

Abstract
Inducing a strong and specific immune response is the hallmark of a successful vaccine. Nanoparticles have emerged as promising vaccine delivery devices to discover and elicit immune responses. Fine-tuning a nanoparticle vaccine to create an immune response with specific antibody and other cellular responses is influenced by many factors such as shape, size, and composition. Peptide amphiphile micelles are a unique biomaterials platform that can function as a modular vaccine delivery system, enabling control over many of these important factors and delivering payloads more efficiently to draining lymph nodes. In this study, the modular properties of peptide amphiphile micelles are utilized to improve an immune response against a Group A Streptococcus B cell antigen (J8). The hydrophobic/hydrophilic interface of peptide amphiphile micelles enabled the precise entrapment of amphiphilic adjuvants which were found to not alter micelle formation or shape. These heterogeneous micelles significantly enhanced murine antibody responses when compared to animals vaccinated with nonadjuvanted micelles or soluble J8 peptide supplemented with a classical adjuvant. The heterogeneous micelle induced antibodies also showed cross-reactivity with wild-type Group A Streptococcus providing evidence that micelle-induced immune responses are capable of identifying their intended pathogenic targets.
Keywords: Group A Streptococcus, J8 peptide, peptide amphiphile micelles, vaccine, biodistribution
Graphical abstract
INTRODUCTION
A growing class of therapeutics leveraging peptides is being studied for both prophylactic and postexposure vaccine applications. Peptides are attractive candidates since they can be precisely designed to contain the minimal epitopes necessary to stimulate an immune response, avoiding common problems associated with killed or attenuated pathogen vaccines such as autoimmunity, risk of infection, and allergic reaction.1,2 The biggest challenge facing peptide-based vaccines is the fact that peptides alone are weak immunogens. To improve peptide immunogenicity, biomaterials-based platforms have been developed.3–8 Despite these advances, these systems have been unable to match or exceed immune responses induced by conventional vaccination approaches employing immune-potentiating molecules termed adjuvants.9–11 An opportunity exists to combine these technologies into a rationally designed peptide vaccine platform that precisely delivers a combination of antigens and adjuvants to better stimulate and control the nature of the immune response.
Peptide amphiphiles (PAs) are a class of peptide-based biomaterials consisting of bioactive peptide head groups conjugated to hydrophobic alkyl tails which self-assemble in aqueous solution into micellar structures. The hydrophobic tails are protected from water in the core of the micelle, and a multivalent display of peptide head groups form the micelle corona. Peptide amphiphile micelles (PAMs) have previously been shown to function as a peptide delivery system for a variety of applications including cancer therapy,12–14 angiogenesis,15,16 osteogenesis,17,18 and atherosclerosis treatment.19,20 In our previous studies, micelles were used as a vaccine delivery vehicle that induced a peptide-specific antibody response.21 Interestingly, PAMs stimulated a stronger antibody response than peptide alone without the use of an adjuvant making it a self-adjuvanting device. Furthermore, while the hydrophobic tails enhance cell uptake by anchoring into cell membranes,22 they do not act as pathogen recognition receptor (PRR) agonists and therefore do not function as molecular adjuvants.21 Rather, it is thought to be the self-assembled, particular-based physical nature of PAMs that affords them immunogenicity. Self-assembly of PAs has the added benefit of being able to facilitate the fabrication of multifunctional heterogeneous micelles through simple mixing of different PAs or other amphiphilic molecules.19 The modular nature of peptide amphiphile micelles provides the opportunity to further enhance and shape their immunogenicity through precise control of their components. Specifically, the incorporation of secondary molecular signals or amphiphilic adjuvants capable of simulating Th-cells or PRRs found on APCs into PAMs has the capacity to yield more robust host immune responses.
Nanoparticle physical characteristics including size and shape have been found to influence immune responses as well as in vivo biodistribution and clearance.4,23 Very small soluble particles readily diffuse and rapidly dilute after subcutaneous injection.4 Larger, intermediate sized colloidal particles have smaller diffusion speeds and efficiently transport to the lymphatic system by convection.4 As size increases to over about 500 nm, the particles become too large for transport and become trapped in the interstitial space.4 Antigenic PAs previously used for vaccine applications self-assemble into long, flexible cylindrical micelles approximately 5–15 nm in diameter and 200 nm to 2 μm in length, after annealing.21 Recognizing that no universal rules exist that can be applied to predict the in vivo behavior of all nanoparticles, it is important that the biodistribution and clearance of nanomaterials be evaluated for each new structure. Given the unique properties of this system and its inherent modularity, understanding biodistribution and clearance profiles can help to further elucidate micelle mechanisms of adjuvanticity and inform the rational design of micelles for vaccination applications.
This paper explores the potential of peptide amphiphile micelles to serve as a modular immunotherapeutic platform. Specifically, a conformationally dependent B cell epitope derived from the M1 surface protein of Group A Streptococcus (GAS) bacteria was used. GAS causes a range of mild to severe ailments, from simple pharyngitis (“strep throat”) to necrotizing fasciitis (flesh-eating disease), as well as postinfection autoimmune diseases like rheumatic heart disease for which an effective vaccine has yet to be developed. Using straightforward chemistry and simple self-assembly, PAMs were designed, tested, and optimized to raise GAS-specific antibody titers.
MATERIALS AND METHODS
Micelle Synthesis
J8 peptide (QAEDKVKQSREAKKQVEKALKQLEDKVQK) was synthesized on Rink amide MBHA resin (Novabiochem) utilizing standard Fmoc solid phase synthesis with the aid of a PS3 Peptide Synthesizer (Protein Technologies, Inc.). The N-terminus was either acetylated using 10× molar excess of acetic anhydride in DMF or covalently coupled to Rhodamine (Rho) fluorophore (Anaspec Inc.) by an amidation reaction yielding Rho-J8. The resulting J8 peptides were treated using a concentrated trifluoroacetic acid solution to deprotect side groups and cleave the peptide from resin. High pressure liquid chromatography with mass spectrometry controlled fraction collection (LCMS; Shimadzu Corp.) utilizing a reversed-phase C8 column (Waters) with a gradient of acetonitrile in Milli-Q water containing 0.1% formic acid was employed to purify J8 peptide. For J8 or Rho-J8 peptide amphiphiles, the hydrophobic moiety dipalmitoylglutamic acid (diC16) was synthesized by a previously established method.24 J8 or Rho-J8 peptide was synthesized similarly to that above, except the C-terminal lysine was protected with DDE instead of Boc, which was used for the other lysines. The peptide was treated with 2% hydrazine in DMF to orthogonally deprotect the C-terminal lysine amine group which was then covalently coupled to diC16 by an amidation reaction yielding J8-diC16 and Rho-J8-diC16 peptide amphiphiles. These peptide amphiphiles were further processed and purified by the same methods as the peptides above. All samples were lyophilized and stored at −20 °C until used. It should be noted that all peptide and peptide amphiphiles were created in a chemical synthesis laboratory using appropriate personal protective equipment to eliminate exposure to biological contaminants.
