

Abstract
Background and Purpose
The effects of lytic stroke therapy in patients with sickle cell anemia (SCA) are unknown, although a recent study suggested that co-existent SCA does not increase the risk of cerebral hemorrhage. This finding calls for systemic analysis of the effects of thrombolytic stroke therapy, first in humanized sickle mice, then in patients. There is also a need for additional predictive markers of SCA-associated vasculopathy.
Methods
We used Doppler ultrasound to examine the carotid artery of Townes sickle mice tested their responses to repetitive-mild hypoxia-ischemia (rmHI) and transient hypoxia-ischemia (tHI)-induced stroke at three or six months of age, respectively. We also examined the effects of tissue-plasminogen activator (tPA) treatment in tHI-injured sickle mice.
Results
Three-month-old SS mice showed elevated resistive index (RI) in the carotid artery and higher sensitivity to rmHI-induced cerebral infarct. Six-month-old SS mice showed greater RI and increased flow velocity without obstructive vasculopathy in the carotid artery. Instead, the cerebral vascular wall in SS mice showed ectopic expression of Plasminogen Activator Inhibitor-1 (PAI-1) and P-Selectin, suggesting a pro-adhesive and pro-thrombotic propensity. Indeed, SS mice showed enhanced leukocyte and platelet adherence to the cerebral vascular wall, broader fibrin deposition, and higher mortality after tHI. Yet, post-tHI treatment with tPA reduced thrombosis and mortality in SS mice.
Conclusions
Sickle mice are sensitive to hypoxia/ischemia-induced cerebral infarct, but benefit from thrombolytic treatment. An increased resistive index in carotid arteries may be an early marker of sickle cell vasculopathy.
Keywords: hypoxia-ischemia, silent cerebral infarct, resistive index, sickle cell mice
INTRODUCTION
Stroke is a common and severe complication of SCA.1 Without preventive treatment, as many as 37% of SCA children develop silent cerebral infarct (SCI) by age 14, and 11% show large overt stroke before age 20.1–3 Overt stroke is often associated with intimal lesions in the carotid arteries, which manifest as obstructions on magnetic resonance angiography or accelerated blood flow in the transcranial Doppler (TCD) measurement.4–7 In contrast, SCI is diagnosed by magnetic resonance imaging (MRI) in patients with SCA who lack clear symptoms, but often have a history of transient ischemic episodes or cognitive impairment.2,3 SCI and overt stroke may share similar pathological underpinnings or could be precipitated by different severities of hypoxia, ischemia, anemia, or infection.2,3
There are two important areas for improvement in the clinical care of sickle cell stroke. For acute ischemic stroke therapy, the 2014 NIH guideline recommends exchange transfusions without mentioning thrombolysis, despite its benefits in general ischemic stroke. However, a recent report showed that co-existent sickle cell disease does not increase the incidence of tPA-induced cerebral hemorrhage.8 This finding calls for systemic evaluation of the effects of lytic therapy in sickle cell stroke. Secondly, because SCI is not associated with increased flow velocity on TCD, additional ultrasonic markers of high-risk children are needed for preventive treatments.9,10
Knock-in/out mice expressing the human α, γ, and sickle-β hemoglobin genes simulate the pathogenesis of SCA and reproduce many of its complications.11 Yet, it remains unclear whether sickle mice develop accelerated blood flow or intimal lesions in the carotid arteries similar to SCA patients. Past studies indicated that sickle mice develop enhanced clot formation and larger infarcts after photoactivated thrombosis, but their responses to lytic therapy are unknown.12–14
In this study, we examined blood flow and pulse waveforms in the carotid artery of sickle mice, and tested their responses to two versions of sub-threshold hypoxia-ischemia (HI) insult at 3 and 6 months of age, respectively. The advantage of the HI model is that its trigger, dual hypoxia-ischemia, is relevant to the pathophysiology of SCA stroke.3 Moreover, since tPA treatment confers protection of tHI in wildtype mice,15,16 we can use this stroke model to assess the risks and benefits of thrombolytic therapy in sickle mice. Our results suggest that sickle mice are more sensitive to HI-induced stroke, but benefit from acute tPA therapy. Finally, elevated resistive index (RI) may be an earlier ultrasonic marker of sickle cell vasculopathy than increased flow velocity.
MATERIALS AND METHODS
The authors are pleased to share detailed analytic methods and data with researchers upon request. All study materials in this study are available through commercial vendors.
Animal Surgery and treatment
Sickle mice were obtained from Jackson Laboratories (Stock #013071) and complete blood counts were measured by a Hemavet 1500 blood analyzer (CDC Technologies, Oxford, CT). Three-month-old male AA and SS mice were subjected to repetitive mild hypoxia-ischemia (rmHI) or transient hypoxia-ischemia (tHI), as previously described with minor modifications.15,16 For rmHI, mice were anesthetized under 2% isofluorane for permanent right common carotid artery occlusion (RCCAO) with a 4.0 silk suture. The animal core temperature was maintained at 37.5 ± 0.5°C. After RCCAO, mice were subjected to 15 min hypoxia (7.5% O2/92.5% N2) each day for three consecutive days. For tHI, 6-month-old male AA, AS and SS mice were anesthetized by 2% isoflurance to undergo transient RCCAO with two releasable knots of 4.0 silk sutures, and exposed to hypoxia (7.5% O2 and 92.5% N2) via a face-mask for 20 min, while the core temperature was maintained at at 37.5 ± 0.5°C. After hypoxia, the knots on the RCCA were released. In wildtype mice, 20 min tHI insult very rarely induces cerebral infarction, and so we refer this insult in this study as sub-threshold. Recombinant human tPA (10 mg/kg) (Activase, Genetech, San Francisco) was injected via the tail vein at 30 min after tHI insult (50% as a bolus and 50% infused over 30 min). Surgical procedures were approved by the Institutional Animal Care and Use Committee and conform to the NIH Guide for Care and Use of Laboratory Animals.
