

Abstract
The 2015 Paediatric European Network for Treatment of AIDS (PENTA) guidelines provide practical recommendations on the management of HIV‐1 infection in children in Europe and are an update to those published in 2009. Aims of treatment have progressed significantly over the last decade, moving far beyond limitation of short‐term morbidity and mortality to optimizing health status for adult life and minimizing the impact of chronic HIV infection on immune system development and health in general. Additionally, there is a greater need for increased awareness and minimization of long‐term drug toxicity. The main updates to the previous guidelines include: an increase in the number of indications for antiretroviral therapy (ART) at all ages (higher CD4 thresholds for consideration of ART initiation and additional clinical indications), revised guidance on first‐ and second‐line ART recommendations, including more recently available drug classes, expanded guidance on management of coinfections (including tuberculosis, hepatitis B and hepatitis C) and additional emphasis on the needs of adolescents as they approach transition to adult services. There is a new section on the current ART ‘pipeline’ of drug development, a comprehensive summary table of currently recommended ART with dosing recommendations. Differences between PENTA and current US and World Health Organization guidelines are highlighted and explained.
Keywords: antiretroviral therapy, child, HIV‐1
1. Introduction
These guidelines apply to children with HIV‐1 infection in Europe.
Thresholds for starting antiretroviral therapy (ART) have changed as continuing improvements in treatment mean that the objectives of ART should increasingly be optimizing health status for a full and productive adult life rather than just survival.
New drugs have been incorporated into the guideline as first‐ and second‐line options.
The 2015 Paediatric European Network for Treatment of AIDS (PENTA) guidelines have been updated from those of 2009 1, and make recommendations based on a shift in aims of treatment away from minimization of short‐term morbidity and mortality towards optimizing immune status and general health for a full and productive adult life. This mirrors the general trend in global treatment guidelines that now include higher CD4 thresholds for ART initiation and an increased number of clinical indications in both adults and children 2, 3, 4, 5, 6.
The aim is to provide a practical guide to treatment rather than a comprehensive review of all the evidence on ART in children. More detailed information for resource‐rich and resource‐poor settings is available from recently updated US 4 and World Health Organization (WHO) 6 paediatric guidelines. Special considerations for children in resource‐limited settings where background rates of concomitant infections and malnutrition are much higher are not considered here, and the reader is referred to WHO guidelines. Differences from the WHO and US Department of Health and Human Services (DHHS) guidelines will be referred to where relevant in the document and are summarized in Table 1.
Table 1.
Comparison of current World Health Organization (WHO), US Department of Health and Human Services (DHHS) and Paediatric European Network for Treatment of AIDS (PENTA) guidelines for antiretroviral therapy (ART) initiation
| WHO 2013 | DHHS 2014 | PENTA 2015 | |
|---|---|---|---|
| < 1 year | All | All | All |
| 1–3 years |
All Prioritize: 1–2 years WHO stage 3/4 CD4 count ≤ 750 cells/μL CD4 percentage ≤ 25% |
CD4 count < 1000 cells/μL CD4 percentage < 25% CDC category B/C HIV VL > 100 000 copies/ml Consider: All |
CD4 count ≤ 1000 cells/μL CD4 percentage ≤ 25% WHO stage 3/4 CDC category B/C Consider: All* |
| 3–5 years |
All Prioritize: WHO stage 3/4 CD4 count ≤ 750 cells/μL CD4 percentage ≤ 25% |
CD4 count < 750 cells/μL CD4 percentage < 25% CDC category B/C HIV VL > 100 000 copies/ml Consider: All |
CD4 count ≤ 750 cells/μL CD4 percentage ≤ 25% WHO stage 3/4 CDC category B/C Consider: HIV VL > 100 000 copies/ml |
| > 5 years |
CD4 ≤ 500 cells/μL Prioritize: WHO stage 3/4 CD4 count ≤ 350 cells/μL |
CD4 count < 500 cells/μL CDC category B/C HIV VL > 100 000 copies/ml Consider: All |
CD4 count ≤ 350 cells/μL WHO stage 3/4 CDC category B/C Consider: CD4 count ≤ 500 cells/μL HIV VL > 100 000 copies/ml |
This table summarizes the main immunological, virological and clinical indications and is not exhaustive.
‘All’, all children irrespective of immunological status; CDC, Centers for Disease Control and Prevention; VL, viral load.
*In children aged 1–3 years consider ART in all especially if VL > 100 000 copies/ml.
Licensing of newer drugs in children and the availability of more appropriate formulations have allowed inclusion of a larger number of drug options at different ages. These drugs, with limited data on long‐term toxicity, are included with the caveat that clinicians should be vigilant for signs of adverse effects and fully counsel children and families regarding what is known and unknown about long‐term use of newer drugs.
Much of the new paediatric data (published since 2009) that have been used to inform the 2015 PENTA guidelines have come from non‐European settings and therefore must be considered with caution when applied to European cohorts. As with previous versions of the guidelines, there remains a paucity of data from randomized controlled trials (RCTs) on which to base European paediatric ART guidelines. Therefore, we continue to rely on cohort studies, extrapolation from adult data, and expert opinion. Wherever possible, children should be entered into clinical trials.
The guidelines have been developed using a similar method to that used for the adult European AIDS Clinical Society (EACS) guidelines 3, 7. A panel of experts reviewed the published literature and formulated the main recommendations. The full PENTA Steering Committee then reviewed and refined the recommendations until consensus was reached. This approach has recently been criticized by some. Latest WHO and British HIV Association (BHIVA) guidelines have now moved to using the structured Grading of Recommendations Assessment, Development and Evaluation (GRADE) system 7. It is the opinion of PENTA that the paucity of RCT evidence means that the GRADE system would not provide sufficient additional benefit to the guidelines' development to justify its use and therefore the writing group has elected to continue using the less formalized system described above.
PENTA guidelines seek to optimize treatment for children in Europe. However, particularly during adolescence, care may need to be individualized. This document should not be seen as a standard for litigation as individualization of case management and departure from this guidance may be necessary and indicated.
Significant changes since the 2009 guidelines include:
decreased frequency of laboratory monitoring in clinically stable children both on and off ART;
consideration of ART initiation in all children aged 1–3 years in order to minimize the risks of disease progression or death;
consideration of ART initiation at higher CD4 thresholds in children > 5 years of age in order to optimize potential for immune reconstitution;
additional clinical indications for ART initiation at all ages;
addition of newer protease and integrase inhibitors to first‐line preferred and alternative third agent options, respectively;
update on specific guidance in the context of hepatitis B and C virus and tuberculosis (TB) coinfection in light of new ART options at younger ages;
a summary of new drugs [including new fixed dose combinations (FDCs)] that can be considered for second‐ and third‐line options and of the ‘pipeline’ of new drugs likely to become available;
an emphasis on the needs of older children and adolescents as they approach transition to adult care;
an updated drug dosing table including all currently recommended licensed antiretroviral drugs for children.
2. Summary of recommendations
3. Diagnosis, baseline investigations and pretreatment monitoring
If a woman is diagnosed with HIV infection, all of her children potentially at risk of infection should be tested for HIV irrespective of age.
Children under 18 months of age at risk of perinatally acquired HIV infection should be tested using blood DNA or RNA polymerase chain reaction (PCR) with subsequent confirmatory repeat PCR if positive.
Post‐exposure prophylaxis (PEP) should be given for 4−6 weeks to all babies born to HIV‐infected mothers according to local guidelines.
Babies born to HIV‐infected mothers should have an HIV RNA PCR test at birth and at least two further separate PCR tests (the first 2 weeks after cessation of PEP, and the second at least 6 weeks after cessation of PEP). For babies with high risk of transmission, an additional PCR test midway through PEP is recommended.
Breast feeding is not recommended. In circumstances in which the mother is choosing to breast feed against recommendations, the baby should have regular screening PCR tests. Two negative HIV RNA PCR tests (2 and 6 weeks after cessation of breast feeding) are required to confirm that the baby is not infected.
Children over 18 months of age can be tested using serological assays with subsequent confirmatory PCR if positive or equivocal.
A negative serological test in children who have had a positive HIV RNA PCR test does not exclude ongoing HIV infection.
A detailed history of any possible previous ART given to the child and/or mother (or other likely source of infection) should be documented.
The genotypic HIV resistance profile should be documented at baseline.
The human leucocyte antigen (HLA) B*5701 genotype should be confirmed negative before using abacavir (ABC).
Clinical assessment should be carried out 3−4‐monthly in children who are stable off ART, with frequency of laboratory monitoring dictated by age, clinical status and proximity to thresholds for ART initiation [minimum 6‐monthly HIV viral load (VL) and CD4 count].
Local guidelines for bacillus Calmette−Guérin (BCG) immunization of babies of HIV‐infected mothers should be followed. HIV‐exposed infants at low risk of HIV transmission (maternal VL < 50 HIV‐1 RNA copies/ml at or after 36 weeks of gestation) with high risk of TB exposure may receive BCG vaccination at birth, prior to definitive diagnosis/exclusion of HIV infection.
4. Prophylaxis against opportunistic infections
Prophylaxis against Pneumocystis jirovecii pneumonia (PJP) should be given to all HIV‐infected infants from age 1 month and to older children with low CD4 counts: in children aged 1–4 years, CD4 count < 500 cells/μL or < 15%; in children aged ≥ 5 years, CD4 count < 200 cells/μl or < 15%. Co‐trimoxazole is the drug of first choice (see drug table in Supplementary Table S1 for dosing).
Routine primary prophylaxis against other infections is not recommended.
5. When to start ART
ART is recommended:
in all children under 1 year of age;
in all children with significant disease [WHO stage 3 or 4 or Centers for Disease Control and Prevention (CDC) stage B or C];
in asymptomatic children over 1 year of age based on age‐specific CD4 count thresholds;
to be initiated before the CD4 count reaches the CD4 treatment threshold;
in those with hepatitis C virus (HCV) or active TB coinfection.
ART should be considered:
in asymptomatic children over 5 years of age at CD4 counts of 350–500 cells/μl, to potentially optimize CD4 count in adulthood;
in children with high VL (> 100 000 copies/ml);
in asymptomatic children aged 1–3 years irrespective of immune status and VL;
in sexually active adolescents, to minimize the risk of onward transmission;
in the presence of any significant HIV‐related clinical symptoms;
in hepatitis B virus (HBV) coinfection irrespective of immune status.
6. Which ART regimen to start as first‐line therapy
Children should start effective (at least three drugs) ART, usually a dual or triple nucleoside reverse transcriptase inhibitor (NRTI) backbone together with either a ritonavir‐boosted protease inhibitor (PI) or a nonnucleoside reverse transcriptase inhibitor (NNRTI).
Children exposed to nevirapine (NVP) during failed prevention of mother‐to‐child transmission (PMTCT) (or in whom perinatal NVP exposure cannot be excluded) should be started on a boosted PI‐containing regimen as transmitted resistance may lead to failure of NVP‐containing ART.
Children aged < 3 years not exposed to NVP during failed PMTCT may be initiated on either NVP or ritonavir‐boosted lopinavir (LPV/r)‐containing ART. We recommend that NVP should be given together with three NRTIs [ABC, lamivudine (3TC) and zidovudine (ZDV)] in all infants and in children aged 1–3 years with VL > 100 000 copies/ml or signs of central nervous system (CNS) involvement as an induction‐maintenance strategy, unless any of these drugs are contraindicated (such as ABC in HLA B*5701‐positive children).
In children aged > 3 years, either NNRTI or boosted PI‐based ART is acceptable for initial therapy. Factors such as availability of age‐appropriate formulations, palatability, dosing frequency and adherence should be considered when choosing NNRTIs or boosted PIs.
The preferred NNRTI is NVP in children aged < 3 years not exposed to NVP during failed PMTCT, and efavirenz (EFV) in children aged > 3 years. The preferred PI in children aged < 6 years is LPV/r, in children aged 6−12 years it is ritonavir‐boosted atazanavir (ATV/r), and in children aged > 12 years it is ATV/r or ritonavir‐boosted darunavir (DRV/r).
Integrase inhibitor (INSTI)‐based ART may be an alternative regimen in children over age 12 years.
The preferred first‐line NRTIs are ABC/3TC in children aged < 12 years, and tenofovir/emtricitabine (TDF/FTC) or ABC/3TC (if VL < 100 000 copies/ml) in children aged > 12 years.
Age, HLA B*5701 genotype, previous drug exposure, resistance profile, coinfections, available formulations and likely adherence should be taken into account when choosing a first‐line regimen.
See Table 4 (later) for details of recommended first‐line ART regimens.
Table 4.
Recommended first‐line antiretroviral therapy (not in the context of hepatitis B virus or tuberculosis coinfection)
| < 1 year | 1–3 years | 3–6 years | 6–12 years | > 12 years | ||
|---|---|---|---|---|---|---|
| Preferred | Third agent |
LPV/r NVP |
LPV/r NVP |
LPV/r EFV |
ATV/r EFV |
ATV/r DRV/r EFV |
| Backbone | ABC*/3TC (+ ZDV if NVP) ‡ | ABC*/3TC (+ ZDV if NVP and CNS involvement or high VL) † | ABC*/3TC | ABC*/3TC |
TDF/FTC
§
ABC*/3TC (if VL < 105 copies/mL) |
|
| Alternative | Third agent | – | – |
NVP DRV/r |
NVP LPV/r DRV/r |
NVP LPV/r RAL** DTG |
| Backbone | ZDV ¶ /3TC | ZDV ¶ /3TC |
ZDV
¶
/3TC TDF/3TC (FTC) |
ZDV
¶
/3TC TDF/3TC (FTC) |
ABC*/3TC |
3TC, lamivudine; ABC, abacavir; ATV/r, ritonavir‐boosted atazanavir; CNS, central nervous system; DRV/r, ritonavir‐boosted darunavir; DTG, dolutegravir; EFV, efavirenz; FTC, emtricitabine; LPV, lopinavir; NVP, nevirapine; RAL, raltegravir; TDF, tenofovir; VL, viral load; ZDV, zidovudine.
*Prior to starting abacavir (ABC), patients should be tested for human leucocyte antigen (HLA) B*5701. If positive, then ABC should not be prescribed. †In children < 3 years, consider adding zidovudine (ZDV) to nevirapine (NVP)‐based regimens if there is a very high viral load (VL) or central nervous system (CNS) involvement until VL has been suppressed for at least 3 months. ‡Four‐drug induction for infants on NVP‐based therapy may be considered until VL has been suppressed for at least 3 months, followed by three‐drug maintenance therapy. §Tenofovir (TDF)/emtricitabine (FTC) is preferred in older children with VL > 100 000 copies/ml. Some clinicians would advocate deferring the use of TDF until after puberty. ¶ZDV should be avoided if possible, apart from the indications described in the above notes. **In rare instances (e.g. transmitted resistance or toxicity), raltegravir (RAL) may be used as first‐line therapy in children < 12 years of age.
7. Adherence and HIV knowledge
Adherence to treatment is paramount and should be discussed at each clinic visit.
Every effort should be made to simplify a regimen to support adherence (e.g. using once‐daily regimens, FDCs, and ‘forgiving’ regimens with higher barriers to resistance). Simple adherence aids should be used when appropriate.
Children should know of their HIV diagnosis before adolescence.
Monitoring for psychological, neurocognitive and mental health issues should be routine, allowing early supportive and therapeutic intervention.
8. Monitoring on ART
The aim of ART is to achieve an undetectable VL (< 50 copies/ml) and CD4 reconstitution.
Laboratory monitoring for drug toxicity should be performed initially within 2–4 weeks of starting a new drug, then at least every 6 months if there are no ongoing toxicity concerns.
After starting ART, VL should be checked early (at around 1 month) to confirm that VL is decreasing (this can coincide with toxicity monitoring).
VL and CD4 count can then be monitored approximately every 3–4 months once the patient has been established on treatment.
Once CD4 cells are reconstituted and VL has been < 50 copies/ml consistently for over 1 year, CD4 parameters can be monitored less frequently (every 6–8 months, i.e. at alternate clinic visits).
-
More frequent clinical and laboratory monitoring is required:
in infancy;
if adherence is poor;
soon after starting or changing therapy (e.g. liver function tests should be performed within 2 weeks);
in the context of ongoing drug toxicity;
when giving medications with significant drug interactions with ART such as antituberculous therapy.
9. Drug toxicities and interactions
Toxicities depend on the individual drugs and ART combination and should be assessed at each clinic visit.
Drug interactions should be considered when starting new medications in a child on ART. Use http://www.hiv‐druginteractions.org/ to check drug interactions and toxicities.
See Table 5 (later) for common ART‐associated toxicities.
Table 5.
Common side effects of antiretroviral therapy in children*
| Toxicities | Antiretrovirals |
|---|---|
| Neuropsychiatric symptoms/insomnia/other CNS symptoms | Efavirenz, raltegravir and atazanavir |
| Neuropathy | Didanosine, stavudine and zidovudine |
| Myopathy | Zidovudine |
| Headache | All antiretrovirals |
| Nausea and vomiting | All antiretrovirals, in particular zidovudine and protease inhibitors |
| Diarrhoea | Protease inhibitors (in particular lopinavir) and didanosine |
| Pancreatitis | Didanosine, stavudine and raltegravir |
| Hepatitis/liver toxicity/liver dysfunction |
All antiretrovirals (in particular nevirapine and didanosine). Indinavir and atazanavir cause hyperbilirubinaemia |
| Renal dysfunction | Tenofovir and atazanavir |
| Bone demineralization/osteopenia/osteoporosis | Combination antiretroviral therapy, especially following initiation, regardless of regimen; in particular, protease inhibitors, tenofovir and stavudine |
| Severe dermatological conditions (SJS/EM major/TEN) | All antiretrovirals, in particular, nevirapine, efavirenz, etravirine, fosamprenavir, abacavir, darunavir, zidovudine, didanosine, boosted lopinavir and atazanavir |
| Rash | All antiretrovirals, in particular NNRTIs |
| Skin hyperpigmentation | Emtricitabine (more prominent in non‐Caucasians) |
| Systemic hypersensitivity reaction | Abacavir, nevirapine and enfuvirtide |
| Lipodystrophy |
All protease inhibitors and efavirenz (lipohypertrophy). Didanosine, stavudine and zidovudine (lipoatrophy) |
| Dyslipidaemia | All protease inhibitors, NRTIs, especially stavudine, and NNRTIs (efavirenz > nevirapine) |
| Glucose intolerance |
NRTI thymidine analogues (stavudine, didanosine and zidovudine). Some protease inhibitors (ritonavir‐boosted lopinavir; less often atazanavir and fosamprenavir) |
| Lactic acidosis | NRTIs, in particular, didanosine and stavudine (enhanced in combination); less commonly zidovudine |
| Granulocytopaenia, neutropaenia and/or anaemia | Zidovudine and fosamprenavir |
| Respiratory symptoms | Abacavir, lamivudine and zidovudine |
CNS, central nervous system; SJS, Stevens−Johnson syndrome; EM, erythema multiforme; TEN, toxic epidermal necrolysis; NRTI, nucleoside reverse transcriptase inhibitor; NNRTI, nonnucleoside reverse transcriptase inhibitor.
*The list is not exhaustive; for a complete list, refer to the summary of product characteristics (http://www.ema.europa.eu/ema/) and DHHS website (http://aidsinfo.nih.gov/drugs).
10. Coinfections
Hepatitis B virus and hepatitis C virus
Liver disease in children with HBV or HCV coinfection should be managed jointly with paediatric experts in viral hepatitis.
HCV coinfection is an indication for starting ART.
For HBV coinfection, if treatment of HIV infection is not indicated and there is no evidence of liver disease, HIV treatment should be considered but may be deferred.
Tuberculosis
All HIV‐infected children exposed to an individual with infectious TB and all children with evidence of latent TB infection should have preventive TB treatment (once active TB disease has been excluded).
In children with active TB disease, TB treatment should be started at TB diagnosis. ART should be started, as soon as practicable, and within 2 and 8 weeks of TB treatment in children with severe and moderate immunosuppression, respectively. ART may be deferred at higher CD4 counts until TB treatment is completed.
There is significant interaction between ART and TB therapy. Therapeutic drug monitoring (TDM), where available, should be used in the context of potential significant interactions.
Children with TB coinfection should be managed in consultation with an expert in the treatment of paediatric TB. A specialist in drug‐resistant TB (DRTB) should be involved in the management of DRTB contacts and cases.
See Table 6 (later) for ART choices in children with TB.
Table 6.
Antiretroviral therapy (ART) regimens for children treated for tuberculosis (TB) coinfection with rifampicin‐containing regimens
| < 3 years | > 3 years | ||
|---|---|---|---|
| Initiating ART | Preferred |
LPV/R*
,
†
+ 2 NRTIs NVP † , ‡ , § + 2 NRTIs |
EFV + 2 NRTIs |
| Alternative | N/A |
LPV/R*
,
†
+ 2 NRTIs DRV/r † + 2 NRTIs ATV/r † + 2 NRTIs |
|
| Already on ART | Preferred |
LPV/R*
,
†
+ 2 NRTIs NVP † , § + 2 NRTIs |
EFV + 2 NRTIs |
| Alternative | 3 NRTIs ¶ |
LPV/R*
,
†
+ 2 NRTIs ATV/r † + 2 NRTIs DRV/r † + 2 NRTIs 3 NRTIs ¶ |
ATV/r, ritonavir‐boosted atazanavir; DRV/r, ritonavir‐boosted darunavir; EFV, efavirenz; LPV, lopinavir; NRTI, nucleoside reverse transcriptase inhibitor; NNRTI, nonnucleoside reverse transcriptase inhibitor; NVP, nevirapine; N/A, not available.
*Superboost lopinavir with increased ritonavir dose (R) to achieve lopinavir/ritonavir ratio of 1:1. Return to regular dosing once rifampicin has been discontinued. †Therapeutic drug monitoring is recommended (where available) to adjust doses. ‡Start nevirapine (NVP) without lead‐in dose and at the maximum recommended dose for age. §Two weeks after starting NVP, consider increasing the NVP maintenance dose by a further 20–30%. Watch for liver toxicity. Return to regular dose once rifampicin has been discontinued. ¶In virally suppressed children.
Opportunistic infections
We recommend that ART should be initiated as early as possible, apart from in the context of cryptococcal meningitis, where evidence in adults has shown that delaying ART may be associated with reduced mortality.
11. When to switch, resistance testing and second and subsequent ART regimens
ART regimens may be changed because of treatment failure, because of toxicity or for simplification.
Virological failure – second and subsequent regimens
Switching to second‐line therapy following virological failure should occur early (VL > 1000 copies/ml) for those failing on combinations including drugs with a low genetic barrier to resistance [NNRTIs or raltegravir (RAL)].
Where there are blips in VL (detectable VL < 400 copies/ml), blood tests should be repeated within 4 weeks to confirm re‐suppression.
Reinforcement of adherence support, as the main reason for treatment failure, should be prioritized. Switching treatment when there are ongoing problems with adherence may lead to loss of efficacy of further classes of ART.
Table 7 (see later) summarizes potential strategies for choosing second‐line therapy. If the suggested options are not applicable, seek expert advice.
Table 7.
Switching to second‐line antiretroviral therapy (ART)
| Age | Failed on first‐line NNRTI backbone | Failed on first‐line boosted PI backbone | ||
|---|---|---|---|---|
| Resistance mutations | No resistance mutations | Resistance mutations | No resistance mutations | |
| All |
Evaluate/support adherence +++ Switch depending on resistance |
Evaluate/support adherence +++ Switch to a more robust/forgiving PI‐based regimen (see below) or Restart same first‐line regimen |
Evaluate/support adherence +++ Switch depending on resistance |
Evaluate/support adherence +++ Switch to a simplified regimen (see below) or Restart same first‐line regimen |
| < 6 years | LPV/r + 2 active NRTIs (e.g. TDFb + ZDV if first‐line ART was ABC + 3TC) | LPV/r + same first‐line NRTIs |
DRV/ra + 2 active NRTIs (e.g. TDFb + ZDV if first‐line ART was ABC + 3TC) or DRV/ra + INSTI + 1 active NRTI |
Simplification options, depending on child's weight (aim for once daily): Boosted PI Simpler formulations Fixed dose combinations if available for the child's age |
| 6–12 years | ATV/r or DRV/r + 2 active NRTIs | ATV/r or DRV/r + same first line NRTIs | ||
| > 12 years | New regimen with at least 3 new active drugs; e.g. boosted PI, NRTI, INSTI, CCR5 inhibitor – seek expert advice | |||
- Present your case to others with expertise in the field (e.g. ‘virtual clinic’). If switching to third‐line or salvage therapy, always seek expert advice.
