inhibits absorption of HN mutants (La Sota-G1, -G2, -G3, and -G12) in a glycan-dependent manner, whereas La Sota-G4 was not sensitive to this inhibitory effect of CG-1B. Moreover, in the internalization assay, the RNA copy numbers in La Sota-G4- and -G12-incubated DF-1 cells were significantly decreased in comparison with parental virus, indicating that loss of G4 and G12 glycans significantly reduced the viral internalization, which may further contribute to the low replication ability of La Sota-G4 and -G12. Loss of G4 N-glycans may result in misfolding of HN protein, and simultaneously eliminate the G12 N-glycans that may lead to improper interaction of HN with F protein (5, 12). Thus, loss of G4 and G12 N-glycans may impair the fusion promotion activity of HN protein, which leads to inefficient internalization of La Sota-G4 and -G12. Besides, similar to the parental virus, CG-1B treatment had no obvious effects on the internalization of all HN mutant viruses (Fig. 8E, panel b).
CG-1B reduces NDV replication in DF-1 cells
DF-1 cells transfected with pCAGGS-FLAG-CG-1B or control plasmid pCAGGS-FLAG were infected with F48E9 and La Sota. In the culture supernatants of both F48E9- and La Sota-infected DF-1 cells, the titers of progeny virus in pCAGGS-FLAG-CG-1B-transfected cells were significantly lower than in pCAGGS-FLAG-transfected and control DF-1 cells (Fig. 8F), indicating that CG-1B inhibits replication of NDV in target cells.
We further examined whether extracellular CG-1B contributed to the inhibition of NDV replication. Levels of endogenous CG-1B expressed in DF-1 cells and FLAG-tagged CG-1B secreted into culture supernatants of transfected DF-1 cells were below the limit of detection (data not shown). Therefore, NDV-infected DF-1 cells were treated with exogenous CG-1B after the adsorption period. CG-1B-treated cells also produced lower viral titers than control cells produced (Fig. 8G). Collectively, our results indicated that the presence of CG-1B in the life cycle of NDV can reduce NDV replication.
CG-1B inhibits cell-surface expression of HN glycoprotein
To examine whether the interaction between CG-1B and HN glycoprotein contributed to inhibition of NDV replication, we further investigated the subcellular localization of CG-1B and HN glycoprotein in NDV-infected cells using confocal laser-scanning microscopy. HN glycoprotein was distributed on the cell membrane surface in pCAGGS-FLAG-transfected and control DF-1 cells; in contrast, CG-1B and HN glycoprotein were co-localized in the cytoplasm of F48E9- and La Sota-infected DF-1 cells (Fig. 9, A and B). HN glycoprotein was also distributed on the cell-membrane surface in DF-1 cells that were treated with CG-1B after adsorption, whereas CG-1B bound and co-localized with HN glycoprotein on the cell surface. Expression of NDV HN glycoprotein on the cell surface was quantified by flow cytometry. As shown in Fig. 9C, panels a and b, surface expression of the HN glycoprotein indicated by mean fluorescence intensities in pDsRED-CG-1B-positive DF-1 cells was significantly lower than in pDsRED-positive DF-1 cells. However, surface expression of HN protein in CG-1B-treated DF-1 cells was comparable with that in control cells (Fig. 9C, panels c and d). These data indicate that intracellular CG-1B reduces cell-surface expression of HN glycoprotein, whereas extracellular CG-1B binds and co-localizes with the cell-surface HN glycoprotein without influencing the cell-surface transport of HN glycoprotein.
Figure 9.

Intracellular distribution and cell-surface expression of HN protein. A and B, intracellular distribution of CG-1B and NDV HN glycoprotein. Distribution of CG-1B and HN glycoprotein was detected in DF-1 cells transfected with pCAGGS-FLAG-CG-1B (panel a) and pCAGGS-FLAG (panel b), and DF-1 cells treated with 7 μm CG-1B after incubation (panel c). Untreated DF-1 cells were used as controls (panel d). NDV-infected cells were fixed at 24 h after infection and then subjected to indirect immunofluorescence to detect CG-1B and NDV HN glycoprotein using rabbit anti-FLAG/His antibody (red) and chicken anti-NDV HN antibody (green). The nuclei were stained with DAPI (blue) in the merged images. The triple-stained cells were observed by Leica SP2 laser confocal microscopy. C, cell-surface expression of HN was determined by flow cytometry. DF-1 cells transfected with pDsRed-CG-1B or pDsRed2-N1 were infected with NDV F48E9 (panel a) and La Sota (panel b) at m.o.i. 0.1. Surface expression of the HN protein in red fluorescence-positive DF-1 cells was assessed by flow cytometry at 24 hpi with an NDV HN-specific monoclonal antibody followed by a FITC-conjugated secondary antibody. In addition, surface expression of the HN protein in F48E9-infected (panel c) and La Sota-infected (panel d) DF-1 cells treated with 7 μm CG-1B after incubation was also detected. Surface immunofluorescence was quantified by fluorescence-activated cell sorter analysis with a Cytomics FC 500 flow cytometer.
Correlation between expression of CG-1B and replication of NDV
LMH cells, which endogenously express CG-1B, CG-1B-overexpression and CG-1B knockdown LMH cells, were infected with La Sota, La Sota-G1, -G2, -G3, -G4, and G12, progeny virus titers in the cell supernatants were determined at the indicated time points. As shown in Fig. 10A, the replication kinetics of La Sota, La Sota-G1, -G2, -G3, -G4, and -G12 in LMH cells were similar to those in chicken embryos and 1-day-old chickens. Moreover, in comparison with LMH cells, the replication levels of La Sota and La Sota-G1, -G2, -G3, and -G12 were significantly declined in CG-1B-overexpression LMH cells (Fig. 10B). In CG-1B knockdown LMH cells, the replication levels of La Sota and La Sota-G1, -G2, -G3, and -G12 were obviously increased compared with those in LMH cells (Fig. 10C). However, the replication level of La Sota-G4, which lacked the N-glycans that are critical for efficient CG-1B binding, was not obviously changed in both CG-1B-overexpression and CG-1B knockdown LMH cells when compared with that in LMH cells (Fig. 10, B and C). This result indicated that the replication level of NDV was correlated with the expression of CG-1B, and the inhibitory effect of CG-1B on NDV replication depended on the interaction with specific N-glycans on HN glycoprotein.
Figure 10.

Replication of NDV was correlated with the expression of CG-1B. A, LMH cells were infected with La Sota, La Sota-G1, -G2, -G3, -G4, and -G12 at m.o.i. 0.1. B, LMH cells were transfected with pCAGGS-FLAG-CG-1B, and the expression of FLAG-tagged CG-1B was assessed by Western blotting (supplemental Fig. 5). At 48 h post-transfection, the CG-1B-overexpression LMH cells were infected with La Sota, La Sota-G1, -G2, -G3, -G4, and -G12 at m.o.i. 0.1. C, knockdown of CG-1B in LMH cells was performed using siRNA that targets CG-1B sequence. The expression of CG-1B was assessed by real-time RT-PCR and Western blotting (supplemental Fig. 5). At 48 h post-transfection, the CG-1B knockdown LMH cells were infected with La Sota, La Sota-G1, -G2, -G3, -G4, and -G12 at m.o.i. 0.1. Cell culture supernatants were harvested at 8-h intervals, and the viral titers were determined by HA assay and expressed as HAU. In LMH cells, the viral titers of La Sota-G1, -G2, -G3, -G4, and -G12 at each time point were compared with that of La Sota. The viral titers of La Sota, La Sota-G1, -G2, -G3, -G4, and -G12 in CG-1B-overexpression and CG-1B knockdown LMH cells at each time point were compared with that in LMH cells, respectively. Each value represents the mean and S.D. Data were compared by using Student's t test. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Discussion
Besides contributing to the immune homeostasis as an important regulatory factor (17, 21), mammalian galectin-1 can also mediate the host–pathogen interaction via binding glycans on the surface of pathogens (8, 9, 24, 29). During infection, mammalian galectin-1 may participate in host antiviral defense or promote the course of virus invasion through specific interaction with glycans on the viral envelope glycoprotein (8, 9, 23, 24, 29, 30). To date, whether CG-1A and CG-1B, which are closely related to human galectin-1 (31, 32), play a role in avian virus infection has not been reported. In this study, we showed that CG-1B reduces NDV infection by inhibiting both adsorption and replication processes of NDV. The inhibitory effect of CG-1B on NDV infection depends on the interaction of CG-1B with specific N-glycans on HN glycoprotein. Our findings suggest for the first time that similar to the mammalian galectin-1, chicken galectin-1B participates at the host antiviral defense via mediating host–virus interactions during NDV infection.
