

Key Points
High CD123 expression increases proliferation and results in enhanced survival in response to low concentration of IL-3 in vitro.
High CD123-expressing LSCs downregulate chemokine receptor expression, affecting niche interactions.
Abstract
High expression of the α chain of the interleukin-3 receptor (IL-3Rα; CD123) is a hallmark of acute myeloid leukemia (AML) leukemic stem cells (LSCs). Elevated CD123 expression is part of the diagnostic immunophenotyping of myeloid leukemia, and higher expression is associated with poor prognosis. However, the biological basis of the poorer prognosis is unclear, and may include heightened IL-3 signaling and non–cell autonomous interactions with the bone marrow (BM) microenvironment. We used TF-1 cells expressing different levels of CD123 and found elevated CD123 levels amplified the proliferative response to exogenous IL-3 and maintained viability in reducing IL-3 concentrations. This was associated with stronger activation of STAT5, Akt, and extracellular signal-regulated kinase 1/2 in vitro. Surprisingly, in vivo e14.5 fetal liver cells transduced with retroviral constructs to express high CD123 failed to engraft in syngeneic recipients. In exploring the underlying mechanism for this, we found that CXCR4, a key molecule involved in LSC/BM interactions, was specifically downregulated in CD123 overexpressing cells in a manner dependent on IL-3 signaling. CXCR4 downregulation was sufficient to alter the chemotactic response of hematopoietic cells to stromal derived factor-1 (SDF-1). Thus, we propose that the overexpression of CD123 in AML LSC dictates their location by altering CXCR4/SDF-1 interaction in the BM, raising the possibility that this mechanism underpins the egress of BM AML LSC and more mature cells into the circulation.
Visual Abstract
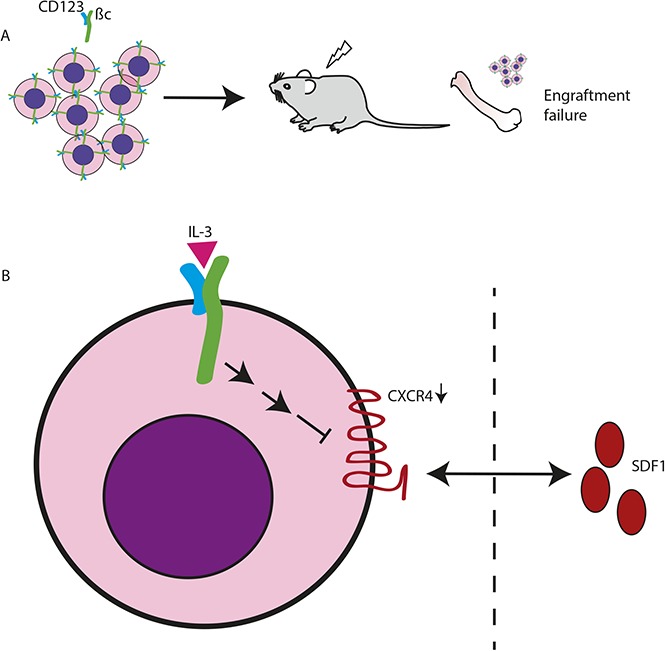
Introduction
Serial transplantation of acute myeloid leukemia (AML) patient cells into immunodeficient mice provided some of the first proof of the existence of leukemic stem cells (LSCs).1-3 The developmental hierarchy of leukemic hematopoietic cells mimics that of normal hematopoiesis; however, LSCs possess enhanced self-renewal activity and a characteristic expression profile of surface markers. One such marker is CD123, the α subunit of the receptor for interleukin-3 (IL-3R).4-6 We and others have shown that CD123 is a biomarker for LSCs,7-11 and monoclonal antibodies targeted against CD123 are currently in clinical trials for AML treatment.12-15 However, the functional consequence of elevated CD123 expression in vitro and in vivo remains unknown. Correlations between higher expression of CD123 on AML cells and increased IL-3–dependent STAT5 activation, increased proliferation, a more primitive immunophenotype, and resistance to apoptosis have all been described,8,16,17 but a cause-and-effect relationship has not been established. Clinically, high CD123 expression in AML is associated with higher blast counts at diagnosis, lower complete remission rates, and lower 5-year survival.16 IL-3 and, to a lesser extent, other cytokines, are an integral part of AML growth and survival, with some reports supporting AML autocrine or paracrine secretion of cytokines.18-20 The specific role of enhanced IL-3R signaling, driven by high CD123 expression, may play a role in driving leukemogenesis; however, compelling evidence has been missing.
IL-3R signaling maintains hematopoietic cell viability and stimulates proliferation.21 IL-3Rs are present on a range of early and more mature hematopoietic progenitors and, within the committed cell populations, on both myeloid and lymphoid lineages.22 IL-3 initially binds to CD123 and subsequently recruits βc (CD131) to form the high-affinity receptor. This results in activation of a series of signaling pathways including the JAK/STAT, Ras-MAPK, and phosphatidylinositol 3-kinase pathways.23 Although the βc subunit is absolutely required for IL-3R signaling, the much shorter cytoplasmic domain of CD123 is also critical for signal transduction. We have previously reported that deletion of the cytoplasmic domain of CD123 does not affect ligand binding, but abolishes IL-3 signaling,24 whereas others have similar findings for the intracellular domain of granulocyte-macrophage colony-stimulating factor receptor-α (GM-CSFRα)25 and IL-5Rα.26 These receptors all contain a conserved membrane proximal proline-rich motif, similar to the Box 1 region of βc. The IL-5Rα is necessary for tyrosine phosphorylation of a number of cellular proteins including βc, SH2/SH3-domain containing proteins, and JAK2 kinase.27,28 Mutation of Pro352 and Pro355 of this motif showed it to be crucial for cell proliferation and activation of JAK2/STAT5.27 This proline-rich motif in GM-CSFRα has been shown to induce phosphatidylinositol 3-kinase signaling.29,30 These mutant receptors thus represent unique opportunities to distinguish between IL-3 binding and signaling.
It is widely believed that hematopoietic stem cells (HSCs), both normal and malignant, reside in specific regions in the bone marrow (BM) in complex multicellular microenvironment referred to as BM niches.31-33 Multiple factors have been reported as being essential for the homing and retention of HSC/LSC into the niche,34 particularly the interaction between CXCR4 and SDF-1,35 and the growth factor receptor c-kit.36 The question of competition between normal and malignant cells for the protective niche environment in the BM is hotly debated. Functional analysis of competition between normal and malignant cells showed that 1 or the other, but not both, can reside in the niche and that the presence of normal HSC can limit the expansion of leukemic cells.37 However, the molecular mechanisms underlying this is not understood.
We present data that elevated CD123 expression enhances IL-3R signaling to promote proliferation and cell viability. We also show, for the first time, that regulation of CXCR4 expression is mediated by IL-3/CD123 signaling. Enhanced IL-3R signaling resulting from high CD123 expression reduced CXCR4 RNA and protein expression. As a consequence, the chemotactic migration of hematopoietic cells toward SDF-1 decreased in vitro. Downregulation of CXCR4 may contribute to failure of CD123-expressing BM cells to engraft in lethally irradiated recipients. Thus, our data support the hypothesis that high CD123 expression plays a pathogenic role in AML and raise the possibility that downregulation of CXCR4 is at least 1 mechanism by which AML cells, particularly high CD123-expressing LSC, leave the BM. This has important implications for therapies in current trials with anti-CD123 antibodies.