- Resistance testing should be undertaken while the patient is still on or recently off a failing regimen. Results will direct second‐ line choices. If testing is performed too late (more than 4 weeks) after cessation of a failing regimen, the results will probably show wild‐type virus. If this is the case or if testing is not available, resistance should be assumed.
- Reinforce adherence support. This is the main reason for treatment failure.
- Simplify regimens where possible (once‐daily and fixed dose combinations). Switching to NNRTI‐based regimens or PI monotherapy is not advised if there are serious adherence issues.
- If resistance results suggest that these second‐ line options in the table would not be effective, seek expert advice.
Seek expert advice if the patient is aged < 3 years and failing LPV/r with mutations and for any child < 2 years old failing first‐line therapy with NRTI mutations.
3TC, lamivudine; ABC, abacavir; ATV/r, ritonavir‐boosted atazanavir; DRV/r, ritonavir‐boosted darunavir; INSTI, integrase inhibitor; LPV/r, ritonavir‐boosted lopinavir; NRTI, nucleoside reverse transcriptase inhibitor; NNRTI, nonnucleoside reverse transcriptase inhibitor; PI, protease inhibitor; NVP, nevirapine; TDF, tenofovir; ZDV, zidovudine.
DRV/r is not licensed for < 3 years of age.
TDF is not licensed for < 2 years of age.
Resistance testing
Resistance testing should be performed prior to switching regimens when there is virological failure. Resistance testing should be undertaken while the patient is still on the failing regimen. If this is not possible, ideally test for resistance within 4 weeks of stopping the failing regimen.
Resistance testing may include reverse transcriptase/protease/integrase/V3 loop/envelope sequencing.
The interpretation of resistance results can be guided by the Stanford HIV Drug Resistance Database (http://hivdb.stanford.edu/).
Substituting single drugs in a failing regimen without prior resistance testing is not recommended.
Simplification
Where possible, regimens should be simplified (once‐daily and fixed dose combinations), but switching to NNRTI‐based regimens or PI monotherapy is not advised if there are adherence issues.
12. Stopping treatment and treatment interruption
Treatment interruptions cannot be routinely recommended and starting ART currently means lifelong therapy.
Judicious use of planned treatment interruptions may be considered in circumstances when ART needs to be stopped such as because of toxicity or adherence difficulties, while the latter is being addressed.
Stopping NNRTIs when HIV is fully suppressed requires a replacement or staggered stop to reduce the risk of developing NNRTI resistance as a result of the longer half‐life of NNRTIs. A replacement stop is preferable.
13. Adolescence, mental health and transition
Adolescents commencing first‐line therapy should be started on boosted PI‐based ART and subsequently switch to NNRTI‐based ART once adherence has been established and VL is consistently < 50 copies/ml.
Multidisciplinary monitoring for signs of psychological distress and mental health disorder should be routine as children progress through adolescence towards transition.
Early and ongoing support from clinical psychologists with specialist paediatric knowledge is recommended.
14. Pipeline and upcoming trials
See Table 8.
Table 8.
The paediatric antiretroviral pipeline
| Compound | Sponsor | Formulation(s) and dose | Status and comments |
|---|---|---|---|
| Nucleotide reverse transcriptase inhibitors | |||
| TAF | Gilead |
Dose to be determined for children Under investigation in adolescents with adult dose as a component of EVG/COBI/FTC/TAF (see below) |
Phase II/III; EVG/COBI/FTC/TAF; treatment‐naïve adolescents aged 12 to < 18 years; enrolling Co‐formulation with FTC under discussion |
| Nonnucleoside reverse transcriptase inhibitors | |||
| ETR | Janssen | Dispersible tablets 25 (scored), 100 mg |
Approved for 6 to18 years Phase I /II; treatment‐experienced children aged 2 months to < 6 years and treatment‐naïve children aged ≥ 2 months to < 2 years; enrolling |
| RPV | Janssen |
Tablet 25 mg Granules 2.5 mg /g |
Phase II; adolescents aged 12 to < 18 years with weight > 32 kg; enrolling Phase I/II; children aged > 2 to < 12 years; planned |
| Protease inhibitors and combinations | |||
| ATV | Bristol‐Myers Squibb (BMS) |
Powder 50 mg sachet under development Capsules 100, 150, 200, 300 mg |
Approved for 3 months and above by FDA Phase III/IIIb; ongoing; RTV‐boosted ATV for treatment‐naïve and ‐experienced children aged 3 months to < 6 years Other studies in children aged up to 11 years ongoing |
| ATV/COBI | Gilead/BMS | Co‐formulated boosted PIs in development | Phase II/III; treatment‐experienced children aged 3 months to < 18 years |
| DRV/COBI | Gilead/ Janssen | ||
| LPV/r | Cipla | 40/10 mg pellets in capsules | Submitted to FDA |
| LPV/r/3TC/ABC or ZDV | DNDi/Cipla | 4‐in‐1 FDC granules | Formulation work ongoing |
| Booster | |||
| COBI | Gilead |
75 mg tablets 20 mg dispersible tablets for oral suspension |
As booster with ATV and DRV Under development as component of EVG/COBI/FTC/TDF and EVG/COBI/FTC/TAF |
| Integrase inhibitors and combinations | |||
| RAL | Merck | Granules for suspension 6 mg/kg (100 mg sachet) |
FDA approval for use in children 4 weeks of age and older Neonate passive PK study ongoing (neonates born to women who received RAL in pregnancy and during labour) Neonate PK and safety study for prophylaxis; ongoing in high‐risk HIV‐exposed neonates from birth to 6 weeks |
| EVG | Gilead | EVG reduced‐strength tablets and suspension in development |
EVG PK; completed; RTV‐boosted; adolescents aged 12 to < 18 years RTV‐boosted EVG to be studied in all age groups |
| EVG/COBI/FTC/TDF (Stribild) | Gilead | Reduced‐strength tablets in development |
Studies underway in treatment‐naïve adolescents aged 12 to < 18 years 6 to < 12 years planned (waiver < 6 years) |
| EVG/COBI/FTC/TAF | Gilead | Reduced‐strength tablets in development |
Studies under way in treatment‐naïve adolescents aged 12 to < 18 years 6 to < 12 years planned (waiver < 6 years) |
| DTG | ViiV Healthcare |
Granule formulation in development Reduced‐strength 10 mg and 25 mg tablets |
Approved for adolescents aged 12 to < 18 years weighing > 40 kg in USA and Europe Phase I/II study; treatment‐naïve and ‐experienced children aged 6 weeks to < 18 years; ongoing Exposures from granules were moderately higher than with tablets and highest with formula milk in HIV‐negative adults |
| DTG/ABC/3TC (572‐Trii) | ViiV |
Paediatric formulation development planned Dosing to be determined |
Dependent on ongoing studies confirming DTG dose in children and ability to establish appropriate dosing ratios for components |
| CCR5 receptor antagonist | |||
| MVC | ViiV | Suspension 20 mg/mL | Phase IV; treatment‐experienced, CCR5 tropic children aged 2 to < 18 years |
3TC, lamivudine; ABC, abacavir; ATV, atazanavir; COBI, cobicistat; DRV, darunavir; DTG, dolutegravir; ETR, etravirine; EVG, elvitegravir; FDA, US Food and Drug Administration; FDC, fixed dose combination; FTC, emtricitabine; LPV/r, lopinavir/ritonavir; RAL, raltegravir; TAF, tenofovir alafenamide; MVC, maraviroc; PI, protease inhibitor; PK, pharmocokinetic; RPV, rilpivirine; RTV, ritonavir; ZDV, zidovudine.
3. Diagnosis, baseline investigations and pretreatment monitoring
If a mother is diagnosed with HIV infection, all children potentially at risk of infection should be tested for HIV irrespective of age.
Children under 18 months of age at risk of perinatally acquired HIV infection should be tested using blood DNA or RNA PCR with subsequent confirmatory repeat PCR if positive.
PEP should be given for 4–6 weeks to all babies born to HIV‐infected mothers according to local guidelines.
Babies born to HIV‐infected mothers should have an HIV RNA PCR test at birth and at least two further separate PCR tests (the first 2 weeks after cessation of PEP, and the second at least 6 weeks after cessation of PEP). For babies at high risk of transmission, an additional PCR test midway through PEP is recommended.
Breast feeding is not recommended. In circumstances in which the mother is choosing to breast feed against recommendations, the baby should have regular screening PCR tests. Two negative HIV RNA PCR tests (2 and 6 weeks after cessation of breast feeding) are required to confirm that the baby is not infected.
Children over 18 months of age can be tested using serological assays with subsequent confirmatory PCR if positive or equivocal.
A negative serological test in children who have had positive HIV RNA PCR does not exclude ongoing HIV infection.
A detailed history of any possible previous ART given to the child and/or mother (or other likely source of infection) should be documented.
The genotypic HIV resistance profile should be documented at baseline.
The HLA B*5701 genotype should be confirmed negative before using ABC.
Clinical assessment should be carried out 3−4‐monthly in children who are stable off ART, with frequency of laboratory monitoring dictated by age, clinical status and proximity to thresholds for ART initiation (minimum 6‐monthly HIV VL and CD4 count).
Local guidelines for BCG immunization of babies of HIV‐infected mothers should be followed. HIV‐exposed infants at low risk of HIV transmission (maternal VL < 50 copies/ml at or after 36 weeks of gestation) with high risk of TB exposure may receive BCG vaccination at birth, prior to definitive diagnosis/exclusion of HIV infection.
3.1. Confirmation of HIV diagnosis
Optimal treatment of HIV infection in children depends upon timely diagnosis. This requires early HIV testing of all infants born to HIV‐infected women and prompt testing of infants or older children at risk or with symptoms or signs of HIV infection. Infants born to women with HIV infection will be HIV antibody positive because of transplacental transfer of maternal antibodies. An HIV PCR test is needed to confirm or exclude the diagnosis. Either HIV RNA or DNA PCR may be used, depending on local availability 8, 9, 10, 11, 12. PEP should be given for 4–6 weeks to all babies born to HIV‐infected mothers according to local guidelines. A PCR test should be performed at birth. At least two positive PCR tests on separate samples from the baby (not the umbilical cord) are required to confirm an HIV diagnosis. A repeat test should be carried out as soon as possible after any positive PCR test in an infant to avoid delay in confirming the diagnosis and initiating treatment. Up to 62% of HIV‐infected neonates may have a negative initial PCR test in the first 48 hours of life 12. PCR tests become more reliable 14–21 days after birth. The purpose of the initial test at birth is thus to allow rapid identification of those that are already positive so that a confirmatory test and treatment can be initiated without delay. Later tests are essential for excluding HIV infection in babies who test negative at birth.
In the absence of breast feeding, at least two separate negative HIV RNA PCRs after stopping PEP are required to confirm that an exposed baby is uninfected (the first 2 weeks after cessation of PEP, and the second at least 6 weeks after cessation of PEP). For babies born with high risk of transmission, an additional PCR test midway through PEP is recommended to allow earlier identification of infected infants. The use of fourth‐generation point‐of‐care antibody/antigen testing for diagnosis of HIV infection in children under 18 months of age is not recommended in view of low sensitivity for distinguishing between HIV infection and positive serology from maternal antibody 13. Some less prevalent HIV subtypes may escape detection by PCR (e.g. HIV A, C‐H and O) 14. Those using PCR for diagnosis should know the sensitivity of the assay being used for the likely subtype being tested for. Expert advice should be sought if there is any doubt about the interpretation of results. However, initiation of ART in infants should not be unduly delayed by referrals to an expert centre.
Babies of HIV‐infected women may subsequently become infected after initial negative tests if they are breast‐fed. Breast feeding is not recommended in high‐income countries, where alternative feeding is safe and practical. If in exceptional circumstances an uninfected baby is breast‐fed, the mother should be on effective ART. Two confirmatory PCR tests, 2 and 6 weeks after cessation of breast feeding and PEP, are required to confirm that the baby is not infected. It is important to note that HIV‐infected children who have commenced ART may become seronegative by standard commercial testing after loss of passively acquired maternal antibodies, especially after early ART initiation (< 3–6 months of age) 15. A negative serological test in this context does not exclude ongoing HIV infection as the HIV DNA PCR test remains positive.
Infants and children > 18 months of age who present with symptoms consistent with HIV infection and unknown maternal HIV status should initially have an HIV antibody test. If this is positive, they will require a confirmatory PCR test. It is recommended that all previously untested children of HIV‐infected women should be tested for HIV whatever their age, as infected children may remain asymptomatic throughout childhood and adolescence 16. All siblings at risk of perinatally acquired infection (irrespective of age) should also have an HIV test. Adult physicians and family doctors should routinely ask all HIV‐infected men and women in their care if they have children and refer those at risk for testing 17.
3.2. Baseline assessments once diagnosis has been confirmed
Once a diagnosis of HIV infection has been confirmed, children should be assessed clinically, including assessment of growth and development, to allow staging of HIV infection according to WHO (or CDC) classifications 4, 6.
To plan future ART, it is important to document whether the child has been exposed to previous ART, in utero, through breast feeding, as PEP or as therapy (e.g. in their country of origin). If available, the antiretroviral history of the mother or other source case should also be documented. In view of the possibility of transmitted drug resistance 18, 19 and unreported prior ART exposure, HIV genotypic resistance testing is recommended at baseline (including reverse transcriptase, protease and integrase resistance testing when available). If available, the results of resistance testing of the source case, as close as possible to the time of transmission, should also be documented. Baseline viral co‐receptor [C‐C chemokine receptor type 5 (CCR5)] tropism testing is not indicated, as CCR5 receptor antagonists are not currently recommended as first‐line therapy. Other baseline investigations after HIV diagnosis include measurements of HIV RNA VL and CD4 lymphocyte percentage and absolute count, tests for other vertically transmitted or postnatally acquired infections [infections with HBV, HCV, toxoplasma, cytomegalovirus (CMV), syphilis, TB and Chagas (in those at risk)], full blood count, haemoglobinopathy screen (in risk groups), bone profile, and tests of liver and renal function. Baseline echocardiography in those at risk of cardiomyopathy should be considered. HLA B*5701 testing, where available, is recommended at baseline prior to starting ABC 20. See baseline investigations at http://www.chiva.org.uk for further guidance.
A full vaccine history should be taken and if necessary serology performed to confirm immunity and to aid in decision making around catch‐up and boosting 21. Local BCG practices for babies of HIV‐infected mothers should be followed. In areas of low TB incidence or if there is high risk of HIV transmission, BCG vaccination should be postponed until an infant is confirmed as HIV negative. HIV‐exposed infants at low risk of HIV transmission (maternal VL < 50 copies/ml at or after 36 weeks of gestation) with high risk of TB exposure may receive BCG vaccination at birth, prior to definitive diagnosis/exclusion of HIV infection 22. The very low risk of HIV transmission with an undetectable maternal VL means that in these circumstances the risk of severe TB from not vaccinating is greater than the risk of HIV‐related BCG complications. In resource‐poor settings with high TB prevalence, BCG vaccination is given to all infants prior to determination of HIV status. Early ART in this context has been shown to minimize the risk of BCG‐related complications 23.
In those presenting outside the neonatal period, a baseline chest radiograph allows assessment for respiratory complications, including lymphoid interstitial pneumonitis and TB. Infants and children with advanced HIV disease should have an ophthalmic examination for evidence of retinitis, and a blood CMV PCR test (if available). Infants and children with evidence of neurological involvement should undergo baseline neuroimaging. Additional baseline immunological assessment for evidence of TB infection is recommended [tuberculin skin test (TST) and/or interferon gamma release assay (IGRA)].
Children not yet requiring ART (see treatment criteria below) should have clinical monitoring at intervals of no longer than 3–4 months. Routine monitoring should include clinical examination and measurement of growth parameters. Monitoring for HIV disease progression and complications using CD4 count/percentage and VL, renal and liver function and urinalysis is recommended on a 6‐monthly basis, and should be performed more frequently in younger children and those approaching treatment thresholds. Annual assessment of neurodevelopment, blood pressure, nutrition (including lipids) and puberty is also recommended. Vitamin D should be assessed and managed according to local guidelines as in HIV‐uninfected children. Less frequent assessment of these parameters has been suggested in adult guidelines 2. However, in view of the possibility of more rapid disease progression in children, the frequency of laboratory monitoring should remain as recommended above for children not receiving ART.
4. Prophylaxis against opportunistic infections
Prophylaxis against Pneumocystis jirovecii should be given to all HIV‐infected infants from age 1 month and to older children with low CD4 counts (see criteria below). Co‐trimoxazole is the drug of first choice (see drug table in Supplementary Table S1 for dosing).
No routine primary prophylaxis against other infections is recommended.
Prophylaxis with co‐trimoxazole is highly effective at preventing potentially life‐threatening infection with P. jirovecii, and also at reducing bacterial infections 24, 25. In view of their susceptibility to severe P. jirovecii infection, all HIV‐infected infants should receive prophylaxis from 4 weeks of age until their first birthday, regardless of CD4 and VL 26. HIV‐exposed babies at high risk for transmission should also commence P. jirovecii prophylaxis at 4 weeks of age and continue until HIV infection has been excluded. Co‐trimoxazole is the first‐choice drug unless contraindicated. Co‐trimoxazole has not previously been recommended before the age of 4 weeks because of the theoretical risk of kernicterus with sulphonamide administration in neonates. Hard evidence for this is lacking 27 and it is unclear what the relative risks of this are compared with the risks of P. jirovecii infection in young infants in whom the diagnosis of HIV infection is made before 4 weeks of age.
Children aged 1 to 4 years should receive prophylaxis against P. jirovecii if they have a CD4 count below 500 cells/μL or 15% of total lymphocyte count. Children aged 5 years and above should receive prophylaxis against P. jirovecii if they have a CD4 count below 200 cells/μl or 15%, and it should be considered when they are approaching these thresholds 28, 29. While intermittent dosing (3 days a week) is sufficient for P. jirovecii prophylaxis, daily co‐trimoxazole prophylaxis according to weight bands, as in the current WHO guidelines 6, simplifies recommendations with the additional benefit of protecting against bacterial infections. This option is also therefore endorsed by these guidelines. Daily co‐trimoxazole prophylaxis may also be considered for children travelling to countries with a high prevalence of bacterial infections and/or malaria, irrespective of their CD4 count/percentage and current treatment status 6, 24, 30, 31. The use of co‐trimoxazole in this context may also have additional benefit as antimalarial prophylaxis 32, although specific malaria prophylaxis appropriate to the regions being visited should always be prescribed.
Once ART has been started and the CD4 count has risen, the risk of P. jirovecii infection decreases 33, 34. Most paediatricians stop co‐trimoxazole in children over 1 year of age living in well‐resourced settings 6 months after CD4 count recovery. The AntiRetroviral Research for Watoto (ARROW) trial has reported additional benefits of continuing co‐trimoxazole prophylaxis after immune reconstitution in children in resource‐poor settings. It is likely that these additional benefits are related to antibacterial, antimalarial and anti‐inflammatory effects. The findings of this trial are therefore less likely to be of relevance in most European settings 30.
Prophylaxis against other infections has been suggested for those with very low CD4 counts. There is insufficient evidence to make any recommendations for routine primary prophylaxis, but specific guidance is available 35. Prophylaxis against TB can be considered for children visiting countries highly endemic for TB (see Section 10 below). The most important means to reduce susceptibility to all opportunistic infections is prompt initiation of ART when indicated.
5. When to start ART
ART is recommended:
in all children under 1 year of age;
in all children with significant disease (WHO stage 3 or 4 or CDC stage B or C);
in asymptomatic children over 1 year of age based on age‐specific CD4 count thresholds;
to be initiated before the CD4 count reaches the CD4 treatment threshold;
in those with HCV or active TB coinfection.
ART should be considered:
in asymptomatic children over 5 years of age at CD4 counts of 350–500 cells/μl, to potentially optimize CD4 count in adulthood;
in children with high VL (> 100 000 copies/ml);
in asymptomatic children aged 1–3 years irrespective of immune status and VL;
in sexually active adolescents, to minimize the risk of onward transmission;
in the presence of any significant HIV‐related clinical symptoms;
in HBV coinfection irrespective of immune status.
5.1. Children under 1 year of age
ART should be started as soon as possible in all HIV‐infected children under 1 year of age irrespective of clinical or immunological status 4, 6. Evidence for this comes from the South African randomized controlled Children with HIV Early Antiretroviral Therapy (CHER) trial 36, 37, which showed a 4‐fold reduction in mortality among asymptomatic infants starting ART before 3 months of age compared with those starting at a CD4 percentage < 25% or WHO stage 3 or 4. In addition, in Europe, a 4‐fold reduction in HIV progression/mortality was observed among infants starting ART at less than 3 months of age compared with later in a large infant cohort meta‐analysis [European Infant Collaboration (EIC)] 38.
Additional CHER trial substudies have added further evidence of the clinical 23, 39, immunological 40 and neurodevelopmental 41 benefits of early ART initiation in infants. Data from European cohorts have also shown that virological, clinical and immunological benefits from early treatment are sustainable outside the trial setting 42, 43. Further analysis of laboratory parameters from the EIC has demonstrated an association between early infant ART, more rapid control of viraemia and a higher CD4 count up until 12 months of age 44.
Universal treatment of all infants with HIV infection, although challenging, is an achievable goal. The risks of drug resistance and early toxicity are markedly outweighed by improvements in short‐term mortality and disease progression, especially prevention of irreversible HIV encephalopathy.
Infants should be reviewed at a minimum of monthly intervals up to 6 months of age in order to increase ART dosing in line with growth. Symptomatic infants presenting with severe illness (including opportunistic infections) should start ART as soon as possible. Debate remains about whether ART should be started immediately or deferred until treatment for the presenting illness has started and the child is clinically stable. There is no evidence to inform this, and it is recommended that ART be started as soon as the child is stable (ideally within 2 weeks of diagnosis). Expert pharmacist advice should be obtained if there is a complex treatment for a coinfection (e.g. TB) as drug interactions may interfere with effectiveness and/or cause side effects. See Section 10 below for further specific information on coinfection.
5.2. Children over 1 year of age – general principles
Starting ART is recommended in all children over 1 year of age with HIV‐related symptoms, and in asymptomatic children with CD4 counts or percentages below or approaching recommended age‐related thresholds. Starting ART should also be considered in those with a high HIV RNA VL (> 100 000 copies/ml), as they are more likely to progress rapidly to symptoms or have a rapid fall in CD4 values 45, 46, 47.
The evidence for clinical benefit of ART in children with AIDS/CDC category C disease is so strong that parental refusal to treat is a child protection issue. CDC clinical category B covers a wide range of disease severity. A retrospective study from the USA demonstrating a significant reduction in rate of progression of disease adds further weight to the recommendation that children with category B or C disease should be treated irrespective of their CD4 count/percentage 48. PENTA now recommends that treatment should be initiated for all children in WHO stage 3 or 4 (CDC category B or C) and considered in all children with HIV‐related symptoms (WHO stage 2; CDC category A).
CD4‐guided treatment thresholds in asymptomatic children are based predominantly on analysis of paediatric and adult cohort data and extrapolation from the adult Simple Trial Comparing Two Strategies for Management of Anti‐Retroviral Therapy (SMART study) 47, 49, 50, 51, 52. Only one randomized trial [Pediatric Randomised Early versus Deferred Initiation in Cambodia and Thailand (PREDICT)] has addressed the question of when to start ART in children over 1 year of age. Thai children aged 1–12 years (median age 6.4 years), with CD4 percentage 15–24%, were randomized to start immediate ART or defer ART until the CD4 percentage dropped below 15%. Rates of progression and death were unexpectedly low in both groups during 144 weeks of follow‐up. As a result, the study was underpowered to detect a difference in the primary endpoint of AIDS‐free survival. Analysis of secondary endpoints did, however, demonstrate better height‐for‐age Z scores in the immediate treatment group 53.
