

Abstract
Viruses coevolve with their hosts to overcome host resistance and gain the upper hand in the evolutionary arms race. Drosophila innubila nudivirus (DiNV) is a double stranded DNA virus, closely related to Oryctes rhinoceros nudivirus (OrNV) and Kallithea virus. DiNV is the first DNA virus found to naturally infect Drosophila and therefore has the potential to be developed as a model for DNA virus immune defense and host/virus coevolution within its well-studied host system. Here we sequence and annotate the genome of DiNV and identify signatures of adaptation, revealing clues for genes involved in host-parasite coevolution. The genome is 155555bp long and contains 107 coding open reading frames (ORFs) and a wealth of AT-rich simple sequence repeats. While synteny is highly conserved between DiNV and Kallithea virus, it drops off rapidly as sequences become more divergent, consistent with rampant rearrangements across nudiviruses. Overall, we show that evolution of DiNV is likely due to adaptation of a very few genes coupled with high gene turnover.
Keywords: DNA virus, Drosophila virus, viral evolution, population genetics
1. Introduction
Baculoviruses and nudiviruses are large double stranded DNA viruses (90-180kbp genomes, 30-300nm virions) that infect a wide array of arthropods (Jehle et al., 2006). They contain between 90 and 180 genes, of which a common set of 20 are key to the activity of the virus. Baculoviruses can usually be characterized by their helically symmetrical, rod-shaped nucleocapsids contained in stable occlusion bodies (known as polyhedra) and a viral encoded RNA polymerase (Jehle et al., 2006; Rohrmann, 2013). These factors allow the viruses to remain stable and infectious in most environmental conditions, and to remain active independent of the host RNA polymerase. Nudiviruses are close relatives of baculoviruses and while they are like other baculoviruses in many ways, they differ in the viral particle shape and that some do not form a baculovirus-like occlusion body (Wang et al., 2006). Currently there very few described nudiviruses and most infect arthropods, including fruit flies (Drosophila), rhinoceros beetles (Oryctes rhinoceros), crane flies (Tipulidae) and tiger prawns (Penaeus) (Burand, 1998; Unckless, 2011; Wang et al., 2012). Bracoviruses are also found as a sister group to nudiviruses. These viruses are symbiotic with their host braconid wasp, making up a component of the parasitoid wasps venom (Bézier et al., 2009).
Though baculoviruses are among the best studied insect DNA viruses, we have limited understanding of how the arthropod immune system has evolved to suppress DNA viruses, and how the viruses in turn have evolved to escape this suppression. Recently, a nudivirus was discovered in the mushroom-feeding Drosophilid species, Drosophila innubila (Unckless, 2011). The Drosophila innubila nudivirus (DiNV) is actually found across a large range of Drosophila species in the new world, varying in frequencies from 3% to ∼60% (Unckless, 2011). DiNV has been shown to reduce the viability of infected flies (infected flies survive 8-17 days post-infection, versus 20-31 days survival in mock infected controls), had significantly shorter lifespans in wild collected flies (median survival of 18 days and 43 days in virus infected and uninfected wild flies respectively) (Unckless, 2011). Infected females also laid significantly fewer eggs compared to uninfected flies (a median of ∼82% fewer off spring than mock infected controls). While not yet characterized in DiNV, other nudiviruses cause swollen, translucent larvae and increased larval deaths in their hosts (Burand, 1998; Payne, 1974). DiNV, like other nudiviruses, is suspected to infect the gut of infected adults and larvae. With the recently discovered Kallithea virus (Webster et al., 2015), DiNV has the potential to be developed into a powerful tool to study host-DNA virus interactions (Unckless, 2011) because of the wealth of resources available for studying the Drosophila innate immune system (Hales et al., 2015; Hoffmann, 2003).
To begin to gain an understanding of the host/virus coevolutionary arms race, we must start with a detailed characterization of the virus itself, including the sequencing, annotation and analysis of the viral genome. Here we sequence the DNA of an individual D. innubila male fly infected with DiNV and use the resulting metagenomic data to report the assembly and annotation of the DiNV genome. As found previously, DiNV is closely related to OrNV and the more recently found Kallithea virus. We find evolution across the genes in DiNV that is consistent with divergence based analyses across other baculoviruses and a population-level analysis of Autographa californica Multiple Nucleoployhedrovirus (AcMNPV) (Hill and Unckless, 2017). These results suggest that very few genes show overlapping signatures of evolution across this diverse group of viruses and that DiNV may be a useful model for understanding the evolution of a pathogenic DNA virus and the corresponding evolution of the host immune system.
2. Methods
2.1. Genome sequencing
Wild Drosophila innubila were captured at the Southwest Research Station in the Chiricahua Mountains between September 8th and 15th, 2016. Baits consisted of store-bought white button mushrooms (Agaricus bisporus) placed in large piles about 30cm in diameter. A sweep net was used to collect the flies over the baits. Flies were sorted by sex and species at the University of Arizona and males were frozen at -80°C before being shipped on dry ice to Lawrence, KS. All D. innubila males were homogenized in 50 microliters of viral buffer (a media meant to preserve viral particles, taken from (Nanda et al., 2008)) and half of the homogenate was used to extract DNA using the Qiagen Gentra Puregene Tissue kit (#158689, Germantown, Maryland, USA). We determined whether flies were infected by PCR screening for two viral genes, P47 and LEF-4 (Supplemental Table 1 for primers and PCR conditions). The amplicons from flies screening positive for DiNV were sequenced (ACGT, Inc., Wheeling, IL, USA) to confirm the identity of the PCR product. One infected individual (ICH01M) was selected for sequencing. We constructed a genomic DNA library consisting of virus, Drosophila and other microbial DNA using a modified version of the Nextera DNA Library Prep kit (#FC-121-1031, Illumina, Inc., San Diego, CA, USA) meant to conserve reagents (Baym et al., 2015). We sequenced the library on one-twentieth of an Illumina HiSeq 2500 System Rapid-Run to generate 14873460 paired-end 150 base-pair reads (available at NCBI accession number SAMN07638923 [to be released upon acceptance of the manuscript]).