To fabricate micelles, J8-diC16 peptide amphiphiles were film cast by dissolving them in methanol and evaporating the solvent using nitrogen as a drying gas. The resulting film was hydrated at 70 °C for 60 min in phosphate-buffered saline (PBS) and allowed to equilibrate overnight. Fluorescent micelles were assembled by dissolving Rho-J8-diC16 and J8-diC16 (25:75 molar ratio) in methanol and prepared by the same methods as J8-diC16 micelles mentioned above. Toll-Like Receptor 4 (TLR4) Agonist Monophosphoryl Lipid A (MPLA, Sigma-Aldrich) or TLR2 Agonist Pam2Cys-SK4 (P2C-SK4 Invivogen) adjuvant supplemented micelles were assembled by combining J8-diC16 with either adjuvant and fabricated with the film cast method as mentioned above. Each micelle formulation contained 12 nmol of J8-diC16 and MPLA and P2C-SK4 were included at 10 mol %.
Micelle Characterization
Micelles were characterized by previously defined methodologies14,19,25 including critical micelle concentration (CMC) analysis and transmission electron microscopy (TEM). CMC was measured by fluorescent sequestration where varying concentrations of J8-diC16 were exposed to 1 mM 1,6-diphenyl-1,3,5-hexatriene (DPH) which greatly increases in fluorescence intensity when trapped within the micelle core. Solutions were prepared and allowed to equilibrate for 1 h prior to fluorescent measurement utilizing a Tecan Infinite 200 plate reader (ex. 350 nm, em. 428 nm). The data were fit with two trend lines which were set equal to one another to determine the fluorescence inflection point (i.e., CMC). Micelle morphology was investigated using negative stain TEM. J8-diC16, J8-diC16/MPLA, and J8-diC16/P2C micelle solutions (1 μL of 200 μM) were allowed to incubate on Formvar-coated copper grids (Ted Pella, Inc.) for 1 min after which excess liquid was wicked away with filter paper. Grids were then washed with Milli-Q water and incubated with aqueous phosphotungstic acid (1 wt %) for 1 min before the solution was wicked away. Samples were allowed to air-dry and then imaged on a FEI Tecnai 12 TEM using an accelerating voltage of 120 kV. For Förster resonance energy transfer (FRET) experiments, three different micelle formulations were made. Micelles with rhodamine only were made to contain unlabeled J8-diC16 with 10% rhodamine labeled J8-diC16 and 10% unlabeled P2C. Micelles with fluorescein only were made to contain unlabeled J8-diC16 and 10% fluorescein labeled P2C. Micelles containing fluorescein and rhodamine were made to contain unlabeled J8-diC16 with 10% rhodamine labeled J8-diC16 and 10% fluorescein labeled P2C. Micelles were excited at 490 nm and emission between 490 and 700 nm was recorded on a Jasco FP-6500 Spectrofluorometer.
Whole Animal and Animal Organ Imaging
Female BALB/c mice 6−8 weeks old were purchased from Charles River and immunized to investigate the biodistribution and trafficking of the peptide and micelle vaccines. Mice were shaved and naired before they were anesthetized with 2% isoflurane in O2 and subcutaneously injected at the nape of the neck with 100 μL of 120 μM Rho-J8/J8 or Rho-J8-diC16/J8-diC16 (25:75 molar ratio) suspended in PBS. Whole body fluorescence imaging was conducted at multiple time points (ex. 570 nm, em. 620 nm, IVIS 200, Xenogen, Caliper Life Sciences, Hopkinton, MA, USA). Micelles were allowed to circulate for up to 24 h before mice were euthanized via CO2 overdose and cervical dislocation, after which the lymph nodes were harvested. Fluorescence imaging of organs was conducted using an IVIS 200, and quantification of the fluorescence signal was achieved via the Living Image software (PerkinElmer, Downers Grove, IL, USA).
Murine Vaccination
Female BALB/c mice 6–8 weeks old were purchased from Charles River and immunized to investigate the capacity for various micelle formulations to induce an antibody-mediated immune response. For control groups, the potent physical adjuvant Incomplete Freund’s Adjuvant (IFA) was used and purchased from Sigma-Aldrich. Micelles utilized for vaccination were fabricated by the film deposition, rehydration, and annealing method outlined above. Mice were vaccinated in the nape of the neck subcutaneously at days 0 (prime), 21 (boost 1), 28 (boost 2), and 35 (boost 3) with one of eight vaccine formulations:
PBS
J8: 12 nmol of J8 peptide
J8 + IFA: 12 nmol of J8 peptide in 50 μL of IFA and 50 μL of PBS
J8 + MPLA: 12 nmol of J8 peptide + 1.33 nmol of MPLA in 100 μL of PBS
J8 + P2C-SK4: 12 nmol of J8 peptide + 1.33 nmol of P2C-SK4 in 100 μL of PBS
J8-diC16: 12 nmol of J8-diC16 in 100 μL of PBS
J8-diC16/MPLA: 12 nmol of J8-diC16 + 1.33 nmol of MPLA in 100 μL of PBS
J8-diC16/P2C-SK4: 12 nmol of J8-diC16 + 1.33 nmol of P2C-SK4 in 100 μL of PBS
Whole blood was collected from saphenous veins prevaccination on days 21, 28, and 35 as well as on day 42 to analyze for J8-specific antibodies induced by the previous round of immunization. The blood was centrifuged at 10 000 rpm for 10 min to separate out red blood cells, and the supernatant serum was harvested and stored at −20 °C until analysis.
