

Summary
The spontaneous deamination of cytosine is a major source of C•G to T•A transitions, which account for half of known human pathogenic point mutations. The ability to efficiently convert target A•T base pairs to G•C therefore could advance the study and treatment of genetic diseases. While the deamination of adenine yields inosine, which is treated as guanine by polymerases, no enzymes are known to deaminate adenine in DNA. Here we report adenine base editors (ABEs) that mediate conversion of A•T to G•C in genomic DNA. We evolved a tRNA adenosine deaminase to operate on DNA when fused to a catalytically impaired CRISPR-Cas9. Extensive directed evolution and protein engineering resulted in seventh-generation ABEs (e.g., ABE7.10), that convert target A•T to G•C base pairs efficiently (~50% in human cells) with very high product purity (typically ≥ 99.9%) and very low rates of indels (typically ≤ 0.1%). ABEs introduce point mutations more efficiently and cleanly than a current Cas9 nuclease-based method, induce less off-target genome modification than Cas9, and can install disease-correcting or disease-suppressing mutations in human cells. Together with our previous base editors, ABEs advance genome editing by enabling the direct, programmable introduction of all four transition mutations without double-stranded DNA cleavage.
The formation of uracil and thymine from the spontaneous hydrolytic deamination of cytosine and 5-methylcytosine, respectively1,2 occurs an estimated 100–500 times per cell per day in humans1 and can result in C•G to T•A mutations, accounting for approximately half of all known pathogenic SNPs (Fig. 1a). The ability to convert A•T base pairs to G•C base pairs at target loci in the genomic DNA of unmodified cells therefore could enable the correction of a substantial fraction of human SNPs associated with disease.
Figure 1. Scope and overview of base editing by an A•T to G•C base editor (ABE).
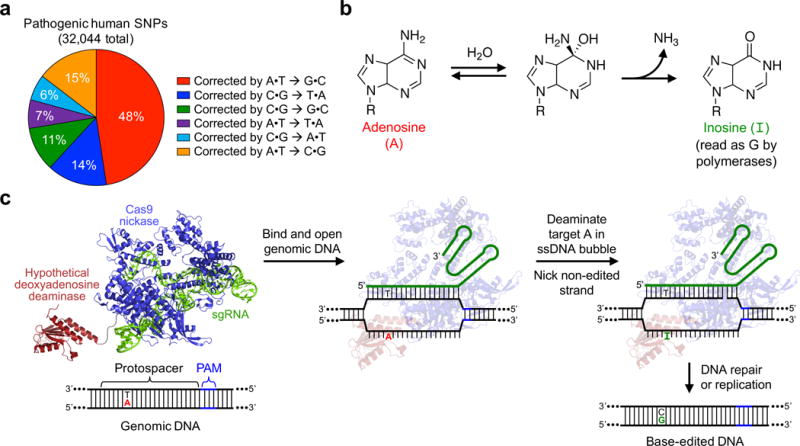
a, Base pair changes required to correct pathogenic human SNPs in the ClinVar database. b, The deamination of adenosine (A) forms inosine (I), which is read as guanosine (G) by polymerase enzymes. c, ABE-mediated A•T to G•C base editing strategy. ABEs contain a hypothetical deoxyadenosine deaminase, which is not known to exist in nature, and a catalytically impaired Cas9. They bind target DNA in a guide RNA-programmed manner, exposing a small bubble of single-stranded DNA. The hypothetical deoxyadenosine deaminase domain catalyzes A to I formation within this bubble. Following DNA repair or replication, the original A•T base pair is replaced with a G•C base pair at the target site.
Base editing is a form of genome editing that enables direct, irreversible conversion of one base pair to another at a target genomic locus without requiring double-stranded DNA breaks (DSBs), homology-directed repair (HDR) processes, or donor DNA templates3–5. Compared with standard genome editing methods to introduce point mutations, base editing can proceed more efficiently3, and with far fewer undesired products such as stochastic insertions or deletions (indels) or translocations3–8.
The most commonly used base editors are third-generation designs (BE3) comprising (i) a catalytically impaired CRISPR-Cas9 mutant that cannot make DSBs, (ii) a single-strand-specific cytidine deaminase that converts C to uracil (U) within a ~5-nucleotide window in the single-stranded DNA bubble created by Cas9, (iii) a uracil glycosylase inhibitor (UGI) that impedes uracil excision and downstream processes that decrease base editing efficiency and product purity5, and (iv) nickase activity to nick the non-edited DNA strand, directing cellular DNA repair processes to replace the G-containing DNA strand3,5. Together, these components enable efficient and permanent C•G to T•A base pair conversion in bacteria, yeast4,9, plants10,11, zebrafish8,12, mammalian cells3–8,13,14, mice8,15,16, and even human embryos17,18. Base editing capabilities have expanded through the development of base editors with different protospacer-adjacent motif (PAM) compatibilities7, narrowed editing windows7, enhanced DNA specificity8, and small-molecule dependence19. Fourth-generation base editors (BE4 and BE4-Gam) further improve editing efficiency and product purity5.
To date, all reported base editors mediate C•G to T•A conversion. In this study, we used protein evolution and engineering to develop a new class of adenine base editors (ABEs) that convert A•T to G•C base pairs in DNA in bacteria and human cells. Seventh-generation ABEs efficiently convert A•T to G•C at a wide range of target genomic loci in human cells efficiently and with a very high degree of product purity, exceeding the typical performance characteristics of BE3. ABEs greatly expand the scope of base editing and, together with previously described base editors, enable programmable installation of all four transitions (C to T, A to G, T to C, and G to A) in genomic DNA.
Results
Evolution of an adenine deaminase that processes DNA
The hydrolytic deamination of adenosine yields inosine (Fig. 1b). Within the constraints of a polymerase active site, inosine pairs with C and therefore is read or replicated as G20. While replacing the cytidine deaminase of an existing base editor with an adenine deaminase could, in theory, provide an ABE (Fig. 1c), no enzymes are known to deaminate adenine in DNA. Although all reported examples of enzymatic adenine deamination occurs on free adenine, free adenosine, adenosine in RNA, or adenosine in mispaired RNA:DNA heteroduplexes21, we began by replacing the APOBEC1 component of BE3 with natural adenine deaminases including E. coli TadA22,23, human ADAR224, mouse ADA25, and human ADAT226 (Supplementary Sequences 1) to test the possibility that these enzymes might process DNA when present at a high effective molarity. Unfortunately, when plasmids encoding these deaminases fused to Cas9 D10A nickase were transfected into HEK293T cells together with a corresponding single guide RNA (sgRNA), we observed no A•T to G•C editing above that of untreated cells (Extended Data Fig. E1 and E2b). These results suggest that the inability of these natural adenine deaminase enzymes to accept DNA precludes their direct use in an ABE.
Given these results, we sought to evolve an adenine deaminase that accepts DNA as a substrate. We developed a bacterial selection for base editing by creating defective antibiotic resistance genes that contain point mutations at critical positions (Supplementary Table 8 and Supplementary Sequences 2). Reversion of these mutations by base editors restores antibiotic resistance. To validate the selection, we used a bacterial codon-optimized version of BE23 (APOBEC1 cytidine deaminase fused to dCas9 and UGI), since bacteria lack nick-directed mismatch repair machinery27 that enables more efficient base editing by BE3. We observed successful rescue of a defective chloramphenicol acetyl transferase (CamR) containing an A•T to G•C mutation at a catalytic residue (H193R) by BE2 and an sgRNA programmed to direct base editing to the inactivating mutation.
Next we adapted the selection plasmid for ABE activity by introducing a C•G to T•A mutation in the CamR gene, creating an H193Y substitution that confers minimal chloramphenicol resistance (Supplementary Table 8 and Supplementary Sequences 2). A•T to G•C conversion at the H193Y mutation should restore chloramphenicol resistance, linking ABE activity to bacterial survival.
Our previously described base editors3,5,7,8 exploit the use of cytidine deaminase enzymes that operate on single-stranded DNA but reject double-stranded DNA. This feature is critical to restrict deaminase activity to a small window of nucleotides within the single-stranded bubble created by Cas9. TadA is a tRNA adenine deaminase22 that converts adenine to inosine (I) in the single-stranded anticodon loop of tRNAArg. E. coli TadA shares homology with the APOBEC enzyme28 used in our original base editors, and some ABOBECs bind single-stranded DNA in a conformation that resembles tRNA bound to TadA28. TadA does not require small-molecule activators (in contrast with ADAR29) and acts on polynucleic acid (unlike ADA25). Based on these considerations, we chose E. coli TadA as the starting point of our efforts to evolve a DNA adenine deaminase.
We created unbiased libraries of ecTadA-dCas9 fusions containing mutations only in the adenine deaminase portion of the construct to avoid altering favorable properties of the Cas9 portion of the editor (Supplementary Table 7). The resulting plasmids were transformed into E. coli harboring the CamR H193Y selection (Fig. 2a and Supplementary Table 8). Colonies surviving chloramphenicol challenge were strongly enriched for TadA mutations A106V and D108N (Fig. 2b). Sequence alignment of E. coli TadA with S. aureus TadA, for which a structure complexed with tRNAArg has been reported30, predicts that the side-chain of D108 hydrogen bonds with the 2’-OH group of the ribose in the U upstream of the substrate A (Fig. 2c). Mutations at D108 likely abrogate this hydrogen bond, decreasing the energetic opportunity cost of binding DNA. DNA sequencing confirmed that all clones surviving the selection showed A•T to G•C reversion at the targeted site in CamR. Collectively, these results indicate that mutations at or near TadA D108 enable TadA to perform adenine deamination on DNA substrates.
Figure 2. Protein evolution and engineering of ABEs.
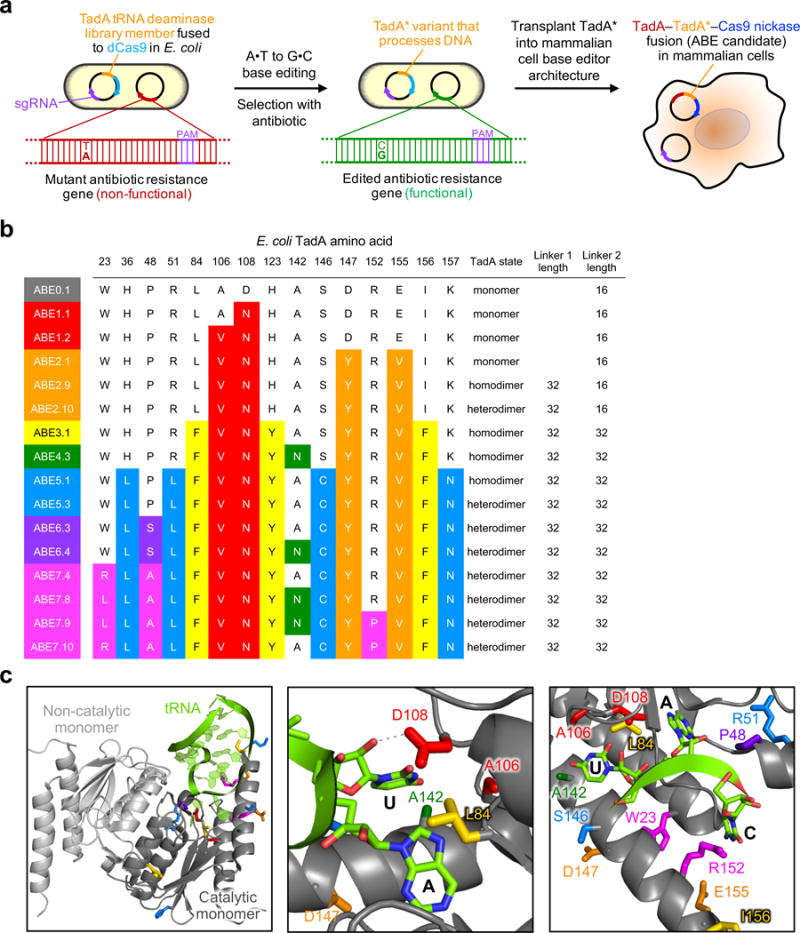
a, Strategy to evolve a DNA deoxyadenosine deaminase starting from TadA. A library of E. coli harbors a plasmid library of mutant ecTadA (TadA*) genes fused to dCas9 and a selection plasmid requiring targeted A•T to G•C mutations to repair antibiotic resistance genes. Mutations from surviving TadA* variants were imported into an ABE architecture for base editing in human cells. b, Genotypes of a subset of evolved ABEs. For a list of 57 evolved TadA* genotypes, see Extended Data Figure E1. The dimerization state (monomer, TadA*–TadA* homodimer, or wild-type TadA–evolved TadA* heterodimer) and linker length (in amino acids) are also listed. c, Three views of the E. coli TadA deaminase (PDB 1Z3A) aligned with S. aureus TadA (not shown) complexed with tRNAArg2 (PDB 2B3J). The UAC anticodon loop of the tRNA is the native substrate of wild-type TadA.
