

Key Points
PF-06747143, a novel CXCR4 antagonist IgG1 Ab, mobilizes malignant cells from the BM and induces their death via Fc-effector function.
PF-06747143 reduces tumor burden in NHL, AML, and MM models, both as a monotherapy or in combination with standard-of-care agents.
Abstract
The chemokine receptor CXCR4 is highly expressed and associated with poor prognosis in multiple malignancies. Upon engagement by its ligand, CXCL12, CXCR4 triggers intracellular signaling pathways that control trafficking of cells to tissues where the ligand is expressed, such as the bone marrow (BM). In hematologic cancers, CXCR4-driven homing of malignant cells to the BM protective niche is a key mechanism driving disease and therapy resistance. We developed a humanized CXCR4 immunoglobulin G1 (IgG1) antibody (Ab), PF-06747143, which binds to CXCR4 and inhibits CXCL12-mediated signaling pathways, as well as cell migration. In in vivo preclinical studies, PF-06747143 monotherapy rapidly and transiently mobilized cells from the BM into the peripheral blood. In addition, PF-06747143 effectively induced tumor cell death via its Fc constant region–mediated effector function. This Fc-mediated cell killing mechanism not only enhanced antitumor efficacy, but also played a role in reducing the duration of cell mobilization, when compared with an IgG4 version of the Ab, which does not have Fc-effector function. PF-06747143 treatment showed strong antitumor effect in multiple hematologic tumor models including non-Hodgkin lymphoma (NHL), acute myeloid leukemia (AML), and multiple myeloma (MM). Importantly, PF-06747143 synergized with standard-of-care agents in a chemoresistant AML patient-derived xenograft model and in an MM model. These findings suggest that PF-06747143 is a potential best-in-class anti-CXCR4 antagonist for the treatment of hematologic malignancies, including in the resistant setting. PF-06747143 is currently in phase 1 clinical trial evaluation (registered at www.clinicaltrials.gov as #NCT02954653).
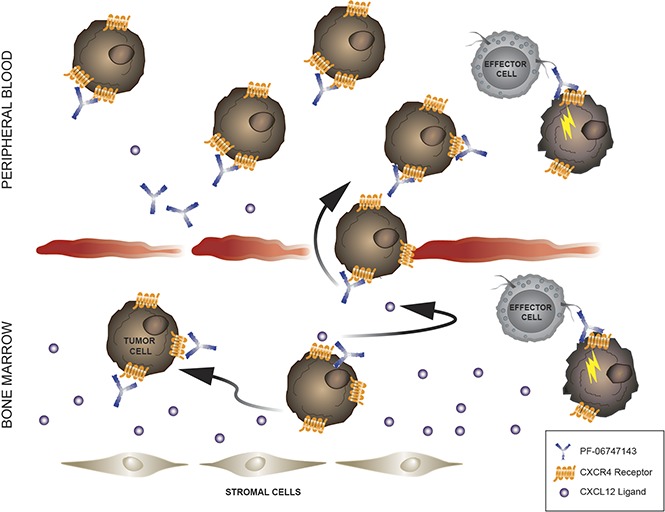
Visual Abstract
Introduction
The 7-transmembrane G-protein coupled chemokine receptor CXCR4, also known as CD184, is normally expressed in various tissues and predominantly in hematopoietic cells.1,2 Moreover, CXCR4 is overexpressed in >75% of cancers, including hematologic malignancies and solid tumors, and its expression correlates with poor prognosis.3-7 The chemokine CXCL12, also known as SDF-1, is the only CXCR4 ligand identified to date. It is highly expressed by mesenchymal stromal cells in the liver, lungs, bone marrow (BM), and lymphatic tissues.8 CXCR4 and CXCL12 have strong chemotactic activity and play a critical role in the cross talk between cancer cells and the local tumor microenvironment.9 Upon ligand binding, CXCR4 triggers signaling pathways that promote malignant cell survival, migration, and invasiveness.10,11 Furthermore, chemotherapy treatment upregulates expression of CXCL12 in BM cells and the receptor in tumor cells.12,13 The collective evidence suggests that the CXCR4 signaling axis protects malignant cells from spontaneous and chemotherapy-induced apoptosis in the BM niche,14-16 promoting resistance and minimal residual disease in hematologic malignancies.17-19
The CXCL12-CXCR4 interaction is crucial for attracting tumor cells to the BM. Thus, disruption of the pathway using CXCR4 antagonists as a way to mobilize tumor cells from the protective BM, and to sensitize them to chemotherapy, has been proposed as an attractive therapeutic approach in hematologic malignancies.13,20-23 In that regard, several CXCR4 antagonists are currently undergoing clinical evaluation. It is important to note that the ability of anti-CXCR4 agents to simply mobilize cells appears to be insufficient to drive antitumor activity. For instance, the CXCR4 peptide antagonists LY251092424 and BKT140/BL8040/TN1400325 induce cell mobilization as monotherapies but failed to reduce tumor burden in clinical trials.25,26 Interestingly, the CXCR4 partial agonist small molecule AMD3100 (Plerixafor; Mozobil), which induces mobilization of leukemic blasts from the BM, is undergoing clinical evaluation in hematologic malignancies in combination with chemotherapy, as a means to eliminate the mobilized cancer cells.18,19,23
