

Abstract
The highly conserved Wnt signalling pathway plays an important role in embryonic development and disease pathogenesis, most notably cancer. The ‘canonical’ or β‐catenin‐dependent Wnt signal initiates at the cell plasma membrane with the binding of Wnt proteins to Frizzled:LRP5/LRP6 receptor complexes and is mediated by the translocation of the transcription co‐activator protein, β‐catenin, into the nucleus. β‐Catenin then forms a complex with T‐cell factor (TCF)/lymphoid enhancer binding factor (LEF) transcription factors to regulate multiple gene programmes. These programmes play roles in cell proliferation, migration, vasculogenesis, survival and metabolism. Mutations in Wnt signalling pathway components lead to constitutively active Wnt signalling that drives aberrant expression of these programmes and development of cancer. It has been a longstanding and challenging goal to develop therapies that can interfere with the TCF/LEF–β‐catenin transcriptional complex. This review will focus on the (i) structural considerations for targeting the TCF/LEF–β‐catenin and co‐regulatory complexes in the nucleus, (ii) current molecules that directly target TCF/LEF–β‐catenin activity and (iii) ideas for targeting newly discovered components of the TCF/LEF–β‐catenin complex and/or downstream gene programmes regulated by these complexes.
Linked Articles
This article is part of a themed section on WNT Signalling: Mechanisms and Therapeutic Opportunities. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.24/issuetoc
Abbreviations
- APC
adenomatous polyposis coli
- CBP
cAMP response element binding protein (CREB)‐binding protein
- DVL
Dishevelled
- LEF
lymphoid enhancer binding factor
- LRP
low‐density lipoprotein receptor‐related protein
- MCT
monocarboxylate transporter
- TCF
T‐cell factor
- WRE
Wnt response element
Introduction
Wnt signalling comprises a set of signal transduction cascades that are highly conserved across many different species including both non‐vertebrates (such as nematodes and fruit flies) and vertebrates (frogs, mice and humans). These signals play important roles not only in cell fate decisions during embryonic development and stem cell homeostasis in somatic niches of normal and injured tissues, but also in diseases such as cancer (Nusse and Varmus, 1982; Bodmer et al., 1987; McMahon and Moon, 1989; Rocheleau et al., 1997). A great deal of work has been performed to uncover the intricacies of the signal transduction steps at the cell surface and the cytoplasm to better understand the normal and dysfunctional activity of Wnt signalling. Recent reviews published elsewhere and in this issue describe our current understanding of these steps in detail (Niehrs and Acebron, 2012; DeBruine et al., 2017; Driehuis and Clevers, 2017; Nusse and Clevers, 2017; van Kappel and Maurice, 2017; Zimmerli et al., 2017).
In brief, the ‘canonical’, or β‐catenin‐dependent Wnt signalling pathway is the primary source of dysregulated transcription in the disease setting. In the normal ‘on‐state’, this signal initiates at the cell surface when Wnts bind to Frizzled:LRP5/LRP6 receptor complexes, and it culminates in the nucleus where it triggers the formation of a powerful transcription‐activating complex (Figure 1). The main mediator of this cell surface‐to‐nucleus signal is β‐catenin, a membrane/cytoplasmic armadillo repeat protein with no ability to promote transcription by binding DNA on its own (Niehrs and Acebron, 2012; Masuda and Ishitani, 2017; Nusse and Clevers, 2017). Instead, through a multitude of protein interactions, β‐catenin moves to the nucleus where DNA‐binding T‐cell factor (TCF)/lymphoid enhancer binding factor (LEF) transcription factors recruit it to promoters and enhancers of Wnt target genes (Cadigan and Waterman, 2012; Masuda and Ishitani, 2017). Once tethered to DNA by TCF/LEFs, β‐catenin recruits co‐activators and other regulatory components to activate transcription of the downstream genes known collectively as Wnt target genes. It is this increased transcription of coordinated sets of genes (i.e. gene programmes) that directs cells to proliferate, self‐renew, differentiate and survive in different tissues and contexts. In normal cells and tissues, feedback inhibition results in this activity occurring only transiently, preventing over‐activation of Wnt target gene transcription. Signal transduction is ‘turned off’ in cells with low or absent Wnt because β‐catenin becomes unstable: it is efficiently tagged in the cytoplasm for ubiquitination by the β‐catenin destruction complex and degraded by proteasomes. However, in diseases (i.e. colon cancer) with genetic mutations of one or more destruction complex components [e.g. adenomatous polyposis coli (APC), AXIN2 and FAM123B/WTX] or inactivating mutation of negative regulators of Wnt/receptor interactions (e.g. RNF43/ZNRF3) or mutations that enhance Wnt signalling (RSPO2 and RSPO3), Wnt signalling is improperly activated. These mutations negate the cytoplasmic feedback controls and create cells with constitutive, high levels of β‐catenin and aberrantly high levels of Wnt target gene transcription that can initiate the transformation of colon epithelia into malignant cells (Cancer Genome Atlas Network et al., 2012; Polakis, 2012; Mazzoni and Fearon, 2014; Nusse and Clevers, 2017).
Figure 1.
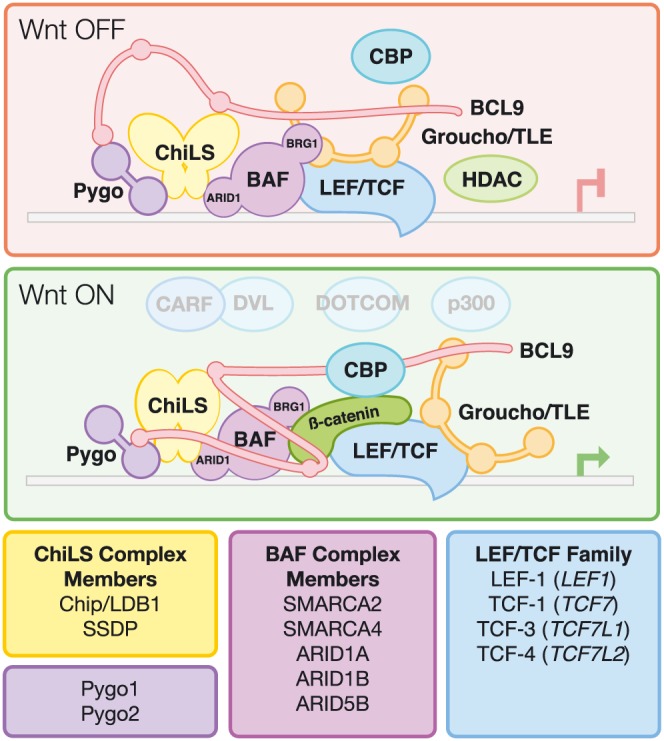
Model of the Wnt enhancesosome. In the absence of nuclear β‐catenin, the Wnt enhancesosome complex (Fiedler et al., 2015) is directly associated with TCF/LEF transcription factors binding to WREs in promoters and enhancers. This complex forms through interactions of BCL9 with Pygopus (PYGO1/PYGO2), the ChiLS complex [Chip/LDB1, SSDP: composed of a dimer of Chip, LDB (an LIM domain‐binding protein) and a tetramer of single‐stranded DNA‐binding protein(SSDP)] and Groucho/TLE [either as a dimer or possible tetramer (Chodaparambil et al., 2014)]. CBP/p300 is also present in the OFF state, although the nature of its binding interaction in this state is less certain. CARF, a newly identified binding partner of DVL2, has been shown to be a positive regulator of Wnt signalling, although its precise binding to β‐catenin remains unknown (He et al., 2017). The chromatin remodelling complex BAF interacts through the ARID1 subunit via directly interacting with ChiLS. The presence of nuclear β‐catenin induces a conformational change in BCL9, allowing CBP/p300/DOTCOM to bind to the c‐terminal end of β‐catenin, and BCL9 binding to the N‐terminal end. Groucho/TLE is inactivated, allowing for transcription of associated Wnt target genes. Figure adapted from van Tienen et al. (2017).