Materials and methods
Patient samples
Peripheral blood (PB) and BM samples from AML patients were obtained after informed consent in accordance with protocols approved by the Human Research Committee of SA Pathology/Royal Adelaide Hospital. See Table 1 for clinical information for patients used in this study. Mononuclear cells were isolated by density gradient centrifugation and cryopreserved for storage. Cells were thawed in Iscove modified Dulbecco medium with 20% fetal calf serum (FCS; Bovogen, Victoria, Australia) and DNase (50 U/mL; Sigma-Aldrich, Australia) and cultured for 24 hours ± 10 ng/mL human IL-3.38
Table 1.
Clinical features of AML patient samples and quantified expression of CD123
| Patient legend | CD123 − IL-3 | CD123 + IL-3 | Age at dx (y) | Sex | FAB | BM blast% | WCC at dx (109/L) | FLT3 mutation | NPM1mutation | Simple cytogenetics | Detailed cytogenetics |
|---|---|---|---|---|---|---|---|---|---|---|---|
| AML363 | NA | NA | 77 | M | M1 | 96 | 46.30 | FLT3-ITD | NPM1 | Normal | Normal |
| AML266 | 157 | 204 | 55 | F | M4 | 39 | 67.10 | FLT3-ITD | Negative | Normal | Normal |
| AML243 | 328 | 503 | 32 | M | M5 | 90 | 55 | Negative | Negative | t(9;11), 11q23, MLL | 46,XY,t(9;11)(p22;q23)[20] |
| AML418 | NA | NA | 81 | M | M5 | 92 | 178 | Negative | Negative | ?MLL, -Y | 45,X,-Y,ins(11;10)(q23;p11.2p11.2)[18]/46,XY[3].ish ins(11;10)(5KMT2Adim+;3KMT2A+,5KMT2A+).nuc ish(5KMT2Ax3,3KMT2Ax2)(5KMT2A con 3KMT2A ×1)(5KMT2Adim con 3KMT2A ×1)[193/200] |
| AML428 | 68 | 74 | 78 | M | M4 | 67 | 68.1 | Negative | Negative | der(11) | 47,XY,+der(11)(11pter->11p15::?::cen::?::11q14-22->11qter)[5]/46,XY[10].nuc ish(KMT2Ax4)[119/200] |
| AML269 | 17 348 | 23 689 | 56 | M | M4 | 75 | 132 | FLT3-ITD | Negative | Normal | Normal |
| AML057 | 163 | 216 | 64 | F | M5 | 36 | 43.3 | Negative | NPM1 | Normal | Normal |
| AML264 | 1 333 | 1 763 | 64 | F | M1 | 100 | 111.1 | Negative | negative | del(16q) | 46,XX,del(16)(q22)[20].nuc ish(CBFBx1)[193/200] |
| NA | 3 317 | 4 995 | 74 | M | NA | 22* | NA | Negative | negative | tri 14 | 47XY, +14 |
| AML380 | 7 294 | 15 328 | 59 | F | M2 | 63 | 119 | FLT3-ITD | NPM1 | Normal | Normal |
| AML419 | 1 807 | 1 577 | 56 | F | M1 | 89 | 57.10 | Negative | Negative | Normal | Normal |
| AML101 | 7 294 | 15 328 | 48 | M | M1 | 99 | 98.10 | Negative | Negative | 46,XY,del(11)(q23)[20].ishdel(11)(5′MLL+,3′MLL-) | 46,XY,del(11)(q23)[20].ish del(11)(5′MLL+,3′MLL-) |
| AML063 | NA | NA | 69 | F | M1 | 98 | 107.20 | FLT3-ITD | Negative | Normal | Normal |
| AML427 | 111 | 257 | 59 | M | M5 | 72 | 8.68 | NA | NA | Normal | Normal |
| AML379 | 857 | 339 | 63 | F | M1 | 90 | 35.20 | Negative | Negative | Normal | Normal |
| AML235 | 3 100 | 3 195 | 28 | F | M2 | 73 | 18.30 | Negative | Negative | t(6;9) | 46,XX,t(6;9)(p23;q34) |
| FL CD123-low (EV) | 277 | 292 | |||||||||
| FL CD123-high-WT | 10 402 | 11 526 |
dx, Diagnosis; F, female; M, male; NA, not available.
Diagnosis from PB blast percentage.
Flow cytometry
Surface antigen expression was determined using antibodies as shown in supplemental Table 1. Live cells were analyzed by flow cytometry using BD LSR Fortessa cell analyzer (BD Biosciences). Analysis of flow cytometry data were performed using FCS Express software (De Novo Software, Glendale, CA).
Cloning and lentiviral/retroviral production
Human CD123 was cloned into the Spe1 and Xho1 sites of pLenti-IRES-eGFP.39 TF-1 cells40 were engineered to overexpress human CD123 (CD123-high-WT), as previously described,39 and sorted on GFP. An empty vector control line (EV/CD123-low) was generated using pLenti-IRES-eGFP.
pMSCV IRES Mus musculus CD123 eGFP or pMSCV IRES CXCR4 mCherry retroviral vectors were transfected into HEK293T cells with the ecotropic pEQ packaging plasmid using Lipofectamine 2000 (Life Technologies). Target cells were infected with the viral supernatant as previously described.41 Quikchange mutagenesis as per manufacturer’s instructions was used to replace the proline residues of CD123 with alanine in the ARIA and APIA constructs.
Cellular studies
Proliferation was assessed using tritiated thymidine incorporation42 and normalized to maximum growth. Survival was determined by Annexin V negative staining.20 For competition, assays cells were set up with known ratios of CD123-high-WT to CD123-low (EV or parental cells). The ratio of the 2 cell types were monitored by flow cytometry for GFP or CD123.43 The rate of ligand binding was determined as previously described.44 For colony assays, 5000 transduced fetal liver (FL) cells were plated in 1 mL methylcellulose (Stem Cell Technologies) supplemented with growth factors. Colonies of 50 or more cells were counted at 7 days postplating. Receptor complex affinity for IL-3 was measured by Scatchard analysis. Cells were incubated with 125I-labeled IL-3 over a concentration range of 10 pmol/L to 10 nmol/L. Dissociation constants were calculated using KELL software.
Immunoprecipitations and immunoblot analysis
Cells were deprived of growth factors and stimulated as shown in the figure legends, lysed in NP-40 lysis buffer45 for immunoprecipitations, and processed as previously described.45 Signals were developed using enhanced chemiluminescence (Amersham Pharmacia, Piscataway, NJ; or West Dura; Pierce, Rockford, IL).
Murine engraftment studies
Animal studies were performed under the institutional guidelines approved by the SA Pathology/Central Adelaide Local Health Network Animal Ethics Committee. FL cells (e14.5) from C57BL/6 CD45.1 (B6.SJL-PtprcaPep3b/BoyJArc) mice infected with viral supernatant were cultured in α-MEM medium (Sigma-Aldrich, St Louis, MO) supplemented with 10% FCS, 50 μM β-mercaptoethanol (Sigma-Aldrich), 10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES; Life Technologies), 100 ng/mL mouse stem cell factor (SCF; produced in-house), 10 ng/mL human IL-6, 50 ng/mL mTpo, and 10 ng/mL mFlt-3L (all from Shenandoah Biotechnology, Warwick, PA). Forty-eight hours after retroviral infection, 3 × 106 murine FL cells were injected into γ-irradiated (9 or 10 Gy) C57BL/6 CD45.2 female mice via tail vein.
