

Abstract
Human immunodeficiency virus (HIV) infection is associated with B-cell malignancies in patients though HIV-1 is not able to infect B-cells. The rate of B-cell lymphomas in HIV-infected individuals remains high even under the combined antiretroviral therapy (cART) that reconstitutes the immune function. Thus, the contribution of HIV-1 to B-cell oncogenesis remains enigmatic. HIV-1 induces oxidative stress and DNA damage in infected cells via multiple mechanisms, including viral Tat protein. We have detected elevated levels of reactive oxygen species (ROS) and DNA damage in B-cells of HIV-infected individuals. As Tat is present in blood of infected individuals and is able to transduce cells, we hypothesized that it could induce oxidative DNA damage in B-cells promoting genetic instability and malignant transformation. Indeed, incubation of B-cells isolated from healthy donors with purified Tat protein led to oxidative stress, a decrease in the glutathione (GSH) levels, DNA damage and appearance of chromosomal aberrations. The effects of Tat relied on its transcriptional activity and were mediated by NF-κB activation. Tat stimulated oxidative stress in B-cells mostly via mitochondrial ROS production which depended on the reverse electron flow in Complex I of respiratory chain. We propose that Tat-induced oxidative stress, DNA damage and chromosomal aberrations are novel oncogenic factors favoring B-cell lymphomas in HIV-1 infected individuals.
Keywords: HIV-1, Tat, Oxidative stress, Mitochondria, DNA damage, B-cell lymphomas
Graphical abstract
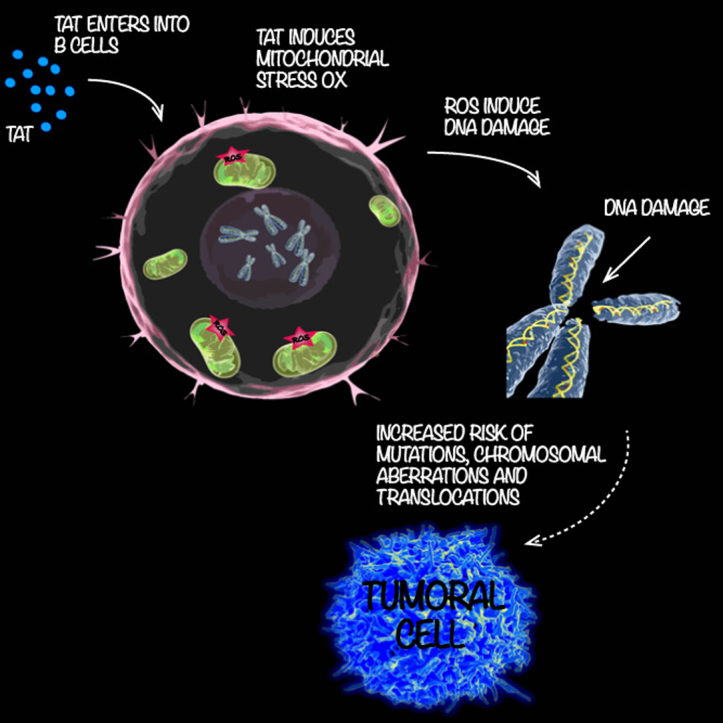
Highlights
-
•
B-cells of HIV-infected individuals exhibit elevated levels of oxidative stress, DNA damage and chromosomal aberrations.
-
•
Purified HIV-1 Tat protein reproduces this effect and induces oxidative stress and DNA damage in B-cells.
-
•
HIV-1 Tat induces mitochondrial oxidative stress and activates NF-kB in B-cells.
-
•
This condition increases the risk of developing chromosomal abnormalities and translocations.
1. Introduction
Despite recent advances in combined antiretroviral therapy (cART), the incidence of cancer remains high in HIV-infected individuals. B-cell lymphomas are the most common types of HIV-related cancer [1]; they also cause significant mortality in HIV-infected patients [2], [3]. HIV-positive subjects have an increased risk to develop specific lymphoma subtypes including diffuse large B-cell lymphoma (DLBCL), Burkitt lymphoma (BL) and Hodgkin lymphoma (HL) [1], [4], [5]. Molecular mechanisms of these lymphomas are quite different. While BL has a defining translocation involving the MYC locus on chromosome 8 and one of the Immunoglobulin gene loci on chromosomes 2, 14 or 22 [6], DLBCL, the most common subtype of non-Hodgkin's lymphoma (NHL), is characterized by several translocations involving the immunoglobulin locus, including t(8;14), t(3;14), and t(14;18) [7], [8]. However, a significant percentage of DLBCLs lack specific genetic abnormalities [9]. HL is characterized by increased genomic instability, even if some chromosomal aberrations and translocations involving the 3q27, 6q15, 7q22, 11q23, 14q32 loci occur with an increased frequency, there are no specific genetic aberrations that are characteristic for malignant transformation [10], [11].
We have recently addressed the link between HIV and BL and have shown that HIV-1 transactivator of transcription (Tat) protein that is released by infected cells into the blood stream, could remodel the B-cell nucleus bringing together the potential translocation partners, the MYC and IGH loci thus increasing the probability of the t(8:14) translocation characteristic of BL [12]. At the same time, an increased occurrence of DLBCL and HL in HIV-infected individuals cannot be explained by the proposed mechanism as these lymphomas are associated with chromosomal translocations that are neither specific nor well defined, though remodeling of the nucleus was observed in HL cells [13]. We have hypothesized that HIV-1 Tat might play a role in oncogenesis of HL and DLBCL via an alternative mechanism(s).
Genome instability results from mutations and chromosomal rearrangements within the genome. These mutations can be the consequence of the accumulation of DNA damage (DD) [14]. There are different exogenous and endogenous sources of DD in the cells [15]; some of this damage is due to DNA exposure to free radicals and the reactive oxygen species (ROS) [16], [17]. Oxidative DNA damage is a major source of mutation load and genomic instability [18], [19] in cells. Double-stranded DNA breaks (DSBs) induced by ROS may be converted into chromosomal translocations [20], [21], [22], [23].
In aerobic cells, ROS are generated during mitochondrial oxidative metabolism as well as in cellular response to UV radiation, xenobiotics, bacterial invasion and viral infection [24]; the mitochondria are thought to be the largest contributors to intracellular ROS production in most cell types [25], [26], [27]. Several enzymes in mitochondria are potentially capable of producing ROS [28] with nicotinamide adenine dinucleotide dehydrogenase (Complex I) playing an important role in this process [29]. ROS participate in cell signaling as secondary messengers, at the same time, overproduction of ROS and the deficiencies in the antioxidant systems leads to oxidative stress (OS) that may induce different OS-related human diseases [30]. ROS can induce oxidative DNA damage, a major source of the mutation load in living organisms, with more than one hundred oxidative DNA adducts identified. They include DNA strand breaks and oxidized base residues [31], [32], [33], [34].
HIV pathogenesis triggers OS via several proteins including the envelope glycoprotein gp120, the Vpr, Nef and Tat proteins [35], [36]. HIV-1 Tat is a small (~12 kDa) hydrophobic protein excreted by HIV-infected cells. Tat can penetrate other cell types, including B-cells [37], [38]. Inside the cell, Tat can activate both viral and cellular genes [39], [40], [41], [42]. Tat induces ROS production by activating NADPH and spermine oxidases in T-cells [43], [44]. It may also induce mitochondrial membrane permeabilization and inactivation of cytochrome c oxidase [45]. ROS may in turn oxidize nuclear DNA leading to oncogenic transformation [19]. Indeed, when expressed in mice, Tat induces B-cell lymphomas [39], [46].
Normally, living organisms tolerate the presence of ROS through the balance with the antioxidant defense system. The antioxidant system controlling ROS in living cells is divided into two groups: enzymatic and non-enzymatic antioxidants. The major intracellular non-enzymatic antioxidants are represented by the thiol groups, the most important is the glutathione (GSH) [47], [48], [49], [50]. GSH concentration is strongly dependent on the organism, cell type and extracellular or intracellular compartments [51], [52]. GSH exists in two forms, oxidized (GSSG) and reduced (GSH). The ratio of GSH/GSSG is used to measure cellular OS [53]. The reduced form of GSH exerts its antioxidant effect by providing an electron (e−) to ROS to neutralize them [54]. The diminution of GSH/GSSG ratio is one of the marker of human cancer (reviewed in [55]) and disease (reviewed in [56]). GSH synthesis is catalyzed by γ-glutamylcysteine synthetase (γ-GCS).
After the disruption of redox homeostasis, OS can trigger the activation of different signaling pathways, and it can be detected by several markers. The nuclear factor kappa B (NF-κB), a versatile transcription factor is one of the potential biomarkers for OS [57]. Interestingly, activation of NF-κB could regulate the redox state in the cells; it can be pro- or antioxidant, depending on the cellular model [58].
Here, for the first time, we have studied the effect of Tat on OS and DD in human B-cells that are not infectable by HIV and found that Tat induced mitochondrial OS in B-cells by modifying the intracellular GSH level and transcriptional deregulation of some OS key regulator genes. This induced abnormal activation of NF-κB and DD in B-cells.
2. Materials and methods
2.1. HIV patients and controls
Whole blood samples were obtained from eight HIV-positive individuals treated in the Department of Clinical Immunology, Hôpital Saint-Louis, Paris, France. In agreement with the French law, this study was registered as a non-interventional study at the Agence Nationale de Sécurité du Médicament (2016-A01316-45) and informed consent was obtained from all subjects. Their clinicopathological characteristics are summarized in Table 1. Blood samples were also obtained from healthy donors through the Etablissement Français du Sang, Rungis, France in accordance with the French national legislation.
Table 1.
Main physiopathological features in the eight individuals infected with HIV and investigated in this study.
| Patients | Test | Effective ART duration | Plasma HIV RNA | CD4+ T cells | Seric Tat concentration |
|---|---|---|---|---|---|
| #, Sex (Age, years) | # | (months) | (log10copies/ml) | (x 108 /L) | (ng/ml) |
| 1, F (48) | 1 | 54 | <1.3 | 428 | 0.40 |
| 2, F (56) | 1 | 100 | <1.3 | 502 | 2.83 |
| 3, M (59) | 1 | 66 | <1.3 | 747 | 1.14 |
| 4, M (47) | 1 | 25 | <1.3 | 417 | 2.91 |
| 5, F (51) | 1 | 102 | <1.3 | 556 | 0.21 |
| 6, M (52) | 1 | 228 | <1.3 | 494 | 0.27 |
| 7, M (33) | 1 | 11 | <1.3 | 619 | 4.06 |
| 8, F (62) | 1 | 73 | <1.3 | 536 | 9.02 |
2.2. Blood sample processing
Peripheral blood mononuclear cells (PBMC) were isolated from blood by Pancoll (PAN biotech) density gradient centrifugation. B-cells were purified by negative cell selection using the B-cell isolation kit II (Macs Milteny Biotec). Purity levels were determined by CD19 (Biolegend) and CD3 (BD Pharmigen) staining followed by FACS analysis. Purified B-cells were cultivated in vitro in the RPMI medium supplemented with 10% heat-inactivated fetal calf serum (FCS) in the presence of 1% penicillin/streptomycin (complete medium). B-cells from HIV-infected individuals were purified in a Biosafety Level 3 (BSL3) laboratory as described above.