In addition to small molecules and peptides, a humanized immunoglobulin G4 (IgG4) antibody (Ab) targeting CXCR4, ulocuplumab, has been evaluated in acute myeloid leukemia (AML), chronic lymphocytic leukemia, and multiple myeloma (MM) phase 1 clinical trials.20,27-29 Therapeutic Ab’s may induce target cell killing via immune-mediated effector functions (Fc-effector function), such as Ab-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC).30 Human Ab subclasses effector function is dictated by their differential binding to Fc receptors in immune effector cells. Human IgG1 and IgG3 Ab’s mediate potent Fc-effector function, whereas human IgG2 and IgG4 Ab’s display little or no Fc-effector function.30 In line with this, ulocuplumab, an IgG4 Ab, was recently reported to have no Fc-effector tumor cell–killing activity.31 In the clinic, this Ab causes cell mobilization, as do other CXCR4 antagonists; however, because of its longer half-life (3 days in humans) and lack of Fc-effector function, the mobilized cells survive and stay in the peripheral blood (PB) for several days, resulting in at least 1 incidence of hyperleukocytosis.28,29 Extensive leukocyte mobilization presents a safety risk associated with respiratory and neurological distress.32
All therapeutic CXCR4 antagonists currently in the clinic have been shown to mobilize cancer cells from the BM niche, facilitating their elimination by chemotherapy and other anticancer drugs. Therefore, we hypothesized that by developing a CXCR4 Ab capable of both mobilizing and rapidly killing CXCR4-expressing cancer cells through Fc-effector function, it would be possible to enhance antitumor activity and mitigate the safety risk associated with prolonged cell mobilization, previously observed with other anti-CXCR4 agents in the clinic. Here we describe the generation of a novel humanized IgG1 CXCR4 antagonist Ab, PF-06747143, that displays those properties. PF-06747143 potently inhibited hematologic tumor growth as a monotherapy and in combination with standard-of-care (SOC) agents in various preclinical models, including chemotherapy-resistant ones. Studies comparing PF-06747143 with the IgG4 version of the Ab showed that the Fc-effector function activity contributed to improved efficacy and prevented prolonged cell mobilization. Based on its unique mechanisms of action, PF-06747143 has broad therapeutic potential in hematologic malignancies.
Methods
Functional assays
The cyclic adenosine monophosphate (cAMP) assay was performed using CHO-K1 cells expressing human CXCR4 (hCXCR4; DiscoveRx) (3 × 105 per well) in triplicates. The cAMP Hunter eXpress GPCR assay kit (DiscoveRx) was used to perform the assay, following the manufacturer’s protocol. Tumor cell migration assays were performed plating 0.5 × 106 cells per well (in quadruplicates) in the top chamber of transwell plates (Corning), in serum free RPMI 1640. CXCL12 (100 ng/mL) was added to the bottom wells. After 24 hours, tumor cells in the bottom well were measured using CellTiter-Glo Luminescent Cell Viability Assay (Promega). The percent of migration inhibition was calculated relative to control (IgG1 Ab)–treated cells, using the following formula: % migration inhibition = (number of cells in PF-06747143–treated group − number of cells in IgG1 control–treated group) × 100/number of cells in IgG1 control–treated group. GraphPad Prism 6.0 was used to plot data and generate dose-response curves used to calculate 50% inhibitory concentration values.