Mice were maintained in sterile conditions and monitored daily. Tissues from surviving mice were analyzed as noted in the figures.
Real-time quantitative PCR
RNA was isolated from samples using the Qiagen RNeasy Plus Micro kit. Reverse transcription was performed using QuantiTect Reverse Transcription kit and real-time polymerase chain reaction (RT-PCR) reactions were carried out using the QuantiTect SYBR Green PCR kit (both Qiagen).
Chemotaxis assay
Chemotaxis assays with murine FL cells were performed using transwell inserts (6.5 mm diameter, 5 μm pore, Corning, NY). A total of 100 μL of chemotaxis buffer (α-MEM + 2% FCS + 50 μM βME+ 10 mM HEPES) containing 5 × 105 cells were loaded into the upper chamber and 600 μL of Chemotaxis buffer alone or with 10 ng/mL SDF-1, 100 ng/mL SDF-1, or 10% FCS were loaded into the bottom well. After 4 hours, 50 μL of Flow Count Fluorospheres (Beckman Coulter) was added to the cells from the bottom chamber and the number of migrated cells/10 000 fluorospheres counted using the BD LSR Fortessa cell analyzer. Migration is calculated as a percentage of the cell count in the input sample/10 000 fluorospheres. For human AML samples, the method was similar, but 96-well plates with 5-μm pore were used. BM cells were thawed, incubated overnight in IL-3 (10 ng/mL), and then plated. Postmigration cells were stained for CD34, CD38, and CD123. Chemotaxis buffer for human cells (RPMI 1640 with 0.5% bovine serum albumin and 20 mM HEPES) was used to dilute SDF-1 (100 ng/mL) and human serum (10% final) as a positive control. Events were collected relative to beads as described previously. Migration of LSC with high vs low CD123 was plotted.
Statistics
Comparisons between 2 groups were made using Student t tests and between multiple groups using analysis of variance. The correlation plots were made using the “Corrplot” package in R.
Results
Elevated expression of CD123 enhances IL-3–dependent proliferation and viability
Elevated expression of CD123 on AML LSC is associated with poorer overall survival and aggressive disease. We hypothesized that higher CD123 expression may increase the sensitivity of these cells to cytokine stimulation, resulting in a pathological response to IL-3 stimulation. The erythroleukemic cell line, TF-1, which requires IL-3 or GM-CSF for survival, was transduced with a lentivirus encoding human CD123 and GFP. This resulted in elevated cell surface expression of CD123, whereas CD131 expression remained unchanged (supplemental Figure 1A). CD123 overexpressing cells (CD123-high-WT) proliferated (Figure 1A) or survived (Figure 1B) in 10 to 100× less human IL-3 than control cells while maintaining normal response to human GM-CSF. In vitro competitive assays showed that the CD123-high-WT cells rapidly outgrew the control cells in IL-3 (Figure 1C). Antiphosphotyrosine western blots showed more tyrosine phosphorylation at lower IL-3 concentrations in CD123-high-WT compared with control cells (Figure 1D). Specific phosphorylation of extracellular signal-regulated kinase 1/2 (Erk1/2), Akt, and STAT5 was also detected at lower concentrations of IL-3, but control and CD123-high-WT were similarly responsive to GM-CSF (Figure 1E). Analysis of 125I-labeled IL-3 binding to the IL-3R showed a significantly more rapid rate of IL-3 ligand binding in CD123-high-WT cells than in CD123-low-WT cells (supplemental Figure 1B). Consistent with this observation, a time course of IL-3 stimulation showed a more rapid phosphorylation of STAT5 in CD123-high-WT cells (supplemental Figure 1C). To confirm that similar enhancement of signaling was seen in murine cells, we transduced murine FL-derived cell lines to overexpress murine CD123 (or an empty vector; supplemental Figure 1D) and then repeated the proliferation (supplemental Figure 1E), in vitro competitive growth assay (supplemental Figure 1F), and phosphorylation of Erk1/2, Akt, and STAT5 (supplemental Figure 1G). Together, these results demonstrate that elevation of CD123 levels sensitizes all cells to IL-3 stimulation, promoting enhanced survival and proliferation.
Figure 1.

Increased expression of CD123 is associated with enhanced proliferation and IL-3R signaling in response to low concentrations of IL-3. TF-1.8 CD123-low (EV) or CD123-high-WT cells were starved of cytokine for 16 hours. A total of 105 cells were stimulated in a titration of human IL-3 (Ai) or GM-CSF (Aii) for 48 hours. Proliferation was measured by the addition of 0.5 µCi of 3H-thymidine/well for the final 5 hours of stimulation. Data are represented as the mean counts per minute ± standard deviation and is representative of 3 experiments. (B) Cells were washed and plated out at 5 × 105 cells/mL in medium containing IL-3 as shown on graph. After 48 hours, cells were costained with annexin V and propidium iodide and analyzed by flow cytometry. The average of 2 experiments each with triplicate samples with the standard error is indicated. (C) Cells were combined in a 99:1 ratio (99% TF-1.8 parental, 1% TF-1.8 CD123-high-WT) and cultures maintained in 0.1 ng/mL of IL-3 (Ci) or GM-CSF (Cii). The relative proportion of each population was monitored by flow cytometry. (D) Cells were starved of cytokine in media containing 0.5% FBS for 16 hours and stimulated with a titration of human IL-3 (Di) or GM-CSF (Dii) for 10 minutes at 37°C. The cells were lysed and human βc was immunoprecipitated, run on sodium dodecyl sulfate-polyacrylamide gel electrophoresis and the phosphorylation of the tyrosine residues on the βc analyzed by western blotting, probing with anti-phospho-tyrosine mAb (clone 4G10). (E) Cells were starved and stimulated with IL-3 (Ei) or GM-CSF (Eii) as before. Whole cell lysates were run on sodium dodecyl sulfate-polyacrylamide gel electrophoresis and western blotted. Blots were probed with antibodies to phosphorylated STAT-5 (Tyr694), Erk1/2 (Thr202/Tyr204), and Akt (Ser473) then reprobed with α-tubulin as a loading control. Statistical analysis: #nonsignificant difference, *P < .05, **P < .01, ***P < .001. Solid blue lines/bars, CD123-low (EV or parental cells); hashed or solid red lines/bars, CD123-high-WT.
High expression of CD123 prevents engraftment of hematopoietic cells in vivo.