2.2. DiNV genome assembly
We used an iterative approach to assemble the DiNV genome. First, we trimmed all Illumina paired-end short reads using sickle (parameters: minimum length = 20, minimum quality = 20) (Joshi and Fass, 2011) and checked our data for any biases, high levels of PCR duplicates or any over represented sequences using FastQC (Andrews, 2010). Ruling out these problems, we then mapped all Illumina paired-end short reads of the infected D. innubila fly ICH01M to a draft D. innubila genome (Robert L. Unckless, unpublished) using BWA MEM (parameters: -M) (Li and Durbin, 2009). Second, we took all unmapped reads and assembled them using Spades (default parameters) (Bankevich et al., 2012). Following this, we identified each contig's closest hit via a BLASTn search to the non-redundant database with an E-value cutoff of 0.0001 (Altschul et al., 1990). Third, we took all contigs, including those with BLAST hits to any nudivirus or baculoviruses, and concatenated these to the draft D. innubila genome. We then re-mapped all reads to a preliminary Drosophila innubila genome, with the putative DiNV contigs attached (BWA mem parameters: -M) and collected all unmapped reads, as well as all reads mapping to the nudivirus or baculovirus contigs. We performed a further assembly using Spades with these reads, and assigning all nudivirus or baculovirus contigs as trusted contigs and all other previously assembled contigs with non-viral hits as untrusted (--trustedcontigs -untrustedcontigs). Finally, we repeated this process one further time, which yielded a 157429bp contig with considerable similarity to nudiviruses. This contig has a mean coverage of 1124, a maximum coverage of 1887 and minimum of 116.
2.3. DiNV validation
We compared our assembled sequence with all known nudiviruses using MAFFT to identify aligned regions (MAFFT parameters: --auto) and its divergence from each other nudivirus (Katoh et al., 2002). We also remapped our short-read data to the Drosophila innubila genome with the viral genome concatenated (BWA MEM -M) (Li and Durbin, 2009) and visualized it using the Integrated Genomics Viewer to identify any inconsistencies that may come with assembling a circular genome (Robinson et al., 2011), including the collapsing of duplicated regions, repeats of genes from the ‘start’ of the sequence onto the ‘end’ of the genome, or large structural rearrangements. While we found no large structural problems or duplication issues, we found inconsistent coverage across the last 1561bp of the sequence. This region showed strong similarity to Serratia liquifaciens. While the median coverage of the genome was 1124, the median coverage of this Serratia portion was 157, suggesting either a misassembly or low frequency insertion.
We used pindel (default parameters) to attempt to identify further structural errors in our genome, but only confirmed our low confidence with the Serratia portion by its high frequency deletion (Ye et al., 2009). We concluded this region was not part of the consensus sequence due to its low coverage versus the rest of the genome and its low frequency found with pindel (0.128), though it may be a segregating horizontal gene transfer. To finally confirm or reject the presence of this Serratia portion, we designed primers across the edge of the Serratia portion and across the start/end of the DiNV sequence, labelled A-F in Supplementary Table 1, along with each primers sequences and PCR conditions. One group of primers (A:C, A:D, B:C, B:D) will generate products if this insertion is present, while a second group (A:E, A:F, B:E, B:F) should generate products if the insertion is absent. Only the second group of PCRs generated products, consistent with the absence of this insertion and a misassembly of the genome. We sequenced the generated PCR products across the ends of DiNV, which confirmed the Serratia misassembly, to NCBI (accession: MF966380).
Because considerable viral genetic variation existed within this individual Drosophila male, we sought to generate a consensus DiNV sequence. To that end, we called high frequency variants using GATK HaplotypeCaller (parameters: --ploidy 10), which we then inserted into the sequence using GATK FastaAlternateReferenceMaker, resulting in a final circular genome, 155555bp long (DePristo et al., 2011). The genome and annotation is available at NCBI accession number MF966379 (to be released upon acceptance of the manuscript).
2.4. DiNV gene identification and content
We identified the gene content of DiNV based on methods used previously (Wang et al., 2012, 2008, 2007; Yang et al., 2014). We predicted methionine-initiated open reading frames (ORFs) encoding 50 amino acids or more and showing minimum overlap using ORF Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html) (Rombel et al., 2002), the putative coding regions were numbered as DiNV ORFs. We first used BLASTP and BLASTN to identify orthologs in a database of all nudivirus ORFs (-evalue 0.0001, downloaded from the NCBI gene database in October 2016) and performed reciprocal BLASTP and BLASTN searches versus Kallithea virus, Oryctes rhinoceros Nudivirus (OrNV) and Gryllus bimaculatus Nudivirus (GrBNV) to confirm the hits found previously.
Following this we confirmed each ORFs annotation via BLASTP and BLASTN to the NCBI non-redundant database using default parameters with an e-value cutoff of 0.0001. We also using BLASTP to identify orthologous ORFs to baculoviruses, using a database of amino acid sequences from Autographa californica multiple Nucleopolyhedrovirus, Bombyx mori Nucleopolyhedrovirus and Helicoverpa armigera single Nucleopolyhedrovirus with an e-value cut-off of 0.001. We found hits for all 20 conserved genes as well as polyhedrin. All ORFs were investigated for characteristic sequence signatures using the conserved domain search tool (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) and Pfam with an E-value cutoff of 1 (Finn et al., 2016), with any identified domains recorded in table S2. Finally, to confirm these results, we used HHpred to identify any conserved protein domains with higher sensitivity (https://toolkit.tuebingen.mpg.de/#/tools/hhpred). We noted the top hit found for each protein, with an e-value cutoff of 1 (Söding et al., 2005).
