

Abstract
X-chromosome inactivation (XCI) epigenetically silences transcription of an X chromosome in females; patterns of XCI are thought to be aberrant in women’s cancers, but are understudied due to statistical challenges. We develop a two-stage statistical framework to assess skewed XCI and evaluate gene-level patterns of XCI for an individual sample by integration of RNA sequence, copy number alteration, and genotype data. Our method relies on allele-specific expression (ASE) to directly measure XCI and does not rely on male samples or paired normal tissue for comparison. We model ASE using a two-component mixture of beta distributions, allowing estimation for a given sample of the degree of skewness (based on a composite likelihood ratio test) and the posterior probability that a given gene escapes XCI (using a Bayesian beta-binomial mixture model). To illustrate the utility of our approach, we applied these methods to data from tumors of ovarian cancer patients. Among 99 patients, 45 tumors were informative for analysis and showed evidence of XCI skewed towards a particular parental chromosome. For 397 X-linked genes, we observed tumor XCI patterns largely consistent with previously identified consensus states based on multiple normal tissue types. However, 37 genes differed in XCI state between ovarian tumors and the consensus state; 17 genes aberrantly escaped XCI in ovarian tumors (including many oncogenes), whereas 20 genes were unexpectedly inactivated in ovarian tumors (including many tumor suppressor genes). These results provide evidence of the importance of XCI in ovarian cancer and demonstrate the utility of our two-stage analysis.
Keywords: Bayesian, mixture model, ovarian cancer, RNA-Seq
INTRODUCTION
In females, X chromosome inactivation (XCI) is a developmentally regulated process that randomly silences transcription of one of the two homologous copies of the X chromosome, resulting in equivalent gene expression dosage with males[Ross, et al. 2005]. Unlike the active copy of the X chromosome (Xa), the inactive X (Xi) forms a compact heterochromatin structure known as the Barr body[Ross, et al. 2005]. This process is epigenetic in nature, dictated by up-regulation of the non-coding RNA encoded by XIST and resulting in cis-acting transcriptional silencing of the chromosome by DNA hypermethylation and enrichment of histone modifications. However, an estimated 9%–14% of X-linked genes escape XCI and are transcribed on both chromosomes, even beyond the pseudo-autosomal regions[Cotton, et al. 2013]. Furthermore, X-linked genes can demonstrate variability in escape status across tissue types, as well as variability across individuals at the gene-level[Cotton, et al. 2015]. Consequently, XCI may contribute to sexual dimorphism and female phenotypic diversity through the female-specific epigenetic regulation of X gene expression.
The plasticity of XCI may also play a role in disease, particularly women’s cancers. Although acquired genomic alterations are often of primary interest in tumorigenesis, recent studies have demonstrated that epigenetic changes also occur in the tumor environment, as reviewed in [Sharma, Kelly, & Jones 2010]. Relevant to XCI, Barr body loss has been observed in ovarian and breast cancer tumors[Barr & Moore 1957; Pageau, Hall, Ganesan, Livingston, & Lawrence 2007]. Although the phenomenon of Barr body loss has largely been attributed to structural alteration via concomitant deletion of the Xi and duplication of Xa, recent breast cell cancer line studies have indicated the epigenetic erosion of XCI and local reactivation of genetic transcription on the Xi[Chaligne, et al. 2015]. Aberrant de-activation of X-linked tumor suppressors and/or re-activation of oncogenes may result in substantial dysregulation of cancer-related gene expression and contribute to tumor progression. In fact, it is thought that tumor suppressor genes that escape from XCI may contribute to the sex bias observed for some cancers[Dunford, et al. 2016]. Moreover, the X chromosome is enriched with hormone-related genes, and disruption of XCI could play a role in hormonal carcinogenesis[Henderson & Feigelson 2000]. However, practical analysis of XCI is complicated by the complex biology, cell and tissue-specificity, and difficulty in obtaining direct measurements, and patterns of potential XCI aberrations in women’s cancers are currently understudied[Chaligne & Heard 2014].
While studying XCI in model organisms is possible via marking maternal and paternal chromosomes[Berletch, Yang, Xu, Carrel, & Disteche 2011], study in humans is more complicated. Outside of cancer, previous methods to study XCI at the gene level in humans have utilized rodent/human cell hybrids, total expression or allelic expression data derived from a microarray, or DNA methylation data[Carrel & Willard 2005; Cotton, et al. 2013; Cotton, et al. 2015]. Single cell sequencing studies have also begun to emerge[Tukiainen, et al. 2016]. These previous approaches have several limitations; for example, reliance on cell hybrids has limited clinical utility. Methods relying on methylation are problematic because it is an indirect method to measure XCI; DNA methylation is one epigenetic mechanism by which XIST silences expression of the Xi, but other epigenetic factors are also involved. The use of microarray-derived expression data represents a more direct approach to measure the genes that are expressed on the Xa; however, these data are subject to potential confounding due to batch effects. Previous studies have also utilized male samples as a comparison population[Cotton, et al. 2015; Sharp, et al. 2011]; however, female-specific diseases such as ovarian cancer necessitate analytical methods that are independent of male samples. Additionally, previous approaches do not provide an estimated probability of escape for a given gene for a given individual. Furthermore, previous methods cannot distinguish XCI from possible copy number deletions or loss of heterozygosity (LOH), where only a single copy of the X chromosome remains. Lastly, previous analyses have focused on a single genomic data type, whereas the process of XCI is inherently multi-omic, involving genetic, epigenetic, and expression components.
The quantification of allele-specific expression (ASE), the relative proportion of transcribed mRNA attributable to each homologous copy of each gene, has successfully been utilized in association studies by correlating proximal genetic variation with allelic imbalance[Sun 2012]. Similar principles used to study these cis-acting expression quantitative trait loci on the autosomes can be extended to characterize gene-level patterns of XCI. For a given cell, mono-allelic expression suggests that the gene undergoes XCI and is not transcribed on the Xi; in contrast, evidence of balanced allelic expression of an X-linked gene indicates that the gene escapes XCI. In a tissue sample consisting of multiple cells, however, the ability to make this distinction is complicated by the potential for cellular heterogeneity. ASE from a tissue sample where the maternal chromosome is inactivated in some cells, but the paternal chromosome is inactivated in others will not be informative for gene-level XCI, as all genes will exhibit relatively balanced allelic expression regardless of XCI status. In contrast, if the Xi is heavily skewed toward a specific parental chromosome, discrimination between active and inactivated genes may be possible via patterns of ASE imbalance. In addition to XCI, ASE imbalance of X-linked genes may also manifest due to overlapping copy number alterations (CNAs); for instance, copy-neutral LOH would lead to only a single distinct allele being expressed within the impacted segment. Furthermore, an approach to study XCI in the tumor needs to be informed by expected XCI patterns in normal tissue. Consequently, chromosome-wide studies of XCI require advanced statistical methods that take these properties into consideration.
To address these concerns, we have developed a novel integrative approach that utilizes allele-specific expression data to directly measure gene-level patterns of XCI for a given tumor sample, relative to expected patterns based on normal tissue, and does not require analogous male samples for comparison (Figure 1). Particularly, in this study, we develop the statistical framework for a two-stage approach for the evaluation of gene-level patterns of XCI that relies on the integration of tumor RNA sequence, CNA, and germline genotype data and apply these methods to study XCI tumor escape patterns in a collection of ovarian cancers. First, we leverage ASE to identify tumors with evidence of the Xi heavily skewed toward one parental chromosome to address potential confounding with XCI heterogeneity. We then estimate the tumor XCI escape status for each gene using baseline patterns informed by consensus states from multiple tissue types. Finally, we discuss our findings and highlight future research directions for investigating XCI in cancer using multi-omic applications.
Figure 1.

Measuring XCI with allele expression. (A) For a given cell, either the maternal or paternal X is inactivated (top); for XCI Subject genes, only the active copy (Xa) is expressed, whereas for XCI Escape Genes, both the Xa and Xi are expressed (bottom). (B) In collections of cells under random XCI, the maternal Xa and paternal Xa are in equal proportions, so genes subject to XCI show no allelic expression imbalance compared to genes escaping XCI (top). Under skewed XCI, either the maternal Xa or paternal Xa is present in higher proportions, leading to allelic expression imbalance for genes subject to XCI (bottom).
METHODS
Study Participants
Study subjects included a total of N = 99 patients from the Mayo Clinic enrolled into an IRB-approved protocol within one year of diagnosis of pathologically confirmed primary invasive epithelial ovarian, fallopian tube, or primary peritoneal cancer between 2000 and 2009 (Table S1). All tissues were snap frozen immediately following surgery and stored at −80°C as a source of RNA; a gynecologic oncologist reviewed each sample to confirm diagnosis and ensure at least 70% tumor content in tumor samples and the absence of tumor in normal samples. Peripheral blood drawn prior to chemotherapy was used as a source of germline DNA.
Tissue RNA Sequencing
RNA was sequenced on an Illumina HiSeq2000 with six samples per lane in two batches. For 15 samples, 500ng RNA was used to generate polyA libraries using the Illumina TruSeq kit, and samples were run with paired end 50bp reads. The remaining 84 samples had 1μg RNA treated with riboZero and libraries were made using the Illumina TruSeq Stranded Total RNA kit, and samples run with 100bp paired end reads.
