

Abstract
Increasing evidence suggests a crucial role of inflammation in cytokine-mediated β-cell dysfunction and death in type 1 diabetes mellitus, although the mechanisms are incompletely understood. Sphingosine 1-phosphate (S1P) is a multifunctional bioactive sphingolipid involved in the development of many autoimmune and inflammatory diseases. Here, we investigated the role of intracellular S1P in insulin-secreting INS1E cells by genetically manipulating the S1P-metabolizing enzyme S1P lyase (SPL). The expression of spl was down-regulated by cytokines in INS1E cells and rat islets. Overexpression of SPL protected against cytokine toxicity. Interestingly, the SPL overexpression did not suppress the cytokine-induced NFκB-iNOS-NO pathway but attenuated calcium leakage from endoplasmic reticulum (ER) stores as manifested by lower cytosolic calcium levels, higher expression of the ER protein Sec61a, decreased dephosphorylation of Bcl-2–associated death promoter (Bad) protein, and weaker caspase-3 activation in cytokine-treated (IL-1β, TNFα, and IFNγ) cells. This coincided with reduced cytokine-mediated ER stress, indicated by measurements of CCAAT/enhancer-binding protein homologous protein (chop) and immunoglobulin heavy chain binding protein (bip) levels. Moreover, cytokine-treated SPL-overexpressing cells exhibited increased expression of prohibitin 2 (Phb2), involved in the regulation of mitochondrial assembly and respiration. SPL-overexpressing cells were partially protected against cytokine-mediated ATP reduction and inhibition of glucose-induced insulin secretion. siRNA-mediated spl suppression resulted in effects opposite to those observed for SPL overexpression. Knockdown of phb2 partially reversed beneficial effects of SPL overexpression. In conclusion, the relatively low endogenous Spl expression level in insulin-secreting cells contributes to their extraordinary vulnerability to proinflammatory cytokine toxicity and may therefore represent a promising target for β-cell protection in type 1 diabetes mellitus.
Keywords: cytokine, diabetes, inflammation, insulin secretion, pancreatic islet, sphingosine-1-phosphate (S1P), S1P-lyase, insulin-secreting cells
Introduction
Type 1 diabetes mellitus (T1DM)5 is an autoimmune disease characterized by a gradual loss of insulin-secreting pancreatic β-cells (1, 2). During T1DM development, activated immune cells generate and secrete in the vicinity of β-cells in the islets a number of inflammatory mediators, which either directly damage the β-cells or act via β-cell-surface receptors to alter their intracellular signaling and metabolism. This leads to β-cell dysfunction and death (1–4). The proinflammatory cytokines IL-1β, TNFα, and, to a lesser extent, IFNγ play a crucial role in the destruction process of β-cells by inducing nitrooxidative stress together with mitochondrial and ER disturbances (3, 5–8). Although recent years have strengthened our knowledge about the role of inflammation in T1DM development (6, 9–11), the inflammatory process within the β-cells is not fully understood.
Sphingolipid metabolism and sphingosine 1-phosphate (S1P) play an important role in the development of several inflammatory and autoimmune disorders (12–16). S1P is a bioactive, signaling sphingolipid, which is found in the circulation and various tissues (14). S1P is involved in innate and adaptive immunity, lymphocyte trafficking, angiogenesis, development, cell growth, apoptosis, carcinogenesis, and the inflammatory response (12–15, 17). S1P can act intracellularly as a second messenger or be released and activate G protein–coupled cell-surface receptors (18). Together, these two signaling mechanisms mediate the manifold biological activities of S1P. Because of its signaling functions, the intracellular concentration of S1P is low and is tightly regulated by the balance between its synthesis and degradation (18). The degradation of S1P into hexadecenal and phosphoethanolamine is the last, irreversible step in sphingolipid metabolism and is controlled by the highly conserved ER enzyme S1P lyase (SPL) (18).
Whereas adverse, proapoptotic effects of ceramide and sphingosine in β-cells and a protective role of S1P in type 2 diabetes mellitus are well established (19–21), contradictory findings have been published on the role of S1P in the context of T1DM. On the one hand, incubations with S1P and its analogues showed protective effects on islet β-cell viability against cytokine-mediated cell death (22). On the other hand, fingolimod, an antagonist of the S1P receptor (FTY720), had protective effects in animal models of T1DM (23, 24). It prevented manifestation of diabetes in the LEW.1AR1-iddm rat (23, 24) and the onset of diabetes in nonobese diabetic mice (25), although S1P was also shown to inhibit the CD4(+) T-cell activation and inflammation in this mouse model (26). The protective effect of fingolimod in animal models of T1DM was strongly linked to its ability to regulate T-cell trafficking (23–25).
SPL is a promising drug target for design of autoimmune and anticancer therapies (27–29). SPL deficiency in mice has been shown to induce systemic inflammation (27). SPL inhibition or knockdown contributes to deleterious inflammatory responses in the brain, heart, colon, and lungs (27, 30, 31) and can cause cardio- and neurotoxicity (32–34). So far, very little is known about the role of SPL in insulin-secreting cells. The present study demonstrates that overexpression of SPL can efficiently prevent cytokine-induced dysfunction and cell death in an NFκB/NO-independent manner by maintenance of calcium homeostasis and prevention of cytokine-induced mitochondrial and ER stress in insulin-secreting cells.
Results
Effects of extracellular S1P in insulin-secreting INS1E cells
Insulin-secreting INS1E cells express S1P receptors (S1Pr2, S1Pr3, and S1Pr5) and S1P transporters (Table 1). The abca1 transporter was predominantly expressed, followed by an ∼10-fold lower expression of the abcc1 transporter and 100-fold lower expression of spns2 (Table 1). Their expression was affected by proinflammatory cytokines. A short incubation of 6 h with the mixture of proinflammatory cytokines (60 units/ml IL-1β, 185 units/ml TNFα, and 14 units/ml IFNγ) led to a transient decrease of transcription (with the exception of S1Pr2), whereas a prolonged incubation of 24 h with cytokines increased it (supplemental Table S1).
Table 1.
S1P receptor and transporter gene expression in insulin-secreting INS1E cells
Total RNA was isolated, and after reverse transcription, real-time RT-PCR was performed. Expression was normalized to the housekeeping gene β-actin. Data are means × 1000 ± S.E., with the number of experiments indicated in parentheses. ND, not detectable.
| Gene | Relative expression |
|---|---|
| S1Pr1 | ND |
| S1Pr2 | 0.1 ±0.02 (4) |
| S1Pr3 | 0.6 ±0.1 (6) |
| S1Pr4 | ND |
| S1Pr5 | 0.2 ±0.02 (6) |
| abca1 | 8 ±1 (4) |
| abcc1 | 0.3 ±0.01 (4) |
| abcg2 | ND |
| spns2 | 0.04 ±0.003 (4) |
The analysis of S1P effects in insulin-secreting INS1E cells revealed no induction of cell viability loss in the nanomolar to 1 μm concentration range (supplemental Table S2), at which S1P acts extracellularly (35). Concentrations above 5 μm, at which S1P acts intracellularly (35), however, caused cell viability loss (supplemental Table S2). Proinflammatory cytokines, IL-1β as well as a mixture of IL-1β, TNFα, and IFNγ, stimulated the activation of caspase-3 (Fig. 1A). At a concentration of 5 μm, S1P did not induce caspase-3 activation (Fig. 1A) but slightly inhibited the proliferation rate (data not shown). S1P (5 μm) inhibited cytokine-induced caspase-3 activation (Fig. 1A) and, again, at higher concentrations potentiated cytokine toxicity (data not shown). Although S1P (5 μm) slightly induced the NFκB activation (124 ± 7% versus 100 ± 3% in untreated cells, p < 0.05), it failed to potentiate cytokine-mediated NFκB activation (701 ± 33% (IL-1β) versus 713 ± 34% (IL-1β + S1P); 504 ± 27% (cytokine mixture) versus 520 ± 327% (cytokine mixture + S1P), n = 8).
Figure 1.

Effects of S1P in insulin-secreting INS1E cells. Insulin-secreting INS1E cells were incubated in the absence or presence of 5 μm S1P for 24 h and thereafter. A, caspase-3 activation was measured by the Caspase3-Glo assay after a 24-h incubation with 600 units/ml IL-1β or a cytokine mixture (60 units/ml IL-1β, 185 units/ml TNFα, 14 units/ml IFNγ) in the absence or presence of S1P. B, insulin secretion from cells incubated with 3, 10, and 30 mm Glc was estimated by RIA. C, cAMP concentration in cells grown at 10 mm Glc in the absence or presence of S1P was measured by cAMP-Glo assay. D, ATP content in cells grown at 10 mm Glc in the absence or presence of S1P was measured by the ATPlite assay. Data are means from 4–6 independent experiments, each performed at least in duplicates. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus untreated or 3 mm Glc; #, p < 0.05 versus cells treated identically but without S1P; ANOVA followed by Bonferroni correction. Error bars, S.E.