Fc-effector function assays
ADCC activity was determined using the NK-92 FcγRIIIA 158V (NK92 158V) cell line (Conkwest) as effector cells. Ab’s were incubated for 4 hours, with tumor cells and effector cells, NK-92 158V at 1:10 ratio in quadruplicates. ToxiLight bioluminescent cytotoxicity assay (Lonza) was used to detect cell lysis. CDC activity was determined using 10 000 Daudi cells per well in duplicates, incubated with 5 μg/mL (33 nM) of Ab’s, in the presence of 2.5% human donor complement (Sigma), for 4 hours. Cytotoxicity was measured using CytoTox 96 nonradioactive kit (Promega).
In vivo efficacy studies
Non-Hodgkin lymphoma (NHL) subcutaneous xenograft model was generated by subcutaneous implantation of 5 × 106 Ramos tumor cells on the right hind limb of SCID beige female mice (Charles River). Tumors were measured by caliper, and volumes were calculated by (length × width2)/2. MM and AML disseminated xenograft tumor models employed tumor cells stably transduced with the luciferase gene. Cells were implanted IV in the tail vein of NSG female mice (Jackson Laboratory). Tumor burden was monitored by bioluminescence imaging (IVIS 200). According to the Institutional Animal Use and Care Committee protocol, mice were euthanized once hind leg paralysis was observed (survival end point). The AML BM012407L PDX model (Jackson Labs) is derived from an AML patient with blasts with the following characteristics: dim CD4, partial CD7, CD11b, CD13, partial CD22, CD33, CD34, CD117, and HLA-DR. FAB-M2, Karyotype 46, XY AML fluorescence in situ hybridization panel negative, and FMS-like tyrosine kinase-3–internal tandem duplication (FLT3-ITD) positive. The patient suffered aggressive disease, underwent 2 cycles of induction chemotherapy, relapsed within 2 months of chemotherapy treatment, and died of refractory AML within 6 months of diagnosis. The AML PDX model was passaged using BM cells collected from donor mice. Tumor burden in PB and BM was determined by flow cytometry using CD45–fluorescein isothiocyanate (1:10 dilution; BD Biosciences), CD33-allophycocyanin (1:20 dilution; eBiosciences), or CXCR4 (CD184)–phycoerythrin (1:30 dilution; BD Biosciences) Ab’s at 4°C for 45 minutes. Isotype-matched IgG nonspecific Ab’s were used as negative controls. Flow cytometry analysis was done in FACSCalibur (Becton Dickinson).
Nonhuman primate (NHP) cell mobilization
Cynomolgus monkeys were administered with Ab’s or vehicle by IV bolus injection. A control group of animals received vehicle (phosphate-buffered saline). Blood samples were collected in K2 EDTA for hematology analysis, and mean absolute white blood cell (WBC) numbers were determined using an Advia 2120 (Siemens) hematology analyzer.
Statistical analysis
Data were analyzed with Prism software (GraphPad Software Inc.). Two-group comparisons were analyzed with a 2-tailed Student t test, and a greater number of groups were analyzed using 1-way analysis of variance with Tukey multiple comparison test. A P value <.05 was considered significant. Survival analyzes were performed using Kaplan-Meier followed by a log-rank (Mantel-Cox) test. All mouse and NHP studies were conducted under a Pfizer Institutional Animal Use and Care Committee approved protocol.
Results
PF-06747143 binds CXCR4 with high selectivity and affinity
Binding specificity of PF-06747143 to hCXCR4 was assessed by flow cytometry. PF-06747143 bound specifically to CHO cells expressing hCXCR4 (CHO-hCXCR4), whereas no binding was observed to cells transfected with empty vector (CHO-parental) (supplemental Figure 1). Consistent with these findings, PF-06747143 bound to the hCXCR4-positive hematologic malignant cells Ramos (99%), OPM-2 (71%), and MV4-11 (80%), but not to hCXCR4-negative TF-1 cells33 (3%) (supplemental Figure 2; supplemental Table 2).
