

Abstract

There is significant interest in developing analytical methods to characterize molecular recognition events between proteins and phosphoinositides, which are a medically important class of carbohydrate-functionalized lipids. Within this scope, one area of high priority involves quantitatively evaluating drug candidates that pharmacologically inhibit protein–phosphoinositide interactions. As full-length proteins are often difficult to produce, establishing methods to study these interactions with shorter, bioactive peptides would be advantageous. Herein, we report an atomic force microscopy (AFM)-based force spectroscopic approach to detect the specific interaction between an amphipathic, α-helical (AH) peptide derived from the hepatitis C virus NS5A protein and its biological target, the phosphatidylinositol (4,5)-bisphosphate [PI(4,5)P2] phosphoinositide receptor. After optimization of the peptide tethering strategy and measurement parameters, the binding specificity of AH peptide for PI(4,5)P2 receptors was comparatively evaluated across a panel of phosphoinositides and the influence of ionic strength on AH–PI(4,5)P2 binding strength was tested. Importantly, these capabilities were translated into the development of a novel experimental methodology to determine the inhibitory activity of a small-molecule drug candidate acting against the AH–PI(4,5)P2 interaction, and extracted kinetic parameters agree well with literature values obtained by conventional biochemical methods. Taken together, our findings provide a nanomechanical basis for explaining the high binding specificity of the NS5A AH to PI(4,5)P2 receptors, in turn establishing an analytical framework to study phosphoinositide-binding viral peptides and proteins as well as a broadly applicable approach to evaluate candidate inhibitors of protein–phosphoinositide interactions.
Phosphoinositides are carbohydrate-functionalized lipids that play important roles in a wide range of biological processes related to signaling, metabolism, and membrane trafficking, among other activities.1−3 Phosphoinositide molecules are characterized by an inositol ring, which can be phosphorylated at one or more ring positions by kinases and phosphatases, and this carbohydrate moiety (known as the inositol phosphate) is linked to diacylglycerol via a phosphodiester linkage.3 There are numerous inositide-binding domains found in proteins that recognize different phosphoinositides on the basis of the molecular structure of the phosphorylated inositol ring, that is, the number and position of the phosphate groups.4,5 Regarded as a key class of protein–carbohydrate interactions, protein–phosphoinositide interactions occur through specific binding between a protein’s binding pocket and a phosphoinositide molecule6 or through more promiscuous multivalent interactions based on electrostatic attraction between a protein surface and several phosphoinositide molecules.7 Unraveling the details of these interactions is highly nuanced, depending on the system under consideration,8 and numerous analytical tools have been developed to monitor phosphoinositide synthesis9 and phosphorylation steps,10 to determine the location and quantity of phosphoinositides in cellular membranes,11,12 and to probe these interactions with real-time monitoring.13−15 The development of new analytical tools to study protein–phosphoinositide interactions remains an important need, especially in terms of evaluating pharmacological strategies to treat phosphoinositide-associated diseases of importance to human health and medicine.16−18
There is growing evidence that phosphoinositides are critically involved in the life cycles of medically important viruses.19 A classic example is the phosphatidylinositol (4,5)-bisphosphate [PI(4,5)P2] lipid, which directs the human immunodeficiency virus type 1 (HIV-1) Gag protein to plasma membranes as part of the process to create new virus particles.20 Recently, it was discovered that PI(4,5)P2 also plays an important role in the life cycle of the hepatitis C virus (HCV) by interacting with the N-terminal amphipathic α-helix (AH) of the HCV nonstructural 5A (NS5A) protein as part of viral genome replication.21−23 In particular, it was reported that the NS5A AH specifically binds intracellular PI(4,5)P2, which is necessary for viral genome replication.24 A pair of positively charged lysine residues flanking a hydrophobic face within the NS5A AH was required for PI(4,5)P2 binding and genome replication, leading to identification of the basic amino acid PI(4,5)P2 pincer (BAAPP) domain.24 While there are other known structural motifs that mediate PI(4,5)P2 binding,25 none of these were present in the NS5A AH. BAAPP domains are not unique to HCV, however, and appear to be widespread among pathogen and even host cell proteins.24 The role of PI(4,5)P2 in the HCV life cycle represents the first example of phosphoinositides mediating viral genome replication. Understanding mechanistic details about the NS5A AH–PI(4,5)P2 interaction can provide insights into the viral life cycle and identify novel antiviral strategies targeting this interaction.
Toward this goal, a quantitative measurement strategy is needed to unravel the factors that contribute to the specificity and strength of the interaction between NS5A AH and PI(4,5)P2 receptors. Developing such capabilities would also be useful for evaluating small-molecule inhibitors and other drug candidates. While a large number of qualitative and semiquantitative methods have been developed for studying protein–carbohydrate interactions in general,26−28 precise determination of binding affinities is difficult to achieve with conventional measurement approaches that rely on ensemble averaging26,29 and typically have large errors.30−32 One promising approach involves direct measurement of the binding forces between individual proteins and carbohydrate moieties by atomic force microscopy (AFM) force spectroscopy.33−35 Touhami et al.36 first reported AFM force spectroscopic measurements of specific lectin–carbohydrate interactions involving the concanavalin A protein, and the corresponding binding forces are known to be weaker than those associated with antibody–antigen interactions.37 Other protein–carbohydrate interactions also have relatively weak binding forces,38 highlighting the importance of multivalency in protein–carbohydrate systems.39 Among phosphoinositides, Malkovskiy et al.40 have measured the interaction between the PLCδ/1PH protein and PI(4,5)P2 molecules and determined similar binding force values. While the feasibility of measuring protein–carbohydrate interactions by AFM force spectroscopy has been demonstrated, important shortcomings remain to be addressed. To date, all relevant studies have utilized full-length proteins, and extending this approach to shorter bioactive peptides, as has been done for peptide–nucleic acid systems,41,42 would greatly advance measurement capabilities, especially for proteins that are difficult to produce. Indeed, recombinant expression and purification of full-length HCV NS5A protein is technically challenging,43 and the N-terminal AH is often not included.44,45 Hence, establishing measurement approaches based on synthetic bioactive peptides would be broadly useful and could provide a tractable means to examine NS5A AH BAAPP domain–phosphoinositide interactions. Another important unrealized opportunity relates to applying these analytical technologies to quantitatively evaluate the efficacy of candidate inhibitors that antagonize protein–phosphoinositide interactions.