2.5. DiNV divergence evolution
We identified genes that may be evolving under positive selection between DiNV and its two closest relatives, Kallithea virus and Oryctes rhinoceros Nudivirus, by comparing the rates of nonsynonymous to synonymous divergence in each of the 85 shared ORFs. We aligned each set of orthologous nucleotide sequences using PRANK (parameters: -codon +F) (Löytynoja, 2014). Using these codon-based alignments, we found codons shared across all genomes and calculated non-synonymous and synonymous divergence using a custom Biopython script. In this script, we parsed the PRANK generated phylip files for each ORF and identified codons present in both genomes. Using the standard codon table, we identified the number of codons with nucleotide substitutions resulting in an amino acid change (non-synonymous), the number of codons with substitutions resulting in no change (synonymous), and the number of possible synonymous and non-synonymous substitutions for all shared codons in each ORF. For each ORF, we used these numbers to find the proportion of non-synonymous substitutions of all possible non-synonymous substitutions (dN), and the proportion of synonymous substitutions of all possible synonymous substitutions (dS), and dN/dS.
Following this we also defined amino acid substitutions as either radical (to an amino acid of a different group based on their side chains) or conservative (to an amino acid with a similar side chain – in the same group) (Smith, 2003). For a broader view of genome-level evolution, we aligned each genome using lastZ to identify blocks of synteny which we visualized using RCircos (Rahmani et al., 2011; Zhang et al., 2013).
We also aligned the nucleotide sequences for the 20 conserved ORFs from all nudiviruses and AcMNPV using MAFFT (Katoh et al., 2002) and concatenated these sequences, we then generated a phylogeny using PhyML (model = GTR, bootstraps = 100, gamma = 4) (Guindon et al., 2010) to place DiNV in the nudivirus phylogeny.
2.6. DiNV population genetics
Because we found considerable within-host DiNV genetic variation, we identified polymorphisms in ICH01M DiNV. For this we used Lofreq (Wilm et al., 2012) and allowed for the detection of indels (Lofreq parameters: indelqual –dindel, call –call-indels –min-mq 20), we considered polymorphisms with a minimum frequency threshold of 0.002, which corresponds to about two-fold coverage of a specific site (Wilm et al., 2012). We also filtered these SNPs for polymorphisms exclusively at synonymous sites.
Using all variation detected with Lofreq (and synonymous variation), we performed a genome wide scan of within host polymorphism to find Watterson's theta, Tajima's pi and Tajima's D across sliding windows and within each gene, using Popoolation (Kofler et al., 2011; Tajima, 1989). We also performed McDonald-Kreitman tests (McDonald and Kreitman, 1991) with either Kallithea virus or OrNV as the outgroup and calculated alpha (the proportion of adaptive substitutions) (Smith and Eyre-Walker, 2002) between each genome and DiNV using a custom Biopython script and the gene codon alignments generated by PRANK previously for the estimation of dN/dS.
We also calculated a simulated neutral expectation of Tajima's pi and Tajima's D for the genome based on a population growth model in ms (Hudson, 2002). We estimated this expectation using both the silent and total estimates of Watterson's theta, the estimated population size from Lofreq (1000) and the median growth rate taken from across a range of viruses (0.48). We then compared our simulated 2.5th and 97.5th quantiles to the observed quantiles for both silent and total polymorphism. We repeated this for exclusively silent polymorphism.
3. Results & Discussion
3.1. DiNV structure and genes
Following an iterative assembly approach, we found the DiNV genome is 155,555bp, making it among the larger genomes for sequenced nudiviruses (Bézier et al., 2015) and slightly larger than its closest relative, the Kallithea virus (152,390bp) (Webster et al., 2015). The DiNV GC content (30%) is also comparable to other nudiviruses which range from 25 to 42% GC (Bézier et al., 2015). We found 107 ORFs (Figure 1A, Supplementary Figure 1, Supplementary Table 2), resulting in a coding density of 71.7%, similar to Kallithea virus, but on the low end of coding densities for nudiviruses and much lower than all other baculoviruses (Bézier et al., 2015; Wang et al., 2012). DiNV, shares 89 (83%) of its ORFs with the other Drosophila nudivirus, Kallithea virus, 85 (79%) ORFs with its next closest relative, OrNV, and 68 (64%) ORFs with GrBNV, and has 16 putatively novel ORFs. Not surprisingly, the 68 ORFs found in all four genomes included all 20 of the core conserved baculovirus ORFs that are necessary for baculovirus function: ORFs associated with late and very late gene transcription (P47, LEF-8, LEF-9, LEF-4, VLF-1, and LEF- 5), replication (DNA polymerase and Helicase), virus structure (P74, PIF-1, PIF-2, PIF-3, AC68, VP91, VP39, 38K, PIF-4/19kda and ODV-E56), and those of unknown function (AC81 and AC92) (Jehle et al., 2006; Wang et al., 2012; Wang and Jehle, 2009). Protein identity of these 20 ORFs between DiNV and Kallithea virus ranges from 16 to 94% (median = 75%), between DiNV and OrNV ranges from 23 to 98% (median = 83%) and between DiNV and GrBNV ranges from 35 to 99% (median = 67%).
Figure 1. DiNV genome and its relation to other nudiviruses.
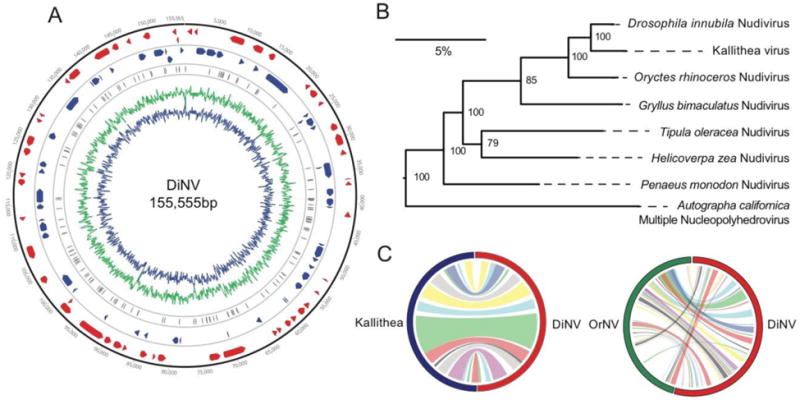
A) DiNV genome map. The genome is 155555bp, containing 107 ORFs. ORFs in one direction are shown in red, while ORFs in the alternate direction are shown in blue and repeat regions are shown in grey. The percent of AT/GC content is show across the genome in green/blue. B) DiNV on a nudivirus maximum-likelihood phylogeny, using nucleotide sequences of the 20 core ORFs found in all baculoviruses. We have also included the baculovirus AcMNPV as an outgroup (Wang et al., 2012). DiNV is a sister genome to Kallithea virus with OrNV as its next closest genome. Each branch point shows the bootstrap support from 100 bootstrap replicates, with a scale bar representing 5% nucleotide divergence. C) DiNV synteny with Kallithea virus and OrNV. Colors are randomly assigned, with extensive blocks of synteny separated by regions with no assignable orthology. Notice that gene order and the size of synteny blocks declines as viruses become more diverged.