The evidence for absolute age‐related CD4 thresholds for starting ART remains as for the PENTA 2009 guidelines, as follows.
-
(1)
Analysis of adult data
Data from the SMART trial clearly showed that adults with CD4 counts between 250 and 350 cells/μL have significantly better outcomes on ART than off ART 51. Adult (US and European) guidelines strongly recommend ART initiation at CD4 cell counts below 350 cells/μl 2, 3, 5, 6.
-
(2)
Comparison of child and adult data
Comparison of the short‐term risks of disease progression in pre‐ART adult seroconverters in the Concerted Action on SeroConversion to AIDS and Death in Europe (CASCADE) cohort collaboration and in children aged 5 years and older in the paediatric HIV Paediatric Prognostic Markers Collaborative Study (HPPMCS) cohort showed that the short‐term risk of disease progression was very similar in young adults (around 20 years old) and children aged 5 years and older 52. Absolute CD4 count, rather than percentage, should therefore be used to determine treatment thresholds in children from the age of 5 years. These should follow the same CD4 threshold recommendations for treatment recommended in adult guidelines.
-
(3)
Analyses of child data
Analyses from the HPPMCS cohort demonstrated that CD4 counts are highly prognostic of disease progression at all ages after infancy 49. However, to obtain a uniform progression risk with the thresholds for adults and children aged 5 years and over, thresholds between 1 and 5 years of age would have to change approximately every 6 months or less. This would require too many age bands for a workable guideline, and the historical data on which progression risks are based are not robust enough to warrant ignoring the importance of practical guidance and the desirability of general concordance with other international guidelines. Therefore, only two age bands between 1 and 5 years have been selected. CD4 count as well as CD4 percentage thresholds should be taken into account. In the HPPMCS data, 10–20% of CD4 percentages and CD4 counts are discordant in terms of ART initiation thresholds adopted for these PENTA guidelines. However, these values are frequently concordant on a subsequent blood sample. If consistent discordance is observed, and particularly if the count is below the threshold although the percentage is not, then initiation of ART is strongly recommended 54.
The prognostic significance of plasma HIV RNA for short‐term risk of disease progression is much weaker than CD4 count or percentage 45, 46, 47, 50. However, ART is recommended in asymptomatic children with VL persistently above 100 000 copies/ml, even if they do not meet CD4 count criteria.
Rapid clinical, virological or immunological failure may occur, but, in general, ART does not need to be started quickly except in infants or in an older child seriously ill with advanced HIV disease. Time spent preparing and educating the family, particularly about adherence, is very important. Starting ART needs to be supported and promoted by the caregivers if it is to succeed. It is preferable not to start ART at the first clinic visit. Older children should preferably know why they are taking treatment, and timing of full or partial naming of HIV diagnosis in relation to starting ART is an important consideration. These CD4 thresholds are for children without coinfection. See Section 10 for guidance in the context of HBV/HCV, TB and opportunistic infections.
5.3. Children aged 1–3 years
As in the previous PENTA guidelines 1, CD4 thresholds for absolute indication to start treatment outside the infant period are based on data extrapolated from adults and analysis of HPPMCS data. The age bands (1−< 3, 3−< 5 and ≥ 5 years) provide harmony with DHHS guidelines (which are also largely based on HPPMCS data) 4 but contrast with new WHO guidelines which group all children aged 1–5 years together. The PENTA 2009 guidelines defined thresholds based on an individual child's risk of progression to AIDS or death over the subsequent 1 year (derived from HPPMCS data using the on‐line PENTA calculator available at http://www.hppmcs.org/). The absolute treatment thresholds aimed to maintain the overall risk of mortality below 2% and the AIDS progression risk below 5% (acknowledging that progression risk is higher and more variable in the first few years after infancy). The ongoing success of ART means that we should continue to expect better clinical outcomes for children, and in 2015 these rates of disease progression are no longer deemed acceptable. Looking more closely at the calculated risks, using a threshold of < 1000 cells/μl and < 25%, between 1 and 3 years, risk of progression at these thresholds increases dramatically at younger ages (Table 2). For these reasons, while we recommend keeping the 2009 CD4 thresholds in this age range, it is also recommended that ART should be considered in all children aged 1–3 years in order to minimize risk of progression and death and to minimize potential deleterious effects of ongoing viral replication on the child's rapidly developing brain and immune system.
Table 2.
Percentage risk of progression to AIDS or death in the next 12 months associated with absolute thresholds at age 1–3 years
| AIDS | Death | |||
|---|---|---|---|---|
| CD4 = 25% | CD4 = 1000 cells/μL | CD4 = 25% | CD4 = 1000 cells/μL | |
| 1 year (%) | 16 | 23 | 4.5 | 6.6 |
| 2 years (%) | 8.8 | 9.4 | 2 | 1.7 |
| 3 years (%) | 6 | 5.1 | 1.2 | 0.6 |
5.4. Children aged 3–5 years
As mentioned above, the levels of risk deemed acceptable in previous versions of the PENTA guidelines (2% mortality and 5% AIDS) should be lowered in the light of the ongoing success of ART, and the absolute CD4 threshold for ART initiation in children aged 3–5 years has been increased to 750 cells/μl and 25% accordingly (Table 3). This is in line with current US and WHO guidance 4, 6.
Table 3.
Percentage risk progression to AIDS or death in the next 12 months associated with absolute thresholds at age 3–5 years at (a) previous thresholds and (b) updated thresholds
| (a) | ||||
|---|---|---|---|---|
| AIDS | Death | |||
| CD4 = 20% | CD4 = 500 cells/μL | CD4 = 20% | CD4 = 500 cells/μL | |
| 3 years (%) | 8.1 | 8.1 | 1.8 | 1.5 |
| 4 years (%) | 6 | 3.7 | 1.2 | 0.4 |
| 5 years (%) | 4.7 | 3.5 | 0.9 | 0.3 |
| (b) | ||||
|---|---|---|---|---|
| AIDS | Death | |||
| CD4 = 25% | CD4 = 750 cells/μL | CD4 = 25% | CD4 = 750 cells/μL | |
| 3 years (%) | 6 | 5.6 | 1.2 | 0.7 |
| 4 years (%) | 4.5 | 3.5 | 0.8 | 0.3 |
| 5 years (%) | 3.6 | 3.4 | 0.6 | 0.3 |
5.5. Children aged > 5 years
For older children, the recommended absolute threshold for ART initiation remains at a CD4 count of 350 cells/μl, in line with current European adult guidelines 2, 3. However, ART should be considered below 500 cells/μl, in line with current US and WHO guidelines, in order to potentially optimize ultimate CD4 count in adulthood (see below). Treatment should certainly be initiated before the CD4 count reaches 350 cells/μl rather than letting it fall below this value. The ongoing Strategic Timing of Antiretroviral Treatment (START) trial comparing ART initiation at > 500 cells/μl versus < 350 cells/μl will report in 2016.
5.6. Other indications for ART initiation irrespective of immunological or virological status
Coinfection with HCV or TB
Autoimmune manifestations (e.g. thrombocytopenia)
Malignancy
Growth or puberty delay
Neurocognitive delay
Prevention of transmission in sexually active adolescents
Pregnancy
Primary infection (e.g. after nosocomial or sexual transmission)
Child and family wish to start treatment (following full discussion of risk/benefit)
5.7. Consideration for starting antiretroviral therapy to optimize immune function in adulthood
The aim of treatment in paediatric HIV infection should extend beyond survival to maximizing long‐term outcomes and quality of life. Current treatment goals should thus include normal growth and physical, pubertal, neurological and psychological development and immune reconstitution, while minimizing long‐term drug toxicity and viral drug resistance. To date, studies with such long‐term outcome data have been lacking, but in the field of immune reconstitution, mathematical models may allow us to begin to use predicted long‐term outcomes for treatment initiation decisions.
A number of studies indicate that, even with good adherence, long‐term immunity may remain suboptimal after starting ART. The reasons for this are likely to be multifactorial and include: a depleted/inadequate immunological memory for childhood infections and vaccinations; destruction/skewing of B‐ and T‐cell repertoires; and persistently low CD4 counts in relation to healthy age‐matched children (reviewed in 55). Mathematical modelling of data from large European and African cohorts indicates that CD4 cell recovery depends strongly on both the age and CD4 count at ART initiation 56, 57. The important predictions from these studies are as follows.
-
1)
Children under 5 years of age have very good potential for recovering their CD4 counts, even when counts are low at ART initiation.
-
2)
In contrast, with every year that passes after the age of 5 years, the potential for long‐term CD4 count recovery to the normal range diminishes (Fig. 1).
Figure 1.
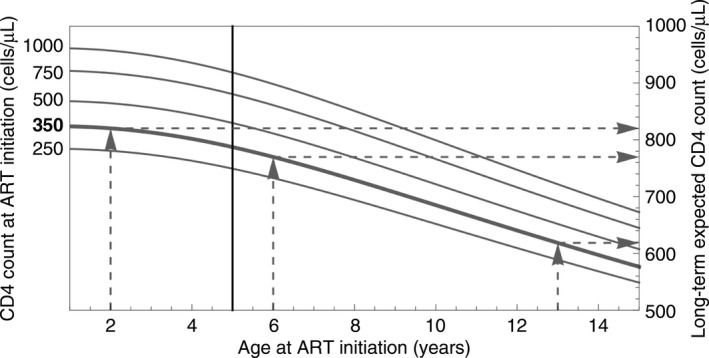
Predicted long‐term CD4 count following antiretroviral therapy (ART) initiation at thresholds of 250, 350, 500, 750 and 1000 cells/μL (curves), using models derived from the AntiRetroviral Research for Watoto (ARROW) study. Delaying treatment in children younger than 5 years (to the left of the vertical line) results in relatively small differences in long‐term expected CD4 count. In contrast, children aged over 5 years (right of the vertical line) are predicted to experience a steady deterioration in long‐term CD4 count as ART is initiated at increasingly older ages (and a constant CD4 threshold). Dashed lines show that a 6‐year‐old delaying treatment until the age of 13 years, with a CD4 count of 350 cells/μL throughout, may expect the long‐term CD4 count to be lowered by 151 cells/μL: from 770 to 619 cells/μL.
Current PENTA guidelines recommended a uniform threshold for children over 5 years old and continuing at the same threshold through adulthood (CD4 count < 350 cells/μl). These mathematical projections raise the concern that this approach may substantially compromise long‐term CD4 recovery in adulthood, particularly for children older than 10 years at ART initiation. The reasons for the effects of age and initial CD4 count on immune reconstitution are only partially understood. They are likely to be attributable to a combination of declining thymic output from its peak at 1 year of age and irreversible immune injury. Whatever the causes, it would appear logical to strive to initiate ART at combinations of age and CD4 counts that are most likely to achieve a better long‐term reconstitution.
The absolute thresholds for ART initiation represent the lowest CD4 thresholds for ART initiation to prevent disease progression; however, these may not be optimal thresholds for CD4 recovery in adulthood. Although Figure 1 represents extrapolations in time and from available data, it can be used as a guide to help indicate how delay in ART initiation may adversely influence eventual CD4 recovery. With current treatment regimens, which are less toxic and easier to take, there is a theoretical case for starting ART at any CD4 count in older children, especially those over 5 years old.
Another long‐term consideration when starting ART in children, irrespective of CD4 count, is the possible benefit of minimizing HIV viral reservoirs in order to optimize the potential for achieving eventual ‘functional HIV cure’. Recent reports in both adults and children have indicated that early and sustained full suppression of HIV may limit viral reservoirs 58, 59 and that a longer cumulative time spent with ongoing viral replication is associated with a larger viral reservoir 60. How this may relate to future ‘cure’ is yet to be determined, but it should also be borne in mind when considering ART initiation in young children who may see the advent of new curative treatments within their lifetime.
We therefore recommend that the CD4 thresholds indicate the lowest limits below which a child should not be allowed to fall before starting treatment. Where resources allow and families are motivated, discussions should be had with children, young people and their families about the option of starting treatment at higher CD4 counts with the aim of optimizing long‐term immune recovery, while always balancing possible long‐term toxicity effects of ART and the potential for viral drug resistance if adherence is poor. With an increase in the number of indications for ART, and in the context of less robust evidence for clinical benefit, optimal adherence is essential so as not to compromise future treatment options though development of resistance to first‐line drugs.
5.8. Comparison of PENTA guidelines with DHHS and WHO guidelines for HIV‐infected children and adults
As can be seen from Table 1, PENTA and US guidelines are generally in agreement; however, DHHS guidelines advise that ART initiation should be considered for any child irrespective of immune and clinical status. Furthermore, the CD4 thresholds are higher for children over 5 years of age. This is in line with recent updates to adult DHHS and WHO guidelines, which give a universal ART threshold of 500 cells/μl, while DHSS recommend that asymptomatic adults should be considered for treatment at any CD4 count. The latter is in part based on cohort data demonstrating a possible benefit with respect to non‐AIDS‐related morbidity for all patients on ART as well as a known decrease in the risk of onward sexual transmission (summarized in 5).
These most recent changes to the adult US guidelines have been debated extensively; the strength of the evidence on which they are based is relatively weak 7, 61, 62. It is the opinion of PENTA that extrapolation of these adult data to guide treatment for children is inadvisable at present. Results of large randomized trials comparing higher versus lower treatment thresholds in adults are expected in 2016 [http://clinicaltrials.gov/show/NCT00867048 (START); http://clinicaltrials.gov/show/NCT00495651 (TEMPRANO)]. This evidence will clarify the wider risks and benefits of starting treatment at higher CD4 counts.
Current European/UK adult guidelines are more conservative 2, 3. The absolute CD4 threshold for ART initiation remains at 350 cells/μl, with a number of exceptions where starting at higher thresholds is indicated (end organ involvement, hepatitis/TB coinfection, pregnancy, discordant couples or primary infection). The PENTA 2015 guidelines are in line with European adult guidance, with the additional option that, for older children, ART initiation at CD4 counts > 350 cells/μl, to optimize immune reconstitution/CD4 count in adulthood, should be considered.
WHO guidelines have harmonized and simplified adult and child recommendations as far as possible, recommending starting ART in all children under 5 years of age, and in older children with CD4 counts < 500 cells/μl. Treatment is recommended for all children under 5 years old for mainly programmatic reasons alongside extrapolation of data from adult cohort studies suggesting clinical benefit to treating irrespective of immune status, while accepting that neither data from PREDICT 53 nor data from a causal modelling study from a large paediatric cohort study 63 indicate clinical benefit. It is suggested that, by simplifying guidance and making treatment universal, access to ART for children will increase in resource‐poor settings where it remains inadequate. This is fortunately not as relevant for the majority of European countries, so for this reason universal treatment is still only recommended by PENTA for those under 1 year of age, for whom the evidence‐based health benefits are incontrovertible.
6. Which ART regimen to start as first‐line therapy
Children should start effective (at least three drugs) ART, usually a dual or triple NRTI backbone together with either a ritonavir‐boosted PI or an NNRTI.
Children exposed to NVP during failed PMTCT (or in whom perinatal NVP exposure cannot be excluded) should be started on a boosted PI‐containing regimen, as transmitted resistance may lead to failure of NVP‐containing ART.
Children aged < 3 years not exposed to NVP during failed PMTCT may be initiated on either NVP or ritonavir‐boosted lopinavir (LPV/r)‐containing ART. We recommend that NVP should be given together with three NRTIs (ABC, 3TC and ZDV) in all infants and in children aged 1–3 years with VL > 100 000 copies/ml or signs of CNS involvement as an induction‐maintenance strategy, unless any of these drugs are contraindicated (such as ABC in HLA B*5701‐positive children).
In children aged > 3 years, either NNRTI or boosted PI‐based ART is acceptable for initial therapy. Factors such as availability of age‐appropriate formulations, palatability, dosing frequency and adherence should be considered when choosing NNRTIs or boosted PIs.
The preferred NNRTI is NVP in children aged < 3 years not exposed to NVP during failed PMTCT, and EFV in children aged > 3 years. The preferred PI in children aged < 6 years is LPV/r, in children aged 6−12 years it is ATV/r, and in children aged > 12 years it is ATV/r or DRV/r.
INSTI‐ based ART may be an alternative regimen in children over age 12 years.
The preferred first‐line NRTIs are ABC/3TC in children aged < 12 years and TDF/FTC or ABC/3TC (if VL < 100 000 copies/ml) in children aged > 12 years.
Age, HLA B*5701 genotype, previous drug exposure, resistance profile, coinfections, available formulations and likely adherence should be taken into account when choosing a first‐line regimen.
See Table 4 for details of recommended first‐line ART regimens.
6.1. General principles of treatment
To achieve long‐term virological suppression requires high levels of ART adherence. Children's doses should be checked for age and weight or surface area at each visit, and this should be done frequently during periods of rapid growth, especially infancy. Doses should be rounded up (not down) to convenient syrup volumes or tablet formulations, and parents should be given careful instructions on dosage, timing, administration, repeating doses if there is vomiting within 1 hour after taking medication, and seeking medical attention rather than discontinuing if drugs are refused or side effects are suspected. Supervised initiation of therapy in hospital or at home with visiting nurses may be appropriate for some children and families, particularly newly diagnosed infants. When drugs show comparable toxicity and efficacy profiles, clinicians should be aware of pricing, drug availability and national policies.
The standard first‐line treatment regimen remains two NRTIs with either an NNRTI or a boosted PI (see Table 4). Although transmitted viral resistance remains rare in children, it may lead to suboptimal response to the first‐line treatment 19. Therefore, pretreatment resistance genotyping should be performed.
6.2. NNRTI or boosted PI for first‐line ART?
Boosted PIs have a higher barrier to viral resistance, but have more potential drug interactions and cause higher rates of dyslipidaemia, while NNRTIs are often more palatable although virological failure frequently results in whole‐class resistance. Recent studies have produced discordant results on whether or not NNRTI‐ and boosted PI‐based regimens are equally effective, especially in the youngest children. This discordance has resulted in conflicting recommendations for first‐line drug regimens and merits consideration.
6.2.1. Infants and young children (< 3 years old)
Infected infants exposed to NVP during failed PMTCT (or in whom perinatal NVP exposure cannot be excluded) should be started on a boosted PI‐containing regimen, as transmitted resistance may lead to failure of NVP‐containing ART 64, 65. LPV/r should not be administered to premature neonates or to term neonates below 2 weeks of postnatal age because of the increased risk of toxicities reported in premature and very young babies 66, 67.
International Maternal Pediatric Adolescent AIDS Clinical Trials (IMPAACT) P1060 suggested better efficacy for LPV/r than for NVP in children aged below 3 years, even without prior NVP exposure. Children randomized to an NVP‐containing regimen were twice as likely as those on an LPV/r‐based regimen to reach a composite endpoint of: virological failure, treatment discontinuation or death at 24 weeks of follow‐up (40.8% versus 19.3%, respectively; P < 0.001) 68. Transmitted NVP resistance, emergence of NVP resistance because of high VL in infancy and possible low NVP levels during lead‐in dosing 69 may have played a role. A composite endpoint at only 24 weeks, and the low‐income setting, make it difficult to extrapolate the results in terms of longer outcomes and generalizability to higher income settings.
In contrast, PENTA 9/ Pediatric AIDS Clinical Trials Group (PACTG) 390 (PENPACT‐1), a randomized trial conducted in resource‐rich settings, showed no difference between first‐line NNRTI – and boosted PI‐based regimens in virological outcomes in children aged 0.1–17.8 years (median age 6.5 years) 70. An underpowered subanalysis by individual drug in children younger than 3 years of age found no difference in virological outcome between children on NVP‐ and LPV/r‐containing ART regimens (G. Tudor‐Williams, unpublished results). In agreement with PENPACT‐1, the Prevention of Malaria and HIV Disease in Tororo, Uganda (PROMOTE) study, a small open‐label RCT in Uganda of children aged 0.4–5.9 (median 3.1) years not exposed to NVP, showed comparable results at 48 weeks in terms of virological suppression, CD4 gain and severe adverse events for NNRTI‐ and LPV/r‐containing ART regimens 71.
The European Pregnancy and Paediatric HIV Cohort Collaboration (EPPICC) study, of children under 1 year of age commencing ART at a median age of 3.6 months, showed no difference in virological suppression or CD4 response between children receiving NNRTI‐based ART (mostly NVP) and those receiving LPV/r‐based ART over a median of 5.9 years of follow‐up. The power to detect small differences was low as comparatively few infants started boosted PI‐based ART. However, in this cohort study, children on an NNRTI‐based four‐drug regimen (NNRTI + 3 NRTIs) had significantly better virological suppression and CD4 gain at 12 months after ART initiation when compared with either PI‐ or NNRTI‐based three‐drug regimens 42. The initial rapid virological suppression and immune reconstitution on an NVP‐based four‐drug regimen compared with a three‐drug regimen have been confirmed in ARROW, although differences in virological/immunological outcomes were not sustained following a switch to three‐drug therapy at 36 weeks from ART initiation 72 (see Section 6.5).
The evaluation of virological outcomes in real‐life clinical settings in the national cohort of HIV‐infected children in the UK/Ireland [Collaborative HIV Paediatric Study (CHIPS)] showed similar rates of VL suppression by 12 months for different first‐line regimens in children aged < 3 years (NVP + 2 NRTIs; NVP + 3 NRTIs; LPV/r + 2 NRTIs) 73. However, three‐drug NVP‐based regimens were associated with faster progression to virological failure long term, while four‐drug NVP‐based regimens had the lowest risk of failure.
In practice, the poor palatability of boosted LPV/r liquid formulation precludes its use in many young children, and NVP remains a first‐line option in infants and young children not exposed to NVP perinatally. NVP has good palatability, high cerebrospinal fluid (CSF) penetration and a favourable lipid profile. Taking the above into consideration, either NVP‐ or LPV/r‐containing ART may be initiated in children younger than 3 years of age. However, in view of the above‐mentioned evidence and the fragility of NVP at high VLs, we recommend that NVP should be accompanied by three NRTIs, as an induction‐maintenance strategy, in infants and young children with VL > 100 000 copies/ml or signs of CNS involvement, including neurodevelopmental delay (Table 4). Earlier viral suppression and greater early CD4 responses with the use of four‐drug induction with a triple NRTI backbone 42, 71 may have potential benefits in terms of reduced viral reservoirs and reduced risk of CNS compartmentalization. Early detection of treatment failure should be picked up by regular monitoring of viral response in the first few months following treatment initiation.
6.2.2. Children > 3 years of age
As mentioned above, PENPACT‐1 found no difference in clinical, virological or immunological outcomes between NNRTI‐ and PI‐based regimens 70. The virological outcome analysis in the CHIPS cohort study showed that in children aged > 3 years overall virological suppression by 12 months was high (> 93%) and there was no difference between NNRTI‐ and PI‐based regimens. However, the progression to virological failure was different between regimens: in the first 2 years on therapy, it was slowest for EFV + 2 NRTIs or NNRTI + 3 NRTIs and fastest for NVP + 2 NRTIs. After 2 years on therapy, the risk was similar for EFV and NVP three‐drug ART, and remained lowest for NNRTI four‐drug ART, although the number of children on the latter regimen was small 73.
Either NNRTI‐ or boosted PI‐containing regimens are acceptable for initial therapy in children above 3 years of age. Issues to consider when choosing an NNRTI or boosted PI regimen include the availability of age‐appropriate formulations, palatability, dosing frequency and adherence. For EFV and NVP, single point mutations in the reverse transcriptase gene rapidly lead to virological failure and whole‐class resistance. Families should be counselled carefully about these issues before starting ART. If predicted adherence is questionable, as is often seen in teenagers, a more forgiving PI‐based regimen should be initiated with a possibility of subsequent simplification to an NNRTI FDC one‐tablet‐once‐daily regimen when virological suppression is sustained and adherence is established.