Herein, we report AFM force spectroscopic measurements for characterizing the interaction between an amphipathic α-helical peptide comprising the NS5A AH BAAPP domain (termed AH peptide) and substrate-bound PI(4,5)P2 receptors, as well as its pharmacological inhibition by a small-molecule drug candidate. A polymer-based tethering strategy was utilized for peptide immobilization to the probe tip, and various measurement parameters were optimized for sensing performance, including the peptide coating density and contact time between the probe tip and substrate. With the optimized measurement settings, the binding specificity of AH BAAPP domain to PI(4,5)P2 receptors was evaluated across a panel of phosphoinositides and the influence of ionic strength on the AH–PI(4,5)P2 binding interaction was tested. Following this approach, we investigated the potential of a small-molecule drug candidate, neomycin, to inhibit the AH–PI(4,5)P2 binding interaction, and we were also able to extract the association constant for the monovalent interaction between neomycin and PI(4,5)P2 receptors. Taken together, our findings establish a measurement platform for evaluating molecular recognition events between short bioactive peptides and phosphoinositide receptors, identify the importance of nonelectrostatic factors in driving the high binding specificity of the NS5A AH BAAPP domain for PI(4,5)P2 receptors, and offer a new experimental approach for evaluating the inhibitory activity and quantitative binding characteristics of small-molecule drug candidates.
Materials and Methods
Materials
Phosphate-buffered saline (PBS, pH 7.4; Life Technologies Corp., Carlsbad, CA), ethanol (Merck Millipore, Billerica, MA), dehydrated toluene (Thermo Fisher Scientific, Waltham, MA), 2-aminoethanethiol (Tokyo Chemical Industry, Tokyo, Japan), N-hydroxysuccinimide-[poly(ethylene glycol)]24-maleimide ester (NHS-PEG24-MAL, 95% purity; Quanta Biodesign, Plain City, OH), m-dPEG24-NHS (NHS-PEG24-OMe, 98% purity; Quanta Biodesign), and polyoxyethylene sorbitan monolaurate (Tween-20, Bio-Rad, Hercules, CA) were used in the experiments. All other laboratory-grade chemicals were obtained from Sigma–Aldrich (St. Louis, MO) unless otherwise noted. Phosphoinositide strips for protein–lipid interaction analysis were obtained from Echelon Biosciences (catalog no. P-6001; Salt Lake City, UT) and contain 100 pmol of each compound in the highly purified lipid form (diC16). The strips were stored at 4 °C in a dark environment per the manufacturer’s instructions. Before experiment, the strips were incubated for 1 h in PBS solution containing 0.1% (v/v) Tween-20 in order to passivate nonspecific hydrophobic interactions and then extensively rinsed with PBS solution while remaining immersed in liquid throughout the subsequent AFM experiments. The AH peptide was obtained from Anaspec Corp. (San Jose, CA), where it was synthesized by conventional fluorenylmethyloxycarbonyl (Fmoc) solid-phase peptide synthesis and purified to >95% by reversed-phase high-performance liquid chromatography. The AH peptide’s amino acid sequence is H-Ser-Gly-Ser-Trp-Leu-Arg-Asp-Val-Trp-Asp-Trp-Ile-Cys-Thr-Val-Leu-Thr-Asp-Phe-Lys-Thr-Trp-Leu-Gln-Ser-Lys-Leu-NH2. All solutions were prepared by using Milli-Q-treated water (Merck Millipore, Billerica, MA).
Tip Functionalization
Rectangular-shaped, single-crystal silicon cantilevers (CSG-01, NT-MDT, Moscow, Russia) with ∼0.03 N/m nominal spring constant were used to measure the peptide–phosphoinositide binding interaction. The as-supplied probe tip was coated with a 3-nm-thick adhesion layer of titanium, followed by a 35-nm-thick gold layer. The coated tip’s radius of curvature is approximately 35 nm. The gold-coated tips were extensively rinsed with ethanol, dried with a stream of nitrogen gas, and then subjected to oxygen plasma treatment (PSD-UV Benchtop UV–ozone cleaner, Novascan, Ames, IA). The oxygen plasma-treated tips were then incubated in 10 mM 2-aminoethanethiol in ethanol for 6 h. After incubation, the tips were washed with pure ethanol solution, dried with a stream of nitrogen gas, and next immersed in anhydrous toluene solution containing 1 mM NHS-PEG24-MAL for 6 h, as previously described.46,47 Afterward, the immersed tips were rinsed with sequential anhydrous toluene and ethanol washing steps and dried with nitrogen gas. Finally, the tips were incubated overnight in a PBS buffer solution containing 52 μM AH peptide, and immediately before each experiment, the functionalized tips were gently rinsed with PBS solution.
Force Spectroscopic Measurements
Measurements were
performed on a commercial AFM instrument (NX-Bio, Park Systems, Suwon,
South Korea), as previously described.48 For force mapping data set acquisition, the resolution was set at
16 × 16 points with a scan area of 10 × 10 μm2, generating 256 force curves per data set. The approach and
retract speeds of the AFM cantilever were set at 1000 nm/s with a
maximum loading force of 3 nN. To quantify the force–distance
curves, the experimentally measured spring constant of each individual
AFM cantilever was determined by the thermal vibration method,49 and the values were typically around 0.043 N/m.