We next investigated whether overexpression of murine CD123 on murine HSC, transplanted into syngeneic recipients, would result in myeloproliferative disease in vivo. E14.5 FL cells infected with retrovirus encoding either an EV or CD123-low or murine CD123 (CD123-high-WT) were injected into lethally irradiated mice (Figure 2A). The donor cells were characterized for expression of CD123 and GFP (Figure 2C) and viability (Figure 2D). Mice engrafted with CD123-high-WT cells died within 10 to 20 days posttransplant, with pale extremities and hypocellularity in the circulation, BM, and lymphoid tissues, consistent with irradiation sickness and indicative of engraftment failure. In contrast, mice infused with control cells survived until the end of the experiment (Figure 2B). At week 6 posttransplant, surviving mice demonstrated high CD45.2/GFP expression in BM, indicating their donor origin (supplemental Figure 2A). To determine whether high CD123 cells could engraft in a competitive transplant model, we transplanted lethally irradiated recipient mice with equal numbers of CD123-low (EV) and CD123-high-WT cells, all expressing GFP. These mice were allowed to engraft over 2 to 8 weeks and analyzed for GFP and CD123 expression. CD123-low (EV)-transduced cells constituted the majority of transplanted cells in the BM and spleen. The number of CD123-high-WT cells in the BM declined over time, such that no transplanted animals had this population by 8 weeks (supplemental Figure 2B). Thus we conclude that cells with high expression of murine CD123 inefficiently engrafted and over time were lost. To determine whether malignant cells underwent the same fate as normal HSC, we transduced FL cells with the HoxA9 gene in combination with either EV or murine CD123-high-WT constructs previously described. Mice engrafted with HoxA9 and murine IL-3 had accelerated disease, whereas mice with HoxA9 and high CD123 showed no difference to HoxA9 alone (Figure 2E). Analysis of white cell count (Figure 2F) and spleen weight (Figure 2G) confirmed that high CD123 had not exacerbated the phenotype. Analysis of the recipient mice showed the CD123-high-WT cells were not detected and engrafted donor AML cells had endogenous receptor levels (supplemental Figure 2C).
Figure 2.

Overexpression of CD123 prevents FL engraftment. (A) Experimental design of engraftment studies: 3 × 106 CD45.1 e14.5 murine FL cells transduced to express either low CD123 (EV) or overexpress CD123 (CD123-high-WT) were transplanted into lethally irradiated recipient mice. (B) Survival of mice engrafted with CD123-low cells (n = 8) or CD123-high-WT cells (n = 8). Mean survival of CD123-high-WT was 10.75 ± 1.16 days; CD123-low mice were humanely euthanized at 6 weeks after engraftment. Solid lines/bars, CD123-low (EV); hashed lines/bars, CD123-high-WT. (C) Flow cytometric analysis for GFP and CD123 expression on input cells injected into mice for engraftment studies (i) CD123-low (EV) and (ii) CD123-high-WT. (D) Viability of murine FL cells transduced to express CD123-low (EV) or CD123-high-WT assessed using Fixable Viability Stain 780 (FVS 780). Red bars, total cell population; blue bars, GFP +ve (transduced) cells only. The average of 3 experiments with the standard error is indicated. (E) Survival of mice engrafted with EV (purple hashed line; n = 6), HoxA9-transduced cells alone (dotted and dashed red line; n = 5), HoxA9 + high CD123 (solid light green line; n = 3), or HoxA9 + IL-3 (solid blue line; n = 6). White blood cell count (F) and spleen weight (G) for mice engrafted with EV, HoxA9-transduced cells alone, HoxA9 + high CD123, or HoxA9 + IL-3 at time of analysis as in panel E.
Proline residues in the cytosolic domain of CD123 are essential for IL-3R signaling.
To separate the binding activity of IL-3 to CD123 from the signaling properties associated with high expression of this receptor subunit, we constructed a mutant that could bind IL-3 with normal affinity and kinetics but not initiate receptor signaling. Previous studies identified proline residues in the membrane proximal region of the α chains of IL-5 and GM-CSF are required for signal transduction from these receptors. The equivalent region in CD123 with the motif 336PRIP339 (Figure 3A) was mutated, replacing both proline residues with alanine. This mutant receptor was expressed on immortalized murine FL cells at levels equivalent to CD123-high-WT and bound IL-3 with similar kinetics (Kd 689pM CD123-WT and 634pM CD123-ARIA). However, the mutant receptor was unable to support proliferation of cells in response to human IL-3 (Figure 3B). Proliferation in response to SCF was unaffected by expression of mutant CD123, indicating cells could still respond to an unrelated cytokine with a functional receptor (supplemental Figure 3A). The CD123-ARIA mutation abolished Akt, Erk1/2, and STAT5 phosphorylation in response to IL-3 stimulation (Figure 3C). We also validated the analogous mutant in murine CD123 (Figure 3D) using the same assays. In murine cells, expression of CD123-high-WT but not the CD123-high-APIA constructs enhanced IL-3–dependent proliferation (Figure 3E) and signal transduction (Figure 3F). Expression profiles for CD123 on the murine cells are shown in supplemental Figure 3B.
Figure 3.

Mutation of 2 intracellular proline residues in CD123 prevents IL-3 signaling. (A) Site-directed mutagenesis of CD123 was used to generate proline to alanine substitutions (336 PRIP 339 → ARIA). (B) Cells expressing huβc and CD123-high-WT or CD123-high-ARIA were starved of cytokine for 6 hours and then stimulated in a titration of human IL-3 for 48 hours. Proliferation was measured by the addition of 0.5 µCi of 3H-thymidine/well for the final 5 hours of stimulation. Data are represented as percentage of maximum proliferation in 100 ng/mL mouse SCF ± standard error of the mean (SEM), n = 3. (C) Cells were starved in media containing 0.5% serum for 6 hours and then stimulated ± 100 ng/mL huIL-3 for 10 minutes at 37°C. Western blotting was as previously described in Figure 1. (D) Site-directed mutagenesis of murine CD123 was used to generate proline to alanine substitutions (365 PPIP 368 → APIA). (E) WT FL cells expressing murine CD123-high-WT or CD123-high-APIA were starved of cytokine and then stimulated with a titration of murine IL-3. Proliferation was measured as previously. Data are represented as percentage of maximum proliferation of the EV line in 1000 ng/mL IL-3 ± SEM, n = 3. (F) Cells were starved and stimulated with a titration of murine IL-3 for 10 minutes at 37°C. Western blotting was as described previously. Statistical analysis: #nonsignificant difference, *P < .05, **P < .01, ***P < .001. Solid blue lines/bars, EV; hashed or solid red lines/bars, CD123-high-WT; dotted or solid green lines/horizontal bars, CD123-high-APIA.
We proceeded to compare cells with high expression of CD123-high-WT and CD123-high-APIA transplanted into irradiated mice to establish whether high CD123 was sufficient to inhibit engraftment, or whether IL-3R signaling was required. Mice were housed for 7 weeks and donor cells in BM, spleen, and PB analyzed by flow cytometry. We monitored transduced cells both by GFP expression (Figure 4A) and total donor cell population using CD45.1 (Figure 4B). Because high GFP expression in the CD123-high-APIA but not CD123-high-WT cells was observed, we restained BM for CD123. Analysis revealed that CD123-high-APIA cells remained in the BM of engrafted mice, but CD123-high-WT cells did not (Figure 4C). Analysis of the PB showed similar distribution of CD45.1 and GFP-expressing cells as the BM (Figures 4D-E). These data show that IL-3R signaling contributes to the failure of engraftment.
Figure 4.