Antibody Response Characterization
An enzyme-linked immunosorbent assay (ELISA) was utilized to determine J8-specific antibody titers. Flat-bottom 96-well EIA microtiter plates (Costar) were coated overnight with 100 μL of 10 μg/mL J8 peptide in sodium bicarbonate coating buffer in each well at 4 °C. The wells were washed with 200 μL of 0.05% Tween 20 in PBS (PBS-T) three times and then blocked with 200 μL of assay diluent (10% FBS in PBS) for 1 h. The blocking solution was removed, and 100 μL of 1:1000 diluted sera samples was added to the top row and then serially diluted 2-fold with assay diluent down the plate. After a 2 h incubation, wells were washed with PBS-T three times and incubated with 100 μL of 1:3000 diluted detection antibody (IgM, IgG, IgA, IgG1, IgG2a, IgG3, or IgG4; Invitrogen) for 1 h. PBS-T was used to wash wells three times after which 100 μL of Ultra TMB-ELISA substrate solution (Pierce) was added to the wells. Plates were allowed to incubate for 15 min in darkness, and then, optical density (OD) was measured for each well at 650 nm using a Tecan Infinite M200 plate reader. End point antibody titers were defined as the greatest serum dilution where OD was at least twice that of normal mouse serum at the same dilution. If end-point titers are not reached with one plate, then additional titrations were utilized until ODs were diluted to background.
Antibodies Binding to M1 Proteins on GAS
Both wild-type and Δemm 5448 GAS were generously provided by Chelsea Stewart and Partho Ghosh at UCSD. Bacteria were fixed to poly-L-lysine coated slides using 4% paraformaldehyde. After blocking the bacteria with 0.5% BSA in PBS, sera from immunized mice were added to the slides at a 1:200 dilution and incubated at room temperature for 2 h. The slides were washed vigorously in 50 mL of PBS in a glass staining jar for 10 min using a stir bar. The bacteria were then incubated for 2 h at room temperature with goat-antimouse IgG F(ab′)2 conjugated to FITC to detect antibodies bound to the bacteria. Controls included secondary antibody only and sera from nai̇̈ve mice (both negative). Fluorescent images were taken by a Zeiss confocal microscope. The same power, pinhole, and gain settings were used for all images.
Statistical Analysis
JMP software (SAS Institute) was used to make comparisons between groups using an ANOVA followed by Tukey’s HSD test to determine pairwise statistically significant differences (p < 0.05). Within the figure graphs, groups that possess different letters have statistically significant differences in mean whereas those that possess the same letter are similar.
RESULTS AND DISCUSSION
Micelles Clear as Fast as Soluble Peptide but Traffic to the Lymph Nodes More Efficiently
Vaccines come in a wide variety of forms, from killed or attenuated pathogens to recombinant subunit or virus-like particles. As the field of immunoengineering has become more sophisticated, subunit antigen and peptide vaccines have emerged as a promising solution to the weaknesses of previous generation vaccines.1,2,9 Modular nanoparticle platforms, and peptide amphiphile micelles in particular, enable the control over many properties that affect vaccine-based immune responses such as size, shape, and composition. While a variety of nanoparticle vaccine systems currently exist,4–8 micelles possess several advantages over other nanoparticle-based systems. Micelles are water-soluble which makes them easy to deliver via injection, comprised of more than 80% peptide by weight, and able to deliver peptides with native peptide secondary structure (e.g., α-helicity). Previous literature has shown that palmitic acid-based moieties can act as PRR agonists which enhance the immunogenicity of linked peptides.26,27 While the exact mechanism responsible for the adjuvanticity of our micelle system is not fully understood, a previous Tirrell lab publication determined that the hydrophobic diC16 moiety did not activate TLR-2 in vitro, despite the chemical structure similarities between diC16 and known TLR-2 stimulants.21 Instead, micellization itself was found to be important, since the codelivery of J8 peptide separated from mock micelles (J8 + diC16−SK4) was unable to induce an immune response.21 Another factor which affects the immune response is where and how immune cells interact with nanoparticles. After a subcutaneous injection, a vaccine may remain at the site of injection to act as a depot or be quickly trafficked via the lymphatic system to interact with immune cells in the lymph nodes. Therefore, in order to further understand the nature of micelle adjuvanticity, their biodistribution and clearance was investigated and compared to free peptide.
In order to ensure that the biodistribution and clearance of micelles could be adequately imaged, 25 mol % of Rho-J8 products was incorporated into the vaccine formulations. Free J8 peptide and J8-diC16 micelles with 25 mol % Rho-J8 and Rho-J8-diC16, respectively, were subcutaneously injected at the nape of the neck. To be consistent and relevant to the vaccine formulations used throughout the previous and current paper, a volume of 100 μL and a concentration of 120 μM were used. After injection, mice were immediately imaged for a zero-minute time point, followed by images at 10, 30, 60, 120, 180, 300, and 360 min (Figure 1A). After 6 h, in vivo imaging confirmed that the vaccine formulations fully diffused away from the initial injection site and no in vivo accumulation of peptide or micelles could be seen. Since no in vivo accumulation could be viewed, the vaccines were presumably either degraded in the subcutaneous space or trafficked to other areas in the body at low enough concentrations to be indiscernible above background on a full mouse imaging scale.
Figure 1.
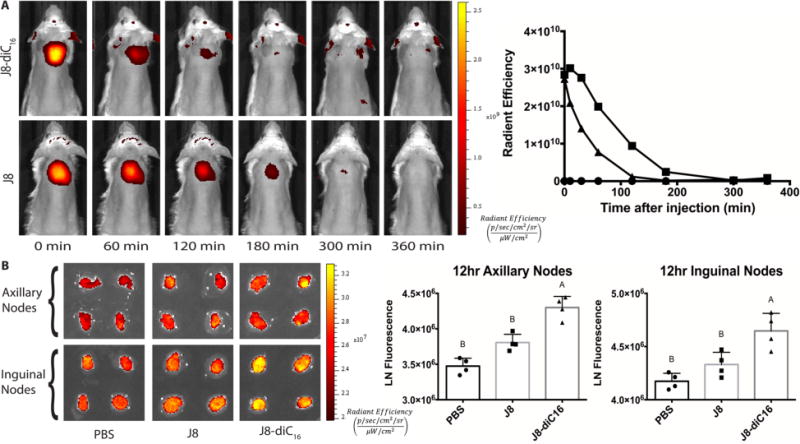
Micelles clear on the same order as free peptide but traffic to the lymph nodes more efficiently. (A) In vivo optical images of BALB/c mice injected with J8-diC16 micelles (top) and free J8 peptide (bottom) with 25 mol % Rho-J8-diC16 or Rho-J8, respectively. Micelle fluorescence clears approximately 1 h faster than free J8 peptide (● = PBS, ■ = J8, and ▲ = J8-diC16). (B) IVIS fluorescence imaging and quantification of excised draining axillary and inguinal lymph nodes from BALB/c mice at 12 h (N = 4 lymph nodes per group). Within a graph, groups that possess different letters have statistically significant differences in mean (p ≤ 0.05) whereas those that possess the same letter are similar (p > 0.05).