Given that the unifying feature of Wnt‐linked cancer is aberrant target gene regulation, it has been a longstanding imperative for at least 20 years to develop a therapy that can interfere with TCF/LEF–β‐catenin‐regulated transcription (Polakis, 2012; Nusse and Clevers, 2017). In particular, there has been strong interest in designing an inhibitor that can act at the nuclear level and effectively target all Wnt‐linked cancers, independently of whether they are caused by receptor mutation or mutations of further downstream signalling components (destruction complex with APC or β‐catenin mutations). As straightforward as this goal is, there have been numerous hurdles: first, the challenge of navigating the large, multi‐component regulatory complex that β‐catenin assembles (some of which is still unknown); second, designing a molecule that disrupts key protein–protein interactions (PPIs) within that complex; and third, finding a pharmacokinetically favourable small molecule that can cross both the plasma membrane and nuclear membrane to disrupt those key interactions. These challenges have been formidable to the degree that despite a tremendous amount of academic and industry resources dedicated to the problem, not a single drug that inhibits TCF/LEF–β‐catenin complexes is currently in clinical use. In addition, since Wnt signalling has a crucial role in stem cell proliferation and homeostasis of multiple organ systems (i.e. intestinal, bone, skin and hair), there has been concern that inhibiting this pathway may result in intolerable toxicities in the intestine or integrity of bone (Kahn, 2014; Nusse and Clevers, 2017). Despite these concerns, several molecules targeting various components of Wnt signalling have been developed and administered in clinical trials with minimal and tolerable toxicities to the gastrointestinal tract, bone, skin and other organs in human patients (El‐Khoueiry et al., 2013; Le et al., 2015; Mita et al., 2016). These molecules have successfully completed Phase I clinical trials (safety and toxicity evaluation) and have recently been approved by the FDA to proceed to Phase II clinical testing (El‐Khoueiry et al., 2013; Le et al., 2015; Mita et al., 2016). Although only one of these molecules undergoing clinical studies acts at the nuclear level (PRI‐724, which is discussed in detail in this review), the collective results of the early phase clinical trials of these Wnt inhibitors are encouraging and provide a proof of principle that it is possible to inhibit the Wnt signalling pathway in human patients without causing severe toxicity. Furthermore, recent advances in our knowledge of gene programmes that Wnt signalling targets for cell transformation, coupled with a better understanding of the composition and actions of TCF/LEF–β‐catenin complexes, have led to the preclinical development of several small molecules that disrupt this activity and downstream effects. These advances are encouraging and suggest that the pace of drug development for Wnt‐linked cancers is accelerating (Kahn, 2014; Zhan et al., 2016; Nusse and Clevers, 2017). This review will focus on the (i) structural considerations for targeting TCF/LEF–β‐catenin and co‐regulatory complexes in the nucleus, (ii) current molecules that directly target TCF/LEF–β‐catenin activity and (iii) ideas for targeting newly discovered components of the TCF/LEF–β‐catenin complex and/or downstream gene programmes regulated by these complexes.
General principles in designing small molecule inhibitors of TCF/LEF–β‐catenin
β‐Catenin is recruited to target genes through direct recruitment interactions with the N‐termini of TCF/LEF transcription factors (Cadigan and Waterman, 2012; Masuda and Ishitani, 2017). There are four mammalian TCF/LEFs (protein names: TCF‐1, TCF‐3, TCF‐4 and LEF‐1; gene names TCF7, TCF7L1, TCF7L2 and LEF1 respectively), and the β‐catenin‐binding domain is one of the most highly conserved features of the transcription factor family (Cadigan and Waterman, 2012; Masuda and Ishitani, 2017). It stands to reason that disrupting this interaction would be a good strategy for interfering with the overabundant TCF/LEF–β‐catenin complexes that drive oncogenic Wnt signalling. However, structural studies of β‐catenin binding to its myriad inhibitor/activator partners quite rightly predicted the challenges in identifying small molecules that can specifically disrupt binding to TCF/LEFs. The N‐terminal ~50 amino acids of TCF/LEFs are intrinsically unstructured until they engage in extensive hydrophobic and salt‐bridge interactions with the armadillo repeat array of β‐catenin (Graham et al., 2000; Sun et al., 2000; Daniels et al., 2001; Poy et al., 2001; Choi et al., 2006). The interactions span from arm repeat 3 through arm repeat 10 – and given that there are only 11 armadillo repeats in β‐catenin, this interaction represents an extensive ‘zipping together’ of the β‐catenin arm repeat array with the N‐terminus of TCF/LEFs. The extensive binding interactions with multiple points of direct contact between β‐catenin and TCF/LEFs make it challenging to design a small molecule inhibitor (Daniels et al., 2001; Xu and Kimelman, 2007; Kahn, 2014). An additional challenge comes from the discovery of a convergence of binding modes for inhibitory and activating partners of β‐catenin; that is, the same armadillo repeats engage in extensive and similar interactions with E‐cadherin and other inhibitory proteins such as APC, AXIN and ICAT (summarized in Daniels et al., 2001). Despite these challenges, there has been some success in identifying small molecule inhibitors, and more recently, success in moving a few of these promising hits through preclinical xenograft studies. Most successes have come chiefly through the development of high throughput screens for molecules that disrupt the transcription regulating activities of TCF/LEF–β‐catenin complexes. These high‐throughput screens most frequently utilize either a TCF/LEF–β‐catenin‐specific luciferase reporter construct (TOPFlash, SuperTOPFlash, etc.) or TCF/LEF–β‐catenin binding ELISA assays to measure modulation in overall Wnt signalling output (Korinek et al., 1997; Lepourcelet et al., 2004; Kahn, 2014). Many screens have utilized cell lines with activated Wnt signalling (i.e. SW480 and HCT116 colon cancer cell lines) in which to perform these assays. It should be noted that although these assays are useful as an initial rapid screening tool, the readouts use artificial promoter constructs containing multiple Wnt response elements (WREs) and, therefore, follow‐up studies are often required to assay for changes in endogenous expression of various Wnt signalling components and targets to further determine the site of action (extracellular, cytoplasmic or intranuclear) in the pathway (Kahn, 2014; Duchartre et al., 2016). Below, we summarize the screens that have used these types of strategies to identify lead compounds that directly target TCF/LEF–β‐catenin activity (Figure 2 and Table 1 ).
Figure 2.
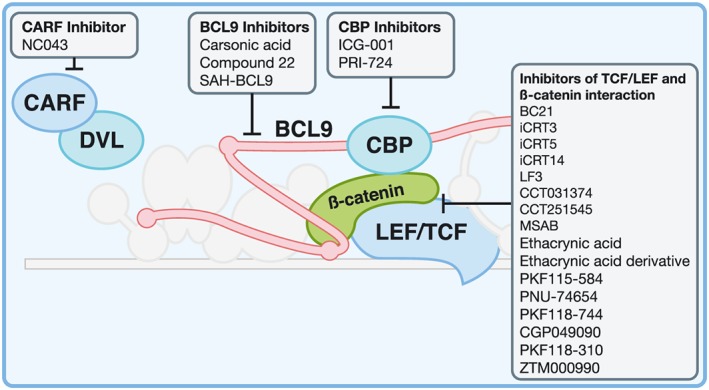
Small molecule inhibitors targeting the Wnt enhancesosome. Detailed in the main text, the inhibitors have been categorized by their target: CBP, interactions between β‐catenin and LEF/TCF members and CARF‐DVL2.
Table 1.