Viral infection can modulate the expression and function of galectin-1, and the complex interplay between galectin-1 and virus may ultimately determine the outcome of infection (7, 8, 19, 24, 29). Although the expression of CG-1B was up-regulated in target organs of chickens infected with both lentogenic La Sota and velogenic F48E9, the outcomes of the clinical illness caused by these two viruses were distinct. La Sota is a lentogenic NDV strain that only replicates in the respiratory tract and induces no apparent clinical signs. In our previous work, we found that the expression of CG-1B was up-regulated in the trachea after infecting with La Sota at 7 and 12 dpi, while the presence of virus was undetectable since 12 dpi (25). The up-regulation of CG-1B may contribute to the virus clearance, as we have clearly demonstrated that CG-1B can inhibit the adsorption and replication of NDV in vitro (Fig. 8). F48E9 is a velogenic NDV strain that can cause systemic infection and result in 100% mortality within 72–96 hpi in chickens (26). In comparison with La Sota, the viral loads in F48E9-infected target organs were significantly higher (data not shown); thus, the high levels of virus replication may stimulate the host to produce CG-1B rapidly (24 hpi). However, the CG-1B produced may be insufficient to inhibit F48E9 infection, as reported in an influenza virus infection model (8). The rapid replication of F48E9 in most organs could induce severe lesions and subsequently decrease expression of CG-1B with time and ultimately result in death of the infected chickens. Our results indicated that NDV infection generally induced up-regulation of CG-1B, whereas the expression change of CG-1B during infection may be associated with the pathogenesis of NDV.
Because the HA activity of NDV is mediated by HN glycoprotein, the binding of CG-1B to NDV and the HI activity of CG-1B on NDV suggest that CG-1B binds to the glycans located on the HN glycoprotein and consequently influences the HA activity of NDV. As expected, the results of affinity chromatography, LC-MS/MS, and Co-IP confirmed that CG-1B interacts with HN glycoprotein of NDV. Galectin-1 can bind to specific N-glycans on Nipah virus F glycoproteins through its CRD to reduce endothelial cell fusion (19). In this study, we demonstrated that although all N-glycans may be involved in the interaction of CG-1B with NDV HN, G4 N-glycans contribute significantly to this interaction. In the course of N-linked glycosylation modification, a high mannose core was attached to the amide nitrogen of asparagine in the motif Asn-Xaa-(Ser/Thr), and then the oligosaccharide side chain was further modified in the endoplasmic reticulum and Golgi to yield high-mannose, hybrid, and complex glycan structures (33). It has been suggested that complex-type glycans arising from the tertiary structure of HIV-1 gp120 glycoprotein, rather than the primary sequence of particular glycans, contribute mostly to its specific recognition by galectin-1 (9). We found that the HN G4 mutant migrated faster than the other single mutant HN proteins in SDS-PAGE, which likely related to the degree of glycosylation of the sites that have been reported in other virus glycoproteins (6, 34, 35). The faster migration rate of the HN G4 mutant indicates the G4 N-glycans are a complex type, higher than other sites, and significantly contribute to the specific interaction with CG-1B, as reported in HIV-1 (9).
The structural analysis showed that the ectodomain of NDV HN glycoprotein consists of a stalk domain and a C-terminal globular head domain (4, 36). The globular head domain of HN glycoprotein is responsible for attachment to receptors (37–40), whereas the interactions of the stalk domain with F protein are associated with fusion activation (41). The N-linked glycosylation site G1 is located in the stalk region, whereas G2, G3, and G4 are located in the globular head domain. Although the N-glycans on the HN protein were not determinants for receptor attachment, the inhibitory effect of extracellular CG-1B on the adsorption mediated by the interaction of CG-1B with specific N-glycans (G4) on the head domain of HN glycoprotein was observed. Galectin-1 can bind to glycans on cell-surface glycoproteins and form galectin–glycoprotein lattices that contribute to the distribution, maturation, and function of glycoproteins on the cell surface (7, 42, 43). Hence, we speculate that during NDV adsorption, CG-1B may specifically cross-link the N-glycans displayed in the HN globular domain to create a viral surface galectin–glycoprotein lattice. This further interferes with the process of cell-receptor recognition mediated by HN glycoprotein through the steric hindrance. Steric hindrance mediated by antibody and improper N-glycosylation modification has been found to contribute to the inhibition of viral receptor-binding activity (44–46). In addition, it is suggested globular head domain of the paramyxovirus receptor-binding protein is dispensable for F activation (41, 47). Therefore, it is reasonable that the interaction of CG-1B with G4 N-glycans located on the head domain does not affect the internalization of NDV (Fig. 8E, panel b).
The N-linked glycans are critical for folding, transport, and biological activities of viral glycoproteins (5, 6, 12, 48). It is reported that G4 is necessary for the correct processing and transport of NDV HN glycoprotein. The cell-surface expression, NA activity, and fusion promotion of G4 mutant HN glycoprotein are significantly decreased (5, 12), and the replication of G4 mutant NDV is delayed (5). In this study, we showed that intracellular CG-1B co-localized with HN glycoprotein in the cytoplasm and reduced the cell-surface expression of HN glycoprotein. Thus, the reduced replication of NDV may be attributed to intracellular CG-1B interacting with G4 N-glycans on the HN glycoprotein, which results in inefficient cell-surface expression of HN glycoprotein and, consequently, the inefficient release of the progeny virus particles in NDV-infected cells. The G4 glycosylation site of HN glycoprotein also plays important roles in the NA activity and the fusogenicity of NDV; thus, whether CG-1B inhibits the spread of progeny virus via binding G4 N-glycans needs to be elucidated.
Extracellular CG-1B binds and co-localizes with HN glycoprotein on the cell surface rather than affecting the distribution and cell-surface expression of HN glycoprotein with reduced NDV production. During the life cycle of NDV, newly synthesized glycoproteins need transport to the infected cell plasma membrane to complete assembly and release of progeny virus. Because galectin-1 can bind to cell-surface glycoproteins and form galectin–glycoprotein lattices (42), we infer that extracellular CG-1B secreted from cells may result in inhibition of NDV replication by interfering with release of progeny virus and preventing adsorption of progeny virus to new target cells during the infectious cycle through specific interaction with HN glycoprotein, rather than interfering with expression and transport of HN glycoprotein in target cells. The anti-NDV activity of CG-1B during NDV infection can be summarized as follows: the interaction between extracellular CG-1B and HN glycoprotein inhibits NDV entry and progeny virus release by generating galectin–HN glycoprotein lattices, and the interaction between intracellular CG-1B and HN glycoprotein may inhibit NDV replication by preventing maturation and function of HN glycoprotein.