Like several other annotated nudiviruses, we also find a polyhedrin/granulin ORF (ORF93, polh/gran), orthologous to the lepidopteran ORF (BLASTp e-value < 0.01); This protein has 97% identity with Kallithea ORF68, 91% identity with OrNV ORF16, 82% identity with GrBNV ORF65, 63% identity with ToNV ORF59 and 58% identity with AcMNPV ORF8. Consistent with previous results (Afonso et al., 2001), we found no evidence of an ortholog to DiNV ORF93 in Culex nigripalpus NPV (BLASTp e-value < 1). It is unclear what role this gene plays in the nudivirus lifecycle, or its function in its atypical occlusion bodies. It is generally thought that polh/gran stabilizes baculovirus virions (Coulibaly et al., 2007; Rohrmann, 2013), so may perform a similar role in the stable formation of virion in nudivruses.
ODV-E56 appears to be duplicated in both DiNV and Kallithea virus, with a novel copy at 5.5kbp (ODV-E56-2) and the original at 122.8kbp. A maximum likelihood phylogeny of ODV-E56 nucleotide sequences from nudiviruses suggests this duplication occurred before the DiNV-Kallithea divergence (Supplementary Figure 2).
The 16 putative ORFs unique to DiNV show no significant difference in GC-content or length from previous described proteins (Mann-Whitney U Test p-value = 0.87, W = 1375). We used PFam and HHpred to identify conserved protein domains in these proteins, among these 16 novel ORFs, 6 have motifs shared with other proteins including a thymidylate synthase, a maturase domain for intron splicing, a T-cell activation factor, a glycosylation protein, a transcription factor domain and a Gastropod egg laying hormone precursor protein domain, while an additional 3 novel ORFs share known motifs with mitochondrial carrier proteins (Supplementary Table 2).
The genome is comprised of 5.1% simple repeats dispersed across 156 regions (Figure 1A in grey, Supplementary Table 2). These repeats are primarily AT-rich (e.g. ATAT, ATTT, TAATTA, TTGATA), contributing to the low GC content seen throughout the genome (the genome is 33.9% GC after removing repeats). When comparing the densities of repeats within and outside of coding regions, we find no significant difference in the density of repeats between regions (Mann Whitney U test W = 1138, p-value 0.9476), and no excess of repeats in the larger non-coding regions (> 1000bp) versus the smaller regions (< 1000bp) (Mann Whitney U test W = 1297, p-value 0.2067).
A nudivirus phylogeny built using the nucleotide sequences of the 20 ORFs shared across all baculoviruses (Figure 1B), shows that DiNV clusters with Kallithea virus and OrNV. Most of the DiNV genome is syntenic with Kallithea virus, with slight differences in gene content and position of ORFs (Figure 1C, Table S2). However, DiNV shows much less gene retention or synteny with OrNV (Figure 1C, Table S2) and we were unable to find regions of synteny for blocks larger than individual genes for more divergent nudiviruses including GrBNV (Table S2). These results are consistent with other nudivirus and baculovirus studies which found both gene content and synteny are poorly conserved (Wang et al., 2012).
3.2. Nudivirus evolution within and between hosts
We suspect that positive selection observed between OrNV and the two Drosophila infecting nudiviruses may be due to adaptation to a new host system. To test this, we looked for signatures of adaptation between genes DiNV shares with both Kallithea virus and OrNV.
We calculated dN/dS, the proportion of non-synonymous substitutions to synonymous substitutions between DiNV and Kallithea virus, and DiNV and OrNV. Most proteins are under purifying selection in both cases (dN/dS < 1), with no ORFs, in either comparison, showing evidence of strong positive selection, suggested by a dN/dS greater than 1. The functional category with the median highest dN/dS are involved in host infection (e.g. VLF-1, PIF-1, PIF-3), suggesting that these genes may be important to adapting to a new host, though this group is not a statistical outlier (81st percentile based on 100000 permutations). As we find no signatures of positive selection, we attempted to identify genes under unconstrained evolution or putative adaptation and identify genes which overlap in several analyses looking for adaptation, hoping to infer which genes are the most likely to be undergoing adaptation within and between hosts. Note again that this analysis was performed based on genetic variation within a single individual. Using an arbitrary threshold of dN/dS > 0.5 for unconstrained evolution/putative selection, Helicase, ODV-E56-2 and a hypothetical protein are the only ORFs to not show signatures of purifying selection in both comparisons (Figure 2A). These results with Helicase are consistent with previous findings which show Helicase is one of the most rapidly evolving genes across baculoviruses and nudiviruses (Hill and Unckless, 2017). Helicase has previously been strongly implicated in host range expansion of baculoviruses (Argaud et al., 1998; Croizier et al., 1994), so adaptive evolution of viruses across differing host species is not unexpected. Only two hypothetical ORFs are above the 0.5 threshold exclusively in the DiNV/Kallithea virus divergence (Figure 2A, black points). Interestingly, twelve genes have dN/dS above 0.5 exclusively between DiNV and OrNV (Figure 2A, orange points). These twelve include LEF-3, GrBNV_gp28-like protein, and ten other hypothetical proteins, including two trypsin-serine proteases and one patatin phospholipase. As expected, most ORFs are under purifying selection, likely because they are close to a fitness optimum, with few changes being adaptive. While Kallithea virus and DiNV are found in similar hosts, OrNV infects a strikingly different host organism, Oryctes rhinoceros. Thus, the higher rate of amino acid substitutions in these ORFs between DiNV and OrNV may be important for adaptation to a new host system.