2.3. B-cell treatment
B-cells were plated at 106 per ml and treated with purified recombinant Tat protein produced by Ablinc and obtained through the NIH AIDS Research and Reagent Program. According to the manufacturer, the Tat protein was >95% pure and purified by heparin-affinity chromatography and reverse phase HPLC which removed endotoxins [59]. If not stated differently, Tat was used for all experiments at a concentration of 250 ng/ml. TatC22 is a mutant Tat protein with a 22 Cysteine → 22 Glycine substitution whose transactivation function was abrogated [60], [61]. Produced by Abcam, the TatC22 recombinant protein was used at 250 ng/ml. Some Tat treatments of B-cells were performed in the presence of the ROS scavenger Tempol at a final concentration of 10 μM (Sigma Aldrich) or N-acetylcystein at 1 mM (NAC; Sigma Aldrich) or the mitochondrial complex I inhibitor Rotenone at 10 μM (Sigma Aldrich) or a specific mitochondria-targeting antioxidant 10-(6’-plastoquinonyl)decyltriphenyl-phosphonium (SkQ1) at 20 ng/ml (synthesized by G.A. Korshunova and N.V. Sumbatyan, Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University) or the mitochondrial uncoupler 2,4-Dinitrophenol (DNP) at 10 µM, or Bay 11-7082, a NF-κB inhibitor, at 1 mM (Calbiochem). B-cells were incubated with all mitochondria-targeting reagents for 1 h prior to Tat treatment.
2.4. Microscope image acquisition and analysis
DHE staining and Comet assay images were acquired using a fluorescent microscope (Scanner Microvision instruments) with a 20X objective. To create a full image of the specimen, images of adjacent fields were stitched together using Cartograph software (Microvision). Images of MitoSOXRed and γH2AX staining were acquired using a TCS SP8 confocal microscope (Leica Microsystems) with a 63X oil immersion objective. Z-stacks were acquired using a frame size of 1024×1024, and 0.5 µm z-steps, with sequential multitrack scanning using the 405, 488 and 543 nm laser wavelengths.
2.5. Immunofluorescence
Purified B-cells were seeded on Poly-L-lysine-coated 15×15 mm coverslips (20 min, 37 °C and 5% CO2). After adhesion, B-cells were washed with 0.3X PBS and fixed in 4% paraformaldehyde (Euromedex) in 0.3X PBS (10 min, RT) followed by 3 washes in 1X PBS. After blocking with 0.5% BSA (Euromedex) in 1X PBS (40 min, RT), cells were stained with mouse monoclonal antibodies against γH2AX (phospho Ser139 H2AX, Active Motif, 1:500) or with rabbit monoclonal antibodies against NF-κB (phosphoRelA/NF-κB p65 protein, R&D Systems, 1:100), or with rabbit monoclonal antibodies against total NF-κB (NF-κB p65 protein, Cell Signaling, 1:400). Incubation with the primary antibodies (2 h, RT) was followed by 3 washes in 1X PBS and by incubation (1 h, RT) with a secondary antibody conjugated to Alexa Fluor-488 (Life Technologies, 1:200).
2.6. Alkaline Comet assay and scoring for DNA damage
The alkaline comet assay was performed using the Trevigen CometAssay kit (Trevigen # 4250050-k) and 20-well CometSlide™ (Trevigen). Briefly, the cells were combined at 105/ml with a molten low melting point agarose (LMAgarose) at 37 °C at the ratio of 1/10 (v/v) and 50 µl were deposed onto a 20-well pre-coated CometSlide™. The agarose was allowed to solidify by placing the slides at 4 °C in the darkness for 15 min. The slides were then immersed in a pre-chilled lysis solution (Trevigen) for 1 h at 4 °C to remove the membrane, cellular proteins and histones. Following lysis, the slides were incubated in a freshly prepared alkaline DNA unwinding solution (200 mM NaOH, 1 mM EDTA pH>13) for 1 h at room temperature. The slides were then subjected to electrophoresis at 21 V for 30 min at 4 °C in the same alkaline solution, washed twice with H2O for 5 min each, washed in 70% ethanol for 5 min, dried at 37 °C for 30 min, and stained with 50 µl of diluted SYBR Green solution (1/10 000 in TE buffer pH 7.5, Trevigen). The slides were allowed to dry completely at room temperature before observation under the epifluorescence microscope (Microvision instruments, using the 488 nm laser wavelengths, 20X zoom). Images of 100 randomly selected non-overlapping cells were captured and analyzed using Tritek CometScore software. A variety of measurements including the percentage of DNA in the tail (expressed as % of total DNA), the tail length (measured from the leading edge of the comet head) and tail moment (the measure of tail length multiplied by tail intensity) were calculated to evaluate the extent of DNA damage. Mean values from at least 100 nuclei were used in calculations. All steps were conducted in the dark to minimize extraneous sources of DNA damage.
2.7. RT-qPCR
For quantitative evaluation of gene transcription levels, total RNA was extracted from purified B-cells using the NucleoSpin® RNA II kit according to the manufacturer's recommendations (Macherey-Nagel). Total RNA (1 µg) was primed in a 20 µl reaction mixture with an oligo (dT) primer and reverse transcriptase (Thermo Scientific). The obtained cDNA was amplified using specific primers and the FastStart Universal SYBR Green Master mix (Sigma-Aldrich). Expression of target genes was analyzed using the 2-ΔΔCt method with normalization against GAPDH and comparison of expression between variously treated cells. For quantification, expression levels were set to 1 in controls. The following primer sequences were used: gamma-glutamylcysteine synthetase heavy subunit (gamma-GCS-HS), forward 5’- AGGCCAGATACCTTTATGATCAGT-3’, reverse 5’- GCTGTCTATTGAGTCATATCGGGATTTAC-3’; gamma-glutamylcysteine synthetase light subunit (gamma-GCS-LS), forward 5’- TGTCTTGGAATGCACTGTATCTCATGC-3’, reverse 5’- TTCAATAGGAGGTGAAGCAATGATCAC-3’; GAPDH forward 5’-CTGCACCACCAACTGCTTAG-3’, reverse 5’-AGGTCCACCACTGACACGTT-3’.
2.8. DHE staining
Superoxide levels were measured using the Dihydroethidium probe (DHE) (Muse oxidative stress kit, Millipore). In the presence of the superoxide anion, DHE is rapidly oxidized to oxyethidium, which binds DNA and emits light in the 570–580 nm ranges when excited at 488 nm. B-cells after appropriate treatments were rapidly washed in 0.3X PBS, fixed in 4% paraformaldehyde (Euromedex) in 0.3X PBS (10 min, RT) and incubated in a DHE-containing ROS staining solution diluted to 1:8000 in a proprietary assay buffer for 15 min in the dark at room temperature while shaking, washed 3 times with 1X PBS, mounted using a DAPI mounting medium (Vector laboratories) and observed under a fluorescent microscope (excitation/emission: 520/610 nm, red fluorescence). Images from adjacent fields were stitched together by using Cartograph software (Microvision) to create one large image of the specimen. The fluorescence of at least 1000 cells per sample was quantified using ImageJ software, normalized to cells number following DAPI staining. After background subtraction using the "rolling ball" algorithm [62], the integrated density value of each cell was measured automatically by the software.
In the graphical representations, the value corresponding to untreated B-cells (used as negative control) was set as 1 and the value of the tested conditions was represented as the fold change compared to the negative control.
2.9. Amplex red assay
H2O2 generation by Tat-treated B-cells was quantified by the Amplex red/horseradish peroxidase assay (Sigma Aldrich). Cells (7.5–10 × 106) in the Dulbecco's phosphate-buffered saline (D-PBS) supplemented with CaCl2 and MgCl2 were incubated with D-glucose (1 mg/ml), horseradish peroxidase (0.5 U/ml; Roche), and Amplex red (50 mM; Sigma Aldrich); the fluorescence was immediately measured in a microplate reader (Victor3; PerkinElmer) at 37 °C for 40 min using excitation at 530 nm and emission at 595 nm. Released H2O2 concentration (nmol/hour*107 cells) was quantified using standard calibration curves [63], [64].
2.10. MitoSOXRed staining
After adhesion on Poly-L-Lysine coated slides, and washing with 1X PBS, B-cells were incubated in darkness with 1.5 ml of 5 µM solution of MitoSOXred (Thermo Fisher) for 20 min at 37 °C. The B-cells were then washed gently three times with warm 1X PBS, stained with Hoechst, and analyzed using confocal (Leica SP8) and fluorescent (Scanner Microvision instruments) microscopes.
2.11. GSH and GSH/GSSG ratio determination
Total glutathione concentrations were measured in B lymphocytes isolated from the blood of healthy donors using GSH/GSSG Ratio Detection Assay Kit II (Abcam #205811), according to the manufacturers’ protocol.
2.12. Flow cytometry
After Tat treatment, cells were washed and resuspended in 1X PBS containing 100 μM monochlorobimane (MClB) (Sigma-Aldrich), to measure the intracellular GSH level [65], [66], [67]. After incubation for 20 min in the dark, cells were washed, resuspended in 1X PBS, and MClB fluorescence was immediately measured using a flow cytometer (CyAn, Beckman Coulter) and analyzed by using Summit V4.3 software.
2.13. Western blot
Nuclear and cytosolic protein fractions were prepared from 108 B-cells using the NE-PER Nuclear and Cytoplasmic Extraction Reagents kit (Thermo Scientific) following the manufacturer's instructions. The proteins were quantified using the Pierce BCA protein assay kit (Thermo Scientific) with absorbance measured at 562 nm with a Nano-Drop 2000c (Thermo Scientific). The protein fractions thus obtained were mixed with the NuPAGE LDS Sample Buffer 4x (Thermo Scientific), denatured (85 °C, 10 min), loaded onto a 4–12% Bis-Tris gel (Thermo Scientific) and run at 190 V for 1 h at RT. Proteins were transferred from the gels onto PVDF membranes (Merck Millipore) using a semi-dry transfer cell (25 V overnight at 4 °C). Blots were revealed with AmidoBlack and after washing with sterile H2O blocked in 5% (w/v) BSA in TBS and 0.1% Tween (1 h, RT). After three washes in TBS and 0.1% Tween (5 min, RT), the membrane was incubated with antibodies against human Phospho-Rel A/NF-κB p65 (R&D Systems, 1:1000), mouse antibodies against α-tubulin (Sigma, 1:1000) or human Topoisomerase II (1:10000, Merck Millipore) for 2 h at RT with gentle rocking. After three washes in 0.1% TBS with 0.1% Tween, the membrane was incubated (1 h, RT) with an anti-rabbit secondary antibody conjugated to horseradish peroxidase (Jackson ImmunoResearch). Blots were revealed using Immobilon Western Chemiluminescent HRP substrate (Millipore) and an ImageQuant Las 4000 mini (GE Healthcare). The obtained bands were then analyzed by density measurement using the ImageJ software. For quantification, protein levels in controls were set to 1.