The glycosylation of viral envelope glycoprotein is associated with viral replication and virulence (6), and host lectins can modulate the virus infection via direct interaction with the glycans distributed on the viral surface. As we have demonstrated that CG-1B can inhibit NDV infection via interaction with specific N-glycans on the HN glycoprotein, in theory, the replication level of the HN mutant that lacked the N-glycans that are responsible for CG-1B binding should be higher than the parental virus. However, the replication levels of all HN mutants both in chicken embryos and 1-day-old chickens were lower than parental virus at different degrees (Fig. 6, A and B). To elucidate the divergent results, we further evaluate the replication levels of parental and HN mutant viruses in LMH cells that endogenously express CG-1B, as well as CG-1B overexpression and CG-1B knockdown LMH cells. We demonstrated that, in CG-1B knockdown LMH cells, the replication levels of La Sota, La Sota-G1, -G2, -G3, and -G12 were significantly higher than that of La Sota-G4 (Fig. 10C). Although the presence of CG-1B in LMH cells inhibited the replication of La Sota, La Sota-G1, -G2, -G3, and -G12, without affecting the replication of La Sota-G4 (Fig. 10A), it ultimately resulted in no considerable differences in replication level among the parental and mutant viruses in LMH cells. Besides, overexpression of CG-1B significantly reduced the replication of La Sota, La Sota-G1, -G2, -G3, and -G12, without affecting the replication of La Sota-G4 (Fig. 10B). These results indicated that there is an inverse correlation between the replication levels of La Sota, La Sota-G1, -G2, -G3, and -G12 and the expression of CG-1B, whereas the replication of La Sota-G4, which lacked the N-glycans that are critical for efficient CG-1B binding, was not significantly affected by the expression of CG-1B. Moreover, the replication kinetics of parental and HN mutant viruses in mouse-origin BHK-21 cells without expressing CG-1B were similar to those in CG-1B knockdown LMH cells (data not shown), indicating the intrinsic low replication ability of La Sota-G4. The N-glycans on the G4 site are closely related to the processing and biological activities of the HN glycoprotein, and loss of G4 N-glycans can reduce NA and fusion activities (5, 12) and decrease internalization efficiency (Fig. 8E, panel b). These changes may be responsible for the decreased replication ability as well as the decline in the virulence of La Sota-G4. As we identified that G4 N-glycans contribute significantly to CG-1B binding to NDV HN glycoprotein, the presence of CG-1B during NDV infection may interfere with the biological function of HN glycoprotein through specific interaction with G4 N-glycans, which results in poor replication efficiency and reduced virulence.
This study demonstrated that NDV can generally induce expression of CG-1B in chicken, and this host factor can inhibit the adsorption and replication of NDV in vitro via specific interaction with N-glycans on the HN glycoprotein. Moreover, NDV initially infects the tracheal epithelium and elicits an inflammatory response (49, 50), and whether CG-1B is involved in recovery from inflammatory responses induced by NDV, as found in influenza virus infection model in mice (8), needs to be elucidated. In consideration of the inhibition effects of CG-1B on adsorption and replication of NDV, CG-1B might participate at various levels of host antiviral defense; and the anti-NDV effects of CG-1B may not be restricted to specific NDV strains, as reported for influenza virus. In summary, CG-1B is of great potential to be developed for a novel antiviral agent for NDV. Moreover, CG-1B may also be explored for targeting other important avian viruses (e.g. infectious bronchitis coronavirus and various subtypes of avian influenza virus), which contain glycoproteins on the virion surface.
Experimental procedures
Cells and viruses
DF-1, BSR-T7/5, and LMH cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) or antibiotics. Velogenic F48E9 and lentogenic La Sota NDV strains were used. All strains were propagated once in 9–11-day-old SPF embryonated chicken eggs. NDV particles in the allantoic fluid of inoculated eggs were confirmed using HA inhibition analysis with specific antibody. Viral titers were determined by inoculation at 10-fold dilutions into groups of five 10-day-old embryonated chicken eggs. The EID50 was calculated by the method of Reed and Muench (51).
Animal experiments
Two groups of 10 White Leghorn SPF chickens (Harbin Veterinary Research Institute, China) were housed in isolators under negative pressure, and food and water were provided ad libitum. At 20 days of age, one group of chickens was inoculated with NDV F48E9 through the intranasal and intraocular routes with a dose of 106 EID50 in 0.1 ml of diluent per chicken. The chickens in the remaining group were mock-inoculated with sterile allantoic fluid as a negative control. This study was approved by the Animal Welfare Committee of Heilongjiang Province, China. All experiments were conducted at appropriate biosafety levels. Five birds from each group were killed humanely at 24 and 48 hpi, and fresh tissue was harvested from the trachea, kidneys, spleen, liver, cecal tonsils, proventriculus, lungs, Harderian glands, and bursa of Fabricius, snap-frozen in liquid nitrogen, and stored at −80 °C for subsequent analysis.
Real-time RT-PCR
Viral load in tissue samples from NDV-F48E9-infected chickens was analyzed as described previously (25). Viral RNA was extracted from the tissue samples of birds from each group using TRIzol reagent (Invitrogen). For detection of the viruses, specific primers and TaqMan probes against NDV RNA were selected (52). Real-time RT-PCR was performed on a LightCycler® 480 real-time PCR system (Roche Diagnostics Deutschland GmbH, Mannheim, Germany) with One-Step PrimeScript RT-PCR kit (TaKaRa, Dalian, China). Samples from chickens that were mock-inoculated with sterile allantoic fluid were used as a negative control. All of the samples were tested in triplicate in each reaction. Standard templates with known concentration and no template negative control were retained for quantitative analysis.
Specific primers for the analysis of mRNA abundance of the CG-1B gene was designed according to the published gene sequences using Beacon Designer software 7.5 (Premier Biosoft International, Palo Alto, CA). Total tissue RNA was extracted from birds of each group at each time point using TRIzol reagent. The real-time RT-PCR and quantitative analyses of the data were determined using the LightCycler 480 real-time PCR system with the relative quantification 2−ΔΔCt model (53). Cp values of target genes in the control group at each time point were used as the calibrator (relative expression = 1), and 18S rRNA gene was used as an internal reference gene (54).
Production of recombinant CG-1B protein
The coding sequence (CDS) of CG-1B gene was amplified from the chicken tracheal RNA. The recombinant plasmid pET-CG-1B, encoding the CG-1B protein fused to a His tag at its C terminus, was constructed by cloning CG-1B CDS into the prokaryotic expression vector pET-30a (+), using NdeI and XhoI restriction enzymes. The scheme for clone construction is shown in supplemental Fig. 1. Histidine-tagged CG-1B protein was expressed in Escherichia coli strain Rosetta (DE3) transformed with the recombinant plasmid. After induction by isopropyl β-d-thiogalactopyranoside, recombinant CG-1B protein was purified using Ni-NTA affinity chromatography under native conditions. The purified CG-1B protein was desalted and concentrated with 5000 MWCO Spin-X UF concentrators (Corning, Costar, NY) and sterilized by filtration through a 0.22-μm filter. The CG-1B protein was identified by SDS-PAGE and Western blotting (supplemental Fig. 1) using anti-His antibody (Sigma). Mock preparations were also prepared using E. coli strain Rosetta (DE3) transformed with pET-30a (+) vector under the same purification conditions.
HA assay
The bioactivities of CG-1B were estimated by HA assay as described previously (23), using PBRCs from SPF chickens. For determining the titers of NDV, serial 2-fold dilutions of NDV in phosphate-buffered saline (PBS) were mixed with 1% PBRCs in V-shaped 96-well plates at room temperature for 30 min. The reciprocal of the highest dilution of the virus capable of causing complete HA was expressed as 1 HAU. The HA test was also used to test the binding between NDV and CG-1B. CG-1B (4 μm) in the presence of 0.1 m β-galactose or mannose was incubated with NDV F48E9 and La Sota containing different HAUs for 1 h at room temperature, followed by addition of 1% PBRCs. After an additional 30-min incubation at room temperature, HA was recorded.