Figure 2. Evolution of DiNV ORFs.
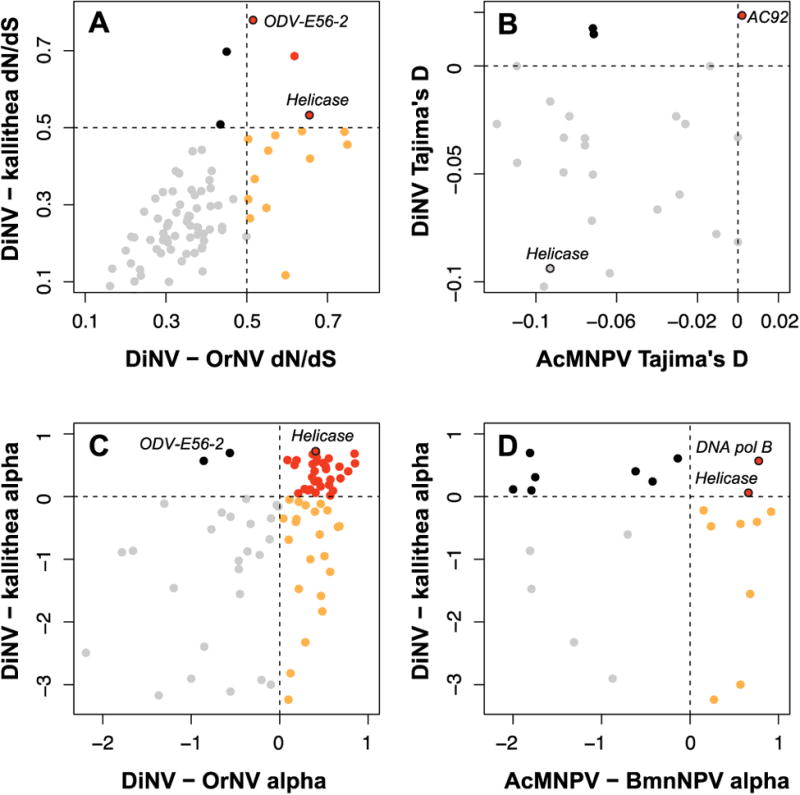
For each comparison, we assigned a cut off, either arbitrary to indicate less constrained purifying selection (in the case of dN/dS) or to indicate natural selection (in the case of alpha and Tajima's D). ORFs above the cutoff in both comparisons are colored red, those above the cutoff in exclusively the OrNV (or AcMNPV/BmNPV) comparison are colored orange, those above the cutoff in exclusively the Kallithea virus comparison are colored black and those below the cut off in both cases are colored grey. A) dN/dS of DiNV ORFs using Kallithea virus and OrNV as paired sequences with an arbitrary cutoff of 0.5 shown (dotted line). Very few genes show adaptive evolution in both comparisons. B) Tajima's D (a measure of selection within a population) for ORFs shared between AcMNPV and DiNV. C) Alpha (the proportion of adaptive amino acid substitutions, from Mcdonald tests) between DiNV – Kallithea and DiNV – OrNV. D) Alpha compared for AcMNPV and DiNV. Only two genes overlap with adaptive substitutions, Helicase and DNA polymerase.
Using the divergence data between DiNV and Kallithea virus or OrNV coupled with polymorphism in DiNV, we calculated the proportion of adaptive substitutions in each gene (alpha) using the McDonald-Kreitman test (McDonald and Kreitman, 1991). This was done using polymorphism found in the virus found in a single host, so it may not necessarily represent the entire population. When Macdonald-Kreitman tests are significant, values of alpha greater than zero indicate that some amino acid substitutions were fixed by natural selection in that gene (Smith and Eyre-Walker, 2002).
Like our dN/dS analysis, we find no genes showing significant levels of adaptation in a McDonald-Kreitman test (Chi-squared test p-value > 0.23 for all genes). Though we find no ORFs showing significant signatures of adaptation in DiNV, we find thirty-five ORFs which have an alpha value greater than 0 in both the comparison using Kallithea virus as an outgroup and OrNV as an outgroup, suggesting these genes have at least one substitution fixed by adaptation (Table S2) (Smith and Eyre-Walker, 2002). Eight of the ORFs with an alpha value greater than zero in these two estimations are among the core 20 baculovirus ORFs (Helicase, 19K, DNA polymerase, P74, VLF-1, Ac92 and PIF-3), as well as polyhedrin/granulin, a ligase and 26 hypothetical proteins (Figure 2C). Consistent with the divergence analysis, we find two only two ORFs (ODV-E56-2 and a hypothetical protein) with potentially adaptive substitutions exclusively between DiNV and kallithea virus, versus 22 ORFs (P47, VP39, VP91, PIF-1, PIF-2, LEF-3, LEF-4, ribosomal reductase 1, ribosomal reductase 2, 61K, AC81 and 11 hypothetical proteins) with potentially adaptive substitutions between DiNV and OrNV. Among the 20 core baculovirus ORFs, only Helicase has an alpha value greater than zero in all tests. This is also consistent when looking across baculoviruses in general (Hill and Unckless, 2017). A similar analysis was performed on a relatively closely related baculovirus, AcMNPV, comparing the results of these two surveys, we find that Helicase and DNA polymerase are the two ORFs with alpha greater than 0 for both the DiNV and AcMNPV analyses (Figure 2D, Supplementary Table 2) (Hill and Unckless, 2017). Helicase has previously been implicated in the extension of host range for a baculovirus (Argaud et al., 1998; Croizier et al., 1994), so putatively selected changes between host species comes as no surprise, however an interpretation of unconstrained changes between similar host species is less plausible (Kang et al., 1998; Maeda et al., 1993). Thus, while our data does not show a significant deviation from neutral evolution for Helicase (or any other gene), the fact that it consistently shows up as potentially under selection is intriguing.