Basal insulin secretion was increased by a 1-h incubation with 5 μm S1P (Fig. 1B). S1P potentiated glucose-induced insulin secretion by a factor of 2 in response to 10 and 30 mm glucose (Fig. 1B). This effect went along with an increased rate of cAMP generation (Fig. 1C). The rate of ATP production, however, was not affected by S1P (Fig. 1D).
Expression of sphingolipid pathway enzymes in insulin-secreting INS1E cells
To estimate the intracellular potential of insulin-secreting INS1E cells for S1P production, an extensive gene expression analysis of enzymes involved in the sphingolipid pathway was performed (supplemental Table S3 and Fig. 2). The results revealed a high capacity for de novo generation of ceramide in the ER, namely high expression levels of serine palmitoyl transferase (spt) and ceramide synthase (cers), providing evidence for a high capacity for conversion of membrane sphingomyelin into ceramide (Fig. 2A). The enzymes involved in the ceramide conversion to sphingosine (ceramidases; CDase) were more strongly expressed than the enzymes responsible for ceramide conversion into sphingomyelin (sphingomyelin synthase; sms). Insulin-secreting INS1E cells expressed sphingosine kinases (sk), which phosphorylate sphingosine to the bioactive lipid S1P. The enzymes responsible for S1P dephosphorylation (S1P phosphatase; spp) or for its irreversible degradation (S1P lyase; spl) were expressed in insulin-secreting cells, with a higher expression of spl (Fig. 2A). The expression of aldh3a2, the enzyme responsible for the degradation of trans-2-hexadecenal (the S1P degradation product) was in a similar range as that of SPL (supplemental Table S3). Thus, in untreated INS1E cells, the expression pattern of sphingolipid pathway enzymes points toward a high rate of ceramide and sphingosine formation and a lower rate of S1P generation with a good detoxification capacity for S1P degradation products.
Figure 2.

Expression of sphingolipid pathway enzymes in insulin-secreting INS1E cells. The gene expression levels of various rat enzymes of the sphingolipid pathway were measured by real-time RT-PCR in control INS1E insulin-secreting cells. A, schematic enzyme gene expression pattern in untreated cells; B, schematic enzyme gene expression changes upon 24-h incubation with a mixture of cytokines (60 units/ml IL-1β, 185 units/ml TNFα, 14 units/ml IFNγ); C, gene expression determined by quantitative RT-PCR, means ± S.E., n = 3–8. CDase, ceramidases; CerS, ceramide synthase; SPT, serine palmitoyl transferase; SMS, sphingomyelin synthetase; SK, sphingosine kinases. Upward arrows, increase; downward arrows, decrease; =, no change in the gene expression level; the number of plus signs indicates the magnitude of gene expression. *, p < 0.05; **, p < 0.01 versus untreated; ANOVA followed by Bonferroni correction. Error bars, S.E.
Upon a prolonged exposure to proinflammatory cytokines (24 h), the expression pattern was changed, as depicted in Fig. 2 (B and C). Ceramide synthesis was induced, providing more substrate for sphingosine formation. In parallel, the expression of the enzymes converting ceramide into sphingomyelin was reduced, whereas spp expression was strongly increased (Fig. 2, B and C). sk1 expression was not affected by proinflammatory cytokines, whereas sk2 expression was increased and spl was down-regulated after a prolonged incubation with cytokines (24 h). Thus, a higher sphingosine generation rate along with increased S1P turnover in the presence of proinflammatory cytokines can be expected (Fig. 2B).
Generation of SPL-overexpressing and knockdown clones of insulin-secreting INS1E cells
All steps in the sphingolipid cascade are reversible with the exception of the last step, the degradation of S1P into ethanolamine phosphate and hexadecenal catalyzed by SPL (Fig. 2). The expression of spl in insulin-secreting INS1E cells was similar to that in rat islets and much lower than in rat liver, but in an intermediate range when compared with other tissues such as heart, intestine, or skeletal muscle (Table 2). Immunofluorescence staining for Spl revealed that this enzyme is localized in the vicinity of the endoplasmic reticulum in insulin-secreting cells (co-staining with the ER marker Pdi; data not shown). The spl gene expression was transiently weakly increased by proinflammatory cytokines in INS1E cells (6 h; 100 ± 7 for untreated, 132 ± 13 for IL-1β, 157 ± 15 for cytokine mixture; n = 6; p < 0.05), followed by a decrease after a 24-h incubation with cytokines, the time point of cytokine toxicity occurrence (Fig. 2, B and C). This was confirmed in rat islets treated with cytokines for 24 h (Fig. 2C).
Table 2.
S1P lyase (spl) gene expression in various rat tissues
Total RNA was isolated from different rat tissues. Real-time RT-PCR was performed to determine rat spl expression. SPL expression was normalized to β-actin. Data are means ± S.E., with the number of experiments provided in parentheses. The value for liver was 0.019 ± 0.002 (arbitrary units) and was set as 100%. ***, p < 0.001 versus liver, ANOVA followed by Bonferroni correction.
| Tissue | Rat spl |
|---|---|
| % | |
| Brain | 11 ±2 (4)*** |
| Heart | 1 ±0.5 (4)*** |
| Intestine | 5 ±1 (4)*** |
| Kidney | 16 ±5 (4)*** |
| Liver | 100 ±13 (12) |
| Skeletal muscle | 0 ±0 (4)*** |
| Spleen | 16 ±1 (4)*** |
| Testis | 2 ±0.5 (4)*** |
| Pancreas | 16 ±5 (8)*** |
| Pancreatic islets | 21 ±3 (10)*** |
| INS1E cells | 16 ±3 (12)*** |
To define the role of intracellular S1P in insulin-secreting cells, SPL was stably overexpressed or suppressed by a use of siRNA technology in INS1E cells (Fig. 3A). Overexpression of SPL cDNA resulted in a 20-fold increase in SPL expression (human SPL gene: 24 ± 4-fold (INS1E-SPL K1) and 6 ± 0.2-fold (INS1E-SPL K2) changes versus INS1E-control cells, n = 3), whereas suppression by a mixture of three siRNAs against spl led to a 60% decrease in SPL expression (rat spl gene: 100 ± 5% (siQ) and 41 ± 6% (siSPL), n = 6) (Fig. 3A). This was confirmed by Western blotting (Fig. 3A). The intracellular S1P concentration in INS1E cells was 0.2 nmol/μg protein (n = 4). SPL overexpression resulted in a 35 ± 4% decrease in intracellular S1P concentration, and Spl suppression resulted in an 18 ± 2% increase in the S1P concentration (n = 4, p < 0.05).
Figure 3.
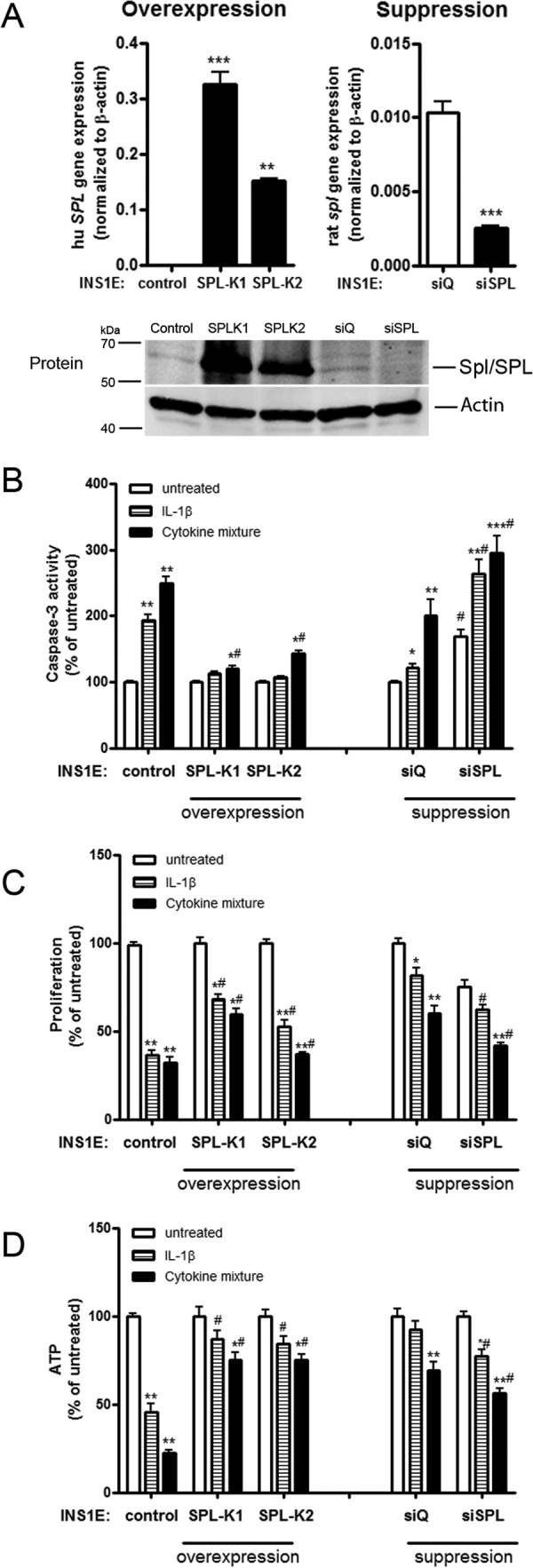
SPL expression and effects on cytokine toxicity. Insulin-secreting control, SPL-overexpressing, or SPL-suppressing INS1E cells were incubated for 24 h with 600 units/ml IL-1β or a cytokine mixture (60 units/ml IL-1β, 185 units/ml TNFα, and 14 units/ml IFNγ). A, expression of SPL was measured by quantitative RT-PCR and Western blotting. B, caspase-3 activation was estimated by Caspase3-Glo assay. C, proliferation of cells was estimated by BrdU ELISA. Data are expressed as a percentage of the values in untreated INS1E-control cells. D, ATP was measured by an ATPlite chemiluminescence assay. Data are means from 4–6 independent experiments, each performed at least in duplicates. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus untreated; #, p < 0.05 versus control cells treated identically; ANOVA followed by Bonferroni correction. Error bars, S.E.