6.3. Which NNRTI?
The choice for the first‐line NNRTI‐based therapy in children is either EFV or NVP. Until recently, only NVP was licensed for children < 3 years of age because of very poor bioavailability of EFV syrup in this age group. The US Food and Drug Administration (FDA) have recently approved EFV capsule sprinkles for children as young as 3 months of age, based only on the results of a paediatric pharmocokinetic (PK) population model including three open‐label paediatric studies and a PK comparison study in adults 74, 75. Variable EFV exposure in young children, as shown in a recent study of EFV PK in children under 3 years old 76, and lack of experience with EFV sprinkle formulation in children may prevent its widespread use. While waiting for the results of further studies, EFV sprinkles should only be considered as an alternative regimen to NVP in this age group and PK monitoring is currently recommended. Pharmacogenetics may be a useful adjunct in the future as testing for cytochrome P450 2B6 (CYP2B6) polymorphisms has been shown to predict adequate dosing 76.
As seen in adult studies 77, two recent retrospective paediatric cohort studies, in resource‐limited and resource‐rich settings, comparing EFV and NVP showed that EFV was associated with superior virological outcomes 78 73, although in the UK/Ireland CHIPS cohort, the benefit of EFV‐based regimens over NVP‐based regimens in terms of virological failure was very modest after 2 years of therapy 73. In the ARROW trial, a nonrandomized comparison of NNRTI‐ based regimens showed favourable short‐term VL suppression with EFV; however, long‐term suppression depended on age, and was better with EFV in children aged < 10 years, and with NVP in those aged > 10 years 79. A recent systematic review and meta‐analysis which included data on nearly 4000 children showed a lower frequency of severe adverse events and treatment discontinuations with EFV than NVP, although EFV use was associated with higher rates of CNS adverse events 80. Overall, these data support the use of EFV as the preferred NNRTI in children aged > 3 years. However, for children with neurodevelopmental and psychiatric comorbidities, NVP may be a better choice in view of potential CNS side effects associated with EFV.
In summary, the preferred NNRTI is NVP in children aged < 3 years, and EFV in children aged > 3 years.
6.4. Which boosted PI?
PI use in children should be ritonavir‐boosted to optimize efficacy. Consideration of which PI should be used is based on balancing pill burden, toxicity, experience, available paediatric data and available formulations for a given age group.
LPV/r [Kaletra (AbbVie, North Chicago, IL, USA)] is the only combined PI/ritonavir liquid or tablet formulation for children and the only PI licensed for children aged < 3 years in Europe. It must be given twice daily 81. ATV/r, a once‐daily PI, is licensed for children aged > 6 years in Europe, and is the preferred boosted PI in this age group. DRV/r is licensed for children aged > 3 years in the USA (treatment‐naïve and treatment‐experienced) and Europe (treatment‐experienced aged > 3 years; treatment‐naïve > 12 years). Paediatric studies with once‐daily administration of DRV/r have not been conducted in children aged 6–12 years. It is therefore recommended that DRV/r be used twice daily in children aged < 12 years while awaiting new data. In view of this, we recommend DRV/r as an alternative first‐line PI in children in this age group (6–12 years), especially when there is known transmitted PI resistance or intolerance precluding use of other PIs. Both ATV/r and DRV/r are acceptable first‐choice PIs in older children (> 6 years old for ATV/r and > 12 years old for DRV/r) because of their favourable virological outcomes and the availability of once‐daily treatment (Table 4). Fosamprenavir/r (fAPV/r), a twice‐daily PI, is licensed for children from age 6 years in Europe. Although fAPV/r is approved by the FDA in children aged > 4 weeks, it is not recommended for children aged < 6 months because of low drug exposure in this age group 82. In view of limited paediatric experience, the adverse effect on lipids, the twice‐daily regimen and meal requirements, it is not recommended for first‐line therapy in children.
Some PIs (ATV and fAPV) may be used unboosted in special circumstances where ritonavir is not tolerated, but this is not recommended because of reduced efficacy. Where possible, doses of unboosted PIs should be monitored by TDM.
In summary, the preferred PI for children less than 6 years old is LPV/r, for children 6–12 years old it is ATV/r, and for children > 12 years old it is ATV/r or DRV/r (Table 4).
6.5. Choice of NRTI backbone
Factors to consider when choosing the dual NRTI combination:
the potential for resistance and cross‐resistance (and hence future therapy options);
tolerability and toxicity;
dosing frequency;
age‐appropriate formulations including FDCs.
3TC and ABC are the preferred first‐line NRTIs in children < 12 years of age. Superiority of ABC over ZDV has previously been reported in children 83. Starting with ABC and 3TC also has the advantage of preserving (and even increasing) susceptibility to ZDV for future options in cases of virological failure, whereas starting on ZDV may lead to accumulation of thymidine analogue‐associated mutations (TAMs) affecting subsequent susceptibility to ABC 84, 85. Both ABC and 3TC can be given once daily to children over 3 months old, which is supported by PK data in PENTA 13 and PENTA 15, and virological outcomes in ARROW 71, 86, 87, 88. 3TC and ABC both have palatable liquid formulations, and scored breakable tablets have recently become available. Children over 30 kg can be given the adult fixed dose combined pill [Kivexa (ViiV Healthcare, Middlesex, UK)]. As a consequence of inferior 96‐week virological efficacy of ABC/3TC compared with TDF/FTC in adults with VL ≥ 100 000 copies/ml 2, an ABC/3TC backbone is recommended as an alternative rather than the preferred backbone in older children with very high VLs (Table 4).
ZDV is recommended as a substitute for ABC in children with the HLA genotype B*5701, as they have an increased risk of severe hypersensitivity reactions to ABC. ZDV is also recommended alongside ABC and 3TC in infants and in young children with high VLs receiving NVP (see Section 6.3 above). It should be given twice daily. It is not recommended as first‐line treatment in other situations, but remains an important component of neonatal PEP.
TDF has been licensed for children aged > 2 years by the FDA and European Medicines Agency (EMA); however, the long‐term risk of renal and bone toxicity remains a possible concern (see Section 9 on drug toxicity). As a result of limited long‐term safety data in young children and unresolved concerns over the effects on bone and kidneys with prolonged use, TDF is recommended as a preferred first‐line NRTI only in older children (aged > 12 years or weight > 35 kg) with no underlying renal insufficiency or other risk factors significantly affecting bone and renal health. Some clinicians prefer to wait until after puberty because of uncertain effects on bone health (Table 4). TDF is also recommended as a first‐line NRTI in all children aged > 2 years with HBV coinfection (see Section 10 on coinfections). It is available in co‐formulated tablets with FTC as mentioned above.
FTC is chemically very similar to 3TC and they are interchangeable. FTC has been co‐formulated with TDF [Truvada (Gilead, Forest City, CA, USA)] and TDF and EFV (Atripla, Bristol‐Myers Squibb, New York, NY, USA and Gilead), which may be used in older children (see below). It is also included with TDF in the more recently developed FDCs based on the second‐generation NNRTI rilpivirine (RPV) [Eviplera (Gilead and Janssen Therapeutics, Titusville, NJ, USA)] and the INSTI elvitegravir (EVG) [Stribild (Gilead)].
Where possible, once‐daily and fixed dose formulations should be used to reduce pill burden and potentially improve adherence.
In view of well‐documented toxicities, stavudine (d4T) is no longer recommended for treatment of HIV‐infected children in Europe. However, its efficacy is acknowledged and it is accepted that, in rare circumstances, it may be the only option available. Didanosine (ddI) is no longer recommended for first‐line therapy because of the high risk of toxicities and the availability of safer options (see Section 9 on toxicity). Children arriving in Europe from other regions on d4T or ddI should be changed to another regimen if possible.
6.6. NRTI‐only regimens
Triple NRTI therapy has been shown to have inferior virological and immunological outcomes and a high risk of selection for resistance mutations in ART‐naïve adult patients 89. The ARROW study showed that children on a three‐NRTI maintenance regimen with 3TC, ABC and ZDV had inferior virological outcomes at week 144 and more ZDV‐related neutropenia, but no difference in disease progression or immunological outcomes when compared to NNRTI plus 3TC and ABC 71. Based on these data, triple‐NRTI regimens are not recommended except in special circumstances when significant drug interactions or toxicity risks prevent the use of NNRTI‐ or PI‐containing ART (e.g. for concomitant TB treatment; see Section 10 below).
6.7. Integrase inhibitors (INSTIs)
RAL, a twice‐daily INSTI, in combination with two NRTIs is now a first‐line options for adults 2, 5 and is licensed for treatment‐experienced children from 4 weeks of age. Dolutegravir (DTG), a second‐generation INSTI, has recently been licensed for treatment‐naïve adults and children > 12 years of age. An international paediatric trial of its use in ART‐naïve and ‐experienced children is planned (the PENTA 20 trial). EVG, a once‐daily INSTI, requires boosting with ritonavir or cobicistat. It is a component of the FDC Stribild and is now recommended as another preferred regimen for ART‐naïve adult patients in the USA and Europe 2, 5.
Given the evidence on the efficacy and safety of RAL and DTG derived from studies of treatment‐naïve adults and treatment‐experienced children 90, 91, RAL‐ and DTG‐containing regimens can now be recommended as alternative first‐line ART in children older than 12 years. In children younger than 12 years, there are no data on the use of either drug in treatment‐naïve children. Their first‐line use should therefore be restricted to specific circumstances (transmitted resistance and intolerance).
7. Adherence and HIV knowledge
Adherence to treatment is paramount and should be discussed at each clinic visit.
Every effort should be made to simplify a regimen to support adherence (e.g. using once‐daily regimens, FDCs and ‘forgiving’ regimens with higher barriers to resistance). Simple adherence aids should be used when appropriate.
Children should know of their HIV diagnosis before adolescence.
Monitoring for psychological, neurocognitive and mental health issues should be routine, allowing early supportive and therapeutic intervention.
Optimal adherence to treatment is of paramount importance for long‐term efficacy of ART, and younger children rely on caregivers to deliver this. Although there are some data on the barriers to and predictors of adherence 92, there are few studies of successful interventions to improve it 93, 94, and there is no gold standard for measuring it. Adherence can be influenced by many factors, including those related to the child/young person (e.g. developmental stage, treatment fatigue and knowledge of status), family and caregivers (e.g. relationship to the child, responsibility for adherence and caregiver beliefs), the antiretroviral regimen (e.g. convenience, palatability, formulation and toxicity), culture and society 95, 96. Some of these factors are outside the control of the treating clinician, but should be acknowledged and addressed. Factors that can be influenced by the medical team include once‐daily medication regimens, side effects, choice of formulation and route of administration (e.g. oral versus gastrostomy). A recent meta‐analysis of RCTs in adults has highlighted that once‐daily regimens and lower pill burden are associated with better adherence, the latter also being associated with better virological suppression 97.
Despite the difficulties and a lack of easy solutions, the issue of adherence should always be addressed nonjudgementally, both before and after starting children on ART. It is acknowledged that adherence issues change with age and that adherence may be particularly difficult in adolescence 98. Despite every effort to support adherence, some HIV‐infected children and young people may have difficulty with taking medications, leading to detectable VL and associated risk of poor health status, development of drug resistance and risk of onward transmission in sexually active adolescents. In these individuals, careful consideration should be given to options such as a switch to a regimen with a higher barrier to resistance, or even treatment interruption alongside ongoing education regarding HIV transmission.
Every effort should be made to simplify adherence to treatment for children and caregivers. Simple aids to adherence should be used where appropriate – including adherence apps, dossette boxes, pill diaries, text messages and phone alarms. The following adherence points should be considered before prescribing a child's antiretroviral regimen.
Is there a once‐daily regimen?
-
What is the most forgiving regimen in terms of:
timing? (Can a dose be missed or late from time to time? Does administration need to be timed with food intake? What timing best suits the family's routine?)
pill count? (Is an FDC available?)
barrier to resistance? (Should a boosted PI backbone be used?)
Are the parents on treatment, and could the same regimen be used for the child?
What are the possible side effects of the regimen – might they reduce adherence (e.g. jaundice with ATV)?
Who is taking responsibility for adherence support – within the family and the medical team?
How is adherence going to be measured (VL, drug levels, pill counts etc.)?
Before a child or young person goes home from the clinic with their new medication, an adherence support plan should be in place, with contact numbers, a review schedule, peer support plan etc. Families should be encouraged to call the clinic team if there are problems, rather than struggle unsupported.
Children's knowledge of their illness should be assessed and an age‐appropriate process of gradual knowledge‐building started. Increasingly, clinicians now address issues of disclosure at an earlier age than previously 99, with awareness that early, general discussions focusing on healthy diet and lifestyle and knowledge about the blood and immune system can provide a useful foundation for later, specific discussions about HIV. It will generally be appropriate for most children to know their HIV status (i.e. for the disease to be named) before adolescence (i.e. from age 9–10 years), although the timing of naming HIV will vary according to the young person's pre‐existing knowledge, maturity and developmental age, and the process can be initiated earlier if deemed appropriate by the family and the multidisciplinary team (reviewed in 100). This process may be delayed in children with significant cognitive/learning difficulties. Once disclosure is complete, adolescents should understand the risks of onward sexual transmission, and safe sex and contraception should be regularly addressed. Giving young people an opportunity to speak with clinic staff on their own is an important part of this process. Appropriate services for adolescents with perinatally acquired HIV infection and the management of transition of their care to adult services are discussed in Section 13.
8. Monitoring on ART
The aim of ART is to achieve an undetectable VL (< 50 copies/ml plasma) and CD4 reconstitution.
Laboratory monitoring for drug toxicity should be performed initially within 2–4 weeks of starting a new drug, then at least every 6–8 months if there are no ongoing toxicity concerns.
After starting ART, VL should be checked early (at around 1 month) to confirm that VL is decreasing (this can coincide with toxicity monitoring).
VL and CD4 count can then be monitored 3−4‐monthly once the patient has been established on treatment.
Once CD4 cells are reconstituted and VL has been < 50 copies/ml consistently for over 1 year, CD4 parameters can be monitored less frequently (every 6–8 months, i.e. at alternate clinic visits).
-
More frequent clinical and laboratory monitoring is required:
in infancy;
if adherence is poor;
soon after starting or changing therapy (e.g. Liver function tests should be performed within 2 weeks);
in the context of ongoing drug toxicity;
when giving medications with significant drug interactions with ART, such as antituberculous therapy.
Clinical and laboratory monitoring requirements for children on ART are similar to those of ART‐naïve children (see Section 3). In addition to the routine clinical, growth, development, urinalysis, vaccination status/immunity and laboratory monitoring, it is important to check specifically for adherence to therapy, side effects and the need for dose modification with changing age and weight. Other current medications should be regularly reviewed, as there is potential for ART to interact with medication obtained from other sources (e.g. oral contraceptive, inhaled steroids and antacids). Other medical teams who may prescribe such medication (e.g. family doctors) should be informed of the potential for drug interactions when children are started on ART. Monitoring needs to be more frequent in infancy and shortly after initiating or changing therapy, but once children are established on treatment and stable, clinic visits can be 3–4‐monthly. In order to minimize disruption to schooling, appointments can be made after school or to coincide with school holidays.
Recent modifications to adult guidelines have recommended less frequent laboratory monitoring in those on long‐term suppressive ART 2. The results of the ARROW trial in children have also shown clinical monitoring to be a safe and effective alternative to laboratory monitoring, in resource‐poor settings 71. This supports a recommendation that monitoring of CD4 count and laboratory tests for drug toxicity can safely be performed less frequently than every 3 months when a child is clinically well, has had VL < 50 copies/ml for over 1 year and is not severely immunosuppressed.
TDM of NNRTIs and PIs is available in several quality‐controlled laboratories in Europe. There are no studies to inform recommendations for routine use of TDM in children, but it may be particularly useful where there is: (1) suspicion of drug toxicity, poor adherence or drug interactions (e.g. TB treatment); (2) failure to suppress viraemia despite good reported adherence; (3) renal or hepatic dysfunction; (4) use of unlicensed dosing regimens 101, 102. More evidence is required regarding the utility of TDM in monitoring use of drugs with highly variable PK (e.g. EFV). TDM may also be considered in infants and neonates, or with new drugs where PK data are less well established. TDM is generally not indicated for NRTIs as intracellular levels of the active metabolite are difficult to measure/interpret and large blood volumes are required.
One effect of starting ART may be immune reconstitution inflammatory syndrome (IRIS). This occurs within a few weeks or months after starting therapy, is often associated with an exaggerated immune response to an underlying opportunistic infection and is not specific to any drug. The diagnosis is clinical and requires the exclusion of active infection and drug toxicity. Symptoms can be severe and may need treatment with anti‐inflammatory drugs (e.g. steroids) and additional therapy for underlying opportunistic infection. In the majority of cases ART should be continued.
9. Drug toxicities and interactions
Toxicities depend on the individual drugs and ART combination and should be assessed at each clinic visit.
Drug interactions should be considered when starting new medications in a child on ART. Use http://www.hiv‐druginteractions.org/ to check drug interactions and toxicities.
See Table 5 for common ART‐associated toxicities.
Drug toxicity has been a major limitation of ART to date, and one of the aims of modern ART regimens is to reduce side effects. Side effects can be acute, appearing early (usually within days to a few weeks after initiation), or late, declaring themselves after prolonged use of particular antiretrovirals.
Acute toxicities can be caused by any antiretroviral and include nausea, diarrhoea, headache, rash [mild to severe dermatological manifestations, such as Stevens−Johnson syndrome (SJS)], liver dysfunction (from asymptomatic elevation of liver enzymes to drug‐induced hepatitis) and allergic reactions (Table 5). If severe events occur, ART should be discontinued with a subsequent replacement of drugs suspected to have caused the reaction (see Section 11 on switching). Some side effects, such as vomiting and diarrhoea, are transient and tend to resolve with time, but close monitoring and supportive/symptomatic treatment may be required.
Early adherence may be affected by common side effects that cause significant disturbance to daily life – for example diarrhoea caused by LPV/r, dysphoria caused by EFV, and nausea or headache caused by ZDV. Patients and their carers should be counselled appropriately prior to the start of ART and necessary support and close liaison should be assured during the first few weeks.
In the longer term, specific organ dysfunction, haematological complications and metabolic disturbances including mitochondrial toxicity, bone mineral loss, lipodystrophy, elevated cholesterol and triglycerides, and altered glucose homeostasis may occur. A recent cohort study of European children reported that nearly half had signs of fat redistribution and nearly a quarter had an abnormal lipid profile 103. Risk factors included advanced HIV disease, and use of d4T, NNRTIs, PIs and triple‐class ART. The relationship between lipodystrophy and ART is complex because of multiple exposures to different antiretrovirals and physiological changes in body composition during childhood and adolescence. Late side effects should be monitored for and addressed appropriately at every visit (see Section 8).
A number of studies have documented increased rates of cardiovascular and cerebrovascular disease in HIV‐infected adults, which appear to be multifactorial and relate both to HIV disease itself and ART, with the greater risk associated with PIs and ABC 104, 105. Several mechanisms have been postulated, including metabolic and lipid derangement (especially related to PI), insulin resistance, direct vascular injury and increased inflammation 106. Some studies in children have shown an increase in markers of cardiovascular risk such as immune activation markers, carotid intima‐media thickness (IMT) and carotid‐radial pulse‐wave velocity 107, 108, 109. Other adverse effects related to specific organ dysfunction, such as CNS disorders, renal abnormalities, hepatitis or bone loss, may also be attributable to either ART or HIV itself. The SMART trial in adults has been important in demonstrating that treatment with ART results in fewer such problems than withholding drugs 51.
9.1. Common toxicities with antiretrovirals used in children
NNRTIs
The main toxicity of EFV is neuropsychological symptoms (e.g. bad dreams, mood swings, drowsiness, dizziness, impaired learning and depression) which anecdotally may be worse in older children (possibly secondary to a reporting bias). This should be borne in mind when considering which drug to use in children with established psychological/neurological disturbance. Other complications associated with EFV are dyslipidaemia, abnormal fat distribution 103 and gynaecomastia.
NVP may be associated with skin rash, hepatitis and SJS, which typically occur within the first few months of exposure. Rash and hepatic dysfunction are less common when a 2‐week half‐dose lead‐in dosage is used. NVP should not be used in children with liver dysfunction or co‐administration of other hepatotoxic drugs. Prolonged‐release tablet formulations are available for children > 6 years old and produce a more even drug exposure, but are not suitable for the 14‐day lead‐in phase when starting NVP. Rash is much less common with EFV than with NVP; however, there are insufficient data to recommend switching from NVP to EFV in cases of dermatological hypersensitivity reactions, as repeat rash after substitution has been reported to occur in more than 12% of adult patients 110.
PIs
PIs are associated with dyslipidaemia and lipodystrophy 103, 111, 112, 113. However, once‐daily PIs, such as ATV/r and DRV/r, in adults tend to cause fewer lipid abnormalities 114, 115, 116. Measures to improve dyslipidemia can include lifestyle modifications (diet and exercise) and change of ART to ‘lipid friendly’ antiretrovirals (NVP, ATV/r, DRV/r and RAL). In rare cases when no effect is seen from ART substitution, lipid‐lowering agents, such as statins, may be used. However, in view of their frequent side effects and drug interactions with antiretrovirals, this ideally should be done in consultation with experts in ART and lipid agents.
The common side effect of ATV in children is hyperbilirubinaemia, which was reported in 45% of treated children in clinical studies 117. It is not associated with elevation of liver enzymes, and often improves with time. In cases when hyperbilirubinaemia is significant and jaundice is noticeable by the patient's peers, consideration should be given to substitution of the drug with an alternative agent.
NRTIs
A rare but clinically important early side effect is ABC hypersensitivity associated with the presence of HLA B*5701.This may be fatal if the drug is reintroduced after a reaction. Possible cardiovascular toxicity of ABC in adults remains controversial. A meta‐analysis of RCTs did not find an increased cardiovascular risk 118 and the biological mechanism of possible cardiovascular toxicity of ABC has not been established. To date, no link to cardiovascular toxicity has been found in ABC‐exposed children.
TDF is associated with bone and renal toxicity, precluding its use as a preferred NRTI in younger children. Studies of TDF‐associated bone toxicity in treatment‐experienced children yielded conflicting results 119. A significant loss of bone mineral density (BMD) was observed in young and pre‐pubertal children, and in those who received higher exposures to TDF because of higher doses or concomitant use of PIs 120, 121. Reassuringly, in most children on TDF‐containing ART, BMD z‐scores after an initial decrease tend to stabilize 121, 122, 123. Although the clinical significance of delayed bone mineralization has not been established, the concern is that suboptimal bone accrual in childhood and failure to achieve expected peak bone mass could result in increased fractures in adulthood. Paediatric data on TDF renal toxicity are also controversial, showing conflicting results from no renal dysfunction to increased rates of proteinuria and hypophosphataemia 124. Extrapolating from adult studies showing that renal dysfunction is associated with increased TDF plasma levels and PI use 125, dosing accuracy and meticulous attention to monitoring for renal dysfunction in children, especially in those who are on concomitant PIs, are of particular importance.
The use of NRTIs, in particular d4T, ddI and to a lesser extent ZDV, is associated with lipoatrophy, peripheral neuropathy, lactic acidosis and other toxicities linked to mitochondrial damage. Noncirrhotic portal hypertension has been reported as a rare complication of exposure to ddI in adults 126, 127, 128 and children 129, 130, may be associated with a genetic predisposition 126 and can become evident after ddI has been discontinued. PENTA does not support the use of d4T or ddI in first‐ or second‐line ART. However, in cases of multiple resistance, if no alternatives are available, ddI may be used to construct a fully active ART regimen. It should be noted that ddI should not be used together with TDF in view of increased toxicity. ZDV, still frequently used in paediatrics, affects bone marrow, causing macrocytic anaemia and neutropenia, and rarely thrombocytopenia or bone marrow suppression with pancytopenia 131. 3TC and FTC are generally well tolerated; however, they can cause allergic reactions and constitutional symptoms.
INSTIs
RAL and DTG have a very good safety profile in adults and are well tolerated, although data on long‐term exposure are more limited. The most common adverse effects reported in adult patients are constitutional symptoms (fatigue, nausea, dizziness, insomnia and headache), rash, diarrhoea, abnormal liver function tests and raised creatinine kinase. Hypersensitivity reactions have been reported and were characterized by rash and, in some cases, hepatic failure.