All force spectroscopic experiments were performed at room temperature
in the appropriate aqueous buffer solution (PBS unless otherwise noted).
For force curve analysis, data organization was conducted by use of
the XEI v1.8.2 software program (Park Systems), and calculation of
the adhesion force and rupture length was done with a custom-written
script in Python (available upon request). Force–distance curves
with a maximum adhesion force between 0 and 1200 pN and rupture length
between 1 and 80 nm were selected for data analysis. The most probable
adhesion forces and rupture lengths were determined by fitting a Gaussian
curve to histogram plots of all rupture events within these criteria,
and the statistical error was estimated as 2.35σ/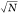 , where σ is the standard
deviation
and N is the number of rupture events in the set.50 The interaction probability was also detected
by classifying specific rupture events as possessing rupture lengths
between 6 and 20 nm, whereas events with shorter or longer rupture
lengths were classified as nonspecific interactions.51
, where σ is the standard
deviation
and N is the number of rupture events in the set.50 The interaction probability was also detected
by classifying specific rupture events as possessing rupture lengths
between 6 and 20 nm, whereas events with shorter or longer rupture
lengths were classified as nonspecific interactions.51
Results and Discussion
Platform Design
A covalent tethering strategy based on a flexible, heterobifunctional cross-linker was utilized to attach AH peptide to the probe tip,46−48 as presented in Figure 1. First, a gold-coated probe tip was functionalized with 2-aminoethanethiol via gold–sulfur dative bonding,52 which resulted in the presentation of free amine groups on the tip surface. Subsequently, the amine-functionalized tip was coated with NHS-PEG24-MAL, where NHS, PEG, and MAL refer to N-hydroxysuccinimide, poly(ethylene glycol) polymer, and maleimide ester, respectively, and the NHS functional group covalently reacts with amine groups on the tip surface. The functionalized tip was then incubated with AH peptide,53 leading to covalent conjugation of the peptide by formation of stable thioether linkages between free maleimide groups on the PEG chains and the cysteine residue of AH peptide. In particular, the cysteine residue has a free sulfhydryl group that forms an irreversible covalent bond with a maleimide group under the experimental conditions.54
Figure 1.

Tethering strategy to functionalize gold-coated AFM probe tip with bioactive AH peptide. (i) A gold-coated AFM tip is used for surface functionalization. (ii) Coating with 2-aminoethanethiol results in presentation of free amine groups on the tip surface. (iii) Coating with NHS-PEG24-MAL is achieved by covalent reaction between N-hydroxysuccinimide and surface-exposed amine groups, with free maleimide esters remaining exposed. (iv) The cysteine residue of AH peptide is conjugated to the PEG-based tether by reaction with a maleimide ester group to form a stable thioether linkage. Note that schematic components are not drawn to scale.
The sensing performance of the peptide-coated probe tips was then evaluated by conducting AFM force spectroscopic measurements on phosphoinositide-coated hydrophobic surfaces containing PI(4,5)P2 molecules (Figure 2a,b). A representative force–distance curve of the AH–PI(4,5)P2 interaction is presented in Figure 2c and shows a rupture length of around 13 nm, which is consistent with the PEG24 spacer’s length of ∼9.4 nm along with the spatial proximity of the cysteine residue relative to the BAAPP domain (∼2 nm) and some degree of unfolding of the peptide’s α-helical character during the retraction step (∼2 nm).46,55 The force required to break the AH–PI(4,5)P2 interaction is around 180 pN, which is within the range of other protein–carbohydrate interactions.36−38 By contrast, when the tip was not functionalized with AH peptide, there was no specific interaction between the tip and PI(4,5)P2 molecules, demonstrating that the detected interaction is mediated by the AH ligand (Figure 2d). The corresponding histograms of individual rupture events, denoted by the associated adhesion force and rupture length, are presented for the two cases, with and without AH pepetide, in Figure 2 panels e and f, respectively. While most individual rupture events exhibited characteristic adhesion force and rupture length pairs, larger forces were observed in some cases and coincident with larger rupture lengths, indicating multiple rupture events and/or nonspecific interactions. The analyzed histogram data were collected from over 100 force–distance curves per experimental set.
Figure 2.

Selective detection of specific AH–PI(4,5)P2 binding events. (a, b) Schematic illustration of AFM cantilever deflection, (a) when an AH peptide-functionalized tip specifically binds to PI(4,5)P2 receptors and (b) when an equivalent tip without AH peptide coating has only weak nonspecific interactions with PI(4,5)P2 receptors on the substrate. (c, d) Representative AFM force–distance curves are presented for the two cases in panels a and b, respectively. (e, f) Scatter plots show all individual binding events, as a function of adhesion force and rupture length, for the two cases presented in panels c and d, respectively. Corresponding histograms are presented from which the most probable values and their corresponding errors were computed.