Most specific amino acid changes between DiNV and OrNV are either to aliphatic or uncharged residues (3592 and 3003 respectively, of 10734 changes), a similar proportion to the standing amino acid types (11035 and 11501 respectively, of 37833 amino acids). One sign that natural selection is driving sequence divergence is if amino acid changes are more likely to be ‘radical’ changes than expected by chance e.g. changing to a different amino acid type (polar-uncharged, polar-acidic, polar-basic, non-polar-aliphatic, non-polar-aromatic and other non-polar). A significant proportion of changes are radical compared to ‘conservative’ changes to similar amino acids (Wilcoxon paired test: W = 40213, p-value = 1.31e-11). However, when categorizing the data by ORF functional group (e.g. replication, transcription, host-infection) or individual ORF, we find no significant excess of radical changes in any ORFs (Wilcoxon paired test p-value > 0.21), with no effect of functional category (p-value > 0.12). Polymorphic amino acid changes seen in the virus are also primarily to aliphatic or uncharged amino acids from any amino acid type, with no difference in the ratio of conserved to radical changes seen at any level (Wilcoxon paired test W < 191 p-value > 0.32).
Evolution within DiNV
Recent adaptive evolution is characterized by reduced DNA polymorphism in the region surrounding the selected locus and an excess of rare mutations compared to the neutral expectation. The Tajima's D statistic allows for the detection of this: a negative Tajima's D is consistent with a recent selection at an ORF due to an excess of low frequency derived polymorphism, while a positive Tajima's D suggests balancing selection and maintained polymorphism (Tajima, 1989). We calculated the per site Tajima's D both using a sliding window approach across the genome of DiNV and by individual ORFs, using SNPs called from the pool of DiNV particles infecting a single individual, ICH01M. Given the evidence for recombination in related viruses (Hill and Unckless, 2017; Rohrmann, 2013), natural selection can leave signatures in specific regions of the genome.
Tajima's D is mostly negative across the viral genome (78 ORFs have Tajima's D < 0), consistent with the fact that the viral population size is much reduced upon initial infection, then increases as the infection proceeds. We simulated the expected Tajima's D in a population growth model using ms (Hudson, 2002), and no ORFs were below the 2.5th quantile of the simulated distribution (-0.149), suggesting no deviation from the neutral expectation, similar to our dN/dS results. Because the detection of sweeps may be affected by the action of direct selection on non-synonymous polymorphism, we also estimated Tajima's D again using only synonymous sites. Again, we find no ORFs are below the 2.5th quantile of the simulated expectation of Tajima's D (-0.153).
Though Tajima's D does not differ from the simulated expectation, we find that Tajima's D is mostly negative, and varies across the genome, consistent with differing signatures of selection across the genome. We consider regions in the lower 2.5 percentile of Tajima's D to be the most likely to have recently undergone selection (Figure 2B, Supplementary Figure 3). These windows include only 5 genes: 2 hypothetical proteins, ODV-E56-2, Helicase and 61K. These ORFs are also in windows below the 2.5th percentile for pairwise diversity (Supplementary Figure 3, Supplementary Table 2). When analyzing only synonymous sites, in windows below the 2.5th empirical percentile for observed synonymous Tajima's D, we only find one ORF, ORF59, a trypsin-serine protease not found in the previous survey (Figure S3, Table S2).
Helicase is involved in the replication of viral DNA, and is found in a strongly conserved gene cluster present in all baculoviruses (Herniou et al., 2003; Hill and Unckless, 2017; Rohrmann, 2013; Wang et al., 2012). Our results suggest that Helicase may be a common target for host suppression, as it contains a conserved domain and is vital to viral replication. This may explain Helicases frequent signatures of unconstrained evolution, positive selection and selective sweeps, as alleles that evolve to escape this suppression are positively selected, resulting in the signatures we observe here (Hill and Unckless, 2017). In fact, previous genetic mapping has found that variation in host range, and ability for host swapping is primarily due to sequence variation in the Helicase sequence (Argaud et al., 1998; Croizier et al., 1994; Miller and Albert Lu, 1997). While Helicase frequently shows signatures of adaptation across baculoviruses (Hill and Unckless, 2017), thus far, ODV-E56 shows putative signatures of selection in only the Drosophila-infecting nudiviruses (the duplicated copy) and in the alphabaculovirus clade (the original copy), a group of viruses limited to closely related lepidoptera hosts. We looked for evidence of gene conversion between both ODV-E56 copies, which could lead to patterns like signatures of adaptation. Apart from the first site, there is no shared polymorphism between the two copies and no evidence of gene conversion.
In some windows across the genome, high values of Tajima's D and pairwise diversity suggest that genetic variation is maintained by balancing selection (Figure 2B, Supplementary Figure 3). We find 14 ORFs have Tajima's D above 0, and 7 are in windows above the 97.5th percentile for the simulated estimate of Tajima's D (0.0527), while only 2 ORFs were in windows above the upper 97.5th percentile of the empirically estimated pairwise diversity and Tajima's D (AC92 and LEF-9). Using only synonymous polymorphism, we again find two ORFs in windows above the 97.5th percentile for both Tajima's D (0.08) and pairwise diversity (AC92 and ORF81, a putative deoxynucleoside kinase). AC92 was also found to have the highest Tajima's D and pairwise diversity estimates in a population of AcMNPV (Hill and Unckless, 2017), suggesting that variation may be being maintained in this ORF in several baculoviruses due to some selective mechanism (Figure 2D). AC92 is a sulfhydryl oxidase, we are uncertain what role this protein plays in baculovirus infection (Rohrmann, 2013). It's possible that variation is maintained in this ORF due to its involvement in multiple functions, where different substitutions are beneficial for the proteins separate functions.
4. Conclusions
The assembly and annotation of the DiNV genome provides the basis for the development of a powerful new model system for the study of host/DNA virus interaction. The structure of the DiNV genome is largely like other nudiviruses but contains a relatively low percent coding content and several regions with repeated arrays. While we find no strong selective signatures between DiNV and its closest relatives, we find several overlaps of unconstrained selection with signatures of adaptation suggesting these genes are key to DiNV infection. Several of the genes in DiNV that show selective signatures are not only under selection since the transition from an ancestral host to Drosophila, but also show signatures of selection in other baculoviruses. This suggests that in baculoviruses and nudiviruses, only a few key genes are consistently evolving in an adaptive arms races with their hosts.