Cells with high expression of WT but not signaling-defective CD123 have impaired engraftment in mice. A total of 3 × 106 CD45.1 e14.5 murine FL cells transduced to express EV (CD123-low) and overexpress CD123 WT or mutant (CD123-high-WT or CD123-high-APIA) were transplanted into lethally irradiated recipient CD45.2 mice and allowed to engraft for 6 weeks. Flow cytometric analysis (A) transduced donor cells in recipient mouse BM using GFP, (B) total donor cells in recipient mouse BM using antibody to CD45.1, and (C) percentage of cells in BM with high expression of CD123. Total donor cell content of recipient mice was analyzed in peripheral blood (D) and spleen (F) and GFP expression of transduced cells in the circulation (E) and spleen (G). Each symbol represents an individual mouse. Statistical analysis: #nonsignificant difference, *P < .05, **P < .01, ***P < .001.
High CD123 expression correlates with reduced levels of CXCR4 mRNA and protein
Next, we sought a mechanism to explain the failure of engraftment of CD123 high cells. We focused on adhesion proteins reported to be required for homing of cells into the BM niche. E14.5 FL cells from C57BL/6 mice, transduced with CD123-high-WT or CD123-high-APIA, were purified and the messenger RNA (mRNA) of the adhesion proteins measured using RT-PCR after IL-3 stimulation. Notably, the chemokine receptor CXCR4 (Cxcr4) showed significant downregulation in response to IL-3 stimulation in CD123-high-WT cells. We observed minimal changes in gene expression in response to IL-3 for integrins β1 (Itgb1), α4 (Itga4), and α5 (Itga5) and PSGL1 (Selplg) (Figure 5A). LFA-1 (Lfa1) was marginally increased in response to IL-3 stimulation, but in cells transduced with EV and CD123-high-WT. The c-kit receptor (c-kit) showed significant downregulation in response to IL-3 stimulation, but also was not restricted to CD123-high-WT cells. Flow cytometric analysis showed surface CXCR4 protein expression was downregulated in the CD123-high-WT cells (Figure 5B). CD123-high-APIA cells showed no downregulation of CXCR4 mRNA (Figure 5C) or protein expression (data not shown) upon IL-3 stimulation. Thus we conclude that enhanced IL-3 signaling in CD123-high-WT cells is associated with downregulation of CXCR4.
Figure 5.

Overexpression of CD123 downregulates CXCR4 gene and protein expression in response to IL-3 and reduces chemotactic responses to SDF-1. Cells were cultured ±10 ng/mL mIL-3 for 48 to 72 hours posttransduction. (A) Gene expression of a panel of adhesion molecules and chemotactic receptors using RT-PCR. Expression is presented as fold change in response to IL-3 ± SEM, n = 3. (B) Flow cytometry analysis of surface CXCR4 expression reveals specific downregulation in cells overexpressing CD123 in response to IL-3 (black, negative; blue, CD123-high-WT no IL-3; red, +CD123-high-WT + IL-3). (C) CXCR4 gene expression changes in response to IL-3 were analyzed in FL containing CD123-low (EV), CD123-high-WT, or CD123-high-APIA. Data were normalized to β-actin and represented as foldchange in response to IL-3 stimulation ± SEM, n = 3. (D) TF-1 cells were cultured in IL-3 or GM-CSF and expression of CXCR4 measured by flow cytometry (black, negative; blue, no IL-3; red, +IL-3). (E) SDF-1–induced migration demonstrates reduced chemotactic response with CD123 overexpression, which can be rescued by addition of muCXCR4. Migration was analyzed using a transwell migration assay and migrated cells were counted using flow cytometry and fluorospheres. Data are normalized to a blank sample and are presented as mean migration relative to 10% FBS ± SEM, n = 3. Statistical analysis: #nonsignificant difference, *P < .05, **P < .01, ***P < .001.
We confirmed the regulation of CXCR4 by CD123 in human cells. TF-1 cells cultured in either IL-3 or GM-CSF for 48 hours were assayed for CXCR4 by flow cytometry (Figure 5D) and RT-PCR (supplemental Figure 5). The downregulation of CXCR4 was specific to stimulation with IL-3. Because GM-CSF and IL-3 signal through the same β-common chain, these data indicate that the differential CXCR4 expression specifically requires CD123.
To determine if this level of downregulation of CXCR4 expression is sufficient to alter the cells response to its ligand SDF-1, we performed chemotaxis assays. E14.5 FL cells, prepared as described previously, were loaded into the top chamber of a transwell with a 5-μm pore size and the chemo-attractant (10 ng/mL or 100 ng/mL SDF-1) or controls (blank or 10% FCS) were loaded into the bottom well. Cells were allowed to migrate for 4 hours before being counted by flow cytometry. Gating on GFP-expressing cells analyzed the transduced population and a migration index was calculated. Although CD123-low and CD123-high-WT cells had equivalent migration in response to 10% FCS, CD123-high-WT cells showed decreased migration toward SDF-1 at both 10 and 100 ng/mL (Figure 5E). The specificity of CXCR4 was tested in CD123-high-WT cells using a retroviral construct encoding for CXCR4. Restoration of CXCR4 expression in CD123-high-WT cells was sufficient to rescue the migration defect (Figure 5E).
Downregulation of CXCR4 is linked to high expression of CD123 in AML patient samples.
To examine the relationship between high IL-3 signals and CXCR4 expression in primary human cells, we incubated PB cells from AML patients for 24 hours in medium with or without IL-3 and isolated RNA. CD123 and CXCR4 relative to β-actin were measured using RT-PCR (Figure 6A). There was a significant reduction in CXCR4 expression upon stimulation with IL-3 in AML patient samples, similar to transduced FL cells. Supporting results were obtained when we examined protein expression (supplemental Figure 6A). We also looked at the correlation of CD123 and CXCR4 expression in AML samples by flow cytometry. We observed strong concordance between CD123 expression in PB and BM, and a similar concordance between CXCR4 expression in blood and BM (supplemental Figure 6B). In a correlation matrix of the mean fluorescence intensity of CD123 and CXCR4 expression as determined by flow cytometry, there was a negative correlation between CD123 and CXCR4 expression (supplemental Figure 6C), although this did not reach statistical significance.
Figure 6.

AML patients with high CD123 show downregulation of CXCR4, which can be rescued by blocking IL-3 signaling. (A) AML samples were cultured in the absence (no IL-3) or presence of 10 ng/mL human IL-3 for 24 hours. RT-PCR was used to analyze CXCR4 gene expression. Data are normalized to β-actin and each line represents a paired measurement in an individual patient. Significance was calculated by performing Wilcoxon matched-pairs signed rank test. (B) AML samples were cultured in the absence (no IL-3) or presence of 10 ng/mL human IL-3 and ± 1 μM of the IL-3 blocking mAb 7G3 or a control IgG2a for 24 hours. RT-PCR was used to analyze CXCR4 gene expression. Data are normalized to β-actin and are presented as foldchange in response to IL-3 and antibody. Significance was calculated using a 2-tailed paired t test. (C) CXCR4 function was compared between CD123-high and CD123-low AML BM cells after 4 hours of migration to SDF-1. Cells were cultured overnight in 10 ng/mL human IL-3 and plated into 96-well transwell plates with SDF-1 (or chemotaxis buffer as a control) in the lower chamber. Migration of specific populations was analyzed using flow cytometric staining for CD34, CD38, and CD123 postmigration. Data are normalized to beads. Statistical analysis: #nonsignificant difference, *P < .05, **P < .01, ***P < .001.