2.14. Preparation of chromosome spreads
Lymphoblastoid cell line RPMI8866 was kindly provided by Joelle Wiels (UMR 8126-CNRS, Villejuif, France). Cells were cultivated in the RPMI medium supplemented with 10% heat inactivated FCS, 1% of L-Glutammine, 1% Sodium Pyruvate, 2% Glucose, Plasmocin 2.5 mg/ml, 1% penicillin/streptomycin (complete medium). They were treated with Tat or Tat together with Tempol at the indicated concentrations or left untreated for 5 days. Chromosome spreads were prepared as previously described [68] and analyzed at the Hôpital Saint Antoine-Paris, France.
2.15. Statistical analyses
Comparisons between more than two averages were performed using the Bonferroni's post-test based on the results generated using the one-way Anova test. The Student t-test (two-tail distribution) was used to compare means between only two samples. All tests were performed using the Graphpad Prism 5 software (Graphpad software).
3. Results
3.1. Tat induces ROS and oxidative DNA damage in B-cells
The HIV-1 Tat protein is present in blood of infected individuals in concentrations ranging from 1 to 500 ng/ml and can penetrate into uninfected cells [69], [70], [71]. We have tested whether Tat could induce ROS production in B-cells. B lymphocytes purified from blood of healthy donors were treated with 250 ng/ml of purified Tat protein for various times and then analyzed for the presence of ROS. To this aim, untreated and Tat-treated B-cells were stained with the ROS sensor Dihydroethidium (DHE) which, in the presence of ROS, is converted to the red fluorescent compound oxyethidium [72], [73]. The integrated density of red fluorescence was quantified automatically using an ImageJ algorithm and normalized to cells numbers, as described in Material and Methods. In Tat-treated cells, ROS level increased with time, reaching a peak at 6 h, compared to the controls (1.8±0.1-fold higher; p<0.001) (Fig. 1A). Representative images of DHE staining of Tat-treated and the control cells are shown in Fig. 1B. A basal level of ROS staining is observed in untreated B-cells, because the production of ROS is common to all aerobic cells [74]. The observed effect of Tat was dose-dependent (Fig. 1C). We next confirmed that Tat induced a significant level of ROS using a complementary approach, Amplex Red assay to detect H2O2 production [63], [64]. The concentration of H2O2 released by the Tat-treated B-cells after 6 h of treatment, was significantly higher than that released by the untreated controls (p<0.05; Fig. 1D).
Fig. 1.

HIV-1 Tat protein induces oxidative DNA damage via ROS production. (A) ROS production in Tat-treated and untreated B-cells. ROS production was detected in human B-cells isolated from peripheral blood of healthy donors and treated with 250 ng/ml Tat for 1, 6 and 12 h by DHE staining as described in Materials and Methods. The Y axis shows the ROS level in treated B-cells compared to the untreated control, for which the obtained basal value was set as 1. At least 1000 cells were analyzed for each condition, and quantified using the ImageJ algorithm normalized to cells number. The experiments were carried out in duplicate. (B) Representative images of DHE-stained B-cells in the presence or absence of Tat for 6 h. The intensity of red fluorescence reflects the level of ROS in B-cells. DAPI is shown in blue and oxyethidium in red. Scale Bar = 10 µm. (C) Tat induces ROS production in a dose-dependent manner. ROS production was detected by DHE staining in cells treated with 15 and 250 ng/ml Tat for 6 h. The Y axis shows the ROS level in treated B-cells compared to the untreated control, for which the obtained basal value was set as 1. The fluorescence intensity was calculated from a minimum of 1000 cells examined for each condition. The experiments were carried out in duplicate. (D) Detection of ROS in Tat-treated cells with the Amplex red assay. The concentration of H2O2 released in nmol/hour*107 cells by B-cells treated with 250 ng/ml Tat and untreated B cells after 6 h is represented on Y axis. The experiments were carried out in duplicate. (E) Tat induces DNA damage in B-cells. The Y axis shows the proportion of B-cells treated with 250 ng/ml Tat for 6 h and exhibiting γH2AX staining compared to the negative control, for which the obtained basal value was set as 1. A minimum of 300 cells were examined per time point. The data are means ± SEM from at least three fields with B-cells from a single donor at every time point except at 6 h, when experiments were repeated on B-cells from three different healthy donors. (F) Kinetic analysis of Tat-induced DNA Damage by Comet Assay. B-cells were treated with 250 ng/ml Tat for 1, 6 and 12 h. The Y axis shows the tail moment representing the level of DNA damage in Tat-treated B-cells compared to the controls, for which the obtained basal value was set as 1. The experiment was performed on B-cells from three different healthy donors. At least 100 nuclei were analyzed in each experiment. (G) ROS scavenger Tempol and the antioxidant NAC inhibit Tat-induced ROS production. The Y axis shows ROS production in B-cells treated with 250 ng/ml Tat alone or with 10 µM Tempol or 1 mM NAC for 6 h compared to the untreated control, for which the obtained basal value was set as 1. The experiment was performed on B-cells from two different healthy donors. (H) ROS scavenger Tempol and the antioxidant NAC inhibit Tat-induced DNA Damage. B-cells were treated either with 250 ng/ml Tat or simultaneously with 250 ng/ml Tat and 10 µM Tempol or 1 mM NAC, fixed at 6 h post-treatment and immunostained for γH2AX. The Y axis shows the proportion of cells exhibiting γH2AX staining compared to the untreated B-cells, for which the obtained basal value was set as 1. The results represent a minimum of 300 cells examined per time point. The experiments were carried out in duplicate. All data are expressed as the mean±SEM. The statistical analyses were carried out by the one-way ANOVA test. The statistical significance was calculated vs. untreated controls. (I) Tat induces chromosomal aberrations in LCL 8866. Cells were treated either with 250 ng/ml Tat or simultaneously with 250 ng/ml Tat and 10 µM Tempol for 5 days and processed as described in Materials and Methods. The Y axis shows the average number of chromosomal aberrations per cell compared to the untreated LCLs, for which the obtained basal value was set as 1. The experiments were carried out in duplicate. All data are expressed as the mean±SEM. The statistical analyses were carried out by the one-way ANOVA test. The statistical significance was calculated vs. untreated controls; ***: p value<0.001; **: 0.001 <p< 0.01; * 0.01 <p<0.5.
A significant share of DNA damage occurring in living cells is caused by ROS [75], [76]. ROS-induced DD can lead to development of pathological conditions, including cancer [30], [77]. We next used γH2AX staining to test whether Tat could induce DSBs in B-cells. Indeed, the number of Tat-treated B-cells showing γH2AX staining was 1.63±0.02-fold higher than in the untreated controls at 6 h (p<0.001; Fig. 1E); this coincided with the maximal ROS production in Tat-treated B-cells. Representative images of the γH2AX staining are shown in Fig. SA. This result was confirmed using the single cell alkaline gel electrophoresis (Comet assay). In this experiment, the level of DNA damage was 2.1±0.2 times higher in Tat-treated B-cells compared to the controls (p<0.001; Fig. 1F, Supplementary Fig. B).
In order to test whether there is a link between Tat-induced ROS and DNA damage, we have next simultaneously treated B-cells with Tat and 10 μM of the ROS scavenger Tempol or 1 mM of the antioxidant 1- N-acetylcysteine (NAC). Tat can induce neither ROS production (Fig. 1G) nor DD (Fig. 1H) in the presence of Tempol or NAC. These results indicate a direct link between Tat-induced ROS production and oxidative DNA damage. We also observed that Tat induced a higher number of chromosomal abnormalities in a lymphoblastoid B-cell line RPMI8866 compared to the untreated control (1.25±0.05 fold; p<0.05). This effect was reduced by Tempol treatment (Fig. 1I).
3.2. OS and DD are present in B-cells from HIV-infected individuals
We next analyzed the OS status and the presence of DD in B-cells isolated from blood of eight different HIV-infected patients. We found that B-cells of HIV-positive subjects had a significantly higher level of OS as compared to the healthy controls (2.2±0.2-fold; p<0.001; Fig. 2A). We next evaluated the level of DD in HIV-infected individuals vs. healthy donors by γH2AX staining and found out that in HIV patients, the level of DD was increased 7.4 ± 0.8-fold on average (p<0.001; Fig. 2B). We have also found that Tat was present in blood of these HIV patients in concentration ranging from 1 to 10 ng/ml (data not shown).
Fig. 2.

HIV-1 induces oxidative DNA damage via ROS production in B-cells from HIV patients. (A) ROS production in B-cells isolated from the blood of healthy donors and HIV-infected individuals detected by DHE staining. The Y axis shows the ROS level in B-cells from HIV-patients compared to that in healthy donors, for which the obtained basal value was set as 1. A minimum of 1000 cells were examined for each sample. (B) DNA damage in B-cells isolated from the blood of healthy donors and HIV patients immunostained for γH2AX. The Y axis shows the proportion of cells from HIV-infected individuals exhibiting γH2AX staining compared to B-cells from healthy donors (negative control), for which the obtained basal value was set as 1. A minimum of 300 cells were examined for each sample. All data are expressed as the mean ± SEM. The statistical analyses were carried out by the one-way ANOVA test. The statistical significance was calculated vs. untreated controls; ***: p value<0.001). Eight different HIV patients were examined in this study (see Table 1).
3.3. Tat induces mitochondrial OS in B-cells
Mitochondria are one of the most important sources of ROS in the cells and it was shown that Tat could perturb the normal mitochondrial function that in turn could regulate the redox state of the cells [78], [79], [80]. To test whether Tat induced mitochondrial ROS production in B-cells, we used rotenone (10 µM), an inhibitor of the mitochondrial complex I, SkQ1 (20 ng/ml), a specific mitochondria-targeting antioxidant [81], [82] and 2,4-dinitrophenol (DNP) (10 µM), an uncoupler of oxidative phosphorylation [83]. We treated B-cells with these different molecules simultaneously with 250 ng/ml Tat for 6 h and observed that Tat-induced ROS production was inhibited in their presence to the control levels, indicating that mitochondria were a source of ROS in Tat-treated cells (Fig. 3A).
Fig. 3.