Optimization of Peptide Coating Density and Contact Time
To clarify how the avidity of AH–PI(4,5)P2 interactions influences the measurement response, we systematically investigated the effect of peptide coating density on the measured adhesion force (Figure 3). This was achieved by coating the tip with a mixture of NHS-PEG24-MAL and a monofunctional PEG cross-linker (NHS-PEG24-OMe) that possesses an N-hydroxysuccinimide functional group and a methoxy (OMe) terminus on its other end. As a result, NHS-PEG24-OMe can attach to the probe tip but there are no remaining functional groups for peptide attachment. In the case of 0 mol % NHS-PEG24-MAL, there was no attached peptide and the resulting most probable adhesion force was 43 pN, which is attributed to weak, nonspecific adhesion events in this system (Figure 3a). On the other hand, at 20 mol % NHS-PEG24-MAL, the most probable adhesion force increased to 86 pN, which is indicative of some specific AH–PI(4,5)P2 interactions beyond force values associated with nonspecific binding events alone. At higher NHS-PEG24-MAL fractions (50% and 100%), the most probable adhesion force reached saturation values around 175–185 pN, indicating a greater fraction of specific AH–PI(4,5)P2 interactions. Importantly, the trend in measured adhesion forces correlated with the probability of specific binding events, which was highest for the 100 mol % NHS-PEG24-MAL case at around 79% (Figure 3b). Collectively, the findings support that the most probable adhesion force and event probability of the AH–PI(4,5)P2 interaction are influenced by the peptide coating density, and for all subsequent studies reported below, the NHS-PEG24-MAL coating density was fixed at 100%.
Figure 3.

Effect of peptide coating density on AH–PI(4,5)P2 binding interaction. (a) Most probable adhesion force and (b) event probability (percentage of specific binding events) for the AH–PI(4,5)P2 binding interaction are expressed as a function of the molar percentage of NHS-PEG24-MAL used for tip coating. The remaining percentage is composed of NHS-PEG24-OMe that does not bind to AH peptides.
Following these observations, we next optimized the time duration (denoted as contact time) during which the probe tip remains in contact with the PI(4,5)P2-coated surface. While the time scale of structural changes within peptides is typically on the order of tens to hundreds of nanoseconds,56 the tethering of peptides in force spectroscopic measurements reduces the degree of spatial freedom, so a sufficiently long contact time is necessary for promoting specific adhesion events. Figure 4 presents representative deflection curves, most probable adhesion forces, and most probable rupture lengths for force spectroscopic measurements conducted at different contact times: 0.1, 1, and 3 s. The deflection curves are shown in Figure 4a–c. At 0.1 s contact time, the most probable adhesion force was 54.0 ± 14.0 pN, which is comparable to the nonspecific binding case and supports that there is insufficient contact time to promote specific binding interactions (Figure 4d). With a 1 s contact time, the most probable adhesion force increased to 179.1 ± 13.1 pN, which is well above the background signal (Figure 4e). In both the 0.1 and 1 s contact time cases, the typical rupture lengths were similar, around 11.4 ± 1.6 and 14.8 ± 1.7 nm, respectively, and consistent with expected rupture lengths in the 10–15 nm range as described. On the other hand, with a 3 s contact time, the most probable adhesion force increased to 248.7 ± 20.3 pN; however, the typical rupture length was significantly larger, around 35.1 ± 2.9 nm, and a large number of multiple rupture events were detected, unlike the predominantly single rupture events observed for shorter contact times (Figure 4f). Therefore, to focus on the specific AH–PI(4,5)P2 interaction, we selected a 1 s contact time for subsequent experiments due to the combination of a suitably large, most probable adhesion force and typical rupture lengths that are consistent with detecting single rupture events in this system. In addition, the dependence of measured adhesion force on contact time supports that the AH–PI(4,5)P2 interaction is relatively slow and likely involves significant changes in the peptide’s molecular structure. These observations are in line with previous biochemical results indicating that the AH peptide undergoes a conformational change upon PI(4,5)P2 binding.24
Figure 4.

Influence of tip–substrate contact time on AH–PI(4,5)P2 binding interaction. Different contact times were tested: 0.1, 1, and 3 s. (a–c) Representative measurement traces show the cantilever deflection as a function of time, including the time span during which tip–substrate contact occurs. (d–f) Adhesion force histograms are presented for the AH–PI(4,5)P2 binding interaction, and the most probable adhesion forces and corresponding errors are presented as well.
Evaluation of Phosphoinositide Binding Specificity
With the optimized measurement settings, we next investigated the specificity of AH binding interaction to PI(4,5)P2 receptors as compared to other phosphoinositide receptors with different degrees of phosphorylation at various ring positions. The tested receptors were PI, PI(3)P, PI(4)P, PI(5)P, PI(3,4)P2, PI(3,5)P2, PI(4,5)P2, and PI(3,4,5)P3. As presented in Figure 5a, the AH peptide demonstrated particularly strong binding to PI(4,5)P2, with a most probable adhesion force around 205 pN in this set. By contrast, there were appreciably weaker interactions between the AH and other phosphoinositide bisphosphates [PI(3,4)P2 and PI(3,5)P2] despite their having similar surface potentials to the negatively charged PI(4,5)P2. The only difference is the geometrical positions of the two phosphate groups on the inositol ring. The most probable adhesion forces for AH binding to PI(3,4)P2 and PI(3,5)P2 receptors were around 97 and 47 pN, respectively. In addition, the most probable adhesion force of AH binding to PI(3,4,5)P3 was 58 pN, whereas binding to other phosphoinositides, PI, PI(3)P, PI(4)P, and PI(5)P, was fully negligible (<40 pN). Altogether, these findings demonstrate that the AH has a specific binding interaction with PI(4,5)P2 receptors that can be selectively detected by force spectroscopic measurements.
Figure 5.

Specificity of AH peptide binding to PI(4,5)P2 receptors. (a) Most probable adhesion forces for AH peptide binding to different phosphoinositide receptors. (b) Effect of ionic strength on most probable adhesion force of AH–PI(4,5)P2 binding interaction. In this experimental series, the buffer solution was 10 mM Tris (pH 7.5), and ionic strength was adjusted by varying the NaCl concentration.