Small molecule inhibitors that disrupt the transcription regulating activities of TCF/LEF–β‐catenin complexes
| Target | Compound name | Screening assay | Clinical trial status | References |
|---|---|---|---|---|
| TCF/LEF and β‐catenin interaction | Ethacrynic acid | TOPFlash | Preclinical | (Lu et al., 2009) |
| Ethacrynic acid derivative | TOPFlash | Preclinical | (Jin et al., 2009) | |
| PKF115‐584 | TCF‐4 and β‐catenin ELISA assay | Preclinical | (Lepourcelet et al., 2004) | |
| PNU‐74654 | TCF‐4 and β‐catenin ELISA assay | Preclinical | (Lepourcelet et al., 2004) | |
| PKF118‐744 | TCF‐4 and β‐catenin ELISA assay | Preclinical | (Lepourcelet et al., 2004) | |
| CGP049090 | TCF‐4 and β‐catenin ELISA assay | Preclinical | (Lepourcelet et al., 2004) | |
| PKF118‐310 | TCF‐4 and β‐catenin ELISA assay | Preclinical | (Lepourcelet et al., 2004) | |
| ZTM000990 | TCF‐4 and β‐catenin ELISA assay | Preclinical | (Lepourcelet et al., 2004) | |
| BC21 | TOPFlash | Preclinical | (Tian et al., 2012) | |
| iCRT3 iCRT5 iCRT14 | dAxin‐dsRNA–induced dTF12 reporter (TOPflash like) | Preclinical | (Gonsalves et al., 2011) | |
| LF3 | TOPFlash | Preclinical | (Fang et al., 2016) | |
| CCT031374 | xNR3 WRE promoter | Preclinical | (Ewan et al., 2010) | |
| MSAB | TOPFlash | Preclinical | (Hwang et al., 2016) | |
| CBP | ICG‐001 | TOPFlash | Preclinical | (Emami et al., 2004) |
| PRI‐724 (second generation of ICG‐001) | – | Phase I, Phase II | (Duchartre et al., 2016) | |
| BCL9 | Carsonic acid | BCL9 peptide fragment and β‐catenin ELISA | Preclinical | (de la Roche et al., 2012) |
| Compound 22 (optimal molecule in series) | Rational design from protein structure database | Preclinical | (Hoggard et al., 2015) | |
| SAH‐BCL9 | Rational design from protein structure database | Preclinical | (Takada et al., 2012) | |
| CARF | NC043 | TOPFlash | Preclinical | (He et al., 2017) |
Compounds targeting the TCF/LEF and β‐catenin interaction
One early class of molecules that was discovered to specifically inhibit Wnt signalling is a class that directly targets the transcription factor LEF1 and interferes with its ability to interact with β‐catenin. Ethacrynic acid (EA) was first discovered when 960 FDA‐approved drugs were screened for the ability to decrease the activity of a luciferase reporter (TOPFlash) in HEK293 cells overexpressing Dishevelled (DVL; Wnt signalling activator) (Lu et al., 2009). The initial screen was complemented by a secondary screen using Wnt‐3a to activate signalling through Frizzled:LRP5/6 receptors on the plasma membrane, as well as by expression of a combination of various other Wnt components such as Wnt‐1/LRP6, Wnt‐3/LRP6, DVL or β‐catenin (Lu et al., 2009). The results revealed that EA could inhibit Wnt signalling activity in a dose‐dependent manner in all activation models suggesting that its molecular action was either targeting β‐catenin or disrupting events further downstream. EA was also found to have selective cytotoxicity towards chronic lymphocytic leukaemia (CLL) cells with an IC50 of 8.56 ± 3 μM compared to 34.97 ± 15.97 μM in normal peripheral blood mononuclear cells (Lu et al., 2009). Furthermore, the authors found that EA could inhibit the expression of Wnt target genes LEF1 and CCND1. Through co‐immunoprecipitation experiments using an antibody to EA, and LEF‐1, EA was found to directly bind to LEF‐1 protein, leading to destabilization of LEF‐1/β‐catenin interactions (Lu et al., 2009). Unfortunately, the IC50 for other cancer cell lines such as colon cancer (SW480 and HCT116) were found to be higher at 68 and 58 μM, respectively, suggesting that this molecule has less potent cytotoxicity in other cancers (Lu et al., 2009). The same group also showed that various amide derivatives of EA could be synthesized and these exhibited a lower IC50 when treating CLL cells (Jin et al., 2009); however, so far, these higher affinity derivatives do not appear to have been developed further.
The Shivdasani group performed a high throughput screen of ~7000 natural compounds using a custom‐designed TCF‐4 and β‐catenin ELISA assay to detect small molecules which could disrupt their association (Lepourcelet et al., 2004). Eight compounds were identified with reproducible IC50 < 10 uM, and of these, six compounds (PKF115‐584, CGP049090, PKF222‐815, PKF118‐744, PKF118 310 and ZTM000990) were found to inhibit TOPFlash reporter activity in the HCT116, human colon cancer cell line (Lepourcelet et al., 2004). Immunoblot experiments showed that these compounds could inhibit both CCND1 and c‐Myc protein expression (Wnt target genes) in a dose‐dependent manner, while cellular β‐catenin protein levels remained intact. Furthermore, the authors also showed that these compounds could inhibit the in vitro proliferation of colon cancer cell lines (HCT116 and HT29) and prostate cancer cell lines (PC‐3 and DU‐145). To date, however, there are no clinical trials using these molecules or next‐generation derivatives.
Using information from co‐crystallization studies of β‐catenin and the β‐catenin‐binding fragment of the TCF‐4 N‐terminus, the An group performed a virtual screen to identify 200 potential compounds from a library of 1990 compounds that could bind to three potential regions within the TCF‐4 and β‐catenin interaction domains that had been previously proposed (Fasolini et al., 2003; Tian et al., 2012). These 200 compounds were then screened in a TOPFlash luciferase reporter assay in the HCT116colon cancer cell line. Of these compounds, BC21 was found to be most effective compound in inhibiting TOPFlash luciferase reporter activity. This compound reduced the endogenous expression of Wnt target genes CCND1 and C‐MYC at the mRNA and protein level with an IC50 of 15 μM. At the time of this publication, however, there is no evidence showing that BC21 has advanced to clinical trials.
The DasGupta group screened 14 977 compounds and selected molecules that could disrupt β‐catenin‐dependent transcription activation of a TOPFlash‐like luciferase reporter (dTF12) in Drosophila Cl8 cells (Gonsalves et al., 2011). Several compounds were identified (iCRT3, iCRT5 and iCRT14), and their mode of action was shown to interfere directly with the binding of TCF‐4 to β‐catenin. The anti‐tumour effects of these compounds were validated using primary human colon cancer specimens in a microspheroid‐based 3D assay for cell survival. The average IC50 across six patient samples was determined to be comparable to other commonly used chemotherapy drugs in colon cancer such as fluorouracil (5‐FU; Gonsalves et al., 2011). Unfortunately, there is no evidence that these compounds have advanced beyond the preclinical stage.
A few years later, the Dale group screened 63,000 compounds in HEK293 cultures with activated Wnt signalling (DVL‐oestrogen receptor fusion protein) to identify small molecules that could inhibit a luciferase reporter assay containing three endogenous Wnt response elements (WREs) in the xNR3 promoter (Ewan et al., 2010). Secondary screens with three additional assays in which Wnt signalling was activated via manipulation at various cellular levels (extracellular: LRP6, cytoplasmic: AXIN2 and intracellular: TCF‐4) confirmed a final set of 20 compounds that demonstrated Wnt inhibition (Ewan et al., 2010). Of these compounds, CCT031374 exhibited the strongest suppression of cell proliferation in human cancer cell lines that are dependent on Wnt signalling (HCT116, SW480, HT29 and SNU475). The related 3‐indolylmethaneamine compound, CCT036477, was effective in blocking Wnt‐dependent phenotypes in zebrafish and Xenopus embryos. The authors did not further characterize which components of the TCF/LEF complex were directly targeted by these compounds, and there is no current evidence that these leads have advanced beyond the preclinical stage. However, a highly related set of 3‐indolylmethaneamine compounds has recently been shown to be effective in blocking the growth of HL‐60 promyelocytic and SKOV‐3 ovarian cancer xenograft tumours in mice (Guthrie et al., 2015).