HIV-1 Tat protein induces mitochondrial OS in B-cells. (A) Tat-induced ROS production is diminished by the mitochondrial ROS production inhibition. B-cells were treated with 250 ng/ml Tat alone or with 10 µM mitochondrial complex I inhibitor rotenone or 20 ng/ml mitochondria-targeting antioxidant SkQ1 or 10 µM uncoupler of oxidative phosphorylation DNP for 6 h and stained with DHE. The Y axis shows ROS production calculated from a minimum of 100 cells examined for each condition and compared to the negative control for which the obtained basal value was set as 1. The experiment was carried out on blood from three different healthy donors. (B) Representative images of MitoSOXRed-stained B-cells. Hoechst is shown in blue (nuclear staining), MitoSOX in red. Scale Bar = 10 µm. (C) Tat induces superoxide anion production in mitochondria of B-cells. B lymphocytes treated with 250 ng/ml Tat alone or with 20 ng/ml SkQ1 were stained with MitoSOXRed and visualized by fluorescence microscopy 6 h after treatment. ROS production was quantified as described in Materials and Methods. The Y axis represents the proportion of cells with MitoSOXRed staining in Tat-treated B-cells compared to the untreated control, for which the obtained basal value was set as 1. The values were calculated from a minimum of 100 cells examined for each condition. The experiment was carried out on blood from two different healthy donors. (D) SkQ1 inhibits Tat-induced DD. B-cells were treated with 250 ng/ml Tat alone or together with 20 ng/ml SkQ1 and then fixed at 6 h post-treatment and immunostained for γH2AX. The Y axis shows the proportion of γH2AX-positive Tat-treated B-cells compared to the untreated controls, for which the obtained basal value was set as 1. The number of γH2AX positive cells was calculated from a minimum of 300 cells examined per experimental point. The experiment was carried out on blood from two healthy donors. All data are expressed as the mean±SEM. The statistical analyses were carried out by the one-way ANOVA test. The statistical significance was calculated vs. untreated controls; ***: p value<0.001.
We further ascertained the mitochondrial nature of Tat-induced ROS using MitoSOXRed, a specific mitochondrial superoxide indicator for living cells. B-cells were treated with 250 ng/ml Tat for 6 h, stained in vivo with the MitoSOXRed, visualized by confocal microscopy and analyzed as described in Materials and Methods. Stronger MitoSOXRed staining was observed in Tat-treated cells compared to the untreated control (Fig. 3B, Supplementary Fig. C). In Tat-treated cells, MitoSOXRed fluorescence representing the level of mitochondrial superoxide anion was 3.7±0.2 times higher than in the control B-cells (p<0.001). Addition of 20 ng/ml of SkQ1 prior to Tat treatment reduced the level of the mitochondrial superoxide to the basal level (1±0.1 relative to the untreated controls; Fig. 3C). These results confirmed the mitochondrial origin of Tat-induced ROS production. We next confirmed that Tat-induced mitochondrial ROS production was implicated in the oxidative DD. We evaluated the number of γH2AX positive B-cells treated with 250 ng/ml of Tat and B-cells treated with Tat and 20 ng/ml of SkQ1. Inhibition of mitochondria-derived ROS by SkQ1 also reduced DD to basal levels thus confirming our hypothesis (Fig. 3D).
3.4. Tat induces glutathione deficiency in B-cells
ROS production is usually kept under control through a finely regulated balance between pro-oxidant and anti-oxidant factors, including GSH [84], [85], [86]. We next quantified the intracellular levels of reduced GSH, the most abundant non-protein thiol, using monochlorobimane (MClB), a cell-permeant probe for the detection of thiol groups. Tat-treated and untreated B-cells were stained by MClB as described in Materials and Methods and analyzed by FACS. We observed depletion of GSH in Tat-treated cells as early as 1 h after the beginning of Tat treatment (p<0.01) (Fig. 4A). We next analyzed the concentration of total GSH (GSH+GSSG) and the GSH/GSSG ratio in B-cells treated with Tat. Both values decreased after Tat treatment (Fig. 4B, C, respectively).
Fig. 4.

Tat induces glutathione deficiency in B-cells. (A) Intracellular glutathione (GSH) staining using monochlorobimane (MClB). B-cells treated with 250 ng/ml of Tat for 1 h and untreated controls were stained with MClB and the amount of emitted fluorescence was analyzed by FACS. The experiment was carried out on blood from two different healthy donors. Total glutathione (GSH) (B) and GSH/GSSG ratio (C) determined using the GSH/GSSG Ratio Detection Assay Kit. B-cells treated with 250 ng/ml of Tat for 1 h and untreated controls were processed following the manufacturer's instructions. The experiment was carried out on blood from two different healthy donors. (D) Analysis of expression of genes encoding for heavy and light subunits of γ-GCS in B-cells treated with Tat. The transcriptional levels were measured by RT-qPCR after 1 h of treatment with 250 ng/ml Tat. Data represent means ± SEM from three different donors and are expressed as fold change normalized vs. GAPDH and compared to untreated controls. (E) Transcriptional activity of Tat is essential for ROS production. B-cells were treated with either 250 ng/ml Tat or its transcriptionally inactive form TatC22. ROS production was measured by DHE staining 6 h after treatment. The Y axis shows ROS production calculated from a minimum of 1000 cells examined for each condition, compared to the untreated B-cells, for which the obtained basal value was set as 1. The experiment was carried out on blood from two different healthy donors. (F) Transcriptional activity of Tat is essential for DNA damage induction. B-cells were treated with 250 ng/ml of either Tat or TatC22 mutant, and then fixed at 6 h post-treatment and immunostained for γH2AX. The number γH2AX-positive cells was calculated from a minimum of 300 cells was examined per time point, compared to B-cells as negative control, for which the obtained basal value was set as 1. The experiment was carried out on blood from three different healthy donors. All data are expressed as the mean ± SEM. The statistical analyses were carried out by the one-way ANOVA test. The statistical significance was calculated vs. untreated controls; ***: p value<0.001; **: 0.001 <p< 0.01; * 0.01 <p<0.5.
As Tat is a transcriptional activator known to regulate hundreds of host cell genes [41], [42], we next tested whether Tat could affect expression of genes involved in GSH synthesis. Indeed, 1 h after addition of Tat to B-cells, the level of expression of the light and heavy subunits of the gamma-glutamylcysteine synthetase (γ-GCS) decreased 3.57±0.06 and 2.7±0.03 fold, respectively (Fig. 4D).
HIV-1 Tat has a pronounced transcriptional activity, but it can also affect host cells by interacting with a multitude of proteins ([87], [88] reviewed in [38]). To prove that transactivation activity of Tat is essential for ROS induction, we treated isolated B-cells with a mutated form of the Tat protein (TatC22) where a cysteine is substituted by a glycine in the position 22. This mutation abolishes the transcriptional activity of Tat [60], [89]. Addition of TatC22 to isolated B-cells from healthy donors induced neither ROS production (Fig. 4E), nor DD (Fig. 4F). TatC22 also failed to downregulate GSH levels and to affect the expression of oxidative stress related genes (data not shown), suggesting that the transcriptional activity of Tat is essential for induction of OS and DD.
3.5. Pro-oxidant effect of Tat-activated NF-κB in B-cells
NF-κB is a versatile transcription factor that is often upregulated and activated in HIV-infected cells [38]. NF-κB controls and regulates several cellular processes including ROS balance [58]; it can also be regulated by OS [90]. When activated, NF-κB p65 subunit relocalizes into the nucleus and may induce pro- or antioxidant pathways, depending on the cellular context [91], [92]. We have first analyzed the presence of nuclear NF-κB p65 in Tat treated or untreated B-cells. We used anti-NF-κB p65 monoclonal antibody to detect by immunofluorescence the nuclear NF-κB, and we found that the number of nuclei with NF-κB-p65 staining was 1.61±0.33 times higher in Tat-treated B-cells as compared to the negative control (p<0.001; Fig. 5A).
Fig. 5.

Tat activates NF-κB in B-cells. (A) Tat induces nuclear translocation of NF-κB in B-cells. The Y axis shows the number of cells exhibiting NF-κB/p65 nuclear staining in B-cells treated with 250 ng/ml of Tat for 6 h, compared to the untreated control, for which the obtained basal value was set as 1. A minimum of 100 cells were analyzed per condition. The experiment was carried out on blood from one healthy donor. (B) Tat-induced nuclear translocation of phosphorylated NF-κB RelA/p65 subunit in normal B-cells. Right panel: Western blot analysis of cytosolic and nuclear fractions from normal B-cells incubated or not with 250 ng/ml Tat for 6 h. α-tubulin was used as cytosolic marker and DNA topoisomerase II as a nuclear marker. Left panel: the bands obtained with blot were quantified by densitometric analysis with ImageJ software. The intensity of phosphorylated RelA/p65 (S536) bands was normalized with the intensity of the controls (α-tubulin and DNA topoisomerase II). The values found for the untreated control B-cells were set as 1 and the fold change in the presence of Tat was calculated. The experiment was carried out on blood from two different healthy donors. (C) NF-κB activation is regulated by OS and Tat transcriptional activity. The Y axis shows the proportion of B-cells treated with 250 ng/ml Tat exhibiting the nuclear staining for the phosphorylated RelA/p65 compared to the untreated control, for which the obtained basal value was set as 1. A minimum of 100 cells was analyzed per condition. The experiment was carried out on blood from two different healthy donors. (D) NF-κB inhibition decreases ROS production in Tat-treated B-cells. B-cells were treated with 250 ng/ml Tat alone or with 1 mM NF-κB inhibitor BAY 11–7082. ROS production was measured by DHE staining 6 h after treatment. The Y axis shows ROS production in Tat-treated B-cells compared to the negative control, for which the obtained basal value was set as 1. A minimum of 1000 cells were examined for each condition. The experiment was carried out on blood from two different healthy donors. (E) NF-κB inhibition leads to the reduction of Tat-induced DD. The Y axis shows the proportion of γH2AX-positive B-cells treated with 250 ng/ml Tat alone or together with 1 mM NF-κB inhibitor Bay 11–7082, for 6 h, compared to the untreated controls, for which the obtained basal value was set as 1. The results were calculated from a minimum of 100 cells examined per condition. The experiment was carried out on blood from two different healthy donors. All data are expressed as the mean±SEM. The statistical analyses were carried out by the one-way ANOVA test. The statistical significance was calculated vs. untreated controls; ***: p value<0.001; **: 0.001 <p< 0.01; * 0.01 <p<0.5.
Phosphorylation of RelA/p65 subunit at serine residues 276, 529, and 536 facilitates its nuclear translocation [93], [94] and is involved in the response to OS [95]. We next tested whether the phosphorylated form of RelA/p65 was translocated into the nuclei in Tat-treated B-cells. We prepared cytosolic and nuclear protein extracts and used Western blot analysis to analyze the relative nuclear vs. cytosolic localization of RelA/p65 subunit. After 6 h of Tat treatment, the nuclear abundance of the phosphorylated RelA/p65 (S536) was 6.4±0.08 fold higher in Tat-treated cells than in the control B-cells (p<0.001; Fig. 5B). Interestingly, the use of Tempol diminished Tat-induced NF-κB activation, suggesting that Tat-induced ROS were essential for this effect (Fig. 5C). Moreover, NF-κB activation by Tat was also dependent on its transactivation activity as TatC22 did not induce nuclear relocalization of NF-κB (Fig. 5C).