The TadA A106V and D108N mutations were incorporated into a mammalian codon-optimized TadA–Cas9 nickase fusion construct that replaces dCas9 with the Cas9 D10A nickase used in BE3 to manipulate cellular DNA repair to favor desired base editing outcomes3, and adds a C-terminal nuclear localization signal (NLS). We designated the resulting TadA*–XTEN–nCas9–NLS construct, where TadA* represents an evolved TadA variant and XTEN is a 16-amino acid linker used in BE33, as ABE1.2. Transfection of plasmids expressing ABE1.2 and sgRNAs targeting six diverse human genomic sites (Extended Data Fig. E2a) resulted in very low, but observable A•T to G•C editing efficiencies (3.2±0.88%; all editing efficiencies are reported as mean±SD of three biological replicates 5 days post-transfection without enrichment for transfected cells unless otherwise noted) at or near protospacer position 5, counting the PAM as positions 21–23 (Fig. 3a). These data confirmed that an ABE capable of catalyzing low levels of A•T to G•C conversion emerged from the first round of protein evolution and engineering.
Figure 3. Evolved ABEs mediate A•T to G•C base editing at human genomic DNA sites.
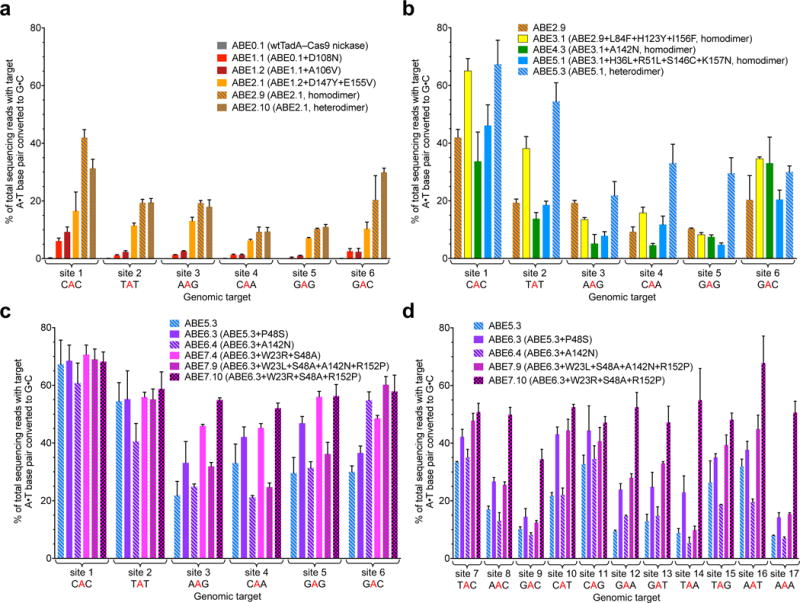
A•T to G•C base editing efficiencies in HEK293T cells of round 1 and round 2 ABEs (a), round 3, round 4, and round 5 ABEs (b), and round 6 and round 7 ABEs (c) at six human genomic DNA sites. d, Editing efficiencies in HEK293T cells of round 6 and round 7 ABEs at an expanded set of human genomic sites. Values and error bars reflect the mean and s.d. of three independent biological replicates performed on different days. Homodimer indicates fused TadA*–TadA*–Cas9 nickase architecture; heterodimer indicates fused wtTadA–TadA*–Cas9 nickase architecture. ABEs in (b) are homodimers except ABE5.3; ABEs in (c) and (d) are all heterodimers.
Improved Deaminase Variants and ABE Architectures
To improve editing efficiencies, we generated an unbiased library of ABE1.2 variants and challenged the resulting TadA*1.2–dCas9 mutants in bacteria with higher concentrations of chloramphenicol than were used in round 1 (Supplementary Tables 7 and 8). From round 2 we identified two new mutations, D147Y and E155V, predicted to lie in a helix adjacent to the TadA tRNA substrate (Fig. 2c). In mammalian cells, ABE2.1 (ABE1.2 + D147Y + E155V) exhibited 2- to 7-fold higher activity than ABE1.2 at the six genomic sites tested, resulting in an average of 11±2.9% A•T to G•C base editing (Fig. 3a).
Next we sought to improve ABE2.1 through additional protein engineering. Fusing the TadA(2.1)* domain to the C-terminus of Cas9 nickase, instead of the N-terminus, abolished editing activity (Extended Data Fig. E2c), consistent with our previous findings with BE33. We also varied linker lengths between TadA(2.1)* and Cas9 nickase. An ABE2 variant (ABE2.6) with a linker twice as long (32 amino acids, (SGGS)2-XTEN-(SGGS)2,) as the linker in ABE2.1 offered modestly higher editing efficiencies, now averaging 14±2.4% across the six genomic loci tested (Extended Data Fig. E2c).
Alkyl adenine DNA glycosylase (AAG) catalyzes the cleavage of the glycosidic bond of inosine in DNA31. To test if inosine excision impedes ABE performance, we created ABE2 variants designed to minimize potential sources of inosine excision. Given the absence of known protein inhibitors of AAG, we attempted to block endogenous AAG from accessing the inosine intermediate by separately fusing to ABE2.1 catalytically inactivated versions of enzymes involved in inosine binding or removal: human AAG (inactivated with a E125Q mutation31), or E. coli Endo V (inactivated with a D35A mutation32). Neither ABE2.1–AAG(E125Q) (ABE2.2) nor ABE2.1–Endo V(D35A) (ABE2.3) exhibited altered A•T to G•C editing efficiencies in HEK293T cells compared with ABE2.1 (Extended Data Fig. E2d). Indeed, ABE2.1 in Hap1 cells lacking AAG failed to increase base editing efficiency or product purity compared with Hap1 cells containing wild-type AAG (Extended Data Fig. E2e). Moreover, ABE2.1 induced virtually no indels (≤ 0.1%) or A•T to non-G•C products (≤ 0.1%) in HEK293T cells, consistent with inefficient excision of inosine (Extended Data Fig. E3). Taken together, these observations suggest that cellular repair of inosine intermediates created by ABEs is inefficient, obviating the need to subvert base excision repair. This situation contrasts with that of BE3 and BE4, which are strongly dependent on inhibiting uracil excision to maximize base editing efficiency and product purity3,5.
As a final ABE2 engineering study, we explored the role of TadA* dimerization on base editing efficiencies. TadA natively operates as a homodimer, with one monomer catalyzing deamination, and the other monomer acting as a docking station for the tRNA substrate30. During selection in E. coli, endogenous TadA likely serves as the non-catalytic monomer. In mammalian cells, we hypothesized that tethering an additional wild-type or evolved TadA monomer might improve base editing by minimizing reliance on intermolecular ABE dimerization. Indeed, co-expression with ABE2.1 of either wild-type TadA or TadA*2.1 (ABE2.7 and ABE2.8, respectively), as well as direct fusion of either evolved or wild-type TadA to the N-terminus of ABE2.1 (ABE2.9 and ABE2.10, respectively), substantially improved editing efficiencies (Fig. 3a and Extended Data Fig. E4a). A fused TadA*–ABE2.1 architecture (ABE2.9) was identified to offer the highest editing efficiencies (averaging 20±3.8% across the six genomic loci, a 7.6±2.6-fold average improvement at each site over ABE1.2) and was used in all subsequent experiments (Fig. 2b and 3a).
Finally, we determined which of the two TadA* subunits within the TadA*–ABE2.1 fusion was responsible for A to I catalysis. We introduced an inactivating E59A mutation22 into either the N-terminal or the internal TadA* monomer of ABE2.9. The variant with an inactivated N-terminal TadA* subunit (ABE2.11) demonstrated comparable editing efficiencies to ABE2.9, whereas the variant with an inactivated internal TadA* subunit (ABE2.12) lost all editing activity (Extended Data Fig. E4a). These results establish that the internal TadA subunit is responsible for ABE deamination activity.
ABEs That Efficiently Edit a Subset of Targets
Next we performed a third round of bacterial evolution starting with TadA*2.1–dCas9 to further increase editing efficiencies. We increased selection stringency by introducing two early stop codons (Q4stop and W15stop) in the kanamycin resistance gene (KanR, aminoglycoside phosphotransferase, Supplementary Table 8 and Supplementary Sequences 2). Each of the mutations requires an A•T to G•C reversion to correct the premature stop codon. We subjected a library of TadA*2.1–dCas9 variants containing mutations in the TadA domain to this higher stringency selection (Supplementary Table 8), resulting in the strong enrichment of three new TadA mutations: L84F, H123Y, and I157F. These mutations were imported into ABE2.9 to generate ABE3.1 (Fig. 2b). In HEK293T cells, ABE3.1 resulted in editing efficiencies averaging 29±2.6% across the six tested sites, a 1.6-fold average increase in A•T to G•C conversion at each site over ABE 2.9, and a 11-fold average improvement over ABE1.2 (Fig. 3b). Using longer (64- or 100-amino acid) linkers between the two TadA monomers, or between TadA* and Cas9 nickase, did not consistently improve editing efficiencies compared to ABE3.1 (Extended Data Fig. E1 and E4b).
Although ABE3.1 mediated efficient base editing at some targets, such as the CAC in site 1 (65±4.2% conversion), for other sites, such as the GAG in site 5, editing efficiencies were much lower (8.3±0.67%) (Fig. 3b). The results from six genomic loci with different sequence contexts surrounding the target A suggested that ABEs from rounds 1–3 strongly preferred target sequence contexts of YAC, where Y = T or C. This preference was likely inherited from the substrate specificity of native E. coli TadA, which deaminates the A in the UAC anticodon of tRNAArg. The utility of an ABE would be greatly limited, however, by such a target sequence restriction.
To overcome this sequence preference, we initiated a fourth evolution campaign focusing mutagenesis at TadA residues predicted to interact with the nucleotides upstream and downstream of the target A30. We subjected TadA*2.1–dCas9 libraries (Supplementary Table 7) containing randomized amino acids at five such positions (E25, R26, R107, A142, and A143) to a new selection in which A•T to G•C conversion of a non-YAC target (GAT, which causes a T89I mutation in the spectinomycin resistance protein) restores antibiotic resistance (Supplementary Table 8 and Supplementary Sequences 2). Surviving bacteria strongly converged on the TadA mutation A142N. Although apparent A•T to G•C base editing efficiency in bacterial cells with TadA*4.3–dCas9 (TadA*3.1+A142N–dCas9) was higher than with TadA*3.1–dCas9 as judged by spectinomycin resistance (Extended Data Fig. E4c), in mammalian cells ABE4.3 exhibited decreased base editing efficiencies (averaging 16±5.8%) compared with ABE3.1 (Fig. 2b and 3b). We hypothesized that the A142N mutation may benefit base editing in a context-dependent manner, and revisited its inclusion in later rounds of evolution (see below).
We performed a fifth round of evolution to increase ABE catalytic performance and broaden target sequence compatibility. We generated a library of TadA*3.1–dCas9 variants containing unbiased mutations throughout the TadA* domain as before (Supplementary Table 7). To favor ABE constructs with faster kinetics, we subjected this library to the CamR H193Y selection with higher doses of chloramphenicol after allowing ABE expression for only half the duration (7 h) of the previous rounds of evolution (~14 h) (Supplementary Table 8). Surprisingly, importing a consensus set of mutations from surviving clones (H36L, R51L, S146C, and K157N) into ABE3.1, creating ABE5.1, decreased overall editing efficiencies in HEK293T cells by 1.7±0.29-fold (Fig. 2b and 3b).
ABE5.1 included seven mutations since our dimerization state experiments on ABE2.1. We speculated that the accumulation of these mutations may impair the ability of the non-catalytic N-terminal TadA subunit to play its structural role in mammalian cells. In E. coli, endogenous wild-type TadA is provided in trans, potentially explaining the difference between bacterial selection phenotypes and mammalian cell editing efficiencies. Therefore, we examined the effect of using wild-type TadA instead of evolved TadA* variants in the N-terminal TadA domain of ABE5 variants. A heterodimeric construct containing wild-type E. coli TadA fused to an internal evolved TadA* (ABE5.3) exhibited greatly improved editing efficiencies compared to homodimeric ABE5.1 with two identical evolved TadA* domains. ABE5.3 editing efficiencies across the six genomic test sites averaged 39±5.9%, with an average improvement at each site of 2.9±0.78-fold relative to ABE5.1 (Fig. 2b and 3b). Importantly, ABE5.3 also showed broadened sequence compatibility that now enabled 22–33% editing of non-YAC targets in sites 3–6 (Fig. 3b).