Supplementary Material
Highlights.
We sequence the genome of DiNV.
Few genes are rapidly evolving between nudiviruses.
We find high rates of gene turnover between DiNV and its closest relatives.
Helicase and ODV-E56 show consistent signatures of adaptation.
Acknowledgments
We thank Brittny Smith for help preparing the library for sequencing the infected fly, Todd Schlenke at the University of Arizona for assistance with fly collections, the Southwest Research Station in Portal, Arizona for allowing us to collect Drosophila and two anonymous reviewers for their helpful comments and recommendations for the presented work. Work for this grant including next generation sequencing at the University of Kansas Genome Sequencing Core Laboratory and other supplies and travel was supported in part by the University of Kansas Center for Molecular Analysis of Disease Pathways (NIH P20 GM103638). This project was supported by a Max Kade Postdoctoral Fellowship to TH and NIH grant 4R00GM114714-02 to RLU.
Abbreviations
- DiNV
Drosophila innubila Nudivirus
- OrNV
Oryctes rhinoceros Nudivirus
- AcMNPV
Autographa californica Multiple Nucleopolyhedrovirus
- ORF
Open reading frame
- ODV-E56
Occlusion derived virus envelope protein 56
- SNP
Single nucleotide polymorphism
- Indel
Insertion/Deletion
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Afonso CL, Tulman ER, Lu Z, Balinsky Ca, Moser Ba, Becnel JJ, Rock DL, Kutish GF. Genome sequence of a baculovirus pathogenic for Culex nigripalpus. J Virol. 2001;75:11157–11165. doi: 10.1128/JVI.75.22.11157-11165.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. doi: http://dx.doi.org/10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Andrews S. FastQC: a quality control tool for high throughput sequence data 2010 [Google Scholar]
- Argaud O, Croizier L, López-Ferber M, Croizier G. Two key mutations in the host-range specificity domain of the p143 gene of Autographa californica nucleopolyhedrovirus are required to kill Bombyx mori larvae. J Gen Virol. 1998;79:931–935. doi: 10.1099/0022-1317-79-4-931. [DOI] [PubMed] [Google Scholar]
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham SON, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MAXA, Pevzner PA. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baym M, Kryazhimskiy S, Lieberman TD, Chung H, Desai MM, Kishony R. Inexpensive multiplexed library preparation for megabase-sized genomes. PLoS One. 2015:1–15. doi: 10.1371/journal.pone.0128036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bézier A, Annaheim M, Herbinière J, Wetterwald C, Gyapay G, Bernard-Samain S, Wincker P, Roditi I, Heller M, Belghazi M, Pfister-Wilhem R, Periquet G, Dupuy C, Huguet E, Volkoff AN, Lanzrein B, Drezen JM. Polydnaviruses of braconid wasps derive from an ancestral nudivirus. Science. 2009;323:926–930. doi: 10.1126/science.1166788. [DOI] [PubMed] [Google Scholar]
- Bézier A, Thézé J, Gavory F, Gaillard J, Poulain J, Drezen J, Herniou A, Nv H, Nv H. The Genome of the Nucleopolyhedrosis-Causing Virus from Tipula oleracea Sheds New Light on the Nudiviridae Family. J Virol. 2015;89:3008–3025. doi: 10.1128/JVI.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burand JP. Nudiviruses. In: Miller LK, Ball LA, editors. The Insect Viruses. Springer; US, Boston, MA: 1998. pp. 69–90. [Google Scholar]
- Coulibaly F, Chiu E, Ikeda K, Gutmann S, Haebel PW, Schulze-Briese C, Mori H, Metcalf P. The molecular organization of cypovirus polyhedra. Nature. 2007;446:97–101. doi: 10.1038/nature05628. [DOI] [PubMed] [Google Scholar]
- Croizier G, Croizier L, Argaud O, Poudevigne D. Extension of Autographa californica nuclear polyhedrosis virus host range by interspecific replacement of a short DNA sequence in the p143 helicase gene. Proc Natl Acad Sci U S A. 1994;91:48–52. doi: 10.1073/pnas.91.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, Salazar GA, Tate J, Bateman A. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016;44:D279–D285. doi: 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performanceof PhyML 3.0. Syst Biol. 2010;59:307–21. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Hales KG, Korey CA, Larracuente AM, Roberts DM. Genetics on the fly: A primer on the drosophila model system. Genetics. 2015;201:815–842. doi: 10.1534/genetics.115.183392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herniou EA, Olszewski JA, Cory JS, Reilly DRO. The genome sequence and evolution of baculoviruses. Annu Rev Entomol. 2003 doi: 10.1146/annurev.ento.48.091801.112756. [DOI] [PubMed] [Google Scholar]
- Hill T, Unckless RL. Baculovirus molecular evolution via gene turnover and recurrent positive selection of key genes. J Virol. 2017 doi: 10.1128/JVI.01319-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann JA. The immune response of Drosophila. Nature. 2003;426:33–38. doi: 10.1038/nature02021. [DOI] [PubMed] [Google Scholar]
- Hudson RR. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics. 2002;18:337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
- Jehle JA, Blissard GW, Bonning BC, Cory JS, Herniou EA, Rohrmann GF, Theilmann DA, Thiem SM, Vlak JM. On the classification and nomenclature of baculoviruses : A proposal for revision Brief Review 1257–1266. 2006 doi: 10.1007/s00705-006-0763-6. [DOI] [PubMed] [Google Scholar]
- Joshi N, Fass J. Sickle: A sliding window, adaptive, quality-based trimming tool for fastQ files 1.33 2011 [Google Scholar]