When comparing the speed with which the fluorescent signal cleared from the injection site, free peptide signal seems to last slightly longer than the micelle formulation signal. Despite this, the rate of clearance is essentially the same magnitude, approximately a few hours. As mentioned previously, a trend exists where small soluble particles readily diffuse from the injection site, intermediate particles diffuse less, which allows better transport to the lymphatic system, and larger particles become trapped in the interstitial space.4 These trends tend to hold true for colloidal particles. The peptide amphiphile micelles formed in this research, on the other hand, self-assemble into long, flexible cylindrical micelles approximately 5–15 nm in diameter and 200 nm to 2 μm in length. On the basis of this size profile and data from other nanoparticle platforms, it was hypothesized that the micelles would get trapped in the interstitial space and act as an antigen depot. On the basis of the live whole animal imaging data, however, it appears that micelles do not stay at the injection site any longer than its free peptide counterpart.
Since no in vivo accumulation could be seen from the whole mouse imaging, draining lymph nodes were imaged to assess the trafficking potential of each formulation. Vaccine formulations were injected subcutaneously, and 12 h later, draining lymph nodes were excised and kept intact to be imaged by IVIS. Figure 1B shows the distribution of the vaccines containing 25 mol % rhodamine within the inguinal and axillary lymph nodes. The micelle formulation showed significantly greater fluorescence in both sets of draining lymph nodes than in either the PBS or free peptide formulation.
Therefore, even though both vaccine formulations seemed to clear from the injection site at the same rate, micelles cleared to the lymph nodes more efficiently than free peptides. Combining this information, it suggests that the long cylindrical micelles may not primarily act as an antigen depot but rather traffic to the lymph nodes to induce an immune response. Given the clearance time frame in relation to free peptide and the lymph node accumulation, it could be that the micelles break down into more intermediately sized particles that can traffic to the lymphatic system. This explanation is supported by the fact that micelles are self-assembled structures held together by weak hydrophobic forces. While these micelles form long cylinders in stable solutions, they could easily reform into smaller micelles in the body. In fact, Liu et al. provided evidence that spherical amphiphilic micelles breakdown and traffic to the lymph nodes by albumin hitchhiking.28 Additionally, micelles readily interact with cells facilitating their internalization, whereas peptides do not readily internalize. Either system could account for trafficking to the lymph nodes, with micelle breakdown indicating acellular lymphatic trafficking and internalization indicating cellular lymphatic trafficking. Additional research is needed to interpret this further.
Design and Self-Assembly of Mixed Micelles
Conventional adjuvants and carrier proteins work, at least in part, by stimulating Th-cells and/or activating innate immune responses via stimulation of PRRs on antigen presenting cells. A strategy for peptide-based vaccines is thus to incorporate defined PRR agonists into the antigen delivery system such that immune cells can interact with both the target peptide antigen and an associated secondary signal molecule. PRR agonists can be either heterogeneously mixed in with a peptide or conjugated directly to the peptide epitope. Previous literature has validated the conjugation approach and indicated that palmitic acid-based moieties can act as TLR2 agonists which enhance the immunogenicity of linked peptides.26,27 While our peptide amphiphile micelle system is structurally similar, a previous Tirrell lab publication determined that the hydrophobic diC16 moiety did not activate TLR-2 in vitro.21 Instead, micellization itself was found to be important, since the codelivery of J8 peptide separated from mock micelles (J8 + diC16−SK4) was unable to induce an immune response.21 Despite this result, we recognize that the exact mechanism responsible for the adjuvanticity of our peptide amphiphile system is not fully understood and further in vivo research is worthwhile. While this is being clarified, a parallel investigation of heterogeneous incorporation of PRR agonists to the micelle system was explored.
Co-assembling TLR agonists into micelles has many advantages compared to covalent linking. Multiple agonists and multiple peptide amphiphile antigens can be incorporated into micelles in a modular fashion. Thus, for different applications, different agonists or peptides could be incorporated by simple mixing. Depending on the application, different agonists, or amounts of agonists, could be incorporated to bias the immune response to a specific response. Taking advantage of the modular nature of PAMs, heterogeneous micelles were made that incorporate different amphiphilic adjuvants to help boost the immune response through increasing antibody titers and/or reducing the number of immunizations required. Heterogeneous J8-diC16 micelles were made by mixing in either MPLA or P2C-SK4. These adjuvants were chosen due to their amphiphilic structures, providing an opportunity to form heterogeneous micelles comprised of antigens and adjuvants. MPLA is also an approved adjuvant for human use.29 Figure 2 shows the chemical structures of each micelle component. Figure 2A–C shows negative stain TEM images of J8-diC16 micelles, 90/10 J8-diC16/MPLA, and 90/10 J8-diC16/P2C-SK4 solutions, each incorporating 10 mol % of one of the secondary signal molecules. Heterogeneous micelles possess the long, cylindrical shape seen with pure J8-diC16 micelles indicating adjuvant entrapment does not affect micelle shape.
Figure 2.
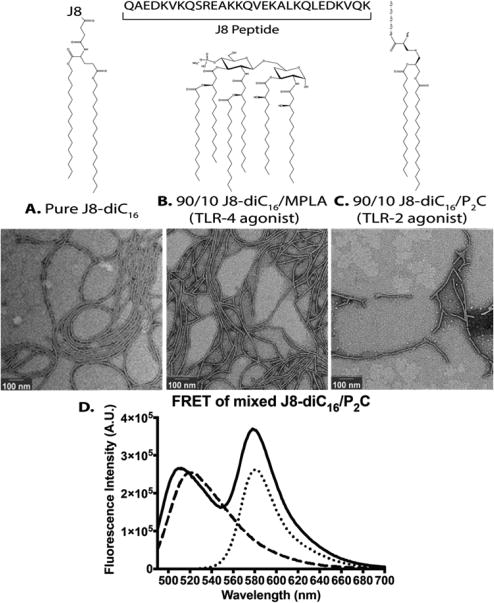
Ampiphilic adjuvants mixed with peptide amphiphiles form heterogeneous micelles. Negative stain TEM images of (A) J8-diC16 and J8-diC16 mixed micelles incorporating 10 mol % of either (B) MPLA or (C) P2C-SK4. Above each image is the structure of the secondary signal molecule. (D) Spectrophotometry revealed that Förster resonance energy transfer occurred between a rhodamine labeled J8-diC16 and a Fluorescin labeled P2C-SK4 (— = Rho-J8-diC16 and P2C-SK4-FL; --- = P2C-SK4-FL; ⋯ = Rho-J8-diC16). The rhodamine peak increases when fluorescein is excited in micelles that contain P2C-SK4-FL and Rho-J8-diC16 compared to micelles that contain Rho-J8-diC16 alone.