Ultrasound imaging
Ultrasound imaging was performed using the Vevo 2100-ultrasound imaging system with a MS-550D probe (FUJIFILM VisualSonics, Toronto, Canada).17 Animals were anesthetized using 2% isoflurane and positioned supine on the heating pad with a heart rate monitor. All images including B-mode imaging for anatomy and doppler measurment of blood flow in the common carotid artery (CCA) and bifurcation were obtained. The peak systolic velocity (PSV) and the resistive index (RI) ([PSV – end diastolic velocity]/PSV)18 of CCA were calculated using the Vevo LAB Workstation software.
Assessment of cerebral infarction
Identification of brain infarcts was performed by in-vivo 2,3,5-Triphenyltetrazolium chloride (TTC) staining 24 h after tHI or rmHI by a lab member blind to the mouse genotype. Brains were sectioned to 0.7 mm thickness slices (8 per brain) and analyzed with the ImageJ 1.4 software (NIH, Bethesda, MD). The infarct size was quantified as the ratio of the infarcted area to the area in contralateral hemisphere.19
Immunohistochemistry and immunoblotting analysis
Immunohistochemistry and immunoblotting were performed as previously described.20 Semi-quantification of immunohistochemistry was performed using the CellSens software (Olympus). The same contrast and threshold criteria were applied to all images to obtain the total intensity of positive staining signals. The value of intensity was normalized to the AA group. The following antibodies were used: rabbit anti-fibrinogen (a gift of Dr. Jay Degen), rabbit anti-GFAP (#AB5804, EMD Millipore, Temecula, CA), rabbit anti-smooth muscle actin (#ab5694, Abcam), rabbit anti-elastin (#ab217356, Abcam, Cambridge, MA), rat anti-p-selectin (#553742, BD Bioscience, San Diego, CA), mouse anti-PAI-1 (#612024, BD Bioscience), and mouse anti-β-actin (#A5441, Sigma-Aldrich, St. Louis, MO).
Intravital fluorescence imaging
Wild type (C57BL/6), AA, and SS mice were subjected to 20-min tHI insults, followed by intravital imaging of cerebral vessel at 4 h recovery, as previously reported.12 Briefly, platelets (1×108) were isolated from a wildtype mouse and stained with 90 μM carboxyfluorescein succinimidyl ester (CFSE), followed by tail-vein injection 5 min before intravital imaging. Circulating leukocytes were further labeled by 0.02 % rhodamine 6G in continuous infusion. Fluorescently labelled leukocytes and platelets were visualized on an inverted microscope (AX70, Olympus, Tokyo, Japan). Intravital images were randomly selected to capture 20 frames/min for 2 min by a CCD camera (CoolSNAP HQ2, Photometrics, Tucson, AZ) and analyzed with the MetaMorph image software. Adherent cells were defined as having a stationary interaction on the venular wall for longer than 3 seconds.
Histological assay of vascular obstruction
Vascular perfusion and blood-brain-barrier (BBB) leakage were evaluated by Evans blue (EB)-albumin as described.15 Briefly, mice received tail-vein injection of 200 μl 2% EB solution at 4 h after tHI. After 10 min for the EB dye circulation, mice were sacrificed and the brains were quickly placed in 4% paraformaldehyde. Fixed brains were sectioned at 100 μm and the fluorescence was observed using a 680 nm emission filter on microscope (BX43, Olympus, Tokyo, Japan).
Statistical analysis
Statistical analysis was performed using one-way ANOVA followed by the Newman-Keuls test or unpaired t-test for two samples (GraphPad Prism software, La Jolla, CA). The mortality analysis in rmHI was performed using the Kaplan-Meier method. The P values of survival rate comparisons in tHI were determined using the contingency table and Chi-square (χ2) test. P values less than 0.05 were considered a significant difference. All values were expressed as mean ± SEM.
RESULTS
Sickle mice are more sensitive to repetitive mild hypoxia/ischemia-induced cerebral infarct
Townes sickle mice complete the fetal-to-adult hemoglobin switch around postnatal day 21.21 By three months of age, male SS mice showed a lower blood hemoglobin level (9.0 ± 0.31 g/dl, n=6) than AA (11.3 ± 0.1 g/dl, n=5) and AS mice (12.8 ± 0.2 g/dl, n=6). Doppler ultrasound revealed a higher resistive index (RI), but normal mean velocity (MV) and peak systolic velocity (PSV) in the carotid artery in SS than AA and AS mice (n=5–6 for each genotype, Figure 1A). The greater RI in SS mice, shown as a deeper PSV-to-end diastolic velocity (EDV) drop in pulse waveforms (Figure 1B), suggests greater resistance to blood flow in the cerebral microvascular bed.18
Figure 1.