Concurrently, we subjected a round 5 library to the non-YAC spectinomycin selection used in round 4. Although no highly enriched or beneficial mutations emerged (Extended Data Fig. E5a), mutations from two genotypes emerging from this selection, N72D + G125A; and P48S + S97C, were included in subsequent library generation steps. In addition, eight heterodimeric wild-type TadA–TadA* ABE5.3 variants (ABE5.5 to ABE5.12) containing 24-, 32-, or 40-residue linkers between the TadA domains or between TadA and Cas9 nickase showed no obvious improvements in base editing efficiency over ABE5.3 (Extended Data Fig. E1 and E5b). Subsequent studies therefore used the ABE5.3 architecture containing heterodimeric wtTadA–TadA*–Cas9 nickase with two 32-residue linkers.
Highly Active ABEs With Broad Sequence Compatibility
A sixth round of evolution aimed to remove any non-beneficial mutations by DNA shuffling and to reexamine mutations from previous rounds of evolution that may benefit ABE performance once liberated from negative epistasis with other mutations. Evolved TadA*–dCas9 variants from rounds 1 through 5 along with wild-type E. coli TadA were shuffled and subjected to the spectinomycin resistance T89I selection (Supplementary Table 8). Two mutations were strongly enriched from this selection: P48S/T and A142N (first seen in round 4). These mutations were added either separately or together to ABE5.3, forming ABE6.1 to ABE6.6 (Extended Data Fig. E1). ABE6.3 (ABE5.3+P48S) resulted in 1.3±0.28-fold higher average editing relative to ABE5.3 at the six genomic sites tested, and an average conversion efficiency of 47±5.8% (Fig. 2b and 3c). P48 is predicted to lie ~5 Å from the substrate adenosine 2’-hydroxyl in the TadA crystal structure (Fig. 2c), and we speculate that mutating this residue to Ser may improve compatibility with a deoxyadenosine substrate. While at most sites ABE6 variants that contained the A142N mutation were less active than ABEs that lack this mutation, editing by ABE6.4 (ABE6.3 + A142N) at site 6, which contains a target A at position 7 in the protospacer, was 1.5±0.13-fold more efficient than editing by ABE6.3, and 1.8±0.16-fold more efficient than editing by ABE5.3 (Fig. 3c). These results suggest that ABEs containing A142N may offer improved editing of adenines closer to the PAM than position 5.
Although six rounds of evolution and engineering yielded substantial improvements, ABE6 editors still suffered from reduced editing efficiencies (~20–40%) at target sequences containing multiple adenines near the targeted A (Fig. 3c). To address this challenge, we performed a seventh round of evolution in which new unbiased libraries of TadA*6–dCas9 variants were targeted to two separate sites in the kanamycin resistance gene: the Q4stop mutation used in round 3 that requires editing TAT, and a new D208N mutation that requires editing TAA (Supplementary Table 7 and 8, Supplementary Sequences 2). Surviving clones contained three enriched sets of mutations: W23L/R, P48A, and R152H/P.
Introducing these mutations separately or in combinations into mammalian cell ABEs (ABE7.1 to ABE7.10), substantially improved editing efficiencies, especially at targets that contain multiple A residues (Fig. 2b, 3c, and 3d, and Extended Data Fig. E1, E6a, and E6b). ABE7.10 edited the six genomic test sites with an average efficiency 58±4.0%, an average improvement at each site of 1.3±0.20-fold relative to ABE6.3 (Fig 3c), and 29±7.4-fold compared to ABE1.2. Although mutational dissection revealed that all three of the new mutations contribute to enhanced editing efficiencies (Extended Data Fig. E1, E6a, and E6b), the R152P substitution is particularly noteworthy, as this residue is predicted to contact the C in the UAC anticodon loop of the tRNA substrate (Fig. 2b and 2c). We speculate that substitution of Arg for Pro abrogates base-specific enzyme:DNA interactions, broadening target sequence compatibility.
Characterization of Late-Stage ABEs
We characterized in-depth the most promising ABEs from rounds 5–7. We chose an expanded set of 17 human genomic targets that place a target A at position 5 or 7 of the protospacer and collectively include all possible NAN sequence contexts (Extended Data Fig. E2a). Overall, we observed strong improvement of A•T to G•C editing efficiencies in HEK293T cells during the progression from ABE5 to ABE7 variants (Fig. 3c and 3d). The base editing efficiency of the most active editor overall, ABE7.10, averaged 53±3.7% at the 17 sites tested, exceeded 50% at 11 of these sites, and ranged from 34–68% (Fig. 3c and 3d). These efficiencies compare favorably to the typical C•G to T•A editing efficiencies of BE33.
Next we further characterized the base editing activity window of late-stage ABEs. We chose a human genomic site containing an alternating 5’-A-N-A-N-A-N-3’ sequence that could be targeted with either of two sgRNAs such that an A would be located either at every odd position (site 18) or at every even position (site 19) from 2 to 9 in the protospacer (Extended Data Fig. E2a). The resulting editing outcomes (Extended Data Fig. E7a), together with an analysis of editing efficiencies at every protospacer position across all 19 sites tested (Extended Data Fig. E7b) suggest that the activity windows of late-stage variants are approximately 4–6 nucleotides wide, from protospacer positions ~4–7 for ABE7.10, and from positions ~4–9 for ABE6.3, ABE7.8, and ABE7.9, counting the PAM as positions 21–23 (Fig. 5). We note that the precise editing window boundaries can vary in a target-dependent manner (Supplementary Table 1), as is the case with BE3 and BE4. We also tested ABE7.8-7.10 in U2OS cells at sites 1–6 and observed similar editing results as in HEK293T cells (Extended Data Fig. E6c), demonstrating that ABE activity is not limited to HEK293T cells.
Figure 5. Comparison of ABE7.10-mediated base editing and Cas9-mediated HDR, and application of ABE7.10 to two disease-relevant SNPs.
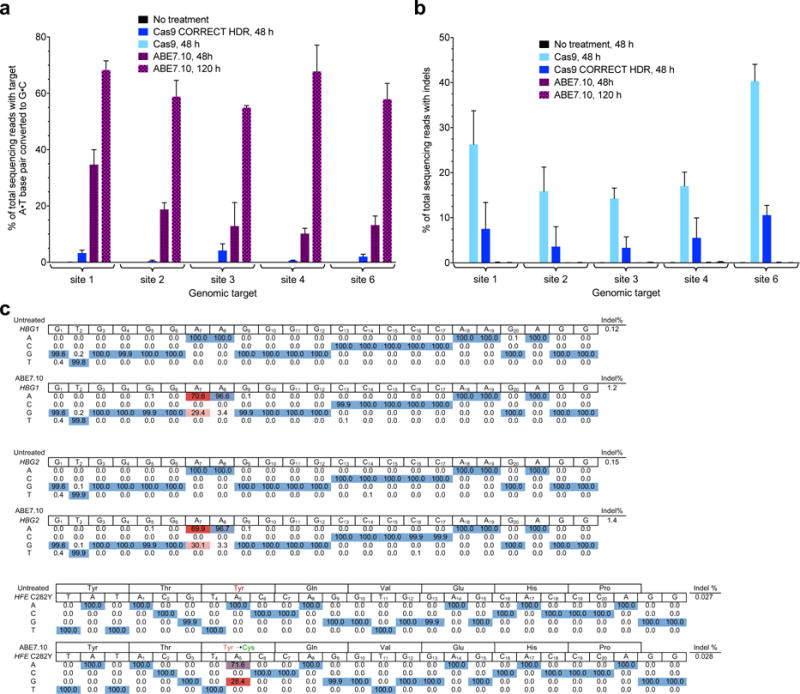
a, A•T to G•C base editing efficiencies in HEK293T cells treated either with ABE7.10, or with Cas9 nuclease and an ssDNA donor template (following the CORRECT HDR method33) targeted to five human genomic DNA sites. b, Indel formation in HEK293T cells treated as described in (a). c, Application of ABE to install a disease-suppressing SNP, or to correct a disease-inducing SNP. Top: ABE7.10-mediated -198T→C mutation (on the strand complementary to the one shown) in the promoter region of HBG1 and HBG2 genes in HEK293T cells. The target A is at protospacer positon 7. Bottom: ABE7.10-mediated reversion of the C282Y mutation in the HFE gene in LCL cells. The target A is at protospacer position 5.
Analysis of individual high-throughput DNA sequencing reads from ABE editing at 6 to 17 genomic sites in HEK293T cells reveals that base editing outcomes at nearby adenines within the editing window are not statistically independent events. The average normalized linkage disequilibrium (LD) between nearby target adenines steadily increased as ABE evolution proceeded (Extended Data Fig. E8), indicating that early-stage ABEs edit nearby adenines more independently, while late-stage ABEs edit nearby adenines more processively. These findings suggest that during the course of evolution, TadA may have evolved kinetic changes that decrease the likelihood of substrate release before additional As within the editing window are converted, resulting in processivity similar to that of BE33.
In contrast to the formation of C to non-T edits and indels that can arise from BE3-mediated base editing of cytidines, ABEs convert A•T to G•C very cleanly in HEK293T and U2OS cells, with indel frequencies and A to non-G editing similar to those of untreated cells (typically ≤ 0.1%) among the 17 genomic NAN sites tested (Fig. 4 and Supplementary Table 1). We recently showed that undesired products of BE3 arise from uracil excision and downstream repair processes5. The remarkable product purity of all tested ABE variants suggests that the activity or abundance of enzymes that remove inosine from DNA may be low compared to the those of UNG, resulting in minimal base excision repair following ABE editing.
Figure 4. Product purity of late-stage ABEs.

Product distributions and indel frequencies at two representative human genomic DNA sites in HEK293T cells treated with ABE7.10 or ABE7.9 and the corresponding sgRNA, or in untreated HEK293T cells. 22,746-111,215 sequencing reads were used at every position.
We compared ABE7.10-catalyzed A•T to G•C editing efficiencies to those of a current Cas9 nuclease-mediated HDR method, CORRECT33. At five genomic loci in HEK293T cells we observed average target point mutation frequencies ranging from 0.47% to 4.2% with 3.3% to 10.6% indels using the CORRECT HDR method under optimized 48-h conditions in HEK293T cells (Fig. 5a). At these same five genomic loci, ABE7.10 resulted in average target mutation frequencies of 10–35% after 48 h, and 55–68% after 120 h (Fig. 5a), with < 0.1% indels (Fig. 5b). The target mutation:indel ratio averaged 0.43 for CORRECT HDR, and > 500 for ABE7.10, representing a > 1,000-fold improvement in product selectivity favoring ABE7.10. While we note that HDR is well-suited to introduce insertions and deletions into genomic DNA, these results demonstrate that ABE7.10 can introduce A•T to G•C point mutations with much higher efficiency and far fewer undesired products than a current Cas9 nuclease-mediated HDR method.
Next we examined off-target editing by ABE7 variants. Since no method yet exists to comprehensively profile off-target activity of ABEs, we assumed that off-target ABE editing primarily occurs at the off-target sites that are edited when Cas9 nuclease is complexed with the same guide RNA, as we and others observed to be the case with BE33,8,34. We treated HEK293T cells with three well-characterized guide RNAs35 and either Cas9 nuclease or ABE7 variants and sequenced the on-target loci and the 12 most active off-target human genomic loci associated with these guide RNAs as identified by the genome-wide GUIDE-Seq method35. The efficiency of on-target indels by Cas9 and the efficiency of on-target base editing by ABE7.10 both averaged 54% (Supplementary Table 2–4). We observed detectable modification (≥ 0.2% indels) by Cas9 nuclease at nine of the 12 (75%) known off-target loci (Fig. 5c and Supplementary Table 2–4). In contrast, when complexed with the same sgRNAs, ABE7.10, ABE7.9, or ABE 7.8 led to ≥ 0.2% off-target base editing at only four of the 12 (33%) known Cas9 off-target sites. Moreover, the nine confirmed Cas9 off-target loci were modified with an average efficiency of 14% indels, while the four confirmed ABE off-target loci were modified with an average of only 1.3% A•T to G•C mutation (Supplementary Table 2–4). Although seven of the nine confirmed Cas9 off-target loci contained at least one A within the ABE activity window, three of these seven off-target loci were not detectably edited by ABE7.8, 7.9, or 7.10. Together, these data strongly suggest that ABE7 variants may be less prone to off-target genome modification than Cas9 nuclease, although a comprehensive, unbiased method of profiling the DNA specificity of ABEs is needed. In addition, we did not detect any apparent ABE-induced A•T to G•C DNA editing outside of on-target or off-target protospacers following ABE treatment.