To understand whether NF-κB played a pro- or antioxidant role in Tat-treated B-cells, we used Bay 11-7082, an inhibitor of IκBα phosphorylation and therefore of NF-κB activation [96]. Neither ROS induction nor DD were observed in B-cells treated simultaneously with 250 ng/ml Tat and 1 mM Bay 11–7082 (Fig. 5D, E, respectively); thus abnormal activation of NF-κB played a pro-oxidant role in Tat-treated B-cells.
4. Discussion
This study addresses a puzzling question of HIV-associated B-cell oncogenesis by linking HIV-1 Tat to oxidative DNA damage (DD) and genomic instability that are important features of cancer cells. HIV-1 does not infect B-cells, but most HIV-related lymphomas are of B-cell origin [1]. Several mechanisms were proposed to explain this phenomenon, the most important being the depletion of T-lymphocytes. Indeed, lower CD4 counts were associated with B-cell lymphomas [97]. While introduction of highly active antiretroviral therapy (HAART) has been associated with a decrease in AIDS-associated malignancies [98], HIV-infected persons still have an increased risk to develop specific lymphoma subtypes including DLBCL, BL and HL [1], [4], [99]. Several HIV proteins have been proposed to promote DD and genomic instability in infected cells (reviewed in [38], [100]). However, their overall oncogenic effect on B-cells is likely to be limited, given that HIV-1 does not infect B-cells. One exception from this is the HIV-1 Tat protein that is secreted into blood by infected cells. Tat is able to transduce B-cells [37]. We have recently shown that Tat could affect expression of several host genes and induce spatial proximity between potential translocation partners in BL, the MYC and IGH gene loci [12]. We have hypothesized that HIV-1 Tat might play a role in oncogenesis of other HIV-related B-cells lymphomas via an alternative mechanism(s), e.g. by inducing DD and genetic instability in B-cells. Indeed, we have shown that B-cells of HIV-infected individuals had a significantly higher level of DD as compared to healthy controls. We have further demonstrated that incubation of HIV-1 Tat alone was sufficient to initiate a cascade of events that eventually resulted in the generation of DSBs. The capacity of Tat to induce an oncogenic phenotype is substantiated by the increased frequency of B-cell lymphomas in mice expressing Tat [39], [46].
Like many viruses, HIV-1 initiates OS in infected cells. However, HIV-1-infected cells may induce OS in uninfected cell types through the release of low molecular weight mediators or secreted proteins, including Tat [101]. The increased levels of ROS, together with the reversal of DD by treatment with antioxidants, including mitochondria-targeting ones, demonstrate that Tat promotes genomic instability via induction of mitochondrial ROS. Previously, high levels of OS were reported in cells infected with HIV-1 [35], [36]. Tat was shown to induce ROS production by activating NADPH and spermine oxidases and mitochondrial membrane permeabilization and inactivation of cytochrome c oxidase in T-cells [43], [44], [45]. Tat can also induce OS by down-regulation of the antioxidant mechanisms. Here we have shown that Tat induced mitochondrial ROS generation and a simultaneous decrease in cellular GSH/GSSG ratio in B-cells. Our data are in agreement with the previous study where Tat was found to reduce the GSH/GSSG ratio in brain endothelial cells [102]. B-cells have a limited uptake capacity for extracellular cystine [103], a rate-limiting substrate for GSH synthesis, as well a low level of gamma-glutamyl transpeptidase (γGT) expression [104], [105]. This could explain the low level of reduced GSH detected in this study and high sensitivity of B-cells to OS. Moreover we have observed downregulation of expression of the two subunits (light and heavy) of the gamma-glutamylcysteine synthetase (γ-GCS) after addition of Tat to B-cells explaining reduction of total glutathione content. In this study, we demonstrated for the first time, the pro-oxidant effect of Tat via depletion of reduced GSH, total GSH and the GSH/GSSG ratio in B-cells.
This in turn triggered NF-κB activation that further enhanced OS and DD. In addition to induction of oxidative DD, ROS may also oxidize other cellular molecules including proteins and lipids thus altering signaling cascades that regulate cell growth, differentiation and apoptosis. Through the induction of ROS, Tat may promote transformation by initiating signaling cascades that are further activated by other factors, for example by NF-κB. Tat can act on several key elements in the signaling pathway of NF-κB. A direct physical interaction between Tat and the IκBα inhibitor has been demonstrated; Tat competes with the IκB, trapping it, leading to the release of NF-κB into nucleus [106]. Moreover, Tat can also interact directly with the p65 subunit of NF-κB; this promotes the p65 DNA-binding affinity and the transcriptional activity [107], [108]. In addition Tat can bind to the enhancer element of the promoter of certain NF-κB-regulated genes promoting the recruitment and the activation of NF-κB [40], [109]. In our case, transcriptional activity of Tat rather that protein-protein interactions was required to activate NF-κB.
Our findings suggest that the induction of DD and genomic instability by ROS in B-cells may be unwanted consequences of the strategy used by HIV to infect T-cells, as ROS as second messengers initiate a cascade for NF-κB activation; NF-κB controls transcription of genes involved HIV replication and this increases the replication of HIV [110]. Conversely, DD produced by ROS favors HIV integration of into the host genome [111].
Immunization with Tat is proposed as one of the HIV vaccination strategies [112]. Our study shows that this approach may have potentially dangerous consequences. The use of antioxidants was proposed as a potential therapy of the HIV infection for many years (reviewed in [113]), although conflicting evidence for their beneficial effect was obtained so far. Here we have shown that Tat induced mitochondrial ROS. New generation of antioxidants specifically targeting mitochondria is currently being developed [114], [115], [116]. Their use may allow for further advances in understanding the processed involved in HIV-induced oncogenesis. The use of recently developed Tat inhibitors [117] may also potentially reduce the occurrence of HIV-related lymphomas. Further studies of the mechanisms linking the HIV-1 Tat, ROS and DD in B-cells are required if potential targets for therapy/prevention are going to be developed.
Acknowledgements
This research was supported by grants from ANRS, INSERM (ENVIBURKITT), La Ligue Contre le Cancer (M27231) to YSV, the Russian Foundation for Basic Research (16-54-16001) to EVS and the Russian Scientific Foundation (14-24-00107) to BC. TT was a recipient of the ANRS fellowship. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments
Competing interests
The authors have declared that no competing interests exist.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2017.11.024.
Appendix A. Supplementary material
(A) Representative images of γH2AX staining of B-cells treated with 250 ng/ml Tat for 6 h. γH2AX is shown in green, DAPI nuclear staining in blue. Scale bar=10 µm. (B) Representative images of the Comet assay of B-cells treated with 250 ng/ml Tat for 6 h. Scale bar=50 µm. (C) Representative images of MitoSOXRed-stained B-cells treated with 250 ng/ml Tat for 6 h. Hoechst nuclear staining is shown in blue, MitoSOX in red. Scale Bar = 10 µm. Supplementary material
References
- 1.Gibson T.M., Morton L.M., Shiels M.S., Clarke C.A., Engels E.A. Risk of non-Hodgkin lymphoma subtypes in HIV-infected people during the HAART era: a population-based study. AIDS. 2014;28:2313–2318. doi: 10.1097/QAD.0000000000000428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Achenbach C.J., Buchanan A.L., Cole S.R., Hou L., Mugavero M.J., Crane H.M., Moore R.D., Haubrich R.H., Gopal S., Eron J.J., Hunt P.W., Rodriguez B., Mayer K., Saag M.S., Kitahata M.M. HIV viremia and incidence of non-Hodgkin lymphoma in patients successfully treated with antiretroviral therapy. Clin. Infect. Dis. 2014;58:1599–1606. doi: 10.1093/cid/ciu076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Howlader N., Shiels M.S., Mariotto A.B., Engels E.A. Contributions of HIV to non-Hodgkin lymphoma mortality trends in the United States. Cancer Epidemiol. Biomark. Prev. 2016;25:1289–1296. doi: 10.1158/1055-9965.EPI-16-0273. [DOI] [PubMed] [Google Scholar]
- 4.Chao C., Xu L., Abrams D., Leyden W., Horberg M., Towner W., Klein D., Tang B., Silverberg M. Survival of non-Hodgkin lymphoma patients with and without HIV infection in the era of combined antiretroviral therapy. AIDS. 2010;24:1765–1770. doi: 10.1097/QAD.0b013e32833a0961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dunleavy K., Wilson W.H. How I treat HIV-associated lymphoma. Blood. 2012;119:3245–3255. doi: 10.1182/blood-2011-08-373738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ribrag V., Camara-Clayette V., Bosq J., Vasstezky Y. Lymphome de Burkitt. EMC - Hématol. 2012;7:1–11. [Google Scholar]
- 7.Kramer M.H., Hermans J., Wijburg E., Philippo K., Geelen E., van Krieken J.H., de Jong D., Maartense E., Schuuring E., Kluin P.M. Clinical relevance of BCL2, BCL6, and MYC rearrangements in diffuse large B-cell lymphoma. Blood. 1998;92:3152–3162. [PubMed] [Google Scholar]
- 8.Iqbal J., Sanger W.G., Horsman D.E., Rosenwald A., Pickering D.L., Dave B., Dave S., Xiao L., Cao K., Zhu Q., Sherman S., Hans C.P., Weisenburger D.D., Greiner T.C., Gascoyne R.D., Ott G., Müller-Hermelink H.K., Delabie J., Braziel R.M., Jaffe E.S., Campo E., Lynch J.C., Connors J.M., Vose J.M., Armitage J.O., Grogan T.M., Staudt L.M., Chan W.C. BCL2 translocation defines a unique tumor subset within the germinal center B-cell-like diffuse large B-cell lymphoma. Am. J. Pathol. 2004;165:159–166. doi: 10.1016/s0002-9440(10)63284-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vose J.M. Current approaches to the management of non-Hodgkin's lymphoma. Semin. Oncol. 1998;25:483–491. [PubMed] [Google Scholar]