Genotyping
Germline DNA was genotyped with Illumina Infinium Beadchips (for all cases, OncoArray-500k; additionally for 81 cases, Human610-Quad; for 18 cases, Omni2.5–8), and a subset of 80 participants were also genotyped using a customized Affymetrix array targeting rare variants[Winham, et al. 2016]. Genotypes were merged at each SNP using the most comprehensive array for variants assayed on multiple platforms. In addition, tumor DNA was genotyped using the Infinium BeadChip (OncoArray-500k) and was utilized for CNA analysis (described below).
Copy Number Aberration Segmentation Analysis
To address potential confounding via structural alterations, we applied OncoSNP[Yau, et al. 2010] to call large segmental (low resolution) tumor CNAs using the OncoArray 500k data. We utilized the paired ‘tumor-normal’ mode, ‘intratumor’ and ‘stromal’ options, and assumed normal contamination levels of 0%–30%.
Quantifying Allele-Specific Expression
RNA sequence reads are ASE-informative only if they overlap one or more expressed heterozygous single nucleotide polymorphisms (eSNPs). First, germline genotypes were chromosomally pre-phased using SHAPEIT2 and imputed using IMPUTE2 with the 1000 Genomes as a background set, resulting in two phased chromosome-wide haplotypes denoted (H1, H2). Imputed genotypes were retained based upon an allele dosage r2 > 0.25, minor allele frequency (MAF) ≥ 1%, and genotype posterior probability > 0.5. To quantify ASE per sample, haplotype-specific BAM files (one each for H1, H2) of the aligned RNA sequence reads were produced for each sample using the asSeq[Sun 2012] package in R 3.0.2, and gene-level ASE read counts were generated using HTSeq version 0.5.3p9 (mode = intersection-nonempty). ASE data for genes that at least partially overlapped with genomic segments exhibiting structural alterations (duplications, deletions, and/or loss of heterozygosity) for a given sample were excluded. Batch effects were assessed by comparing per-sample median ASE across genotyping and RNA sequencing batches.
Statistical Model Assumptions
For a given sample with observed ASE data for G total X-linked genes, let (Yg1, Yg2) represent the bivariate vector of ASE reads mapped to the two phased X chromosome haplotypes (H1, H2) for gene g ∈ {1, …, G} and define Ng = Yg1 + Yg2. Let GE and GI respectively denote the sample-specific subsets of genes that are transcriptionally active (i.e., escaping) and inactive on the Xi. We assume Yg1 follows a binomial distribution with size Ng and success rate pg, such that
| (1) |
where we assume pg = 0.5 for all g ∈ GE regardless of XCI skewness, while pg ≠ 0.5 for g ∈ GI when the sample corresponds to skewed XCI. Due to random phase switch errors from statistical phasing of genotypes[Delaneau, Zagury, & Marchini 2013], we assume the Yg1 reads mapped to H1 are overall equally likely to correspond to either parental chromosome across genes; we also assume Yg1 is subject to extra-binomial variation[Pickrell, et al. 2010]. To accommodate over-dispersion and unknown X homolog membership, we assume pg follows a mixture of beta distributions (Figure 2), such that
Figure 2.
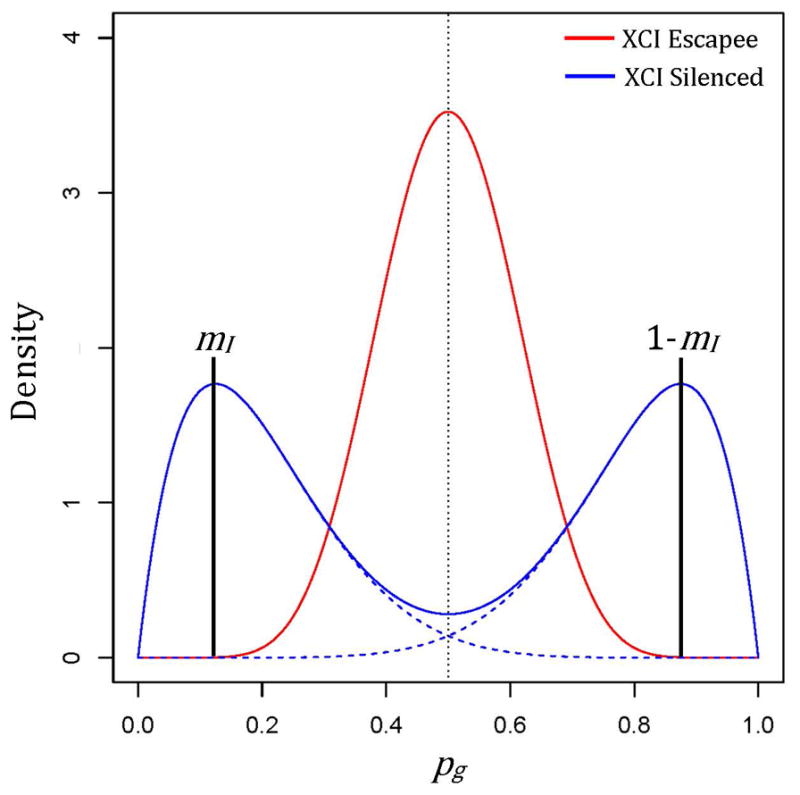
Depiction of beta-mixtures corresponding to underlying allelic expression ratios (AERs) for genes escaping XCI (red) or silenced by XCI (blue), with degree of skewness (mI) indicated by vertical black lines at the respective modes of the component symmetric beta distributions (dashed blue lines).
| (2) |
where Beta(a, b) is defined as and B(a, b) is the beta function. Note that fpg(p|g ∈ GE) reduces to Beta(aE, aE), unimodal and symmetric about 0.5. For modeling purposes, we define Yg = min(Yg1, Yg2) as the ASE of the least expressed allele and assume Yg follows a folded distribution[Porzio & Ragozini 2009]. Conditional on XCI gene set membership, the probability Yg = y for can be defined as
| (3) |
where δ(y, Ng) = 1 if y = Ng − y and 0 otherwise, and (a, b) is equivalent to (aI, bI) or (aE, aE) for GI and GE, respectively. To simplify prior specification and improve interpretation, we adopt a mode-dispersion parameterization (m, θ) of the beta distribution Beta(a, b), with mode and dispersion parameter θ = a + b, for m ∈ [0,0.5] and θ ≥ 2. Mode parameter simplifies to a fixed constant for genes in GE while mI ≤ 0.5 indicates skewed XCI. Note that mI can be interpreted as a measure of sample skewness.
Consensus XCI Status Across Multiple Tissues
To leverage prior knowledge of genes that escape XCI, we applied the results of Balaton et al.[Balaton, Cotton, & Brown 2015]. In brief, the authors combined analysis results across three prior studies [Carrel & Willard 2005; Cotton, et al. 2013; Cotton, et al. 2015] that examined XCI across 27 tissue types using multiple genomic approaches (cell hybrids, gene expression, DNA methylation). Based on the patterns of XCI across these three studies, genes were categorized into eight possible consensus states: escape, mostly escape, variable escape, mostly variable escape, subject, mostly subject, discordant, and no call (Table S2). As these studies were based on tissue types available in both sexes, XCI patterns for female-specific tissues, including ovarian epithelium, were not available.
Stage 1: XCI Skewness Testing
ASE from a tissue sample will not be informative for gene-level XCI if the Xa is sufficiently heterogeneous in representation by maternal and paternal chromosomes, since all genes will exhibit relatively balanced allelic expression ratios (i.e., 0.5 = mE ≈ mI). In contrast, if the Xa is heavily skewed toward a specific parental chromosome, discrimination between escape and inactive genes may be possible via downstream model-based clustering. To identify samples demonstrating evidence of skewed XCI, we evaluate allelic expression ratio imbalances of the subset of genes a priori likely to be inactive on the Xi based upon consensus XCI gene status calls from Balaton et al.[Balaton, Cotton, & Brown 2015], denoted , which encompass genes declared to be “subject” or “mostly subject” to XCI. We similarly define subset for genes declared to be “escaping” or “mostly escaping” XCI. All remaining XCI consensus states (i.e., discordant, variable escape, mostly variable escape, no call) are treated equally as unknown.
Under the null condition of random (non-skewed) XCI, the distribution of ASE reads for genes is comparable to genes , equivalent to testing H0: mI = 0.5 versus H1 : mI ≤ 0.5. For each sample, we define Y to be the vector of observed Yg for genes and derive the maximum likelihood estimates (m̂I, θ̂I) using box-constrained numerical optimization of the log-likelihood function, ℓ(mI, θI; Y) = Σg ln(Pr(Yg; Ng, mI, θI)) under the null and alternative hypotheses, with Pr(Yg) = 1 for genes corresponding to Ng = 0. We then compute a likelihood ratio test (LRT) statistic Λ = −2(ℓ(m̂I, θ̂I; Y) − ℓ(0.5, θ̂I; Y)). Since the null condition mI = 0.5 is on the boundary of the support for mI, we estimate the null distribution of Λ by computing Λb for b = 1, …, B parametric bootstrap samples under the fitted null model[Mclachlan 1987]. The LRT P-value is computed as .
Stage 2: XCI Escape Gene Analysis
Conditional on sample XCI skewness, we expect the two mixture components comprising fpg(p)to be identifiable, corresponding to the latent class categories of ‘inactive’ or ‘escape’. We opt for a Bayesian approach to inference and fit the model using Markov Chain Monte Carlo (MCMC) methods. We assume a flat Uniform(0,0.5) prior distribution on mI. We additionally assume θI ≥ θE by modeling , and set priors and π(θI) to vague Gamma distributions. Finally, we left-truncate π(θE) at 3.0 to ensure unimodal distributions for pg.