Virion-binding assay
An ELISA-based method was used to evaluate the binding between CG-1B and NDV. First, 100 μl of 2-fold serially diluted La Sota and F48E9 were used to coat 96-well high-binding polystyrene plates overnight at 4 °C. Uninfected SPF chicken embryo allantoic fluid was used as a negative control. The free binding site was blocked with 5% bovine serum albumin (BSA) at 37 °C for 1 h. Anti-NDV La Sota antiserum (1:500) was added, followed by incubation at room temperature for 1 h. Horseradish peroxidase (HRP)-conjugated goat anti-chicken antibody was added to each well and incubated at room temperature for 1 h, and the plates were developed with tetramethylbenzidine substrate solution. The reaction was stopped with 2 n H2SO4 after incubation for 5 min at room temperature, and the absorption was measured at 630 nm. Washing was performed five times between all steps with PBS containing 0.1% Tween 20. Based on the OD of 2-fold serially diluted NDVs, the standard curves of ELISA were established.
CG-1B (2 μg) was diluted in 0.05 m carbonate/bicarbonate buffer, pH 9.6, and used to coat 96-well high-binding polystyrene plates (Corning) overnight at 4 °C. The free binding site was blocked with 5% BSA at room temperature for 4 h. Control wells were coated with mock-purified preparations. A total of 100 μl of 6 log2 HAU of NDV F48E9 or La Sota, diluted in 1% BSA/PBS, was added and incubated overnight at 4 °C. Uninfected SPF chicken embryo allantoic fluid and BSA were used as negative controls. The incubation of anti-La Sota antiserum and HRP-conjugated goat anti-chicken antibody and the color development were carried out as described above. To assess the specificity and glycan-dependent effects of interactions between CG-1B and NDVs, 0.1 m β-galactose or mannose was added to wells before addition of NDV. The amount of virions bound to CG-1B was calculated based on the linear standard curves.
Microscale affinity chromatography
CG-1B affinity columns were prepared using NHS-activated agarose (Thermo Fisher Scientific, Waltham, MA) with some modifications (55). Purified CG-1B and mock preparations were coupled to NHS-activated agarose by inoculation for 2 h at room temperature; the free binding site was blocked with 0.1 m Tris, pH 8.5. Viral protein extracts were prepared from NDV-infected allantoic fluids using lysis buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 0.25% sodium deoxycholate, and 2 mm EDTA) containing 1 mm PMSF and protease inhibitor mixture (Sigma). The viral proteins were applied to the CG-1B column and incubated for 2 h at room temperature. After washing five times with PBS, bound viral proteins were eluted with 0.1 m β-galactose in PBS. Samples were subjected to SDS-PAGE, and gels were stained with Coomassie Blue R-350 (GE Healthcare), and protein bands were excised from the gels and subjected to LC-MS/MS identification. Data were searched by GPS Explorer (version 3.6) with the search engine MASCOT (version 2.1). The parameters for data searching were set as follows: Uniprot database (550,552 sequences; 196,472,675 residues), and the viruses taxonomy (14,776 sequences); trypsin cleavage (one missed cleavage allowed), carbamidomethylation of cysteine as a fixed modification, peptide mass tolerance set at 15 ppm, and fragment tolerance set at 20 milli-mass units. The criterion for successful identification of a protein was the individual ion scores of >21, which indicated identity or extensive homology (p < 0.05). The eluted viral proteins were identified by Western blotting analysis with mouse anti-NDV HN monoclonal antibody. Mock preparation applied to microscale affinity chromatography under the same conditions was retained as negative control.
Co-IP
CG-1B was incubated with NDV viral lysates for 1 h at 4 °C and pre-cleared with protein A/G-agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA). The lysates were immunoprecipitated with anti-His or anti-HN monoclonal antibody, and precipitated proteins were captured on protein A/G-agarose beads and washed three times with lysis buffer. Bound proteins were separated by SDS-PAGE, and precipitated NDV viral protein and CG-1B were detected with anti-NDV HN and anti-His monoclonal antibodies, respectively. To confirm the specificity of the interaction between HN and CG-1B, the Co-IP assay was performed with anti-His antibody in the absence of CG-1B. In addition, normal mouse IgG2 was used as the isotype control antibody for His and HN monoclonal antibody, which both belong to the IgG2 subclass.
Generation of HN glycosylation site mutant NDVs
The CDS of the NP, P, and L genes were amplified from La Sota cDNA using specific primers, and the optimal Kozak sequence (GCCACC) was included (Table 2). To generate the helper plasmids, the amplified fragments were then cloned into pCI-neo (Promega, Madison, WI) between the XbaI and NotI restriction enzyme sites, and the obtained plasmids were designated as pCI-NP-K, pCI-P-K, and pCI-L-K, respectively (supplemental Fig. 2A). Construction of plasmid pNDFL containing full-length cDNA of NDV La Sota has been described previously (56). To introduce HN glycosylation site mutations into pNDFL, an NotI–SpeI fragment containing four glycosylation sites of the HN gene was amplified from pNDFL and cloned into pMD18-T (TaKaRa). Site-directed mutagenesis was performed on the cDNA clone of the HN gene to generate a cluster of HN mutants through overlap PCR using specific primers (Table 3). The HN glycosylation site mutants were named with a G (glycosylation site) and the number of the glycosylation site mutated (1–4 and 12). To eliminate functional glycosylation sites, the asparagine residue in the N(X)T/S sequence motif was mutated to glutamine. Previous studies have indicated that G1 and G2 N-glycans in HN protein play essential roles in modulating the biological activities of HN protein, and replication, fusogenicity, and virulence of NDV are significantly affected by simultaneously eliminating the G1 and G2 N-linked glycans (5, 12). Therefore, the double mutant, G12, was also generated to evaluate the potential roles of G12 in the interaction with CG-1B. This double HN mutant was obtained by mutating the second glycosylation site on the cDNA clone G1 with the specific primers for constructing the G2 cDNA clone. All HN mutants were sequenced in their entirety to confirm the presence of these introduced mutations. The mutagenized NotI–SpeI fragments were excised from pMD18-T and inserted into the NotI–SpeI site of pNDFL. The resulting mutated full-length cDNA plasmids were designed as pNDFL-G1, -G2, -G3, -G4, and -G12, respectively (supplemental Fig. 2B).
Table 2.
Primers used for construction of helper plasmids
| Gene | Forward primera (5′–3′) | Reverse primera (5′–3′) |
|---|---|---|
| NP | GCTCTAGAGCCACCATGTCTTCCGTATTTGATGAGb | ATTTGCGGCCGCTCAATACCCCCAGTCGGTGTC |
| P | GCTCTAGAGCCACCATGGCCACCTTTACAGATGCb | ATTTGCGGCCGCTTAGCCATTTAGAGCAAGGC |
| L | GCTCTAGAGCCACCATGGCGAGCTCCGGTCCTGAAAGGb | ATTTGCGGCCGCTTAAGAGTCACAGTTACTGTAATA |
a Restriction enzyme sites are shown in bold.
b Kozak sequence is underlined.
Table 3.
Primers used for construction of the HN glycosylation site mutant cDNA
| Primer name | Primer sequence (5′–3′) | Fragment region |
|---|---|---|
| LaFull-F | TAGGCGCCATTATTGGCGGTGTG | 4896–8134a |
| LaFull-R | TGGCTTCTCTAACCCCGTCATC | |
| LaG1-F | GGAGCTGCACAGAACAGTGGGTb | 6757–6778 |
| LaG1-R | ACCCACTGTTCTGTGCAGCTCC | |
| LaG2-F | AAGCGATACCAGGACACATGCCC | 7423–7445 |
| LaG2-R | GGGCATGTGTCCTGGTATCGCTT | |
| LaG3-F | CCTATGACAGTCAGCCAGAAAAC | 7693–7715 |
| LaG3-R | GTTTTCTGGCTGACTGTCATAGG | |
| LaG4-F | CTTCTATAGACAGCACACCTTGC | 7842–7864 |
| LaG4-R | GCAAGGTGTGCTGTCTATAGAAG |
a Nucleotide positions of PCR-amplified fragment in La Sota are shown.
b Letters underlined and italics represent mutated nucleotides, which changed the corresponding amino acids.