Clinical trials using therapeutic monoclonal anti-CD123 antibodies are ongoing. To test the effect of anti-CD123 monoclonal antibody (mAb) on CXCR4 expression in AML cells, we cultured mononuclear cells from AML patient samples with 10 ng/mL human IL-3, with or without 1 μM of the IL-3 blocking mAb 7G3 or control IgG2a antibody for 24 hours. CXCR4 expression was measured by RT-PCR (Figure 6B). Specifically blocking CD123 prevented CXCR4 downregulation in response to IL-3 stimulation in primary AML cells. The functional consequence of the downregulation of CXCR4 on AML cells was confirmed using migration to SDF-1. BM cells from 2 patients were cultured overnight in IL-3 (10 ng/mL). Cells were analyzed by flow cytometry following 4 hours of migration. The population of cells with higher CD123 (and therefore lower CXCR4) showed reduced migration toward SDF-1 compared with those cells with lower CD123 (Figure 6C).
Discussion
AML is a highly heterogeneous disease with poor prognosis. There is no common genetic lesion in AML for therapeutic targeting. Recently, the α subunit of the receptor for IL-3 (CD123) has been identified as a marker of AML LSC in all subclasses of AML.7-11 Beyond being a biomarker of AML LSC, we propose that expression of CD123 may play a critical role in disease. It has been widely believed that HSC and LSC have similar interactions with the BM niche, which protects the cells from hematologic stress and, in patients, chemotherapeutic agents. However, this is challenged in a recent report from Hawkins and colleagues46 who suggest a more dynamic situation in which leukemic cells are actively dividing and exiting/entering the marrow during and following chemotherapy. The concept of stem cell occupancy of the BM niche is addressed by Boyd and colleagues who found, using competitive reconstitution assays, that normal HSC consistently outperformed their malignant counterparts for niche residency.47
We used in vitro and in vivo analyses to reveal that an unexpected function for CD123, which contributes to the hematopoietic stem/progenitor cell:BM niche interaction in AML, is the CD123/CXCR4 axis. Previously, IL-3 has been widely used in combination with other cytokines for the culture and expansion of hematopoietic stem/progenitor cells in vitro; however, studies have shown that culture in IL-3 (and other cytokines) impairs the engraftment of these cells into recipient mice.48,49 Exposure to high levels of IL-3 is associated with increased progenitor mobilization out of the BM,50 whereas clinically, IL-3 has been used in combination with granulocyte CSF to potentiate progenitor mobilization for stem cell transplantation.51
The engraftment failure of CD123-expressing cells, whether they were normal hematopoietic cells or transformed by HoxA9 expression, was unexpected. One might anticipate that enhanced proliferation and survival derived from active IL-3R signaling would translate into effective engraftment or more rapid onset of AML. Instead, CD123 overexpressing cells could not be found in transplanted mice.
We exploited a CD123 mutant unable to activate IL-3R signaling to explore the mechanisms of the engraftment failure of CD123-high-WT cells. Mutation of 2 intracellular proline residues generates a receptor that binds IL-3 with normal affinity, but does not activate signaling through the Ras/MAPK, JAK/STAT, or Akt pathways. Our observation that CD123-high-WT cells could not repopulate sublethally irradiated mice, but that animals injected with CD123-high-APIA reconstituted as efficiently as those given CD123-low (EV) cells, clearly established that the enhanced IL-3–induced signaling is responsible for the engraftment defect.
Enhanced IL-3R signaling downregulated CXCR4. CXCR4 has been extensively reported to be essential for HSC homing32,52-55 and is the only functional chemokine receptor on hematopoietic progenitor cells.56 The CXCR4/SDF-1 interaction is the focus of early targeted therapy developments in AML because these factors are produced by all cell types in the BM lining.34,57-59 The role of CXCR4 in the competition between HSC and LSC for niche occupancy was investigated using antagonists of the CXCR4-CXCL12 interaction. These agents displaced the leukemic cells, demonstrating the importance of this interaction in their retention in the BM niche.47 This was confirmed separately in a study showing constitutive CXCR4 signaling results in retention of quiescent stem cells in the niche.60
Mononuclear cells from AML patients also downregulated CXCR4 in response to exogenous stimulation with IL-3. Engagement of the IL-3R by IL-3 was essential for the reduction of CXCR4 and reduced migration toward SDF-1 in chemotaxis assays. The CD123-blocking mAb 7G343 confirmed the specificity for IL-3 in this CD123:CXCR4 relationship. Antibodies directed against CD123 are in clinical trials worldwide both because it was thought they may reduce LSC survival (by inhibiting IL-3R signaling) and by driving antibody-dependent cellular cytotoxicity killing of LSC. Our data point to a complex interaction between IL-3R signaling and the BM niche, which may influence the potential clinical impact of anti-CD123 antibodies. Our model suggests that proliferating malignant cells with normal CD123 expression retain CXCR4 expression that is sufficient to maintain cells in the BM niche. However, as CD123 expression increases, as in AML LSC, CXCR4 is reduced, which may result in the release of cells from the niche, while cells retain IL-3R signaling in lower IL-3 concentrations. Our results support the notion that CD123 overexpression regulates CXCR4. One may speculate that a combination of CXCR4 inhibition and CD123 antibodies would release malignant LSC still resident in a chemoprotective niche, enhancing the effectiveness of anti-CD123 mAb or other drugs.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors acknowledge Tim Hercus for generation of growth factors, Rebecca Wright and Bill Panagopoulos for technical assistance, Duncan McKenzie for advice with the migration assays, Daniel Thomas for critical review of the manuscript, Stuart Pitson for the pLenti-IRES-eGFP construct, staff from the SA Pathology Animal Care Facility and the Detmold Facility for flow cytometry service, Saumya Samaraweera for collating the patient information, and Josef Ngyuen for irradiation of mice.
This work was supported by project grants from the National Health and Medical Research Council (1048071) (P.G.E. and H.S.R.), (1081376) (G.B.), (1071897) (A.F.L.); and the Cancer Australia/Leukaemia Foundation (1010336) (A.F.L. and H.S.R.). H.S.R. is supported by the Peter Nelson Leukaemia Research Fund. N.L.W. was supported by a Leukaemia Foundation of Australia PhD scholarship. G.B. is supported by a Victorian Cancer Agency Mid-Career fellowship. This work was made possible through Victorian State Government Operational Infrastructure Support, and L.E.W. Carty Charitable Fund and Ian Rollo Currie Estate Foundation for infrastructure support.
Authorship
Contribution: H.S.R., N.L.W., G.B., A.F.L., and P.G.E. designed experiments, interpreted the data, and wrote the paper; R.J.D. provided patient characteristics; and H.S.R., N.L.W., G.B., C.M., M.K.P., M.D., and J.J.S. performed some of the experiments.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Hayley S. Ramshaw, Centre for Cancer Biology, University of South Australia and SA Pathology, Frome Rd, Adelaide, SA 5000, Australia; e-mail: hayley.ramshaw@unisa.edu.au.