Although additional studies are needed to examine possible RNA editing by ABEs, we observed no elevated adenine mutation rate in abundant mRNAs or in RNAs containing regions of homology to the native tRNA substrate of E. coli TadA in ABE7.10-treated HEK293T cells compared to untreated cells (Extended Data Fig. E9), nor any apparent ABE toxicity in bacterial or human cells under the conditions used in this study. We speculate that the evolved mutations at TadA residues known to interact with the ribose 2’-hydroxyl (Fig. 2c), the fused Cas9 nickase, or ABE nuclear localization may impede RNA editing.
Installation of Disease-Relevant Mutations With ABE
Finally, we tested the potential of ABEs to introduce disease-suppressing mutations and to correct pathogenic mutations in human cells. Mutations in β-globin genes cause a variety of blood diseases. Humans with the rare benign condition HPFH (hereditary persistence of fetal hemoglobin) are resistant to some β-globin diseases including sickle-cell anemia. In certain patients, this phenotype is mediated by mutations in the promoters of the γ-globin genes HBG1 and HBG2 that enable sustained expression of fetal hemoglobin, which is normally silenced in humans around birth36. We designed an sgRNA that programs ABE to simultaneously mutate -198T to C in the promoter driving HBG1 expression, and -198T to C in the promoter driving HBG2 expression, by placing the target A•T base pair at protospacer position 7. These mutations are known to confer British-type HPFH and enable fetal hemoglobin production in adults37. ABE7.10 installed the desired T•A to C•G mutations in the HBG1 and HBG2 promoters with 29% and 30% efficiency, respectively, in HEK293T cells (Fig. 5c, Extended Data Fig. E10).
The iron storage disorder hereditary hemochromatosis (HHC) is an autosomal recessive genetic disorder commonly caused by a G to A mutation at nucleotide 845 in the human HFE gene, resulting in a C282Y substitution in the HFE protein that leads to excessive iron absorption and potentially life-threatening elevation of serum ferritin38. We transfected DNA encoding ABE7.10 and a guide RNA that places the target A at protospacer position 5 into an immortalized lymphoblastoid cell line (LCL) harboring the HFE C282Y genomic mutation. Due to the extreme resistance of LCL cells to transfection, we isolated transfected cells and measured editing efficiency by HTS of their genomic DNA. We observed the clean conversion of the Tyr282 codon to Cys282 in 28% of sequencing reads from transfected cells, with no evidence of undesired editing or indels at the on-target locus (Fig. 5c). Although much additional research is needed to develop these and other ABE editing strategies into potential future clinical therapies for diseases with a genetic component, including the development of ABEs that accept a wide variety of PAMs7, these examples demonstrate the potential of ABEs to correct disease-driving mutations, and to install mutations known to suppress genetic disease phenotypes, in human cells.
In summary, seven rounds of evolution and engineering transformed a protein with no ability to deaminate adenine at target loci in DNA (wild-type TadA–dCas9) into forms that edit DNA weakly (ABE1s and ABE2s), variants that edit limited subsets of sites efficiently (ABE3s, ABE4s, and ABE5s), and, ultimately, highly active ABEs with broad sequence compatibility (ABE6s and ABE7s). We recommend ABE7.10 for general A•T to G•C base editing. When the target A is at protospacer positions 8–10, ABE7.9, ABE7.8, or ABE6.3 may offer higher editing efficiencies than ABE7.10, although conversion efficiencies at these positions are typically lower than at protospacer positions 4–7. The development of ABEs greatly expands the capabilities of base editing and the fraction of pathogenic SNPs that can be addressed by genome editing without introducing DSBs (Fig. 1a). Together with BE33 and BE45, these ABEs advance the field of genome editing by enabling the direct installation of all four transition mutations at target loci in living cells with a minimum of undesired byproducts.
Methods
General methods
DNA amplification was conducted by PCR using Phusion U Green Multiplex PCR Master Mix (ThermoFisher Scientific) or Q5 Hot Start High-Fidelity 2× Master Mix (New England BioLabs) unless otherwise noted. All mammalian cell and bacterial plasmids generated in this work were assembled using the USER cloning method as previously described39 and starting material gene templates were synthetically accessed as either bacterial or mammalian codon-optimized gBlock Gene Fragments (Integrated DNA Technologies). All sgRNA expression plasmids were constructed by a 1-piece blunt-end ligation of a PCR product containing a variable 20-nt sequence corresponding to the desired sgRNA targeted site. Primers and templates used in the synthesis of all sgRNA plasmids used in this work are listed in Supplementary Table 5. All mammalian ABE constructs, sgRNA plasmids and bacterial constructs were transformed and stored as glycerol stocks at −80°C in Mach1 T1R Competent Cells (Thermo Fisher Scientific), which are recA−. Molecular Biology grade, Hyclone water (GE Healthcare Life Sciences) was used in all assays and PCR reactions. All vectors used in evolution experiments and mammalian cell assays were purified using ZympPURE Plasmid Midiprep (Zymo Research Corportion), which includes endotoxin removal. Antibiotics used for either plasmid maintenance or selection during evolution were purchased from Gold Biotechnology.
Generation of bacterial TadA* libraries (evolution rounds 1–3, 5, and 7)
Briefly, libraries of bacterial ABE constructs were generated by two-piece USER assembly of a PCR product containing a mutagenized E. coli TadA gene and a PCR product containing the remaining portion of the editor plasmid (including the XTEN linker, dCas9, sgRNA, selectable marker, origin of replication, and promoter). Specifically, mutations were introduced into the starting template (Supplementary Table 7) in 8 × 25 μL PCR reactions containing 75 ng-1.2 μg of template using Mutazyme II (Agilent Technologies) following the manufacturer’s protocol and primers NMG-823 and 824 (Supplementary Table 6). After amplification, the resulting PCR products were pooled and purified from polymerase and reaction buffer using a MinElute PCR Purification Kit (Qiagen). The PCR product was treated with Dpn1 (NEB) at 37°C for 2 h to digest any residual template plasmid. The desired PCR product was subsequently purified by gel electrophoresis using a 1% agarose gel containing 0.5 μg/mL ethidium bromide. The PCR product was extracted from the gel using the QIAquick Gel Extraction Kit (Qiagen) and eluted with 30 μL of H2O. Following gel purification, the mutagenized ecTadA DNA fragment was amplified with primers NMG-825 and NMG-826 (Supplementary Table 6) using Phusion U Green Multiplex PCR Master Mix (8 × 50 μL PCR reactions, 66 °C annealing, 20-s extension) in order to install the appropriate USER junction sequences onto the 5’ and 3’ end of the fragment. The resulting PCR product was purified by gel electrophoresis. Next, the backbone of the bacterial base editor plasmid template (Supplementary Table 7), was amplified with primers NMG-799 and NMG-824 (Supplementary Table 6) and Phusion U Green Multiplex PCR Master Mix (100 μL per well in a 98-well PCR plate, 5–6 plates total, Tm 66 °C, 4.5-min extension) following the manufacturer’s protocol. Each PCR reaction was combined with 300 mL of PB DNA binding buffer (Qiagen) and 25 mL of the solution was loaded onto a HiBind DNA Midi column (Omega Bio-Tek). Bound DNA was washed with 5 column volumes of PE wash buffer (Qiagen) and the DNA fragment was eluted with 800 μL of H2O per column. Both DNA fragments were quantified using a NanoDrop 1000 Spectrophotometer (Themo Fisher Scientific).
TadA* libraries were assembled following a previously reported USER assembly procedure39 with the following conditions: 0.22 pmol of ecTadA mutagenized DNA fragment 1, 0.22 pmol of plasmid backbone fragment 2, 1 U of USER (Uracil-Specific Excision Reagent, New England Biolabs) enzyme, and 1 U of DpnI enzyme (New England Biolabs) per 10 μL of USER assembly mixture were combined in 50 mM potassium acetate, 20 mM Tris-acetate, 10 mM magnesium acetate, 100 μg/mL BSA at pH7.9 (1× CutSmart Buffer, New England Biolabs). Generally, each round of evolution required ~1 mL of USER assembly mixture (22 nmol of each DNA assembly fragment) which was distributed into 10-μL aliquots across multiple 8-well PCR strips. The reactions were warmed to 37 °C for 60 min, then heated to 80 °C for 3 min to denature the two enzymes. The assembly mixture was slowly cooled to 12 °C at 0.1 °C/s in a thermocycler to promote annealing of the freshly generated ends of the two USER junctions.
With a library of constructs in hand, we removed denatured enzymes and reaction buffer from the assembly mixture by adding 5 vol of PB buffer (Qiagen) to the assembly reaction mixture and binding the material onto a MinElute column (480 μL per column). ABE hybridized library constructs were eluted in 30 μL of H2O per column and 2 μL of this eluted material was added to 20 μL of NEB 10-beta electrocompotent E. coli and electroporated with a Lonza 4D-Nucleofector System using bacterial program 5 in a 16-well Nucleocuvette strip. A typical round of evolution used ~300 electroporations to generate 5–10 million colony forming units (cfu). Freshly electroporated E. coli were recovered in 200 mL pre-warmed Davis Rich Media (DRM) at 37 °C, and incubated with shaking at 200 rpm in a 500-mL vented baffled flask for 15 min before carbenicillin (for plasmid maintenance) was added to 30 μg/mL. The culture was incubated at 37 °C with shaking at 200 rpm for 18 h. The plasmid library was isolated with a ZympPURE Plasmid Midiprep kit following manufacturer’s procedure (50 mL culture per DNA column), except the plasmid library was eluted in 200 μL pre-warmed water per column. Evolution rounds 1–3, 5 and 7 followed this procedure in order to generate the corresponding library with minor variations (Supplementary Table 7).
Generation of site-saturated bacterial TadA* library (evolution round 4)
Mutagenesis at Arg24, Glu25, Arg107, Ala142, and Ala143 of ecTadA was achieved using ecTadA*(2.1)-dCas9 as a template and amplifying with appropriately designed degenerate NNK-containing primers (Supplementary Table 6). Briefly, ecTadA*(2.1)-dCas9 template was amplified separately with two sets of primers: NMG-1197 + NMG-1200, and NMG-1199 + NMG-1200, using Phusion U Green Multiplex PCR Master Mix, forming PCR product 1 and PCR product 2 respectively. Both PCR products were purified individually using PB binding buffer and a MiniElute column and eluted with 20 μL of H2O per 200 μL of PCR reaction. In a third PCR reaction, 1 μL of PCR product 1 and 1 μL PCR product 2 were combined with exterior, uracil-containing primers NMG-1202 and NMG-1197, and amplified by Phusion U Green Multiplex PCR Master Mix to form the desired extension-overlap PCR product with flanking uracil-containing USER junctions. In a fourth PCR reaction, ecTadA*(2.1)-dCas9 was amplified with NMG-1201 and NMG-1198 to generate the backbone DNA fragment for USER assembly. After DpnI digestion and gel purification of both USER assembly fragments, the extension-overlap PCR product (containing the desired NNK mutations in ecTadA) was incorporated into the ecTadA*(2.1)-dCas9 backbone by USER assembly as described above. The freshly generated NNK library was transformed into NEB 10-beta electrocompotent E. coli and the DNA was harvested as described above.