SPL overexpression protects against cytokine-mediated inhibition of cell proliferation and caspase-3 activation in insulin-secreting INS1E cells.
Proinflammatory cytokines stimulated caspase-3 activation in control insulin-secreting INS1E cells (Fig. 3B). The cytokine-induced caspase-3 activation was significantly lower in INS1E-SPL cells (Fig. 3B). The spl suppression by siRNA resulted in a potentiation of cytokine toxicity (Fig. 3B). Cyclosporine A, a blocker of the ABCA1 S1P transporter (36), potentiated cytokine-mediated caspase-3 activation and reversed the protective effects of SPL overexpression (data not shown). The most pronounced potentiating effect of cyclosporine A was observed in INS1E cells treated with siRNA against spl (data not shown).
SPL overexpression partially protected against cytokine-mediated reduction of cell proliferation (Fig. 3C). Again, an opposite effect was observed by spl suppression (Fig. 3C).
SPL overexpression does not affect the cytokine-mediated NFκB-iNOS-NO pathway and nitrooxidative stress
Next, we examined the key mechanisms involved in cytokine toxicity toward insulin-secreting cells, namely the activation of the master transcription factor NFκB and the iNOS pathway (Fig. 4, A and B). The untreated SPL-overexpressing INS1E cells showed a lower level of the NFκB activation as compared with control cells (80 ± 6% versus 100 ± 3% in INS1E-control cells, n = 8). In line with this, spl suppression resulted in a slightly increased level of the NFκB activation in untreated cells (123 ± 14%, n = 4).
Figure 4.

Mechanisms of cytokine toxicity and SPL expression. Insulin-secreting control, SPL-overexpressing, or SPL-suppressing INS1E cells were incubated for 24 h with 600 units/ml IL-1β or a cytokine mixture (60 units/ml IL-1β, 185 units/ml TNFα, and 14 units/ml IFNγ). A, NFκB activation was measured by a SEAP-reporter gene assay. B, iNOS protein expression was analyzed by Western blotting; a representative blot of four independent experiments is shown. C, accumulated nitrite was measured by a Griess assay. D, reactive oxygen species formation was estimated by dichlorofluorescein fluorescence. E, bip gene expression. F, chop gene expression measured by real-time RT-PCR. G, intracellular Ca2+ (at 10 mm Glc) was measured by the fluorescence sensor pCASE12 expressed either in the cytoplasm or in mitochondria. H, P-Bad and tBad, representative Western blots from n = 6 and the ratio of P-Bad/tBad from the densitometry measurements of all blots. I, Sec61a, a representative Western blot from n = 4 and densitometry measurements. J, Phb2 after 24-h cytokine incubation, a representative Western blot from n = 6. K, Phb2 knockdown (siPhb2) confirmation by Western blotting. L, ATP after Phb2 knockdown was measured by an ATPlite assay. M, caspase-3 activation after Phb2 knockdown was estimated by a Caspase3-Glo assay. Data are means from 3–6 independent experiments, each performed at least in duplicates. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus untreated; #, p < 0.05 versus control cells treated identically; §, p < 0.05; §§, p < 0.01 versus the same cell clone incubated with siQ treated identically; ANOVA followed by Bonferroni correction. Error bars, S.E.
Although providing a significant protection against cytokine-induced caspase-3 activation, SPL overexpression did not reduce the cytokine-mediated NFκB activation, iNOS protein expression, or nitrite accumulation (Fig. 4, A–C). No significant effect of SPL overexpression or suppression on reactive oxygen species generation upon exposure to cytokines was observed (Fig. 4D).
SPL overexpression reduces cytokine-induced ER stress
Proinflammatory cytokines induced an ER stress response in insulin-secreting INS1E cells. The expression of chop was significantly enhanced, whereas the expression of bip was down-regulated in INS1E-control cells incubated with 600 units/ml IL-1β or a cytokine mixture (Fig. 4, E and F). Exposure of insulin-secreting INS1E cells to a nontoxic concentration of S1P (5 μm) did not modify the cytokine-induced expression level of the ER stress marker gene chop (data not shown). Interestingly, overexpression of SPL reduced cytokine-mediated chop gene expression (Fig. 4F). In cells with a knockdown of spl, the effects of cytokines on chop expression were more pronounced than those observed in control INS1E cells (Fig. 4F). SPL overexpression prevented the cytokine-mediated reduction of bip expression, whereas spl suppression showed a mild potentiating effect on cytokine-mediated bip decrease (Fig. 4E).
SPL overexpression regulates Ca2+ homeostasis and Bcl2-associated death promotor (Bad) phosphorylation in INS1E cells
To analyze the intracellular calcium levels, we employed the genetically encoded fluorescence sensor pCASE12, which allows a direct detection of Ca2+ concentration changes in a physiological range inside living cells in various subcellular compartments. Interestingly, we observed that changes in Spl expression can influence intracellular Ca2+ (Fig. 4G). SPL-overexpressing cells had lower cytosolic Ca2+ levels when compared with control cells (Fig. 4G). On the contrary, spl suppression resulted in a significant increase of the cytosolic Ca2+ pool (Fig. 4G). The mitochondrial Ca2+ level was mildly decreased by SPL overexpression, whereas spl suppression resulted in a significant rise (Fig. 4G). The protein expression of Serca2b (sarcoplasmic/endoplasmic reticulum calcium ATPase; ER Ca2+ pump) was not affected by SPL overexpression (data not shown); however, SPL-overexpressing cells had a higher protein expression of Sec61a (ER peptide and calcium transport protein subunit α1), also in the presence of cytokines (Fig. 4I). Because an increase in cytosolic Ca2+ can lead to dephosphorylation of Bad, we measured the expression of P-Bad in INS1E cells overexpressing SPL (Fig. 4H). The experiments revealed that the basal and cytokine-mediated protein expression of P-Bad was higher in INS1E-SPL K1 cells as compared with INS1E-control cells (Fig. 4H). The total Bad protein expression was similar in control and SPL-overexpressing INS1E cells (Fig. 4H). The ratio of P-Bad/Bad in cytokine-treated cells was significantly higher in INS1E-SPL cells than in control INS1E cells (Fig. 4H).
SPL overexpression increases the expression of mitochondrial prohibitin 2 (Phb2) and prevents the cytokine-mediated ATP drop
Interestingly, INS1E cells overexpressing SPL were protected against a cytokine-dependent drop in ATP production (Fig. 3D). Suppression of spl potentiated the toxic cytokine effects on ATP content (Fig. 3D). To uncover the mechanism underlying the SPL overexpression effects on cytokine-mediated ATP changes, we measured the expression of two mitochondrial chaperones, mimitin and prohibitin 2, that are involved in the regulation of ATP formation and glucose-induced insulin secretion (GSIS) (37, 38). Both proteins were up-regulated in untreated INS1E-SPL cells (data not shown), and because Phb2 was expressed on a higher level than mimitin, we explored the role of Phb2 in the protective effect of SPL overexpression on ATP in INS1E cells. The Phb2 protein expression was stronger in untreated INS1E-SPL cells as compared with INS1E-control cells (122 ± 14% versus 100% in INS1E-control cells, n = 4; Fig. 4J). The Phb2 expression remained significantly higher also upon cytokine exposure in SPL-overexpressing cells (Fig. 4J).
Knockdown of phb2 in SPL-overexpressing INS1E cells resulted in a partial reduction of ATP content in cytokine-treated cells and boosted the cytokine-mediated caspase-3 activation (Fig. 4, K–M).