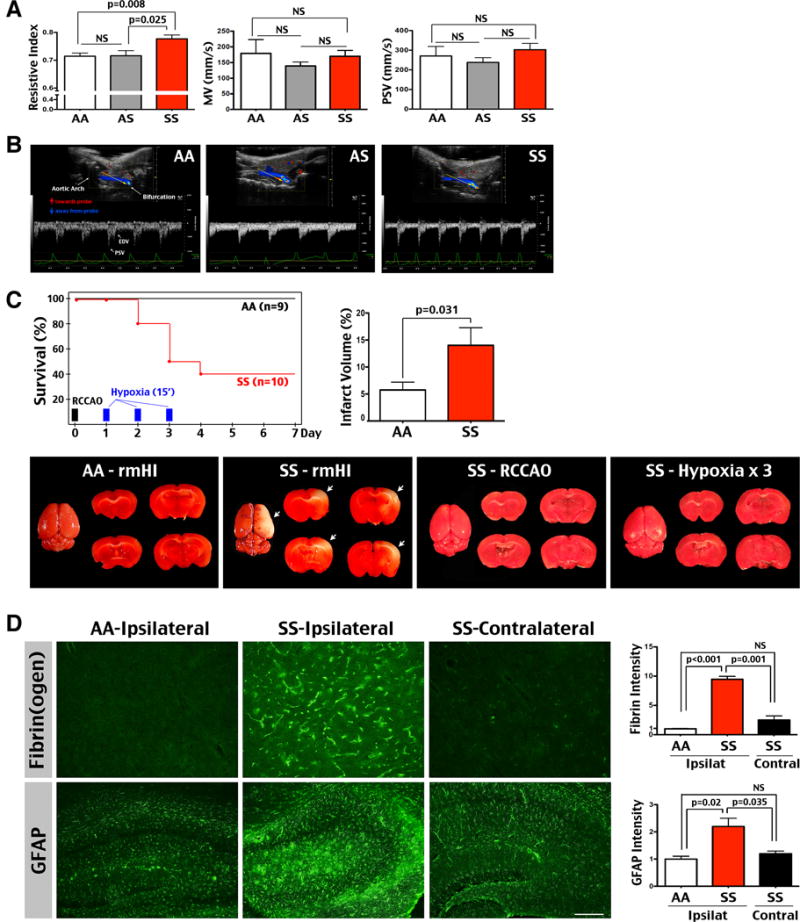
Three-month-old SS mice are sensitive to repeated mild hypoxia-ischemic (HI)-induced cerebral infarct. (A) 3-month-old SS mice had a significantly increased resistive index (RI; SS: 0.78±0.0; AA: 0.72±0.01; AS: 0.72±0.02) in the carotid artery, despite similar mean velocity (MV; SS: 170.5±18.59 mm/sec; AA: 179.6±43.99; AS: 139.2±12.62) and peak systolic velocity (PSV; SS: 302.7±32.42 mm/sec; AA: 270.6±48.79; AS: 238±23.94) compare with AA and AS mice (n=6 for AA and AS; n=5 for SS mice). Shown are mean ± SEM. p-values were determined by unpaired t-test. NS: p>0.05. (B) Typical Doppler waveforms in the carotid artery of AA, AS, and SS mice. The yellow arrows indicate the direction of ultrasonic transducer. Red arrows are the direction of blood flow towards the transducer, while blue arrows, flow away from the transducer. EDV: End-Diastolic Velocity. (C) SS mice showed an accumulated death rate of 20% at day 2, 50% at day 3, and 60% from day 4 till day 7 after unilateral right common carotid artery occlusion (RCCAO) and daily exposure to 15-min hypoxia for 3 consecutive days (n=10). In contrast, no AA mice died after repetitive mild HI (rmHI) insult (n=9). Furthermore, neither RCCAO nor daily exposure to 15-min hypoxia for three days caused mortality or cerebral infarction in SS (n=3 for each condition) or AA mice (data not shown). The SS mice that survived rmHI injury showed larger infarct than AA mice by TTC stain (AA: 5.75±1.45%; SS: 14.03±3.27%). Shown are the mean ± SEM. p-values were determined by unpaired t-test. (D) The SS mice that survived rmHI also showed greater fibrin(ogen) deposition and GFAP-immunoreactivity than AA mice (n=4 for SS and AA mice). The intensity of immunostaining was quantified and compared by t-test. Scale bar: 100 μm
To simulate the chronic circulatory insults in SCI (hypoxia and ischemia confronted with anemia)3, we subjected three-month-old male AA and SS mice to repetitive mild hypoxia-ischemia (rmHI) that consists of unilateral right common carotid artery occlusion (RCCAO) and daily 15 min exposure to 7.5% oxygen for 3 days. No AA mice died from the rmHI insult (n=9), though SS mice showed the cumulative death rate of 20% after the first episode of hypoxia, 50% after the second episode, and 60% after the third episode of hypoxia and without further death for 3 days (n=10, Figure 1C). The surviving SS mice on day 7 exhibited larger cerebral infarct than AA mice (14% versus 5.7% of contralateral hemisphere, p=0.031 by t-test, Figure 1C). In contrast, neither ischemia (RCCAO) nor hypoxia (daily exposure to 15 min hypoxia for 3 days) triggered mortality or cerebral infarct in either AA or SS mice (n=3 for each genotype-challenge, Figure 1C). These results suggest unique pathological effects of combined hypoxia-ischemia on sickle mice. Immunostaining showed greater fibrin(ogen) deposition and glial fibrillary acidic protein (GFAP)-positive astrogliosis in the ipsilateral hemisphere of surviving SS mice, but not in the contralateral hemisphere or rmHI-injured AA mouse brains (the immunostaining intensity was quantified for comparison with t-test, n=4 for each genotype, Figure 1D). We also examined the response to one episode of 20 min tHI, but found no mortality or cerebral infarct in either AA or SS mice (data not shown). These results suggest that three-month-old SS mice already develop a higher sensitivity to rmHI-induced cerebral infarct and mortality, correlated with an elevated RI in the carotid artery.
Sickle mice show pro-coagulant, but not proliferative-obstructive vasculopathy
To test whether older sickle mice develop accelerated carotid blood flow like SCA patients at risk for overt stroke,6 we compared the carotid artery flow velocity and pulse waveforms in 6-month-old male sickle and control mice. At this age, SS mice retained a similar degree of anemia to 3-month-old mice (hemoglobin: 9.47 g/dl compared to 12 g/dl in AA and AS mice), but developed a greater MV, PSV, and RI than AA mice (n=7 for AA and n=9 for SS mice, Figure 2A). AS mice showed higher MV than AA mice, but lower RI than SS mice (n=8 for AS mice, Figure 2A). The increased RI was also shown as a deeper PSV-to-EDV drop in 6-month-old SS mice (Figure 2B). Thus, six-month-old SS mice developed greater flow velocity and persistently elevated RI, despite a similar degree of anemia to 3-month-old mice.
Figure 2.