In 2016, the Birchmeier group completed an ELISA screen of 16 000 synthetic compounds to identify molecules that could inhibit the binding of β‐catenin to a TCF4‐derived peptide (Fang et al., 2016). Amongst the compounds screened, LF3 was found to have the strongest selective, disruptive activity. Further studies showed that it could inhibit TOPFlash reporter activity as well as endogenous protein interactions between β‐catenin and TCF‐4 or LEF‐1 (Fang et al., 2016). This compound reduced the protein and mRNA expression of Wnt target genes AXIN2, C‐MYC and CCND1 in a dose‐dependent manner, and it also inhibited cell proliferation of HCT116 and HT29 colon cancer cell lines with an approximate IC50 of 30 μM (Fang et al., 2016). Importantly, an in vivo xenograft mouse model using SW480 colon cancer cells showed that LF3 strongly inhibited tumour growth by approximately 40% at 40 days (study endpoint), suggesting that at least in mice, this compound has anti‐tumour effects. Interestingly, LF3‐treated tumours also appeared to be more differentiated compared to untreated control tumours. It was encouraging that the authors did not find any signs of obvious systemic toxicity, such as weight loss, and it leaves potential for future study using this drug in clinical development.
Also recently, the S.W. Lee group performed a screen of 22 000 compounds for their ability to inhibit TOPFlash reporter activity in HCT116 cells (Hwang et al., 2016). Methyl 3‐{[(4‐methyl‐ phenyl)sulfonyl]amino}benzoate (MSAB) was identified as the best candidate molecule, and further validation showed that it inhibits the proliferation of many Wnt‐dependent cell lines but not Wnt‐independent cell lines such as breast epithelial cells and human dermal fibroblasts. In vivo studies showed that MSAB inhibits tumour growth of xenografts from HCT116, HT115 and H23 cell lines, and in addition, it inhibits the endogenous expression of β‐catenin at the protein level (Hwang et al., 2016). Furthermore, the authors also showed how this compound could inhibit the expression of Wnt target genes AXIN2 and C‐MYC at both the protein and mRNA level (Hwang et al., 2016). Mechanistic studies revealed that MSAB binds directly to β‐catenin and promotes its degradation by preventing its binding to the TCF/LEF transcription factors (Hwang et al., 2016). The in vivo xenograft mouse models showed no obvious signs of systemic toxicity upon administration of the drug, which is encouraging and shows potential for future clinical studies.
Compounds targeting Wnt co‐factors
The identification of a complete list of co‐regulatory factors that assemble with TCF/LEFs at WREs in promoters and enhancers associated with β‐catenin‐dependent Wnt signalling has been an active area of research for many years. Early genetic studies in Drosophila and subsequent confirmation in other model systems identified a few of these players including the direct association of Groucho/TLE transcription co‐repressors with TCF/LEFs in the absence of Wnt signalling (Levanon et al., 1998; Brantjes et al., 2001) and cAMP response element binding protein (CREB)‐binding protein (CBP)/p300 transcription co‐activators in the presence of active Wnt signalling (Waltzer and Bienz, 1998; Hecht et al., 2000; Sun et al., 2000; Takemaru and Moon, 2000). In addition, the Drosophila co‐regulators Pygopus and Legless were discovered to have mammalian homologues (PYGO1, PYGO2 and BCL9, B9L respectively) (Willis et al., 1998; Kramps et al., 2002; Thompson et al., 2002; Townsley et al., 2004). Other studies have identified additional regulatory complexes, such as the DOTCOM complex, that contribute to β‐catenin regulation of transcription (Mohan et al., 2010), but complexes such as these are more broadly acting and are not necessarily dedicated to β‐catenin‐TCF/LEF complexes (Cadigan and Waterman, 2012). More recently, there have been exciting advances in identifying the specific complex of proteins established by TCF/LEFs on chromatin and understanding how that complex is modified by Wnt signalling and β‐catenin (Chodaparambil et al., 2014; Fiedler et al., 2015; van Tienen et al., 2017) (Figure 1). The old model of β‐catenin displacing Groucho/TLE repressors from TCF/LEFs to activate gene expression clearly needs refinement as current studies suggest that co‐repressors and co‐activators might exist simultaneously. The entry of β‐catenin into the complex modifies pre‐existing, structurally based interactions and activities to exert transcription activating functions. The insertion of small molecules that selectively disrupt the activation process is a challenge and rather complex to identify and define mechanistically.
Some of the newly identified TCF/LEF–β‐catenin complex associating factors contain enzymatic or scaffolding activity and have been exploited as potential targets for small molecule inhibitors. Of these co‐factors, the scaffolding, PPI between BCL9 and β‐catenin has been found to be an effective target for small molecule inhibition by various groups. The Bienz group used a customized BCL9 peptide fragment (homology domain 2) and β‐catenin‐binding ELISA assay to screen through a library of 46 250 compounds and found that carnosic acid (CA), which is a natural occurring component in rosemary, had the highest and most specific inhibitory activity with a K i value of 3.3 ± 1.8 μM (de la Roche et al., 2012). This compound could also inhibit the transcription of the Wnt target gene AXIN2 in cells with high levels of Wnt signalling such as LiCl‐treated HeLa cells, SW480 cells and HCT116 cells. In addition, CA was found to decrease TOPFlash luciferase reporter activity by ~90% in SW480 cells compared to the DMSO‐treated control. Although this study did not investigate if CA could inhibit tumour growth in mice, there have been ongoing studies by other groups to determine if this molecule and other natural components of rosemary may have anti‐neoplastic benefits.
The Carrosco group used a rational target design approach to design a synthetic peptide which could interfere with the binding of the α‐helical HD2 (homology domain 2) of BCL9 (residues 351 to 374) and surface groove formed by α helices 2 and 3 of the armadillo repeat 1 of β‐catenin (Sampietro et al., 2006; Takada et al., 2012). The investigators mutated the residues at the BCL9 binding interface (H358A or R359A) and designed a series of cell permeable peptides (SAH‐BCL9, stabilized α helix of BCL9 peptides), which could inhibit the ability of BCL9 to bind to β‐catenin (Takada et al., 2012). Of these peptides, SAH‐BCL9B was found to be the most effective at targeting β‐catenin and could selectively disrupt the BCL9/ β‐catenin complex in a dose‐dependent manner, as measured by ELISA and immunoprecipitation in two different human colon cancer cell lines (Colo320 and DLD‐1) (Takada et al., 2012). This molecule inhibited Wnt signalling in a dose‐dependent manner in Colo320 colon cancer cells, as measured by decreased TOPFlash luciferase reporter activity and mRNA levels of various Wnt target genes (AXIN2, LGR5, LEF1, VEGF‐A and C‐MYC) (Takada et al., 2012). Furthermore, SAH‐BCL9B inhibited tumour growth, angiogenesis and metastasis in mouse xenograft models of colon cancer (Colo320) and multiple myeloma (INA‐6) (Takada et al., 2012). The administration of SAH‐BCL9B did not appear to cause any obvious systemic toxicity such as cytopaenia or severe weight loss (Takada et al., 2012). These findings are encouraging and demonstrate potential for future clinical studies.