SPL overexpression does not modulate basal GSIS but partially protects against cytokine-mediated inhibition of GSIS
Control insulin-secreting INS1E cells showed a concentration-dependent glucose responsiveness of insulin secretion (Fig. 5A). SPL overexpression or suppression did not significantly influence GSIS in INS1E cells (Fig. 5A). Interestingly, SPL overexpression partially protected against cytokine-mediated loss of GSIS (Fig. 5B). On the other hand, spl suppression aggravated the inhibitory effects of cytokines on GSIS (Fig. 5B). Insulin content in control INS1E cells was 28 ± 3 ng/μg DNA and was somewhat higher in INS1E-SPL cells (33 ± 3 ng/μg DNA (INS1E-SPL K1) versus 42 ± 3 ng/μg DNA (INS1E-SPL K2)). Knockdown of phb2 did not significantly affect cytokine-mediated effects on GSIS (data not shown).
Figure 5.

Effects of SPL expression on insulin secretion. Insulin-secreting control, SPL-overexpressing, or SPL-suppressing INS1E cells were incubated with 3, 10, or 30 mm glucose (Glc), after which insulin secretion was measured. A, insulin secretion from cells grown in 3, 10, or 30 mm Glc. B, effects of cytokines (24-h incubation with either 600 units/ml IL-1β or a cytokine mixture consisting of 60 units/ml IL-1β, 185 units/ml TNFα, and 14 units/ml IFNγ) on insulin secretion at 10 mm Glc. Data are means from 4–6 independent experiments, each performed in duplicate. *, p < 0.05; **, p < 0.01 versus control conditions; #, p < 0.05 versus control clone treated identically; ANOVA followed by Bonferroni correction. Error bars, S.E.
Discussion
Inflammation plays an important role in the autoimmune process responsible for β-cell dysfunction and death in T1DM (1, 6, 9, 10, 39). The extraordinary vulnerability of β-cells to proinflammatory cytokines is caused by their weak intracellular antioxidative and anti-inflammatory defense mechanisms and leads to the induction of nitrooxidative stress with parallel mitochondrial and ER disturbances (3, 5–8). Cytokines induce the generation of a variety of inflammatory mediators in β-cells (1–4, 11), among which the formation of ceramide from the plasma membrane sphingomyelin has been linked to TNFα toxicity and inhibition of insulin transcription (22, 40). TNFα is the major cytotoxic proinflammatory cytokine released from the islet infiltrating immune cells in T1DM (41).
Many studies have linked disturbances in the sphingolipid metabolism and S1P milieu to autoimmune and inflammatory diseases (12–16). In the present study, we observed that the sphingolipid pathway enzymes were expressed in a similar fashion in insulin-secreting cells as in other cell types (12–16, 42). Proinflammatory cytokines shifted the pathway toward generation of proapoptotic ceramide and sphingosine and a differential expression of the enzymes involved in S1P generation, dephosphorylation, and degradation. These data are in line with earlier reports from various β-cell lines (43, 44). Whereas short-term incubations with cytokines decreased expression of S1P transporters, longer incubations resulted in an enhanced expression of both S1P transporters and receptors. This is in accordance with the lack of S1P release from INS1 cells upon short-term incubation with IL-1β and TNFα as shown before (43). It suggests that upon cytokine exposure, accumulation of S1P might take place, especially because SPL expression is reduced upon a prolonged incubation with cytokines as we showed here in INS1E cells and rat islets. The expression of sk2 in INS1E cells was stronger than that of sk1, confirming earlier observations (43). sk2-mediated S1P generation is associated in many cell types with the proapoptotic effect of S1P (32, 45–47). Both IL-1β and TNFα increase Sk activity, with the predominance of Sk2, in INS1 cells and rat islets (43). Thus, in insulin-secreting cells, S1P is predominantly formed via Sk2, which is suggestive of its proapoptotic function (Fig. 6, A and B).
Figure 6.

The mechanisms of cytokine-induced dysfunction and death of insulin-secreting cells mediated by intracellular S1P. A, control conditions. B, Cytokine incubation. A, pancreatic β-cells are characterized by a relatively high endogenous expression of the proapoptotic SK2, which generates S1P. S1P is well metabolized upon control conditions as a result of an adequate expression of sphingolipid pathway enzymes, together with a parallel presence of S1P transporters (T) and receptors (R). This keeps S1P at a low concentration and enables the proper function and viability of β-cells and participates in Ca2+ homeostasis. B, proinflammatory cytokines, IL-1β, TNFα, and IFNγ, acting via their specific extracellular receptors, stimulate the expression and activity of SK2, resulting in an increased rate of S1P generation in ER, mitochondria, and nucleus. In parallel, cytokines decrease the expression of S1P transporters and SPL, leading to the accumulation of S1P within the cell. The transcription factor CHOP, which participates in β-cell apoptosis, is activated, and the chaperone protein BIP is down-regulated in the ER. BIP is a partner protein for the polypeptide-transporting channel SEC61 (which binds to its SEC61a1 subunit), and the decreased expression of BIP results in the failure of SEC61 channel closure and leakage of Ca2+ from the ER lumen into the cytoplasm. Elevation of cytosolic Ca2+ activates calpain, leading to dephosphorylation of BAD. This results in cytochrome c release from the mitochondrial membrane and caspase-3/7 activation. A parallel increase in Ca2+ within mitochondria disturbs mitochondrial function. S1P produced inside the nucleus regulates the expression level of a variety of genes probably by histone deacetylase modifications. S1P accumulation results in a reduction of prohibitin 2 and mimitin expression, the two mitochondrial proteins involved in ATP generation. As a consequence, the ATP content is reduced, leading to disturbances in glucose-induced insulin secretion. A reduced ATP content potentiates the proapoptotic effect of intracellular S1P. Thus, intracellular S1P is a novel and important second messenger and regulatory molecule involved in cytokine-mediated β-cell dysfunction and death.
Insulin-secreting cells respond to extracellular S1P via cell-surface S1P receptors (22, 45, 48), the expression of which we confirmed here in INS1E cells. We showed that the S1Pr2 expression was strongly increased by cytokines. S1P at nanomolar concentrations has been shown to protect pancreatic islets against cytokine toxicity (22) and preserves GSIS in islets from diabetes animal models in vitro (22, 45, 48). In the present study, S1P at low concentrations was not toxic and was slightly protective against cytokine-mediated caspase-3 activation in insulin-secreting INS1E cells without inhibition of the NFκB signaling pathway, thus confirming earlier studies (22). However, when used at high concentrations (>5 μm), S1P was toxic to insulin-secreting cells, leading to the activation of caspase-3 and NFκB.6 S1P at low concentrations is known to act via cell-surface receptor activation, whereas when present at high concentrations, much above the optimal receptor-activating concentrations of extracellular S1P (49), it acts intracellularly and was shown to induce deleterious effects (35).
In view of the contradictory reports on the role of S1P under T1DM conditions, we studied the role of intracellularly generated S1P in insulin-secreting cells exposed to proinflammatory cytokines. The analysis was performed by genetic manipulations of SPL expression, the ER-localized enzyme catalyzing the breakdown of S1P to phosphoethanolamine and hexadecenal (18). We showed that the expression of spl in insulin-secreting cells and rat islets is in the intermediate range as compared with other tissues, with levels being 15–20% of the spl expression in liver. Because the expression level of spl did not differ between rat pancreatic islets and the insulin-secreting INS1E cell line, we used in the present study INS1E cells as model β-cells.
Changes of the spl expression level revealed an association between intracellular S1P and cytokine-mediated dysfunction and apoptosis of insulin-secreting INS1E cells (Fig. 6). We observed a reduced cytokine-mediated caspase-3 activation and proliferation inhibition in INS1E-SPL cells, whereas spl knockdown potentiated cytotoxic effects of cytokines. Our data are in accordance with results in the mouse knockout model of Spp2, the enzyme dephosphorylating S1P to sphingosine (hence decreasing the S1P intracellular concentration), in which a worsening of β-cell function was reported (50). Interestingly, SPL overexpression resulted in a parallel 2-fold reduction of the proapoptotic sk2 expression,7 an effect that might be mediated by reduced S1P availability for inhibition of HDAC1/2 (13, 51) by SPL overexpression.
The cytokine-mediated NFκB-iNOS-NO pathway activation is a crucial event in cytokine toxicity to rat insulin-secreting cells (3, 52). Interestingly, although providing protection against cytokine-mediated cell death, SPL overexpression did not inhibit the cytokine-mediated NFκB-iNOS-NO pathway in INS1E cells. A similar observation has been made in insulin-secreting INS1E cells overexpressing mimitin, where the protection against cytokine toxicity via suppression of mitochondrial stress was also achieved without concomitant inhibition of the NFκB-iNOS-NO pathway (37). Thus, the protective effect of SPL overexpression did not depend on the inhibition of cytokine-mediated nitrooxidative stress, explaining the relatively weaker protection against cytokine toxicity as compared with the strategies involving nitrooxidative stress inhibition (7, 53).