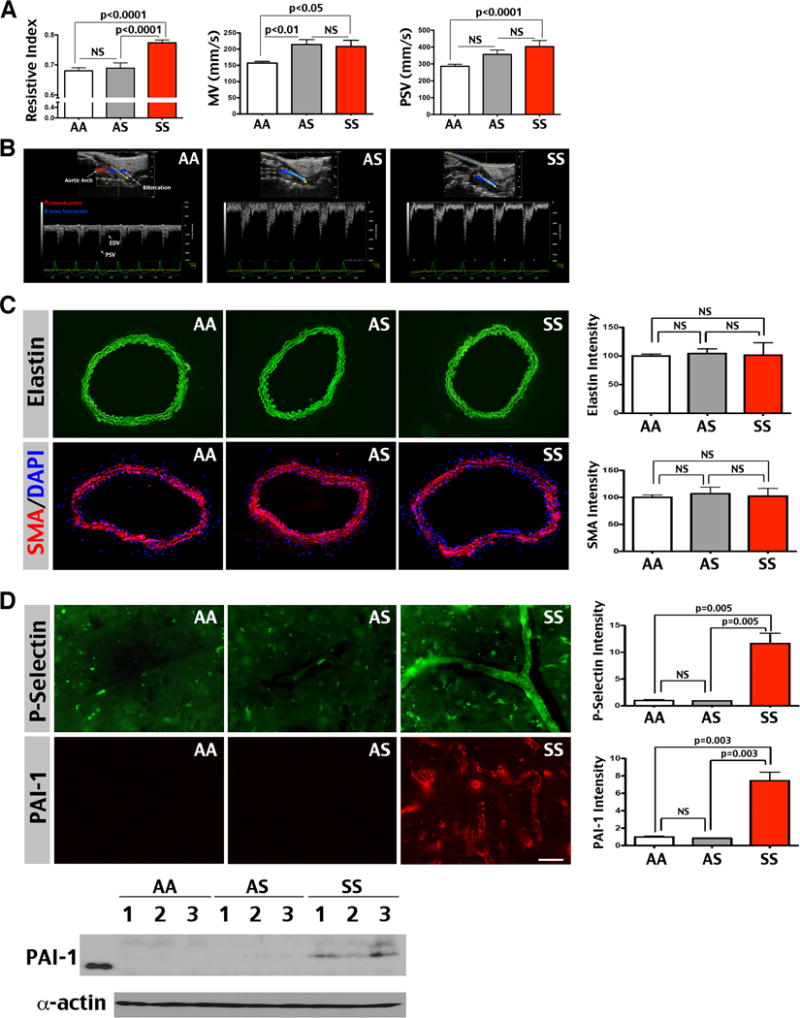
Six-month-old SS mice show increased resistive index (RI) and flow velocity (MV and PSV), but no sign for proliferative-obstructive vasculopathy in the carotid artery. (A) Six-month-old SS mice (n=9) showed a greater RI (0.77±0.0; AA: 0.68±0.01; AS: 0.69±0.02), MV (208.3±18.94 mm/sec; AA: 157±5.41; AS: 214.3±15.46), and PSV (403±35.69 mm/sec; AA: 286.1±12.36; AS: 356.4±25.99) in the carotid artery compared than AA (n=7) and AS (n=8) mice. Shown are mean ± SEM. The p-values were determined by one-way ANOVA and post hoc Newman-Keuls test. NS: p>0.05. (B) Typical Doppler pulse-waveforms in the carotid artery of AA, AS, and SS mice. (C) The carotid arteries of AA, AS, and SS mice showed comparable intensity for the internal elastic membrane (green) and anti-smooth muscle actin labeling (SMA, red), counter-stained by DAPI (blue, n=3 for each genotype). The immunostaining intensity was quantified and compared by t-test. Note that SS mice showed no evidence of duplication-interruption of the internal elastic lamina or hypertrophy of peri-vascular smooth muscle that was detected in SCA patients with overt stroke.4,5 (D) Immunofluorescence labeling showed ectopic P-Selectin (a marker for endothelial activation) and Plasminogen Activator Inhibitor-1 (PAI-1) expression in the cerebral blood vessel wall of SS mice, but not in AA or AS mice (n=3 for each genotype). The intensity of immunostaining was quantified for comparison by t-test. Likewise, immunoblotting showed consistent anti-PAI-1 signals in SS, but not AA or AS mouse brains. Shown in immunoblot are 3 mice for each genotype. Scale bar: 40 μm in C, 50 μm in D.
Significantly increased TCD flow velocity in SCA patients (>200 cm/s) is considered a sign of large-vessel obstructive vasculopathy, including duplication of the internal elastic membrane and expansion of the smooth muscle ring.4–6 Surprisingly, we found no differences in the elastic laminae and smooth muscle actin (SMA) expression in the carotid artery between 6-month-old AA, AS, and SS mice (immunostaining signals were quantified and compared by t-test, n=3 for each genotype, Figure 2C). Neither was there evidence for intimal hypertrophy in SS mice. Instead, the cerebral blood vessel wall showed ectopic expression of P-Selectin and Plasminogen Activator Inhibitor 1 (PAI-1), correlated with positive PAI-1 signals in SS mouse brains by immunoblot (immunostaining signals were quantified and compared by t-test, n=3 for each genotype, Figure 2D). These results, similar to the reports of ectopic P-Selectin and PAI-1 expression in the pulmonary endothelium13,22, suggest a pro-adhesive, pro-coagulant endothelial phenotype in sickle mice.
Sickle mice are sensitive to hypoxia/ischemia-induced leukocyte and platelet aggregation
To test the pro-coagulant propensity in SS mice, we used intravital microscopy to compare the flow of leukocytes (labeled by Rhodamine 6G) and platelets (labeled by carboxyfluorescein succinimidyl ester, CFSE) in the cerebral blood vessels in 3-month-old AA and SS mice.12 This analysis showed more leukocytes—but not platelets—adhered to the cerebral venular wall in the absence of insult in SS mice (the attached leukocytes and platelets are indicated by arrows; p-value is determined by t-test, n=3–4 for each genotype, Figure 3A and Supplementary Videos 1–6). These results suggest a baseline of enhanced leukocyte-endothelium interaction in the SS mouse brains.
Figure 3.