Using a similar approach, the Ji group was able to identify a small molecule, 4′‐fluoro‐N‐phenyl‐[1,1′‐biphenyl]‐3‐carboxamide that could inhibit the BCL9/β‐catenin PPI (Hoggard et al., 2015). They then used this molecule as a generic scaffold to design 30 small molecule inhibitors that were more specific for the BCL9/β‐catenin PPI (Hoggard et al., 2015). These molecules were then tested against CA (see above), and compound 22 was found to be most effective at inhibiting BCL9/β‐catenin PPI at 2.1 ± 0.41 μM (Hoggard et al., 2015). Compound 22 was found to inhibit the endogenous transcription of several Wnt target genes (AXIN2, LGR5, LEF1 and CCND1) by qRT‐PCR in a triple negative breast cancer cell line (MDA‐MB‐231) in a dose‐dependent manner (Hoggard et al., 2015). The authors also showed that compound 22 inhibited cell proliferation in colorectal (SW480, HCT116 and HT29) and triple negative breast cancer cell lines (MDA‐MB‐231 and MDA‐MB‐436) with an IC50 of ~2–3 μM (Hoggard et al., 2015). However, the authors did not study this molecule further in an in vivo model, and there is no current evidence that these compounds have advanced beyond the preclinical stage of development.
The Li group used a TOPFlash reporter assay in HEK293 cells treated with Wnt‐3a to screen a library of 4000 compounds. They identified the small molecule NC043 as having the highest inhibitory activity of TOPflash reporter activity and endogenous mRNA levels of Wnt target genes (AXIN2 and SURVIVIN) in Wnt‐dependent colon cancer cell lines (SW480 and Caco‐2) (Wang et al., 2011). This molecule also inhibited tumour growth in a SW480 colon cancer xenograft mouse model in a dose‐dependent manner by up to ~70%. A detailed follow‐up study with the goal of determining the inhibitory mechanism of NC043 found that this molecule covalently binds to CARF (collaborator of ARF), a protein with context‐dependent effects on proliferation (He et al., 2017). Through a series of biochemical and proteomic studies, CARF was found to interact directly with DVL in the nucleus and to potentiate the formation of the TCF/LEF–β‐catenin complex and transcription of Wnt target genes (He et al., 2017). The authors concluded that NC043 could covalently bind to CARF and disrupt its interaction with DVL, which then led to inhibition of β‐catenin‐dependent Wnt signalling in the nucleus. These findings show potential for future clinical studies.
Finally, the molecule that has advanced the most along the bench‐to‐bedside path is a compound that disrupts the interaction of β‐catenin and a ubiquitous transcription co‐activator protein named CBP (for CREB binding protein). To discover this compound, the Kahn group screened a small molecule library of 5000 molecules for the ability to inhibit TOPFlash in SW480 colon cancer cells (Emami et al., 2004). From this library, three molecules showed promising Wnt inhibition, and of these, ICG‐001 was the most potent with an IC50 of 3 μM (Emami et al., 2004). An affinity assay with a biotinylated version of ICG‐001 found that it interacted only with CBP and not its highly related paralogue p300. Further studies using ICG‐001 synthesized with the radioactive isotope C14 found that it specifically inhibited CBP (Emami et al., 2004). The authors demonstrated that ICG‐001 acted as a competitive inhibitor of the β‐catenin : CBP interaction, and when applied in cell culture, could inhibit the expression of Wnt target genes CCND1 and SURVIVIN at the mRNA and protein levels (Emami et al., 2004). Furthermore, the authors showed that ICG‐001 could selectively inhibit the growth of human colon cancer cells (SW480 and HCT116) both in vitro and in vivo (Emami et al., 2004). Prism Pharmaceuticals® recently developed a second generation β‐catenin : CBP inhibitor, PRI‐724, and prepared this molecule for clinical studies in human patients. In a Phase Ia clinical study, PRI‐724 exhibited an acceptable toxicity profile and it could decrease SURVIVIN and BRC5 expression in circulating tumour cells (El‐Khoueiry et al., 2013). Of the 18 patients who were enrolled and treated in the Phase Ia clinical trial, adverse effects were limited to hyperbilirubinaemia (two patients, 11%), diarrhoea (two patients; 11%), hypophosphataemia (two patients, 11%), nausea (one patient, 6%), fatigue (one patient, 6%), anorexia (one patient, 6%), thrombocytopaenia (one patient, 6%) and elevated alkaline phosphatase (one patient, 6%) (El‐Khoueiry et al., 2013). Most recently, PRI‐724 has successfully completed Phase Ib and is proceeding to Phase II clinical trials (NCT02413853) to test the efficacy of this agent in combination with standard cytotoxic chemotherapy and bevacizumab in metastatic colon cancer patients (McWilliams et al., 2015; Duchartre et al., 2016).
As discussed at the beginning of this section, the TCF/LEF–β‐catenin transcriptional complex has been found to be tightly regulated in a context‐dependent manner by different co‐regulatory factors that assemble at WREs in promoters and enhancers of target genes (Figure 1) (Levanon et al., 1998; Waltzer and Bienz, 1998; Hecht et al., 2000; Sun et al., 2000; Takemaru and Moon, 2000; Brantjes et al., 2001; Chodaparambil et al., 2014; Fiedler et al., 2015; van Tienen et al., 2017). These findings have led to a ‘Wnt enhanceosome model’ (Fiedler et al., 2015). In this model, multiple proteins assemble at WREs throughout the genome and use BCL9/B9L as scaffolding proteins to establish an enhanceosome complex. It is this enhanceosome complex that captures newly stabilized, nuclear‐localized β‐catenin. The binding of β‐catenin then triggers structural rearrangements to BCL9, negates TLE repressive functions and activates the TCF/LEF–β‐catenin transcriptional complex and transcription of Wnt target genes (Chodaparambil et al., 2014; Fiedler et al., 2015; van Tienen et al., 2017). Based on this model, one potential strategy for future drug development would be to design small molecule inhibitors that disrupt key protein‐protein interactions that drive the activity of the Wnt enhanceosome complex.
Targeting downstream gene programmes regulated by TCF/LEF–β‐catenin
In addition to directly inhibiting the Wnt signalling pathway itself, downstream Wnt target gene programmes regulated by TCF/LEF–β‐catenin complexes also show potential as therapeutic targets. Interfering with specific Wnt‐controlled gene programmes allows for the targeting of key activities that cancer cells exploit and a strategy that might minimize adverse effects in non‐malignant cells. Wnt signalling has been shown to play key roles in cell proliferation, survival and migration (metastasis), all of which are cellular phenotypes driven by aberrantly active Wnt gene programmes that could prove to be effective drug targets in tumours (Reya and Clevers, 2005; Barker et al., 2009; Nguyen et al., 2009). However, Wnt target genes that drive proliferation such as C‐MYC and CCND1 are often viewed as un‐druggable since they lack enzymatic activity that can be inhibited and because they are required in non‐malignant cells (Musgrove et al., 2011; Rennoll and Yochum, 2015; Koh et al., 2016). Instead, recent efforts have started to investigate whether C‐MYC binding factors and/or downstream C‐MYC targets could be inhibited (Rennoll and Yochum, 2015; Koh et al., 2016). Drug development focused on inhibitors of cyclin‐dependent kinases (CDK) CDK4 and CDK6, rather than directly targeting CCND1, has been successful and led to two FDA‐approved drugs (palbociclib and ribociclib) for patients with advanced breast cancer (Finn et al., 2015, 2016; Turner et al., 2015; Hortobagyi et al., 2016). Whether these drugs are also effective in Wnt‐linked cancer such as colon cancer is a potentially promising possibility that should be explored.