SPL overexpression/suppression did not significantly modulate glucose-induced insulin secretion in INS1E cells in the absence of cytokines. Nevertheless, SPL overexpression provided a mild protection against cytokine-mediated reduction of GSIS. This protective effect of SPL overexpression correlated with a prevention of cytokine-mediated ATP reduction and a superior expression of the mitochondrial chaperone Phb2, a highly conserved mitochondrial protein, which regulates mitochondrial assembly and respiration and is crucial for mitochondrial structure integrity (54). phb2 knockout mice develop diabetes due to impaired β-cell function and reduced GSIS (38). We observed that the protein expression of Phb2 in INS1E-SPL cells was higher in SPL-overexpressing cells and was not decreased by proinflammatory cytokines. Suppression of phb2 decreased ATP content and potentiated caspase-3 activation in the presence of cytokines, an effect that was more pronounced in the case of INS1E-SPL cells. Suppression of phb2 failed, however, to inhibit the protective effect of SPL overexpression on GSIS in cytokine-treated cells, suggesting the involvement of another mechanism regulating SPL-mediated effects on GSIS. Thus, SPL overexpression prevented cytokine-mediated reduction of ATP content and GSIS in insulin-secreting INS1E cells at least partially by increasing the expression of the mitochondrial Phb2.
S1P induces Ca2+ mobilization from ER and increases cytosolic Ca2+ (18), which we confirmed here in insulin-secreting INS1E cells. We observed that spl suppression resulted in a significant increase of Ca2+ in cytosol and mitochondria, which correlated with greater cytokine toxicity. Ca2+ modulates pancreatic β-cell function and fate (55, 56). A prolonged elevation of cytosolic Ca2+ activates calpains, the Ca2+-dependent nonlysosomal cysteine proteases, which play a key role in the activation of caspases and proapoptotic members of the Bcl2-protein cascade, like BAD (57). Such a mechanism has also been reported in insulin-secreting MIN6 cells treated with TNFα + IFNγ (56). We observed greater Bad phosphorylation in the INS1E-SPL cells, which were characterized by lower cytosolic Ca2+ and reduced cytokine-mediated caspase-3 activation. Thus, the protective effect of SPL overexpression correlated with decreased cytosolic Ca2+, increased Phb2 expression, and maintenance of Bad phosphorylation.
In pancreatic β-cells, cytokines deplete the ER Ca2+ stores mostly by a NO-dependent mechanism involving SERCA2b (55, 58). However, in the case of SPL overexpression, a NO-independent mechanism of controlling Ca2+ homeostasis must be involved, because SPL overexpression did not influence the NFκB-iNOS-NO pathway and did not affect cytokine-mediated serca2b expression. We showed that SPL overexpression enhanced basal and cytokine-mediated expression of the Sec61 polypeptide transporter, which has been recently shown to be responsible for controlling of Ca2+ leakage from ER by a Bip-dependent mechanism (59). This indicates an excellent polypeptide translocation capacity in INS1E-SPL cells and is in line with higher insulin content and a mild protection against cytokine-mediated GSIS inhibition in INS1E-SPL cells. The Sec61a1 transporter requires for its proper function a guarding by the ER chaperone protein BIP (60), which was expressed at a higher level in SPL-overexpressing cells. A higher bip expression in INS1E-SPL cells can additionally contribute to the maintenance of ER Ca2+ homeostasis by a recently described mechanism involving PERK-regulated ER-plasma membrane contact site formation and refilling of the Ca2+ stores in the ER (61). Thus, SPL overexpression prevents Ca2+ leakage from ER and protects Ca2+ homeostasis by maintaining the adequate Bip and Sec61a expression upon cytokine incubation.
In the present study, we showed that SPL overexpression resulted in a reduction of chop and an increase of bip expression upon cytokine exposure. Reduction of cytokine-mediated ER stress by SPL overexpression is in line with the observation made in the Spp2 knockout mice, where increased intracellular S1P levels correlated with a stronger activation of ER stress in pancreatic β-cells (50).
SPL deficiency participates in the development and progression of immune and neurological disorders, cancer as well as lung and heart inflammation (27, 30–32, 34, 62). Our study indicates that the relatively low expression level of SPL compared with the expression pattern of other sphingolipid pathway enzymes in insulin-secreting cells contributes to their extraordinary vulnerability against proinflammatory cytokines. Accumulation of Sk2-derived S1P in ER and mitochondria as a result of low expression of Spl boosts cytokine-induced β-cell dysfunction and death independently from the NFκB-iNOS-NO pathway (Fig. 6). Because TNFα, the main proinflammatory cytokine present in the endocrine pancreas during T1DM development (41), does not activate the NFκB-iNOS-NO pathway in β-cells, the proposed mechanism might be of particular relevance for its toxicity. It has recently been shown that SPL deficiency can be successfully overcome in colon cancer by oral administration of plant-type sphingolipids called sphingadiens (34) or by delivery of bacterial SPL as in the case of fibrosis therapy (63). Thus, SPL may be a new promising target for β-cell protection in T1DM.
Experimental procedures
Chemicals
BiothermTM Taq polymerase was from GeneCraft (Münster, Germany). Hybond N nylon membranes and the ECL detection system were from Amersham Biosciences (Freiburg, Germany). S1P ELISA was from Echelon Bioscience Inc. (Salt Lake City, UT), and pCASE vectors were from Evrogen (Moscow, Russia). Cytokines were from Promocell (Heidelberg, Germany). All other reagents were from Sigma Chemicals (Munich, Germany).
Animals and tissues
Pancreatic islets and other tissues were from 250–300-g adult male Lewis rats bred in our institution according to the German animal law. Islets were isolated by collagenase digestion, separated by Ficoll gradient, and hand-picked under a stereomicroscope. Isolated islets were incubated overnight in 5 mm Glc fully supplemented RPMI medium as described earlier (37). Thereafter, islets were incubated with IL-1β (600 U/ml) or cytokine mixture (60 units/ml IL-1β, 185 units/ml TNFα, and 14 units/ml IFNγ). RNA was isolated as described below.
Cell culture and cytokine incubations
Insulin-secreting INS1E cells were cultured as described (64) in fully supplemented RPMI medium, with 10 mm glucose, 10% (v/v) FCS, penicillin, and streptomycin, in a humidified atmosphere at 37 °C and 5% CO2. Cells were regularly checked for mycoplasma contamination, and only mycoplasma-free cells were used in this study. For tests, cells were incubated with 600 units/ml IL-1β or the cytokine mixture for 24 h.
Overexpression of SPL in insulin-secreting cells
The human SPL cDNA (pcDNA3.1-hSPL vector) (65) was stably overexpressed in insulin-secreting INS1E cells using the LipofectamineTM transfection method. Positive clones were selected under G418, and SPL expression levels were confirmed by real-time RT-PCR measurements and Western blotting.
Suppression of spl and phb2 in insulin-secreting cells
INS1E cells were transfected with rat spl siRNA (SPL08, -10, and -12) (Santa Cruz Biotechnology, Heidelberg, Germany) or rat phb2 stealth siRNA (RSS340509) (Invitrogen) using Lipofectamine RNAiMAX (ThermoFisher Scientific), and silencing was verified by Western blotting.
Proliferation assay
The proliferation rate of INS1E cells was quantified by a colorimetric method using the Cell Proliferation BrdU-ELISA (Roche, Mannheim, Germany). Cells were seeded at a concentration of 40,000 cells/well in 96-well microtiter plates and allowed to attach for 24 h. Thereafter, cells were incubated with the chemical compounds for 24 h. The proliferation assay was performed as described (9). Absorbance was measured at 450 nm (reference wavelength 650 nm). Data were expressed as a percentage of untreated cells.
Insulin secretion and content
Insulin secretion and content in control and genetically modified insulin-secreting INS1E cell clones were measured by radioimmunoassay (RIA) as described (66). Cells were seeded at a density of 500,000 cells/well onto 6-well plates 2 days before the addition of test compounds. A 24-h incubation with the test compounds was followed by a 1-h incubation without glucose and a 2-h stimulation with glucose (3, 10, or 30 mm) or KCl (25 mm) in the absence or presence of the chemical compounds. Thereafter, supernatants and cell pellets were collected for RIA. Insulin values were normalized to the DNA content of the incubated cells measured by a PicoGreen assay.