Förster resonance energy transfer (FRET) was used to demonstrate that P2C and J8-diC16 reside in the same self-assembled heterogeneous micelles and do not segregate into a mixed population of different micelles. J8-diC16 and P2C-SK4 were labeled with rhodamine (Rho) and fluorescein (FL), respectively, which act as a FRET pair. When fluorescein is excited and is in close proximity to rhodamine, fluorescein nonradiatively transfers energy to rhodamine, causing rhodamine to become excited and emit light. Since the Förster distance (R0) for fluorescein and rhodamine is 5.5 nm, the molecules must be less than 11 nm apart (2 × R0), for FRET to occur which is achievable with PAMs.
When P2C-SK4-FL is coassembled with Rho-J8-diC16, the fluorescein peak has a distinct blue shift compared to the P2C-SK4-FL on its own when excited at 490 nm (Figure 2D). FRET occurs more efficiently at the wavelengths that rhodamine absorbs. Since rhodamine has a peak excitation of 552 nm, the higher wavelengths of fluorescein decrease more than the lower wavelengths, causing the blue shift in the fluorescein peak. FRET is further demonstrated by the increase in the rhodamine peak at 580 nm.
Heterogeneous Micelles Improve Antibody Response
To assess the ability of heterogeneous micelles to enhance antibody titers, mice were immunized with formulations comprised of 12 nmol J8-diC16 and 10 mol % TLR agonist. For controls, mice were also immunized with only J8-diC16 and mixed formulations of 12 nmol of free J8 peptide and 10 mol % TLR agonist or IFA (Figure S1; see full formulations in Materials and Methods). Harvested serum samples were analyzed by ELISA to determine J8-specific antibody isotype titers (IgM, IgG, and IgA). Figure 3 compares IgM and IgG titers induced by the heterogeneous micelles after each immunization with the titers of the pure J8-diC16 micelles and the peptides in IFA. All micelle vaccines were able to induce appreciable IgM titers which were all found to be significantly higher than J8 in IFA after the first boost. No vaccine treatment tested induced an IgA response (data not shown).
Figure 3.
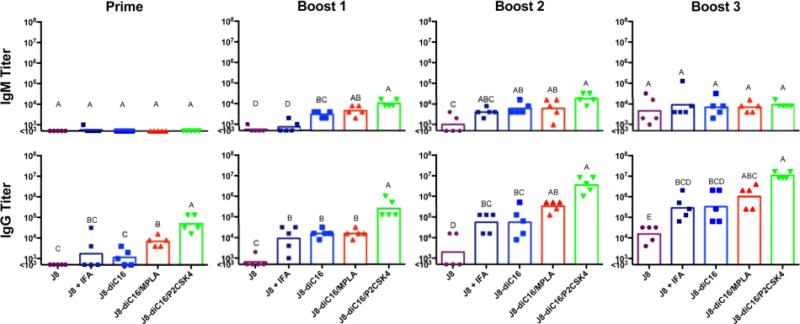
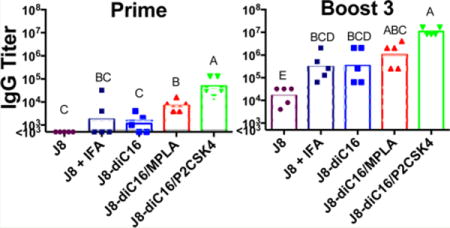
Mixed peptide amphiphile micelles induced strong antibody isotype responses in vivo. J8-specific antibody titers induced by J8 + IFA and J8-diC16 micelles were compared to titers stimulated by mixed micelles containing 10% of either MPLA or P2C-SK4. Each secondary signal incorporated into the micelles resulted in an enhanced antibody response. While IgA titers were assessed, no mouse produced above background levels. Each data point represents one mouse (N = 5); bars represent the geometric mean. Within a graph, groups that possess different letters have statistically significant differences in mean (p ≤ 0.05) whereas those that possess the same letter are similar (p > 0.05).
IgG titers started showing strong responses after just one immunization. The responses were particularly high with the heterogeneous micelles incorporating TLR agonists. After just one immunization with 90/10 J8-diC16/MPLA, antibodies titers were a full order of magnitude higher than that of the micelles alone or soluble peptides in IFA. An even greater response was seen with heterogeneous micelles incorporating P2C-SK4 where a single immunization stimulated titers two full magnitudes higher than that of micelles alone or soluble peptide in IFA. Immunization with heterogeneous micelles incorporating P2C-SK4 stimulated the same J8 specific antibody levels as those seen after two boosts with both J8 peptide in IFA and J8-diC16 micelles. Compared to titers after one boost with pure micelles, one boost with the heterogeneous MPLA micelles resulted in titers that were the same order of magnitude, while titers with the heterogeneous P2C-SK4 micelles were one and a half times higher in magnitude. Antibody titers for heterogeneous MPLA micelles end with a max average titer slightly higher than boost, three titers of both pure micelles and J8 + IFA. Titers from mixed P2C-SK4 micelles leveled off after 2 boosts, with a max average titer exceeding all the other micelles by approximately 1 order of magnitude. Overall, the addition of either MPLA or P2C-SK4 to J8-diC16 micelles stimulates the production of higher antibody titers with fewer doses when compared to the pure J8 micelles alone and the soluble J8 peptide in IFA.
Further investigation into the specific nature of the induced IgG response revealed that it was strongly dominated by IgG1 subtype with IgG3 also being produced in response to heterogeneous micelle vaccination (Figures 4 and S2). A small yet significant level of IgG1 was seen after the prime immunization for the heterogeneous micelle vaccines but not for any other formulation. One boost with heterogeneous micelles containing P2C-SK4 stimulated the same J8-specific IgG1 antibody levels as seen after three boosters with the J8 peptide in IFA and J8-diC16 micelles. Compared to titers after one boost with J8 peptide in IFA or micelles alone, one boost with heterogeneous micelles incorporating P2C-SK4 resulted in IgG1 titers that were one and a half times higher in magnitude. Similar to total IgG, IgG1 antibody titers for heterogeneous P2C-SK4 micelles leveled off after boost two, with a max average titer exceeding that of all other micelles by approximately 1 order of magnitude. IgG1 subtype antibody has been found to protect against GAS infections by bacterial opsonization and macrophage engulfment.30 Interesting IgG3 titers were produced throughout the course of the prime-boost immunization schedule. Again, the heterogeneous micelle formulations produced the highest IgG3 responses, remaining between one and three levels of magnitude higher than J8 peptide in IFA or pure micelle. The micelle with MPLA rose to a titer of approximately 105 after the first boost and essentially remained level through to the end of the boost schedule. IgG3 mixed micelle formulations with P2C peaked before the end of the immunization schedule. Titers rose to approximately 105 after the first boost, then increased to titers greater than 106 after the second boost, and finally decreased back to titers of 105 again after the third boost. Finally, for IgG2a, while a few mice vaccinated with J8 + IFA had above background titers, J8-diC16 micelles induced no appreciable response. Antigen/adjuvant micelles, on the other hand, did raise small but appreciable IgG2a titers. IgG4 was also assayed for, but no titers were observed (data not shown).