Transfection and recovery of mutant NDVs were carried out using reverse genetics as described previously (56, 57). To rescue La Sota-G1, BSR T7/5 cells stably expressing phage T7 RNA polymerase grown to 80% confluence in six-well plates were co-transfected with 8 μg of DNA, consisting of a mixture of pNDFL-G1 with pCI-NP-K, pCI-P-K, and pCI-L-K helper plasmids, at a ratio of 4:2:1:1 using Lipofectamine 2000 (Invitrogen). At 6 h post-transfection, the cells were washed three times with DMEM and cultured in DMEM supplemented with 10% FBS and 1 μg/ml tosylphenylalanyl chloromethyl ketone (TPCK)-treated trypsin (Sigma). After 3 days, the culture supernatant and cells were harvested and inoculated into the allantoic cavities of 9–11-day-old embryonated SPF eggs. NDV particles in the allantoic fluid of inoculated eggs were confirmed using HA and HA inhibition analysis with specific antibodies. Rescue of La Sota-G2, -G3, -G4, and -G12 was also performed by the same procedures.
Biological characterization of HN mutant NDVs
RNA was extracted from the recovered mutant viruses by using TRIzol reagent (Invitrogen). RT-PCR was performed using PrimeScript One-step RT-PCR kit (TaKaRa) with primers LaFull-F and LaFull-R. The amplified DNA fragments were sequenced to confirm the presence of the introduced mutations in the HN mutant viruses. To confirm the loss of carbohydrates and evaluate the expression level of HN glycoprotein in mutant viruses, viral proteins were extracted from La Sota-, La Sota-G1-, G2-, G3-, G4-, and G12-infected allantoic fluids containing equivalent HAU. The protein samples were subjected to 12% SDS-PAGE and Western blotting using an anti-HN monoclonal antibody to evaluate the expression of HN glycoprotein. The NP protein was also detected with anti-NP monoclonal antibody to normalize the expression level of HN glycoprotein.
The multicycle growth kinetics of La Sota, La Sota-G1, -G2, -G3, -G4, and -G12 were determined in SPF embryonated chicken eggs. We inoculated 9-day-old embryonated SPF eggs with 0.5 HAU of La Sota and rescued La Sota-G1, -G2, -G3, -G4, and -G12. Five eggs receiving each virus were harvested at 24-h intervals, and the viral titers were determined by HA assay and expressed as HAU. Replication efficiency of the parental and mutant viruses in vivo was determined in SPF chickens. One hundred and twenty 1-day-old SPF White Leghorn chicks were divided into six groups of 20 birds each and housed in different isolators with negative pressure. Chickens assigned to groups 1–6 were inoculated with La Sota, La Sota-G1, -G2, -G3, -G4, or -G12, by oculonasal application at 1 day of age, with a dose of log 106 EID50/100 μl per chick. Five birds from each group were killed humanely at 1, 3, 6, and 12 dpi, and fresh tissue samples of trachea, lungs, and brain were quickly harvested. Viral load in tissues was analyzed using real-time RT-PCR as described above. Serum HI antibody titers were determined by HI assay. In addition, the virulence of La Sota-G1, -G2, -G3, -G4, and -G12 was determined by the MDT test in 9-day-old SPF embryonated chicken eggs.
Determination of the ability of HN mutant NDVs to bind to CG-1B
First, His-tagged La Sota HN protein was expressed in Expi293F cells and purified using Ni-NTA affinity chromatography under native conditions. The HN protein was identified by SDS-PAGE and Western blotting (supplemental Fig. 3) using anti-HN antibody. Then HN proteins with known concentrations were used to coat 96-well polystyrene plates overnight at 4 °C. The bound protein was detected with anti-HN monoclonal antibody. HRP-conjugated anti-mouse antibody was used as secondary antibody. The block, antibody incubation, washing, and color development were carried out as described under “Virion binding assay.” Based on the OD values, the standard curve of HN protein was established. Then, to determine the HN levels of parental and HN mutant viruses, 100 μl of 2-fold serially diluted La Sota, La Sota-G1, -G2, -G3, -G4, and -G12 was used to coat 96-well plates overnight at 4 °C. Uninfected SPF chicken embryo allantoic fluid was used as a negative control. Anti-HN monoclonal antibody was used as primary antibody, and HRP-conjugated anti-mouse antibody was used as secondary antibody. The block, antibody incubation, washing, and color development were carried out as described under “Virion binding assay.” The amounts of HN protein in NDVs were calculated based on the linear standard curves.
To determine the ability of parental and HN mutant viruses binding CG-1B, La Sota, La Sota-G1, -G2, -G3, -G4, and -G12 containing equivalent HN proteins were subjected to ELISA, as described under “Virion binding assay.” The relative binding efficiency of NDV HN mutants was calculated as the number of HN mutant virions detected/number of parental virions detected. Co-IP assay was performed as described above to further examine the ability of HN mutants to bind to CG-1B. To determine the binding efficiency of HN mutants to CG-1B, the starting materials used for Co-IP assay were contained as the internal control to normalize the quantitative data. The relative binding efficiency of HN mutant proteins was calculated as the amount of precipitated HN mutant protein/amount of precipitated parental HN protein. Bands of proteins were quantitated by densitometry using ImageJ software.
Adsorption and internalization assays
The adsorption and internalization assays were performed as described previously with some modification (29, 58). For the adsorption assays, DF-1 cells were seeded in 12-well plates and infected with NDV F48E9 or La Sota at a multiplicity of infection (m.o.i.) of 1 in the presence or absence of 7 μm CG-1B and incubated for 1 h at 4 °C. To distinguish whether the effects of CG-1B were mediated by binding to NDVs or the target cells, F48E9 and La Sota were pre-incubated with 7 μm CG-1B for 1 h at 37 °C, and then the NDV/CG-1B mixture was added to DF-1 cells and incubated for 1 h at 4 °C. Meanwhile, DF-1 cells were pre-incubated with 7 μm CG-1B for 1 h at 37 °C. After washing three times with PBS, cells were incubated with F48E9 or La Sota for 1 h at 4 °C. The inoculum was removed, and the cells were washed twice with cold PBS. The cells were harvested and submitted to total viral RNA extraction and quantification of viral load via real-time RT-PCR.
For the NDV internalization assays, DF-1 cells were seeded in 12-well plates and incubated with NDV F48E9 or La Sota at m.o.i. 1. After incubation for 1 h at 4 °C, the inoculum was removed, and cells were washed twice with cold PBS. The cells were incubated with DMEM containing 7 μm CG-1B or with only DMEM as a negative control for a further 1 h at 37 °C. The cells were washed with PBS and treated with citrate buffer for 1 min to inactivate the adsorbed but not internalized virus. Finally, cells were washed twice with PBS to remove citrate buffer and submitted to subsequent viral RNA extraction and quantification of viral load.
The adsorption and internalization assays were also performed using La Sota, La Sota-G1, -G2, -G3, -G4, and -G12 as described above. To assess the specificity of effects of CG-1B on the adsorption or internalization process of NDV, the medium was supplemented with 0.1 m β-galactose during CG-1B incubation. The remaining steps were carried out as described above.
Viral replication assay
Expression of endogenous CG-1B in DF-1 cells was below the limit of detection (data not shown); thus, recombinant plasmid encoding CG-1B protein fused to a FLAG tag at its C terminus was constructed by cloning CG-1B CDS into the eukaryotic expression vector pCAGGS-FLAG, using ClaI and XhoI restriction enzymes (supplemental Fig. 4). pCAGGS-FLAG-CG-1B was transfected into DF-1 cells using the TurboFect transfection reagent (Thermo Fisher Scientific). Expression of pCAGGS-FLAG-CG-1B was confirmed by indirect immunofluorescence and Western blotting (Fig. 7F) using rabbit anti-FLAG polyclonal antibody (Sigma). DF-1 cells transfected with pCAGGS-FLAG-CG-1B or pCAGGS-FLAG and untransfected DF-1 cells were infected with NDV F48E9 or La Sota at m.o.i. 0.1. Moreover, DF-1 cells were infected with NDV F48E9 or La Sota at m.o.i. 0.1. After incubation, the inoculum was removed; the cells were washed three times with PBS, and the cells were supplemented with DMEM containing 5% FBS and 7 μm CG-1B. At 16 and 24 hpi, titers of F48E9 produced in the culture supernatants of infected cells were determined by the Reed-Muench method in DF-1 cells (51). At 24 and 48 hpi, titers of La Sota were determined by HA test. TPCK-treated trypsin (1 μg/ml) was supplemented into the culture supernatant to ensure replication of La Sota virus.