Generation of DNA-shuffled bacterial TadA* library (evolution round 6)
DNA shuffling was achieved by a modified version of the nucleotide exchange and excision technology (NExT) DNA shuffling method40. Solutions of 10 mM each of dATP, dCTP, dGTP and dTTP/dUTP (3 parts dUTP: 7 parts dTTP) were freshly prepared. Next, the TadA* fragment was amplified from 20 fmol of a pool of TadA*-XTEN-dCas9 bacterial constructs isolated from evolution rounds 1–5 in equimolar concentrations using Taq DNA Polymerase (NEB), primers NMG-822 and NMG-823 (Supplementary Table 6), and 400 μM each of dATP, dCTP, dGTP, and dUTP/dTTP (3:7) in 1× ThermoPol Reaction Buffer (Tm 63 °C, 1.5-min extension time). The freshly generated uracil-containing DNA library fragment was purified by gel electrophoresis and extracted with QIAquick Gel Extraction Kit (Qiagen), eluting with 20 μL of H2O per extraction column. The purified DNA product was digested with 2 U of USER enzyme per 40 μL in 1× CutSmart Buffer at 37 °C and monitored by analytical agarose gel electrophoresis until digestion was complete. The reaction was quenched with 10 vol of PN1 binding buffer (Qiagen) when the starting material was no longer observed (typically 3–4 h at 37 °C). Additional USER enzyme was added to the reaction if needed. The digested material was purified with QiaexII kit (Qiagen) using the manufacturer’s protocol and the DNA fragments were eluted in 50 μL of pre-warmed H2O per column.
The purified shuffled TadA* fragment was reassembled into full-length TadA*-XTEN-dCas9 product by an internal primer extension procedure. The eluted digested DNA fragments (25 μL) were combined with 4 U of Vent Polymerase (NEB), 800 μM each of dATP, dCTP, dGTP, and dTTP, 1 U of Taq DNA polymerase in 1× ThermoPol Buffer supplemented with 0.5 mM MgSO4. The thermocycler program for the reassembly procedure was the following: 94 °C for 3 min, 40 cycles of denaturation at 92 °C for 30 s, annealing over 60 s at increasing temperatures starting at 30 °C and adding 1 °C per cycle (cooling ramp = 1 °C/s), and extension at 72 °C for 60 s with an additional 4 s per cycle, ending with one final cycle of 72 °C for 10 min. The reassembled product was amplified by PCR with the following conditions: 15 μL of unpurified internal assembly was combined with 1 μM each of USER primers NMG-825 and NMG-826, 100 μL of Phusion U Green Multiplex PCR Master Mix and H2O to a final volume of 200 μL, 63 °C annealing, extension time of 30 s. The PCR product was purified by gel electrophoresis and assembled using thhe USER method into the corresponding ecTadA*-XTEN-dCas9 backbone with corresponding flanking USER junctions generated from amplification of the backbone with USER primers NMG-799 and NMG-824 as before. The library of evolution 6 constructs was isolated using a ZymoPURE Plasmid Midiprep kit following the manufacturer’s procedure following transformation of the hybridized library into NEB 10-beta electrocompotent E. coli.
Bacterial evolution of TadA variants
The previously described strain S103041 was used in all evolution experiments and an electrocompotent version of the bacteria was prepared as previously described39 harboring the appropriate selection plasmid specific to each round of evolution (Supplementary Table 7). Briefly, 2 μL of freshly generated TadA* library (300–600 ng/μL) prepared as described above was added to 22 μL of freshly prepared electrocompotent S1030 cells containing the target selection plasmid and electroporated with a Lonza 4D-Nucleofector System using bacterial program 5 in a 16-well Nucleocuvette strip. A typical selection used 5–10 × 106 cfu. After electroporation, freshly transformed S1030 cells were recovered in a total of 250 mL of pre-warmed DRM media at 37 °C shaking at 200 rpm for 15 min. Following this brief recovery incubation, carbenicillin was added to a final concentration of 30 μg/mL to maintain the library plasmid, along with the appropriate antibiotic to maintain the selection plasmid; see Supplementary Table 7 for the list of selection conditions including the antibiotics used for each round. Immediately following the addition of the plasmid maintenance antibiotics, 100 mM of L-Arabinose was added to the culture to induce translation of TadA*–dCas9 fusion library members, which were expressed from the PBAD promoter. The culture was grown to saturation at 37 °C with shaking at 200 rpm for 18 h, except the incubation time for evolution round 5 was only 7 h).
Library members were challenged by plating 10 mL of the saturated culture onto each of four 500-cm2 square culture dishes containing 1.8% agar-2xYT, 30 μg/mL of plasmid maintenance antibiotics, and a concentration of the selection antibiotic pre-determined to be above the MIC of the S1030 strain harboring the antibiotic alone (Supplementary Table 8). Plates were incubated at 37 °C for 2 days and ~500 surviving colonies were isolated. The TadA* genes from these colonies were amplified by PCR with primers NMG-822 and NMG-823 (Supplementary Table 6) and submitted for DNA sequencing. Concurrently, the colonies were inoculated separately into 1-mL DRM cultures in a 96-deep well plate and grown overnight at 37 °C, 200 r.p.m. Aliquots (100 μL) of each overnight culture were pooled, the plasmid DNA was isolated, and the TadA* genes were amplified with USER primers NMG-825 and NMG-826 (Supplementary Table 6). The TadA* genes were subcloned back into the plasmid backbone (containing the XTEN linker–dCas9, and appropriate guide RNAs) with the USER assembly protocol described above. This enriched library was transformed into the appropriate S1030 (+selection plasmid) electrocompotent cells, incubated with maintenance antibiotic and L-Ara and re-challenged with the selection condition. After 2-day incubation, 300–400 surviving clones were isolated as described above and their TadA* genes were sequenced. Mutations arising from each selection round were imported into mammalian ABE constructs and tested in mammalian cells as described below.
General mammalian cell culture conditions
HEK293T (ATCC CRL-3216) and U2OS (ATTC HTB-96) were purchased from ATCC and cultured and passaged in Dulbecco’s Modified Eagle’s Medium (DMEM) plus GlutaMax (ThermoFisher Scientific) supplemented with 10% (v/v) fetal bovine serum (FBS). Hap1 (Horizon Discovery, C631) and Hap1 AAG-cells (Horizon Discovery, HZGHC001537c002) were maintained in Iscove’s Modified Dulbecco’s Medium (IMDM) plus GlutaMax (ThermoFisher Scientific) supplemented with 10% (v/v) FBS. Lymphoblastoid cell lines (LCL) containing a C282Y mutation in the HFE gene (Coriell Biorepository, GM14620) were maintained in Roswell Park Memorial Institute Medium 1640 (RPMI-1640) plus GlutaMax (ThermoFisher Scientific) supplemented with 20% FBS. All cell types were incubated, maintained, and cultured at 37 °C with 5% CO2. Cell lines were authenticated by the suppliers and tested negative for mycoplasma.
HEK293T tissue culture transfection protocol and genomic DNA preparation
HEK293T cells grown in the absence of antibiotic were seeded on 48-well poly-D-lysine coated plates (Corning). 12–14 h post-seeding, cells were transfected at approximately 70% confluency with 1.5 μL of Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s protocols and 750 ng of ABE plasmid, 250ng of sgRNA expression, and 10 ng of a GFP expression plasmid (Lonza). Unless otherwise stated, cells were cultured for 5 days, with a media change on day 3. Media was removed, cells were washed with 1× PBS solution (Thermo Fisher Scientific), and genomic DNA was extracted by addition of 100 μL freshly prepared lysis buffer (10 mM Tris-HCl, pH 7.0, 0.05% SDS, 25 μg/mL Proteinase K (ThermoFisher Scientific)) directly into each well of the tissue culture plate. The genomic DNA mixture was transferred to a 96-well PCR plate and incubated at 37 °C for 1 h, followed by an 80 °C enzyme denaturation step for 30 min. Primers used for mammalian cell genomic DNA amplification are listed in Supplementary Table 9.
Nucleofection of HAP1 and HAP1 AAG− cells and genomic DNA extraction
HAP1 and HAP1 AAG− cells were nucleofected using the SE Cell Line 4D-Nucleofector X Kit S according to the manufacturer’s protocol. Briefly, 4 × 105 cells were nucleofected with 300 ng of ABE plasmid and 100 ng of sgRNA expression plasmid using the 4D-Nucleofector program DZ-113 and cultured in 250 μL of media in a 48-well poly-D-lysine coated culture plate for 3 days. DNA was extracted as described above.
Nucleofection of U2OS cells and genomic DNA extraction
U2OS cells were nucleofected using the SG Cell Line 4D-Nucleofector X Kit (Lonza) according to the manufacture’s protocol. Briefly, 1.25 × 105 cells were nucleofected in 20 μL of SG buffer along with 500 ng of ABE plasmid and 100 ng of sgRNA expression plasmid using the 4D-Nucleofector program EH-100 in a 16-well Nucleocuvette strip (20 μL of cells per well). Freshly nucleofected cells were transferred into 250 μL of media in a 48-well poly-D-lysine coated culture plate. Cells were incubated for 5 days and media was changed every day. DNA was extracted as described above.
Electroporation of LCL HFE C828Y cells
LCL cells were electroporated using a Gene Pulser Xcell Electroporater (BioRad) and 0.4 cm gap Gene Pulser electroporation cuvettes (BioRad). Briefly, 1 × 107 LCL cells were resuspended in 250 μL RPMI-160 plus GlutaMax. To this media was added 65 μg of plasmid expressing ABE7.10, GFP, and the corresponding sgRNA targeting the C282Y mutation in the HFE gene. The mixture was added to a pre-chilled 0.4 cm gap electroporation cuvette and the cell/DNA mixture was incubated in the cuvette on ice for 10 min. Cells were pulsed at 250 V and 950 μF for 3 ms. Cells were transferred back on ice for 10 min, then transferred to 15 mL of pre-warmed RPMI-160 supplemented with 20% FBS in a T-75 flask. The next day, an additional 5 mL of media was added to the flask and cells were left to incubate for a total of 5 days. After incubation, cells were isolated by centrifugation, resuspended in 400 μL of media, filtered through a 40 μm strainer (Thermo Fisher Scientific), and sorted for GFP fluorescence using an FACSAria III Flow Cytometer (Becton Dickenson Biosciences). GFP-positive cells were collected in a 1.5-mL tube containing 500 μL of media. After centrifugation, the media was removed and cells were washed twice with 600 μL of 1× PBS (Thermo Fisher Scientific). Genomic DNA was extracted as described above.
Comparison between ABE 7.10 and homology directed repair using the ‘CORRECT’ method42
HEK293T cells grown in the absence of antibiotic were seeded on 48-well poly-D-lysine coated plates (Corning). After 12–14 h, cells were transfected at ~70% confluency with 750 ng of Cas9 or base editor plasmid, 250 ng of sgRNA expression plasmid, 1.5 μL of Lipofectamine 3000 (Thermo Fisher Scientific), and for HDR assays 0.7 μg of single-stranded donor DNA template (100 nt, PAGE-purified from IDT) according to the manufacturer’s instructions. 100-mer single-stranded oligonucleotide donor templates are listed in Supplementary Table 10.
Genomic DNA was harvested 48 h post-transfection (as described by Tessier-Lavigne et. al. during the development of the CORRECT method42) using the Agencourt DNAdvance Genomic DNA isolation Kit (Beckman Coulter) according to the manufacturer’s instructions. A size-selective DNA isolation step ensured that there was no risk of contamination by the single-stranded donor DNA template in subsequent PCR amplification and sequencing steps. We re-designed amplification primers to ensure there was minimal risk of amplifying donor oligo template.
High-throughput DNA sequencing (HTS) of genomic DNA samples
Genomic sites of interest were amplified by PCR with primers containing homology to the region of interest and the appropriate Illumina forward and reverse adapters (Supplementary Table 9). Primer pairs used in this first round of PCR (PCR 1) for all genomic sites discussed in this work can be found in Supplementary Table 9. Specifically, 25 μL of a given PCR 1 reaction was assembled containing 0.5 μM of each forward and reverse primer, 1 μL of genomic DNA extract and 12.5 μL of Phusion U Green Multiplex PCR Master Mix. PCR reactions were carried out as follows: 95 °C for 2 min, then 30 cycles of [95 °C for 15 s, 62 °C for 20 s, and 72 °C for 20 s], followed by a final 72 °C extension for 2 min. PCR products were verified by comparison with DNA standards (Quick-Load 100 bp DNA ladder) on a 2% agarose gel supplemented with ethidium bromide. Unique Illumina barcoding primer pairs were added to each sample in a secondary PCR reaction (PCR 2). Specifically, 25 μL of a given PCR 2 reaction was assembled containing 0.5 μM of each unique forward and reverse illumina barcoding primer pair, 2 μL of unpurified PCR 1 reaction mixture, and 12.5 μL of Q5 Hot Start High-Fidelity 2× Master Mix. The barcoding PCR 2 reactions were carried out as follows: 95 °C for 2 min, then 15 cycles of [95 °C for 15 s, 61 °C for 20 s, and 72 °C for 20 s], followed by a final 72 °C extension for 2 min. PCR products were purified by electrophoresis with a 2% agarose gel using a QIAquick Gel Extraction Kit, eluting with 30 μL of H2O. DNA concentration was quantified with the KAPA Library Quantification Kit-Illumina (KAPA Biosystems) and sequenced on an Illumina MiSeq instrument according to the manufacturer’s protocols.