References
- 1.Hope KJ, Jin L, Dick JE. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat Immunol. 2004;5(7):738-743. [DOI] [PubMed] [Google Scholar]
- 2.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730-737. [DOI] [PubMed] [Google Scholar]
- 3.Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645-648. [DOI] [PubMed] [Google Scholar]
- 4.Florian S, Sonneck K, Hauswirth AW, et al. Detection of molecular targets on the surface of CD34+/CD38-- stem cells in various myeloid malignancies. Leuk Lymphoma. 2006;47(2):207-222. [DOI] [PubMed] [Google Scholar]
- 5.Zhang P, Iwasaki-Arai J, Iwasaki H, et al. Enhancement of hematopoietic stem cell repopulating capacity and self-renewal in the absence of the transcription factor C/EBP alpha. Immunity. 2004;21(6):853-863. [DOI] [PubMed] [Google Scholar]
- 6.Brandts CH, Berdel WE, Serve H. Oncogenic signaling in acute myeloid leukemia. Curr Drug Targets. 2007;8(2):237-246. [DOI] [PubMed] [Google Scholar]
- 7.Testa U, Riccioni R, Diverio D, Rossini A, Lo Coco F, Peschle C. Interleukin-3 receptor in acute leukemia. Leukemia. 2004;18(2):219-226. [DOI] [PubMed] [Google Scholar]
- 8.Riccioni R, Pelosi E, Riti V, Castelli G, Lo-Coco F, Testa U. Immunophenotypic features of acute myeloid leukaemia patients exhibiting high FLT3 expression not associated with mutations. Br J Haematol. 2011;153(1):33-42. [DOI] [PubMed] [Google Scholar]
- 9.Yalcintepe L, Frankel AE, Hogge DE. Expression of interleukin-3 receptor subunits on defined subpopulations of acute myeloid leukemia blasts predicts the cytotoxicity of diphtheria toxin interleukin-3 fusion protein against malignant progenitors that engraft in immunodeficient mice. Blood. 2006;108(10):3530-3537. [DOI] [PubMed] [Google Scholar]
- 10.Jordan CT, Upchurch D, Szilvassy SJ, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia. 2000;14(10):1777-1784. [DOI] [PubMed] [Google Scholar]
- 11.Jin L, Lee EM, Ramshaw HS, et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell. 2009;5(1):31-42. [DOI] [PubMed] [Google Scholar]
- 12.Busfield SJ, Biondo M, Wong M, et al. Targeting of acute myeloid leukemia in vitro and in vivo with an anti-CD123 mAb engineered for optimal ADCC. Leukemia. 2014;28(11):2213-2221. [DOI] [PubMed] [Google Scholar]
- 13.Lee EM, Yee D, Busfield SJ, et al. Efficacy of an Fc-modified anti-CD123 antibody (CSL362) combined with chemotherapy in xenograft models of acute myelogenous leukemia in immunodeficient mice. Haematologica. 2015;100(7):914-926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frolova O, Benito J, Brooks C, et al. SL-401 and SL-501, targeted therapeutics directed at the interleukin-3 receptor, inhibit the growth of leukaemic cells and stem cells in advanced phase chronic myeloid leukaemia. Br J Haematol. 2014;166(6):862-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tettamanti S, Biondi A, Biagi E, Bonnet D. CD123 AML targeting by chimeric antigen receptors: A novel magic bullet for AML therapeutics? OncoImmunology. 2014;3(May):e28835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Testa U, Riccioni R, Militi S, et al. Elevated expression of IL-3Ralpha in acute myelogenous leukemia is associated with enhanced blast proliferation, increased cellularity, and poor prognosis. Blood. 2002;100(8):2980-2988. [DOI] [PubMed] [Google Scholar]
- 17.Graf M, Hecht K, Reif S, Pelka-Fleischer R, Pfister K, Schmetzer H. Expression and prognostic value of hemopoietic cytokine receptors in acute myeloid leukemia (AML): implications for future therapeutical strategies. Eur J Haematol. 2004;72(2):89-106. [DOI] [PubMed] [Google Scholar]
- 18.Nowak R, Oelschlägel U, Gurth H, et al. Relations between IL-3-induced proliferation and in vitro cytokine secretion of bone marrow cells from AML patients. Cytokine. 1999;11(6):435-442. [DOI] [PubMed] [Google Scholar]
- 19.Testa U, Torelli GF, Riccioni R, et al. Human acute stem cell leukemia with multilineage differentiation potential via cascade activation of growth factor receptors. Blood. 2002;99(12):4634-4637. [DOI] [PubMed] [Google Scholar]
- 20.Guthridge MA, Powell JA, Barry EF, et al. Growth factor pleiotropy is controlled by a receptor Tyr/Ser motif that acts as a binary switch. EMBO J. 2006;25(3):479-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guthridge MA, Stomski FC, Thomas D, et al. Mechanism of activation of the GM-CSF, IL-3, and IL-5 family of receptors. Stem Cells. 1998;16(5):301-313. [DOI] [PubMed] [Google Scholar]
- 22.Blalock WL, Weinstein-Oppenheimer C, Chang F, et al. Signal transduction, cell cycle regulatory, and anti-apoptotic pathways regulated by IL-3 in hematopoietic cells: possible sites for intervention with anti-neoplastic drugs. Leukemia. 1999;13(8):1109-1166. [DOI] [PubMed] [Google Scholar]
- 23.Hercus TR, Dhagat U, Kan WLT, et al. Signalling by the βc family of cytokines. Cytokine Growth Factor Rev. 2013;24(3):189-201. [DOI] [PubMed] [Google Scholar]
- 24.Barry SC, Korpelainen E, Sun Q, et al. Roles of the N and C terminal domains of the interleukin-3 receptor alpha chain in receptor function. Blood. 1997;89(3):842-852. [PubMed] [Google Scholar]
- 25.Matsuguchi T, Zhao Y, Lilly MB, Kraft AS. The cytoplasmic domain of granulocyte-macrophage colony-stimulating factor (GM-CSF) receptor alpha subunit is essential for both GM-CSF-mediated growth and differentiation. J Biol Chem. 1997;272(28):17450-17459. [DOI] [PubMed] [Google Scholar]
- 26.Cornelis S, Plaetinck G, Devos R, et al. Detailed analysis of the IL-5-IL-5R alpha interaction: characterization of crucial residues on the ligand and the receptor. EMBO J. 1995;14(14):3395-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kouro T, Kikuchi Y, Kanazawa H, et al. Critical proline residues of the cytoplasmic domain of the IL-5 receptor alpha chain and its function in IL-5-mediated activation of JAK kinase and STAT5. Int Immunol. 1996;8(2):237-245. [DOI] [PubMed] [Google Scholar]
- 28.Takaki S, Kanazawa H, Shiiba M, Takatsu K. A critical cytoplasmic domain of the interleukin-5 (IL-5) receptor alpha chain and its function in IL-5-mediated growth signal transduction. Mol Cell Biol. 1994;14(11):7404-7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dhar-Mascareno M, Chen J, Zhang RH, Cárcamo JM, Golde DW. Granulocyte-macrophage colony-stimulating factor signals for increased glucose transport via phosphatidylinositol 3-kinase- and hydrogen peroxide-dependent mechanisms. J Biol Chem. 2003;278(13):11107-11114. [DOI] [PubMed] [Google Scholar]
- 30.Perugini M, Brown AL, Salerno DG, et al. Alternative modes of GM-CSF receptor activation revealed using activated mutants of the common beta-subunit. Blood. 2010;115(16):3346-3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Winkler IG, Barbier V, Nowlan B, et al. Vascular niche E-selectin regulates hematopoietic stem cell dormancy, self renewal and chemoresistance. Nat Med. 2012;18(11):1651-1657. [DOI] [PubMed] [Google Scholar]