RNA isolation, cDNA synthesis, and real-time RT-PCR
Total RNA from insulin-secreting cell clones or rat tissues was obtained using the RNeasy kit (Qiagen, Hilden, Germany). The quality of the total RNA was verified by agarose gel electrophoresis. RNA was quantified spectrophotometrically at 260/280 nm. Thereafter, 2 μg of RNA were reverse transcribed into cDNA using a random hexamer primer (Life Technologies) and RevertAid H Minus M-MuLV reverse transcriptase (Thermo Fisher Scientific, Braunschweig, Germany). QuantiTect SYBR GreenTM technology (Qiagen), which uses a fluorescent dye that binds only double-stranded DNA, was employed. The reactions were performed on a ViiA7 real-time PCR system (Life Technologies) with the following protocol: 50 °C for 2 min, 95 °C for 10 min, and 40 cycles comprising a melting step at 95 °C for 15 s, an annealing step at 58 °C for 60 s, and an extension step at 72 °C for 30 s. The quality of reactions was controlled by analysis of melting curves. Each sample was amplified as triplicate. Data normalization was performed with qBasePlus (Biogazelle, Zwijnaarde, Belgium) against the geometric mean of the housekeeping gene β-actin. The primer sequences are given in supplemental Table S4.
Western blot analyses
Cells were homogenized in ice-cold PBS containing protease inhibitors (Roche) using short bursts (Braun-Sonic 125 Homogenizer, Quigley-Rochester, Rochester, NY). Protein content was determined by the BCA assay (Pierce). For Western blotting, 40 μg of total protein was resolved by SDS-PAGE and then electroblotted onto membranes. Immunodetection was performed using specific primary antibodies (goat anti-SP-lyase T-20 (sc-51431, lot A1808 Santa Cruz Biotechnology), goat anti-NOS2 (sc-649, lot C2414, Santa Cruz Biotechnology), rabbit anti-phospho-BAD (Ser-112) (catalog no. 9291, lot 8, Cell Signaling), rabbit anti-total BAD sc-943 (C-20) (Santa Cruz Biotechnology), mouse monoclonal anti-Phb2 (sc-133094, lot LO413, Santa Cruz Biotechnology), rabbit anti-Sec61a (NB120-15575, lot QD217867, Novus), goat anti-actin C11 (sc1615, lot C1616, Santa Cruz Biotechnology)), as described (37). Pictures were captured by the INTASR chemiluminescence detection system (Intas Science Imaging Instruments, Göttingen, Germany). The intensity of bands was quantified through densitometry with Gel-Pro Analyzer version 4.0 software (Media Cybernetics, Silver Spring, MD).
ATP measurements
ATP content was determined using the ATPlite Detection Assay System (PerkinElmer Life Sciences, Zaventem, Belgium), as described previously (66). Cells were seeded at a density of 40,000 cells/well onto black 96-well plates 24 h before the addition of the chemical compounds. After incubation with test compounds, cells were cultured in the absence of glucose for 1 h, followed by a 2-h incubation with glucose and/or chemical compounds. Cells were then lysed and used for ATP concentration measurements as described (66).
cAMP measurements
The cAMP concentration was measured by the cAMP-GloTM assay kit according to the manufacturer's instructions (Promega, Mannheim, Germany). Cells were seeded at a density of 5,000 cells/well onto white 96-well plates 24 h before the addition of chemical compounds. After incubation with test compounds, cells were cultured in the absence of glucose for 1 h and followed by a 2-h incubation with glucose and/or chemical compounds. Cells were then lysed and used for cAMP concentration measurements as described (66).
NFκB reporter gene assay
20,000 cells/well were seeded onto 96-well plates 24 h before transient transfection was performed with jetPRIMETM transfection reagent (HiSS Diagnostics, Freiburg, Germany). Test compounds were added 24 h after transfection and for 24 h. The pSEAP-NF-κB construct was used as described (37). Secreted alkaline phosphatase was measured using the Phospha-LightTM system kit (Applera, Darmstadt, Germany).
Nitrite measurements
Nitrite accumulation after a 24-h incubation with cytokines was determined spectrophotometrically at 562 nm by the Griess reaction assay as described (37).
Caspase-3/7 assay
Cells were seeded at a density of 10,000 cells/well onto 96-well plates and allowed to attach for 24 h before test compounds were added. Activation of caspase-3 was quantified by the CaspaseGlo-3/7 kit (Promega, Mannheim, Germany) according to the manual's instruction. Samples were analyzed using the GloMax®Multi detection system (Promega), and data were analyzed by Instinct software (Letterkenny, Co. Donegal, Ireland). Data are expressed as the percentage of cells without exposure to test compounds.
S1P ELISA
The S1P-coated 96-well microtiter plates blocked to reduce nonspecific binding were used. Samples were prepared using lysis buffer: 20 mm PIPES, 150 mm NaCl, 1 mm EGTA, 1% (v/v) Triton X-100, 1.5 mm MgCl2, 0.1% SDS, 1 mm sodium orthovanidate, 1× protease inhibitor mixture (without EDTA), pH 7. The S1P-ELISA (Echelon, ImTec Diagnostics, Antwerpen, Belgium) was performed according to the manual's instructions. The absorbance at 450 nm was measured, and the concentration of S1P in the samples was determined by comparison with the standard curve.
Calcium measurements
The levels of intracellular calcium were estimated using the genetically encoded fluorescence sensor pCASE12 (Evrogen), which allows direct detection of Ca2+ concentration changes in a physiological range inside living cells in various subcellular compartments. Constructs enabling cytosolic (pCASE12-Cyto) and mitochondrial (pCASE12-Mito) Ca2+ measurements were transiently transfected using JetPrime technology. At 48 h after transfection, test compounds were added, and fluorescence measurements were performed. pCASE12 fluorescence intensity was analyzed using a CellR/Olympus IX81 inverted microscope system equipped with a Cellcubator (UPLSAPO ×60, 1.35 numerical aperture oil immersion objective, GFP filter (485-nm excitation and 520-nm emission), fluorescence intensity of 12.5%) (Olympus, Hamburg, Germany). The intensity of fluorescence in the region of interest was measured for each condition in at least 100 cells.
Reactive species detection by dichlorofluorescein fluorescence
To detect overall oxidative and nitrosative stress, cells were seeded onto 96-well coated black plates. Before the addition of test compounds, cells were preincubated with 10 μm dichlorodihydrofluorescein diacetate (Invitrogen, Karlsruhe, Germany) for 40 min at 37 °C. Plates were analyzed at 480/520-nm excitation/emission using the fluorescence reader Victor2 1420 Multilabel Counter (PerkinElmer Life Sciences). Each condition was analyzed at least in duplicate. Data were expressed as a percentage of untreated cells.
Data analysis
All data are expressed as means ± S.E. Statistical analyses were performed using the Prism analysis program (GraphPad, San Diego, CA).
Author contributions
C. H. performed experiments, analyzed data, and participated in the manuscript writing; K. T. performed experiments, analyzed data, and revised the manuscript; J. D. S. provided genetic constructs, interpreted data, and revised the manuscript; S. L. discussed the data and revised the manuscript; E. G.-C. conceived and supervised the project, designed and performed experiments, analyzed data, and wrote the manuscript. All authors read and accepted the final version of the manuscript.
Supplementary Material
Acknowledgments
INS1E cells were a kind gift of Prof. Claes B. Wollheim (Lund University, Lund, Sweden). The excellent technical assistance of M. Funck, M. Wirth, and C. Schwab is gratefully acknowledged.
This work was supported by the Innovative Medicines Initiative Joint Undertaking under grant agreement 155005 (IMIDIA), resources of which are composed of a financial contribution from the European Union's Seventh Framework Programme (FP7/2007–2013) and EFPIA companies' in kind contribution. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Tables S1–S4.
C. Hahn, unpublished observations.
E. Gurgul-Convey, unpublished results.
- T1DM
- type 1 diabetes mellitus
- BAD
- Bcl2-associated death promotor
- P-BAD
- phosphorylated BAD
- BIP
- immunoglobulin heavy chain-binding protein
- CHOP
- CCAAT/enhancer-binding protein homologous protein
- ER
- endoplasmic reticulum
- GSIS
- glucose-induced insulin secretion
- iNOS
- inducible NO synthase
- NFκB
- nuclear factor κB
- SK
- sphingosine kinase
- S1P
- sphingosine 1-phosphate
- SPL
- S1P lyase
- PERK
- protein kinase R-like endoplasmic reticulum kinase.