General HTS data analysis
Sequencing reads were demultiplexed in MiSeq Reporter (Illumina). Alignment of amplicon sequences to a reference sequence was performed as previously described using a Matlab script with improved output format (Supplementary Note 1). In brief, the Smith-Waterman algorithm was used to align sequences without indels to a reference sequence; bases with a quality score less than 30 were converted to ‘N’ to prevent base miscalling as a result of sequencing error. Indels were quantified separately using a modified version of a previously described Matlab script in which sequencing reads with more than half the base calls below a quality score of Q30 were filtered out (Supplementary Note 2). Indels were counted as reads which contained insertions or deletions of greater than or equal to 1 bp within a 30-bp window surrounding the predicted Cas9 cleavage site.
Due to homology in the HBG1 and HBG2 loci, primers were designed that would amplify both loci within a single PCR reaction. In order to computationally separate sequences of these two genomic sites, sequencing experiments involving this amplicon were processed using a separate Python script (Supplementary Note 3). Briefly, reads were disregarded if more than half of the base calls were below Q30, and base calls with a quality score below Q30 were converted to ‘N’. HBG1 or HBG2 reads were identified as having an exact match to a 37-bp sequence containing two SNPs that differ between the sites. A base calling and indel window were defined by exact matches to 10-bp flanking sequences on both sides of a 43-bp window centered on the protospacer sequence. Indels were counted as reads in which this base calling window was ≥ 1 bp different in length. This Python script yields output with identical quality (estimated base calling error rate of < 1 in 1,000), but in far less time due to the absence of an alignment step.
To calculate the total number of edited reads as a proportion of the total number of successfully sequenced reads, the fraction of edited reads as measured by the alignment algorithm were multiplied by [1 – fraction of reads containing an indel].
RNA isolation from HEK293T cells and analysis
HEK293T cells were plated and a subset were transfected with ABE 7.10 as described above and incubated for five days before being removed from the plate using TrypLE Express (Thermo Fisher Scientific) and pelleted. RNA was extracted using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. cDNA was generated from the isolated RNA using the ProtoScript II First Strand cDNA Synthesis Kit (New England Biolabs) according to the manufacturer’s instructions with a mixture of random primers and Oligo-dT primers. Amplification of the cDNA for high-throughput sequencing was performed to the top of the linear range using qPCR as described above. The number of cycles for each amplicon was determined empirically for each primer pair: for B-actin, B-catenin, GAPDH, Reticulocalbin: 29 cycles; for MN1 and RSLD1: 25 cycles. High-throughput sequencing of the amplicons was performed as described above. Sequences were aligned to the reference sequence for each RNA, obtained from the NCBI.
Linkage disequlilbrium analysis
A custom Python script (Supplementary Note 4) was used to assess editing probabilities at the primary target A (P1) at the secondary target A (P2), and at both the primary and secondary target As (P1,2). Linkage disequilibrium (LD) was then evaluated as P1,2 – (P1 × P2). LD values were normalized with a normalization factor of Min(P1(1 – P2), (1 – P1)P2). This normalization which controls for allele frequency and yields a normalized LD value from 0 to 1.
Data availability
High-throughput sequencing data have been deposited in the NCBI Sequence Read Archive database under accession code SRP119577. Plasmids encoding ABE6.3, ABE7.8, ABE7.9, and ABE7.10 are available from Addgene.
Extended Data
Extended Data Figure E1. Genotypes of 57 ABEs described in this work.

Mutations are colored based on the round of evolution in which they were identified.
Extended Data Figure E2. Base editing efficiencies of additional early-stage ABE variants.
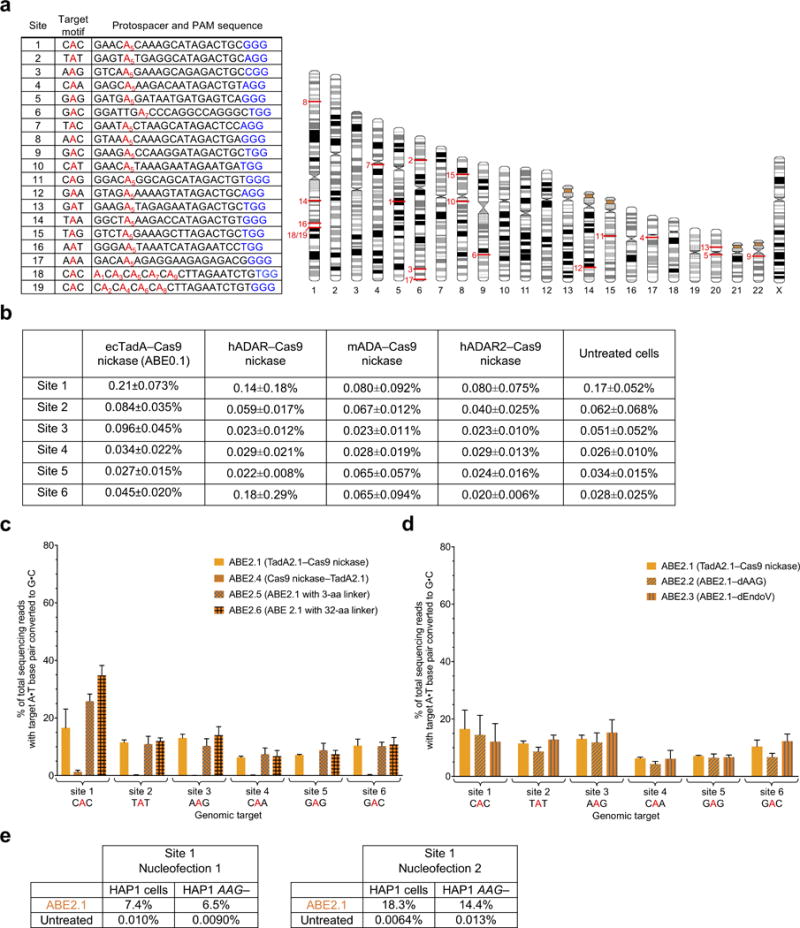
a, Table of 19 human genomic DNA test sites (left) with corresponding locations on human chromosomes (right). The sequence context (target motif) of the edited A in red is shown for each site. PAM sequences are shown in blue. b, A•T to G•C base editing efficiencies in HEK293T cells of various wild-type RNA adenine deaminases fused to Cas9 nickase at six human genomic target DNA sites. Values reflect the mean and standard deviation of three biological replicates performed on different days. c, A•T to G•C base editing efficiencies in HEK293T cells of ABE2 editors with altered fusion orientations and linker lengths at six human genomic target DNA sites. d, A•T to G•C base editing efficiencies in HEK293T cells at six human genomic target DNA sites of ABE2 editors fused to catalytically inactivated alkyl-adenosine glycosylase (AAG) or endonuclease V (EndoV), two proteins that bind inosine in DNA. e, A•T to G•C base editing efficiencies of ABE2.1 in HAP1 cells at site 1 with or without AAG. Values and error bars in (b) and (c) reflect the mean and s.d. of three independent biological replicates performed on different days.
Extended Data Figure E3. High-throughput DNA sequencing analysis of HEK293T cells treated with ABE2.1 and sgRNAs targeting each of six human genomic sites.
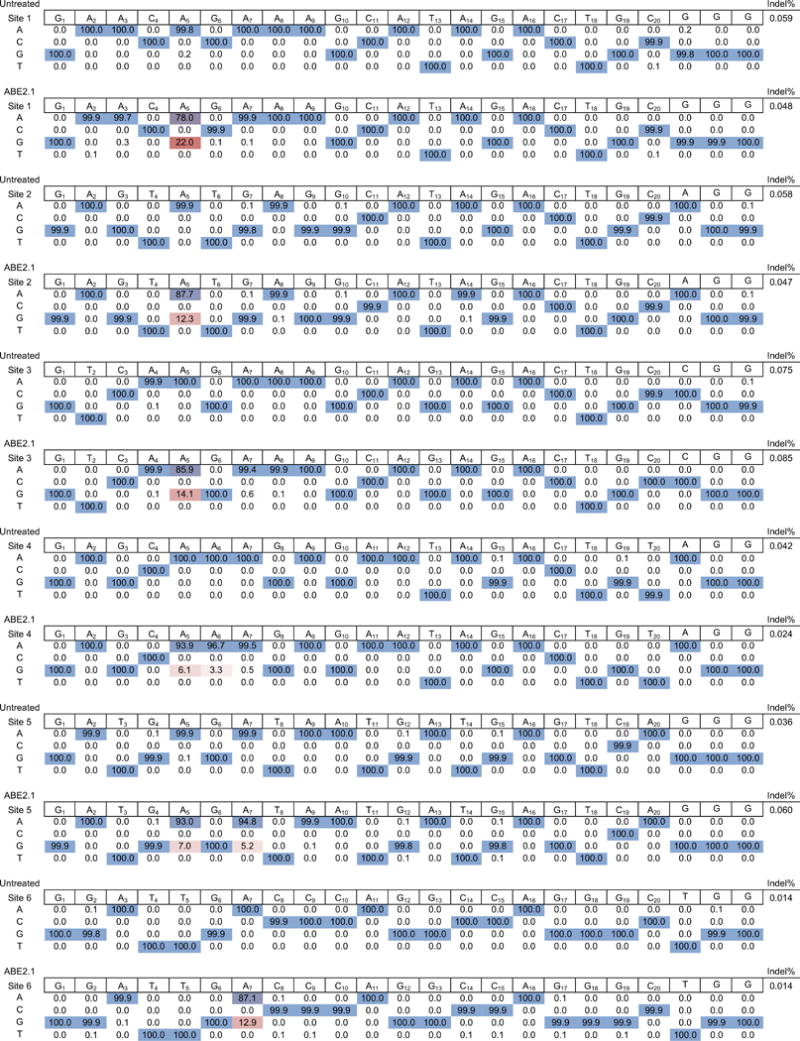
One representative replicate is shown. Data from untreated HEK293T cells are shown for comparison.
Extended Data Figure E4. Base editing efficiencies of additional ABE2 and ABE3 variants, and the effect of adding A142N to TadA*–dCas9 on antibiotic selection survival in E. coli.
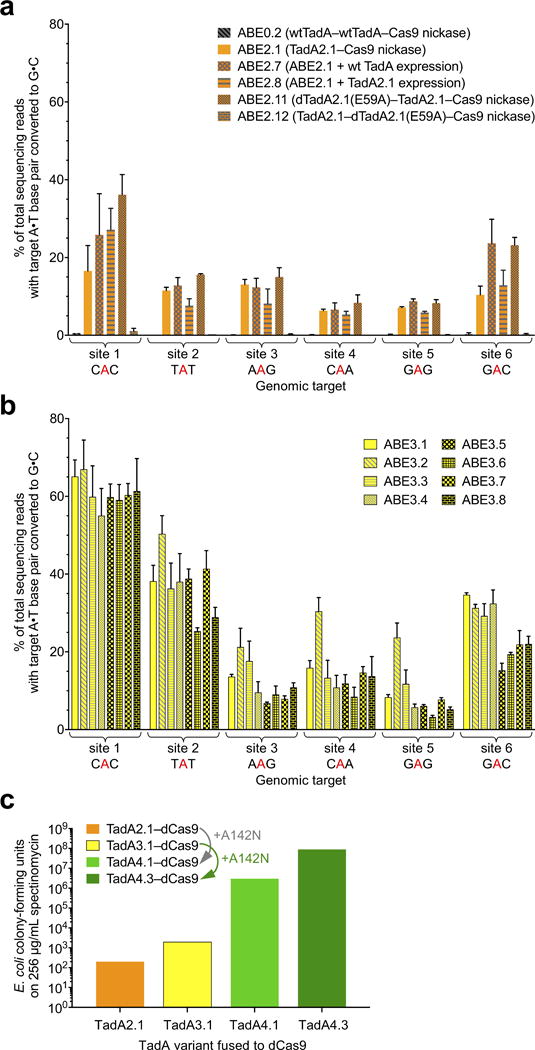
a, A•T to G•C base editing efficiencies in HEK293T cells at six human genomic target DNA sites of ABE2 variants with different engineered dimeric states. A control ABE variant containing two wild-type TadA domains and no evolved TadA* domains (ABE0.2) did not result in A•T to G•C editing at the six genomic sites tested, confirming that dimerization alone is insufficient to mediate ABE activity. b, A•T to G•C base editing efficiencies in HEK293T cells at six human genomic target DNA sites of ABE3.1 variants differing in their dimeric state (homodimer of TadA*–TadA*–Cas9 nickase, or heterodimer of wild-type TadA–TadA*–Cas9 nickase), in the length of the TadA–TadA linker, and in the length of the TadA–Cas9 nickase linker. See Extended Data Figure E1 for ABE genotypes and architectures. c, Colony-forming units on 2xYT agar with 256 μg/mL of spectinomycin of E. coli cells expressing an sgRNA targeting the I89T defect in the spectinomycin resistance gene and a TadA*-dCas9 editor lacking or containing the A142N mutation identified in evolution round 4. Successful A•T to G•C base editing at the target site restores spectinomycin resistance. Values and error bars in (a) and (b) reflect the mean and s.d. of three independent biological replicates performed on different days.