Angiogenesis, which plays an important role in tumour development and maintenance, is another gene programme driven by Wnt signalling (Zhang et al., 2001; Gore et al., 2011). Wnt signalling triggers angiogenesis through activation of VEGFA and the chemokine IL‐8 as well as glycolysis (Zhang et al., 2001; Masckauchán et al., 2005; Schmidt et al., 2009; Gore et al., 2011; Pate et al., 2014). This process has been successfully targeted leading to the development of multiple anti‐angiogenesis agents that are FDA approved and widely used in the clinic (Goel et al., 2012; Jain, 2014). One of the most commonly used anti‐angiogenesis agents is bevacizumab (Avastin®), a VEGF monoclonal antibody that was approved by the FDA in 2004, and is now used in the treatment of multiple cancers known to have overactive Wnt signalling (glioblastoma, colon and non‐small cell lung cancer) (Hurwitz et al., 2004; Sandler et al., 2006; Vredenburgh et al., 2007; Aghajanian et al., 2012).
Recent work has shown that Wnt signalling can also play a direct role in regulating cancer cell metabolism (Lee et al., 2012, 2017; Pate et al., 2014; Sherwood et al., 2014; Sprowl‐Tanio et al., 2016). For example, Wnt signalling controls cancer cell metabolism via the regulation of pyruvate dehydrogenase kinase 1 and monocarboxylate transporter 1 (MCT1/SLC16A1) (Pate et al., 2014; Sprowl‐Tanio et al., 2016). Regulation of both these genes results in cancer cells favouring aerobic glycolysis or Warburg metabolism, which potentiates a unique therapeutic target in Wnt‐high cancers. Additionally, mathematical modelling and in vitro assays show that a combination therapy targeting Wnt signalling and glycolytic activity has a synergistic effect (Lee et al., 2017). Currently, there are at least a dozen cancer metabolism drugs in clinical trials, some of which are promising – such as those targeting isocitrate dehydrogenase (IDH1) (Agios®), glutaminase inhibitor CB‐839 from Calithera®, and fatty acid synthase inhibitor TVB‐2640 from 3‐V Biosciences® (Garber, 2016; Mullard, 2016). Also in early phase clinical trials is AZD3965 from AstraZeneca®, which inhibits the Wnt target and lactate transporter MCT‐1/SLC16A1 (Sprowl‐Tanio et al., 2016). Currently, the biggest hurdle facing clinical trials for many of these metabolism inhibitors are toxicity issues and a lack of appropriate biomarkers to predict disease outcomes (Garber, 2016; Mullard, 2016). These considerations aside efforts to targets cancer metabolism activities that are driven by Wnt signalling could be a promising new therapeutic goal.
Conclusion
In this review, we have summarized recent developments in inhibiting Wnt signalling in the nucleus as a cancer therapy. We focused particularly on inhibitors targeting transcription regulation by interfering with β‐catenin, TCF/LEFs and CBP and also briefly highlighted efforts to target downstream gene programmes that are important for cancer development and progression. Targeting Wnt signalling in the nucleus, and in particular the transcriptional machinery that it directs, is therapeutically difficult, and to date, there has been only one agent (PRI‐724) to proceed into Phase II clinical trials. Continued research into Wnt at the nuclear level and the downstream gene programmes exploited by cancer cells (i.e. proliferation, migration and metabolism) will provide a better understanding of the potential future therapeutic targets, which can hopefully benefit cancer patients.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a, 2015b, 2015c, 2015d).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported by NIH grants (CA096878 and CA108697), California CRCC award CRR‐17‐429379, P30‐CA062203 NIH grant to the Center for Complex Biological Systems and a P30CA062203 to the UC Irvine Chao Family Comprehensive Cancer Center. Y.L. and A.N.H. are supported by CA‐T32 009054 from the National Cancer Institute. The work of G.T.C. and M.L.W. was supported by NIH Grant CA200298. A.N.H. and M.L.W. are supported by NIH grant CA177651. A.N.H. is supported by NSF grant DGE‐1321846. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Science Foundation, National Cancer Institute or the National Institutes of Health.
Lyou, Y. , Habowski, A. N. , Chen, G. T. , and Waterman, M. L. (2017) Inhibition of nuclear Wnt signalling: challenges of an elusive target for cancer therapy. British Journal of Pharmacology, 174: 4589–4599. doi: 10.1111/bph.13963.
Contributor Information
Yung Lyou, Email: ylyou@uci.edu.
Marian L Waterman, Email: mlwaterm@uci.edu.
References
- Aghajanian C, Blank SV, Goff BA, Judson PL, Teneriello MG, Husain A et al (2012). OCEANS: a randomized, double‐blind, placebo‐controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum‐sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J Clin Oncol 30: 2039–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al (2015d). The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M et al (2009). Crypt stem cells as the cells‐of‐origin of intestinal cancer. Nature 457: 608–611. [DOI] [PubMed] [Google Scholar]
- Bodmer WF, Bailey CJ, Bodmer J, Bussey HJ, Ellis A, Gorman P et al (1987). Localization of the gene for familial adenomatous polyposis on chromosome 5. Nature 328: 614–616. [DOI] [PubMed] [Google Scholar]
- Brantjes H, Roose J, van De Wetering M, Clevers H (2001). All TCF HMG box transcription factors interact with Groucho‐related co‐ repressors. Nucleic Acids Res 29: 1410–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadigan KM, Waterman ML (2012). TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb Symp Quant Biol 4: 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network , Muzny DM, Bainbridge MN, Chang K, Dinh HH, Drummond JA et al (2012). Comprehensive molecular characterization of human colon and rectal cancer. Nature 487: 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodaparambil JV, Pate KT, Hepler MRD, Tsai BP, Muthurajan UM, Luger K et al (2014). Molecular functions of the TLE tetramerization domain in Wnt target gene repression. EMBO J 33: 719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H‐J, Huber AH, Weis WI (2006). Thermodynamics of beta‐catenin‐ligand interactions: the roles of the N‐ and C‐terminal tails in modulating binding affinity. J Biol Chem 281: 1027–1038. [DOI] [PubMed] [Google Scholar]
- Daniels DL, Eklof Spink K, Weis WI (2001). beta‐catenin: molecular plasticity and drug design. Trends Biochem Sci 26: 672–678. [DOI] [PubMed] [Google Scholar]
- DeBruine Z, Xu E, Melcher K (2017). Assembly and architecture of the Wnt/β‐catenin signalosome. Br J Pharmacol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Roche M, Rutherford TJ, Gupta D, Veprintsev DB, Saxty B, Freund SM et al (2012). An intrinsically labile α‐helix abutting the BCL9‐binding site of β‐catenin is required for its inhibition by carnosic acid. Nat Commun 3: 680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driehuis E, Clevers H (2017). WNT signalling events near the cell membrane and their pharmacological targeting for the treatment of cancer. Br J Pharmacol 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchartre Y, Kim Y‐M, Kahn M (2016). The Wnt signaling pathway in cancer. Crit Rev Oncol Hematol 99: 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Khoueiry AB, Ning Y, Yang N, Cole S, Kahn M, Zoghbi M et al (2013). A phase I first‐in‐human study of PRI‐724 in patients (pts) with advanced solid tumors. J Clin Oncol 31 no pagination. [Google Scholar]