Confocal laser-scanning microscopy
DF-1 cells in a 30-mm cell culture dish were transfected with pCAGGS-FLAG-CG-1B or pCAGGS-FLAG. Twenty four hours after transfection, cells were inoculated with NDV F48E9 or La Sota at m.o.i. 0.1. One hour later, the viral inoculum was removed, and the infected cells were washed three times with PBS and supplemented with DMEM containing 5% FBS. TPCK-treated trypsin (1 μg/ml) was supplemented into the culture supernatant of DF-1 cells infected with La Sota. Alternatively, untransfected DF-1 cells were infected with NDV F48E9 or La Sota at m.o.i. 0.1. After incubation, the inoculum was removed by washing three times with PBS. The cells were supplemented with DMEM containing 5% FBS and 7 μm CG-1B. At 24 hpi, infected cells were fixed with 4% paraformaldehyde for 30 min, washed three times with PBS, and permeabilized with 0.1% Triton X-100 for 15 min. The cells were incubated with rabbit anti-FLAG/His and chicken anti-HN antibody overnight at 4 °C and then with anti-rabbit IgG-TRITC antibody and anti-chicken IgY (IgG)-FITC antibody (Sigma). After washing with PBS, the cells were stained with 4,6-diamidino-2-phenylindole for 20 min and examined using a Leica SP2 confocal system (Leica Microsystems, Wetzlar, Germany).
Flow cytometry
The coding region removing the termination codon of CG-1B was amplified by PCR from pCAGGS-FLAG-CG-1B. The resulting PCR product was digested with EcoRI and BamHI and subcloned upstream of the coding region of red fluorescent protein (RFP) in the pDsRed2-N1 plasmid. The recombinant plasmid encoding RFP-tagged CG-1B protein was designated as pDsRed-CG-1B. DF-1 cells transfected with pDsRed-CG-1B or pDsRed2-N1 were infected with NDV F48E9 or La Sota at m.o.i. 0.1. Alternatively, DF-1 cells were infected with NDV F48E9 or La Sota at m.o.i. 0.1. After incubation, the inoculum was removed by washing three times with PBS. The cells were supplemented with DMEM containing 5% FBS and 7 μm CG-1B. TPCK-treated trypsin (1 μg/ml) was supplemented into the culture supernatant to ensure replication of La Sota virus. At 24 hpi, the cells were washed twice with PBS and then collected from the dish with PBS containing 50 mm EDTA. After washing three times with PBS, the cells were stained with NDV HN monoclonal antibody and incubated at 4 °C for 1 h. The cells were washed three times with PBS and stained with anti-mouse IgG-FITC/antibody for 1 h at 4 °C. Cells were washed three times with PBS and resuspended in PBS with 1% BSA for analysis by flow cytometry. The fluorescence of 10,000 cells was measured with Cytomics TM FC 500 flow cytometer (Beckman Coulter, Fullerton, CA).
Replication of NDV in CG-1B-deficient and overexpressed cells
The small interference RNAs (siRNA) that specifically recognize the sequence of CG-1B mRNA (GenBankTM accession no. NM_205495; siCG-1B 5′-GUG AUG UGA ACC UCA UUG U-3′) and a control siRNA (5′-GCA CUU GAU ACA CGUGUA A-3′), which does not have a specific target site in chicken cells, were used. LMH cells endogenous expressing CG-1B were transfected with siRNAs using an N-TER nanoparticle siRNA transfection system (Sigma) according to the manufacturer's instructions. Besides, LMH cells were transfected with pCAGGS-FLAG-CG-1B as described under “Viral replication assay.” For validation of CG-1B knockdown and overexpression, see supplemental Fig. 5. To study the effect of CG-1B expression on the replication of NDV, 48 h after transfection, the siRNA and pCAGGS-FLAG-CG-1B-transfected LMH cells were infected with La Sota, La Sota-G1, -G2, -G3, -G4, and -G12 at m.o.i. 0.1. Cell culture supernatants were harvested at 8-h intervals, and the viral titers were determined by HA assay and expressed as HAU.
SDS-PAGE and Western blotting
The protein samples were subjected to 12% SDS-PAGE and then transferred to nitrocellulose membranes. After blocking overnight at 4 °C in blocking buffer (5% skinned milk, 0.1% Tween 20 in PBS), the membranes were incubated with corresponding primary antibodies for 1 h at 37 °C. The membranes were washed three times in washing buffer (0.1% Tween 20 in PBS) and then incubated with HRP-conjugated secondary antibodies for 1 h at 37 °C. Following three further washes, detection was performed using ECL Western blotting substrate (Pierce).
Statistical analysis
Data are expressed as mean ± S.D. Student's t test was performed using GraphPad Prism for Windows version 5 (GraphPad Software, La Jolla, CA).
Author contributions
J. S., Z. H., and S. L. designed the experiments and wrote the manuscript; J. S., Z. H., T. Q., and R. Z. performed the experiments; J. S., Z. H., T. Q., R. Z., and S. L. analyzed the data. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
This work was supported by the National Natural Science Foundation of China Grant 31502088, Special Fund for Agro-scientific Research in the Public Interest Grant 201303033, Natural Science Foundation of Heilongjiang Province Grant QC2015038, and the China Agriculture Research System Grant CARS-40-K18. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S5.
- NDV
- Newcastle disease virus
- CG-1B
- chicken galectin (CG)-1B
- HN
- hemagglutinin–neuraminidase
- NA
- neuraminidase
- DC-SIGN
- dendritic cell-specific ICAM-3 grabbing non-integrin
- SP-A and SP-D
- surfactant associated protein-A and -D
- HA
- SPF, specific pathogen-free
- hpi
- hour post-inoculation
- dpi
- day post-inoculation
- PBRC
- peripheral blood red cell
- PBS
- phosphate-buffered saline
- HAU
- hemagglutinating unit
- Co-IP
- co-immunoprecipitation
- MDT
- mean death time
- m.o.i.
- multiplicity of infection
- NP
- nucleoprotein
- P
- phosphoprotein
- F
- fusion
- CRD
- carbohydrate recognition domain
- HI
- hemagglutination inhibition
- Ni-NTA
- nickel-nitrilotriacetic acid
- RFP
- red fluorescent protein
- TPCK
- tosylphenylalanyl chloromethyl ketone
- TRITC
- tetramethylrhodamine isothiocyanate
- CDS
- coding sequence.