- 10.Cabannes E., Khan G., Aillet F., Jarrett R.F., Hay R.T. Mutations in the IkBa gene in Hodgkin's disease suggest a tumour suppressor role for IkappaBalpha. Oncogene. 1999;18:3063–3070. doi: 10.1038/sj.onc.1202893. [DOI] [PubMed] [Google Scholar]
- 11.Pasqualucci L., Neumeister P., Goossens T., Nanjangud G., Chaganti R.S., Kuppers R., Dalla-Favera R. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature. 2001;412:341–346. doi: 10.1038/35085588. [DOI] [PubMed] [Google Scholar]
- 12.D. Germini, T. Tsfasman, M. Klibi, R. El-Amine, A. Pichugin, O.V. Iarovaia, C. Bilhou-Nabera, F. Subra, Y. Bou Saada, A. Sukhanova, D. Boutboul, M. Raphaël, J. Wiels, S.V. Razin, S. Bury-Moné, E. Oksenhendler, M. Lipinski, Y.S. Vassetzky, HIV Tat induces a prolonged MYC relocalization next to IGH in circulating B-cells, Leukemia 31, 2017, 2515–2522, 〈http://dx.doi.org/10.1038/leu.2017.106〉. [DOI] [PubMed]
- 13.Guffei A., Sarkar R., Klewes L., Righolt C., Knecht H., Mai S. Dynamic chromosomal rearrangements in Hodgkin's lymphoma are due to ongoing three-dimensional nuclear remodeling and breakage-bridge-fusion cycles. Haematologica. 2010;95:2038–2046. doi: 10.3324/haematol.2010.030171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tubbs A., Nussenzweig A. Endogenous DNA damage as a source of genomic instability in Cancer. Cell. 2017;168:644–656. doi: 10.1016/j.cell.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Helleday T., Eshtad S., Nik-Zainal S. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 2014;15:585–598. doi: 10.1038/nrg3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jena N.R. DNA damage by reactive species: mechanisms, mutation and repair. J. Biosci. 2012;37:503–507. doi: 10.1007/s12038-012-9218-2. [DOI] [PubMed] [Google Scholar]
- 17.Hayyan M., Hashim M.A., Alnashef I.M. Superoxide ion: generation and chemical implications. Chem. Rev. 2016;116:3029–3085. doi: 10.1021/acs.chemrev.5b00407. [DOI] [PubMed] [Google Scholar]
- 18.Barzilai A., Yamamoto K.I. DNA damage responses to oxidative stress. DNA Repair. 2004;3:1109–1115. doi: 10.1016/j.dnarep.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 19.Klaunig J.E., Kamendulis L.M., Hocevar B.A. Oxidative stress and oxidative damage in carcinogenesis. Toxicol. Pathol. 2010;38:96–109. doi: 10.1177/0192623309356453. [DOI] [PubMed] [Google Scholar]
- 20.Nambiar M., Raghavan S.C. How does DNA break during chromosomal translocations? Nucleic Acids Res. 2011;39:5813–5825. doi: 10.1093/nar/gkr223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karoui H., Hogg N., Fréjaville C., Tordo P., Kalyanaraman B. Characterization of sulfur-centered radical intermediates formed during the oxidation of thiols and sulfite by peroxynitrite: ESR-spin trapping and oxygen uptake studies. J. Biol. Chem. 1996;271:6000–6009. doi: 10.1074/jbc.271.11.6000. [DOI] [PubMed] [Google Scholar]
- 22.Hakim O., Resch W., Yamane A., Klein I., Kieffer-Kwon K.-R., Jankovic M., Oliveira T., Bothmer A., Voss T.C., Ansarah-Sobrinho C., Mathe E., Liang G., Cobell J., Nakahashi H., Robbiani D.F., Nussenzweig A., Hager G.L., Nussenzweig M.C., Casellas R. DNA damage defines sites of recurrent chromosomal translocations in B lymphocytes. Nature. 2012;484:69–74. doi: 10.1038/nature10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iarovaia O.V., Rubtsov M., Ioudinkova E., Tsfasman T., Razin S.V., Vassetzky Y.S. Dynamics of double strand breaks and chromosomal translocations. Mol. Cancer. 2014;13:249. doi: 10.1186/1476-4598-13-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ray P.D., Huang B.-W., Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012;24:981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murphy M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ott M., Gogvadze V., Orrenius S., Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis. 2007;12:913–922. doi: 10.1007/s10495-007-0756-2. [DOI] [PubMed] [Google Scholar]
- 27.Zorov D.B., Juhaszova M., Sollott S.J. Mitochondrial ROS-induced ROS release: an update and review. Biochim. Biophys. Acta - Bioenerg. 2006;1757:509–517. doi: 10.1016/j.bbabio.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 28.del M., Idelchik P.S., Begley U., Begley T.J., Melendez J.A. Mitochondrial ROS control of cancer. Semin. Cancer Biol. 2017;47 doi: 10.1016/j.semcancer.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hancock J.T., Desikan R., Neill S.J. Role of reactive oxygen species in cell signalling pathways. Biochem. Biomed. Asp. Oxid. Modif. 2001:345–350. doi: 10.1042/0300-5127:0290345. [DOI] [PubMed] [Google Scholar]
- 30.Poprac P., Jomova K., Simunkova M., Kollar V., Rhodes C.J., Valko M. Targeting free radicals in oxidative stress-related human diseases. Trends Pharmacol. Sci. 2017;38:592–607. doi: 10.1016/j.tips.2017.04.005. [DOI] [PubMed] [Google Scholar]
- 31.Demple B., Harrison L. Repair of oxidative damage to DNA: enzymology and biology. Annu. Rev. Biochem. 1994;63:915–948. doi: 10.1146/annurev.bi.63.070194.004411. [DOI] [PubMed] [Google Scholar]
- 32.Dizdaroglu M. Oxidative damage to DNA in mammalian chromatin. Mutat. Res. DNAging. 1992;275:331–342. doi: 10.1016/0921-8734(92)90036-o. [DOI] [PubMed] [Google Scholar]
- 33.Lu A.L., Li X., Gu Y., Wright P.M., Chang D.Y. Repair of oxidative DNA damage: mechanisms and functions. Cell Biochem. Biophys. 2001;35:141–170. doi: 10.1385/CBB:35:2:141. [DOI] [PubMed] [Google Scholar]
- 34.von Sonntag C. New aspects in the free-radical chemistry of pyrimidine nucleobases. Free Radic. Res Commun. 1987;2:217–224. doi: 10.3109/10715768709065286. [DOI] [PubMed] [Google Scholar]
- 35.Banerjee A., Zhang X., Manda K.R., Banks W.A., Ercal N. HIV proteins (gp120 and Tat) and methamphetamine in oxidative stress-induced damage in the brain: potential role of the thiol antioxidant N-acetylcysteine amide. Free Radic. Biol. Med. 2010;48:1388–1398. doi: 10.1016/j.freeradbiomed.2010.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ivanov A.V., Valuev-Elliston V.T., Ivanova O.N., Kochetkov S.N., Starodubova E.S., Bartosch B., Isaguliants M.G. Oxidative stress during HIV infection: mechanisms and consequences. Oxid. Med. Cell. Longev. 2016;2016:1–18. doi: 10.1155/2016/8910396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Romani B., Engelbrecht S., Glashoff R.H. Functions of Tat: the versatile protein of human immunodeficiency virus type 1. J. Gen. Virol. 2010;91:1–12. doi: 10.1099/vir.0.016303-0. [DOI] [PubMed] [Google Scholar]
- 38.Musinova Y.R., Sheval E.V., Dib C., Germini D., Vassetzky Y.S. Functional roles of HIV-1 Tat protein in the nucleus. Cell. Mol. Life Sci. 2016;73:589–601. doi: 10.1007/s00018-015-2077-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kundu R.K., Sangiorgi F., Wu L.Y., Pattengale P.K., Hinton D.R., Gill P.S., Maxson R. Expression of the human immunodeficiency virus-Tat gene in lymphoid tissues of transgenic mice is associated with B-cell lymphoma. Blood. 1999;94:275–282. [PubMed] [Google Scholar]
- 40.Dandekar D.H., Ganesh K.N., Mitra D. HIV-1 Tat directly binds to NFkappaB enhancer sequence: role in viral and cellular gene expression. Nucleic Acids Res. 2004;32:1270–1278. doi: 10.1093/nar/gkh289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marban C., Su T., Ferrari R., Li B., Vatakis D., Pellegrini M., Zack J.A., Rohr O., Kurdistani S.K. Genome-wide binding map of the HIV-1 Tat protein to the human genome. PLoS One. 2011;6:e26894. doi: 10.1371/journal.pone.0026894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reeder J.E., Kwak Y.-T., McNamara R.P., Forst C.V., D’Orso I. HIV Tat controls RNA Polymerase II and the epigenetic landscape to transcriptionally reprogram target immune cells. Elife. 2015;4:e08955. doi: 10.7554/eLife.08955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Capone C., Cervelli M., Angelucci E., Colasanti M., Macone A., Mariottini P., Persichini T. A role for spermine oxidase as a mediator of reactive oxygen species production in HIV-Tat-induced neuronal toxicity. Free Radic. Biol. Med. 2013;63:99–107. doi: 10.1016/j.freeradbiomed.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 44.Gu Y., Wu R.F., Xu Y.C., Flores S.C., Terada L.S. HIV Tat activates c-Jun amino-terminal kinase through an oxidant-dependent mechanism. Virology. 2001;286:62–71. doi: 10.1006/viro.2001.0998. [DOI] [PubMed] [Google Scholar]
- 45.Lecoeur H., Borgne-Sanchez A., Chaloin O., El-Khoury R., Brabant M., Langonné A., Porceddu M., Brière J.-J., Buron N., Rebouillat D., Péchoux C., Deniaud A., Brenner C., Briand J.-P., Muller S., Rustin P., Jacotot E. HIV-1 Tat protein directly induces mitochondrial membrane permeabilization and inactivates cytochrome c oxidase. Cell Death Dis. 2012;3:e282. doi: 10.1038/cddis.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Curreli S., Krishnan S., Reitz M., Lunardi-Iskandar Y., Lafferty M.K., Garzino-Demo A., Zella D., Gallo R.C., Bryant J. B cell lymphoma in HIV transgenic mice. Retrovirology. 2013;10:92. doi: 10.1186/1742-4690-10-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Masella R., Di Benedetto R., Varì R., Filesi C., Giovannini C. Novel mechanisms of natural antioxidant compounds in biological systems: involvement of glutathione and glutathione-related enzymes. J. Nutr. Biochem. 2005;16:577–586. doi: 10.1016/j.jnutbio.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 48.Shan X.Q., Aw T.Y., Jones D.P. Glutathione-dependent protection against oxidative injury. Pharmacol. Ther. 1990;47:61–71. doi: 10.1016/0163-7258(90)90045-4. [DOI] [PubMed] [Google Scholar]