Fusion and entry inhibitors
These two classes of drug are not recommended for first‐line use and are discussed in more detail in Section 11. The CCR5 receptor antagonist maraviroc (MVC) is not licensed for patients under the age of 18 years. Paediatric studies on safety and efficacy in children are ongoing 132. In adults, MVC is generally well tolerated but has been associated with hepatic toxicity, severe skin and hypersensitivity reactions and postural hypotension. The HIV fusion inhibitor enfuvirtide (T20) is almost universally associated with injection site reactions but is otherwise well tolerated apart from mild constitutional symptoms.
For detailed side effects of individual medicines, useful resources include the Electronic Medicines Compendium (http://www.medicines.org.uk/emc/), DHHS website (http://aidsinfo.nih.gov/drugs) and EMA website (http://www.ema.europa.eu/ema/). For all drugs, the effects of prolonged use for decades remain to be seen, and this will be of greater significance as more children start ART at an earlier age.
9.2. Drug interactions
It is widely known that drug−drug interactions are important in HIV treatment in adults; this is true for children as well, although the frequency and type of comorbidities and related co‐medication may be different. ART can both be affected by drug interactions (i.e. the plasma concentrations of the ART may be changed) and/or be the cause of a drug interaction (i.e. the ART influences the plasma concentration of another drug). Increased toxicity or therapy failure may occur.
Many of the drug interactions occur as the result of drug‐induced modification of cytochrome P450 (CYP450) enzyme activity, although other systems, such as membrane transporters [e.g. P‐glycoprotein (P‐gp) or UDP‐glucuronyltransferase (UGT)], may be relevant too.
ART as the cause of a drug interaction
There are a few general rules to remember: PIs are generally inhibitors of CYP450 3A (CYP3A and other isoenzymes) or P‐gp and can lead to increased levels of co‐administered medication. This is, for instance, the case with inhaled corticosteroids, oral contraceptives, benzodiazepines, statins, antidepressants, etc. Both ritonavir and cobicistat are specifically used together with PIs or EVG for their ability to strongly inhibit liver enzymes and increase blood concentration of their co‐administered antiretroviral. The NNRTIs NVP, EFV and etravirine (ETR) are enzyme inducers. They generally reduce the plasma levels of any agents metabolized via the CYP450 pathways with potentially suboptimal therapeutic response.
Drug interactions affecting ART
The greatest risk for ART in terms of drug interaction is when antiretroviral drugs are combined with other enzyme inducers such as rifampicin, carbamazepine and phenytoin. Lower plasma concentrations of the ART can lead to the development of resistance and treatment failure.
Management of drug interactions
In the large majority of cases, drug interactions can be managed by selecting the appropriate co‐medication or change in ART. TDM of the ART or co‐medication might be helpful. We recommend referring to http://www.hiv‐druginteractions.org to assess potential drug interactions with ART, and that a pharmacist is included in the multidisciplinary team.
10. Coinfections
10.1. HBV and HCV
Untreated HIV infection increases the progression of liver disease in HBV or HCV coinfection.
HBV and HCV coinfections both increase the risk of hepatotoxicity with ART (especially NVP).
Drugs used to treat HBV may select for resistant HIV and vice versa.
Liver disease in children with HBV or HCV coinfection should be managed jointly with paediatric experts in viral hepatitis.
HCV coinfection is an indication for starting ART.
For HBV coinfection, if treatment of HIV is not indicated and there is no evidence of liver disease, HIV treatment should be considered but may be deferred.
10.1.1. Screening for viral hepatitis
Baseline screening
All children with HIV infection should be screened for viral hepatitis when they first present. Screening should include the following:
-
(a)
Testing for coinfection, past infection or immunity to HBV. This should include the following markers: HBV surface antigen (HBsAg), HBV core antibody (HBcAb) and HBV surface antibody (HBsAb). Children diagnosed with HBV coinfection should be screened for hepatitis delta virus (HDV) with HDV immunoglobulin M (IgM) and IgG and HDV RNA PCR.
-
(b)
Testing for coinfection with HCV. This should include HCV antibody and HCV RNA PCR when available, as rates of 3–13% of HCV‐seronegative infection have been reported in HIV‐infected adult and paediatric cohorts 133, 134, 135. HCV antigen might be used by some laboratories in screening algorithms. As data on the use of this marker in paediatric infection are lacking, HCV RNA is currently preferred.
-
(c)
Testing for immunity to hepatitis A virus (HAV) infection (HAV IgG).
Vaccination for hepatitis A and B of seronegative children should be recommended if a child is found to be seronegative and levels of HBV antibody should be assessed regularly and booster vaccinations or repeated courses given as required 21. Annual screening for HCV is recommended in adolescence and adults in case risk factors exist such as use of recreational drugs and/or sexual exposure.
Screening of infants exposed to HBV or HCV
When the mother is coinfected with HBV, screening of the exposed infant should include HBsAg, HBsAb and HBcAb at around 14–15 months of age following the last (fourth) vaccination at 1 year of age. Infants born to women coinfected with HCV should be screened for evidence of HCV. In most centres this screening includes a combination of PCR for detection of HCV RNA in infancy as well as HCV antibody at 12–18 months when maternal antibody is expected to wane. HCV RNA‐positive results in infancy do not equate to long‐term infection, and follow‐up is required as a small proportion of HCV‐monoinfected children will undergo spontaneous clearance of infection within the first 3–5 years of life 136, 137. Published data are sparse on the rates of spontaneous HCV clearance in HIV coinfected children.
Investigation of deranged liver function tests
In the case of unexplained deranged liver function tests and/or liver disease, viral hepatitis should be considered, including HAV, HBV, HCV, CMV and Epstein−Barr virus (EBV) infection. Screening should ideally include: HAV IgM and IgG, HBsAg, HBcAb, HBV DNA, HCV RNA, HEV IgM and IgG, HEV RNA, CMV IgM and IgG, and EBV serology. Alternative diagnoses for persistent transaminitis, such as nonalcoholic fatty liver disease, autoimmune hepatitis, Wilson's disease, alpha‐1 antitrypsin deficiency, coeliac disease and muscular dystrophy, should be considered.
HBV or HCV coinfection is a risk factor for hepatocellular carcinoma (HCC). HCC is rare in childhood and more data are required to evaluate the best approach to monitoring for this complication. Adult guidelines recommend monitoring for HCC with 6–12‐monthly serum alpha‐fetoprotein and liver ultrasound in all HBV‐coinfected patients and in HCV‐coinfected patients with cirrhosis 138, 139. In the absence of better monitoring methods, this guidance is endorsed for children and adolescents.
10.1.2. HBV coinfection
In adults, there is a well‐documented interaction between HIV and HBV infection, with increased rates of liver disease. Long‐term follow‐up data for coinfected children are sparse but, in view of the concern about an increased risk of liver disease progression in adult life, ART should be considered in coinfected children regardless of clinical stage or CD4 cell count. 3TC, FTC and TDF have activity against HIV and HBV. They therefore should only be administered as part of fully active ART in order not to select for HIV resistance. In addition, treatment with 3TC or FTC without TDF may quickly select HBV resistance to these drugs and also reduce sensitivity to entecavir, thus compromising future HBV treatment options 140. Combinations of TDF with FTC or 3TC have been shown to increase HBV viral clearance in HBV‐coinfected adults 139. We recommend that two active drugs against HBV (TDF/3TC or TDF/FTC) should be given, unless this is not possible because of prior treatment that has selected for resistant HBV strains. In children > 2 years of age, we recommend starting TDF/3TC or TDF/FTC. In children with HBV infection under 2 years of age, for whom TDF is not yet licensed, if treatment for HBV infection is not required (see treatment algorithm for paediatric patients with chronic hepatitis B infection 141), there are two options: either starting (1) 3TC/FTC‐sparing ART (unless HLA B*5701 positive this would be ABC/ZDV) in order to avoid 3TC/FTC monotherapy and selection for HBV resistance or (2) TDF‐containing ART (off‐licence TDF in this age group) with careful monitoring of TDF bone and renal toxicity.
Children with evidence of past HBV infection (HBsAg negative and HBcAb positive) are at risk of reactivation and ideally ART should include an agent active against HBV. The clinical implications of the rare phenomenon of occult hepatitis B (HBsAg negative, with low replication in the liver and plasma; usually HBV DNA well below 1000 IU/ml) are not clear. These patients should also be treated with ART that contains an agent active against HBV.
Some anti‐HBV drugs (such as entecavir and telbivudine) have partial activity against HIV, which is insufficient to fully suppress HIV but is sufficient to select for resistance mutations. These drugs should not be used to treat HBV infection in HIV‐coinfected patients without an accompanying fully suppressive ART regimen. In an HBV‐coinfected child on ART who develops TDF toxicity or intolerance, the options are very limited. Extrapolating from adult guidelines 139, entecavir as an add‐on drug to fully suppressive ART can be used in older children with TDF intolerance. Children with HBV coinfection who discontinue anti‐HBV antivirals are at risk of HBV reactivation and need to be monitored closely with clinical reviews and liver function tests. Seek expert advice for the appropriate management of HBV‐coinfected children.
10.1.3. HCV coinfection
HCV coinfection also increases the risk of liver disease, but long‐term follow‐up data for coinfected children are sparse. No anti‐HIV ART drugs are effective against HCV. In older children and adults, HCV coinfection is associated with more rapid HCV progression 142, 143 and may also have an adverse effect on HIV progression 142, and therefore early HIV treatment is recommended. Currently in children only combination treatment with pegylated interferon (PEG‐IFN) and ribavirin can be used to treat HCV infection. New direct‐acting antiviral agents (DAAs) against HCV have been approved for treatment of liver disease in adults, and phase III trials of many other compounds are under way. It is anticipated that PEG‐IFN‐free regimens will be available for adults and children in the near future, particularly for genotype 1 and 4 infection.
In children with HCV coinfection, debate is ongoing as to whether clinicians should treat HCV infection immediately or wait for better treatment options. Some experts prefer starting treatment earlier as there is better treatment response and tolerability of PEG‐IFN and ribavirin in children compared with adults 144, 145. This may be especially relevant for patients with HCV genotypes 2 and 3, for whom sustained virological response with PEG‐IFN and ribavirin over 24 weeks is much better than for genotype 1 (sustained virological response in HCV‐monoinfected children 89–93% 146, 147. However, PEG‐IFN has an adverse effect on growth in children, and the risk−benefit ratio should be carefully considered during growth spurts 148. Some specific interactions to consider include:
ribavirin enhances intracellular phosphorylation of ddI; fatal lactic acidosis has been described, and therefore ddI should be avoided;
ribavirin and ZDV both may cause anaemia;
ABC may reduce ribavirin efficacy;
d4T may cause mitochondrial (liver) toxicity;
ATV may increase hyperbilirubinaemia, but there is no clear evidence that this is worse in HCV‐coinfected children.
Seek expert advice for the appropriate management of HCV‐coinfected patients.
10.2. TB coinfection
All HIV‐infected children exposed to an individual with infectious TB and all children with evidence of latent TB infection should have preventive TB treatment (once active TB disease has been excluded).
In children with active TB disease, TB treatment should be started at TB diagnosis. ART should be started as soon as practicable, and within 2 and 8 weeks of TB treatment in children with severe and moderate immunosuppression, respectively. ART may be deferred at higher CD4 counts until TB treatment is completed.
There is significant interaction between ART and TB therapy. TDM, where available, should be used in the context of potential significant interactions.
Children with TB coinfection should be managed in consultation with an expert in the treatment of paediatric TB. A specialist in DRTB should be involved in the management of DRTB contacts and cases.
See Table 6 for ART choices in children with TB.
HIV‐infected children are at increased risk of acquiring TB infection and progression from latent to active TB compared with HIV‐negative children 149, 150. In the combination ART era, in both high and low TB prevalence countries, TB incidence is decreasing, but remains substantially higher in HIV‐positive children 151, 152, 153, 154.
10.2.1. Management of children with TB exposure and latent TB infection
All HIV‐infected children should be screened for TB infection at HIV diagnosis with clinical examination, TST or IGRA and chest X‐ray (see Section 3). Preventive TB treatment has been shown to effectively reduce the incidence of TB disease in HIV‐positive children exposed to an infectious TB source case 155. We recommend that all HIV‐infected children exposed to an individual with infectious TB and all those with evidence of latent TB infection (positive TST or IGRA) and no clinical or radiological signs suggestive of TB disease should have preventive TB treatment.
A Cochrane review of preventive regimens in HIV‐infected individuals aged over 13 years found no difference in incidence of TB disease between a 6‐month regimen with isoniazid monotherapy and 3 months of treatment with co‐administered isoniazid and rifampicin. However, the combination regimen was associated with more discontinuations because of adverse effects 156. A few paediatric studies have shown that a 3‐month regimen of isoniazid and rifampicin treatment is safe and effective 157, 158. Therefore, we recommend that children with HIV infection who have been exposed to TB or have latent TB infection and who are not receiving ART can be treated with either 3 months of isoniazid and rifampicin or 6−9 months of isoniazid (according to local policy in HIV‐uninfected children); for those on ART, in view of drug interactions, the preferred preventive treatment is 6–9 months of isoniazid. For children exposed to drug‐resistant TB, preventive therapy should be decided on an individual basis in consultation with a TB expert. Preventive treatment should only be administered once active TB has been excluded. Children with nonspecific chest X‐ray changes, and no improvement after the course of treatment for their suspected underlying respiratory illness (lymphocytic interstitial pneumonia, chest infection), should be treated for possible TB disease (see below).
10.2.2. TB disease in HIV‐infected children
There are particular difficulties relating to TB diagnosis, drug interactions and immune reconstitution disease in HIV‐infected children coinfected with TB. Negative results of a TST or IGRA cannot be used to rule out TB disease 159, 160. Every effort should be made to make a confirmatory microbiological diagnosis but this is often not possible. New technologies such as Genexpert (Cepheid, California, USA) can expedite diagnosis while providing early information on rifampicin sensitivity 161. Anti‐TB treatment should always be started at TB diagnosis. All HIV‐infected children diagnosed with TB should also be started on ART; however, the optimal timing of ART initiation depends on the degree of immunocompromise. Adult RCTs showed significant reduction in mortality and progression to AIDS with earlier ART in patients with CD4 counts < 50 cells/μl (Starting ART at 3 Points in TB (SAPIT) 162, Cambodian Early versus Late Introduction of Antiretrovirals (CAMELIA) 163 and Immediate Versus Deferred Start of Anti‐HIV Therapy in HIV‐Infected Adults Being Treated for Tuberculosis (STRIDE) 164). A retrospective study in South African children, most of whom were severely immunocompromised with median CD4 percentage < 12%, also showed that delay of ART for longer than 2 months was associated with increased mortality and worse virological response 165. Therefore, children with severe immunosuppression should start ART within 2 weeks of beginning TB treatment, and those with moderate immunosuppression within 8 weeks. Data in adults suggest that delayed ART in patients with no or mild immunocompromise is not associated with worse outcomes. The potential benefits of delayed ART in this group are decreased pill burden and drug interactions, which may lead to better drug tolerance and adherence, as well as reduced occurrence of IRIS. There are insufficient data in children with no or mild immunocompromise but, based on the results of adult studies, delaying ART until TB treatment is well tolerated (or even completed) can be considered. The optimal time for ART initiation in the context of TB meningitis remains to be determined.
In the absence of TB drug resistance, standard TB treatment with isoniazid, rifampicin, pyrazinamide and ethambutol is recommended. Rifampicin, a potent CYP3A4 inducer, has significant interactions with other medicines metabolized through CYP450 enzymes, reducing their blood levels. Considerable interaction occurs when rifampicin is co‐administered with NVP or PIs, whereas interaction with EFV is less significant and achieving therapeutic levels is possible without dose alteration. If available, rifabutin may be used instead of rifampicin to reduce drug interactions. In settings where TDM is available, dose adjusting of antiretrovirals and rifampicin/rifabutin is recommended. The choice of ART in children co‐treated for TB depends on the child's age, whether the child is receiving ART or starting ART, history of previous ART exposure and availability of TDM (Table 6).
In ART‐naïve children less than 3 years of age on TB therapy, it is recommended to start with LPV/r‐based ART with added ritonavir to achieve an LPV/ritonavir ratio of 1:1 (superboosting) 166, 167. Alternatively, ART‐naïve children under 3 years of age can be started on NVP‐based ART but without lead‐in dose and at the maximum recommended dose (200 mg/m2 twice daily). A PK study in children aged < 3 years co‐treated for TB with rifampicin showed suboptimal exposure to NVP on a 300–400 mg/m2 daily dose 168. In order to obtain better exposure in younger children, the maintenance dose can be further increased by 20–30% 2 weeks after starting treatment (H. Lyall and S. Welch, unpublished data). The doses of both rifampicin and NVP can be further adjusted with the results of TDM. NVP should be decreased back to a regular 300–400 mg/m2 daily dose 1–2 weeks after stopping rifampicin. In settings where TDM is not available, superboosted LPV/r‐based ART is a preferable option in children under 3 years of age initiating ART. Recently, EFV sprinkles were approved by the FDA for children aged 3 months to 3 years 74, 75. Further PK, efficacy and safety studies in children < 3 years old receiving anti‐TB treatment are under way. Until the results are available, the use of EFV sprinkles in younger children receiving treatment for TB cannot be widely recommended.
In children aged below 3 years who are already on ART, the options for adjusting doses of antiretrovirals discussed above are applicable. A recent RCT 71 showed that a triple‐NRTI maintenance regimen (ABC + 3TC + ZDV) is immunologically and clinically similar to NNRTI‐based ART and can be valuable in children with controlled HIV infection who develop TB, and is an alternative regimen.
For children aged over 3 years initiating ART, the preferred regimen is EFV‐based ART. Children aged > 3 years who have been receiving NVP should be switched to EFV. If EFV cannot be used (because of NNRTI resistance, neurocognitive problems or suboptimal predicted adherence), an alternative option is superboosted LPV/r as above or other PI‐based ART. For PI‐based ART, TDM should be used where available.
Children aged over 3 years who have been receiving PI‐based ART can continue on their regimen with adjustment of PI doses as above, or they can be switched to EFV. A triple‐NRTI regimen is another alternative option.
RAL has been shown to be an effective option in the treatment of HIV/TB‐coinfected adults 169. There are no data available for coinfected children. RAL cannot therefore be routinely recommended as a treatment option at present; however, its use can be considered with specialist advice and TDM. Data on the use of DTG in this context are awaited. The duration of TB treatment depends on the response to TB, which in turn depends on the degree of immunocompromise and the extent of TB disease. There are no comparative studies suggesting that the duration of TB treatment should be prolonged in HIV‐infected children. In uncomplicated TB, the duration of treatment should be the same as in non‐HIV‐infected children; however, if at the end of the treatment there is an incomplete response, then TB treatment may be extended 170. Adherence, drug levels, drug resistance and IRIS should be adequately addressed.
Drug‐resistant TB should be managed in conjunction with an expert. It should be treated with a TB regimen chosen according to bacterial drug resistance and national TB guidelines. Care needs to be taken to anticipate potential drug interactions and cumulative toxicities between ART and the TB regimen; the choice of ART may be simpler in TB regimens that do not contain rifampicin.
TB‐associated immune reconstitution inflammatory syndrome (TB‐IRIS) is not uncommon in children starting ART 171, 172, 173, 174, 175. TB‐IRIS usually develops within 3 months of starting ART, with higher risk in those patients with advanced HIV disease, low pre‐ART CD4 count and shorter interval to starting ART after initiation of TB treatment 175, 176, 177. ART should be continued, but steroids may be necessary to manage IRIS. Further details on management of TB in HIV‐infected children can be found in the WHO and The International Union Against Tuberculosis and Lung Disease (IUATLD) guidelines 6, 170, 178, 179.
10.2.3. Opportunistic infections
The management of children presenting with opportunistic infections can be complex, especially when a child presents very unwell. These patients should ideally be managed on a case‐by‐case basis. Generally it is recommended that ART should be initiated as early as possible, apart from in the context of cryptococcal meningitis, where a single RCT in adults has shown that delaying ART may be associated with reduced mortality 180.
11. When to switch, resistance testing and second and subsequent ART regimens
ART regimens may be changed because of treatment failure or toxicity, or during successful treatment for simplification.
11.1. Virological failure – second and subsequent regimens
Switching to second‐line therapy following virological failure should occur early (VL > 1000 copies/ml) for those failing on combinations including drugs with a low genetic barrier to resistance (NNRTIs or RAL).
Where there are blips in VL (detectable VL < 400 copies/ml), blood tests should be repeated within 4 weeks to confirm re‐suppression.
Reinforcement of adherence support, as the main reason for treatment failure, should be prioritized. Switching treatment when there are ongoing problems with adherence may lead to loss of efficacy of further classes of ART.
Table 7 summarizes potential strategies for choosing second‐line therapy. If the suggested options are not applicable, seek expert advice.
Virological failure is almost always attributable to poor adherence. In adult practice, ART is switched early if there is detectable viraemia, because of the risk of accumulation of further resistance mutations, which may make subsequent regimens less effective. PENPACT‐1 randomized ART‐naïve children to two NRTIs with either an NNRTI or a boosted PI. Those experiencing virological failure were randomized to switch to second‐line therapy at VL > 1000 copies/ml or > 30 000 copies/ml. Delayed switching (VL > 30 000 copies/ml) on NNRTI‐based ART was not associated with poorer clinical outcome but was associated with increased rates of NRTI resistance. Rates of NNRTI resistance were similar irrespective of VL at the time of switching, with NNRTI resistance being acquired early in virological failure. Children failing therapy on boosted PIs showed very low rates of PI and NRTI resistance irrespective of the timing of the switch to second‐line therapy. PENPACT‐1 is the only paediatric RCT addressing the timing of the switch to second‐line therapy 70. Accumulation of NRTI resistance with delayed switching following VL failure on NNRTI‐based ART has also been reported in paediatric cohort studies; however, the impact of this resistance on clinical outcome has not been addressed 181, 182.
Considering the length of time they are likely to require treatment, it is important that children have not used up all ART options before adulthood. Sequencing of newer classes of antiretroviral drugs should take into account the CCR5 receptor antagonists (e.g. MVC), which are only active against viral populations that use the CCR5 co‐receptor for cell entry. The proportion of perinatally infected adolescents with R5 variants, for whom MVC is a potential option, is highly variable within cross‐sectional studies 183, 184. Co‐receptor tropism should be performed within 3 months of the proposed treatment switch that includes MVC for those with detectable viraemia. Paediatric studies of MVC are ongoing in treatment‐experienced children aged 2–18 years (http://clinicaltrials.gov/show/NCT00791700).
Of note, as in adults, single VL blips (single VL values > 50 but < 400 copies/ml that subsequently return to < 50 copies/ml) do not predict subsequent virological failure, but these should always be followed up as soon as possible, to make sure that they are not the beginning of significant viral rebound 185.
In children, switching therapy after virological failure should only be considered when adherence has been reviewed. Failure of an NNRTI‐based regimen is often a result of viral drug resistance following poor adherence, and switching to a boosted PI is appropriate. In resource‐rich settings, those failing NNRTI‐based regimens should not continue on NNRTI‐based ART, to avoid the accumulation of further NRTI and NNRTI mutations which will impact on future ART options. Two strategies are available; either a direct switch to PI‐based therapy or a short period off ART, if clinical status and CD4 count permit, while adherence is further addressed, followed by PI‐based ART.
Failure of a boosted PI‐based regimen is more likely to be caused by poor adherence than resistance. Children failing PI‐based regimens, without documented resistance mutations, may continue their current regimen while adherence is addressed. Simplified regimens using FDCs and once‐daily PIs should be considered. A switch to NNRTI‐based second‐line therapy is likely to result in rapid development of resistance to the new drugs and is not recommended if adherence has not been addressed. If resistance is detected then switching to an alternative PI without overlapping resistance is the preferred option, with the addition of an agent from a newer class (INSTI or CCR5 receptor antagonist) dependent on the resistance profile but aiming for three active agents.
3TC and ABC may be switched to ZDV and TDF, the latter now licensed for patients from 2 years of age. For infants with first‐line failure, ddI may be substituted for TDF; however, recent case reports of noncirrhotic portal hypertension in HIV‐infected adolescents following prolonged exposure to ddI are of concern 126, 127, 128, 129, 130. Therefore, ddI exposure should be kept to a minimum with substitution of an alternative agent at the earliest opportunity.