To better understand the role of electrostatic forces in phosphoinositide binding, the effect of ionic strength on the AH–PI(4,5)P2 binding interaction was systematically investigated by varying the NaCl concentration between 0 and 250 mM NaCl (Figure 5b). With increasing ionic strength, the most probable adhesion force decreased from 331.0 ± 16.6 pN at 0 mM NaCl to 143.6 ± 10.5 pN at 250 mM NaCl. These findings are consistent with charge-shielding arguments whereby the surface potentials of positively charged lysine residues in the BAAPP domain and negatively charged phosphate groups in the phosphoinositide molecules become attenuated at higher ionic strengths. As a result, the magnitude of the attractive electrostatic force is expected to become weaker with increasing ionic strength (shorter Debye length).57 While a significant decrease in the adhesion force is observed due to charge-shielding effects, it should be emphasized that the resulting magnitude is still significantly greater than those corresponding to nonspecific interactions of AH with other phosphoinositide receptors when tested under equivalent solution conditions. Hence, the findings reinforce that the AH–PI(4,5)P2 binding interaction is strongly influenced by nonelectrostatic forces, and the relative contribution of electrostatic forces to the total interaction force depends on the ionic strength. As mentioned earlier, these findings are consistent with the fact that the AH undergoes a conformational change upon PI(4,5)P2 binding, and initial electrostatic attraction between positively charged amino acid residues in the BAAPP domain and negatively charged phosphate groups of the PI(4,5)P2 molecule is the first step in this process. As with other protein–phosphoinositide interactions, it should be remarked that optimal hydrogen-bonding contacts between amino acid residues and the two phosphate groups on the phosphoinositide molecule likely contribute to the high binding specificity.58
Evaluation of Small-Molecule Drug Inhibitor
Our foregoing observations establish that the AFM measurement platform is capable of detecting and quantifying PI(4,5)P2 binding by the NS5A AH ligand in a highly specific manner. We next asked if the AH–PI(4,5)P2 binding interaction could serve as a competitive probe to measure the association constant of phosphoinositide-binding small molecules that might be promising drug candidates. Indeed, as the force spectroscopic measurement is probing interactions between AH ligands and PI(4,5)P2 receptor pairs, it would be expected that occupancy of a fraction of receptor sites by a phosphoinositide-binding small molecule might influence the measurement response (Figure 6a). Such capabilities would be useful for quantitative biochemical analysis of phosphoinositide–small-molecule interactions as well as for evaluating potential antiviral drug candidates that interfere with AH–PI(4,5)P2 binding.
Figure 6.

Evaluation of a small-molecule drug candidate that inhibits AH–PI(4,5)P2 binding. (a) Schematic illustration of neomycin inhibitor binding to PI(4,5)P2 molecules, thereby decreasing the number of available receptor sites. (b) Representative adhesion force maps obtained for AH–PI(4,5)P2 binding interaction in the presence of selected neomycin concentrations as indicated. Maps were obtained in 2 μm × 2 μm with a lateral resolution of 90 nm for each pixel spot. (c) Effect of neomycin concentration on the most probable adhesion force of AH–PI(4,5)P2 binding. Dashed line represents the most probable adhesion force for control experiment in the absence of neomycin. (d) Event probability of specific AH–PI(4,5)P2 binding interaction as a function of bulk neomycin concentration. (e) Percentage of AH–PI(4,5)P2 binding inhibition as a function of bulk neomycin concentration. The EC50 value was computed from a 4PL fit.
To explore this possibility, we investigated how neomycin, an aminoglycoside antibiotic that is known to bind PI(4,5)P2 receptors,59 affects AH–PI(4,5)P2 binding. For these experiments, various concentrations of free neomycin (100 nM–1 mM) were incubated with the phosphoinositide array for 1 h to allow equilibration of free and bound neomycin prior to each AFM experiment, and then adhesion force measurements were conducted under equilibrated solution conditions. Representative adhesion force maps (2 μm × 2 μm) obtained in the presence of different neomycin concentrations show the force magnitudes obtained at different pixel positions, indicating a transition from high adhesion forces corresponding to strong specific binding at low neomycin concentrations to weak nonspecific interactions at higher neomycin concentrations60 (Figure 6b). A low concentration of neomycin (100 nM) did not affect the binding interaction, and the recorded measurement values in this case were comparable to conventional AH–PI(4,5)P2 binding (adhesion force of ∼225 pN). On the other hand, higher concentrations of neomycin (1–10 μM) exhibited dose-dependent inhibition of AH–PI(4,5)P2 binding. With increasing neomycin concentration across this range, the most probable adhesion force decreased from 194.2 ± 11.1 to 64.5 ± 14.3 pN (Figure 6c). Above this concentration range (≥100 μM neomycin), the measured adhesion forces for AH–PI(4,5)P2 binding reached a stable minimum value that was comparable to the magnitude of nonspecific adhesion events (∼48 pN). Hence, competitive binding of neomycin to PI(4,5)P2 receptors inhibited specific AH–PI(4,5)P2 binding due to neomycin molecules occupying PI(4,5)P2 receptor sites. The sharp transition in adhesion force data as a function of neomycin concentration suggests that the measurement approach may be probing single-molecule binding events.