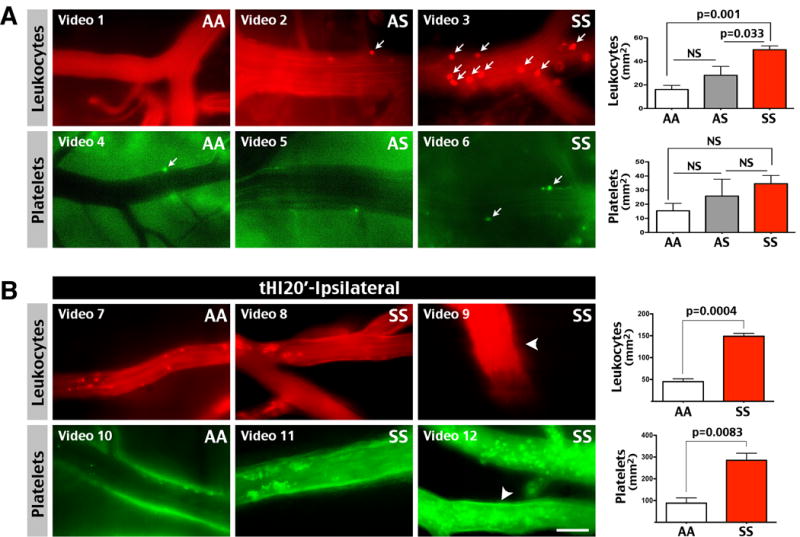
SS mice had increased sensitivity to transient hypoxia/ischemia (tHI)-induced platelet and leukocyte adherence to the cerebral vascular wall. (A) More leukocytes (labeled by IV infusion of Rhodamine 6G), and to a lesser degree platelets (ex-vivo labeled by CFSE and IV-transferred to the recipient mice), were adhered to the cerebral venular wall in 3-month-old SS mice than AA or AS littermates. The p-values were determined by t-test (n=3–4 for each genotype). Adherent cells were indicated by arrows. Also labeled are the corresponding on-line supplementary videos. (B) At 4 h after 20 min tHI insult, 6-month-old SS mice showed enhanced leukocyte and platelet adherence to the cerebral venule wall than AA mice (n=3 for each). Shown are representative intravital images and supplementary videos. Arrowheads indicate leukocyte or platelet aggregates. The numbers of adhered leukocytes and platelets was compared by unpaired t-test. Scale bar: 50 μm.
Next, we compared the flow of leukocytes and platelets at 4 h after 20 min tHI insult in 6-month-old AA and SS mice. At 4 h post-tHI, platelets and leukocytes flowed smoothly in cerebral blood vessels in AA mice (Figure 3B). In contrast, leukocytes and platelets showed a sluggish flow or aggregates in the venules of ipsilateral hemisphere in SS mice (Figure 3B and Supplementary Videos 7–12; aggregates are indicated by arrowheads). No aggregates of leukocytes or platelets were found on the contralateral hemisphere of SS mice at 4 h after 20 min tHI insult (data not shown). Quantification confirmed more leukocytes and platelets adhered to the cerebral venular wall at 4 h after 20 min tHI in the SS than AA mice (p=0.0004 and 0.0083, respectively, Figure 3B, n=3 for each genotype). These data suggest that sickle mice have a higher sensitivity to tHI-induced platelet and leukocyte aggregation.
SS mice are more sensitive to tHI-induced mortality and infarct, but respond to tPA therapy
To assess the consequences of tHI-induced platelet and leukocyte aggregation, we used fibrin(ogen) immunostaining and intravenous Evans blue (EB) dye injection to compare thrombosis and vascular perfusion, respectively, at 4 h after 20 min tHI between 6-month-old AA and SS mice. This analysis showed greater fibrin deposition and reduction of vascular perfusion in the ipsilateral cortex in SS mice, but not in contralateral cortex or tHI-challenged AA mouse brains (Figure 4A, B, shown are representative results in n=3 for each condition). Interestingly, intravenous tPA treatment at 0.5 h post-tHI reduced fibrin deposition and improved vascular perfusion in SS mice (Figure 4B; p<0.05 by one-way ANOVA, n=3 for each genotype). These results suggest that tPA treatment attenuates thrombotic brain injury in SS mice.
Figure 4.
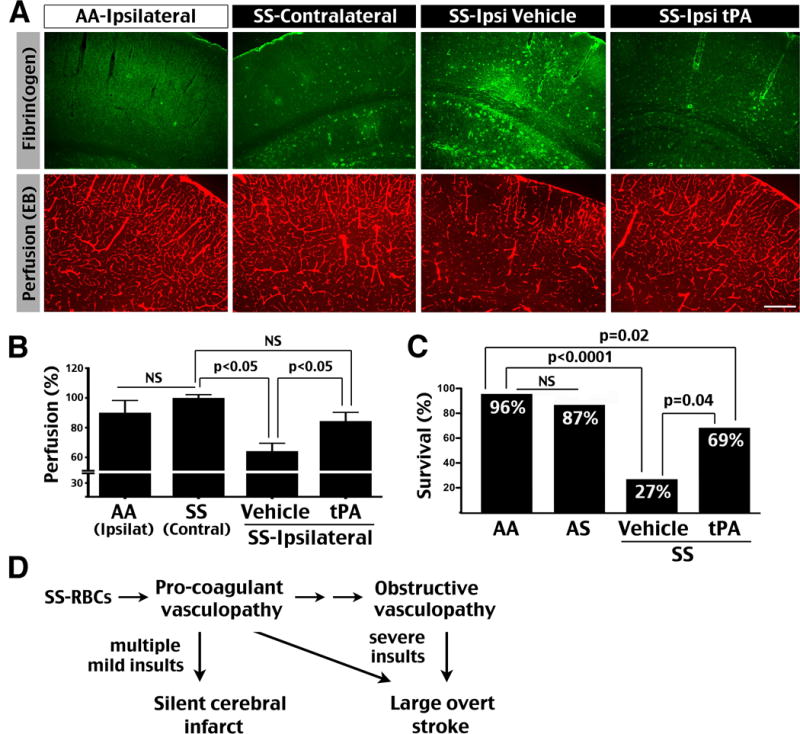
SS mice are hypersensitive to tHI-induced blood clotting and mortality, but respond to the tPA treatment. (A) Challenged by a subthreshold (20 min) tHI insult, 6-month-old SS mice showed impaired distribution of tail vein-injected Evans blue dye, correlated with more intense fibrin(ogen) deposition in the ipsilateral hemisphere at 4 h recovery compared with contralateral hemisphere and tHI-challenged AA mouse brains (n=3 for each). In contrast, the SS mice that received intravenous tPA treatment (10 mg/kg, 50% as bolus and 50% as infusion for 30 min at 0.5 h post tHI) exhibited minimal fibrin(ogen) deposition and improved cerebral vascular perfusion (n=3). Scale bar: 200 μm. (B) Comparison of cerebral vascular perfusion using the Evans blue dye fluorescence quantified by one-way ANOVA and Newman-Keuls post-test. NS: p>0.05. (C) At 24 h after 20 min tHI insult, the survival rate in AA mice was 96% (23/24), 87% in AS mice (7/8), 27 % in vehicle-treated SS mice (3/11), and 69% in tPA-treated SS mice (9/13). No obvious infarction was noted in the brains of surviving mice in either genotype. Using the survival rate in AA mice as the expectancy, the survival rates in AS and tPA-treated SS mice were comparable (NS, by chi-square test), but vehicle-treated SS mice showed significant reduction of survival (p<0.0001). Moreover, the tPA-treated SS mice showed improved survival than vehicle-treated SS mice (p=0.04), although still less than AA mice (p=0.02). (D) Our data suggest the progression from pro-coagulant to obstructive vasculopathy in sickle cell anemia, as well as different precipitating conditions for silent cerebral infarct and large overt stroke.