Figure 4.
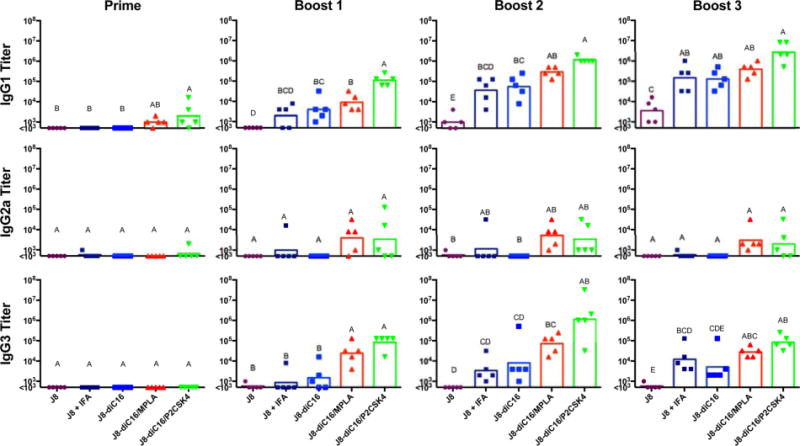
Heterogeneous peptide amphiphile micelles induced strong IgG1 and IgG3 antibody subtype responses in vivo. The strong IgG response induced by J8 peptide in IFA or J8-diC16 vaccines was found to be predominantly comprised of the IgG1 subtype. Heterogeneous micelles, on the other hand, produced a different antibody response than the controls comprised mostly of IgG1 with some IgG3 and a small amount of IgG2a. The modular addition of adjuvants has therefore allowed for the immune response to be controlled. Also, no mouse produced above background levels of IgG4 titers. Each point represents one mouse (N = 5); bars represent the geometric mean. Within a graph, groups that possess different letters have statistically significant differences in mean (p ≤ 0.05) whereas those that possess the same letter are similar (p > 0.05).
These results provide considerable evidence that the modularity of peptide amphiphile micelles can be used to tune corresponding immune responses toward desired applications. There are 11 known TLR subtypes31 where stimulation of different TLRs results in secretion of distinct cytokine signaling molecules influencing the ensuing immune response.32,33 For example, TLR4 agonists are known to promote a Th1 response which is best for intracellular viral and bacterial infections, while TLR2 agonists promote a Th2 response which is best for extracellular bacteria, parasites, and toxins.34 It is possible that specific TLR agonists can be incorporated into a vaccine formulation for the purpose of biasing the immune response. Caution should be taken when using strong TLR agonists, however, as broad activation of innate immunity can sometimes lead to chronic inflammation and tissue damage.35 Unlike the all-in-one peptide vaccines described above that have specific TLR agonists as part of the peptide scaffold, the PA micelle platform has the advantage of modularity.
In an attempt to explain the improved immune responses from Figures 3 and 4, it may be relevant to revisit the micelle trafficking results from Figure 1. Combining these results, we suggest that the enhanced lymph node trafficking capacity of micelles is maintained when heterogeneous antigen/adjuvant micelles are subcutaneously injected, resulting in the codelivery of antigens and adjuvants which dramatically improves the immune response activation in the lymph node. In fact, the importance of lymph node antigen presentation has previously been shown necessary to achieve strong and durable responses.36
Micelles Induce Antibodies That Identify M1 Protein on GAS Bacteria
The ELISAs discussed above demonstrate that the antibodies stimulated by the different micelle formulations, as well as those stimulated by the J8 peptide in IFA, are capable of binding to synthetic J8 peptide. To assess the therapeutic potential of these antibodies, immunocytochemistry was used to determine the capacity of the antibodies to recognize J8 peptide within the context of M1 protein on the surface of actual GAS bacteria. Wild-type GAS bacteria (M1 strain 5448) were fixed on poly-L-lysine-coated glass slides using paraformaldehyde. After adding sera from the immunized mice to the bacteria on the slides, an antimouse IgG F(ab′)2 fragment fluorescently labeled with FITC was used to identify antibodies from the mice that were bound to the bacteria. Similar staining was done using a mutant GAS strain (Δemm 5448) that does not express M1 protein. Figure 5 illustrates the binding of antibodies from mice immunized with 90/10 J8-diC16/MPLA micelles to the wild-type bacteria. Antibodies from mice immunized with all other micelle and peptide formulations were also capable of binding to the wild-type GAS (data not shown). However, none of the antibodies were capable of labeling the mutant bacteria, indicating that the antibodies are specifically recognizing the native M1 protein on the surface of GAS.
Figure 5.
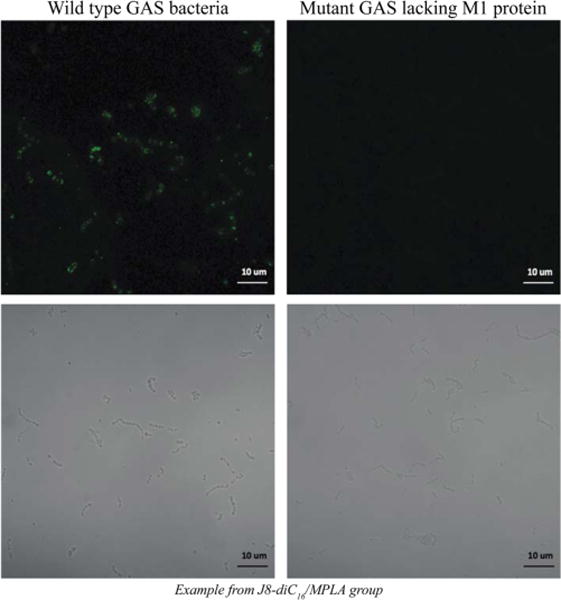
Confocal images showing that antibodies from immunized mice are capable of binding to wild-type GAS but not to mutant GAS lacking surface expression of M1 protein. All images were taken with a Zeiss LSM 700. The fluorescent image is shown on top of its corresponding white light image for each sample.