Third and subsequent regimens are more complicated and need to take into account all previous drug histories and cumulative resistance mutations as well as the current regimen. This will always require expert virologist input. While virological failure with triple class exposure was reported in 12% of children in Europe 186, rates of triple class resistance are variable, ranging from 12 to 32% and increasing with age 187, 188. Construction of effective third‐line and subsequent regimens, ideally with at least two and preferably three fully active agents, requires expert advice. Paediatric experience of combinations that include INSTIs 189, T20 190, ETR 191, 192, MVC 132 and DRV/r is accumulating.
For patients where a suppressive regimen cannot be constructed with currently available ART, newer agents should be obtained (and may be used off‐label with expert advice) through named patient and expanded access programmes and clinical trials. Single agents should not be added to nonsuppressive regimens because of the risk of accumulating further mutations impacting on future treatment options. In this situation, nonsuppressive ART, including 3TC or FTC, may be continued to prevent further immunological decline.
As discussed above (Section 6), RAL and DTG have recently been licensed for paediatric use. ETR, a second‐generation, twice‐daily NNRTI, is licensed for treatment‐experienced children aged > 6 years. It is not fully cross‐resistant with NVP and EFV, but once more than two NNRTI mutations are present its efficacy significantly decreases 193. RPV, a once‐daily NNRTI, is licensed for patients > 18 years old. It is available as once‐daily FDC with TDF and FTC (Eviplera). Although it has fewer CNS side effects than EFV, a smaller pill size, and the possibility of co‐administration with oral contraceptives, the significant disadvantages of increased virological failure rates in patients with VL > 100 000 copies/ml and CD4 count < 200 cells/μL, higher overall rates of resistance mutations compared with EFV‐containing regimens (phase 3 TMC278‐C209 and TMC278‐C215 trials; FDA pooled analysis 194), and administration with a meal requirement preclude its use as a first‐line drug in most situations, but it can be considered in specific circumstances (e.g. patient preference or substitution for EFV intolerance). In young people with fully suppressed HIV and a regular meal pattern (absorption requires ingestion with a meal) 195, Eviplera may be considered for simplification, for example where there is intolerance to EFV‐based therapy. Ongoing studies in children will provide important information on the place of ETR and RPV in paediatric treatment strategies [IMPAACT P1090 (http://clinicaltrials.gov/show/NCT01504841); IMPAACT P1111 (http://clinicaltrials.gov/show/NCT01975012)].
Table 7 summarizes potential strategies for choosing second‐line therapy.
11.2. Resistance testing
Resistance testing should be performed prior to switching regimens when there is virological failure. Resistance testing should be undertaken while the patient is still on the failing regimen. If this is not possible, ideally test for resistance within 4 weeks of stopping the failing regimen.
Resistance testing may include reverse transcriptase/protease/integrase/V3 loop/envelope sequencing.
The interpretation of resistance results can be guided by the Stanford HIV Drug Resistance Database (http://hivdb.stanford.edu/).
Substituting single drugs in a failing regimen without prior resistance testing is not recommended.
Adding or substituting single drugs in a failing ART regimen risks giving the new drug as effective monotherapy, which may result in rapid development of further resistance. It is therefore recommended that all changes in therapy with detectable viraemia be preceded by a resistance test. Ideally, resistance testing should be performed on a sample taken while the patient was still on the old regimen, or within a few weeks of stopping, when mutant quasispecies of HIV are most likely to be detected. Standard resistance testing includes sequencing of reverse transcriptase and protease; however, those failing on INSTIs and T20 require sequencing of integrase and envelope, respectively. Children failing on CCR5 receptor inhibitors require tropism assay/V3 loop sequencing to assess whether dominant viral variant remains R5‐tropic. Expert opinion should be sought in interpreting resistance genotypes. Guidance relating to specific mutations is beyond the scope of these guidelines. The Stanford database (http://hivdb.stanford.edu/) is a useful, regularly updated resource. Resistance testing results should be discussed with an expert centre. If no local expertise is available then we recommend seeking advice from the PENTA network.
11.3. Simplification
Where possible, regimens should be simplified (once‐daily, fixed dose combinations), but switching to NNRTI‐based regimens or PI monotherapy is not advised if there are adherence issues.
Treatment simplification may involve reducing the number of drugs or tablets as a child becomes older, changing from twice‐ to once‐daily therapy, or changing from a boosted PI‐based regimen to an NNRTI‐based regimen once viral suppression is achieved and adherence is assured. Simplification should not be carried out with detectable viraemia because of the risk of selecting for resistance mutations. Simplification is much easier as children reach 35−40 kg as adult co‐formulations are available and once‐daily options include the following.
Three‐drug FDCs:
EFV, TDF and FTC (Atripla);
RPV, TDF and FTC (Eviplera);
EVG, cobicistat, TDF and FTC (Stribild).
Dual NRTI combinations:
TDF and FTC (Truvada);
ABC and 3TC (Kivexa).
Children virologically suppressed on twice‐daily boosted LPV‐based ART may benefit from simplification to once‐daily boosted PIs (ATV/r or DRV/r) when they are able to swallow larger tablets, typically reducing daily pill burden from five to three pills for those over 40 kg. Switching to once‐daily boosted PIs has been associated with improved lipid profiles in adult studies and now this has been demonstrated in a paediatric population 196.
Children suppressed on ART may also switch to reduce the likelihood of ART toxicity. The Nevirapine Resistance Study (NEVEREST), a randomized trial of switching NVP‐exposed infants from suppressive LPV/r‐ to NVP‐based ART (after at least 3 months of VL < 400 copies/ml), showed that this strategy can be successful in infants without evidence of transmitted NVP resistance on conventional testing 64, 197. Children switched to NVP had less long‐term dyslipidaemia and subcutaneous fat loss 112. Furthermore, the recently reported results of the NEVEREST‐3 study 198 indicate that switching children fully suppressed on LPV/r to EFV‐based ART is a safe and effective strategy in the context of prophylactic NVP exposure.
High rates of virological failure (without development of resistance limiting future treatment options) were reported in the Protease Inhibitor Versus Ongoing Triple‐therapy (PIVOT) trial of PI monotherapy in adults 199. Reducing the number of drugs below a standard three‐drug regimen is not currently recommended in children outside clinical trials. The lack of CNS penetration of such regimens is of potential concern for children with HIV encephalopathy 190, 200, 201.
12. Stopping treatment and treatment interruptions
Treatment interruptions cannot be routinely recommended and starting ART currently means lifelong therapy.
Judicious use of planned treatment interruptions may be considered in circumstances when ART needs to be stopped such as because of toxicity or adherence difficulties, while the latter is being addressed.
Stopping NNRTIs when HIV is fully suppressed requires a replacement or staggered stop to reduce the risk of developing NNRTI resistance as a result of the longer half‐life of NNRTIs. A replacement stop is preferable.
It is now clear that immune activation is a key component of HIV immunopathogenesis, and is characterized by polyclonal B‐ and T‐cell activation, increased T‐cell turnover, high levels of apoptosis and raised proinflammatory cytokines and chemokines. Immune activation drives CD4 decline, immunosenescence and immune exhaustion. The aetiology of HIV‐related immune activation is multifactorial but includes immune responses to HIV and other reactivated chronic viral infections, direct T‐cell activation by HIV components, depletion of CD4 regulatory T cells and translocation of microbial products across the gut mucosa 202.
Many studies have shown that levels of immune activation are reduced but not normalized by ART, and it is the ongoing inflammation that may underlie the noninfectious complications of HIV infection such as heart and brain disease. Indeed, the SMART trial, a large randomized CD4 cell count‐driven treatment interruption study in adults, was stopped early because of higher rates of non‐AIDS‐related complications and deaths (cardiovascular, liver and renal) in the treatment interruption arm as a result of presumed increased levels of immune activation off ART 51. Largely because of the SMART trial results, treatment interruptions are no longer recommended in adult patients.
In view of these studies in adults, studying treatment interruptions in children has been difficult. However, earlier ART initiation in children and commencement of ART in all those diagnosed during infancy increase the importance of addressing the question of whether children can undergo periods off ART safely. There are many reasons why children may have superior responses to intermittent therapy than adults. These include the much lower short‐term risk of cardiovascular complications in children, and the benefit of a more active thymus, which facilitates improved capacity for immune reconstitution.
In a paediatric feasibility trial, PENTA 11, assessing clinical, immunological and virological data for planned treatment interruption (PTI) in older children with a median age at entry of 9 years, there were no deaths or serious clinical events in the treatment interruption arm 203. By 2 years after the end of the trial, when the majority of children were back on ART, there was no difference in immunological recovery or viral suppression in the treatment interruption and continuous arms 204. Independent predictors of attaining a higher CD4 percentage after re‐start of ART were higher baseline CD4 percentage prior to PTI and higher CD4 percentage prior to re‐start of ART. Although the PENTA 11 trial showed some evidence of immune reactivation during PTI 205, this did not translate into clinical events. Reassuringly, there was no difference in height or weight gain and no difference in developmental test scores between the two arms. In addition, most carers and children reported better quality of life during PTI, and the clinicians were more likely to re‐start ART after PTI with simpler regimens and FDCs 204.
Can treatment be interrupted after starting early ART in infants? The CHER trial in South Africa showed that it is safe to stop ART at 1 or 2 years of age where clinical, virological and immunological parameters do not meet criteria for ART. However, most of the children who stopped ART at 1 or 2 years of age needed to re‐start in a median time of 33 weeks or 70 weeks, respectively 36. Nevertheless, nearly a third of children who stopped ART at 2 years of age were still off ART at the end of the 6‐year trial. Unfortunately, the study had no comparator continuous treatment arm to ascertain whether it is more advantageous to stay on continuous ART beyond 2 years of age. A small randomized trial in young Kenyan children, with a median age of 30 months, comparing continuous versus interrupted treatment after 2 years of ART, was stopped early because a high proportion required to re‐start ART: 66% by 3 months post interruption and 86% by 18 months 206. So, at present, ART interruption cannot be routinely recommended even after ART has been started in infancy.
Despite the recommendation for continuous therapy, unplanned treatment interruptions in children as a result of adherence difficulties or side effects of ART are common 207. Interruption of treatment occurred in nearly a quarter of children and young people in the Adolescent Master Protocol study in the USA 207. In the light of such frequent unplanned treatment interruptions, strategically planned treatment interruptions may be preferable. Where possible, this should be done safely to avoid development of resistance to classes of drugs that could be used again in the future. The options are a ‘staggered stop’, where the NRTI backbone is continued for at least 7–10 days after stopping the NNRTI to cover the ‘tail’, or a ‘replacement stop’, where the NNRTI is replaced by a boosted PI before discontinuing the PI and NRTIs simultaneously. There are no randomized comparisons of these strategies; however, evaluation of emergent resistance mutations in the SMART trial showed higher rates of follow‐up resistance mutations in patients undertaking a simultaneous or staggered stop compared with a replacement stop (although this did not reach statistical significance) 208. A replacement stop is therefore preferable. The EFV ‘tail’ may be longer compared with that of NVP because of the longer half‐life, and therefore the replacement stop may need to be longer (21 days of the PI and NRTIs) 209.
Further studies of treatment interruptions in adults and children may give better understanding of their place in the management of HIV infection in children and young people. The results of the Botswana/Baylor Antiretroviral Assessment trial as well as a 5‐year follow‐up on immunological and neurodevelopmental outcomes in PENTA 11 are awaited. Further research is needed to ascertain whether PTI can reduce unplanned interruptions in older children and young adults and be advantageous in terms of reduction of resistance, preservation of future treatment options and improvement of quality of life.
Might the case be different following early ART for primary infection? The results of the Short Pulse Anti Retroviral Therapy at HIV Seroconversion (SPARTAC) trial in adults, comparing short‐term ART initiated during primary HIV infection (ART for 48 weeks or 12 weeks) versus standard of care (no ART), showed that a 48‐week course resulted in delayed decline of CD4 count and lower VL off treatment as compared with standard care. Disappointingly, time off treatment was not significantly longer than the 48‐week treatment period and the increase in CD4 count on long‐term ART was similar across all three groups. In contrast to the SMART trial, there were no adverse effects on clinical outcomes 51, 210. Adult case series from prospective observational cohort studies and the results of the Agence Nationale de Recherche sur le SIDA (ANRS) Virological and Immunological Studies in Controllers after Treatment Interruption (VISCONTI) study suggest that some patients with different genetic characteristics and characteristic CD8 cell response to HIV, after prolonged treatment of primary HIV infection, can control HIV replication post treatment interruption for at least several years 211, 212. It appears that restricting the pool of HIV‐infected cells by very early treatment might decrease long‐lived viral reservoirs 213, which may be essential for successful control without therapy 212.
In addition to these adult studies, there is a case report of ART‐free ‘HIV remission’ for over 2 years in a child who was started on ART at 36 hours after birth 214, and a more recent report of an infected infant on continuous ART treatment from 4 hours of age with undetectable proviral DNA by 6 days of life 215. Although the first patient has now been reported as having detectable VL and has commenced ART 216, these reports together provide a proof of concept that in some individuals, after early treatment of primary infection, a prolonged treatment interruption may be possible with preservation of control of HIV replication. The phenomenon of post‐treatment HIV controllers is rare and treatment interruption cannot be applied to routine management of children. Extensive research is being undertaken to identify correlates for prolonged remission in post‐treatment controllers.
In summary, routine treatment interruptions cannot be recommended at present and starting ART currently means lifelong therapy. In circumstances where unplanned treatment interruption is unavoidable, it needs to be done safely, as outlined above. Strategic treatment interruption may be considered in individual circumstances of problematic adherence in order to avoid resistance and preserve future treatment options.
13. Adolescence, mental health and transition
Improved survival produced by ART, high rates of uptake of antenatal HIV screening and successful interventions reducing mother‐to‐child transmission have resulted in an aging European paediatric population, with many children born with HIV infection now transitioning to adult care. Transition has been defined as ‘the planned purposeful process that addresses the medical, psychosocial and educational/vocational needs of adolescents and young adults with chronic physical and medical conditions as they move from child‐centred to adult‐oriented health care systems’ 217. In chronic diseases of childhood no single model of transition has been shown to be superior, although planned transition programmes improve attendance, disease control, self‐management and patient and carer satisfaction. Data are emerging on the transition preferences of HIV‐infected adolescents, with lack of confidence in negotiating adult services, stigma associated with HIV and fear of ending lifelong patient−carer relationships identified as barriers to transition 218, 219, 220. Integrated paediatric and adult care in an age‐specific environment, increasing autonomy, patient‐centred timing of transition and comprehensive management explanations facilitate transition to adult care.
Data suggest that adolescents have poorer adherence to ART when compared with younger children 221. In adolescents commencing first‐line therapy, a boosted PI ‐based regimen potentially reduces the risk of accumulating resistance mutations in the event of virological failure; however, such regimens have a higher pill burden than the FDCs based on NNRTIs or EVG which have a lower genetic barrier to resistance. Adolescents who suppress on a boosted PI‐based regimen can subsequently simplify to an FDC once adherence has been established.
It is increasingly acknowledged that HIV‐infected young people have relatively high rates of mental health disorder 98, 222, 223. Whether these are higher than in other patient groups with chronic medical conditions, HIV‐exposed but uninfected siblings or well‐matched healthy controls is yet to be fully determined 222, 224, 225, 226. Nevertheless, it is essential that the multidisciplinary team at least monitor for symptoms and signs of psychological distress and mental health disorder, as children progress into adolescence and young adult life. Early and ongoing support from clinical psychologists with specialist paediatric knowledge is recommended. The possibility of other interventions including the wider family and peer support should also be considered. Negotiating adolescence with any chronic disease may be difficult, but to do so with one potentially transmissible to future partners and offspring before one has explored one's own sexuality adds a layer of complexity, often compounded by stigma and secrecy associated with HIV.
14. Pipeline and upcoming trials
There are considerable incentives and/or penalties from regulatory agencies to ensure that any new drug that might be of benefit to children must be studied in this population. This is mandatory with both the EMA, which enforces penalties for companies that do not provide a paediatric investigational plan as part of their application (or request a waiver), and the FDA, which also extends 6‐month patent protection to companies that perform requested paediatric studies (voluntary).
Companies must include PK data for all age groups of children, efficacy, tolerability, and differences in side effects. They must have stability and palatability data for formulations and demonstrate that they are able to achieve PK targets associated with efficacy in adults. Most paediatric development programmes take a staggered approach, starting with the older cohorts of children and working down in age. The studies are conducted in children as soon as there are sufficient data from studies in adults. The current paediatric antiretroviral pipeline is shown in Table 8; greater detail can be found in the annually updated i‐Base/TAG Pipeline Report (available at http://www.pipelinereport.org). Planned PENTA studies will look at two new drugs for children in the integrase inhibitor class: DTG and EVG.
Supplementary table
A supplementary table is provided with a practical guide to current PENTA ART drug dosing recommendations (Supplementary Table S1).
Supporting information
Table S1 PENTA dosing recommendations for currently licensed ART.
Acknowledgements
The authors would like to thank Deepak Patel for his contribution to the development of the drug‐dosing table. PENTA activities leading to production of this guideline have received funding from the European Union Seventh Framework Programme (FP7/2007‐2013) under grant agreement n° 260694.
References
- 1. PENTA Steering Committe , Welch S, Sharland M et al PENTA 2009 guidelines for the use of antiretroviral therapy in paediatric HIV‐1 infection. HIV Med 2009; 10: 591–613. [DOI] [PubMed] [Google Scholar]
- 2. Williams I, Churchill D, Anderson J et al British HIV Association guidelines for the treatment of HIV‐1‐positive adults with antiretroviral therapy 2012 (Updated November 2013. All changed text is cast in yellow highlight.). HIV Med 2014; 15 (Suppl 1): 1–85. [DOI] [PubMed] [Google Scholar]
- 3. European AIDS Clinical Society . EACS guidelines. 2013. Available at http://www.eacsociety.org/Portals/0/Guidelines_Online_131014.pdf (accessed June 2014).
- 4. Department of Health and Humas Services . Panel on antiretroviral therapy and medical management of HIV‐infected children. Guidelines for the use of antiretroviral agents in pediatric HIV infection. 2014. Available at http://aidsinfo.nih.gov/contentfiles/lvguidelines/pediatricguidelines.pdf (accessed June 2014).
- 5. Department of Health and Humas Services . Guidelines for the use of antiretroviral agents in HIV‐1‐infected adults and adolescents. 2014. Available at http://aidsinfo.nih.gov/contentfiles/lvguidelines/adultandadolescentgl.pdf (accessed June 2014).
- 6. WHO . Consolidated guidelines on the use of antiretroviral drugs for treating and preventing HIV infection: recommendations for a public health approach. 2013. Available at http://apps.who.int/iris/bitstream/10665/85321/1/9789241505727_eng.pdf?ua=1 (accessed June 2014). [PubMed]
- 7. Sabin CA, Cooper DA, Collins S, Schechter M. Rating evidence in treatment guidelines: a case example of when to initiate combination antiretroviral therapy (cART) in HIV‐positive asymptomatic persons. AIDS 2013; 27: 1839–1846. [DOI] [PubMed] [Google Scholar]
- 8. Nesheim S, Palumbo P, Sullivan K et al Quantitative RNA testing for diagnosis of HIV‐infected infants. J Acquir Immune Defic Syndr 2003; 32: 192–195. [DOI] [PubMed] [Google Scholar]
- 9. Dunn DT, Brandt CD, Krivine A et al The sensitivity of HIV‐1 DNA polymerase chain reaction in the neonatal period and the relative contributions of intra‐uterine and intra‐partum transmission. AIDS 1995; 9: F7–11. [DOI] [PubMed] [Google Scholar]
- 10. Delamare C, Burgard M, Mayaux MJ et al HIV‐1 RNA detection in plasma for the diagnosis of infection in neonates. The French Pediatric HIV Infection Study Group. J Acquir Immune Defic Syndr Hum Retrovirol 1997; 15: 121–125. [DOI] [PubMed] [Google Scholar]
- 11. Lee BE, Plitt SS, Jayaraman GC et al Use of quantitative HIV RNA detection for early diagnosis of HIV infection in infants and acute HIV infections in Alberta, Canada. J Clin Microbiol 2012; 50: 502–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Burgard M, Blanche S, Jasseron C et al Performance of HIV‐1 DNA or HIV‐1 RNA tests for early diagnosis of perinatal HIV‐1 infection during anti‐retroviral prophylaxis. J Pediatr 2012; 160: 60–66. e61. [DOI] [PubMed] [Google Scholar]
- 13. Bhowan K, Sherman GG. Performance of the first fourth‐generation rapid human immunodeficiency virus test in children. Pediatr Infect Dis J 2013; 32: 486–488. [DOI] [PubMed] [Google Scholar]
- 14. Swanson P, de Mendoza C, Joshi Y et al Impact of human immunodeficiency virus type 1 (HIV‐1) genetic diversity on performance of four commercial viral load assays: LCx HIV RNA Quantitative, AMPLICOR HIV‐1 MONITOR v1.5, VERSANT HIV‐1 RNA 3.0, and NucliSens HIV‐1 QT. J Clin Microbiol 2005; 43: 3860–3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Payne H, Mkhize N, Morris L et al Routine Antibody tests have no place in determining HIV status after early ART: evidence from CHER. Conference on Retrovirus and Opportunistic Infections . Boston, USA, 2014.
- 16. Judd A, Ferrand RA, Jungmann E et al Vertically acquired HIV diagnosed in adolescence and early adulthood in the United Kingdom and Ireland: findings from national surveillance. HIV Med 2009; 10: 253–256. [DOI] [PubMed] [Google Scholar]
- 17. British HIV Association, Children's HIV Association . ‘Don't foget the children’ Guidance for the HIV testing ofchildren with HIV‐positive parents. 2009. Available at http://www.bhiva.org/documents/Guidelines/Dont%20Forget%20the%20Children/DFTC.pdf (accessed June 2014).
- 18. Delaugerre C, Chaix ML, Blanche S et al Perinatal acquisition of drug‐resistant HIV‐1 infection: mechanisms and long‐term outcome. Retrovirology 2009; 6: 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wittkop L, Gunthard HF, de Wolf F et al Effect of transmitted drug resistance on virological and immunological response to initial combination antiretroviral therapy for HIV (EuroCoord‐CHAIN joint project): a European multicohort study. Lancet Infect Dis 2011; 11: 363–371. [DOI] [PubMed] [Google Scholar]
- 20. Mallal S, Phillips E, Carosi G et al HLA‐B*5701 screening for hypersensitivity to abacavir. N Engl J Med 2008; 358: 568–579. [DOI] [PubMed] [Google Scholar]
- 21. Menson EN, Mellado MJ, Bamford A et al Guidance on vaccination of HIV‐infected children in Europe. HIV Med 2012; 13: 333–336. e331–314. [DOI] [PubMed] [Google Scholar]
- 22. Taylor GP, Clayden P, Dhar J et al British HIV Association guidelines for the management of HIV infection in pregnant women 2012. HIV Med 2012; 13 (Suppl 2): 87–157. [DOI] [PubMed] [Google Scholar]
- 23. Rabie H, Violari A, Duong T et al Early antiretroviral treatment reduces risk of bacille Calmette‐Guerin immune reconstitution adenitis. Int J Tuberc Lung Dis 2011; 15: 1194–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. WHO . Guidelines on co‐trimoxazole prophylaxis for HIV‐related infections among children, adolescents and adults: recommendations for a public health approach. 2006. Available at http://www.who.int/hiv/pub/guidelines/ctxguidelines.pdf (accessed June 2014).