Extended Data Figure E5. Base editing efficiencies of additional ABE5 variants.
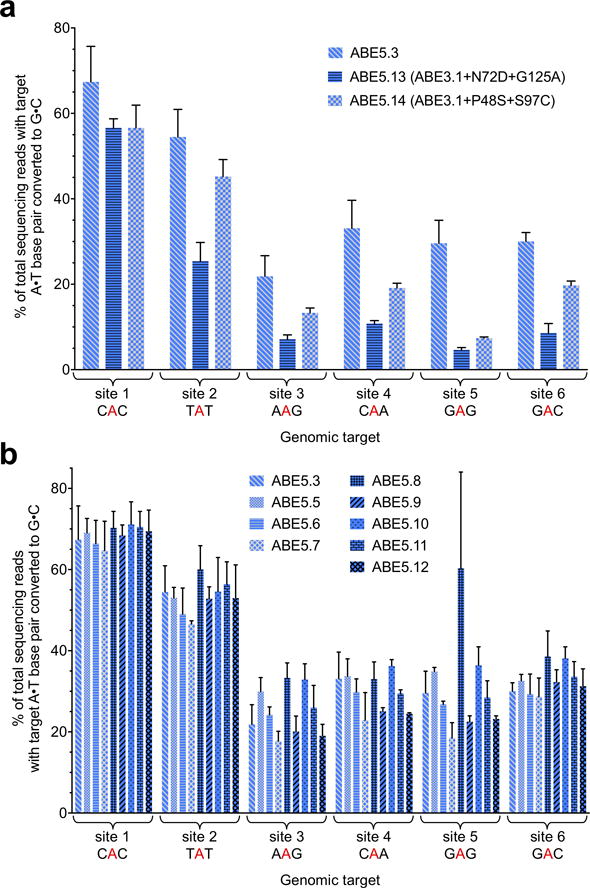
a, A•T to G•C base editing efficiencies in HEK293T cells at six human genomic target DNA sites of two ABE3.1 variants with two pairs of mutations isolated from spectinomycin selection of the round 5 library. b, A•T to G•C base editing efficiencies in HEK293T cells at six human genomic target DNA sites of ABE5 variants with different linker lengths. See Extended Data Figure E1 for ABE genotypes and architectures. Values and error bars reflect the mean and s.d. of three independent biological replicates performed on different days.
Extended Data Figure E6. Base editing efficiencies of ABE7 variants at 17 genomic sites.
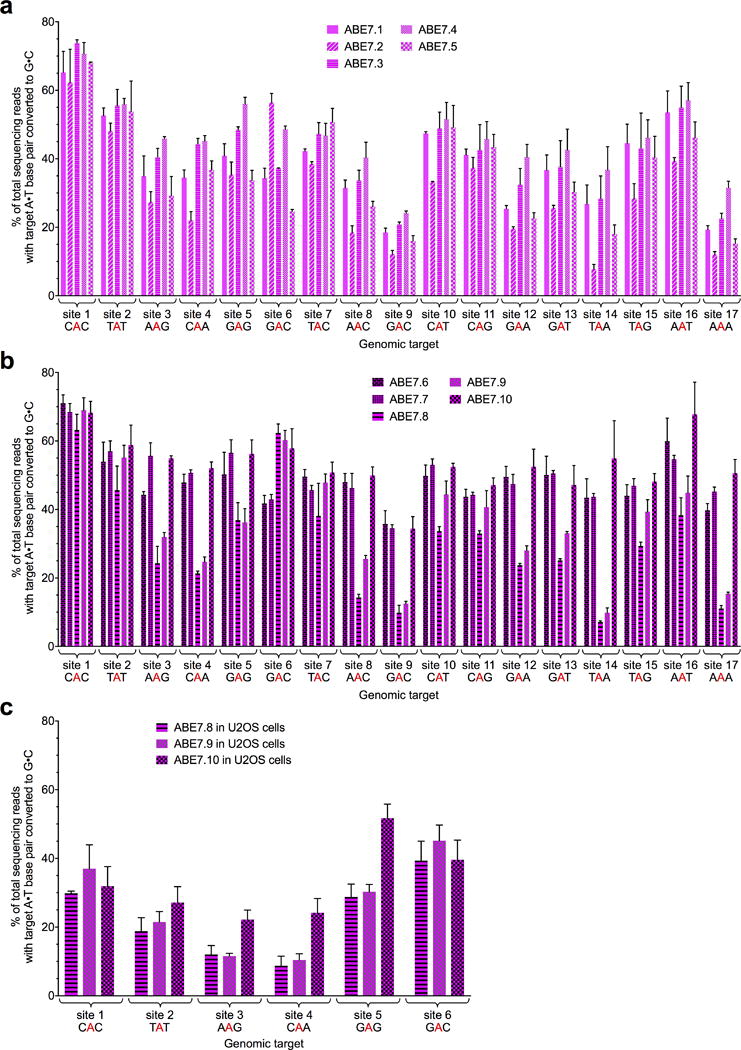
A•T to G•C base editing efficiencies in HEK293T cells at 17 human genomic target DNA sites of ABE7.1-7.5 (a), and ABE7.6-7.10 (b). See Extended Data Figure E1 for ABE genotypes and architectures. c, A•T to G•C base editing efficiencies in U2OS cells at six human genomic target DNA sites of ABE7.8-7.10. The lower editing efficiencies observed in U2OS cells compared with HEK293T cells are consistent with transfection efficiency differences between the two cell lines; we observed transfection efficiencies of ~40–55% in U2OS cells under the conditions used in this study, compared to ~65–80% in HEK293T cells. Values and error bars reflect the mean and s.d. of three independent biological replicates performed on different days.
Extended Data Figure E7. Activity window of late-stage ABEs.
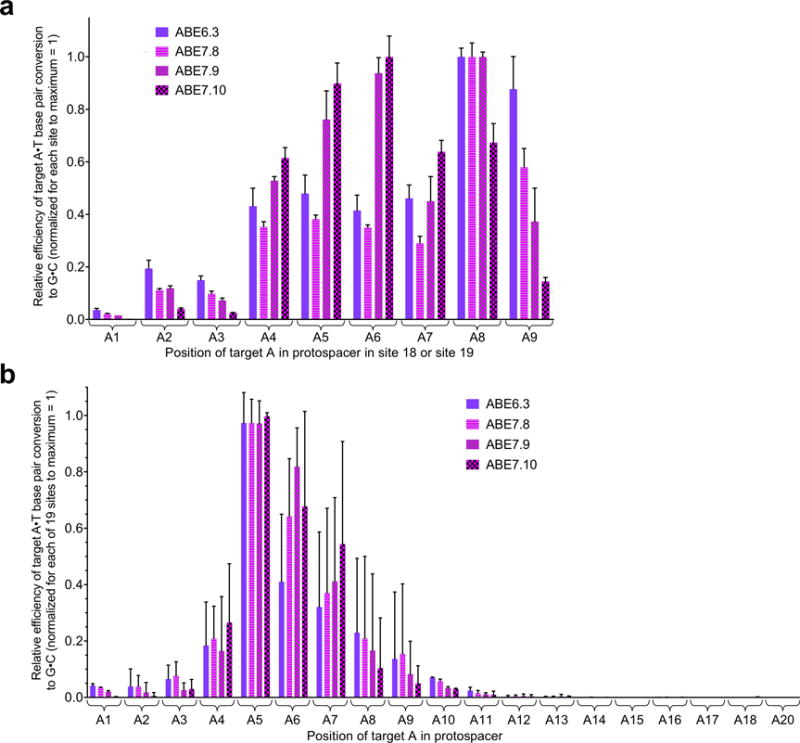
a, Relative A•T to G•C base editing efficiencies in HEK293T cells of late-stage ABEs at protospacer positions 1–9 in two human genomic DNA sites that together place an A at each of these positions. Values are normalized to the maximum observed efficiency at each of the two sites for each ABE = 1. b, Relative A•T to G•C base editing efficiencies in HEK293T cells of late-stage ABEs at protospacer positions 1–18 and 20 across all 19 human genomic DNA sites tested. Values are normalized to the maximum observed efficiency at each of the 19 sites for each ABE = 1. For (a) and (b), values and error bars reflect the mean and s.d. of three independent biological replicates performed on different days.
Extended Data Figure E8. Rounds of evolution and engineering increased ABE processivity.
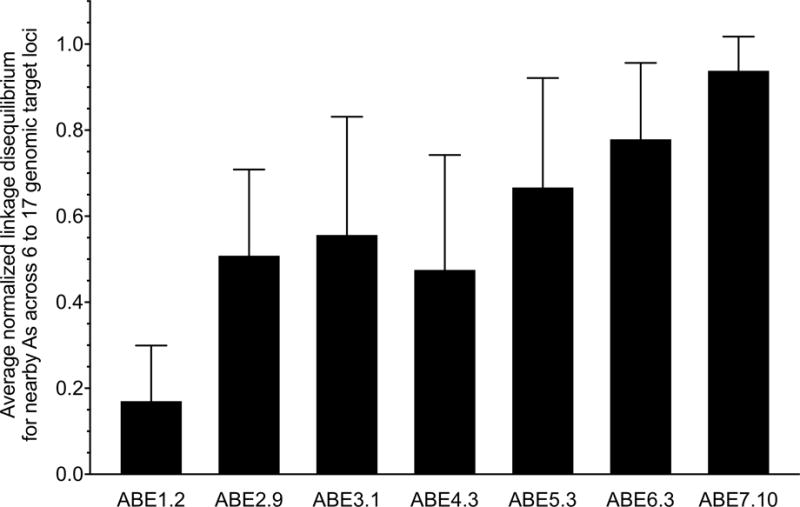
The calculated mean normalized linkage disequilibrium (LD) between nearby target As at 6 to 17 human genomic target DNA sites for the most active ABEs emerging from each round of evolution and engineering. Higher LD values indicate that an ABE is more likely to edit an A if a nearby A in the same DNA strand (the same sequencing read) is also edited. LD values are normalized from 0 to 1 in order to be independent of editing efficiency. Values and error bars reflect the mean and s.d. of normalized LD values from three independent biological replicates performed on different days.
Extended Data Figure E9. Analysis of cellular RNAs in ABE7.10-treated cells compared with untreated cells.