Given the recent peptide amphiphile micelle success in vivo, it is not farfetched to believe that clinical implementation of this vaccine platform could occur in the near future. While there are many factors that affect the clinical translation of a technology, we will quickly touch on some potential advantages and limitations of the micelle platform. In addition to the already discussed advantage of platform modularity and the systemic lymph node trafficking, another advantage is the relative ease with which scale-up could occur. Since the technology required to synthesize peptides cost effectively has already been developed, the addition of a tail by the same chemistry as adding an amino acid can believably be achieved without much more effort.37 Before clinical translation does occur, however, more basic science is needed, including testing these amphiphiles in a more genetically diverse population of mice to better represent human populations. Additionally, effects of storage conditions (time/temperature) on and kinetics of micelles should be thoroughly studied in preparation for commercial application.
CONCLUSIONS
Previously, we have shown that peptide amphiphile micelles can be utilized as a self-adjuvanting vaccine delivery vehicle to induce an antigen-specific antibody response. This research expands upon the micelle vaccine concept and demonstrates how peptide amphiphile modularity can be utilized to improve corresponding responses. It was revealed that micelle formulations cleared from the injection site at a similar rate to the soluble J8 peptide but trafficked to the lymph node more efficiently than soluble peptide. The amphiphilic and modular nature of peptide amphiphiles enabled the precise addition of amphiphilic adjuvants, which did not disrupt the formation of cylindrical micelles. When delivered subcutaneously to mice, heterogeneous micelles induced a stronger IgG1 antibody response than seen with a conventional gold-standard vaccine formulation. These experiments taken together provide convincing evidence that heterogeneous micelles can enhance lymph node codelivery of antigens and adjuvants leading to the dramatically improved antibody response observed.
Supplementary Material
Acknowledgments
The authors gratefully thank Chelsea Stewart and Partho Ghosh at UCSD for the strains of GAS and also Matt Kade for the help in synthesizing and purifying the fluorescein labeled adjuvant.
Funding
The authors gratefully acknowledge support from start-up funds from the University of California, Berkeley, and the University of Chicago, as well as research funding from the University of Chicago Institute for Translational Medicine (CTSA UL1 TR000430). B.D.U. wishes to acknowledge this work was supported in part by funding through the Pharmaceutics Division of the PhRMA Foundation (#00052243), the University of Missouri Research Board (#3784), and start-up funds supplied by the University of Missouri. Research reported in this publication was also supported by the National Institute of Biomedical Imaging and Bioengineering of the National Institutes of Health under Award Number T32EB009412.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsbiomaterials.6b00422.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interest.
References
- 1.Purcell AW, McCluskey J, Rossjohn J. More than one reason to rethink the use of peptides in vaccine design. Nat Rev Drug Discovery. 2007;6(5):404–414. doi: 10.1038/nrd2224. [DOI] [PubMed] [Google Scholar]
- 2.Kristensen D, Chen D, Cummings R. Vaccine stabilization: Research, commercialization, and potential impact. Vaccine. 2011;29(41):7122–7124. doi: 10.1016/j.vaccine.2011.05.070. [DOI] [PubMed] [Google Scholar]
- 3.Wen Y, Collier JH. Supramolecular peptide vaccines: tuning adaptive immunity. Curr Opin Immunol. 2015;35:73–79. doi: 10.1016/j.coi.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Irvine DJ, Swartz MA, Szeto GL. Engineering synthetic vaccines using cues from natural immunity. Nat Mater. 2013;12(11):978–990. doi: 10.1038/nmat3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swartz MA, Hirosue S, Hubbell JA. Engineering Approaches to Immunotherapy. Sci Transl Med. 2012;4(148):148rv9–148rv9. doi: 10.1126/scitranslmed.3003763. [DOI] [PubMed] [Google Scholar]
- 6.Leleux J, Roy K. Micro and Nanoparticle-Based Delivery Systems for Vaccine Immunotherapy: An Immunological and Materials Perspective. Adv Healthcare Mater. 2013;2(1):72–94. doi: 10.1002/adhm.201200268. [DOI] [PubMed] [Google Scholar]
- 7.Sahdev P, Ochyl LJ, Moon JJ. Biomaterials for Nanoparticle Vaccine Delivery Systems. Pharm Res. 2014;31(10):2563–2582. doi: 10.1007/s11095-014-1419-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Acar H, Srivastava S, Chung EJ, Schnorenberg MR, Barrett JC, LaBelle JL, Tirrell M. Self-Assembling Peptide-Based Building Blocks in Medical Applications. Adv Drug Delivery Rev. 2016:1–57. doi: 10.1016/j.addr.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coffman RL, Sher A, Seder RA. Vaccine Adjuvants: Putting Innate Immunity to Work. Immunity. 2010;33(4):492–503. doi: 10.1016/j.immuni.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petrovsky N, Aguilar JC. Vaccine adjuvants: Current state and future trends. Immunol Cell Biol. 2004;82:488–496. doi: 10.1111/j.0818-9641.2004.01272.x. [DOI] [PubMed] [Google Scholar]
- 11.Reed SG, Orr MT, Fox CB. Key roles of adjuvants in modern vaccines. Nat Med. 2013;19(12):1597–1608. doi: 10.1038/nm.3409. [DOI] [PubMed] [Google Scholar]
- 12.Missirlis D, Krogstad DV, Tirrell M. Internalization of p5314–29Peptide Amphiphiles and Subsequent Endosomal Disruption Results in SJSA-1 Cell Death. Mol Pharmaceutics. 2010;7(6):2173–2184. doi: 10.1021/mp100193h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Standley SM, Toft DJ, Cheng H, Soukasene S, Chen J, Raja SM, Band V, Band H, Cryns VL, Stupp SI. Induction of Cancer Cell Death by Self-assembling Nanostructures Incorporating a Cytotoxic Peptide. Cancer Res. 2010;70(8):3020–3026. doi: 10.1158/0008-5472.CAN-09-3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Black M, Trent A, Kostenko Y, Lee JS, Olive C, Tirrell M. Self-Assembled Peptide Amphiphile Micelles Containing a Cytotoxic T-Cell Epitope Promote a Protective Immune Response In Vivo. Adv Mater. 2012;24(28):3845–3849. doi: 10.1002/adma.201200209. [DOI] [PubMed] [Google Scholar]