- Kang W, Tristem M, Maeda S, Crook NE, O'Reilly DR. Identification and Characterization of the Cydia Pamonella Granulovirus Cathepsin and Chitanase Genes. J Gen Virol. 1998;79:2283–2292. doi: 10.1099/0022-1317-79-9-2283. [DOI] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–66. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofler R, Orozco-terWengel P, de Maio N, Pandey RV, Nolte V, Futschik A, Kosiol C, Schlötterer C. Popoolation: A toolbox for population genetic analysis of next generation sequencing data from pooled individuals. PLoS One. 2011;6 doi: 10.1371/journal.pone.0015925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löytynoja A. Phylogeny-aware alignment with PRANK. In: Russell DJ, editor. Multiple Sequence Alignment Methods. Humana Press; Totowa, NJ: 2014. pp. 155–170. [DOI] [PubMed] [Google Scholar]
- Maeda S, Kamita SG, Kondo A. Host range expansion of Autographa californica nuclear polyhedrosis virus (NPV) following recombination of a 0.6-kilobase-pair DNA fragment originating from Bombyx mori NPV. J Virol. 1993;67:6234–6238. doi: 10.1128/jvi.67.10.6234-6238.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/350055a0. [DOI] [PubMed] [Google Scholar]
- Miller LK, Albert Lu. The molecular basis of baculovirus host range, in: The Viruses. 1997:217–235. [Google Scholar]
- Nanda S, Jayan G, Voulgaropoulou F, Sierra-Honigmann AM, Uhlenhaut C, McWatters BJP, Patel A, Krause PR. Universal virus detection by degenerate-oligonucleotide primed polymerase chain reaction of purified viral nucleic acids. J Virol Methods. 2008;152:18–24. doi: 10.1016/j.jviromet.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne CC. The isolation and characterization of a virus from Oryctes rhinoceros. J Gen Virol. 1974;25:105–116. doi: 10.1099/0022-1317-25-1-105. [DOI] [PubMed] [Google Scholar]
- Rahmani AM, Liljeberg P, Plosila J, Tenhunen H. LastZ: An Ultra Optimized 3D Networks-on-Chip Architecture, in: 2011 14th Euromicro Conference on Digital System Design; 2011. pp. 173–180. [DOI] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, WInckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nature. 2011;29:24–26. doi: 10.1038/nbt0111-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrmann GF. Baculovirus Molecular Biology. 2013;211 doi:NBK114593. [PubMed] [Google Scholar]
- Rombel IT, Sykes KF, Rayner S, Johnston SA. ORF-FINDER: a vector for high-throughput gene identification. Gene. 2002;282:33–41. doi: 10.1016/s0378-1119(01)00819-8. doi: https://doi.org/10.1016/S0378-1119(01)00819-8. [DOI] [PubMed] [Google Scholar]
- Smith NGC. Are Radical and Conservative Substitution Rates Useful Statistics in Molecular Evolution? J Mol Evol. 2003;57:467–478. doi: 10.1007/s00239-003-2500-z. [DOI] [PubMed] [Google Scholar]
- Smith NGC, Eyre-Walker A. Adaptive protein evolution in Drosophila. Nature. 2002;415:1022–1024. doi: 10.1038/4151022a. [DOI] [PubMed] [Google Scholar]
- Söding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:244–248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. doi:PMC1203831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unckless RL. A DNA Virus of Drosophila. PLoS One. 2011;6:e26564. doi: 10.1371/journal.pone.0026564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Bininda-emonds ORP, Jehle Ja. Nudivirus Genomics and Phylogeny. Viral Genomes - Mol Struct Divers Gene Expr Mech Host-Virus Interact. 2012;1:33–52. doi: 10.5772/27793. [DOI] [Google Scholar]
- Wang Y, Jehle Ja. Nudiviruses and other large, double-stranded circular DNA viruses of invertebrates: new insights on an old topic. J Invertebr Pathol. 2009;101:187–93. doi: 10.1016/j.jip.2009.03.013. [DOI] [PubMed] [Google Scholar]
- Wang Y, Kleespies RG, Huger AM, Jehle Ja. The genome of Gryllus bimaculatus nudivirus indicates an ancient diversification of baculovirus-related nonoccluded nudiviruses of insects. J Virol. 2007;81:5395–5406. doi: 10.1128/JVI.02781-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kleespies RG, Ramle MB, Jehle JA. Sequencing of the large dsDNA genome of Oryctes rhinoceros nudivirus using multiple displacement amplification of nanogram amounts of virus DNA. J Virol Methods. 2008;152:106–108. doi: 10.1016/j.jviromet.2008.06.003. [DOI] [PubMed] [Google Scholar]
- Wang Y, van Oers MM, Crawford MA, Vlak MJ, Jehle AJ. Genomic analysis of Oryctes rhinoceros virus reveals genetic relatedness to Heliothis zea virus 1. Arch Virol. 2006;152:519–531. doi: 10.1007/s00705-006-0872-2. [DOI] [PubMed] [Google Scholar]
- Webster CL, Waldron FM, Robertson S, Crowson D, Ferrari G, Quintana JF, Brouqui JM, Bayne EH, Longdon B, Buck AH, Lazzaro BP, Akorli J, Haddrill PR, Obbard DJ. The discovery, distribution, and evolution of viruses associated with Drosophila melanogaster. PLoS Biol. 2015;13:e1002210. doi: 10.1371/journal.pbio.1002210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilm A, Poh P, Aw K, Bertrand D, Hui G, Yeo T, Ong SH, Wong CH, Khor CC, Petric R, Hibberd ML, Nagarajan N. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012;40:11189–11201. doi: 10.1093/nar/gks918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YT, Lee DY, Wang Y, Hu JM, Li WH, Leu JH, Chang GD, Ke HM, Kang ST, Lin SS, Kou GH, Lo CF. The genome and occlusion bodies of marine Penaeus monodon nudivirus (PmNV, also known as MBV and PemoNPV) suggest that it should be assigned to a new nudivirus genus that is distinct from the terrestrial nudiviruses. BMC Genomics. 2014;15:628. doi: 10.1186/1471-2164-15-628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel : a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Meltzer P, Davis S. RCircos : an R package for Circos 2D track plots. 2013 doi: 10.1186/1471-2105-14-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.