Absent of any prior knowledge, we may define flat independent prior probabilities of a gene g ∈ GE, ηg, such that ηg = 0.5. Alternatively, we may leverage the consensus XCI states for the genes of interest to assign gene-specific informative priors. For our purposes, we assigned ‘subject’, ‘mostly subject’, ‘mostly escape’, and ‘escape’ statuses ηg values of 0.2, 0.3, 0.7, and 0.8, respectively. All other consensus states were classified collectively as unknown and assigned a prior of ηg = 0.5. Under either prior definition, our measure of interest is the posterior probability of escape for gene g and sample j, PPjg = P(g ∈ GjE|Yj):
| (4) |
Data Simulations - Skewness
To generate realistic XCI ASE data, we based our simulations on autosomal ASE data from chromosome 22, derived from available normal tissue samples. These data provide biological distributions of allelic expression for genes not undergoing epigenetic silencing due to XCI (i.e. the null distribution of balanced allelic expression) and allow us to minimize distributional assumptions. Briefly, consider an autosomal gene with observed values of (Yg1, Yg2), (yg1, yg2), and ng = yg1 + yg2. To simulate data from a hypothetical gene g ∈ GE with total ASE of , we sampled reads from H1 and H2 at the corresponding proportions yg1/ng and yg2/ng. For g ∈ GI with some true skewness level mI, these proportions were respectively multiplied by and prior to sampling. Data were generated such that an observation (yg1, yg2) was randomly selected across all normal samples and genes that satisfied ng ≥ 100 for each simulated expression result.
To evaluate Type I error and power for the skewness test under a variety of conditions, we considered skewness values of m ∈ (0.1,0.2,0.3,0.4,0.5), ASE expression levels of Ng ∈ (10,20,30), and total number of genes evaluated L ∈ (10,20,30), such that mI = 0.5 defines the null condition of random XCI. For each unique simulation condition, a total of 1000 replications were run, with all testing results declared significant at an α-level of 0.05. The number of parametric bootstrap samples B was fixed at 500 in all settings. Power for our skewness test is also dependent upon the accuracy of when selecting a subset of genes for testing. In practice, GI and may not be equivalent for a variety of reasons, including underlying XCI differences by tissue type as well as discordant XCI gene activity within a given sample. To additionally evaluate robustness to the accidental inclusion of escape genes in the skewness test due to sample-specific differences between and GI, we considered contamination of 20% of the tested genes to be simulated as truly in GE. We evaluated power under the same conditions above for mI ∈ (0.1,0.2,0.3,0.4).
Data Simulations – Escape Gene Analysis
The same simulation principles as described above for skewness testing were applied to generate simulated XCI data for XCI escape gene analysis. To generate ASE profiles for analysis, we simulated data under conditions Ng ∈ (10,20,30) and mI ∈ (0.1,0.2,0.3). For a given sample, ASE data were generated for G = 100 hypothetical genes with a fixed XCI escape gene proportion of 15% (i.e., 15 genes in GE). All analyses were conducted using the Bayesian software Stan[Carpenter, et al. 2015] via the rstan package and the statistical software R 3.3.1, and convergence was evaluated using standard diagnostics and visual inspection of posterior samples from two independent chains of 2500 iterations after a burn-in of 2500. PPg for each eligible gene was calculated based on equation (4) using Monte Carlo integration over the combined joint posterior sample of the model parameters.
To investigate the influence of informative priors, we considered two scenarios: lack and presence of discordant genes (i.e. genes inconsistent with their assumed prior state). For the former, we set the XCI consensus state for the genes in GE to be five each for ‘escape’, ‘mostly escape’ and ‘unknown’. For the remaining 85 genes in GI, 30 were designated as ‘subject’, 15 as ‘mostly subject’, and 40 as ‘unknown’. These proportions approximate those observed in the XCI consensus state data. To simulate the presence of discordant genes, we selected 5 ‘subject’ or ‘mostly subject’ genes to be truly in GE, and 5 ‘escape’ or ‘mostly escape’ genes to be truly in GI. Performance was evaluated based on the ability of PPg to discriminate between true XCI-E and XCI-S genes, estimated by classification accuracy, sensitivity, and specificity of escape gene identification using a threshold of PPjg > 0.5.
Extensions to Multi-sample Analysis
In practice, many genes may have low expression for a given sample, and the expected number of informative XCI escape genes per sample could be small. To improve inference, we extend the defined model above in (1) and (2) to a multi-sample approach to borrow information across samples and genes, where GjI and GjE respectively represent sample-specific active and inactive gene subsets for sample j ∈ 1, … J. To accommodate differences in XCI skewness across samples, we define
| (5) |
For XCI escape genes, we assume θjE~Gamma(aθE, bθE) and fpjg(p|g ∈ GjE) = Beta(0.5, θjE). For some prior probability that g ∈ GjE, ηg, we define the probability Yjg = y as
| (6) |
for Θ = {ηg, mjI, θjI, θjE}.
To leverage existing biological knowledge of patterns of XCI while simultaneously borrowing information across samples, we model ηg using probit regression. Let X denote a G × 4 design matrix where the columns of X are binary dummy variables for previously-defined consensus XCI states of ‘subject’, ‘mostly subject’, ‘mostly escape’, and ‘escape’ status. We define Φ−1(η) = β0 + Xβ + ε, where correspond to independent and identically distributed Gaussian random gene effects with variance and Φ−1(·) is the inverse cumulative density function of the standard Gaussian distribution. The posterior probability of escape for gene g and sample j, PPjg = P(g ∈ GjE|Yj) can then be derived as:
Priors for mjI are independent flat Uniform(0,0.5) distributions. An improper prior is applied for σG and a vague independent Gaussian prior N(0,10) for β0. Priors on β preserve the ordinal nature of the consensus state categories, such that π(β1)~N(0,10), , and for k = 2,3,4. Hyperpriors on aθE and bθE are informative priors of π(aθE)~Gamma(10,1) and π(bθE)~Gamma(10,10) based on estimates for autosomal genes of eight available normal tissue samples. We also assume θjI ≥ θjE by modeling and corresponding to a vague Gamma distribution.
Application to Ovarian Cancer
To investigate patterns of XCI in ovarian cancer, we applied our two-stage XCI analytical methods to all ovarian tumor samples that met our inclusion criteria (Figure 3). Samples were declared significantly skewed at a Bonferroni-adjusted alpha level of 0.05, with the adjustment factor determined by the total number of tested samples. Skewed samples were then carried forward for genic XCI escape analyses. All samples with ≥ 5 genes in and Ng ≥ 10 were considered to have sufficient data for XCI skewness testing. All observations corresponding to Njg ≥ 10 were considered to have sufficient gene-level information for genic XCI escape inference, and classification of XCI status of a given gene was determined via a simple decision criterion (i.e., g ∈ GjE if PPjg > 0.5).
Figure 3.

Study Flowchart.
Using our multi-sample approach, we again apply MCMC methods to fit the model to the data using two independent chains of 5000 iterations after a burn-in of 5000. To investigate the influence of the various prior definitions on the final results, we ran single-sample analyses using flat and informative priors in addition to our multi-sample approach.
RESULTS
Skewness Test Simulations
As anticipated, statistical power to detect skewed XCI samples was correlated with increased degree of skewness, increased ASE levels, and higher total number of genes tested (Table I). Our test also demonstrated conservative performance with respect to Type I error control, with estimated false positive proportions near 0.01 at the 0.05 α level. This is likely due to distributional differences between the assumed beta-binomial distribution and the true ASE count data, since we applied a parametric bootstrap approach to characterize the null distribution. However, overall statistical power was high for a large proportion of realistic data conditions, with nearly 100% power to detect instances of skewness when mI ≤ 0.2 under the various levels of L and Ng under consideration. We also found our method to be robust to the accidental inclusion of true escape genes, particularly when mI was low (Table S3). We only observed a substantive loss in power for mI ≥ 0.3 when 20% of the included genes were simulated to be escaping XCI, with absolute power differences compared to our original simulations (without escape gene contamination) ranging from 0.08 to 0.27.
Table I.
Type I error and power of the XCI skewness test under various data conditions.
| Type I Error | Power | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Ng | L | m = 0.50 | m = 0.40 | m = 0.30 | m = 0.20 | m = 0.10 |
| 10 | 10 | 0.013 | 0.073 | 0.484 | 0.955 | 1.000 |
| 20 | 0.006 | 0.084 | 0.718 | 1.000 | 1.000 | |
| 30 | 0.006 | 0.101 | 0.848 | 1.000 | 1.000 | |
| 20 | 10 | 0.010 | 0.079 | 0.678 | 0.999 | 1.000 |
| 20 | 0.016 | 0.107 | 0.901 | 1.000 | 1.000 | |
| 30 | 0.007 | 0.117 | 0.966 | 1.000 | 1.000 | |
| 30 | 10 | 0.016 | 0.109 | 0.777 | 0.999 | 1.000 |
| 20 | 0.019 | 0.128 | 0.963 | 1.000 | 1.000 | |
| 30 | 0.017 | 0.139 | 0.989 | 1.000 | 1.000 | |
Escape Gene Analysis Simulations
Performance for escape gene classification was improved as level of skewness and number of reads increased (Table II). Accuracy for classifying escape genes was very high when the informative prior was correctly specified, ranging from 0.72 to 0.94 based on the number of reads and level of sample skewness. Furthermore, the method is robust to prior misspecification, with a reduction in accuracy of only 0.01–0.06 when an informative prior with 10% error was used. The method also performed well with an uninformative prior, especially for highly skewed samples with a large number of reads (accuracy=0.90, sensitivity=0.98, specificity=0.98). Performance was lower for samples with a low level of skewness (m=0.3), driven primarily by lower levels of specificity for escape gene detection.