- Emami KH, Nguyen C, Ma H, Kim DH, Jeong KW, Eguchi M et al (2004). A small molecule inhibitor of beta‐catenin/CREB‐binding protein transcription [corrected]. Proc Natl Acad Sci U S A 101: 12682–12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewan K, Pajak B, Stubbs M, Todd H, Barbeau O, Quevedo C et al (2010). A useful approach to identify novel small‐molecule inhibitors of Wnt‐dependent transcription. Cancer Res 70: 5963–5973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L, Zhu Q, Neuenschwander M, Specker E, Wulf‐Goldenberg A, Weis WI et al (2016). A small‐molecule antagonist of the β‐catenin/TCF4 interaction blocks the self‐renewal of cancer stem cells and suppresses tumorigenesis. Cancer Res 76: 891–901. [DOI] [PubMed] [Google Scholar]
- Fasolini M, Wu X, Flocco M, Trosset JY, Oppermann U, Knapp S (2003). Hot spots in Tcf4 for the interaction with ??‐catenin. J Biol Chem 278: 21092–21098. [DOI] [PubMed] [Google Scholar]
- Fiedler M, Graeb M, Mieszczanek J, Rutherford TJ, Johnson CM, Bienz M (2015). An ancient Pygo‐dependent Wnt enhanceosome integrated by Chip/LDB‐SSDP. Elife 4: https://doi.org/10.7554/eLife.09073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO et al (2015). The cyclin‐dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first‐line treatment of oestrogen receptor‐positive, HER2‐negative, advanced breast cancer (PALOMA‐1/TRIO‐18): a randomised phase 2 study. Lancet Oncol 16: 25–35. [DOI] [PubMed] [Google Scholar]
- Finn RS, Martin M, Rugo HS, Jones S, Im S‐A, Gelmon K et al (2016). Palbociclib and letrozole in advanced breast cancer. N Engl J Med 375: 1925–1936. [DOI] [PubMed] [Google Scholar]
- Garber K (2016). Cancer anabolic metabolism inhibitors move into clinic. Nat Biotechnol 34: 794–795. [DOI] [PubMed] [Google Scholar]
- Goel S, Wong AH, Jain RK (2012). Vascular normalization as a therapeutic strategy. Cold Spring Harb Perspect Med 2: 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonsalves FC, Klein K, Carson BB, Katz S, Ekas LA, Evans S (2011). An RNAi‐based chemical genetic screen identifies three small‐molecule inhibitors of the Wnt/wingless signaling pathway. Proc Natl Acad Sci U S A 108: 5954–5963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore AV, Swift MR, Cha YR, Lo B, McKinney MC, Li W et al (2011). Rspo1/Wnt signaling promotes angiogenesis via Vegfc/Vegfr3. Development 138: 4875–4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham TA, Weaver C, Mao F, Kimelman D, Xu W (2000). Crystal structure of a beta‐catenin/Tcf complex. Cell 103: 885–896. [DOI] [PubMed] [Google Scholar]
- Guthrie ML, Sidhu PS, Hill EK, Horan TC, Nandhikonda P, Teske KA et al (2015). Antitumor activity of 3‐indolylmethanamines 31B and PS121912. Anticancer Res 35: 6001–6007. [PMC free article] [PubMed] [Google Scholar]
- He X, Zhang W, Yan C, Nie F, Li C, Liu X et al (2017). Chemical biology reveals CARF as a positive regulator of canonical Wnt signaling by promoting TCF/β‐catenin transcriptional activity. Cell Discov 3: 17003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht A, Vleminckx K, Stemmler MP, van Roy F, Kemler R (2000). The p300/CBP acetyltransferases function as transcriptional coactivators of beta‐catenin in vertebrates. EMBO J 19: 1839–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoggard LR, Zhang Y, Zhang M, Panic V, Wisniewski JA, Ji H (2015). Rational design of selective small‐molecule inhibitors for β‐catenin/B‐cell lymphoma 9 protein‐protein interactions. J Am Chem Soc 137: 12249–12260. [DOI] [PubMed] [Google Scholar]
- Hortobagyi GN, Stemmer SM, Burris HA, Yap Y‐S, Sonke GS, Paluch‐Shimon S et al (2016). Ribociclib as first‐line therapy for HR‐positive, advanced breast cancer. N Engl J Med 375: 1738–1748. [DOI] [PubMed] [Google Scholar]
- Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W et al (2004). Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 350: 2335–2342. [DOI] [PubMed] [Google Scholar]
- Hwang S‐Y, Deng X, Byun S, Lee C, Lee S‐J, Suh H et al (2016). Direct targeting of β‐catenin by a small molecule stimulates proteasomal degradation and suppresses oncogenic Wnt/β‐catenin signaling. Cell Rep 16: 28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain RK (2014). Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell 26: 605–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin G, Lu D, Yao S, Wu CC, Liu JX, Carson DA et al (2009). Amide derivatives of ethacrynic acid: synthesis and evaluation as antagonists of Wnt/beta‐catenin signaling and CLL cell survival. Bioorg Med Chem Lett 19: 606–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn M (2014). Can we safely target the WNT pathway? Nat Rev Drug Discov 13: 513–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kappel EC, Maurice MM (2017). Molecular regulation and pharmacological targeting of the β‐catenin destruction complex. Br J Pharmacol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh CM, Sabò A, Guccione E (2016). Targeting MYC in cancer therapy: RNA processing offers new opportunities. Bioessays 38: 266–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW et al (1997). Constitutive transcriptional activation by a beta‐catenin‐Tcf complex in APC−/− colon carcinoma. Science 275: 1784–1787. [DOI] [PubMed] [Google Scholar]
- Kramps T, Peter O, Brunner E, Nellen D, Froesch B, Chatterjee S et al (2002). Wnt/wingless signaling requires BCL9/legless‐mediated recruitment of pygopus to the nuclear beta‐catenin‐TCF complex. Cell 109: 47–60. [DOI] [PubMed] [Google Scholar]
- Le PN, McDermott JD, Jimeno A (2015). Targeting the Wnt pathway in human cancers: therapeutic targeting with a focus on OMP‐54F28. Pharmacol Ther 146: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Chen GT, Edwards RA, Wang K, Waterman ML, Lowengrub J (2017). Mathematical modeling links Wnt signaling to emergent patterns of metabolism in colon cancer. Rev 13: 912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Jeon HM, Ju MK, Kim CH, Yoon G, Han SI et al (2012). Wnt/snail signaling regulates cytochrome c oxidase and glucose metabolism. Cancer Res 72: 3607–3617. [DOI] [PubMed] [Google Scholar]
- Lepourcelet M, Chen Y‐NP, France DS, Wang H, Crews P, Petersen F et al (2004). Small‐molecule antagonists of the oncogenic Tcf/beta‐catenin protein complex. Cancer Cell 5: 91–102. [DOI] [PubMed] [Google Scholar]
- Levanon D, Goldstein RE, Bernstein Y, Tang H, Goldenberg D, Stifani S et al (1998). Transcriptional repression by AML1 and LEF‐1 is mediated by the TLE/Groucho corepressors. Proc Natl Acad Sci U S A 95: 11590–11595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Liu JX, Endo T, Zhou H, Yao S, Willert K et al (2009). Ethacrynic acid exhibits selective toxicity to chronic lymphocytic leukemia cells by inhibition of the Wnt/β‐catenin pathway. PLoS One 4: e8294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masckauchán TNH, Shawber CJ, Funahashi Y, Li C‐M, Kitajewski J (2005). Wnt/β‐catenin signaling induces proliferation, survival and interleukin‐8 in human endothelial cells. Angiogenesis 8: 43–51. [DOI] [PubMed] [Google Scholar]
- Masuda T, Ishitani T (2017). JB special review – Wnt signaling: biological functions and its implications in diseases: context‐dependent regulation of the β‐catenin transcriptional complex supports diverse functions of Wnt/β‐catenin signaling. J Biochem 161: 9–17. [DOI] [PubMed] [Google Scholar]