References
- 1. Roep B. O., and Peakman M. (2011) Diabetogenic T lymphocytes in human type 1 diabetes. Curr. Opin. Immunol. 23, 746–753 [DOI] [PubMed] [Google Scholar]
- 2. van Belle T. L., Coppieters K. T., and von Herrath M. G. (2011) Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol. Rev. 91, 79–118 [DOI] [PubMed] [Google Scholar]
- 3. Mandrup-Poulsen T. (2003) Apoptotic signal transduction pathways in diabetes. Biochem. Pharmacol. 66, 1433–1440 [DOI] [PubMed] [Google Scholar]
- 4. Rabinovitch A., and Suarez-Pinzon W. L. (2003) Role of cytokines in the pathogenesis of autoimmune diabetes mellitus. Rev. Endocr. Metab. Disord. 4, 291–299 [DOI] [PubMed] [Google Scholar]
- 5. Cnop M., Welsh N., Jonas J. C., Jörns A., Lenzen S., and Eizirik D. L. (2005) Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes 54, S97–S107 [DOI] [PubMed] [Google Scholar]
- 6. Coppieters K. T., and von Herrath M. G. (2014) The type 1 diabetes signature: hardwired to trigger inflammation? Diabetes 63, 3581–3583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gurgul-Convey E., Mehmeti I., Lortz S., and Lenzen S. (2011) Cytokine toxicity in insulin-producing cells is mediated by nitro-oxidative stress-induced hydroxyl radical formation in mitochondria. J. Mol. Med. 89, 785–798 [DOI] [PubMed] [Google Scholar]
- 8. Eizirik D. L., Miani M., and Cardozo A. K. (2013) Signalling danger: endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. Diabetologia 56, 234–241 [DOI] [PubMed] [Google Scholar]
- 9. Gurgul-Convey E., and Lenzen S. (2010) Protection against cytokine toxicity through endoplasmic reticulum and mitochondrial stress prevention by prostacyclin synthase overexpression in insulin-producing cells. J. Biol. Chem. 285, 11121–11128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hotamisligil G. S. (2006) Inflammation and metabolic disorders. Nature 444, 860–867 [DOI] [PubMed] [Google Scholar]
- 11. Morgan N. G., Leete P., Foulis A. K., and Richardson S. J. (2014) Islet inflammation in human type 1 diabetes mellitus. IUBMB Life 66, 723–734 [DOI] [PubMed] [Google Scholar]
- 12. Alvarez S. E., Milstien S., and Spiegel S. (2007) Autocrine and paracrine roles of sphingosine-1-phosphate. Trends Endocrinol. Metab. 18, 300–307 [DOI] [PubMed] [Google Scholar]
- 13. Aoki M., Aoki H., Ramanathan R., Hait N. C., and Takabe K. (2016) Sphingosine-1-phosphate signaling in immune cells and inflammation: roles and therapeutic potential. Mediators Inflamm. 2016, 8606878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Degagné E., and Saba J. D. (2014) S1pping fire: sphingosine-1-phosphate signaling as an emerging target in inflammatory bowel disease and colitis-associated cancer. Clin. Exp. Gastroenterol. 7, 205–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Spiegel S., and Milstien S. (2011) The outs and the ins of sphingosine-1-phosphate in immunity. Nat. Rev. Immunol. 11, 403–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Candido K., Soufi H., Bandyopadhyay M., and Dasgupta S. (2016) Therapeutic impact of sphingosine 1-phosphate receptor signaling in multiple sclerosis. Mini Rev. Med. Chem. 16, 547–554 [DOI] [PubMed] [Google Scholar]
- 17. Hait N. C., Oskeritzian C. A., Paugh S. W., Milstien S., and Spiegel S. (2006) Sphingosine kinases, sphingosine 1-phosphate, apoptosis and diseases. Biochim. Biophys. Acta 1758, 2016–2026 [DOI] [PubMed] [Google Scholar]
- 18. Spiegel S., and Milstien S. (2003) Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 4, 397–407 [DOI] [PubMed] [Google Scholar]
- 19. Mastrandrea L. D., Sessanna S. M., Del Toro A., and Laychock S. G. (2010) ATP-independent glucose stimulation of sphingosine kinase in rat pancreatic islets. J. Lip. Res. 51, 2171–2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Qi Y., Chen J., Lay A., Don A., Vadas M., and Xia P. (2013) Loss of sphingosine kinase 1 predisposes to the onset of diabetes via promoting pancreatic beta-cell death in diet-induced obese mice. FASEB J. 27, 4294–4304 [DOI] [PubMed] [Google Scholar]
- 21. Véret J., Bellini L., Giussani P., Ng C., Magnan C., and Le Stunff H. (2014) Roles of sphingolipid metabolism in pancreatic beta cell dysfunction induced by lipotoxicity. J. Clin. Med. 3, 646–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Laychock S. G., Sessanna S. M., Lin M. H., and Mastrandrea L. D. (2006) Sphingosine 1-phosphate affects cytokine-induced apoptosis in rat pancreatic islet beta-cells. Endocrinology 147, 4705–4712 [DOI] [PubMed] [Google Scholar]
- 23. Jörns A., Akin M., Arndt T., Terbish T., Zu Vilsendorf A. M., Wedekind D., Hedrich H. J., and Lenzen S. (2014) Anti-TCR therapy combined with fingolimod for reversal of diabetic hyperglycemia by beta cell regeneration in the LEW.1AR1-iddm rat model of type 1 diabetes. J. Mol. Med. 92, 743–755 [DOI] [PubMed] [Google Scholar]
- 24. Jörns A., Rath K. J., Terbish T., Arndt T., Meyer Zu Vilsendorf A., Wedekind D., Hedrich H. J., and Lenzen S. (2010) Diabetes prevention by immunomodulatory FTY720 treatment in the LEW.1AR1-iddm rat despite immune cell activation. Endocrinology 151, 3555–3565 [DOI] [PubMed] [Google Scholar]
- 25. Maki T., Gottschalk R., Ogawa N., and Monaco A. P. (2005) Prevention and cure of autoimmune diabetes in nonobese diabetic mice by continuous administration of FTY720. Transplantation 79, 1051–1055 [DOI] [PubMed] [Google Scholar]
- 26. Srinivasan S., Bolick D. T., Lukashev D., Lappas C., Sitkovsky M., Lynch K. R., and Hedrick C. C. (2008) Sphingosine-1-phosphate reduces CD4+ T-cell activation in type 1 diabetes through regulation of hypoxia-inducible factor short isoform I.1 and CD69. Diabetes 57, 484–493 [DOI] [PubMed] [Google Scholar]
- 27. Allende M. L., Bektas M., Lee B. G., Bonifacino E., Kang J., Tuymetova G., Chen W., Saba J. D., and Proia R. L. (2011) Sphingosine-1-phosphate lyase deficiency produces a pro-inflammatory response while impairing neutrophil trafficking. J. Biol. Chem. 286, 7348–7358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bektas M., Allende M. L., Lee B. G., Chen W., Amar M. J., Remaley A. T., Saba J. D., and Proia R. L. (2010) Sphingosine 1-phosphate lyase deficiency disrupts lipid homeostasis in liver. J. Biol. Chem. 285, 10880–10889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kunkel G. T., Maceyka M., Milstien S., and Spiegel S. (2013) Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat. Rev. Drug Discov. 12, 688–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Veltman M., Stolarczyk M., Radzioch D., Wojewodka G., De Sanctis J. B., Dik W. A., Dzyubachyk O., Oravecz T., de Kleer I., and Scholte B. J. (2016) Correction of lung inflammation in a F508del CFTR murine cystic fibrosis model by the sphingosine-1-phosphate lyase inhibitor LX2931. Am. J. Physiol. Lung Cell Mol. Physiol. 311, L1000–L1014 [DOI] [PubMed] [Google Scholar]
- 31. Harris C. M., Mittelstadt S., Banfor P., Bousquet P., Duignan D. B., Gintant G., Hart M., Kim Y., and Segreti J. (2016) Sphingosine-1-phosphate (S1P) lyase inhibition causes increased cardiac S1P levels and bradycardia in rats. J. Pharmacol. Exp. Ther. 359, 151–158 [DOI] [PubMed] [Google Scholar]
- 32. Hagen N., Van Veldhoven P. P., Proia R. L., Park H., Merrill A. H. Jr., and van Echten-Deckert G. (2009) Subcellular origin of sphingosine 1-phosphate is essential for its toxic effect in lyase-deficient neurons. J. Biol. Chem. 284, 11346–11353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mitroi D. N., Deutschmann A. U., Raucamp M., Karunakaran I., Glebov K., Hans M., Walter J., Saba J., Gräler M., Ehninger D., Sopova E., Shupliakov O., Swandulla D., and van Echten-Deckert G. (2016) Sphingosine 1-phosphate lyase ablation disrupts presynaptic architecture and function via an ubiquitin-proteasome mediated mechanism. Sci. Rep. 6, 37064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Degagné E., Pandurangan A., Bandhuvula P., Kumar A., Eltanawy A., Zhang M., Yoshinaga Y., Nefedov M., de Jong P. J., Fong L. G., Young S. G., Bittman R., Ahmedi Y., and Saba J. D. (2014) Sphingosine-1-phosphate lyase downregulation promotes colon carcinogenesis through STAT3-activated microRNAs. J. Clin. Invest. 124, 5368–5384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang F., Nobes C. D., Hall A., and Spiegel S. (1997) Sphingosine 1-phosphate stimulates rho-mediated tyrosine phosphorylation of focal adhesion kinase and paxillin in Swiss 3T3 fibroblasts. Biochem. J. 324, 481–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nagao K., Maeda M., Mañucat N. B., and Ueda K. (2013) Cyclosporine A and PSC833 inhibit ABCA1 function via direct binding. Biochim. Biophys. Acta 1831, 398–406 [DOI] [PubMed] [Google Scholar]