- 32.Boulais PE, Frenette PS. Making sense of hematopoietic stem cell niches. Blood. 2015;125(17):2621-2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Calvi LM, Link DC. The hematopoietic stem cell niche in homeostasis and disease. Blood. 2015;126(22):2443-2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rashidi A, DiPersio JF. Targeting the leukemia-stroma interaction in acute myeloid leukemia: rationale and latest evidence. Ther Adv Hematol. 2016;7(1):40-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tavor S, Petit I, Porozov S, et al. CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res. 2004;64(8):2817-2824. [DOI] [PubMed] [Google Scholar]
- 36.Lo Celso C, Scadden DT. The haematopoietic stem cell niche at a glance. J Cell Sci. 2011;124(Pt 21):3529-3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Glait-Santar C, Desmond R, Feng X, et al. Functional niche competition between normal hematopoietic stem and progenitor cells and myeloid leukemia cells. Stem Cells. 2015;33(12):3635-3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hercus TR, Barry EF, Dottore M, et al. High yield production of a soluble human interleukin-3 variant from E. coli with wild-type bioactivity and improved radiolabeling properties. PLoS One. 2013;8(8):e74376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barrett JM, Parham KA, Pippal JB, et al. Over-expression of sphingosine kinase-1 enhances a progenitor phenotype in human endothelial cells. Microcirculation. 2011;18(7):583-597. [DOI] [PubMed] [Google Scholar]
- 40.Kitamura T, Tojo A, Kuwaki T, et al. Identification and analysis of human erythropoietin receptors on a factor-dependent cell line, TF-1. Blood. 1989;73(2):375-380. [PubMed] [Google Scholar]
- 41.Brumatti G, Salmanidis M, Kok CH, et al. HoxA9 regulated Bcl-2 expression mediates survival of myeloid progenitors and the severity of HoxA9-dependent leukemia. Oncotarget. 2013;4(11):1933-1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lopez AF, Dyson PG, To LB, et al. Recombinant human interleukin-3 stimulation of hematopoiesis in humans: loss of responsiveness with differentiation in the neutrophilic myeloid series. Blood. 1988;72(5):1797-1804. [PubMed] [Google Scholar]
- 43.Sun Q, Woodcock JM, Rapoport A, et al. Monoclonal antibody 7G3 recognizes the N-terminal domain of the human interleukin-3 (IL-3) receptor alpha-chain and functions as a specific IL-3 receptor antagonist. Blood. 1996;87(1):83-92. [PubMed] [Google Scholar]
- 44.Woodcock JM, McClure BJ, Stomski FC, Elliott MJ, Bagley CJ, Lopez AF. The human granulocyte-macrophage colony-stimulating factor (GM-CSF) receptor exists as a preformed receptor complex that can be activated by GM-CSF, interleukin-3, or interleukin-5. Blood. 1997;90(8):3005-3017. [PubMed] [Google Scholar]
- 45.Guthridge MA, Stomski FC, Barry EF, et al. Site-specific serine phosphorylation of the IL-3 receptor is required for hemopoietic cell survival. Mol Cell. 2000;6(1):99-108. [PubMed] [Google Scholar]
- 46.Hawkins ED, Duarte D, Akinduro O, et al. T-cell acute leukaemia exhibits dynamic interactions with bone marrow microenvironments. Nature. 2016;538(7626):518-522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boyd AL, Campbell CJV, Hopkins CI, et al. Niche displacement of human leukemic stem cells uniquely allows their competitive replacement with healthy HSPCs. J Exp Med. 2014;211(10):1925-1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peters SO, Kittler ELW, Ramshaw HS, Quesenberry PJ. Ex vivo expansion of murine marrow cells with interleukin-3 (IL-3), IL-6, IL-11, and stem cell factor leads to impaired engraftment in irradiated hosts. Blood. 1996;87(1):30-37. [PubMed] [Google Scholar]
- 49.Yonemura Y, Ku H, Hirayama F, Souza LM, Ogawa M. Interleukin 3 or interleukin 1 abrogates the reconstituting ability of hematopoietic stem cells. Proc Natl Acad Sci USA. 1996;93(9):4040-4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nicolini FE, Cashman JD, Hogge DE, Humphries RK, Eaves CJ. NOD/SCID mice engineered to express human IL-3, GM-CSF and Steel factor constitutively mobilize engrafted human progenitors and compromise human stem cell regeneration. Leukemia. 2004;18(2):341-347. [DOI] [PubMed] [Google Scholar]
- 51.Geissler K, Peschel C, Niederwieser D, et al. Potentiation of granulocyte colony-stimulating factor-induced mobilization of circulating progenitor cells by seven-day pretreatment with interleukin-3. Blood. 1996;87(7):2732-2739. [PubMed] [Google Scholar]
- 52.Kucia M, Reca R, Miekus K, et al. Trafficking of normal stem cells and metastasis of cancer stem cells involve similar mechanisms: pivotal role of the SDF-1-CXCR4 axis. Stem Cells. 2005;23(7):879-894. [DOI] [PubMed] [Google Scholar]
- 53.Burger JA, Peled A. CXCR4 antagonists: targeting the microenvironment in leukemia and other cancers. Leukemia. 2009;23(1):43-52. [DOI] [PubMed] [Google Scholar]
- 54.Lai C-Y, Yamazaki S, Okabe M, et al. Stage-specific roles for CXCR4 signaling in murine hematopoietic stem/progenitor cells in the process of bone marrow repopulation. Stem Cells. 2014;32(7):1929-1942. [DOI] [PubMed] [Google Scholar]
- 55.Lam BS, Adams GB. Hematopoietic stem cell lodgment in the adult bone marrow stem cell niche. Int J Lab Hematol. 2010;32(6 Pt 2):551-558. [DOI] [PubMed] [Google Scholar]
- 56.Wright DE, Bowman EP, Wagers AJ, Butcher EC, Weissman IL. Hematopoietic stem cells are uniquely selective in their migratory response to chemokines. J Exp Med. 2002;195(9):1145-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bernasconi P, Farina M, Boni M, Dambruoso I, Calvello C. Therapeutically targeting SELF-reinforcing leukemic niches in acute myeloid leukemia: a worthy endeavor? Am J Hematol. 2016;91(5):507-517. [DOI] [PubMed] [Google Scholar]
- 58.Cho BS, Zeng Z, Mu H, et al. Antileukemia activity of the novel peptidic CXCR4 antagonist LY2510924 as monotherapy and in combination with chemotherapy [published correction appears in Blood. 2015;126(8):1049]. Blood. 2015;126(2):222-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cooper TM, Sison EAR, Baker SD, et al. A phase 1 study of the CXCR4 antagonist plerixafor in combination with high-dose cytarabine and etoposide in children with relapsed or refractory acute leukemias or myelodysplastic syndrome: a Pediatric Oncology Experimental Therapeutics Investigators’ Consortium study (POE 10-03) [published online ahead of print 14 April 2017]. Pediatr Blood Cancer. doi:10.1002/pbc.26414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cordeiro Gomes A, Hara T, Lim VY, et al. Hematopoietic stem cell niches produce lineage-instructive signals to control multipotent progenitor differentiation. Immunity. 2016;45(6):1219-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.