- 15.Mammadov R, Mammadov B, Toksoz S, Aydin B, Yagci R, Tekinay AB, Guler MO. Heparin Mimetic Peptide Nanofibers Promote Angiogenesis. Biomacromolecules. 2011;12(10):3508–3519. doi: 10.1021/bm200957s. [DOI] [PubMed] [Google Scholar]
- 16.Webber MJ, Tongers J, Newcomb CJ, Marquardt K-T, Bauersachs J, Losordo DW, Stupp SI. Supramolecular nanostructures that mimic VEGF as a strategy for ischemic tissue repair. Proc Natl Acad Sci USA. 2011;108(33):13438–13443. doi: 10.1073/pnas.1016546108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anderson JM, Kushwaha M, Tambralli A, Bellis SL, Camata RP, Jun H-W. Osteogenic Differentiation of Human Mesenchymal Stem Cells Directed by Extracellular Matrix-Mimicking Ligands in a Biomimetic Self-Assembled Peptide Amphiphile Nanomatrix. Biomacromolecules. 2009;10(10):2935–2944. doi: 10.1021/bm9007452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mata A, Geng Y, Henrikson KJ, Aparicio C, Stock SR, Satcher RL, Stupp SI. Bone regeneration mediated by biomimetic mineralization of a nanofiber matrix. Biomaterials. 2010;31(23):6004–6012. doi: 10.1016/j.biomaterials.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mlinar LB, Chung EJ, Wonder EA, Tirrell M. Active targeting of early and mid-stage atherosclerotic plaques using self-assembled peptide amphiphile micelles. Biomaterials. 2014;35(30):8678–8686. doi: 10.1016/j.biomaterials.2014.06.054. [DOI] [PubMed] [Google Scholar]
- 20.Peters D, Kastantin M, Kotamraju VR, Karmali PP, Gujraty K, Tirrell M, Ruoslahti E. Targeting atherosclerosis by using modular, multifunctional micelles. Proc Natl Acad Sci USA. 2009;106(24):9815–9819. doi: 10.1073/pnas.0903369106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trent A, Ulery BD, Black MJ, Barrett JC, Liang S, Kostenko Y, David NA, Tirrell MV. Peptide Amphiphile Micelles Self-Adjuvant Group A Streptococcal Vaccination. AAPS J. 2015;17(2):380–388. doi: 10.1208/s12248-014-9707-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Missirlis D, Teesalu T, Black M, Tirrell M. The Non-Peptidic Part Determines the Internalization Mechanism and Intracellular Trafficking of Peptide Amphiphiles. PLoS One. 2013;8(1):e54611. doi: 10.1371/journal.pone.0054611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Champion JA, Katare YK, Mitragotri S. Particle shape: A new design parameter for micro- and nanoscale drug delivery carriers. J Controlled Release. 2007;121(1–2):3–9. doi: 10.1016/j.jconrel.2007.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berndt P, Fields GB, Tirrell M. Synthetic Lipidation of Peptides and Amino Acids: Monolayer Structure and Properties. J Am Chem Soc. 1995;117(37):9515–9522. [Google Scholar]
- 25.Kastantin M, Ananthanarayanan B, Karmali P, Ruoslahti E, Tirrell M. Effect of the Lipid Chain Melting Transition on the Stability of DSPE-PEG(2000) Micelles. Langmuir. 2009;25(13):7279–7286. doi: 10.1021/la900310k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu X, Ramos TV, Gras-Masse H, Kaplan BE, BenMohamed L. Lipopeptide epitopes extended by an N-palmitoyllysine moiety increase uptake and maturation of dendritic cells through a Toll-like receptor-2 pathway and trigger a Th1-dependent protective immunity. Eur J Immunol. 2004;34(11):3102–3114. doi: 10.1002/eji.200425166. [DOI] [PubMed] [Google Scholar]
- 27.Abdel-Aal A-BM, Al-Isae K, Zaman M, Toth I. Simple synthetic toll-like receptor 2 ligands. Bioorg Med Chem Lett. 2011;21(19):5863–5865. doi: 10.1016/j.bmcl.2011.07.102. [DOI] [PubMed] [Google Scholar]
- 28.Liu H, Moynihan KD, Zheng Y, Szeto GL, Li AV, Huang B, Van Egeren DS, Park C, Irvine DJ. Structure-based programming of lymph-node targeting in molecular vaccines. Nature. 2014;507(7493):519–522. doi: 10.1038/nature12978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alving CR, Peachman KK, Rao M, Reed SG. Adjuvants for human vaccines. Curr Opin Immunol. 2012;24(3):310–315. doi: 10.1016/j.coi.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zaman M, Abdel-Aal A-BM, Fujita Y, Phillipps KSM, Batzloff MR, Good MF, Toth I. Immunological Evaluation of Lipopeptide Group A Streptococcus (GAS) Vaccine: Structure-Activity Relationship. PLoS One. 2012;7(1):e30146–e30147. doi: 10.1371/journal.pone.0030146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2004;17(1):1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- 32.Kadowaki N, Ho S, Antonenko S, de Waal Malefyt R, Kastelein RA, Bazan F, Liu Y-J. Subsets of Human Dendritic Cell Precursors Express Different Toll-like Receptors and Respond to Different Microbial Antigens. J Exp Med. 2001;194(6):863–869. doi: 10.1084/jem.194.6.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reed SG, Bertholet S, Coler RN, Friede M. New horizons in adjuvants for vaccine development. Trends Immunol. 2009;30(1):23–32. doi: 10.1016/j.it.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 34.Agrawal S, Agrawal A, Doughty B, Gerwitz A, Blenis J, Van Dyke T, Pulendran B. Cutting Edge: Different Toll-Like Receptor Agonists Instruct Dendritic Cells to Induce Distinct Th Responses via Differential Modulation of Extracellular Signal-Regulated Kinase-Mitogen-Activated Protein Kinase and c-Fos. J Immunol. 2003;171(10):4984–4989. doi: 10.4049/jimmunol.171.10.4984. [DOI] [PubMed] [Google Scholar]
- 35.Piccinini AM, Midwood KS. DAMPening Inflammation by Modulating TLR Signalling. Mediators Inflammation. 2010;2:1–21. doi: 10.1155/2010/672395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andorko JI, Hess KL, Jewell CM. Harnessing Biomaterials to Engineer the Lymph Node Microenvironment for Immunity or Tolerance. AAPS J. 2015;17(2):323–338. doi: 10.1208/s12248-014-9708-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bray BL. Large-scale manufacture of peptide therapeutics by chemical synthesis. Nat Rev Drug Discovery. 2003;2:587–593. doi: 10.1038/nrd1133. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.