- 25. Chintu C, Bhat GJ, Walker AS et al Co‐trimoxazole as prophylaxis against opportunistic infections in HIV‐infected Zambian children (CHAP): a double‐blind randomised placebo‐controlled trial. Lancet 2004; 364: 1865–1871. [DOI] [PubMed] [Google Scholar]
- 26. Dunn D, Newell M, Ades T, Peckham C, Maria A. CD4 T cell count as predictor of Pneumocystis carinii pneumonia in children born to mothers infected with HIV. European Collaborative Study Group. BMJ 1994; 308: 437–440. [PMC free article] [PubMed] [Google Scholar]
- 27. Thyagarajan B, Deshpande SS. Cotrimoxazole and neonatal kernicterus: a review. Drug Chem Toxicol 2014; 37: 121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. WHO . Antiretroviral therapy of HIV infection in infants and children: towards universal access. 2006. Available at http://www.who.int/hiv/pub/guidelines/art/en/index.html (accessed June 2014). [PubMed]
- 29. Caldwell M, Oxtoby M, Simonds R, Rogers M. 1994 Revised classification system for human immunodeficiency virus infection in children less than 13 years of age. MMWR Morb Mortal Wkly Rep 1994; 43 (RR‐12): 1–10. [Google Scholar]
- 30. Bwakura‐Dangarembizi M, Kendall L, Bakeera‐Kitaka S et al A randomized trial of prolonged co‐trimoxazole in HIV‐infected children in Africa. N Engl J Med 2014; 370: 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. WHO . Co‐trimoxazole prophylaxis for HIV‐exposed and HIV‐infects infants and children. 2009. Available at http://www.who.int/hiv/pub/paediatric/cotrimoxazole.pdf?ua=1 (accessed June 2014).
- 32. Manyando C, Njunju EM, D'Alessandro U, Van Geertruyden JP. Safety and efficacy of co‐trimoxazole for treatment and prevention of Plasmodium falciparum malaria: a systematic review. PLoS ONE 2013; 8: e56916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Urschel S, Ramos J, Mellado M et al Withdrawal of Pneumocystis jirovecii prophylaxis in HIV‐infected children under highly active antiretroviral therapy. AIDS 2005; 19: 2103–2108. [DOI] [PubMed] [Google Scholar]
- 34. Nachman S, Gona P, Dankner W et al The rate of serious bacterial infections among HIV‐infected children with immune reconstitution who have discontinued opportunistic infection prophylaxis. Pediatrics 2005; 115: e488–e494. [DOI] [PubMed] [Google Scholar]
- 35. Siberry GK, Abzug MJ, Nachman S et al Guidelines for the prevention and treatment of opportunistic infections in HIV‐exposed and HIV‐infected children: recommendations from the National Institutes of Health, Centers for Disease Control and Prevention, the HIV Medicine Association of the Infectious Diseases Society of America, the Pediatric Infectious Diseases Society, and the American Academy of Pediatrics. Pediatr Infect Dis J 2013; 32 (Suppl 2): i–KK4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cotton MF, Violari A, Otwombe K et al Early time‐limited antiretroviral therapy versus deferred therapy in South African infants infected with HIV: results from the children with HIV early antiretroviral (CHER) randomised trial. Lancet 2013; 382: 1555–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Violari A, Cotton MF, Gibb DM et al Early antiretroviral therapy and mortality among HIV‐infected infants. N Engl J Med 2008; 359: 2233–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Goetghebuer T, Haelterman E, Le Chenadec J et al Effect of early antiretroviral therapy on the risk of AIDS/death in HIV‐infected infants. AIDS 2009; 23: 597–604. [DOI] [PubMed] [Google Scholar]
- 39. Hainline C, Taliep R, Sorour G et al Early Antiretroviral Therapy reduces the incidence of otorrhea in a randomized study of early and deferred antiretroviral therapy: evidence from the Children with HIV Early Antiretroviral Therapy (CHER) Study. BMC Res Notes 2011; 4: 448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Madhi SA, Adrian P, Cotton MF et al Effect of HIV infection status and anti‐retroviral treatment on quantitative and qualitative antibody responses to pneumococcal conjugate vaccine in infants. J Infect Dis 2010; 202: 355–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Laughton B, Cornell M, Grove D et al Early antiretroviral therapy improves neurodevelopmental outcomes in infants. AIDS 2012; 26: 1685–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Judd A, European Pregnancy and Paediatric HIV Cohort Collaboration (EPPICC) study group in EuroCoord . Early antiretroviral therapy in HIV‐1‐infected infants, 1996–2008: treatment response and duration of first‐line regimens. AIDS 2011; 25: 2279–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chiappini E, Galli L, Tovo PA et al Five‐year follow‐up of children with perinatal HIV‐1 infection receiving early highly active antiretroviral therapy. BMC Infect Dis 2009; 9: 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Goetghebuer T, Le Chenadec J, Haelterman E et al Short‐ and long‐term immunological and virological outcome in HIV‐infected infants according to the age at antiretroviral treatment initiation. Clin Infect Dis 2012; 54: 878–881. [DOI] [PubMed] [Google Scholar]
- 45. Palumbo PE, Raskino C, Fiscus S et al Predictive value of quantitative plasma HIV RNA and CD4+ lymphocyte count in HIV‐infected infants and children. JAMA 1998; 279: 756–761. [DOI] [PubMed] [Google Scholar]
- 46. Mofenson LM, Korelitz J, Meyer WA, 3rd et al The relationship between serum human immunodeficiency virus type 1 (HIV‐1) RNA level, CD4 lymphocyte percent, and long‐term mortality risk in HIV‐1‐infected children. National Institute of Child Health and Human Development Intravenous Immunoglobulin Clinical Trial Study Group. J Infect Dis 1997; 175: 1029–1038. [DOI] [PubMed] [Google Scholar]
- 47. Dunn D, HIV Paediatric Prognostic Markers Collaborative Study Group . Short‐term risk of disease progression in HIV‐1‐infected children receiving no antiretroviral therapy or zidovudine monotherapy: a meta‐analysis. Lancet 2003; 362: 1605–1611. [DOI] [PubMed] [Google Scholar]
- 48. Sturt AS, Halpern MS, Sullivan B, Maldonado YA. Timing of antiretroviral therapy initiation and its impact on disease progression in perinatal human immunodeficiency virus‐1 infection. Pediatr Infect Dis J 2012; 31: 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. HIV Paediatric Prognostic Markers Collaborative Study Group . Predictive value of absolute CD4 cell count for disease progression in untreated HIV‐1‐infected children. AIDS 2006; 20: 1289–1294. [DOI] [PubMed] [Google Scholar]
- 50. HIV Paediatric Prognostic Markers Collaborative Study Group . Use of total lymphocyte count for informing when to start antiretroviral therapy in HIV‐infected children: a meta‐analysis of longitudinal data. Lancet 2005; 366: 1868–1874. [DOI] [PubMed] [Google Scholar]
- 51. Strategies for Management of Antiretroviral Therapy (SMART) Study Group , El‐Sadr WM, Lundgren JD et al CD4+ count‐guided interruption of antiretroviral treatment. N Engl J Med 2006; 355: 2283–2296. [DOI] [PubMed] [Google Scholar]
- 52. Dunn D, Woodburn P, Duong T et al Current CD4 cell count and the short‐term risk of AIDS and death before the availability of effective antiretroviral therapy in HIV‐infected children and adults. J Infect Dis 2008; 197: 398–404. [DOI] [PubMed] [Google Scholar]
- 53. Puthanakit T, Saphonn V, Ananworanich J et al Early versus deferred antiretroviral therapy for children older than 1 year infected with HIV (PREDICT): a multicentre, randomised, open‐label trial. Lancet Infect Dis 2012; 12: 933–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. HIV Paediatric Prognostic Markers Collaborative Study Group , Dunn DT, Castro H et al Discordance between CD4 cell count and CD4 cell percentage: implications for when to start antiretroviral therapy in HIV‐1 infected children. AIDS 2010; 24: 1213–1217. [DOI] [PubMed] [Google Scholar]
- 55. Cotugno N, Douagi I, Rossi P, Palma P. Suboptimal immune reconstitution in vertically HIV infected children: a view on how HIV replication and timing of HAART initiation can impact on T and B‐cell compartment. Clin Dev Immunol 2012; 2012: 805151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Picat MQ, Lewis J, Musiime V et al Predicting patterns of long‐term CD4 reconstitution in HIV‐infected children starting antiretroviral therapy in sub‐Saharan Africa: a cohort‐based modelling study. PLoS Med 2013; 10: e1001542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lewis J, Walker AS, Castro H et al Age and CD4 count at initiation of antiretroviral therapy in HIV‐infected children: effects on long‐term T‐cell reconstitution. J Infect Dis 2012; 205: 548–556. [DOI] [PubMed] [Google Scholar]
- 58. Buzon MJ, Martin‐Gayo E, Pereyra F et al Long‐term antiretroviral treatment initiated in primary HIV‐1 infection affects the size, composition and decay kinetics of the reservoir of HIV‐1 infected CD4 T cells. J Virol 2014; 88: 10056–10065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Persaud D, Palumbo PE, Ziemniak C et al Dynamics of the resting CD4(+) T‐cell latent HIV reservoir in infants initiating HAART less than 6 months of age. AIDS 2012; 26: 1483–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Avettand‐Fenoel V, Blanche S, Le Chenadec J et al Relationships between HIV disease history and blood HIV‐1 DNA load in perinatally infected adolescents and young adults: the ANRS‐EP38‐IMMIP study. J Infect Dis 2012; 205: 1520–1528. [DOI] [PubMed] [Google Scholar]
- 61. Lundgren JD, Babiker AG, Gordin FM, Borges AH, Neaton JD. When to start antiretroviral therapy: the need for an evidence base during early HIV infection. BMC Med 2013; 11: 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Franco RA, Saag MS. When to start antiretroviral therapy: as soon as possible. BMC Med 2013; 11: 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schomaker M, Egger M, Ndirangu J et al When to start antiretroviral therapy in children aged 2–5 years: a collaborative causal modelling analysis of cohort studies from southern Africa. PLoS Med 2013; 10: e1001555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Coovadia A, Abrams EJ, Stehlau R et al Reuse of nevirapine in exposed HIV‐infected children after protease inhibitor‐based viral suppression: a randomized controlled trial. JAMA 2010; 304: 1082–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Palumbo P, Lindsey JC, Hughes MD et al Antiretroviral treatment for children with peripartum nevirapine exposure. N Engl J Med 2010; 363: 1510–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. McArthur MA, Kalu SU, Foulks AR et al Twin preterm neonates with cardiac toxicity related to lopinavir/ritonavir therapy. Pediatr Infect Dis J 2009; 28: 1127–1129. [DOI] [PubMed] [Google Scholar]
- 67. Simon A, Warszawski J, Kariyawasam D et al Association of prenatal and postnatal exposure to lopinavir‐ritonavir and adrenal dysfunction among uninfected infants of HIV‐infected mothers. JAMA 2011; 306: 70–78. [DOI] [PubMed] [Google Scholar]
- 68. Violari A, Lindsey JC, Hughes MD et al Nevirapine versus ritonavir‐boosted lopinavir for HIV‐infected children. N Engl J Med 2012; 366: 2380–2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Penazzato M, Prendergast A, Tierney J, Cotton M, Gibb D. Effectiveness of antiretroviral therapy in HIV‐infected children under 2 years of age. Cochrane Database Syst Rev 2012; (7): CD004772. [DOI] [PubMed] [Google Scholar]
- 70. PENPACT‐1 (PENTA 9/PACTG 390) Study Team , Babiker A, Castro nee Green H et al First‐line antiretroviral therapy with a protease inhibitor versus non‐nucleoside reverse transcriptase inhibitor and switch at higher versus low viral load in HIV‐infected children: an open‐label, randomised phase 2/3 trial. Lancet Infect Dis 2011; 11: 273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ruel TD, Kakuru A, Ikilezi G et al Virologic and immunologic outcomes of HIV‐infected Ugandan children randomized to lopinavir/ritonavir or nonnucleoside reverse transcriptase inhibitor therapy. J Acquir Immune Defic Syndr 2014; 65: 535–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. ARROW Trial team , Kekitiinwa A, Cook A et al Routine versus clinically driven laboratory monitoring and first‐line antiretroviral therapy strategies in African children with HIV (ARROW): a 5‐year open‐label randomised factorial trial. Lancet 2013; 381: 1391–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Duong T, Judd A, Collins I et al Long‐term virological outcome in children on antiretroviral therapy in the UK and Ireland. AIDS 2014; 28: 2395–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bertz R, Tafoya E, Chapel S et al Population pharmacokinetics of efavirenz in pediatric patients to inform dosing in children ≥ 3 months of age. 14th International Workshop on Clinical Pharmacology of HIV Therapy . The Netherlands, 2013.
- 75. Food and Drug Administration . Sustiva prescribing information. 2013. Available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/020972s046,021360s035lbl.pdf (accessed June 2014).
- 76. Moore CB, Capparelli E, Samson P. et al, eds. CYP2B6 Polymorphisms Challenge Generalized FDA Efavirenz Dosing Guidelines in Children < 3 Yrs. Conference on Retrovirus and Opportunistic Infections . Boston, USA, 2014.
- 77. Pillay P, Ford N, Shubber Z, Ferrand RA. Outcomes for efavirenz versus nevirapine‐containing regimens for treatment of HIV‐1 infection: a systematic review and meta‐analysis. PLoS ONE 2013; 8: e68995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lowenthal ED, Ellenberg JH, Machine E et al Association between efavirenz‐based compared with nevirapine‐based antiretroviral regimens and virological failure in HIV‐infected children. JAMA 2013; 309: 1803–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kekitiinwa A, Spyer M, Katuramu R et al Virologic Response To Efavirenz vs Nevirapine‐Containing ART in the ARROW Trial. Conference on Retrovirus and Opportunistic Infections . Boston, USA, 2014.
- 80. Shubber Z, Calmy A, Andrieux‐Meyer I et al Adverse events associated with nevirapine and efavirenz‐based first‐line antiretroviral therapy: a systematic review and meta‐analysis. AIDS 2013; 27: 1403–1412. [DOI] [PubMed] [Google Scholar]
- 81. Lyall H. Final results of koncert: a randomized noninferiority trial of QD vs BD LPV/r dosing in children. Conference on Retrovirus and Opportunistic Infections . Boston, USA, 2014.
- 82. Food and Drug Administration . Lexiva prescribing information. 2012. Available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/021548s031,022116s015lbl.pdf (accessed June 2014).
- 83. Green H, Gibb DM, Walker AS et al Lamivudine/abacavir maintains virological superiority over zidovudine/lamivudine and zidovudine/abacavir beyond 5 years in children. AIDS 2007; 21: 947–955. [DOI] [PubMed] [Google Scholar]
- 84. Harrison L, Gibb D, Fiscus S et al HIV‐1 resistance after randomized virologic switch at 1000 or 30,000 c/ml in children. Conference on Retrovirus and Opportunistic Infections . Boston, USA, 2014.
- 85. Gibb DM, Walker AS, Kaye S et al Evolution of antiretroviral phenotypic and genotypic drug resistance in antiretroviral‐naive HIV‐1‐infected children treated with abacavir/lamivudine, zidovudine/lamivudine or abacavir/zidovudine, with or without nelfinavir (the PENTA 5 trial). Antivir Ther 2002; 7: 293–303. [PubMed] [Google Scholar]
- 86. Musiime V, Kendall L, Bakeera‐Kitaka S et al Pharmacokinetics and acceptability of once‐ versus twice‐daily lamivudine and abacavir in HIV type‐1‐infected Ugandan children in the ARROW Trial. Antivir Ther 2010; 15: 1115–1124. [DOI] [PubMed] [Google Scholar]
- 87. Paediatric European Network for Treatment of AIDS (PENTA) . Pharmacokinetic study of once‐daily versus twice‐daily abacavir and lamivudine in HIV type‐1‐infected children aged 3‐<36 months. Antivir Ther 2010; 15: 297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. LePrevost M, Green H, Flynn J et al Adherence and acceptability of once daily Lamivudine and abacavir in human immunodeficiency virus type‐1 infected children. Pediatr Infect Dis J 2006; 25: 533–537. [DOI] [PubMed] [Google Scholar]
- 89. DART Virology Group and Trial Team . Virological response to a triple nucleoside/nucleotide analogue regimen over 48 weeks in HIV‐1‐infected adults in Africa. AIDS 2006; 20: 1391–1399. [DOI] [PubMed] [Google Scholar]
- 90. Viani RM, Alvero C, Fenton T et al Safety, pharmacokinetics and efficacy of dolutegravir in treatment‐experienced HIV+ children. Conference on Retrovirus and Opportunistic Infections . Boston, USA, 2014.
- 91. Viani RM, Alvero C, Fenton T et al Safety and efficacy of dolutegravir in HIV treatment‐experienced adolescents: 48‐week results. Conference on Retrovirus and Opportunistic Infections . Boston, USA, 2014.
- 92. MacDonell K, Naar‐King S, Huszti H, Belzer M. Barriers to medication adherence in behaviorally and perinatally infected youth living with HIV. AIDS Behav 2013; 17: 86–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Letourneau EJ, Ellis DA, Naar‐King S et al Multisystemic therapy for poorly adherent youth with HIV: results from a pilot randomized controlled trial. AIDS Care 2013; 25: 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bain‐Brickley D, Butler LM, Kennedy GE, Rutherford GW. Interventions to improve adherence to antiretroviral therapy in children with HIV infection. Cochrane Database Syst Rev 2011; (12): CD009513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Simoni JM, Montgomery A, Martin E et al Adherence to antiretroviral therapy for pediatric HIV infection: a qualitative systematic review with recommendations for research and clinical management. Pediatrics 2007; 119: e1371–e1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Haberer J, Mellins C. Pediatric adherence to HIV antiretroviral therapy. Curr HIV/AIDS Rep 2009; 6: 194–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Nachega JB, Parienti JJ, Uthman OA et al Lower pill burden and once‐daily antiretroviral treatment regimens for HIV infection: a meta‐analysis of randomized controlled trials. Clin Infect Dis 2014; 58: 1297–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Koenig LJ, Nesheim S, Abramowitz S. Adolescents with perinatally acquired HIV: emerging behavioral and health needs for long‐term survivors. Curr Opin Obstet Gynecol 2011; 23: 321–327. [DOI] [PubMed] [Google Scholar]
- 99. Butler AM, Williams PL, Howland LC et al Impact of disclosure of HIV infection on health‐related quality of life among children and adolescents with HIV infection. Pediatrics 2009; 123: 935–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Saunders C. Disclosing HIV status to HIV positive children before adolescence. Br J Nurs 2012; 21: 663–669. [DOI] [PubMed] [Google Scholar]
- 101. Burger DM. The role of therapeutic drug monitoring in pediatric HIV/AIDS. Ther Drug Monit 2010; 32: 269–272. [DOI] [PubMed] [Google Scholar]
- 102. Rakhmanina NY, van den Anker JN, Soldin SJ et al Can therapeutic drug monitoring improve pharmacotherapy of HIV infection in adolescents? Ther Drug Monit 2010; 32: 273–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Alam N, Cortina‐Borja M, Goetghebuer T et al Body fat abnormality in HIV‐infected children and adolescents living in Europe: prevalence and risk factors. J Acquir Immune Defic Syndr 2012; 59: 314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Bavinger C, Bendavid E, Niehaus K et al Risk of cardiovascular disease from antiretroviral therapy for HIV: a systematic review. PLoS ONE 2013; 8: e59551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Islam FM, Wu J, Jansson J, Wilson DP. Relative risk of cardiovascular disease among people living with HIV: a systematic review and meta‐analysis. HIV Med 2012; 13: 453–468. [DOI] [PubMed] [Google Scholar]
- 106. Martinez E, Larrousse M, Gatell JM. Cardiovascular disease and HIV infection: host, virus, or drugs? Curr Opin Infect Dis 2009; 22: 28–34. [DOI] [PubMed] [Google Scholar]
- 107. Charakida M, Loukogeorgakis SP, Okorie MI et al Increased arterial stiffness in HIV‐infected children: risk factors and antiretroviral therapy. Antivir Ther 2009; 14: 1075–1079. [DOI] [PubMed] [Google Scholar]
- 108. Sainz T, Alvarez‐Fuente M, Navarro ML et al Subclinical atherosclerosis and markers of immune activation in HIV‐infected children and adolescents: the CaroVIH Study. J Acquir Immune Defic Syndr 2014; 65: 42–49. [DOI] [PubMed] [Google Scholar]
- 109. Di Biagio A, Rosso R, Maggi P et al Inflammation markers correlate with common carotid intima‐media thickness in patients perinatally infected with human immunodeficiency virus 1. J Ultrasound Med 2013; 32: 763–768. [DOI] [PubMed] [Google Scholar]
- 110. Mehta U, Maartens G. Is it safe to switch between efavirenz and nevirapine in the event of toxicity? Lancet Infect Dis 2007; 7: 733–738. [DOI] [PubMed] [Google Scholar]
- 111. Barlow‐Mosha L, Eckard AR, McComsey GA, Musoke PM. Metabolic complications and treatment of perinatally HIV‐infected children and adolescents. J Int AIDS Soc 2013; 16: 18600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Arpadi S, Shiau S, Strehlau R et al Metabolic abnormalities and body composition of HIV‐infected children on Lopinavir or Nevirapine‐based antiretroviral therapy. Arch Dis Child 2013; 98: 258–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Rhoads MP, Lanigan J, Smith CJ, Lyall EG. Effect of specific ART drugs on lipid changes and the need for lipid management in children with HIV. J Acquir Immune Defic Syndr 2011; 57: 404–412. [DOI] [PubMed] [Google Scholar]
- 114. Souza SJ, Luzia LA, Santos SS, Rondo PH. Lipid profile of HIV‐infected patients in relation to antiretroviral therapy: a review. Rev Assoc Med Bras 2013; 59: 186–198. [DOI] [PubMed] [Google Scholar]
- 115. Orkin C, DeJesus E, Khanlou H et al Final 192‐week efficacy and safety of once‐daily darunavir/ritonavir compared with lopinavir/ritonavir in HIV‐1‐infected treatment‐naive patients in the ARTEMIS trial. HIV Med 2013; 14: 49–59. [DOI] [PubMed] [Google Scholar]
- 116. Lu CL, Lin YH, Wong WW et al Outcomes of switch to atazanavir‐containing combination antiretroviral therapy in HIV‐1‐infected patients with hyperlipidemia. J Microbiol Immunol Infect 2011; 44: 258–264. [DOI] [PubMed] [Google Scholar]
- 117. European Medicines Agency . Reyataz: product information. 2014. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Product_Information/human/000494/WC500056380.pdf (accessed June 2014).
- 118. Cruciani M, Zanichelli V, Serpelloni G et al Abacavir use and cardiovascular disease events: a meta‐analysis of published and unpublished data. AIDS 2011; 25: 1993–2004. [DOI] [PubMed] [Google Scholar]
- 119. Puthanakit T, Siberry GK. Bone health in children and adolescents with perinatal HIV infection. J Int AIDS Soc 2013; 16: 18575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Purdy JB, Gafni RI, Reynolds JC, Zeichner S, Hazra R. Decreased bone mineral density with off‐label use of tenofovir in children and adolescents infected with human immunodeficiency virus. J Pediatr 2008; 152: 582–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Gafni RI, Hazra R, Reynolds JC et al Tenofovir disoproxil fumarate and an optimized background regimen of antiretroviral agents as salvage therapy: impact on bone mineral density in HIV‐infected children. Pediatrics 2006; 118: e711–e718. [DOI] [PubMed] [Google Scholar]
- 122. WHO . Use of tenofovir in HIV‐infected children and adolescents: a public health perspective. 2012. Available at http://apps.who.int/iris/bitstream/10665/70944/1/9789241503822_eng.pdf?ua=1 (accessed June 2014).
- 123. Sirisanthana V. 48‐week safety of tenofovir when administered according to weight‐band dosing in HIV+ children > 15 kg as part of a once‐daily HAART regimen. Conference on Retroviruses and Opportunistic Infections . Atlanta, USA, 2013.