Table II.
Simulation results for XCI escape gene analysis. Mean sensitivity, specificity, and accuracy are calculated across the 100 replications and defined with respect to identifying escape genes based upon PPjg > 0. Performance measures for discordant genes under the informative prior are based upon the subset of 10 genes with inaccurate prior definitions.
| Flat Prior | Informative Prior | Informative Prior with Errors | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||
| Overall | Discordant Genes | ||||||||||||
|
| |||||||||||||
| Ng | mI | Acc | Sens | Spec | Acc | Sens | Spec | Acc | Sens | Spec | Acc | Sens | Spec |
| 10 | 0.30 | 0.48 | 0.82 | 0.48 | 0.72 | 0.69 | 0.69 | 0.67 | 0.64 | 0.68 | 0.20 | 0.20 | 0.19 |
| 0.20 | 0.72 | 0.80 | 0.72 | 0.81 | 0.80 | 0.80 | 0.77 | 0.77 | 0.77 | 0.44 | 0.56 | 0.31 | |
| 0.10 | 0.75 | 0.93 | 0.75 | 0.86 | 0.84 | 0.84 | 0.83 | 0.90 | 0.82 | 0.66 | 0.79 | 0.52 | |
| 20 | 0.30 | 0.64 | 0.75 | 0.64 | 0.77 | 0.87 | 0.75 | 0.73 | 0.68 | 0.74 | 0.28 | 0.36 | 0.21 |
| 0.20 | 0.76 | 0.88 | 0.76 | 0.86 | 0.94 | 0.85 | 0.82 | 0.84 | 0.82 | 0.48 | 0.69 | 0.26 | |
| 0.10 | 0.86 | 0.97 | 0.86 | 0.92 | 0.98 | 0.90 | 0.90 | 0.96 | 0.89 | 0.76 | 0.93 | 0.59 | |
| 30 | 0.30 | 0.68 | 0.77 | 0.76 | 0.78 | 0.88 | 0.76 | 0.75 | 0.69 | 0.75 | 0.26 | 0.35 | 0.16 |
| 0.20 | 0.80 | 0.92 | 0.92 | 0.87 | 0.96 | 0.86 | 0.84 | 0.89 | 0.83 | 0.50 | 0.75 | 0.25 | |
| 0.10 | 0.90 | 0.98 | 0.98 | 0.94 | 0.98 | 0.93 | 0.92 | 0.97 | 0.91 | 0.83 | 0.95 | 0.72 | |
Data Application – Ovarian Cancer
We applied our two-stage approach to the ASE data estimated from the ovarian cancer dataset. We did not observe any differences in median ASE due to genotyping platform (p610=p2.5M=0.44, pexome=0.15) or RNAseq batch (p=0.99).
CNA filtering
As CNAs are common in ovarian cancer, a large number of tumor samples were excluded due to CNAs overlapping the genes in , the set of genes likely to be inactive on the Xi based on prior literature, and were excluded from further analysis. Tumors from 52 patients exhibited overlapping CNAs for all X-linked genes with expression data or lacked ASE data for genes in after CNA segment filtering, and were excluded from further analysis (Figure 3). Many of these excluded samples exhibited aneuploidy, or short or long arm aberrations typical of ovarian cancer[Cheng, et al. 1996; Huang, et al. 2012].
Skewness testing
After filtering based on CNAs, 47 tumor samples were adequate for skewness testing, of which 45 demonstrated significantly skewed XCI after correction for multiple testing (Table S4). Maximum likelihood estimates (from skewness testing) and posterior medians (XCI escape analysis) of mjI were highly concordant across the tumor samples (Spearman ρ = 0.858), and indicated trends toward extremely skewed XCI, with m̂jI < 0.05 for 34/45 samples under both analyses (Figure 4).
Figure 4.

Scatterplot of point estimates of mjI from the skewness test (x-axis) compared with posterior medians from the XCI escape analyses (y-axis) for all 45 skewed XCI ovarian samples.
Escape gene analysis
In total, G = 397 X chromosome genes were evaluated for XCI escape in at least one tumor sample, of which 318 (80.1%) corresponded to a priori information of escape status in X. The number of post-filtering ASE gene measurements analyzed per tumor sample ranged from 8 to 142 genes, with a median of 81, while the number of informative samples per gene ranged from 1 to 38 (median = 6). The final data were highly sparse due in combination to low total gene expression and/or extensive CNA overlap in remaining tumor samples, with a missingness rate of approximately 84% across all samples and genes. For all 16 samples with sufficient reads to evaluate XCI escape status for XIST, allelic expression ratios were close to zero (Supplemental Figure 1) and estimated posterior probabilities of escape suggested monoallelic expression as expected (PPjg < 0.05), providing a proof of principle for our methods. No genes resided in the pseudo-autosomal region after filtering, so could not be evaluated for expected high predicted probabilities of escape.
In comparing posterior escape probabilities across the three analyses, results were overall highly concordant (Table S5). The Spearman correlation coefficients of the PPjg across all samples and genes ranged from 0.61 to 0.86, with the multi-sample results and the flat prior single-sample results being the most correlated. Classifications of XCI escape status based upon a decision threshold of 0.50 were also highly similar, with Cohen’s kappa statistics ranging from 0.57 to 0.67. The multi-sample analysis tended to produce more extreme posterior escape probabilities, with approximately 60% of PPjg values < 0.01 or > 0.99. In contrast, the flat and informative prior single-sample analyses, respectively, corresponded to 15.5% and 40% of PPjg values that were similarly extreme. This is likely due to strong prior escape probabilities being inferred by consistent gene-level results across the analyzed samples in the multi-sample analysis. Incidentally, these strong priors would also require greater evidence to identify sporadic instances of discordant gene-level XCI. Results below are reported for the multi-sample analysis using an informative prior.
Concordance with XCI consensus states
Examination of results from the escape gene analysis revealed that genes in demonstrated highly concordant escape analysis results across samples and were estimated to be subject to XCI; similarly, many genes in were estimated to escape from XCI (Figure 5A). Example gene-level XCI escape analysis results comparing allelic expression ratios to PPjg for 38 observations of the gene BGN, encoding biglycan, are presented in Figure 5B, which illustrates a high correlation between allelic expression ratio and posterior probability of escape; while BGN is thought to undergo XCI, many tumor samples showed evidence of escaping XCI while some tumor tissue samples exhibited mono-allelic expression (Table III). Similarly, Figure 5C shows high correlation between allelic expression ratios and PPjg for the gene USP9X, a putative tumor suppressor which is thought to escape XCI, but many tumor samples show evidence of silencing (Table IV). Figure 5D displays the posterior escape probabilities for all genes in and for which at least 10 samples were considered informative for XCI escape analyses (i.e., Njg ≥ 10), separated by XCI consensus states.
Figure 5.




(A) Stacked barplots of XCI escape analysis results for tumor tissue samples, separated by XCI consensus status. ‘E’=escape, ‘VE’=variable escape, and ‘S’=subject, as in Balaton et al. (B) Scatterplot of AERs (x-axis) compared to XCI posterior escape probabilities (PPjg) for XCI subject gene BGN. Sizes of plotted points are indicative of total number of allele-specific reads (Njg). (C) Scatterplot of AERs (x-axis) compared to XCI posterior escape probabilities (PPjg) for XCI escape gene USP9X. Sizes of plotted points are indicative of total number of allele-specific reads (Njg). (D) Tileplot of the posterior escape probabilities organized by XCI consensus state (Escape, Mostly Escape, Mostly Subject, Subject), with each column corresponding to a unique sample. Coloration is correspondent to calculated PPjg, with gradient of cyan-to-magenta corresponding to the spectrum of low-to-high values of PPjg on the scale of 0.00 – 1.00. For samples where there were no data for a given gene, the tile is left blank.
Table III.