Moreover, 20 min tHI caused significantly greater mortality in 6-month-old SS mice (27% survival at 24 h recovery, n=11) than in AS mice (87%, n=8) or AA mice (96%, n=24; p<0.0001 compared with SS mice by chi-square test, Figure 4C). In contrast, the survival rate was more than doubled to 69% in SS mice that received tPA at 0.5 h post-tHI, (n=13; p=0.04 compared to vehicle-treated SS mice), although still lower than that in AA mice (p=0.02 by chi-square test). No obvious infarction was noted in the brains of surviving mice in either genotype. While cerebral infarct may not be the only cause of tHI-induced mortality in SS mice, these results suggest that thrombolysis protects SS mice against transient hypoxic-ischemic injury.
DISCUSSION
Humanized sickle mice are important tools for the development of many novel treatments of SCA, including the prevention of pain crises with anti-P-Selectin antibody (Crizanlizumab) and the prolongation of SS-RBC half-life by GBT-440.23,24 However, there are few stroke models in sickle mice.12–14 Our results suggest two stroke models based on hypoxic-ischemic insults in sickle mice that may relate to SCA patients, in whom the same insult often leads to an imbalance of oxygen consumption and oxygen delivery in the brain.1–3 We showed that 3-month-old SS mice already developed an higher sensitivity to repetitive mild hypoxia in the face of unilateral flow restriction due to carotid ligation (the rmHI model), leading to higher mortality and larger cerebral infarct. In contrast, neither unilateral carotid ligation nor repeated exposures to hypoxia caused cerebral infarct in 3-month-old SS mice, suggesting unique pathogenic effects of combined hypoxia-ischemia. By 6 months of age, SS mice exhibited greater perfusion defects, fibrin deposition, and higher mortality in response to transient hypoxic-ischemic insult (the tHI model) than AA mice. These two models may facilitate mechanistic and therapeutic studies of sickle cell stroke.
Interestingly, only increased resistive index (RI), but not greater peak systolic velocity (PSV) or mean velocity (MV), was developed in the carotid artery of 3-month-old SS mice with a higher sensitivity to rmHI-induced mortality and cerebral infarct. By six months of age, the RI remained elevated, but SS mice also developed greater PSV and MV similar to the flow alterations in SCA patients at higher risk for stroke. The differences in flow velocities between 3 and 6 months in SS mice were not due to progressive anemia, as the hemoglobin levels at these two ages were similar. Of note, RI is determined by the resistance in the distal microvascular bed, but attenuated by the stiffness of the large input vessel.18 Thus, RI may manifest biphasic changes with an initial rise due to the increasing downstream resistance and a secondary decline secondary to stenosis of the input vessel.18 There are several potential factors for a greater carotid RI in SCA, including impaired SS-RBC deformability, the opening of small arterioles to increase the basal CBF, and enhanced blood cell-endothelium adhesion.25 Given that only elevated RI was associated with sensitivity to rmHI-induced infarct in 3-month-old SS mice, our results suggest that RI is a more sensitive indicator of stroke propensity in SCA than the flow velocity.
Interestingly, greater RI has been used as a marker of vasculopathy in other clinical contexts. In obstetrics, high RI in the umbilical artery is an accepted marker of intrauterine growth restriction (IUGR) due to placental vasculopathy.26 Greater RI in renal Doppler sonography in SCA patients is also used as an early sign of reno-vascular pathologies.27 Yet, reduction of RI in the ophthalamic artery was suggested to indicate high risk for overt stroke in SCA patients.28 The Stroke Prevention Trial in Sickle Cell Anemia (STOP) trial did not include RI as a criterion to assess the stroke risk, but extremely dampened Doppler waveforms with decreased flow velocity (therefore low RI) was considered a sign of severe stenosis of cerebral vessels, a late stage vasculopathy in SCA.7 In light of our results showing that increased carotid artery RI in young SS mice signifies higher sensitivity to HI-induced cerebral infarct, future studies are warranted to compare the progression of elevated RI and flow velocity with regard to the risk for overt or silent cerebral infarct in SCA children.
Importantly, 6-month-old SS mice did not exhibit proliferative or stenotic changes in carotid arteries like those implicated in overt stroke in SCA,4,5 despite increased sensitivity to tHI-induced mortality and infarct. Similarly, the link between overt stroke and large cerebral vessel pathology is far from absolute in human SCA, as Rothman et al. identified large-vessel intimal lesions at autopsy in only half of cerebral infarcts from older SCA patients (averaged 21.3 years of age at death versus 12.2 years of age in vascular pathology-free patients).5 Further, as many as 31% of stroke patients in the Stroke With Transfusion Changing to Hydroxyurea (SWiTCH) trial were negative for vascular stenosis on magnetic resonance angiography, despite a higher flow velocity on TCD.29 Hence, the progression of sickle cell vasculopathy and stroke complications may be viewed as a continuum of injury resulting from an imbalance between oxygen demand and supply in the context of cerebral blood flow that is already maximized by compensatory mechanisms (Figure 4D).30 Such imbalance can be precipitated by a variety or events that increase oxygen demand (e.g. fever) or reduce oxygen delivery (e.g. hypoxia, ischemia, exacerbation of anemia, and large vessel stenosis). Viewed in this light, our stroke models recapitulate the hypoxic-ischemic events that may produce large or smaller infarct depending on the severity of insults and the age of sickle mice.