- 49.Lushchak V.I., Lushchak V.I. Glutathione homeostasis and functions: potential targets for medical interventions. J. Amino Acids. 2012;2012:1–26. doi: 10.1155/2012/736837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Valko M., Rhodes C.J., Moncol J., Izakovic M., Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 51.Deponte M. The incomplete glutathione puzzle: just guessing at numbers and figures? Antioxid. Redox Signal. 2017;27:1130–1161. doi: 10.1089/ars.2017.7123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Deponte M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta - Gen. Subj. 2013;1830:3217–3266. doi: 10.1016/j.bbagen.2012.09.018. [DOI] [PubMed] [Google Scholar]
- 53.Zitka O., Skalickova S., Gumulec J., Masarik M., Adam V., Hubalek J., Trnkova L., Kruseova J., Eckschlager T., Kizek R. Redox status expressed as GSH:GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol. Lett. 2012;4:1247–1253. doi: 10.3892/ol.2012.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kaplowitz N. The importance and regulation of hepatic glutathione. Yale J. Biol. Med. 1981;54:497–502. [PMC free article] [PubMed] [Google Scholar]
- 55.Gamcsik M., Kasibhatla M., Teeter S., Colvin O. Glutathione levels in human tumors. Biomarkers. 2012;17:671–691. doi: 10.3109/1354750X.2012.715672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ballatori N., Krance S.M., Notenboom S., Shi S., Tieu K., Hammond C.L. Glutathione dysregulation and the etiology and progression of human diseases. Biol. Chem. 2009;390:191–214. doi: 10.1515/BC.2009.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van den Berg R., Haenen G.R.M.M., van den Berg H., Bast A. Transcription factor NF-κB as a potential biomarker for oxidative stress. Br. J. Nutr. 2001;86:S121. doi: 10.1079/bjn2001340. [DOI] [PubMed] [Google Scholar]
- 58.Morgan M.J., Liu Z. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011;21:103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dudley A.C. Likelihood for False Positives Using Bacterially-Expressed Recombinant Proteins in Anti-Angiogenesis Studies. Cancer Biol. Ther. 2006;5:406. doi: 10.4161/cbt.5.4.2651. [DOI] [PubMed] [Google Scholar]
- 60.Garcia J.A., Harrich D., Pearson L., Mitsuyasu R., Gaynor R.B. Functional domains required for tat-induced transcriptional activation of the HIV-1 long terminal repeat. EMBO J. 1988;7:3143–3147. doi: 10.1002/j.1460-2075.1988.tb03181.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rossi C., Balboni P.G., Betti M., Marconi P.C., Bozzini R., Grossi M.P. Inhibition of HIV-1 replication by a Tat transdominant negative mutant in human peripheral blood lymphocytes from healthy donors and HIV-1-infected patients. Gene. Ther. 1997;4:1261–1269. doi: 10.1038/sj.gt.3300522. [DOI] [PubMed] [Google Scholar]
- 62.Daugaard M., Nitsch R., Razaghi B., McDonald L., Jarrar A., Torrino S., Castillo-Lluva S., Rotblat B., Li L., Malliri A., Lemichez E., Mettouchi A., Berman J.N., Penninger J.M., Sorensen P.H. Hace1 controls ROS generation of vertebrate Rac1-dependent NADPH oxidase complexes. Nat. Commun. 2013;4:1–13. doi: 10.1038/ncomms3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dmitriev P., Bou Saada Y., Dib C., Ansseau E., Barat A., Hamade A., Dessen P., Robert T., Lazar V., Louzada R.A.N., Dupuy C., Zakharova V., Carnac G., Lipinski M., Vassetzky Y.S. DUX4-induced constitutive DNA damage and oxidative stress contribute to aberrant differentiation of myoblasts from FSHD patients. Free Radic. Biol. Med. 2016;99:244–258. doi: 10.1016/j.freeradbiomed.2016.08.007. [DOI] [PubMed] [Google Scholar]
- 64.Carré A., Louzada R.A.N., Fortunato R.S., Ameziane-El-Hassani R., Morand S., Ogryzko V., de Carvalho D.P., Grasberger H., Leto T.L., Dupuy C. When an intramolecular disulfide bridge governs the interaction of DUOX2 with Its partner DUOXA2. Antioxid. Redox Signal. 2015;23:724–733. doi: 10.1089/ars.2015.6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Narasimhan M., Rathinam M., Patel D., Henderson G., Mahimainathan L. Astrocytes prevent ethanol induced apoptosis of Nrf2 depleted neurons by maintaining {GSH} homeostasis. Open J. Apoptosis. 2012;1:9. doi: 10.4236/ojapo.2012.12002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vene R., Delfino L., Castellani P., Balza E., Bertolotti M., Sitia R., Rubartelli A. Redox remodeling allows and controls B-cell activation and differentiation. Antioxid. Redox Signal. 2010;13:1145–1155. doi: 10.1089/ars.2009.3078. [DOI] [PubMed] [Google Scholar]
- 67.Chen W.Y., Chang F.R., Huang Z.Y., Chen J.H., Wu Y.C., Wu C.C. Tubocapsenolide A, a novel withanolide, inhibits proliferation and induces apoptosis in MDA-MB-231 cells by thiol oxidation of heat shock proteins. J. Biol. Chem. 2008;283:17184–17193. doi: 10.1074/jbc.M709447200. [DOI] [PubMed] [Google Scholar]
- 68.Germini D., Saada Y.B., Tsfasman T., Osina K., Robin C., Lomov N., Rubtsov M., Sjakste N., Lipinski M., Vassetzky Y. A one-step PCR-based assay to evaluate efficiency and precision of genomic DNA-editing tools. Mol. Ther. - Methods Clin. Dev. 2017;5:43–50. doi: 10.1016/j.omtm.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xiao H., Neuveut C., Tiffany H.L., Benkirane M., a Rich E., Murphy P.M., Jeang K.T. Selective CXCR4 antagonism by Tat: implications for in vivo expansion of coreceptor use by HIV-1. Proc. Natl. Acad. Sci. USA. 2000;97:11466–11471. doi: 10.1073/pnas.97.21.11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhong Y., Hennig B., Toborek M. Intact lipid rafts regulate HIV-1 Tat protein-induced activation of the Rho signaling and upregulation of P-glycoprotein in brain endothelial cells. J. Cereb. Blood Flow. Metab. 2010;30:522–533. doi: 10.1038/jcbfm.2009.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rayne F., Debaisieux S., Bonhoure A., Beaumelle B. HIV-1 Tat is unconventionally secreted through the plasma membrane. Cell Biol. Int. 2010;34:409–413. doi: 10.1042/CBI20090376. [DOI] [PubMed] [Google Scholar]
- 72.Sorescu D., Griendling K.K. Reactive oxygen species, mitochondria, and NAD(P)H oxidases in the development and progression of heart failure. Congest. Heart Fail. 2002;8:132–140. doi: 10.1111/j.1527-5299.2002.00717.x. [DOI] [PubMed] [Google Scholar]
- 73.Sasaki M., Ozawa Y., Kurihara T., Noda K., Imamura Y., Kobayashi S., Ishida S., Tsubota K. Neuroprotective effect of an antioxidant, lutein, during retinal inflammation. Investig. Ophthalmol. Vis. Sci. 2009;50:1433–1439. doi: 10.1167/iovs.08-2493. [DOI] [PubMed] [Google Scholar]
- 74.Peroja P., Pasanen A.K., Haapasaari K.-M., Jantunen E., Soini Y., Turpeenniemi-Hujanen T., Bloigu R., Lilja L., Kuittinen O., Karihtala P. Oxidative stress and redox state-regulating enzymes have prognostic relevance in diffuse large B-cell lymphoma. Exp. Hematol. Oncol. 2012;1:2. doi: 10.1186/2162-3619-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marnett L.J. Oxyradicals and DNA damage. Carcinogenesis. 2000;21:361–370. doi: 10.1093/carcin/21.3.361. [DOI] [PubMed] [Google Scholar]
- 76.Maser R.S., Monsen K.J., Nelms B.E., Petrini J.H. hMre11 and hRad50 nuclear foci are induced during the normal cellular response to DNA double-strand breaks. Mol. Cell. Biol. 1997;17:6087–6096. doi: 10.1128/mcb.17.10.6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Evans M.D., Dizdaroglu M., Cooke M.S. Oxidative DNA damage and disease: induction, repair and significance. Mutat. Res. - Rev. Mutat. Res. 2004;567:1–61. doi: 10.1016/j.mrrev.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 78.Martinez S.S., Campa A., Li Y., Fleetwood C., Stewart T., Ramamoorthy V., Baum M.K. Low plasma zinc is associated with higher mitochondrial oxidative stress and faster liver fibrosis development in the Miami adult studies in HIV cohort. J. Nutr. 2017;147:556–562. doi: 10.3945/jn.116.243832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Masiá M., Padilla S., Fernández M., Barber X., Moreno S., Iribarren J.A., Portilla J., Peña A., Vidal F., Gutiérrez F. CoRIS, contribution of oxidative stress to non-AIDS events in HIV-infected patients. J. Acquir. Immune Defic. Syndr. 2017;75:e36–e44. doi: 10.1097/QAI.0000000000001287. [DOI] [PubMed] [Google Scholar]
- 80.Rodríguez-Mora S., Mateos E., Moran M., Martín M.Á., López J.A., Calvo E., Terrón M.C., Luque D., Muriaux D., Alcamí J., Coiras M., López-Huertas M.R. Intracellular expression of Tat alters mitochondrial functions in T cells: a potential mechanism to understand mitochondrial damage during HIV-1 replication. Retrovirology. 2015;12:78. doi: 10.1186/s12977-015-0203-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Antonenko Y.N., Avetisyan A.V., Bakeeva L.E., Chernyak B.V., Chertkov V.A., Domnina L.V., Ivanova O.Y., Izyumov D.S., Khailova L.S., Klishin S.S., Korshunova G.A., Lyamzaev K.G., Muntyan M.S., Nepryakhina O.K., Pashkovskaya A.A., Pletjushkina O.Y., Pustovidko A.V., Roginsky V.A., Rokitskaya T.I., Ruuge E.K., Saprunova V.B., Severina I.I., Simonyan R.A., Skulachev I.V., Skulachev M.V., Sumbatyan N.V., Sviryaeva I.V., Tashlitsky V.N., Vassiliev J.M., Vyssokikh M.Y., Yaguzhinsky L.S., Zamyatnin A.A., Skulachev V.P. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 1. Cationic plastoquinone derivatives: synthesis and in vitro studies. Biochemistry. 2008;73:1273–1287. doi: 10.1134/s0006297908120018. [DOI] [PubMed] [Google Scholar]
- 82.Zakharova V.V., Pletjushkina O.Y., Zinovkin R.A., Popova E.N., Chernyak B.V. Mitochondria-targeted antioxidants and uncouplers of oxidative phosphorylation in treatment of the Systemic Inflammatory Response Syndrome (SIRS) J. Cell. Physiol. 2017;232:904–912. doi: 10.1002/jcp.25626. [DOI] [PubMed] [Google Scholar]
- 83.Cunha F.M., Caldeira da Silva C.C., Cerqueira F.M., Kowaltowski A.J. Mild mitochondrial uncoupling as a therapeutic strategy. Curr. Drug Targets. 2011;12:783–789. doi: 10.2174/138945011795528778. [DOI] [PubMed] [Google Scholar]