Based on the adhesion force data, the probability of specific binding events was determined; it ranged from 84% at 0.1 μM neomycin to 20% at 1000 μM neomycin (Figure 6d). From these data, an efficacy curve of neomycin’s inhibitory activity was constructed as a function of drug concentration (Figure 6e). The 0% and 100% efficacy levels were defined as the event probability corresponding to typical AH–PI(4,5)P2 binding (∼79%) and the event probability of no specific interaction (NHS-PEG24-MAL without AH peptide) (∼15%), respectively. Following this approach, the 50% efficacy concentration (EC50) of neomycin was determined to be 2.05 μM. As neomycin binds to PI(4,5)P2 in a 1:1 stoichiometric ratio,61 the EC50 value corresponds to the bulk concentration of neomycin at which 50% of the PI(4,5)P2 receptor sites are occupied. From this result, we can calculate the apparent association constant, Ka, which equals the reciprocal of the neomycin concentration that binds 50% of PI(4,5)P2 receptor sites. The calculated Ka value of 4.88 × 105 M–1 agrees well with literature values obtained by conventional biochemical methods (1–7 × 105 M–1).62,63 Of note, Živković et al.64 previously attempted to use AFM force spectroscopy to measure the effect of 100 mM neomycin inhibitor on peptide–RNA binding interactions, but they still detected some specific peptide–RNA binding events despite the neomycin concentrations being well in excess (by a factor of 5 × 104) of the corresponding dissociation constant. From this viewpoint, our findings reveal for the first time that, by utilizing the AH–PI(4,5)P2 binding interaction as a competitive probe, AFM force spectroscopic measurements can distinguish the extent of neomycin’s inhibitory activity at drug concentrations 5 orders of magnitude lower than previously observed in other AFM force spectroscopic systems and provide quantitative readouts of equilibrium binding constants for phosphoinositide–small-molecule interactions.
Conclusion
In summary, we have established an AFM force spectroscopic platform to quantitatively measure molecular-level interactions between amphipathic peptides and phosphoinositide molecules. The measurement platform was able to detect highly specific binding events while avoiding false positives from weaker nonspecific interactions, even when phosphoinositide molecules in the two cases have nearly identical physicochemical properties. In the present context of exploring the NS5A AH BAAPP domain, these findings provided a nanomechanical basis for explaining the high binding specificity of NS5A AH to PI(4,5)P2 receptors, including the importance of nonelectrostatic factors. Importantly, these capabilities could also be translated into the development of a novel experimental methodology to determine the inhibitory activity of small-molecule drug candidates acting against the AH–PI(4,5)P2 interaction. Proof-of-concept experiments with the neomycin inhibitor demonstrated that this small molecule acts via competitive binding to PI(4,5)P2 receptors, and the measured association constant for the neomycin–PI(4,5)P2 interaction agreed well with literature values obtained by conventional biochemical methods. In light of these demonstrated capabilities for force spectroscopic profiling, there is enormous potential for utilizing this measurement approach to explore protein–phosphoinositide interactions in membranous environments, to understand how amino acid residues within the BAAPP domain influence PI(4,5)P2 binding specificity and related biological activities, and to further characterize drug candidates that act against either PI(4,5)P2 receptors or AH ligands. As AFM force spectroscopic experiments require significant time for performing the measurements and completing data analysis, they offer a complementary tool to traditional, ensemble-average assay formats and provide a highly focused approach with advanced capabilities to quantitatively characterize individual biomacromolecular interactions with statistically rich information. Looking forward, this work contributes an analytical framework to study phosphoinositide-binding peptides and proteins as well as a broadly applicable approach to evaluate candidate pharmacological inhibitors.
Acknowledgments
This work was supported by the National Research Foundation of Singapore through a Competitive Research Programme grant (NRF-CRP10-2012-07) and a Proof-of-Concept grant (NRF2015NRF-POC0001-19), Nanyang Technological University through a start-up grant (M4080751.070), and the National Institutes of Health (RO1 AI099245 and U19AI109662). J.A.J. is the recipient of a postdoctoral research fellowship award from the American Liver Foundation.
Author Contributions
▽ S.-O.K. and J.A.J, contributed equally to this work.
The authors declare no competing financial interest.
References
- McLaughlin S.; Murray D. Nature 2005, 438, 605–611. 10.1038/nature04398. [DOI] [PubMed] [Google Scholar]