Summary of XCI analysis results for genes thought to undergo XCI with strong evidence of escape (PP>0.90) from XCI in at least one ovarian tumor sample.
| Gene | Cytoband (Ensembl)a | Total number of tumors evaluated | Proportion of tumors escaping XCI (N escaping/N total)b | Consensus State[Balaton, Cotton, & Brown 2015] | Known Oncogene or Tumor Suppressor (reference) |
|---|---|---|---|---|---|
| CLCN4 | Xp22.2 | 9 | 13% (1/8) | Mostly Sc | |
| WWC3 | Xp22.2 | 11 | 14% (1/7) | Mostly S | |
| CXorf23 | Xp22.12 | 6 | 50% (2/4) | Sd | |
| DMD | Xp21.1 | 27 | 33% (3/9) | S | |
| CXorf36 | Xp11.3 | 19 | 50% (4/8) | S | Oncogene[Wang, et al. 2014] |
| OPHN1 | Xq12 | 14 | 63% (5/8) | S | |
| BRWD3 | Xq21.1 | 15 | 22% (2/9) | S | |
| SH3BGRL | Xq21.1 | 11 | 11% (1/9) | Mostly S | Oncogene/tumor suppressor[Muniz Lino, et al. 2014; Wang, et al. 2015] |
| DOCK11 | Xq24 | 18 | 7% (1/14) | Mostly S | |
| IL13RA1 | Xq24 | 10 | 14% (1/7) | Mostly S | |
| ELF4 | Xq26.1 | 19 | 8% (1/13) | S | Oncogene/tumor suppressor[Yao, et al. 2007] |
| FHL1 | Xq26.3 | 14 | 17% (1/6) | Mostly S | |
| SLITRK4 | Xq27.3 | 6 | 25% (1/4) | S | Oncogene[Davidson, et al. 2014] |
| BGN | Xq28 | 32 | 67% (12/18) | S | |
| GABRE | Xq28 | 9 | 29% (2/7) | S | |
| TAZ | Xq28 | 8 | 50% (2/4) | S | Oncogene[Chen, Xie, Huang, & Yang 2016; Moroishi, Hansen, & Guan 2015] |
| VMA21 | Xq28 | 11 | 11% (1/9) | S |
as denoted by Ensembl GRCh38
Tumors are defined as escaping XCI if PPjg>0.90, subject to XCI if PPjg<0.10, or otherwise were not called.
Mostly subject’ consensus state defined by previous literature suggests these genes are subject to XCI in most tissues
‘Subject’ consensus state defined by previous literature suggests these genes are subject to XCI in almost all tissues
Table IV.
Summary of XCI analysis results for genes thought to escape XCI with strong evidence of undergoing XCI (PP<0.10) from XCI in at least one ovarian tumor sample.
| Gene | Cytoband (Ensembl)a | Total number of tumors evaluated | Escaping XCI (PPjg>0.5) | Subject to XCI (PPjg<0.5) | Proportion of samples undergoing XCI (N undergoing/N total)b | Consensus State[Balaton, Cotton, & Brown 2015] | Known Oncogene or Tumor Suppressor (reference) |
|---|---|---|---|---|---|---|---|
| ARSD | Xp22.33 | 25 | 11 | 3 | 21% (3/14) | Mostly Ec | |
| KAL1 | Xp22.31 | 14 | 1 | 6 | 86% (6/7) | Mostly E | Tumor suppressor[Liu, Cao, Chen, Xu, & Zhang 2015; Tanaka, et al. 2015] |
| NLGN4X | Xp22.31 | 19 | 8 | 2 | 20% (2/10) | Mostly E | |
| CTPS2 | Xp22.2 | 9 | 0 | 9 | 100% (9/9) | Mostly E | Platinum sensitivity[Gamazon, et al. 2011] |
| GEMIN8 | Xp22.2 | 11 | 1 | 4 | 80% (4/5) | Mostly E | |
| GPM6B | Xp22.2 | 12 | 3 | 7 | 70% (7/10) | Mostly E | |
| OFD1 | Xp22.2 | 4 | 0 | 1 | 100% (1/1) | Mostly E | |
| RBBP7 | Xp22.2 | 2 | 0 | 2 | 100% (2/2) | Mostly E | Tumor suppressor[Li, et al. 2016] [Chaligne, et al. 2015] |
| TCEANC | Xp22.2 | 9 | 0 | 7 | 100% (7/7) | Mostly E | Tumor suppressor with BRCA1[Hill, et al. 2014] |
| TRAPPC2 | Xp22.2 | 23 | 10 | 3 | 23% (3/13) | Mostly E | Tumor suppressor[Tokunaga, et al. 2013] |
| TXLNG | Xp22.2 | 16 | 8 | 1 | 11% (1/9) | Ed | |
| EIF1AX | Xp22.12 | 21 | 4 | 11 | 73% (11/15) | E | |
| EIF2S3 | Xp22.11 | 18 | 1 | 4 | 80% (4/5) | E | |
| CXorf38 | Xp11.4 | 12 | 1 | 8 | 89% (8/9) | E | |
| DDX3X | Xp11.4 | 17 | 9 | 1 | 10% (1/10) | E | Tumor suppressor[Bol, Xie, & Raman 2015; Dunford, et al. 2016] |
| USP9X | Xp11.4 | 23 | 3 | 6 | 67% (6/9) | Mostly E | Tumor suppressor[Jo, Kim, Yoo, & Lee 2016; Perez-Mancera, et al. 2012] |
| IQSEC2 | Xp11.22 | 3 | 1 | 2 | 67% (2/3) | Mostly E | |
| KDM5C | Xp11.22 | 8 | 0 | 8 | 100% (8/8) | Mostly E | Tumor suppressor[Dalgliesh, et al. 2010; Dunford, et al. 2016] |
| RPS4X | Xq13.1 | 13 | 10 | 1 | 9% (1/11) | Mostly E | Prognostic marker[Tsofack, et al. 2013] |
| NAP1L3 | Xq21.32 | 3 | 0 | 2 | 100% (2/2) | Mostly E |
as denoted by Ensembl GRCh38
Tumors are defined as escaping XCI if PPjg>0.90, subject to XCI if PPjg<0.10, or otherwise were not called.
Consensus state defined by previous literature suggests these genes escape XCI in almost all tissues
Consensus state defined by previous literature suggests these genes escape XCI in most tissue
At the sample-level, individual samples also demonstrated high levels of concordance between the most likely genic escape status and the consensus states; on average, 10.3% of a priori ‘escape/mostly escape’ or ‘subject/mostly subject’ XCI genes were discordant for a given sample (Table S6). Of the discordant genes for a given sample, 58.4% were prior XCI escape genes now classified as subject to XCI.
Discordant genes
To isolate a high-confidence subset of genes with potentially aberrant XCI states across the tumor samples compared to the consensus state, we re-classified gene escape status under greater stringency (g ∈ GjE if PPjg >0.90, g ∈ GjI if PPjg < 0.10) and identified the subset of genes that corresponded to a gene in or and ≥1 tumor sample was predicted to be contrary to that state based on the stringent definition (i.e., PPjg > 0.90 and ). This analysis identified 37 discrepant genes, including 41 observations for 17 unique genes in with evidence of genes in tumor samples putatively escaping XCI (Table III). Review of the allele-specific read counts for these genes revealed allelic expression ratios near 0.5, and 5 genes (CXorf36, SH3BGRL, ELF4, SLITRK4, TAZ) have previously been associated with oncogenic properties, and many of these genes reside on the ‘q arm’ of the X. The expression of many of these genes are positively correlated with XIST expression (p<0.05 for all but DMD, ELF4, FHL1, and GABRE; data not shown). Additionally, 88 tumor observations of 20 unique genes corresponded to genes classified as subject to XCI in the tumor despite belonging to (Table 4). 7 genes (KAL1, RBBP7, TCEANC, TRAPPC2, DDX3X, USP9X, KDM5C) have been previously associated with tumor suppression, and two (CTPS2, RPS4X) have associations with treatment response or prognosis, and many of these genes reside on the ‘p arm’ of the X. The expression of all of these were positively associated with XIST expression (p<0.05; data not shown). The full gene-level results are presented in Table S7.
For many of the genes not classified into or based on consensus calls across multiple tissue types, our results suggest consistent XCI status in ovarian tumor tissue. For example, many genes previously classified as variable escape across multiple normal tissue types showed strong evidence of undergoing XCI in more than 10 ovarian tumor samples, such as HCFC1, MED14, PLS3, SHROOM2. Furthermore, genes previously labeled as ‘discordant’ across tissues (where the gene was consistently escaping in one tissue but not all tissues) also showed consistent evidence of undergoing XCI in more than 10 ovarian tumor samples, specifically DIAPH2, PIR, PNMA3, REPS2, and TBL1X. Lastly, among genes that previously had undetermined XCI status, we observed sufficient allele-specific expression in more than 10 ovarian tumor samples, providing evidence to determine XCI status in ovarian tissue. In particular, FAM127C and TMSB4X show evidence of undergoing XCI, whereas LOC389906 and TLR8 show evidence of escape from XCI and variable escape across ovarian tumor tissues, respectively.
DISCUSSION
We have developed a framework for the multi-omic analysis of XCI, have evaluated the performance of our method in simulation studies, and have applied our method to estimate genes that escape XCI in ovarian cancer. Integrating transcriptome sequence data, genome-wide genotyping, and copy-number segmentation results, we also characterized transcriptional XCI patterns in ovarian carcinoma samples with X-linked allelic expression data. In general, we observed allelic expression patterns consistent with the consensus gene-level XCI classifications, particularly genes subject to XCI. Across tumors, most samples corresponded to predicted active Xi transcription for only a small proportion of genes that were a priori anticipated to be inactive, indicative of sporadic XCI reactivation for discordant genes. Similarly, only a small proportion of genes that were a priori anticipated to escape XCI were predicted to be inactivated for most tumor samples, suggesting either deactivation of Xi, or possibly tissue-specific differences in XCI patterns.