Although SS mice did not reproduce proliferative vasculopathy previously identified in SCA patients, they showed endothelial activation (e.g. P-Selectin expression) and enhanced leukocyte- endothelium interactions, especially after tHI insults. This finding, together with the recent report of anti-P-Selectin antibody attenuating painful crisis in SCA patients,23 suggests the possibility that P-Selectin antagonist might have a role in sickle stroke therapy. Moreover, we showed ectopic PAI-1 expression in cerebral blood vessels in SS mice, consistent with the reported induction of this pro-coagulant factor in pulmonary endothelial cells.22 Our data suggest that pro-adhesive, pro-coagulant endothelial activation occurs in the cerebral vasculature, which may contribute to higher sensitivity to tHI-induced infarction and mortality in SS mice. Of note, a limitation of our study is the high mortality rate of sickle mice in rmHI or tHI stroke, which is consistent with the behaviors of sickle mice in the focal photothrombosis model (40% mortality in sickle mice compared with no death in stroked wildtype mice)11, presumably due to the fragility of these mutant mice. We cannot exclude the possibility that tHI may also induce thrombosis in other organs (e.g. pulmonary embolism) to contribute to greater mortality in SS mice.
Nonetheless, a key finding of this study is that tPA treatment attenuates tHI-induced cerebral perfusion deficits and mortality in SS mice, suggesting the possibility that lytic therapy may benefit SCA patients with acute ischemic stroke. Although thrombolysis is a mainstay in acute treatment of stroke due to atherosclerosis and embolic events, there has been reluctance to adopt this therapy in SCA stroke, due to concerns of cerebral hemorrhage from aneurysm or large-vessel vasculopathy. For example, SCA was a safety exclusion criterion in the Thrombolysis in Pediatric Stroke trial (TIPS; NCT01591096). However, a recent analysis of the Get With The Guideline-Stroke (GWTG-Stroke) data indicated that co-existent sickle cell disease did not increase the incidence of cerebral hemorrhage after lytic stroke therapy (p=0.4502).8 This finding, coupled with our preclinical data of tPA-mediated benefits in tHI stroke in sickle mice, highlights the need to carefully assess the effects of lytic stroke therapy in SCA, first in animal models, then in patients.
CONCLUSIONS
Humanized sickle mice exhibit pro-coagulant vasculopathy and enhanced leukocyte and platelet adherence to vascular endothelium after HI insult, but without evidence of proliferative stenosis in large cerebral vessels. Higher resistive index is an early ultrasonic marker for increased sensitivity to HI-induced stroke in sickle mice. Thrombolysis attenuates HI-triggered mortality and cerebral infarct in sickle mice. Further studies to test the effects of lytic stroke therapy in children with SCA warrant considerations.
Supplementary Material
{kind=link}
Acknowledgments
We thank Drs. Neil Granger and Felicity Gavins and Ms. Janice Russell for demonstrating the intravital microscopy procedures; Dr. Hui-Wen Yao and Ms. Prasanthi Chappa for help with some experimental procedures. The ultrasonic measurements were performed at the Children’s Healthcare of Atlanta and Emory University’s Animal Physiology Core.
SOURCES OF FUNDING:
This study was supported by the National Institute of Health grants (NS084744, NS095064, NS093446, & HD080429 to C-Y. K., HL111659 to D.R.A, and HL117721 to C.H.J.) and an American Heart Association grant (17SDG33700003 to Y-Y.S.).
Footnotes
DISCLOSURES:
C.H.J. is a consultant to Global Blood Therapeutics, Inc. No other disclosures.
References
- 1.Adams RJ. Toward a stroke-free childhood in sickle cell disease: the 2013 Sherman Lecture. Stroke. 2013;44:2930–2934. doi: 10.1161/STROKEAHA.113.001312. [DOI] [PubMed] [Google Scholar]
- 2.Yawn BP, Buchanan GR, Afenyi-Annan AN, Ballas SK, Hassell KL, James AH, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 2014;312:1033–1048. doi: 10.1001/jama.2014.10517. [DOI] [PubMed] [Google Scholar]
- 3.DeBaun MR, Armstrong FD, McKinstry RC, Ware RE, Vichinsky E, Kirkham FJ. Silent cerebral infarcts: a review on a prevalent and progressive cause of neurologic injury in sickle cell anemia. Blood. 2012;119:4587–4596. doi: 10.1182/blood-2011-02-272682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boros L, Thomas C, Weiner WJ. Large cerebral vessel disease in sickle cell anaemia. J Neurol Neurosurg Psychiatry. 1976;39:1236–1239. doi: 10.1136/jnnp.39.12.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rothman SM, Fulling KH, Nelson JS. Sickle cell anemia and central nervous system infarction: a neuropathological study. Annals of Neurology. 1986;20:684–690. doi: 10.1002/ana.410200606. [DOI] [PubMed] [Google Scholar]
- 6.Adams RJ, Nichols FT, Figueroa R, McKie V, Lott T. Transcranial Doppler correlation with cerebral angiography in sickle cell disease. Stroke. 1992;23:1073–1077. doi: 10.1161/01.str.23.8.1073. [DOI] [PubMed] [Google Scholar]
- 7.Adams RJ. TCD in sickle cell disease: an important and useful test. Pediatric Radiology. 2005;35:229–234. doi: 10.1007/s00247-005-1409-7. [DOI] [PubMed] [Google Scholar]
- 8.Adams RJ, Cox M, Ozark SD, Kanter J, Schulte PJ, Xian Y, et al. Coexistent Sickle Cell Disease Has No Impact on the Safety or Outcome of Lytic Therapy in Acute Ischemic Stroke: Findings From Get With The Guidelines-Stroke. Stroke. 2017;48:686–691. doi: 10.1161/STROKEAHA.116.015412. [DOI] [PubMed] [Google Scholar]