- Di Paolo G.; De Camilli P. Nature 2006, 443, 651–657. 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- Balla T. Physiol. Rev. 2013, 93, 1019–1137. 10.1152/physrev.00028.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutateladze T. G. Nat. Chem. Biol. 2010, 6, 507–513. 10.1038/nchembio.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond G. R.; Fischer M. J.; Anderson K. E.; Holdich J.; Koteci A.; Balla T.; Irvine R. F. Science 2012, 337, 727–730. 10.1126/science.1222483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyvönen M.; Macias M. J.; Nilges M.; Oschkinat H.; Saraste M.; Wilmanns M. EMBO J. 1995, 14, 4676–4685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H.; Hakala M.; Lappalainen P. Biophys. J. 2010, 98, 2327–2336. 10.1016/j.bpj.2010.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffucci T.; Falasca M. FEBS Lett. 2001, 506, 173–179. 10.1016/S0014-5793(01)02909-X. [DOI] [PubMed] [Google Scholar]
- Yang L.; Nasu Y.; Hattori M.; Yoshimura H.; Kanno A.; Ozawa T. Anal. Chem. 2013, 85, 11352–11359. 10.1021/ac402278f. [DOI] [PubMed] [Google Scholar]
- Tabaei S. R.; Guo F.; Rutaganira F. U.; Vafaei S.; Choong I.; Shokat K. M.; Glenn J. S.; Cho N.-J. Anal. Chem. 2016, 88, 5042. 10.1021/acs.analchem.6b01293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai T.; Shu Q.; Hou J.; Liu P.; Niu L.; Guo X.; Liu C. C.; Yang F. Anal. Chem. 2015, 87, 513–521. 10.1021/ac503224j. [DOI] [PubMed] [Google Scholar]
- Idevall-Hagren O.; De Camilli P. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2015, 1851, 736–745. 10.1016/j.bbalip.2014.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Wilkop T.; Han J. H.; Dong Y.; Linman M. J.; Cheng Q. Anal. Chem. 2008, 80, 6397–6404. 10.1021/ac800845w. [DOI] [PubMed] [Google Scholar]
- Taylor J. D.; Linman M. J.; Wilkop T.; Cheng Q. Anal. Chem. 2009, 81, 1146–1153. 10.1021/ac8023137. [DOI] [PubMed] [Google Scholar]
- Baumann M. K.; Swann M. J.; Textor M.; Reimhult E. Anal. Chem. 2011, 83, 6267–6274. 10.1021/ac2009178. [DOI] [PubMed] [Google Scholar]
- Bunney T. D.; Katan M. Nat. Rev. Cancer 2010, 10, 342–352. 10.1038/nrc2842. [DOI] [PubMed] [Google Scholar]
- Hakim S.; Bertucci M. C.; Conduit S. E.; Vuong D. L.; Mitchell C. A. In Phosphoinositides and Disease; Current Topics in Microbiology and Immunology, Vol. 362; Springer: Dordrecht, The Netherlands, 2012; pp 247–314; DOI: 10.1007/978-94-007-5025-8_12. [DOI] [PubMed] [Google Scholar]
- Angulo I.; Vadas O.; Garçon F.; Banham-Hall E.; Plagnol V.; Leahy T. R.; Baxendale H.; Coulter T.; Curtis J.; Wu C.; et al. Science 2013, 342, 866–871. 10.1126/science.1243292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strating J. R.; van Kuppeveld F. J. Curr. Opin. Cell Biol. 2017, 47, 24–33. 10.1016/j.ceb.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A.; Ablan S. D.; Lockett S. J.; Nagashima K.; Freed E. O. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 14889–14894. 10.1073/pnas.0405596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul D.; Madan V.; Bartenschlager R. Cell Host Microbe 2014, 16, 569–579. 10.1016/j.chom.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elazar M.; Cheong K. H.; Liu P.; Greenberg H. B.; Rice C. M.; Glenn J. S. J. Virol. 2003, 77, 6055–6061. 10.1128/JVI.77.10.6055-6061.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho N.-J.; Cheong K. H.; Lee C.; Frank C. W.; Glenn J. S. J. Virol. 2007, 81, 6682–6689. 10.1128/JVI.02783-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho N.-J.; Lee C.; Pang P. S.; Pham E. A.; Fram B.; Nguyen K.; Xiong A.; Sklan E. H.; Elazar M.; Koytak E. S.; et al. Gastroenterology 2015, 148, 616–625. 10.1053/j.gastro.2014.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olety B.; Veatch S. L.; Ono A. J. Virol. 2015, 89, 7861–7873. 10.1128/JVI.00794-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan K.; Lemmon M. A. Methods 2006, 39, 122–133. 10.1016/j.ymeth.2006.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busse R. A.; Scacioc A.; Hernandez J. M.; Krick R.; Stephan M.; Janshoff A.; Thumm M.; Kühnel K. Autophagy 2013, 9, 770–777. 10.4161/auto.23978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccato L.; Chicanne G.; Nahoum V.; Pons V.; Payrastre B.; Gaits-Iacovoni F.; Viaud J. Sci. Signaling 2016, 9, rs2–rs2. 10.1126/scisignal.aad4337. [DOI] [PubMed] [Google Scholar]
- Rosenhouse-Dantsker A.; Logothetis D. E. Pfluegers Arch. 2007, 455, 45–53. 10.1007/s00424-007-0291-6. [DOI] [PubMed] [Google Scholar]
- Rebecchi M.; Peterson A.; McLaughlin S. Biochemistry 1992, 31, 12742–12747. 10.1021/bi00166a005. [DOI] [PubMed] [Google Scholar]
- Lemmon M. A.; Ferguson K. M.; O’Brien R.; Sigler P. B.; Schlessinger J. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 10472–10476. 10.1073/pnas.92.23.10472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson C. G.; James R. D.; Bigman C. S.; Shepard D. A.; Abdiche Y.; Katsamba P. S.; Myszka D. G.; Prestwich G. D. Bioconjugate Chem. 2005, 16, 1475–1483. 10.1021/bc050197q. [DOI] [PubMed] [Google Scholar]