- 124. Bhimma R, Purswani MU, Kala U. Kidney disease in children and adolescents with perinatal HIV‐1 infection. J Int AIDS Soc 2013; 16: 18596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Fernandez‐Fernandez B, Montoya‐Ferrer A, Sanz AB et al Tenofovir nephrotoxicity: 2011 update. AIDS Res Treat 2011; 2011: 354908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Vispo E, Cevik M, Rockstroh JK et al Genetic determinants of idiopathic noncirrhotic portal hypertension in HIV‐infected patients. Clin Infect Dis 2013; 56: 1117–1122. [DOI] [PubMed] [Google Scholar]
- 127. Scourfield A, Waters L, Holmes P et al Non‐cirrhotic portal hypertension in HIV‐infected individuals. Int J STD AIDS 2011; 22: 324–328. [DOI] [PubMed] [Google Scholar]
- 128. Kovari H, Ledergerber B, Peter U et al Association of noncirrhotic portal hypertension in HIV‐infected persons and antiretroviral therapy with didanosine: a nested case‐control study. Clin Infect Dis 2009; 49: 626–635. [DOI] [PubMed] [Google Scholar]
- 129. Giacomet V, Vigano A, Penagini F et al Splenomegaly and variceal bleeding in a ten‐year‐old HIV‐infected girl with noncirrhotic portal hypertension. Pediatr Infect Dis J 2012; 31: 1059–1060. [DOI] [PubMed] [Google Scholar]
- 130. Scherpbier HJ. Antiretroviral therapy associated liver disease in 4 HIV‐1 infected adolescent girls. EASL Monothematic Conference: HIV and the liver . London, UK, 2012.
- 131. European Medicines Agency . Combivir: product information. 2013. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Product_Information/human/000190/WC500032326.pdf (accessed June 2014).
- 132. Giaquinto C, Keet L, Fortuny C et al Safety and efficacy of maraviroc in CCR5‐tropic HIV‐1‐infected children aged 2 to < 18 years. 7th IAS Conference on HIV Pathogenesis, Treatment and Prevention . Kuala Lumpur, Malaysia, 2013.
- 133. Abad C, Fortuny C, Almendros P et al Progressive liver disease in patients with vertically acquired HIV/HCV co‐infection in Spain. 5th International Workshop on HIV Paediatrics . Kuala Lumpur, Malaysia, 2013.
- 134. Chamie G, Bonacini M, Bangsberg DR et al Factors associated with seronegative chronic hepatitis C virus infection in HIV infection. Clin Infect Dis 2007; 44: 577–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. George SL, Gebhardt J, Klinzman D et al Hepatitis C virus viremia in HIV‐infected individuals with negative HCV antibody tests. J Acquir Immune Defic Syndr 2002; 31: 154–162. [DOI] [PubMed] [Google Scholar]
- 136. Farmand S, Wirth S, Loffler H et al Spontaneous clearance of hepatitis C virus in vertically infected children. Eur J Pediatr 2012; 171: 253–258. [DOI] [PubMed] [Google Scholar]
- 137. Bortolotti F, Verucchi G, Camma C et al Long‐term course of chronic hepatitis C in children: from viral clearance to end‐stage liver disease. Gastroenterology 2008; 134: 1900–1907. [DOI] [PubMed] [Google Scholar]
- 138. Department of Health and Human Services . Guidelines for the prevention and treatment of opportunistic infections in HIV‐infected adults and adolescents:recommendations from the Centers for Disease Control and Prevention, the National Institutes of Health, and the HIV Medicine Association of the Infectious Diseases Society of America. 2013. Available at http://aidsinfo.nih.gov/contentfiles/lvguidelines/AdultandAdolescentGL.pdf (accessed June 2014).
- 139. Wilkins E, Nelson M, Agarwal K et al British HIV Association guidelines for the management of hepatitis viruses in adults infected with HIV 2013. HIV Med 2013; 14 (Suppl 4): 1–71. [DOI] [PubMed] [Google Scholar]
- 140. Ghany MG, Doo EC. Antiviral resistance and hepatitis B therapy. Hepatology 2009; 49: S174–S184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Sokal EM, Paganelli M, Wirth S et al Management of chronic hepatitis B in childhood: ESPGHAN clinical practice guidelines: consensus of an expert panel on behalf of the European Society of Pediatric Gastroenterology, Hepatology and Nutrition. J Hepatol 2013; 59: 814–829. [DOI] [PubMed] [Google Scholar]
- 142. Operskalski EA, Kovacs A. HIV/HCV co‐infection: pathogenesis, clinical complications, treatment, and new therapeutic technologies. Curr HIV/AIDS Rep 2011; 8: 12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Claret‐Teruel G, Noguera‐Julian A, Esteva C et al Impact of human immunodeficiency virus coinfection on the progression of mother‐to‐child transmitted hepatitis C virus infection. Pediatr Infect Dis J 2011; 30: 801–804. [DOI] [PubMed] [Google Scholar]
- 144. Mohan N, Gonzalez‐Peralta RP, Fujisawa T et al Chronic hepatitis C virus infection in children. J Pediatr Gastroenterol Nutr 2010; 50: 123–131. [DOI] [PubMed] [Google Scholar]
- 145. Mack CL, Gonzalez‐Peralta RP, Gupta N et al NASPGHAN practice guidelines: diagnosis and management of hepatitis C infection in infants, children, and adolescents. J Pediatr Gastroenterol Nutr 2012; 54: 838–855. [DOI] [PubMed] [Google Scholar]
- 146. Sokal EM, Bourgois A, Stephenne X et al Peginterferon alfa‐2a plus ribavirin for chronic hepatitis C virus infection in children and adolescents. J Hepatol 2010; 52: 827–831. [DOI] [PubMed] [Google Scholar]
- 147. Wirth S, Ribes‐Koninckx C, Calzado MA et al High sustained virologic response rates in children with chronic hepatitis C receiving peginterferon alfa‐2b plus ribavirin. J Hepatol 2010; 52: 501–507. [DOI] [PubMed] [Google Scholar]
- 148. Wirth S. Current treatment options and response rates in children with chronic hepatitis C. World J Gastroenterol 2012; 18: 99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Hesseling AC, Cotton MF, Jennings T et al High incidence of tuberculosis among HIV‐infected infants: evidence from a South African population‐based study highlights the need for improved tuberculosis control strategies. Clin Infect Dis 2009; 48: 108–114. [DOI] [PubMed] [Google Scholar]
- 150. Marais BJ, Graham SM, Cotton MF, Beyers N. Diagnostic and management challenges for childhood tuberculosis in the era of HIV. J Infect Dis 2007; 196 (Suppl 1): S76–S85. [DOI] [PubMed] [Google Scholar]
- 151. Jensen J, Alvaro‐Meca A, Micheloud D, Diaz A, Resino S. Reduction in mycobacterial disease among HIV‐infected children in the highly active antiretroviral therapy era (1997–2008). Pediatr Infect Dis J 2012; 31: 278–283. [DOI] [PubMed] [Google Scholar]
- 152. Li N, Manji KP, Spiegelman D et al Incident tuberculosis and risk factors among HIV‐infected children in Tanzania. AIDS 2013; 27: 1273–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Edmonds A, Lusiama J, Napravnik S et al Anti‐retroviral therapy reduces incident tuberculosis in HIV‐infected children. Int J Epidemiol 2009; 38: 1612–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Martinson NA, Moultrie H, van Niekerk R et al HAART and risk of tuberculosis in HIV‐infected South African children: a multi‐site retrospective cohort. Int J Tuberc Lung Dis 2009; 13: 862–867. [PMC free article] [PubMed] [Google Scholar]
- 155. Schaaf HS, Cotton MF, Boon GP, Jeena PM. Isoniazid preventive therapy in HIV‐infected and ‐uninfected children (0–14 years). S Afr Med J 2013; 103: 714–715. [DOI] [PubMed] [Google Scholar]
- 156. Akolo C, Adetifa I, Shepperd S, Volmink J. Treatment of latent tuberculosis infection in HIV infected persons. Cochrane Database Syst Rev 2010; (1): CD000171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Spyridis NP, Spyridis PG, Gelesme A et al The effectiveness of a 9‐month regimen of isoniazid alone versus 3‐ and 4‐month regimens of isoniazid plus rifampin for treatment of latent tuberculosis infection in children: results of an 11‐year randomized study. Clin Infect Dis 2007; 45: 715–722. [DOI] [PubMed] [Google Scholar]
- 158. Bright‐Thomas R, Nandwani S, Smith J, Morris JA, Ormerod LP. Effectiveness of 3 months of rifampicin and isoniazid chemoprophylaxis for the treatment of latent tuberculosis infection in children. Arch Dis Child 2010; 95: 600–602. [DOI] [PubMed] [Google Scholar]
- 159. Sester M, Sotgiu G, Lange C et al Interferon‐gamma release assays for the diagnosis of active tuberculosis: a systematic review and meta‐analysis. Eur Respir J 2011; 37: 100–111. [DOI] [PubMed] [Google Scholar]
- 160. Chiappini E, Bonsignori F, Accetta G et al Interferon‐gamma release assays for the diagnosis of Mycobacterium tuberculosis infection in children: a literature review. Int J Immunopathol Pharmacol 2012; 25: 335–343. [DOI] [PubMed] [Google Scholar]
- 161. Bunsow E, Ruiz‐Serrano MJ, Lopez Roa P et al Evaluation of GeneXpert MTB/RIF for the detection of Mycobacterium tuberculosis and resistance to rifampin in clinical specimens. J Infect 2014; 68: 338–343. [DOI] [PubMed] [Google Scholar]
- 162. Abdool Karim SS, Naidoo K, Grobler A et al Integration of antiretroviral therapy with tuberculosis treatment. N Engl J Med 2011; 365: 1492–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Blanc FX, Sok T, Laureillard D et al Earlier versus later start of antiretroviral therapy in HIV‐infected adults with tuberculosis. N Engl J Med 2011; 365: 1471–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Havlir DV, Kendall MA, Ive P et al Timing of antiretroviral therapy for HIV‐1 infection and tuberculosis. N Engl J Med 2011; 365: 1482–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Yotebieng M, Van Rie A, Moultrie H et al Effect on mortality and virological response of delaying antiretroviral therapy initiation in children receiving tuberculosis treatment. AIDS 2010; 24: 1341–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Frohoff C, Moodley M, Fairlie L et al Antiretroviral therapy outcomes in HIV‐infected children after adjusting protease inhibitor dosing during tuberculosis treatment. PLoS ONE 2011; 6: e17273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Ren Y, Nuttall JJ, Egbers C et al Effect of rifampicin on lopinavir pharmacokinetics in HIV‐infected children with tuberculosis. J Acquir Immune Defic Syndr 2008; 47: 566–569. [DOI] [PubMed] [Google Scholar]
- 168. Oudijk JM, McIlleron H, Mulenga V et al Pharmacokinetics of nevirapine in HIV‐infected children under 3 years on rifampicin‐based antituberculosis treatment. AIDS 2012; 26: 1523–1528. [DOI] [PubMed] [Google Scholar]
- 169. Grinsztejn B, De Castro N, Arnold V et al Raltegravir for the treatment of patients co‐infected with HIV and tuberculosis (ANRS 12 180 Reflate TB): a multicentre, phase 2, non‐comparative, open‐label, randomised trial. Lancet Infect Dis 2014; 14: 459–467. [DOI] [PubMed] [Google Scholar]
- 170. International Union Against Tuberculosis and Lung Disease, WHO . Guidance for national tuberculosis and HIV programmes on the management of tuberculosis in HIV‐infected children: recommendations for a public health approach. 2010. Available at http://www.theunion.org/what‐we‐do/publications/english/pub_tbhivchildfinaldoc_webversion2.pdf (accessed June 2014).
- 171. Gkentzi D, Tebruegge M, Tudor‐Williams G et al Incidence, spectrum and outcome of immune reconstitution syndrome in HIV‐infected children following initiation of antiretroviral therapy. Pediatr Infect Dis J 2014; 33: 953–958. [DOI] [PubMed] [Google Scholar]
- 172. Shah I. Immune Reconstitution Syndrome in HIV‐1 infected children – a study from India. Indian J Pediatr 2011; 78: 540–543. [DOI] [PubMed] [Google Scholar]
- 173. Walters E, Cotton MF, Rabie H et al Clinical presentation and outcome of tuberculosis in human immunodeficiency virus infected children on anti‐retroviral therapy. BMC Pediatr 2008; 8: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Zampoli M, Kilborn T, Eley B. Tuberculosis during early antiretroviral‐induced immune reconstitution in HIV‐infected children. Int J Tuberc Lung Dis 2007; 11: 417–423. [PubMed] [Google Scholar]
- 175. Puthanakit T, Oberdorfer P, Akarathum N et al Immune reconstitution syndrome after highly active antiretroviral therapy in human immunodeficiency virus‐infected thai children. Pediatr Infect Dis J 2006; 25: 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Laureillard D, Marcy O, Madec Y et al Paradoxical tuberculosis‐associated immune reconstitution inflammatory syndrome after early initiation of antiretroviral therapy in a randomized clinical trial. AIDS 2013; 27: 2577–2586. [DOI] [PubMed] [Google Scholar]
- 177. Meintjes G, Lawn SD, Scano F et al Tuberculosis‐associated immune reconstitution inflammatory syndrome: case definitions for use in resource‐limited settings. Lancet Infect Dis 2008; 8: 516–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. WHO . Guidance for national tuberculosis programmes on the management of tuberculosis in children. 2006. Available at http://whqlibdoc.who.int/hq/2006/WHO_HTM_TB_2006.371_eng.pdf?ua=1 (accessed June 2014). [PubMed]
- 179. WHO . Antiretroviral therapy for HIV infection in infants and children: towards universal access. 2010. Available at http://whqlibdoc.who.int/publications/2010/9789241599801_eng.pdf?ua=1 (accessed June 2014). [PubMed]
- 180. Makadzange AT, Ndhlovu CE, Takarinda K et al Early versus delayed initiation of antiretroviral therapy for concurrent HIV infection and cryptococcal meningitis in sub‐saharan Africa. Clin Infect Dis 2010; 50: 1532–1538. [DOI] [PubMed] [Google Scholar]
- 181. Wamalwa DC, Lehman DA, Benki‐Nugent S et al Long‐term virologic response and genotypic resistance mutations in HIV‐1 infected Kenyan children on combination antiretroviral therapy. J Acquir Immune Defic Syndr 2013; 62: 267–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Ruel TD, Kamya MR, Li P et al Early virologic failure and the development of antiretroviral drug resistance mutations in HIV‐infected Ugandan children. J Acquir Immune Defic Syndr 2011; 56: 44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Briz V, Garcia D, Mendez‐Lagares G et al High prevalence of X4/DM‐tropic variants in children and adolescents infected with HIV‐1 by vertical transmission. Pediatr Infect Dis J 2012; 31: 1048–1052. [DOI] [PubMed] [Google Scholar]
- 184. Frange P, Briand N, Veber F et al CCR5 antagonists: a therapeutic option in HIV‐1 perinatally infected children experiencing virologic failure? AIDS 2012; 26: 1673–1677. [DOI] [PubMed] [Google Scholar]
- 185. Lee KJ, Shingadia D, Pillay D et al Transient viral load increases in HIV‐infected children in the U.K. and Ireland: what do they mean? Antivir Ther 2007; 12: 949–956. [PubMed] [Google Scholar]
- 186. Pursuing Later Treatment Options II (PLATO II) project team for the Collaboration of Observational HIV Epidemiological Research Europe (COHERE) , Castro H, Judd A et al Risk of triple‐class virological failure in children with HIV: a retrospective cohort study. Lancet 2011; 377: 1580–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. de Mulder M, Yebra G, Navas A et al High drug resistance prevalence among vertically HIV‐infected patients transferred from pediatric care to adult units in Spain. PLoS ONE 2012; 7: e52155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Collaborative HIV paediatric Study . CHIPS annual report 2012/2013. 2013. Available at http://www.chipscohort.ac.uk/summary_data.asp. (accessed June 2014).
- 189. Briz V, Leon‐Leal JA, Palladino C et al Potent and sustained antiviral response of raltegravir‐based highly active antiretroviral therapy in HIV type 1‐infected children and adolescents. Pediatr Infect Dis J 2012; 31: 273–277. [DOI] [PubMed] [Google Scholar]
- 190. Palladino C, Briz V, Gonzalez‐Tome MI et al Short communication: evaluation of the effect of enfuvirtide in 11 HIV‐1 vertically infected pediatric patients outside clinical trials. AIDS Res Hum Retroviruses 2010; 26: 301–305. [DOI] [PubMed] [Google Scholar]
- 191. Tudor‐Williams G, Cahn P, Chokephaibulkit K et al Etravirine in treatment‐experienced, HIV‐1‐infected children and adolescents: 48‐week safety, efficacy and resistance analysis of the phase II PIANO study. HIV Med 2014; 15: 513–524. [DOI] [PubMed] [Google Scholar]
- 192. Briz V, Palladino C, Navarro M et al Etravirine‐based highly active antiretroviral therapy in HIV‐1‐infected paediatric patients. HIV Med 2011; 12: 442–446. [DOI] [PubMed] [Google Scholar]
- 193. Ruxrungtham K, Pedro RJ, Latiff GH et al Impact of reverse transcriptase resistance on the efficacy of TMC125 (etravirine) with two nucleoside reverse transcriptase inhibitors in protease inhibitor‐naive, nonnucleoside reverse transcriptase inhibitor‐experienced patients: study TMC125‐C227. HIV Med 2008; 9: 883–896. [DOI] [PubMed] [Google Scholar]
- 194. Food and Drug Administration . Complera prescribing information. 2013. Available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/202123s003lbl.pdf (accessed June 2014).
- 195. European Medicines Agency . Eviplera: product information. 2014. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Product_Information/human/002312/WC500118802.pdf (accessed June 2014).
- 196. Xiang N, James M, Walters S, Bamford A, Foster C. Improved serum cholesterol in paediatric patients switched from suppressive lopinavir‐based therapy to boosted darunavir or atazanavir. HIV Med 2014; 15: 635–636. [DOI] [PubMed] [Google Scholar]
- 197. Kuhn L, Coovadia A, Strehlau R et al Switching children previously exposed to nevirapine to nevirapine‐based treatment after initial suppression with a protease‐inhibitor‐based regimen: long‐term follow‐up of a randomised, open‐label trial. Lancet Infect Dis 2012; 12: 521–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Coovadia A, Abrams E, Strehlau R et al Virologic efficacy of efavirenz maintenance therapy in nevirapine prophylaxis‐exposed children. Conference on Retrovirus and Opportunistic Infections . Boston, USA, 2014.
- 199. Paton N, Stohr W, Arenas‐Pinto A, Dunn D, Group PT. Randomised controlled trial of a PI monotherapy switch strategy for long‐term HIV management. Conference on Retroviruses and Opportunistic Infections . Boston, USA, 2014.
- 200. Neth O, Falcon‐Neyra L, Ruiz‐Valderas R et al Simplified human immunodeficiency virus maintenance therapy in virologically suppressed children with Ritonavir‐boosted protease inhibitor monotherapy. Pediatr Infect Dis J 2011; 30: 917. [DOI] [PubMed] [Google Scholar]
- 201. Bunupuradah T, Kosalaraksa P, Puthanakit T et al Monoboosted lopinavir/ritonavir as simplified second‐line maintenance therapy in virologically suppressed children. AIDS 2011; 25: 315–323. [DOI] [PubMed] [Google Scholar]
- 202. Klatt NR, Funderburg NT, Brenchley JM. Microbial translocation, immune activation, and HIV disease. Trends Microbiol 2013; 21: 6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Paediatric European Network for Treatment of A . Response to planned treatment interruptions in HIV infection varies across childhood. AIDS 2010; 24: 231–241. [DOI] [PubMed] [Google Scholar]
- 204. Bunupuradah T, Duong T, Compagnucci A et al Outcomes after reinitiating antiretroviral therapy in children randomized to planned treatment interruptions. AIDS 2013; 27: 579–589. [DOI] [PubMed] [Google Scholar]
- 205. Klein N, Sefe D, Mosconi I et al The immunological and virological consequences of planned treatment interruptions in children with HIV infection. PLoS ONE 2013; 8: e76582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Wamalwa D, Benki‐Nugent S, Langat A et al Treatment interruption in infants following 24 months of empiric ART: Kenya. Conference on Retroviruses and Opportunistic Infections . Seattle, USA, 2012.
- 207. Siberry GK, Patel K, Van Dyke RB et al CD4+ lymphocyte‐based immunologic outcomes of perinatally HIV‐infected children during antiretroviral therapy interruption. J Acquir Immune Defic Syndr 2011; 57: 223–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Fox Z, Phillips A, Cohen C et al Viral resuppression and detection of drug resistance following interruption of a suppressive non‐nucleoside reverse transcriptase inhibitor‐based regimen. AIDS 2008; 22: 2279–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Cressey TR, Green H, Khoo S et al Plasma drug concentrations and virologic evaluations after stopping treatment with nonnucleoside reverse‐transcriptase inhibitors in HIV type 1‐infected children. Clin Infect Dis 2008; 46: 1601–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. SPARTAC Trial Investigators , Fidler S, Porter K et al Short‐course antiretroviral therapy in primary HIV infection. N Engl J Med 2013; 368: 207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Lodi S, Meyer L, Kelleher AD et al Immunovirologic control 24 months after interruption of antiretroviral therapy initiated close to HIV seroconversion. Arch Intern Med 2012; 172: 1252–1255. [DOI] [PubMed] [Google Scholar]
- 212. Saez‐Cirion A, Bacchus C, Hocqueloux L et al Post‐treatment HIV‐1 controllers with a long‐term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog 2013; 9: e1003211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Ananworanich J, Puthanakit T, Suntarattiwong P et al Reduced markers of HIV persistence and restricted HIV‐specific immune responses after early antiretroviral therapy in children. AIDS 2014; 28: 1015–1020. [DOI] [PubMed] [Google Scholar]
- 214. Persaud D, Gay H, Ziemniak C et al Absence of detectable HIV‐1 viremia after treatment cessation in an infant. N Engl J Med 2013; 369: 1828–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Persaud D, Deveikis A, Gay H et al Very early combination antiretroviral therapy in perinatal HIV infection: two case studies. Conference on Retrovirus and Opportunistic Infections . Boston, USA, 2014.
- 216. National Institiue of Allergy and Infectious Diseases . ‘Mississippi Baby’ now has detectable HIV, researchers find. 2014. Available at http://www.niaid.nih.gov/news/newsreleases/2014/Pages/MississippiBabyHIV.aspx (accessed June 2014).
- 217. Department of Health . Transition: getting it right for young people. 2006. Available at http://webarchive.nationalarchives.gov.uk/20130107105354/http://www.dh.gov.uk/prod_consum_dh/groups/dh_digitalassets/@dh/@en/documents/digitalasset/dh_4132149.pdf (accessed June 2014).
- 218. Wiener LS, Kohrt BA, Battles HB, Pao M. The HIV experience: youth identified barriers for transitioning from pediatric to adult care. J Pediatr Psychol 2011; 36: 141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Bundock H, Fidler S, Clarke S et al Crossing the divide: transition care services for young people with HIV‐their views. AIDS Patient Care STDS 2011; 25: 465–473. [DOI] [PubMed] [Google Scholar]
- 220. Vijayan T, Benin AL, Wagner K, Romano S, Andiman WA. We never thought this would happen: transitioning care of adolescents with perinatally acquired HIV infection from pediatrics to internal medicine. AIDS Care 2009; 21: 1222–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Khan M, Song X, Williams K et al Evaluating adherence to medication in children and adolescents with HIV. Arch Dis Child 2009; 94: 970–973. [DOI] [PubMed] [Google Scholar]
- 222. Mellins CA, Brackis‐Cott E, Leu CS et al Rates and types of psychiatric disorders in perinatally human immunodeficiency virus‐infected youth and seroreverters. J Child Psychol Psychiatry 2009; 50: 1131–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Domek GJ. Facing adolescence and adulthood: the importance of mental health care in the global pediatric AIDS epidemic. J Dev Behav Pediatr 2009; 30: 147–150. [DOI] [PubMed] [Google Scholar]
- 224. Mellins CA, Elkington KS, Leu CS et al Prevalence and change in psychiatric disorders among perinatally HIV‐infected and HIV‐exposed youth. AIDS Care 2012; 24: 953–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Malee KM, Tassiopoulos K, Huo Y et al Mental health functioning among children and adolescents with perinatal HIV infection and perinatal HIV exposure. AIDS Care 2011; 23: 1533–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Gadow KD, Chernoff M, Williams PL et al Co‐occuring psychiatric symptoms in children perinatally infected with HIV and peer comparison sample. J Dev Behav Pediatr 2010; 31: 116–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 PENTA dosing recommendations for currently licensed ART.