- 9.DeBaun MR, Gordon M, McKinstry RC, Noetzel MJ, White DA, Sarnaik SA, et al. Controlled trial of transfusions for silent cerebral infarcts in sickle cell anemia. N Engl J Med. 2014;371:699–710. doi: 10.1056/NEJMoa1401731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hankins JS, McCarville MB, Rankine-Mullings A, Reid ME, Lobo CL, Moura PG, et al. Prevention of conversion to abnormal transcranial Doppler with hydroxyurea in sickle cell anemia: A Phase III international randomized clinical trial. Am J Hematol. 2015;90:1099–1105. doi: 10.1002/ajh.24198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.LC Wu, Sun CW, Ryan TM, Pawlik KM, Ren J, Townes TM. Correction of sickle cell disease by homologous recombination in embryonic stem cells. Blood. 2006;108:1183–1188. doi: 10.1182/blood-2006-02-004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gavins FN, Russell J, Senchenkova EL, De Almeida Paula L, Damazo AS, Esmon CT, et al. Mechanisms of enhanced thrombus formation in cerebral microvessels of mice expressing hemoglobin-S. Blood. 2011;117:4125–4133. doi: 10.1182/blood-2010-08-301366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaul DK, Hebbel RP. Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice. J Clinical Investigation. 2000;106:411–420. doi: 10.1172/JCI9225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luo W, Su EJ, Wang J, Wang H, Guo C, Pawar A, et al. Increased stroke size following MCA occlusion in a mouse model of sickle cell disease. Blood. 2014;123:1965–1967. doi: 10.1182/blood-2014-01-549717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun YY, Morozov YM, Yang D, Li Y, Dunn RS, Rakic P, et al. Synergy of combined tPA-edaravone therapy in experimental thrombotic stroke. PloS One. 2014;9:e98807. doi: 10.1371/journal.pone.0098807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun YY, Kuan CY. A Thrombotic Stroke Model Based On Transient Cerebral Hypoxia-ischemia. J Vis Exp. 2015:e52978. doi: 10.3791/52978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams R, Needles A, Cherin E, Zhou YQ, Henkelman RM, Adamson SL, et al. Noninvasive ultrasonic measurement of regional and local pulse-wave velocity in mice. Ultrasound Med Biol. 2007;33:1368–1375. doi: 10.1016/j.ultrasmedbio.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 18.Adamson SL. Arterial pressure, vascular input impedance, and resistance as determinants of pulsatile blood flow in the umbilical artery. Eur J Obstet Gynecol Reprod Biol. 1999;84:119–125. doi: 10.1016/s0301-2115(98)00320-0. [DOI] [PubMed] [Google Scholar]
- 19.Sun YY, Yang D, Kuan CY. Mannitol-facilitated perfusion staining with 2,3,5-triphenyltetrazolium chloride (TTC) for detection of experimental cerebral infarction and biochemical analysis. Journal of Neuroscience Methods. 2012;203:122–129. doi: 10.1016/j.jneumeth.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun YY, Li Y, Wali B, Lee J, Heinmiller A, Abe K, et al. Prophylactic Edaravone Prevents Transient Hypoxic-Ischemic Brain Injury: Implications for Perioperative Neuroprotection. Stroke. 2015;46:1947–1955. doi: 10.1161/STROKEAHA.115.009162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McConnell SC, Huo Y, Liu S, Ryan TM. Human globin knock-in mice complete fetal-to-adult hemoglobin switching in postnatal development. Molecular and Cellular Biology. 2011;31:876–883. doi: 10.1128/MCB.00725-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patel N, Sundaram N, Yang M, Madigan C, Kalra VK. Malik P Placenta growth factor (PlGF), a novel inducer of plasminogen activator inhibitor-1 (PAI-1) in sickle cell disease (SCD) Journal of Biological Chemistry. 2010;285:16713–16722. doi: 10.1074/jbc.M110.101691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ataga KI, Kutlar A, Kanter J, Liles D, Cancado R, Friedrisch J, et al. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N Engl J Med. 2017;376:429–439. doi: 10.1056/NEJMoa1611770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oksenberg D, Dufu K, Patel MP, Chuang C, Li Z, Xu Q, et al. GBT440 increases haemoglobin oxygen affinity, reduces sickling and prolongs RBC half-life in a murine model of sickle cell disease. Br J Haematol. 2016;175:141–153. doi: 10.1111/bjh.14214. [DOI] [PubMed] [Google Scholar]
- 25.Lipowsky HH. Microvascular rheology and hemodynamics. Microcirculation. 2005;12:5–15. doi: 10.1080/10739680590894966. [DOI] [PubMed] [Google Scholar]
- 26.Yoshizato T, Satoh S. Morphological and functional evaluation of normal and abnormal fetal growth by ultrasonography. J Med Ultrason. 2009;36:105–117. doi: 10.1007/s10396-009-0224-4. [DOI] [PubMed] [Google Scholar]
- 27.Taori KB, Chaudhary RS, Attarde V, Dhakate S, Sheorain V, Nimbalkar P, et al. Renal Doppler indices in sickle cell disease: early radiologic predictors of renovascular changes. American Journal of Roentgenology. 2008;191:239–242. doi: 10.2214/AJR.07.3125. [DOI] [PubMed] [Google Scholar]
- 28.Seibert JJ, Miller SF, Kirby RS, Becton DL, James CA, Glasier CM, et al. Cerebrovascular disease in symptomatic and asymptomatic patients with sickle cell anemia: screening with duplex transcranial Doppler US–correlation with MR imaging and MR angiography. Radiology. 1993;189:457–466. doi: 10.1148/radiology.189.2.8105505. [DOI] [PubMed] [Google Scholar]
- 29.Helton KJ, Adams RJ, Kesler KL, Lockhart A, Aygun B, Driscoll C, et al. Magnetic resonance imaging/angiography and transcranial Doppler velocities in sickle cell anemia: results from the SWiTCH trial. Blood. 2014;124:891–898. doi: 10.1182/blood-2013-12-545186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prohovnik I, Hurlet-Jensen A, Adams R, De Vivo D, Pavlakis SG. Hemodynamic etiology of elevated flow velocity and stroke in sickle-cell disease. J Cereb Blood Flow Metab. 2009;29:803–810. doi: 10.1038/jcbfm.2009.6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.