References
- 1. Huang Z., Panda A., Elankumaran S., Govindarajan D., Rockemann D. D., and Samal S. K. (2004) The hemagglutinin–neuraminidase protein of Newcastle disease virus determines tropism and virulence. J. Virol. 78, 4176–4184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander D. (2000) Newcastle disease and other avian paramyxoviruses. Rev. Sci. Tech. 19, 443–462 [DOI] [PubMed] [Google Scholar]
- 3. Iorio R. M., Field G. M., Sauvron J. M., Mirza A. M., Deng R., Mahon P. J., and Langedijk J. P. (2001) Structural and functional relationship between the receptor recognition and neuraminidase activities of the Newcastle disease virus hemagglutinin–neuraminidase protein: receptor recognition is dependent on neuraminidase activity. J. Virol. 75, 1918–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yuan P., Swanson K. A., Leser G. P., Paterson R. G., Lamb R. A., and Jardetzky T. S. (2011) Structure of the Newcastle disease virus hemagglutinin–neuraminidase (HN) ectodomain reveals a four-helix bundle stalk. Proc. Natl. Acad. Sci. U.S.A. 108, 14920–14925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Panda A., Elankumaran S., Krishnamurthy S., Huang Z., and Samal S. K. (2004) Loss of N-linked glycosylation from the hemagglutinin–neuraminidase protein alters virulence of Newcastle disease virus. J. Virol. 78, 4965–4975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vigerust D. J., and Shepherd V. L. (2007) Virus glycosylation: role in virulence and immune interactions. Trends Microbiol. 15, 211–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Garner O. B., Aguilar H. C., Fulcher J. A., Levroney E. L., Harrison R., Wright L., Robinson L. R., Aspericueta V., Panico M., Haslam S. M., Morris H. R., Dell A., Lee B., and Baum L. G. (2010) Endothelial galectin-1 binds to specific glycans on Nipah virus fusion protein and inhibits maturation, mobility, and function to block syncytia formation. PLoS Pathog. 6, e1000993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang M. L., Chen Y. H., Wang S. W., Huang Y. J., Leu C. H., Yeh N. C., Chu C. Y., Lin C. C., Shieh G. S., Chen Y. L., Wang J. R., Wang C. H., Wu C. L., and Shiau A. L. (2011) Galectin-1 binds to influenza virus and ameliorates influenza virus pathogenesis. J. Virol. 85, 10010–10020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. St-Pierre C., Manya H., Ouellet M., Clark G. F., Endo T., Tremblay M. J., and Sato S. (2011) Host-soluble galectin-1 promotes HIV-1 replication through a direct interaction with glycans of viral gp120 and host CD4. J. Virol. 85, 11742–11751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aguilar H. C., Matreyek K. A., Filone C. M., Hashimi S. T., Levroney E. L., Negrete O. A., Bertolotti-Ciarlet A., Choi D. Y., McHardy I., Fulcher J. A., Su S. V, Wolf M. C., Kohatsu L., Baum L. G., and Lee B. (2006) N-Glycans on Nipah virus fusion protein protect against neutralization but reduce membrane fusion and viral entry. J. Virol. 80, 4878–4889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shirato K., Miyoshi H., Goto A., Ako Y., Ueki T., Kariwa H., and Takashima I. (2004) Viral envelope protein glycosylation is a molecular determinant of the neuroinvasiveness of the New York strain of West Nile virus. J. Gen. Virol. 85, 3637–3645 [DOI] [PubMed] [Google Scholar]
- 12. McGinnes L. W., and Morrison T. G. (1995) The role of individual oligosaccharide chains in the activities of the HN glycoprotein of Newcastle disease virus. Virology 212, 398–410 [DOI] [PubMed] [Google Scholar]
- 13. Davis C. W., Nguyen H. Y., Hanna S. L., Sánchez M. D., Doms R. W., and Pierson T. C. (2006) West Nile virus discriminates between DC-SIGN and DC-SIGNR for cellular attachment and infection. J. Virol. 80, 1290–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tassaneetrithep B., Burgess T. H., Granelli-Piperno A., Trumpfheller C., Finke J., Sun W., Eller M. A., Pattanapanyasat K., Sarasombath S., Birx D. L., Steinman R. M., Schlesinger S., and Marovich M. A. (2003) DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J. Exp. Med. 197, 823–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ji X., Olinger G. G., Aris S., Chen Y., Gewurz H., and Spear G. T. (2005) Mannose-binding lectin binds to Ebola and Marburg envelope glycoproteins, resulting in blocking of virus interaction with DC-SIGN and complement-mediated virus neutralization. J. Gen. Virol. 86, 2535–2542 [DOI] [PubMed] [Google Scholar]
- 16. Ng W. C., Tate M. D., Brooks A. G., and Reading P. C. (2012) Soluble host defense lectins in innate immunity to influenza virus. J. Biomed. Biotechnol. 2012, 732191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vasta G. R. (2009) Roles of galectins in infection. Nat. Rev. Microbiol. 7, 424–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang R. Y., Rabinovich G. A., and Liu F. T. (2008) Galectins: structure, function and therapeutic potential. Expert Rev. Mol. Med. 10, e17. [DOI] [PubMed] [Google Scholar]
- 19. Levroney E. L., Aguilar H. C., Fulcher J. A., Kohatsu L., Pace K. E., Pang M., Gurney K. B., Baum L. G., and Lee B. (2005) Novel innate immune functions for galectin-1: galectin-1 inhibits cell fusion by Nipah virus envelope glycoproteins and augments dendritic cell secretion of proinflammatory cytokines. J. Immunol. 175, 413–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cedeno-Laurent F., Opperman M., Barthel S. R., Kuchroo V. K., and Dimitroff C. J. (2012) Galectin-1 triggers an immunoregulatory signature in Th cells functionally defined by IL-10 expression. J. Immunol. 188, 3127–3137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cedeno-Laurent F., and Dimitroff C. J. (2012) Galectin-1 research in T cell immunity: past, present and future. Clin. Immunol. 142, 107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Motran C. C., Molinder K. M., Liu S. D., Poirier F., and Miceli M. C. (2008) Galectin-1 functions as a Th2 cytokine that selectively induces Th1 apoptosis and promotes Th2 function. Eur. J. Immunol. 38, 3015–3027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mercier S., St-Pierre C., Pelletier I., Ouellet M., Tremblay M. J., and Sato S. (2008) Galectin-1 promotes HIV-1 infectivity in macrophages through stabilization of viral adsorption. Virology 371, 121–129 [DOI] [PubMed] [Google Scholar]
- 24. Garner O. B., Yun T., Pernet O., Aguilar H. C., Park A., Bowden T. A., Freiberg A. N., Lee B., and Baum L. G. (2015) Timing of galectin-1 exposure differentially modulates nipah virus entry and syncytium formation in endothelial cells. J. Virol. 89, 2520–2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sun J., Han Z., Shao Y., Cao Z., Kong X., and Liu S. (2014) Comparative proteome analysis of tracheal tissues in response to infectious bronchitis coronavirus, Newcastle disease virus, and avian influenza virus H9 subtype virus infection. Proteomics 14, 1403–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ge J., Deng G., Wen Z., Tian G., Wang Y., Shi J., Wang X., Li Y., Hu S., Jiang Y., Yang C., Yu K., Bu Z., and Chen H. (2007) Newcastle disease virus-based live attenuated vaccine completely protects chickens and mice from lethal challenge of homologous and heterologous H5N1 avian influenza viruses. J. Virol. 81, 150–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hogenkamp A., Isohadouten N., Reemers S. S., Romijn R. A., Hemrika W., White M. R., Tefsen B., Vervelde L., van Eijk M., Veldhuizen E. J., and Haagsman H. P. (2008) Chicken lung lectin is a functional C-type lectin and inhibits haemagglutination by influenza A virus. Vet. Microbiol. 130, 37–46 [DOI] [PubMed] [Google Scholar]
- 28. Reading P. C., Bozza S., Gilbertson B., Tate M., Moretti S., Job E. R., Crouch E. C., Brooks A. G., Brown L. E., Bottazzi B., Romani L., and Mantovani A. (2008) Antiviral activity of the long chain pentraxin PTX3 against influenza viruses. J. Immunol. 180, 3391–3398 [DOI] [PubMed] [Google Scholar]