The two-stage methods developed here to assess XCI skewness and gene-level XCI escape status for individual tumor samples have many advantages compared to previous approaches to estimate XCI escape genes. Previous approaches have been statistically simplistic, and have not utilized formal statistical tests or sophisticated probability models, and instead have implemented thresholds of allelic imbalance for escape status classification. We provide a statistical framework for modeling allele-specific expression data in the context of X chromosome inactivation based on known biological properties. Because most genes undergo silencing yet some escape inactivation, we assume a mixture distribution to account for the expected distributions under both of these states. Due to the over-dispersion typically present in expression data, we assume a beta-binomial model for this mixture distribution. Our framework is flexible, and could easily be extended as more is known about X chromosome biology; for example, ‘variable escape’ mixture state could be include, or the choice of reference XCI status could be updated based on domain or tissue-specific knowledge. Furthermore, this method could be applied to any study of XCI with available RNAseq and genotype data, and is not limited to studies of ovarian cancer. For studies of phenotypes affecting both males and females, the method could be extended to include male samples, and future directions involve incorporating total expression data to improve inference.
Using this biologically-driven mixture distribution to model the XCI process and account for two possible XCI states, we first developed a formal statistical test for skewness (based on a likelihood ratio test), allowing us to determine not only which samples exhibit skewed XCI, but also quantify the level of that skewness for a given sample. Furthermore, our second stage escape gene analysis provides estimates of the posterior probability of escape for each gene, for each individual sample; such estimates have not been provided with other approaches, and can aid in follow-up studies to connect the degree of escape from XCI with clinical characteristics. Because the process of XCI occurs in multiple latent states (e.g., escape and silenced) where prior knowledge about these states is available for many genes and a posterior probability of state membership is desired, the Bayesian paradigm is a natural fit for XCI escape gene analysis, which has not previously been utilized.
Additionally, our integrative genomic approach based on multiple genomic data types uses a direct measure of XCI (allele-specific expression data), as opposed to indirect measures such as total expression data or methylation data, and can estimate the degree of XCI dysregulation independent from copy number alterations. Furthermore, the use of ASE data mitigates the presence of batch effects, as the proportion of reads mapping to a particular allele is not dependent on the total coverage. Most importantly, our approach does not rely on analogous male samples for comparison, which are not available for female-specific disorders (such as gynecological cancers)—the phenotypes for which study of XCI is often most relevant. While in this study we applied these methods to a study of ovarian cancer, these methods are not cancer-specific and would be applicable to any study of XCI in females.
We evaluated the performance of our proposed two-stage approach in a series of simulations studies. We examined the type I error and power of the stage 1 test for skewness, and observed that the type I error of the likelihood ratio test is somewhat conservative, likely due to the use of a bootstrap approximation. However, the conservative nature of the test is offset by the extremely high power of the approach to detect moderate to high levels of skewness (consistent with what was observed in the ovarian tumor data). We then evaluated the classification performance of our approach to detect genes that escape from XCI. Accuracy, sensitivity, and specificity were high, especially under the use of an informative prior derived from the consensus states published in prior literature, although the method was also robust to the choice of a flat prior or misspecification of an informative prior, with performance similar for highly skewed samples with even low coverage.
In the stage 1 test for skewness, we observed a large degree of skewed XCI across tumors. These results are consistent with the notion that the clonal nature of tumors would correspond to relative homogeneity of the Xi in the corresponding tissue sample, and underscore the viability of exploring XCI patterns in tumor expression data. Although not evaluated here, a high degree of skewness may also be observed in normal ovarian tissue, because silencing of the inactive X occurs early in development at the time of gastrulation[van den Berg, et al. 2009], prior to the differentiation of Müllerian tissues. Furthermore, the high degree of tumor skewness is also consistent with observed increased levels of skewed XCI in lymphocytes of ovarian cancer patients compared to controls[Buller, Sood, Lallas, Buekers, & Skilling 1999; Lose, Duffy, Kay, Kedda, & Spurdle 2008], although subsequent studies have not observed this phenomenon in blood[Manoukian, et al. 2013]. This also suggests a lack of patients in our study with substantial chromosome-wide reactivation of the inactive X.
In the stage 2 analysis of gene-level XCI escape status, we identified 17 genes escaping XCI contrary to consensus patterns of normal XCI, indicating potential somatic alterations to XCI status, many of which have noted associations with oncogenic properties in the literature. Expression of most of these genes is positively correlated with XIST expression. Oncogenes showing evidence of unexpected escape from XCI in ovarian tumors highlights their potential for involvement in ovarian tumor initiation and progression due to increased gene dosage. Four tumors indicated potential aberrant XCI escape of CXorf36, which is highly expressed in ovarian tissue and encodes a protein of unknown function. A recent study of renal carcinoma indicated upregulated expression and 5′ regional hypomethylation of CXorf36, while similar results were observed in a study of polycystic ovarian syndrome[Wang, et al. 2014]. SLITRK4 is a member of the SLITRK transmembrane protein family that modulate neurite outgrowth, and overexpression of SLITRK4 mRNA and protein products have been noted in primary uterine leiomyosarcomas[Davidson, et al. 2014]. ELF4, also known as MEF, is an ETS-family transcriptional activator that has been associated with both tumor suppression and oncogenic function, and ELF4 overexpression has been shown to increase the oncogenic properties of ovarian cancer cells[Yao, et al. 2007]. Similarly, SH3BGRL demonstrates oncogenic activity in mice but tumor suppressive behavior in humans[Wang, et al. 2015], with upregulation of SH3BGRL protein observed in triple-negative breast cancer, a subtype of breast cancer with many biological, epidemiological and etiological similarities to high-grade serous ovarian cancer [Muniz Lino, et al. 2014]. Furthermore, tafazzin, encoded by TAZ, has been shown to promote the epithelial-mesenchymal transition and progression and tumorigenesis of many cancer types, including ovarian cancer[Chen, Xie, Huang, & Yang 2016; Moroishi, Hansen, & Guan 2015].
In contrast to the above listed genes, some tumor samples indicated XCI silencing of 20 escape genes, including many potential tumor suppressors. This high level of tumor suppressor genes that exhibit unanticipated silencing in ovarian tumors is noteworthy because of their potential for involvement in ovarian tumor initiation and progression due to reduced gene dosage. For instance, 6 tumors exhibited aberrant deactivation of KAL1, Kallman Syndrome sequence 1 gene, has been attributed tumor suppressive properties in studies of squamous cell carcinoma[Liu, Cao, Chen, Xu, & Zhang 2015] and hepatocellular carcinoma[Tanaka, et al. 2015]. RBBP7 protein RBBP7 (retinoblastoma binding protein 7) has been shown to have tumor suppressive properties by interacting with NKX6.1 in repressing vimentin and N-cadherin expression[Li, et al. 2016]. Notably, aberrant silencing of X-linked genes has previously been observed in studies of XCI in breast tumor cell lines[Chaligne, et al. 2015], indicating epigenetic modification of XCI is possible in either direction and may act as a mechanism of disrupting tumor suppressor activity. TCEANC is thought to interact with BRCA1 in ovarian tumor suppression[Hill, et al. 2014], and this gene was aberrantly silenced in seven samples. TRAPPC2 protein, trafficking protein complex 2, also known as MIP-2A, may inhibit tumor proliferation and could be a potential drug target[Tokunaga, et al. 2013]. DDX3X, encoding DEAD-box helicase 3 X-linked, is a putative tumor suppressor, and has also been associated with escape from XCI that could contribute to male sex bias for some cancers[Bol, Xie, & Raman 2015; Dunford, et al. 2016]. USP9X, encoding ubiquitin specific peptidase 9 X-linked, has been linked to tumor suppression in pancreatic and colorectal cancer[Jo, Kim, Yoo, & Lee 2016; Perez-Mancera, et al. 2012]. Interestingly, all eight samples evaluated for KDM5C showed evidence of inactivation. KDM5C encodes a histone lysine demethylase (KDM), and studies have suggested KDMs may act as both oncogenes and tumor suppressors for a variety of cancers[Cloos, Christensen, Agger, & Helin 2008]. KDM5C contributes to ovarian development[Sun, et al. 2016], is a candidate tumor suppressor in renal cancer[Dalgliesh, et al. 2010], and escape from XCI of KDM5C is also associated with cancer male bias[Dunford, et al. 2016].
Furthermore, tumor-specific profiles of XCI may not only provide additional information in identifying novel cancer genes in ovarian cancer, but could prove clinically useful in personalized medicine. Analyses of XIST expression in ovarian carcinoma tissues demonstrated significant correlations with chemotherapeutic response[Huang, et al. 2002]. In our study, two prior escape genes that showed evidence of silencing in the ovarian tumor (CTPS2 and RPS4X) have previously been shown to be associated with treatment response and prognosis. In cell lines, increased expression of CTPS2 was associated with reduced platinum sensitivity (carboplatin and cisplatin)[Gamazon, et al. 2011], and low expression of RPS4X was associated with poor prognosis in serous ovarian cancer [Tsofack, et al. 2013]. Furthermore, TRAPPC2 and DDX3X have also been identified as possible drug targets[Bol, Xie, & Raman 2015; Tokunaga, et al. 2013].
Interestingly, we also observed regional patterns regarding discrepant XCI states outside of the pseudo-autosomal regions. Many of the genes that indicated potential aberrant re-activation compared to the consensus state (including those with oncogenic properties) are located on the short (p) arm of the X chromosome, whereas many of the genes showing evidence of aberrant silencing compared to the consensus state (including many with tumor suppressive properties) are located on the long (q) arm of the X chromosome. This suggests that re-activation may be more likely on the p arm and de-activation may be more common on the q-arm, although further studies are needed to validate this finding and determine its significance.