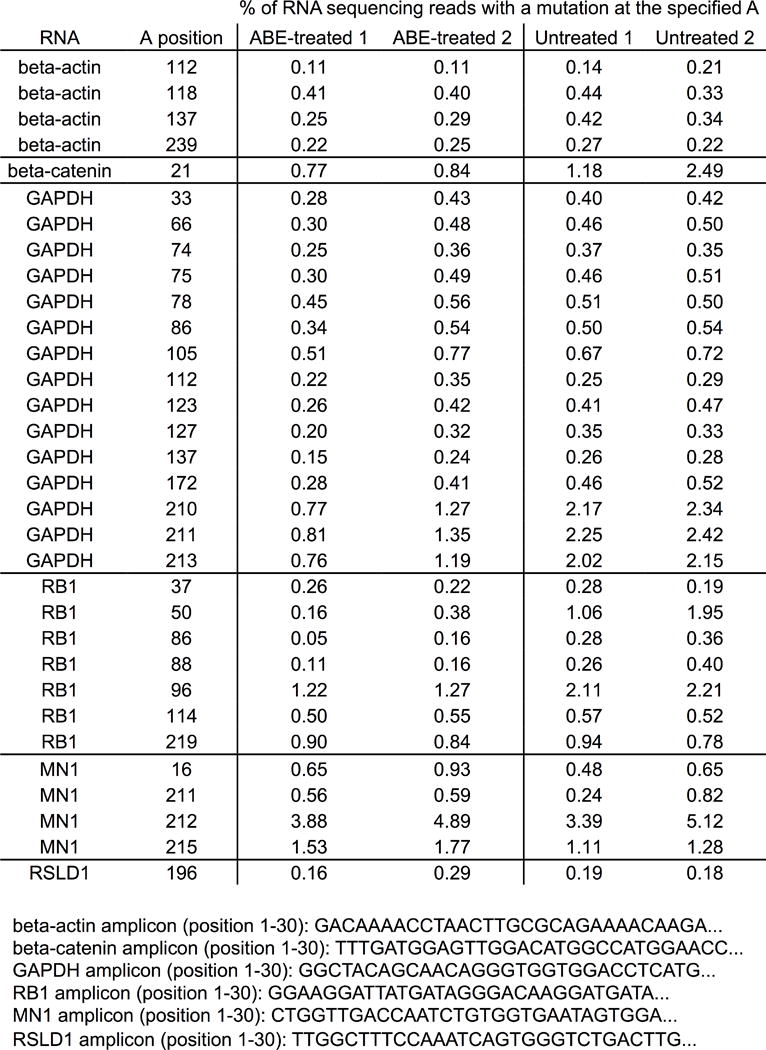
RNA from HEK293T cells treated with ABE7.10 and the sgRNA targeting site 1, or from untreated HEK293T cells, was isolated and reverse transcribed into cDNA. The cDNAs corresponding to four abundant cellular RNAs (encoding beta-actin, beta-catenin, GAPDH, and RB1), and two cellular RNAs with sequence homology to the tRNA anticodon loop that is the native substrate of E. coli TadA (encoding MN1 and RSLD1), were amplified and analyzed by HTS. Within each amplicon, the mutation frequency at each adenine position for which the mutation rate was ≥ 0.2% is shown for two ABE7.10-treated biological replicates (ABE-treated 1 and ABE-treated 2) and for two untreated biological replicates (untreated 1 and untreated 2). The start of each amplicon is shown.
Extended Data Figure E10. High-throughput DNA sequencing analysis of HEK293T cells treated with five late-stage ABE variants and an sgRNA targeting -198T in the promoter of HBG1 and HBG2.

One representative replicate is shown of DNA sequences at the HBG1 (a) and HBG2 (b) promoter targets. ABE-mediated base editing installs a -198T→C mutation on the strand complementary to the one shown in the sequencing data tables. Data from untreated HEK293T cells are shown for comparison.
Supplementary Material
Acknowledgments
This work was supported by DARPA HR0011-17-2-0049, U.S. NIH RM1 HG009490, R01 EB022376, and R35 GM118062, and HHMI. A.C.K. and D.I.B. were Ruth L. Kirchstein National Research Service Awards Postdoctoral Fellows (F32 GM 112366 and F32 GM106621, respectively). M.S.P. was an NSF Graduate Research Fellow and was supported by training grant T32 GM008313. We thank Zack Niziolek for technical assistance. N.M.G. thanks A. E. Martin for his encouragement.
Footnotes
Online Content
Methods, along with any additional Extended Data display items, are available in the online version of the paper; references unique to these sections appear only in the online paper.
Supplementary Information is available in the online version of the paper.
Author contributions
N.M.G designed the research, performed all evolution experiments, conducted human cell experiments, analyzed data, and wrote the manuscript. A.C.K assisted with experimental design and human cell experiments and analyzed data. H.A.R. performed HDR and off-target experiments. M.S.P. performed computational data analyses and developed HTS processing scripts. A.H.B contributed to selection design and evolution strategy. D.I.B. assisted with cloning of late-stage ABEs. D.R.L designed and supervised the research and wrote the manuscript. All of the authors contributed to editing the manuscript.
The authors declare competing financial interests: N.M.G., A.C.K., and D.R.L. have filed patent applications on this work. D.R.L. is a consultant and co-founder of Editas Medicine, Beam Therapeutics, and Pairwise Plants, companies that use genome editing technologies. Readers are welcome to comment on the online version of the paper.
References
- 1.Krokan HE, Drabløs F, Slupphaug G. Uracil in DNA–occurrence, consequences and repair. Oncogene. 2002;21:8935–8948. doi: 10.1038/sj.onc.1205996. [DOI] [PubMed] [Google Scholar]
- 2.Lewis CA, Crayle J, Zhou S, Swanstrom R, Wolfenden R. Cytosine deamination and the precipitous decline of spontaneous mutation during Earth’s history. Proc Natl Acad Sci USA. 2016;113:8194–8199. doi: 10.1073/pnas.1607580113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nishida K, et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science. 2016;353 doi: 10.1126/science.aaf8729. [DOI] [PubMed] [Google Scholar]
- 5.Komor AC, et al. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci Adv. 2017;3:eaao4774. doi: 10.1126/sciadv.aao4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Komor AC, Badran AH, Liu DR. Editing the genome without double-stranded DNA breaks. ACS Chem Biol. 2017 doi: 10.1021/acschembio.7b00710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim YB, et al. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat Biotechnol. 2017;35:371–376. doi: 10.1038/nbt.3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rees HA, et al. Improving the DNA specificity and applicability of base editing through protein engineering and protein delivery. Nat Commun. 2017;8:15790. doi: 10.1038/ncomms15790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Satomura A, et al. Precise genome-wide base editing by the CRISPR nickase system in yeast. Sci Rep. 2017;7:2095. doi: 10.1038/s41598-017-02013-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu Y, Zhu J-K. Precise Editing of a Target Base in the Rice Genome Using a Modified CRISPR/Cas9 System. Mol Plant. 2017;10:523–525. doi: 10.1016/j.molp.2016.11.013. [DOI] [PubMed] [Google Scholar]
- 11.Zong Y, et al. Precise base editing in rice, wheat and maize with a Cas9-cytidine deaminase fusion. Nat Biotechnol. 2017;35:438–440. doi: 10.1038/nbt.3811. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, et al. Programmable base editing of zebrafish genome using a modified CRISPR-Cas9 system. Nat Commun. 2017;8:118. doi: 10.1038/s41467-017-00175-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Billon P, et al. CRISPR-mediated base editing enables efficient disruption of eukaryotic genes through induction of STOP codons. Mol Cell. 2017;67:1068–1079.e1064. doi: 10.1016/j.molcel.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuscu C, et al. CRISPR-STOP: gene silencing through base-editing-induced nonsense mutations. Nat Methods. 2017;14:710–712. doi: 10.1038/nmeth.4327. [DOI] [PubMed] [Google Scholar]
- 15.Kim K, et al. Highly efficient RNA-guided base editing in mouse embryos. Nat Biotechnol. 2017;35:435–437. doi: 10.1038/nbt.3816. [DOI] [PubMed] [Google Scholar]
- 16.Chadwick AC, Wang X, Musunuru K. In vivo base editing of PCSK9 (proprotein convertase subtilisin/kexin Type 9) as a therapeutic alternative to genome editing. Arterioscler Thromb Vasc Biol. 2017;37:1741–1747. doi: 10.1161/ATVBAHA.117.309881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liang P, et al. Correction of β-thalassemia mutant by base editor in human embryos. Protein & Cell. 2017;12:61–12. doi: 10.1007/s13238-017-0475-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li G, et al. Highly efficient and precise base editing in discarded human tripronuclear embryos. Protein & Cell. 2017;532:289–284. doi: 10.1007/s13238-017-0458-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang W, Hu JH, Liu DR. Aptazyme-embedded guide RNAs enable ligand-responsive genome editing and transcriptional activation. Nat Commun. 2017;8:15939. doi: 10.1038/ncomms15939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yasui M, et al. Miscoding properties of 2’-deoxyinosine, a nitric oxide-derived DNA Adduct, during translesion synthesis catalyzed by human DNA polymerases. J Mol Biol. 2008;377:1015–1023. doi: 10.1016/j.jmb.2008.01.033. [DOI] [PubMed] [Google Scholar]
- 21.Zheng Y, Lorenzo C, Beal PA. DNA editing in DNA/RNA hybrids by adenosine deaminases that act on RNA. Nucleic Acids Res. 2017;45:3369–3377. doi: 10.1093/nar/gkx050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim J, et al. Structural and kinetic characterization of Escherichia coli TadA, the wobble-specific tRNA deaminase. Biochemistry. 2006;45:6407–6416. doi: 10.1021/bi0522394. [DOI] [PubMed] [Google Scholar]
- 23.Wolf J, Gerber AP, Keller W. tadA, an essential tRNA‐specific adenosine deaminase from Escherichia coli. EMBO J. 2002;21:3841–3851. doi: 10.1093/emboj/cdf362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matthews MM, et al. Structures of human ADAR2 bound to dsRNA reveal base-flipping mechanism and basis for site selectivity. Nat Struct Mol Biol. 2016;23:426–433. doi: 10.1038/nsmb.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grunebaum E, Cohen A, Roifman CM. Recent advances in understanding and managing adenosine deaminase and purine nucleoside phosphorylase deficiencies. Curr Opin Allergy Clin Immunol. 2013;13:630–638. doi: 10.1097/ACI.0000000000000006. [DOI] [PubMed] [Google Scholar]
- 26.Gerber AP, Keller W. An adenosine deaminase that generates inosine at the wobble position of tRNAs. Science. 1999;286:1146–1149. doi: 10.1126/science.286.5442.1146. [DOI] [PubMed] [Google Scholar]
- 27.Fukui K. DNA mismatch repair in eukaryotes and bacteria. J Nucleic Acids. 2010;2010 doi: 10.4061/2010/260512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi K, et al. Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat Struct Mol Biol. 2017;24:131–139. doi: 10.1038/nsmb.3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Macbeth MR, et al. Inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science. 2005;309:1534–1539. doi: 10.1126/science.1113150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Losey HC, Ruthenburg AJ, Verdine GL. Crystal structure of Staphylococcus aureus tRNA adenosine deaminase TadA in complex with RNA. Nat Struct Mol Biol. 2006;13:153–159. doi: 10.1038/nsmb1047. [DOI] [PubMed] [Google Scholar]
- 31.Lau AY, Wyatt MD, Glassner BJ, Samson LD, Ellenberger T. Molecular basis for discriminating between normal and damaged bases by the human alkyladenine glycosylase, AAG. Proc Natl Acad Sci USA. 2000;97:13573–13578. doi: 10.1073/pnas.97.25.13573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sebastian Vik E, et al. Endonuclease V cleaves at inosines in RNA. Nat Commun. 2013;4:2271. doi: 10.1038/ncomms3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paquet D, et al. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature. 2016;533:125–129. doi: 10.1038/nature17664. [DOI] [PubMed] [Google Scholar]
- 34.Kim D, et al. Genome-wide target specificities of CRISPR RNA-guided programmable deaminases. Nat Biotechnol. 2017;35:475–480. doi: 10.1038/nbt.3852. [DOI] [PubMed] [Google Scholar]
- 35.Tsai SQ, et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Traxler EA, et al. A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat Med. 2016;22:987–990. doi: 10.1038/nm.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wienert B, et al. KLF1 drives the expression of fetal hemoglobin in British HPFH. Blood. 2017;130:803–807. doi: 10.1182/blood-2017-02-767400. [DOI] [PubMed] [Google Scholar]
- 38.Alexander J, Kowdley KV. HFE-associated hereditary hemochromatosis. Genet Med. 2009;11:307–313. doi: 10.1097/GIM.0b013e31819d30f2. [DOI] [PubMed] [Google Scholar]
- 39.Badran AH, et al. Continuous evolution of Bacillus thuringiensis toxins overcomes insect resistance. Nature. 2016;533:58–63. doi: 10.1038/nature17938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Müller KM, et al. Nucleotide exchange and excision technology (NExT) DNA shuffling: a robust method for DNA fragmentation and directed evolution. Nucleic Acids Res. 2005;33:e117–e117. doi: 10.1093/nar/gni116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Esvelt KM, Carlson JC, Liu DR. A system for the continuous directed evolution of biomolecules. Nature. 2011;472:499–503. doi: 10.1038/nature09929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kwart D, Paquet D, Teo S, Tessier-Lavigne M. Precise and efficient scarless genome editing in stem cells using CORRECT. Nat Protocols. 2017;12:329–354. doi: 10.1038/nprot.2016.171. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
High-throughput sequencing data have been deposited in the NCBI Sequence Read Archive database under accession code SRP119577. Plasmids encoding ABE6.3, ABE7.8, ABE7.9, and ABE7.10 are available from Addgene.