- Mazzoni SM, Fearon ER (2014). AXIN1 and AXIN2 variants in gastrointestinal cancers. Cancer Lett 355: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon AP, Moon RT (1989). Ectopic expression of the proto-oncogene int‐1 in Xenopus embryos leads to duplication of the embryonic axis. Cell 58: 1075–1084. [DOI] [PubMed] [Google Scholar]
- McWilliams RR, Ko AH, Chiorean EG, Kwak EL, Lenz H‐J, Nadler PI et al (2015). A phase Ib dose‐escalation study of PRI‐724, a CBP/beta‐catenin modulator, plus gemcitabine (GEM) in patients with advanced pancreatic adenocarcinoma (APC) as second‐line therapy after FOLFIRINOX or FOLFOX. J Clin Oncol 33: (suppl); abstr e15270. [Google Scholar]
- Mita MM, Becerra C, Richards DA, Mita AC, Shagisultanova E, Osborne CRC et al (2016). Phase 1b study of WNT inhibitor vantictumab (VAN, human monoclonal antibody) with paclitaxel (P) in patients (pts) with 1st‐ to 3rd‐line metastatic HER2‐negative breast cancer (BC). J Clin Oncol 34: 2516.27269942 [Google Scholar]
- Mohan M, Herz H‐M, Takahashi Y‐H, Lin C, Lai KC, Zhang Y et al (2010). Linking H3K79 trimethylation to Wnt signaling through a novel Dot1‐containing complex (DotCom). Genes Dev 24: 574–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullard A (2016). Cancer metabolism pipeline breaks new ground. Nat Rev Drug Discov 15: 735–737. [DOI] [PubMed] [Google Scholar]
- Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL (2011). Cyclin D as a therapeutic target in cancer. Nat Rev Cancer 11: 558–572. [DOI] [PubMed] [Google Scholar]
- Nguyen H, Merrill BJ, Polak L, Nikolova M, Rendl M, Shaver TM et al (2009). Tcf3 and Tcf4 are essential for long‐term homeostasis of skin epithelia. Nat Genet 41: 1068–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehrs C, Acebron SP (2012). Mitotic and mitogenic Wnt signalling. EMBO J 31: 2705–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusse R, Clevers H (2017). Wnt/β‐catenin signaling, disease, and emerging therapeutic modalities. Cell 169: 985–999. [DOI] [PubMed] [Google Scholar]
- Nusse R, Varmus HE (1982). Many Tumors Induced by the Mouse Mammary Tumor Virus Contain a Provirus Integrated in the Same Region of the Host Genome. Cell 31: 99–109. [DOI] [PubMed] [Google Scholar]
- Pate KT, Stringari C, Sprowl‐Tanio S, Wang K, TeSlaa T, Hoverter NP et al (2014). Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J 33: 1454–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polakis P (2012). Wnt signaling in cancer. Cold Spring Harb Perspect Biol 4 pii: a008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poy F, Lepourcelet M, Shivdasani RA, Eck MJ (2001). Structure of a human Tcf4‐beta‐catenin complex. Nat Struct Biol 8: 1053–1057. [DOI] [PubMed] [Google Scholar]
- Rennoll S, Yochum G (2015). Regulation of MYC gene expression by aberrant Wnt/β‐catenin signaling in colorectal cancer. World J Biol Chem 6: 290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reya T, Clevers H (2005). Wnt signalling in stem cells and cancer. Nature 434: 843–850. [DOI] [PubMed] [Google Scholar]
- Rocheleau CE, Downs WD, Lin R, Wittmann C, Bei Y, Cha YH et al (1997). Wnt signaling and an APC‐related gene specify endoderm in early C. elegans embryos. Cell 90: 707–716. [DOI] [PubMed] [Google Scholar]
- Sampietro J, Dahlberg CL, Cho U, Hinds TR, Kimelman D, Xu W (2006). Crystal structure of a β‐catenin/BCL9/Tcf4 complex. Mol Cell 24: 293–300. [DOI] [PubMed] [Google Scholar]
- Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A et al (2006). Paclitaxel‐carboplatin alone or with bevacizumab for non‐small‐cell lung cancer. N Engl J Med 355: 2542–2550. [DOI] [PubMed] [Google Scholar]
- Schmidt M, Sievers E, Endo T, Lu D, Carson D, Schmidt‐Wolf IGH et al (2009). Targeting Wnt pathway in lymphoma and myeloma cells. Br J Haematol 144: 796–798. [DOI] [PubMed] [Google Scholar]
- Sherwood V, Chaurasiya SK, Ekström EJ, Guilmain W, Liu Q, Koeck T et al (2014). WNT5A‐mediated ß‐catenin‐independent signalling is a novel regulator of cancer cell metabolism. Carcinogenesis 35: 784–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: Towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprowl‐Tanio S, Habowski AN, Pate KT, McQuade MM, Wang K, Edwards RA et al (2016). Lactate/pyruvate transporter MCT‐1 is a direct Wnt target that confers sensitivity to 3‐bromopyruvate in colon cancer. Cancer Metab 4: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Kolligs FT, Hottiger MO, Mosavin R, Fearon ER, Nabel GJ (2000). Regulation of beta ‐catenin transformation by the p300 transcriptional coactivator. Proc Natl Acad Sci 97: 12613–12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada K, Zhu D, Bird GH, Sukhdeo K, Zhao J‐J, Mani M et al (2012). Targeted disruption of the BCL9/β‐catenin complex inhibits oncogenic Wnt signaling. Sci Transl Med 4: 148ra117–148ra117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemaru KI, Moon RT (2000). The transcriptional coactivator CBP interacts with beta‐catenin to activate gene expression. J Cell Biol 149: 249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson B, Townsley F, Rosin‐Arbesfeld R, Musisi H, Bienz M (2002). A new nuclear component of the Wnt signalling pathway. Nat Cell Biol 4: 367–373. [DOI] [PubMed] [Google Scholar]
- Tian W, Han X, Yan M, Xu Y, Duggineni S, Lin N et al (2012). Structure‐based discovery of a novel inhibitor targeting the beta‐catenin/Tcf4 interaction. Biochemistry 51: 724–731. [DOI] [PubMed] [Google Scholar]
- van Tienen LM, Mieszczanek J, Fiedler M, Rutherford TJ, Bienz M (2017). Constitutive scaffolding of multiple Wnt enhanceosome components by Legless/BCL9. Elife 6: 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsley FM, Thompson B, Bienz M (2004). Pygopus residues required for its binding to Legless are critical for transcription and development. J Biol Chem 279: 5177–5183. [DOI] [PubMed] [Google Scholar]
- Turner NC, Ro J, André F, Loi S, Verma S, Iwata H et al (2015). Palbociclib in hormone‐receptor–Positive advanced breast cancer. N Engl J Med 373: 209–219. [DOI] [PubMed] [Google Scholar]
- Vredenburgh JJ, Desjardins A, Herndon JE, Marcello J, Reardon DA, Quinn JA et al (2007). Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol 25: 4722–4729. [DOI] [PubMed] [Google Scholar]
- Waltzer L, Bienz M (1998). Drosophila CBP represses the transcription factor TCF to antagonize Wingless signalling. Nature 395: 521–525. [DOI] [PubMed] [Google Scholar]
- Wang W, Liu H, Wang S, Hao X, Li L (2011). A diterpenoid derivative 15‐oxospiramilactone inhibits Wnt/β‐catenin signaling and colon cancer cell tumorigenesis. Cell Res 21: 730–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis TG, Zalcberg IR, Coignet LJ, Wlodarska I, Stul M, Jadayel DM et al (1998). Molecular cloning of translocation t(1;14)(q21;q32) defines a novel gene (BCL9) at chromosome 1q21. Blood 91: 1873–1881. [PubMed] [Google Scholar]
- Xu W, Kimelman D (2007). Mechanistic insights from structural studies of beta‐catenin and its binding partners. J Cell Sci 120: 3337–3344. [DOI] [PubMed] [Google Scholar]
- Zhan T, Rindtorff N, Boutros M (2016). Wnt signaling in cancer. Oncogene 4 pii: a008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Gaspard JP, Chung DC (2001). Regulation of vascular endothelial growth factor by the Wnt and K‐ras pathways in colonic neoplasia. Cancer Res 61: 6050–6054. [PubMed] [Google Scholar]
- Zimmerli D, Hausmann G, Cantù C, Basler K (2017). Pharmacological interventions in the Wnt pathway: inhibition of Wnt secretion versus disrupting the protein‐protein interfaces of nuclear factors. Br J Pharmacol 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]