There may also be relevant genes of interest to ovarian cancer that do not correspond to active or inactive XCI consensus states. TBL1X, denoted “discordant” in Balaton et al., was recently identified by Chaligne et al.[Chaligne, et al. 2015] to aberrantly reactivate on the Xi in breast cancer cell line ZR-75-1, resulting in increased gene and protein expression dosage. In our analyses, 23 tumors had available TBLIX ASE data for XCI escape analysis, of which two corresponded to posterior escape probabilities > 0.99. These two observations of TBLIX escaping XCI may be attributable to either somatic alteration in otherwise silenced Xi alleles, or normal inter-sample variability of XCI for TBLIX in ovarian epithelial cells. These findings highlight the importance of tissue-specific expectations of gene-level XCI status as well as paired tumor-normal tissue analyses in cancer studies of XCI, which may better inform likely instances of aberrant XCI activity. These analyses relied on consensus XCI states across multiple tissues for tumor comparisons, but due to potential tissue-specificity of ovarian XCI patterns, future studies to examine XCI states in normal ovarian tissue are warranted.
In addition to lack of paired normal expression data to characterize normal ovarian XCI patterns, there are other limitations to this study. For instance, gene-level inference was limited to only a fraction of the total expressed X-linked genes due to the additional data burden of RNA-Seq reads overlapping eSNPs, and many genes corresponded to zero read counts for ASE. Additional integration of available total expression may make the inclusion of more genes in XCI analysis possible; however, analytical complications arise with the correlation structure of gene co-expression as well as substantial transcriptomic dysregulation in the tumor environment. Furthermore, the detection of skewed XCI samples is dependent upon the assumption that the XCI status of the majority of presumably inactive genes is preserved, as the skewness testing we conducted cannot discriminate between complete reactivation of the Xi and high cellular heterogeneity of XCI. Consequently, tumor samples that failed skewness testing may alternatively correspond to full epigenetic erosion of the Barr body. Moreover, allelic imbalance for a particular gene could be driven by factors unrelated to XCI, such as cis-acting regulatory genetic variation, and genes indicating potential aberrant deactivation on the Xi in tumor samples would require confirmation with paired normal tissue to exclude this possibility. Further integration of epigenetic markers, such as promoter DNA methylation and histone modifications, may provide supporting evidence of genic XCI status and strengthen multi-omic XCI analyses. Finally, our analysis strategies were dependent upon assumed normal copy number states for the assessed genes, and analytical methods that could incorporate the presence of CNAs and LOH would be of both biological and clinical interest. Future evaluation of XCI in cancer would especially benefit from single-cell analyses, which would be able to circumvent issues of uncertainty attributable to potential cellular homogeneity of Xi as well as normal tissue contamination.
CONCLUSIONS
We demonstrate a novel framework that shows dysregulation of XCI in individual ovarian tumors for a number of genes across the X chromosome. While general patterns of XCI in ovarian cancers were largely consistent with previously identified XCI consensus states, we identified discrepant genic XCI status classifications for a number of X-linked genes, including many with potential biological relevance to tumorigenesis and tumor suppression. These results provide initial evidence of potential somatic alteration to XCI in ovarian cancer and highlight an additional mechanism of gene expression dysregulation. Future studies using paired tumor-normal tissue analysis, multi-omic integration of epigenetic data, and single-cell sequencing may further elucidate the degree of susceptibility of XCI to dysregulation as well as its role in the risk, progression, and treatment resistance of ovarian cancer.
Supplementary Material
Histogram of allelic expression ratios for XIST, for 16 samples with a sufficient number of XIST allele-specific reads.
Acknowledgments
FUNDING This work was supported by funding from the National Institute of Health Office of Women’s Health Research (Building Interdisciplinary Careers in Women’s Health Award K12-HD065987) and the Fraternal Order of the Eagles Cancer Research Fund, as well as R01-CA122443, P50-CA136393, 4R00CA184415-02 and P30-CA15083.
Footnotes
CONFLICT OF INTEREST: The authors declare no potential conflicts of interest.
AUTHOR CONTRIBUTIONS SJW conceived of the study. SJW and NBL designed the study, developed the methods, analyzed the data, and drafted the manuscript. ZCF and MCL prepared and analyzed the data, and drafted the manuscript. KRK, BLF, and ELG generated the data, provided interpretation, and drafted the manuscript. KL and SG provided interpretation and drafted the manuscript.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This study was approved by the Mayo Clinic Institutional Review Board and all patients provided informed consent.
AVAILABILITY OF DATA AND MATERIAL The datasets used during the current study are available from the authors upon reasonable request.
COMPETING INTERESTS The authors do not declare any competing interests.
References
- Balaton BP, Cotton AM, Brown CJ. Derivation of consensus inactivation status for X-linked genes from genome-wide studies. Biology of sex differences. 2015;6:35. doi: 10.1186/s13293-015-0053-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr ML, Moore KL. Chromosomes, sex chromatin, and cancer. Proceedings Canadian Cancer Conference. 1957;2:3–16. [PubMed] [Google Scholar]
- Berletch JB, Yang F, Xu J, Carrel L, Disteche CM. Genes that escape from X inactivation. Hum Genet. 2011;130(2):237–45. doi: 10.1007/s00439-011-1011-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bol GM, Xie M, Raman V. DDX3, a potential target for cancer treatment. Mol Cancer. 2015;14:188. doi: 10.1186/s12943-015-0461-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buller RE, Sood AK, Lallas T, Buekers T, Skilling JS. Association between nonrandom X-chromosome inactivation and BRCA1 mutation in germline DNA of patients with ovarian cancer. J Natl Cancer Inst. 1999;91(4):339–46. doi: 10.1093/jnci/91.4.339. [DOI] [PubMed] [Google Scholar]
- Carpenter B, Gelman A, Hoffman M, Lee D, Goodrich B, Betancourt M, Brubaker MA, Guo J, Li P, Riddell A. Stan: a probabilistic programming language. Journal of Statistical Software. 2015 doi: 10.18637/jss.v076.i01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434(7031):400–4. doi: 10.1038/nature03479. [DOI] [PubMed] [Google Scholar]
- Chaligne R, Heard E. X-chromosome inactivation in development and cancer. FEBS letters. 2014;588(15):2514–22. doi: 10.1016/j.febslet.2014.06.023. [DOI] [PubMed] [Google Scholar]
- Chaligne R, Popova T, Mendoza-Parra MA, Saleem MA, Gentien D, Ban K, Piolot T, Leroy O, Mariani O, Gronemeyer H, et al. The inactive X chromosome is epigenetically unstable and transcriptionally labile in breast cancer. Genome research. 2015;25(4):488–503. doi: 10.1101/gr.185926.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Xie J, Huang P, Yang Z. Overexpression of TAZ promotes cell proliferation, migration and epithelial-mesenchymal transition in ovarian cancer. Oncol Lett. 2016;12(3):1821–1825. doi: 10.3892/ol.2016.4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng PC, Gosewehr JA, Kim TM, Velicescu M, Wan M, Zheng J, Felix JC, Cofer KF, Luo P, Biela BH, et al. Potential role of the inactivated X chromosome in ovarian epithelial tumor development. J Natl Cancer Inst. 1996;88(8):510–8. doi: 10.1093/jnci/88.8.510. [DOI] [PubMed] [Google Scholar]
- Cloos PA, Christensen J, Agger K, Helin K. Erasing the methyl mark: histone demethylases at the center of cellular differentiation and disease. Genes & development. 2008;22(9):1115–40. doi: 10.1101/gad.1652908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotton AM, Ge B, Light N, Adoue V, Pastinen T, Brown CJ. Analysis of expressed SNPs identifies variable extents of expression from the human inactive X chromosome. Genome biology. 2013;14(11):R122. doi: 10.1186/gb-2013-14-11-r122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotton AM, Price EM, Jones MJ, Balaton BP, Kobor MS, Brown CJ. Landscape of DNA methylation on the X chromosome reflects CpG density, functional chromatin state and X-chromosome inactivation. Human molecular genetics. 2015;24(6):1528–39. doi: 10.1093/hmg/ddu564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, Davies H, Edkins S, Hardy C, Latimer C, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463(7279):360–3. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson B, Abeler VM, Forsund M, Holth A, Yang Y, Kobayashi Y, Chen L, Kristensen GB, Shih Ie M, Wang TL. Gene expression signatures of primary and metastatic uterine leiomyosarcoma. Human pathology. 2014;45(4):691–700. doi: 10.1016/j.humpath.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaneau O, Zagury JF, Marchini J. Improved whole-chromosome phasing for disease and population genetic studies. Nature methods. 2013;10(1):5–6. doi: 10.1038/nmeth.2307. [DOI] [PubMed] [Google Scholar]