- 84.Liao B.-C., Hsieh C.-W., Lin Y.-C., Wung B.-S. The glutaredoxin/glutathione system modulates NF-κB activity by glutathionylation of p65 in cinnamaldehyde-treated endothelial cells. Toxicol. Sci. 2010;116:151–163. doi: 10.1093/toxsci/kfq098. [DOI] [PubMed] [Google Scholar]
- 85.Lu S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009;30:42–59. doi: 10.1016/j.mam.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Staal F.J. Glutathione and HIV infection: reduced reduced, or increased oxidized? Eur. J. Clin. Invest. 1998;28:194–196. doi: 10.1046/j.1365-2362.1998.00268.x. [DOI] [PubMed] [Google Scholar]
- 87.Van Duyne R., Kehn-Hall K., Carpio L., Kashanchi F. Cell-type-specific proteome and interactome: using HIV-1 Tat as a test case. Expert Rev. Proteom. 2009;6:515–526. doi: 10.1586/epr.09.73. [DOI] [PubMed] [Google Scholar]
- 88.Gautier V.W., Gu L., O’Donoghue N., Pennington S., Sheehy N., Hall W.W. In vitro nuclear interactome of the HIV-1 Tat protein. Retrovirology. 2009;6:47. doi: 10.1186/1742-4690-6-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ruben S., Perkins A., Purcell R., Joung K., Sia R., Burghoff R., Haseltine W.A., Rosen C.A. Structural and functional characterization of human immunodeficiency virus tat protein. J. Virol. 1989;63:1–8. doi: 10.1128/jvi.63.1.1-8.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li N., Karin M. Is NF-kappaB the sensor of oxidative stress? FASEB J. 1999;13:1137–1143. [PubMed] [Google Scholar]
- 91.Anrather J., Racchumi G., Iadecola C. NF-κB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J. Biol. Chem. 2006;281:5657–5667. doi: 10.1074/jbc.M506172200. [DOI] [PubMed] [Google Scholar]
- 92.Hoesel B., Schmid J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer. 2013;12:86. doi: 10.1186/1476-4598-12-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Takada Y., Singh S., Aggarwal B.B. Identification of a p65 peptide that selectively inhibits NF-κ B activation induced by various inflammatory stimuli and its role in down-regulation of NF-κB-mediated gene expression and up-regulation of apoptosis. J. Biol. Chem. 2004;279:15096–15104. doi: 10.1074/jbc.M311192200. [DOI] [PubMed] [Google Scholar]
- 94.Mattioli I., Sebald A., Bucher C., Charles R.-P., Nakano H., Doi T., Kracht M., Schmitz M.L. Transient and selective NF-kappa B p65 serine 536 phosphorylation induced by T cell costimulation is mediated by I kappa B kinase beta and controls the kinetics of p65 nuclear import. J. Immunol. 2004;172:6336–6344. doi: 10.4049/jimmunol.172.10.6336. [DOI] [PubMed] [Google Scholar]
- 95.Song C., Mitter S.K., Qi X., Beli E., Rao H.V., Ding J., Ip C.S., Gu H., Akin D., Dunn W.A., Bowes Rickman C., Lewin A.S., Grant M.B., Boulton M.E., Boulton M.E. Oxidative stress-mediated NFκB phosphorylation upregulates p62/SQSTM1 and promotes retinal pigmented epithelial cell survival through increased autophagy. PLoS One. 2017;12:e0171940. doi: 10.1371/journal.pone.0171940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Strickson S., Campbell D.G., Emmerich C.H., Knebel A., Plater L., Ritorto M.S., Shpiro N., Cohen P. The anti-inflammatory drug BAY 11-7082 suppresses the MyD88-dependent signalling network by targeting the ubiquitin system. Biochem. J. 2013;451:427–437. doi: 10.1042/BJ20121651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bower M., Fisher M., Hill T., Reeves I., Walsh J., Orkin C., Phillips A.N., Bansi L., Gilson R., Easterbrook P., Johnson M., Gazzard B., Leen C., Pillay D., Schwenk A., Anderson J., Porter K., Gompels M., Sabin C.A. for the U.C.S. UK CHIC Steering Committee, CD4 counts and the risk of systemic non-Hodgkin's lymphoma in individuals with HIV in the UK. Haematologica. 2009;94:875–880. doi: 10.3324/haematol.2008.002691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stebbing J., Gazzard B., Mandalia S., Teague A., Waterston A., Marvin V., Nelson M., Bower M. Antiretroviral treatment regimens and immune parameters in the prevention of systemic AIDS-related non-Hodgkin's lymphoma. J. Clin. Oncol. 2004;22:2177–2183. doi: 10.1200/JCO.2004.11.097. [DOI] [PubMed] [Google Scholar]
- 99.Jin Z.-Y., Liu X., Ding Y.-Y., Zhang Z.-F., He N. Cancer risk factors among people living with HIV/AIDS in China: a systematic review and meta-analysis. Sci. Rep. 2017;7:4890. doi: 10.1038/s41598-017-05138-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ryan E., Hollingworth R., Grand R. Activation of the DNA damage response by RNA viruses. Biomolecules. 2016;6:2. doi: 10.3390/biom6010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mollace V., Nottet H.S., Clayette P., Turco M.C., Muscoli C., Salvemini D., Perno C.F. Oxidative stress and neuroAIDS: triggers, modulators and novel antioxidants. Trends Neurosci. 2001;24:411–416. doi: 10.1016/s0166-2236(00)01819-1. [DOI] [PubMed] [Google Scholar]
- 102.Price T.O., Ercal N., Nakaoke R., Banks W.A. HIV-1 viral proteins gp120 and Tat induce oxidative stress in brain endothelial cells. Brain Res. 2005;1045:57–63. doi: 10.1016/j.brainres.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 103.Banjac A., Perisic T., Sato H., Seiler A., Bannai S., Weiss N., Kölle P., Tschoep K., Issels R.D., Daniel P.T., Conrad M., Bornkamm G.W. The cystine/cysteine cycle: a redox cycle regulating susceptibility versus resistance to cell death. Oncogene. 2008;27:1618–1628. doi: 10.1038/sj.onc.1210796. [DOI] [PubMed] [Google Scholar]
- 104.Karp D.R., Shimooku K., Lipsky P.E. Expression of γ-glutamyl transpeptidase protects Ramos B cells from oxidation-induced cell death. J. Biol. Chem. 2001;276:3798–3804. doi: 10.1074/jbc.M008484200. [DOI] [PubMed] [Google Scholar]
- 105.Täger M., Ittenson A., Franke A., Frey A., Gassen H.G., Ansorge S. Gamma-Glutamyl transpeptidase-cellular expression in populations of normal human mononuclear cells and patients suffering from leukemias. Ann. Hematol. 1995;70:237–242. doi: 10.1007/BF01784042. [DOI] [PubMed] [Google Scholar]
- 106.Puca A., Fiume G., Palmieri C., Trimboli F., Olimpico F., Scala G., Quinto I. IκB-α represses the transcriptional activity of the HIV-1 Tat transactivator by promoting its nuclear export. J. Biol. Chem. 2007;282:37146–37157. doi: 10.1074/jbc.M705815200. [DOI] [PubMed] [Google Scholar]
- 107.Demarchi F., d’Adda di Fagagna F., Falaschi A., Giacca M. Activation of transcription factor NF-kappaB by the Tat protein of human immunodeficiency virus type 1. J. Virol. 1996;70:4427–4437. doi: 10.1128/jvi.70.7.4427-4437.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu J., Perkins N.D., Schmid R.M., Nabel G.J. Specific NF-kappa B subunits act in concert with Tat to stimulate human immunodeficiency virus type 1 transcription. J. Virol. 1992;66:3883–3887. doi: 10.1128/jvi.66.6.3883-3887.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fiume G., Vecchio E., De Laurentiis A., Trimboli F., Palmieri C., Pisano A., Falcone C., Pontoriero M., Rossi A., Scialdone A., Fasanella Masci F., Scala G., Quinto I. Human immunodeficiency virus-1 Tat activates NF-κB via physical interaction with IκB-α andp65. Nucleic Acids Res. 2012;40:3548–3562. doi: 10.1093/nar/gkr1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nakamura H., Masutani H., Yodoi J. Redox imbalance and its control in HIV infection. Antioxid. Redox Signal. 2002;4:455–464. doi: 10.1089/15230860260196245. [DOI] [PubMed] [Google Scholar]
- 111.Koyama T., Sun B., Tokunaga K., Tatsumi M., Ishizaka Y. DNA damage enhances integration of HIV-1 into macrophages by overcoming integrase inhibition. Retrovirology. 2013;10:21. doi: 10.1186/1742-4690-10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ensoli F., Cafaro A., Casabianca A., Tripiciano A., Bellino S., Longo O., Francavilla V., Picconi O., Sgadari C., Moretti S., Cossut M.R.P., Arancio A., Orlandi C., Sernicola L., Maggiorella M.T., Paniccia G., Mussini C., Lazzarin A., Sighinolfi L., Palamara G., Gori A., Angarano G., Di Pietro M., Galli M., Mercurio V.S., Castelli F., Di Perri G., Monini P., Magnani M., Garaci E., Ensoli B. HIV-1 Tat immunization restores immune homeostasis and attacks the HAART-resistant blood HIV DNA: results of a randomized phase II exploratory clinical trial. Retrovirology. 2015;12:33. doi: 10.1186/s12977-015-0151-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Aquaro S., Scopelliti F., Pollicita M., Perno C.F. Oxidative stress and HIV infection: target pathways for novel therapies? Futur. HIV Ther. 2008;2:327–338. [Google Scholar]
- 114.Brenza T.M., Ghaisas S., Ramirez J.E.V., Harischandra D., Anantharam V., Kalyanaraman B., Kanthasamy A.G., Narasimhan B. Neuronal protection against oxidative insult by polyanhydride nanoparticle-based mitochondria-targeted antioxidant therapy. Nanomed. Nanotechnol. Biol. Med. 2017;13:809–820. doi: 10.1016/j.nano.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kirkpatrick D.L., Powis G. Clinically evaluated cancer drugs inhibiting redox signaling. Antioxid. Redox Signal. 2017;26:262–273. doi: 10.1089/ars.2016.6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Feniouk B.A., Skulachev V.P. Cellular and molecular mechanisms of action of mitochondria-targeted antioxidants. Curr. Aging Sci. 2017;10:41–48. doi: 10.2174/1874609809666160921113706. [DOI] [PubMed] [Google Scholar]
- 117.Mousseau G., Kessing C.F., Fromentin R., Trautmann L., Chomont N., Valente S.T. The Tat inhibitor didehydro-cortistatin a prevents HIV-1 reactivation from latency. MBio. 2015;6:e00465. doi: 10.1128/mBio.00465-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Representative images of γH2AX staining of B-cells treated with 250 ng/ml Tat for 6 h. γH2AX is shown in green, DAPI nuclear staining in blue. Scale bar=10 µm. (B) Representative images of the Comet assay of B-cells treated with 250 ng/ml Tat for 6 h. Scale bar=50 µm. (C) Representative images of MitoSOXRed-stained B-cells treated with 250 ng/ml Tat for 6 h. Hoechst nuclear staining is shown in blue, MitoSOX in red. Scale Bar = 10 µm. Supplementary material