- Cappella B.; Dietler G. Surf. Sci. Rep. 1999, 34, 1–104. 10.1016/S0167-5729(99)00003-5. [DOI] [Google Scholar]
- Butt H.-J.; Cappella B.; Kappl M. Surf. Sci. Rep. 2005, 59, 1–152. 10.1016/j.surfrep.2005.08.003. [DOI] [Google Scholar]
- Marszalek P. E.; Dufrêne Y. F. Chem. Soc. Rev. 2012, 41, 3523–3534. 10.1039/c2cs15329g. [DOI] [PubMed] [Google Scholar]
- Touhami A.; Hoffmann B.; Vasella A.; Denis F. A.; Dufrêne Y. F. Langmuir 2003, 19, 1745–1751. 10.1021/la026145w. [DOI] [Google Scholar]
- Lekka M.; Laidler P.; Dulińska J.; Łabędź M.; Pyka G. Eur. Biophys. J. 2004, 33, 644–650. 10.1007/s00249-004-0412-5. [DOI] [PubMed] [Google Scholar]
- Hanley W.; McCarty O.; Jadhav S.; Tseng Y.; Wirtz D.; Konstantopoulos K. J. Biol. Chem. 2003, 278, 10556–10561. 10.1074/jbc.M213233200. [DOI] [PubMed] [Google Scholar]
- Yu K.; Creagh A. L.; Haynes C. A.; Kizhakkedathu J. N. Anal. Chem. 2013, 85, 7786–7793. 10.1021/ac401306b. [DOI] [PubMed] [Google Scholar]
- Malkovskiy A.; Wagh D.; Longo F.; Rajadas J. Analyst 2015, 140, 4558–4565. 10.1039/C5AN00498E. [DOI] [PubMed] [Google Scholar]
- Sewald N.; Wilking S. D.; Eckel R.; Albu S.; Wollschläger K.; Gaus K.; Becker A.; Bartels F. W.; Ros R.; Anselmetti D. J. Pept. Sci. 2006, 12, 836–842. 10.1002/psc.820. [DOI] [PubMed] [Google Scholar]
- Chung J. W.; Shin D.; Kwak J. M.; Seog J. J. Mol. Recognit. 2013, 26, 268–275. 10.1002/jmr.2269. [DOI] [PubMed] [Google Scholar]
- Huang L.; Sineva E. V.; Hargittai M. R.; Sharma S. D.; Suthar M.; Raney K. D.; Cameron C. E. Protein Expression Purif. 2004, 37, 144–153. 10.1016/j.pep.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Love R. A.; Brodsky O.; Hickey M. J.; Wells P. A.; Cronin C. N. J. Virol. 2009, 83, 4395–4403. 10.1128/JVI.02352-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellinghuisen T. L.; Marcotrigiano J.; Rice C. M. Nature 2005, 435, 374–379. 10.1038/nature03580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai Y.; Okabe K.-I.; Sekiguchi H.; Hayashi T.; Hara M. Langmuir 2011, 27, 2478–2483. 10.1021/la104178h. [DOI] [PubMed] [Google Scholar]
- Mochizuki M.; Oguchi M.; Kim S.-O.; Jackman J. A.; Ogawa T.; Lkhamsuren G.; Cho N.-J.; Hayashi T. Langmuir 2015, 31, 8006–8012. 10.1021/acs.langmuir.5b01691. [DOI] [PubMed] [Google Scholar]
- Kim S.-O.; Jackman J. A.; Mochizuki M.; Yoon B. K.; Hayashi T.; Cho N.-J. Phys. Chem. Chem. Phys. 2016, 18, 14454–14459. 10.1039/C6CP01168C. [DOI] [PubMed] [Google Scholar]
- Matei G.; Thoreson E.; Pratt J.; Newell D.; Burnham N. Rev. Sci. Instrum. 2006, 77, 083703. 10.1063/1.2336115. [DOI] [Google Scholar]
- Schwesinger F.; Ros R.; Strunz T.; Anselmetti D.; Güntherodt H.-J.; Honegger A.; Jermutus L.; Tiefenauer L.; Plückthun A. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 9972–9977. 10.1073/pnas.97.18.9972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams N. B.; Vasilev C.; Brindley A. A.; Hunter C. N. J. Am. Chem. Soc. 2016, 138, 6591–6597. 10.1021/jacs.6b02827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love J. C.; Estroff L. A.; Kriebel J. K.; Nuzzo R. G.; Whitesides G. M. Chem. Rev. 2005, 105, 1103–1170. 10.1021/cr0300789. [DOI] [PubMed] [Google Scholar]
- Cho N.-J.; Cho S.-J.; Cheong K. H.; Glenn J. S.; Frank C. W. J. Am. Chem. Soc. 2007, 129, 10050–10051. 10.1021/ja0701412. [DOI] [PubMed] [Google Scholar]
- Mattson G.; Conklin E.; Desai S.; Nielander G.; Savage M.; Morgensen S. Mol. Biol. Rep. 1993, 17, 167–183. 10.1007/BF00986726. [DOI] [PubMed] [Google Scholar]
- Lantz M. A.; Jarvis S. P.; Tokumoto H.; Martynski T.; Kusumi T.; Nakamura C.; Miyake J. Chem. Phys. Lett. 1999, 315, 61–68. 10.1016/S0009-2614(99)01201-4. [DOI] [Google Scholar]
- Heinz H.; Farmer B. L.; Pandey R. B.; Slocik J. M.; Patnaik S. S.; Pachter R.; Naik R. R. J. Am. Chem. Soc. 2009, 131, 9704–9714. 10.1021/ja900531f. [DOI] [PubMed] [Google Scholar]
- Denisov G.; Wanaski S.; Luan P.; Glaser M.; McLaughlin S. Biophys. J. 1998, 74, 731–744. 10.1016/S0006-3495(98)73998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson K. M.; Lemmon M. A.; Schlessinger J.; Sigler P. B. Cell 1995, 83, 1037–1046. 10.1016/0092-8674(95)90219-8. [DOI] [PubMed] [Google Scholar]
- Schacht J. J. Neurochem. 1976, 27, 1119–1124. 10.1111/j.1471-4159.1976.tb00318.x. [DOI] [PubMed] [Google Scholar]
- Kim I. H.; Lee M. N.; Ryu S. H.; Park J. W. Anal. Chem. 2011, 83, 1500–1503. 10.1021/ac102695e. [DOI] [PubMed] [Google Scholar]
- Reid D.; Gajjar K. J. Biol. Chem. 1987, 262, 7967–7972. (http://www.jbc.org/content/262/17/7967.full.pdf). [PubMed] [Google Scholar]
- Gabev E.; Kasianowicz J.; Abbott T.; McLaughlin S. Biochim. Biophys. Acta, Biomembr. 1989, 979, 105–112. 10.1016/0005-2736(89)90529-4. [DOI] [PubMed] [Google Scholar]
- Arbuzova A.; Martushova K.; Hangyás-Mihályné G.; Morris A. J.; Ozaki S.; Prestwich G. D.; McLaughlin S. Biochim. Biophys. Acta, Biomembr. 2000, 1464, 35–48. 10.1016/S0005-2736(99)00243-6. [DOI] [PubMed] [Google Scholar]
- Živković J.; Janssen L.; Alvarado F.; Speller S.; Heus H. A. Soft Matter 2012, 8, 2103–2109. 10.1039/C1SM06563G. [DOI] [Google Scholar]