- Dunford A, Weinstock DM, Savova V, Schumacher SE, Cleary JP, Yoda A, Sullivan TJ, Hess JM, Gimelbrant AA, Beroukhim R, et al. Tumor-suppressor genes that escape from X-inactivation contribute to cancer sex bias. Nat Genet. 2016 doi: 10.1038/ng.3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamazon ER, Im HK, O'Donnell PH, Ziliak D, Stark AL, Cox NJ, Dolan ME, Huang RS. Comprehensive evaluation of the contribution of X chromosome genes to platinum sensitivity. Molecular cancer therapeutics. 2011;10(3):472–80. doi: 10.1158/1535-7163.MCT-10-0910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson BE, Feigelson HS. Hormonal carcinogenesis. Carcinogenesis. 2000;21(3):427–33. doi: 10.1093/carcin/21.3.427. [DOI] [PubMed] [Google Scholar]
- Hill SJ, Rolland T, Adelmant G, Xia X, Owen MS, Dricot A, Zack TI, Sahni N, Jacob Y, Hao T, et al. Systematic screening reveals a role for BRCA1 in the response to transcription-associated DNA damage. Genes & development. 2014;28(17):1957–75. doi: 10.1101/gad.241620.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang KC, Rao PH, Lau CC, Heard E, Ng SK, Brown C, Mok SC, Berkowitz RS, Ng SW. Relationship of XIST expression and responses of ovarian cancer to chemotherapy. Molecular cancer therapeutics. 2002;1(10):769–76. [PubMed] [Google Scholar]
- Huang RYJ, Chen GB, Matsumura N, Lai HC, Mori S, Li J, Wong MK, Konishi I, Thiery JP, Goh L. Histotype-specific copy-number alterations in ovarian cancer. BMC Med Genomics. 2012;5:47. doi: 10.1186/1755-8794-5-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo YS, Kim MS, Yoo NJ, Lee SH. USP9X, a Putative Tumor Suppressor Gene, Exhibits Frameshift Mutations in Colorectal Cancers. Pathol Oncol Res. 2016 doi: 10.1007/s12253-016-0140-z. [DOI] [PubMed] [Google Scholar]
- Li HJ, Yu PN, Huang KY, Su HY, Hsiao TH, Chang CP, Yu MH, Lin YW. NKX6.1 functions as a metastatic suppressor through epigenetic regulation of the epithelial-mesenchymal transition. Oncogene. 2016;35(17):2266–78. doi: 10.1038/onc.2015.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Cao W, Chen W, Xu L, Zhang C. Decreased expression of Kallmann syndrome 1 sequence gene (KAL1) contributes to oral squamous cell carcinoma progression and significantly correlates with poorly differentiated grade. J Oral Pathol Med. 2015;44(2):109–14. doi: 10.1111/jop.12206. [DOI] [PubMed] [Google Scholar]
- Lose F, Duffy DL, Kay GF, Kedda MA, Spurdle AB. Skewed X chromosome inactivation and breast and ovarian cancer status: evidence for X–linked modifiers of BRCA1. J Natl Cancer Inst. 2008;100(21):1519–29. doi: 10.1093/jnci/djn345. [DOI] [PubMed] [Google Scholar]
- Manoukian S, Verderio P, Tabano S, Colapietro P, Pizzamiglio S, Grati FR, Calvello M, Peissel B, Burn J, Pensotti V, et al. X chromosome inactivation pattern in BRCA gene mutation carriers. Eur J Cancer. 2013;49(5):1136–41. doi: 10.1016/j.ejca.2012.10.013. [DOI] [PubMed] [Google Scholar]
- Mclachlan GJ. On Bootstrapping the Likelihood Ratio Test Statistic for the Number of Components in a Normal Mixture. Applied Statistics-Journal of the Royal Statistical Society Series C. 1987;36(3):318–324. [Google Scholar]
- Moroishi T, Hansen CG, Guan KL. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer. 2015;15(2):73–9. doi: 10.1038/nrc3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muniz Lino MA, Palacios-Rodriguez Y, Rodriguez-Cuevas S, Bautista-Pina V, Marchat LA, Ruiz-Garcia E, Astudillo-de la Vega H, Gonzalez-Santiago AE, Flores-Perez A, Diaz-Chavez J, et al. Comparative proteomic profiling of triple-negative breast cancer reveals that up-regulation of RhoGDI-2 is associated to the inhibition of caspase 3 and caspase 9. Journal of proteomics. 2014;111:198–211. doi: 10.1016/j.jprot.2014.04.019. [DOI] [PubMed] [Google Scholar]
- Pageau GJ, Hall LL, Ganesan S, Livingston DM, Lawrence JB. The disappearing Barr body in breast and ovarian cancers. Nat Rev Cancer. 2007;7(8):628–33. doi: 10.1038/nrc2172. [DOI] [PubMed] [Google Scholar]
- Perez-Mancera PA, Rust AG, van der Weyden L, Kristiansen G, Li A, Sarver AL, Silverstein KA, Grutzmann R, Aust D, Rummele P, et al. The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature. 2012;486(7402):266–70. doi: 10.1038/nature11114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Marioni JC, Pai AA, Degner JF, Engelhardt BE, Nkadori E, Veyrieras JB, Stephens M, Gilad Y, Pritchard JK. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature. 2010;464(7289):768–72. doi: 10.1038/nature08872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porzio GC, Ragozini G. On the stochastic ordering of folded binomials. Statistics & Probability Letters. 2009;79(9):1299–1304. [Google Scholar]
- Ross MT, Grafham DV, Coffey AJ, Scherer S, McLay K, Muzny D, Platzer M, Howell GR, Burrows C, Bird CP, et al. The DNA sequence of the human X chromosome. Nature. 2005;434(7031):325–37. doi: 10.1038/nature03440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31(1):27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AJ, Stathaki E, Migliavacca E, Brahmachary M, Montgomery SB, Dupre Y, Antonarakis SE. DNA methylation profiles of human active and inactive X chromosomes. Genome Res. 2011;21(10):1592–600. doi: 10.1101/gr.112680.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W. A statistical framework for eQTL mapping using RNA-seq data. Biometrics. 2012;68(1):1–11. doi: 10.1111/j.1541-0420.2011.01654.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YX, Zhang YX, Zhang D, Xu CM, Chen SC, Zhang JY, Ruan YC, Chen F, Zhang RJ, Qian YQ, et al. XCI-escaping gene KDM5C contributes to ovarian development via downregulating miR-320a. Human genetics. 2016 doi: 10.1007/s00439-016-1752-9. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Kanda M, Sugimoto H, Shimizu D, Sueoka S, Takami H, Ezaka K, Hashimoto R, Okamura Y, Iwata N, et al. Translational implication of Kallmann syndrome-1 gene expression in hepatocellular carcinoma. Int J Oncol. 2015;46(6):2546–54. doi: 10.3892/ijo.2015.2965. [DOI] [PubMed] [Google Scholar]
- Tokunaga M, Shiheido H, Tabata N, Sakuma-Yonemura Y, Takashima H, Horisawa K, Doi N, Yanagawa H. MIP-2A is a novel target of an anilinoquinazoline derivative for inhibition of tumour cell proliferation. PLoS One. 2013;8(9):e76774. doi: 10.1371/journal.pone.0076774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsofack SP, Meunier L, Sanchez L, Madore J, Provencher D, Mes-Masson AM, Lebel M. Low expression of the X-linked ribosomal protein S4 in human serous epithelial ovarian cancer is associated with a poor prognosis. BMC Cancer. 2013;13:303. doi: 10.1186/1471-2407-13-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tukiainen T, Villani A-C, Yen A, Rivas MA, Marshall JL, Satija R, Aguirre M, Gauthier L, Fleharty M, Kirby A, et al. Landscape of X chromosome inactivation across human tissues. bioRxiv. 2016 doi: 10.1038/nature25993. [DOI] [PubMed] [Google Scholar]
- van den Berg IM, Laven JSE, Stevens M, Jonkers I, Galjaard R-J, Gribnau J, Hikke van Doorninck J. X Chromosome Inactivation Is Initiated in Human Preimplantation Embryos. American Journal of Human Genetics. 2009;84(6):771–779. doi: 10.1016/j.ajhg.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Liu B, Al-Aidaroos AQ, Shi H, Li L, Guo K, Li J, Tan BC, Loo JM, Tang JP, et al. Dual-faced SH3BGRL: oncogenic in mice, tumor suppressive in humans. Oncogene. 2015 doi: 10.1038/onc.2015.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XX, Wei JZ, Jiao J, Jiang SY, Yu DH, Li D. Genome-wide DNA methylation and gene expression patterns provide insight into polycystic ovary syndrome development. Oncotarget. 2014;5(16):6603–10. doi: 10.18632/oncotarget.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winham SJ, Pirie A, Chen YA, Larson MC, Fogarty ZC, Earp MA, Anton-Culver H, Bandera EV, Cramer D, Doherty JA, et al. Investigation of Exomic Variants Associated with Overall Survival in Ovarian Cancer. Cancer Epidemiol Biomarkers Prev. 2016;25(3):446–54. doi: 10.1158/1055-9965.EPI-15-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao JJ, Liu Y, Lacorazza HD, Soslow RA, Scandura JM, Nimer SD, Hedvat CV. Tumor promoting properties of the ETS protein MEF in ovarian cancer. Oncogene. 2007;26(27):4032–7. doi: 10.1038/sj.onc.1210170. [DOI] [PubMed] [Google Scholar]
- Yau C, Mouradov D, Jorissen RN, Colella S, Mirza G, Steers G, Harris A, Ragoussis J, Sieber O, Holmes CC. A statistical approach for detecting genomic aberrations in heterogeneous tumor samples from single nucleotide polymorphism genotyping data. Genome biology. 2010;11(9):R92. doi: 10.1186/gb-2010-11-9-r92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Histogram of allelic expression ratios for XIST, for 16 samples with a sufficient number of XIST allele-specific reads.