- 37. Hanzelka K., Skalniak L., Jura J., Lenzen S., and Gurgul-Convey E. (2012) Effects of the novel mitochondrial protein mimitin in insulin-secreting cells. Biochem. J. 445, 349–359 [DOI] [PubMed] [Google Scholar]
- 38. Supale S., Thorel F., Merkwirth C., Gjinovci A., Herrera P. L., Scorrano L., Meda P., Langer T., and Maechler P. (2013) Loss of prohibitin induces mitochondrial damages altering beta-cell function and survival and is responsible for gradual diabetes development. Diabetes 62, 3488–3499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Robertson R. P. (1998) Dominance of cyclooxygenase-2 in the regulation of pancreatic islet prostaglandin synthesis. Diabetes 47, 1379–1383 [DOI] [PubMed] [Google Scholar]
- 40. Taha T. A., Mullen T. D., and Obeid L. M. (2006) A house divided: ceramide, sphingosine, and sphingosine-1-phosphate in programmed cell death. Biochim. Biophys. Acta 1758, 2027–2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jörns A., Arndt T., Meyer zu Vilsendorf A., Klempnauer J., Wedekind D., Hedrich H. J., Marselli L., Marchetti P., Harada N., Nakaya Y., Wang G. S., Scott F. W., Gysemans C., Mathieu C., and Lenzen S. (2014) Islet infiltration, cytokine expression and beta cell death in the NOD mouse, BB rat, Komeda rat, LEW.1AR1-iddm rat and humans with type 1 diabetes. Diabetologia 57, 512–521 [DOI] [PubMed] [Google Scholar]
- 42. Saba J. D., and de la Garza-Rodea A. S. (2013) S1P lyase in skeletal muscle regeneration and satellite cell activation: exposing the hidden lyase. Biochim. Biophys. Acta 1831, 167–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mastrandrea L. D., Sessanna S. M., and Laychock S. G. (2005) Sphingosine kinase activity and sphingosine-1 phosphate production in rat pancreatic islets and INS-1 cells: response to cytokines. Diabetes 54, 1429–1436 [DOI] [PubMed] [Google Scholar]
- 44. Zhu Q., Shan X., Miao H., Lu Y., Xu J., You N., Liu C., Liao D. F., and Jin J. (2009) Acute activation of acid ceramidase affects cytokine-induced cytotoxicity in rat islet beta-cells. FEBS Lett. 583, 2136–2141 [DOI] [PubMed] [Google Scholar]
- 45. Jessup C. F., Bonder C. S., Pitson S. M., and Coates P. T. (2011) The sphingolipid rheostat: a potential target for improving pancreatic islet survival and function. Endocr. Metab. Immune Disord. Drug Targets 11, 262–272 [DOI] [PubMed] [Google Scholar]
- 46. Maceyka M., Sankala H., Hait N. C., Le Stunff H., Liu H., Toman R., Collier C., Zhang M., Satin L. S., Merrill A. H. Jr., Milstien S., and Spiegel S. (2005) SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J. Biol. Chem. 280, 37118–37129 [DOI] [PubMed] [Google Scholar]
- 47. Taha T. A., Hannun Y. A., and Obeid L. M. (2006) Sphingosine kinase: biochemical and cellular regulation and role in disease. J. Biochem. Mol. Biol. 39, 113–131 [DOI] [PubMed] [Google Scholar]
- 48. Japtok L., Schmitz E. I., Fayyaz S., Krämer S., Hsu L. J., and Kleuser B. (2015) Sphingosine 1-phosphate counteracts insulin signaling in pancreatic beta-cells via the sphingosine 1-phosphate receptor subtype 2. FASEB J. 29, 3357–3369 [DOI] [PubMed] [Google Scholar]
- 49. Hla T., Lee M. J., Ancellin N., Paik J. H., and Kluk M. J. (2001) Lysophospholipids–receptor revelations. Science 294, 1875–1878 [DOI] [PubMed] [Google Scholar]
- 50. Taguchi Y., Allende M. L., Mizukami H., Cook E. K., Gavrilova O., Tuymetova G., Clarke B. A., Chen W., Olivera A., and Proia R. L. (2016) Sphingosine-1-phosphate phosphatase 2 regulates pancreatic islet beta-cell endoplasmic reticulum stress and proliferation. J. Biol. Chem. 291, 12029–12038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Maceyka M., and Spiegel S. (2014) Sphingolipid metabolites in inflammatory disease. Nature 510, 58–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Eizirik D. L., and Darville M. I. (2001) Beta-cell apoptosis and defense mechanisms: lessons from type 1 diabetes. Diabetes 50, S64–S69 [DOI] [PubMed] [Google Scholar]
- 53. Gurgul E., Lortz S., Tiedge M., Jörns A., and Lenzen S. (2004) Mitochondrial catalase overexpression protects insulin-producing cells against toxicity of reactive oxygen species and proinflammatory cytokines. Diabetes 53, 2271–2280 [DOI] [PubMed] [Google Scholar]
- 54. Strub G. M., Paillard M., Liang J., Gomez L., Allegood J. C., Hait N. C., Maceyka M., Price M. M., Chen Q., Simpson D. C., Kordula T., Milstien S., Lesnefsky E. J., and Spiegel S. (2011) Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 25, 600–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ramadan J. W., Steiner S. R., O'Neill C. M., and Nunemaker C. S. (2011) The central role of calcium in the effects of cytokines on beta-cell function: implications for type 1 and type 2 diabetes. Cell Calcium 50, 481–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chang I., Cho N., Kim S., Kim J. Y., Kim E., Woo J. E., Nam J. H., Kim S. J., and Lee M. S. (2004) Role of calcium in pancreatic islet cell death by IFN-γ/TNF-α. J. Immun. 172, 7008–7014 [DOI] [PubMed] [Google Scholar]
- 57. Smith M. A., and Schnellmann R. G. (2012) Calpains, mitochondria, and apoptosis. Cardiovasc. Res. 96, 32–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cardozo A. K., Ortis F., Storling J., Feng Y. M., Rasschaert J., Tonnesen M., Van Eylen F., Mandrup-Poulsen T., Herchuelz A., and Eizirik D. L. (2005) Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic beta-cells. Diabetes 54, 452–461 [DOI] [PubMed] [Google Scholar]
- 59. Zimmermann R. (2016) Components and mechanisms of import, modification, folding, and assembly of immunoglobulins in the endoplasmic reticulum. J. Clin. Immunol. 36, 5–11 [DOI] [PubMed] [Google Scholar]
- 60. Schäuble N., Lang S., Jung M., Cappel S., Schorr S., Ulucan O., Linxweiler J., Dudek J., Blum R., Helms V., Paton A. W., Paton J. C., Cavalié A., and Zimmermann R. (2012) BiP-mediated closing of the Sec61 channel limits Ca2+ leakage from the ER. EMBO J. 31, 3282–3296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. van Vliet A. R., Giordano F., Gerlo S., Segura I., Van Eygen S., Molenberghs G., Rocha S., Houcine A., Derua R., Verfaillie T., Vangindertael J., De Keersmaecker H., Waelkens E., Tavernier J., Hofkens J., et al. (2017) The ER stress sensor PERK coordinates ER-plasma membrane contact site formation through interaction with filamin-A and F-actin remodeling. Mol. Cell 65, 885–899.e6 [DOI] [PubMed] [Google Scholar]
- 62. Hagen N., Hans M., Hartmann D., Swandulla D., and van Echten-Deckert G. (2011) Sphingosine-1-phosphate links glycosphingolipid metabolism to neurodegeneration via a calpain-mediated mechanism. Cell Death Differ. 18, 1356–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Huwiler A., Bourquin F., Kotelevets N., Pastukhov O., Capitani G., Grütter M. G., and Zangemeister-Wittke U. (2011) A prokaryotic S1P lyase degrades extracellular S1P in vitro and in vivo: implication for treating hyperproliferative disorders. PLoS One 6, e22436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ximenes H. M., Lortz S., Jörns A., and Lenzen S. (2007) Triiodothyronine (T3)-mediated toxicity and induction of apoptosis in insulin-producing INS-1 cells. Life Sci. 80, 2045–2050 [DOI] [PubMed] [Google Scholar]
- 65. Reiss U., Oskouian B., Zhou J., Gupta V., Sooriyakumaran P., Kelly S., Wang E., Merrill A. H. Jr., and Saba J. D. (2004) Sphingosine-phosphate lyase enhances stress-induced ceramide generation and apoptosis. J. Biol. Chem. 279, 1281–1290 [DOI] [PubMed] [Google Scholar]
- 66. Gurgul-Convey E., Hanzelka K., and Lenzen S. (2012) Mechanism of prostacyclin-induced potentiation of glucose-induced insulin secretion. Endocrinology 153, 2612–2622 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.