- 29. Toledo K. A., Fermino M. L., Andrade Cdel C., Riul T. B., Alves R. T., Muller V. D., Russo R. R., Stowell S. R., Cummings R. D., Aquino V. H., and Dias-Baruffi M. (2014) Galectin-1 exerts inhibitory effects during DENV-1 infection. PLoS ONE 9, e112474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rajasagi N. K., Suryawanshi A., Sehrawat S., Reddy P. B., Mulik S., Hirashima M., and Rouse B. T. (2012) Galectin-1 reduces the severity of herpes simplex virus-induced ocular immunopathological lesions. J. Immunol. 188, 4631–4643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. López-Lucendo M. F., Solís D., Sáiz J. L., Kaltner H., Russwurm R., André S., Gabius H. J., and Romero A. (2009) Homodimeric chicken galectin CG-1B (C-14): crystal structure and detection of unique redox-dependent shape changes involving inter-and intrasubunit disulfide bridges by gel filtration, ultracentrifugation, site-directed mutagenesis, and peptide mass fingerprinting. J. Mol. Biol. 386, 366–378 [DOI] [PubMed] [Google Scholar]
- 32. Kaltner H., Kubler D., Lopez-Merino L., Lohr M., Manning J. C., Lensch M., Seidler J., Lehmann W. D., Andre S., Solis D., and Gabius H. J. (2011) Toward comprehensive analysis of the galectin network in chicken: unique diversity of galectin-3 and comparison of its localization profile in organs of adult animals to the other four members of this lectin family. Anat. Rec. 294, 427–444 [DOI] [PubMed] [Google Scholar]
- 33. Kornfeld R., and Kornfeld S. (1985) Assembly of asparagine-linked oligosaccharides. Annu. Rev. Biochem. 54, 631–664 [DOI] [PubMed] [Google Scholar]
- 34. Ng D. T., Hiebert S. W., and Lamb R. A. (1990) Different roles of individual N-linked oligosaccharide chains in folding, assembly, and transport of the simian virus 5 hemagglutinin–neuraminidase. Mol. Cell Biol. 10, 1989–2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shakin-Eshleman S. H., Remaley A. T., Eshleman J. R., Wunner W. H., and Spitalnik S. L. (1992) N-Linked glycosylation of rabies virus glycoprotein. Individual sequons differ in their glycosylation efficiencies and influence on cell-surface expression. J. Biol. Chem. 267, 10690–10698 [PubMed] [Google Scholar]
- 36. El Najjar F., Schmitt A. P., and Dutch R. E. (2014) Paramyxovirus glycoprotein incorporation, assembly and budding: a three way dance for infectious particle production. Viruses 6, 3019–3054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Connaris H., Takimoto T., Russell R., Crennell S., Moustafa I., Portner A., and Taylor G. (2002) Probing the sialic acid binding site of the hemagglutinin–neuraminidase of Newcastle disease virus: identification of key amino acids involved in cell binding, catalysis, and fusion. J. Virol. 76, 1816–1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Crennell S., Takimoto T., Portner A., and Taylor G. (2000) Crystal structure of the multifunctional paramyxovirus hemagglutinin–neuraminidase. Nat. Struct. Biol. 7, 1068–1074 [DOI] [PubMed] [Google Scholar]
- 39. Zaitsev V., von Itzstein M., Groves D., Kiefel M., Takimoto T., Portner A., and Taylor G. (2004) Second sialic acid binding site in Newcastle disease virus hemagglutinin–neuraminidase: implications for fusion. J. Virol. 78, 3733–3741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bousse T. L., Taylor G., Krishnamurthy S., Portner A., Samal S. K., and Takimoto T. (2004) Biological significance of the second receptor binding site of Newcastle disease virus hemagglutinin–neuraminidase protein. J. Virol. 78, 13351–13355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bose S., Song A. S., Jardetzky T. S., and Lamb R. A. (2014) Fusion activation through attachment protein stalk domains indicates a conserved core mechanism of paramyxovirus entry into cells. J. Virol. 88, 3925–3941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Garner O. B., and Baum L. G. (2008) Galectin-glycan lattices regulate cell-surface glycoprotein organization and signalling. Biochem. Soc. Trans. 36, 1472–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu S. D., Tomassian T., Bruhn K. W., Miller J. F., Poirier F., and Miceli M. C. (2009) Galectin-1 tunes TCR binding and signal transduction to regulate CD8 burst size. J. Immunol. 182, 5283–5295 [DOI] [PubMed] [Google Scholar]
- 44. Irie T., and Kawai A. (2002) Studies on the different conditions for rabies virus neutralization by monoclonal antibodies #1–46-12 and #7–1-9. J. Gen. Virol. 83, 3045–3053 [DOI] [PubMed] [Google Scholar]
- 45. Nason E. L., Wetzel J. D., Mukherjee S. K., Barton E. S., Prasad B. V., and Dermody T. S. (2001) A monoclonal antibody specific for reovirus outer-capsid protein σ3 inhibits sigma1-mediated hemagglutination by steric hindrance. J. Virol. 75, 6625–6634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen M., Shi C., Kalia V., Tencza S. B., Montelaro R. C., and Gupta P. (2001) HIV gp120 V(1)/V(2) and C(2)-V(3) domains glycoprotein compatibility is required for viral replication. Virus Res. 79, 91–101 [DOI] [PubMed] [Google Scholar]
- 47. Bose S., Zokarkar A., Welch B. D., Leser G. P., Jardetzky T. S., and Lamb R. A. (2012) Fusion activation by a headless parainfluenza virus 5 hemagglutinin–neuraminidase stalk suggests a modular mechanism for triggering. Proc. Natl. Acad. Sci. U.S.A. 109, E2625–E2634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Iorio R. M., Glickman R. L., and Sheehan J. P. (1992) Inhibition of fusion by neutralizing monoclonal antibodies to the haemagglutinin-neuraminidase glycoprotein of Newcastle disease virus. J. Gen. Virol. 73, 1167–1176 [DOI] [PubMed] [Google Scholar]
- 49. Seal B. S., King D. J., and Sellers H. S. (2000) The avian response to Newcastle disease virus. Dev. Comp. Immunol. 24, 257–268 [DOI] [PubMed] [Google Scholar]
- 50. Brown C. C., Sullivan L., Dufour-Zavala L., Kulkarni A., Williams S., Susta L., Zhang J., and Sellers H. (2013) Comparing presence of avian paramyxovirus-1 through immunohistochemistry in tracheas of experimentally and naturally infected chickens. Avian Dis. 57, 36–40 [DOI] [PubMed] [Google Scholar]
- 51. Reed L. J., and Muench H. (1938) A simple method of estimating 50% end points. Am. J. Epidemiol. 27, 493–497 [Google Scholar]
- 52. Wise M. G., Suarez D. L., Seal B. S., Pedersen J. C., Senne D. A., King D. J., Kapczynski D. R., and Spackman E. (2004) Development of a real-time reverse-transcription PCR for detection of newcastle disease virus RNA in clinical samples. J. Clin. Microbiol. 42, 329–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-(ΔΔCT) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 54. Radonić A., Thulke S., Mackay I. M., Landt O., Siegert W., and Nitsche A. (2004) Guideline to reference gene selection for quantitative real-time PCR. Biochem. Biophys. Res. Commun. 313, 856–862 [DOI] [PubMed] [Google Scholar]
- 55. Birnbaum M. E., Berry R., Hsiao Y. S., Chen Z., Shingu-Vazquez M. A., Yu X., Waghray D., Fischer S., McCluskey J., Rossjohn J., Walz T., and Garcia K. C. (2014) Molecular architecture of the αβ T cell receptor-CD3 complex. Proc. Natl. Acad. Sci. U.S.A. 111, 17576–17581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Peeters B. P., de Leeuw O. S., Koch G., and Gielkens A. L. (1999) Rescue of Newcastle disease virus from cloned cDNA: evidence that cleavability of the fusion protein is a major determinant for virulence. J. Virol. 73, 5001–5009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang X., Liu H., Liu P., Peeters B. P., Zhao C., and Kong X. (2013) Recovery of avirulent, thermostable Newcastle disease virus strain NDV4-C from cloned cDNA and stable expression of an inserted foreign gene. Arch. Virol. 158, 2115–2120 [DOI] [PubMed] [Google Scholar]
- 58. Koishi A. C., Zanello P. R., Bianco É. M., Bordignon J., and Nunes Duarte dos Santos C. (2012) Screening of Dengue virus antiviral activity of marine seaweeds by an in situ enzyme-linked immunosorbent assay. PLoS ONE 7, e51089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.