PF-06747143 binding to hCXCR4 and cynomolgus monkey CXCR4 was evaluated in CXCR4-expressing human Raji and HSC-F (transformed cynomolgus monkey fetal splenocytes) cells by flow cytometry. The 50% effective concentrations (EC50s) for hCXCR4 and cynomolgus monkey CXCR4 were 3.05 and 2.59 nM, respectively, indicating similar binding affinities to CXCR4 protein of both species (supplemental Figure 3).
PF-06747143 inhibits CXCL12-induced signaling pathways and cell migration
We next analyzed how PF-06747143 affects CXCR4-driven cellular functions. CXCL12 binding to CXCR4 inhibits adenyl cyclase and intracellular cAMP levels. Consistent with CXCR4 antagonism, PF-06747143 dose-dependently induced cAMP in the presence of CXCL12 (EC50 of 7.75 nM) (Figure 1A). Furthermore, in accordance with the important role CXCR4 and its ligand, CXCL12, play in migration of tumor cells, PF-06747143 inhibited CXCL12-induced migration of Ramos cells, with an 50% inhibitory concentration (IC50) of 0.24 nM (Figure 1B). Potent migration inhibition by PF-06747143 was also observed in other cell lines such as AML MV4-11 (supplemental Figure 4). Taken together, these results show that PF-06747143 functionally antagonizes ligand-induced activity of CXCR4.
Figure 1.

PF-06747143 blocks CXCL12-induced pathways. (A) cAMP assay was performed in CHO-K1 cells transfected with hCXCR4 (CHO-K1-CXCR4), incubated with CXCL12 at its EC80 (1.25 nM) and PF-06747143 or IgG1 control Ab. Experiments were performed in triplicates. Bars represent standard error of the mean (SEM). (B) Migration assay was performed in Ramos human NHL cells incubated in transwell chambers for 24 hours with PF-06747143 or IgG1 control Ab. CXCL12 (100 ng/mL) was used a chemoattractant in the bottom chamber. Data are shown as mean % migration inhibition. Experiment was performed in quadruplicates. Bars represent SEM.
PF-06747143 induces cell death through its Fc-effector function (ADCC and CDC)
To evaluate the role of Fc-effector functions in ADCC and CDC assays, the variable domains of PF-06747143 parental Ab, m15, were cloned into human IgG1 (m15-IgG1) and human IgG4 (m15-IgG4) Ab backbones. Rituximab, a component of the SOC treatment regimen for NHL, was used as positive control because its clinical activity depends on both ADCC and CDC.34 In the ADCC assay in the Ramos NHL cell line, in the presence of NK92 158V effector cells, m15-IgG1 showed strong cytotoxicity, with an IC50 of 124 pM, when compared with no significant activity with m15-IgG4 (Figure 2A). This is in agreement with the expected diminished ADCC activity of the IgG4 Ab. In addition, PF-06747143 exerted similar cytotoxicity as m15-IgG1 with an IC50 of 115 pM (Figure 2A), confirming that the parent Ab and its humanized derivative have similar ADCC properties. Moreover, both CXCR4 IgG1 Ab’s showed similar ADCC activity to the CD20 Ab rituximab. ADCC-induced cell death was also observed in MV4-11 (AML) and OPM-2 (MM) cell lines treated with PF-06747143 (Figure 2B).
Figure 2.

PF-06747143 induces malignant cell death by Fc-effector function activity. ADCC activity was evaluated by incubating 100 nM PF-06747143, m15-IgG1, m15-IgG4, rituximab, or respective negative control Ab’s for 4 hours in the presence of NK92 158V effector killer cells (effector:target cell ratio 10:1) with tumor target cells: Ramos (A) and MV4-11 and OPM-2 (B). Cell lysis was measured by ToxiLight bioluminescent cytotoxicity assay. Experiments were performed in quadruplicates. Bars represent SEM. CDC was assessed by incubating Daudi NHL target cells in the presence of 2.5% human complement for 4 hours. Experiments were performed in duplicates. Bars represent standard deviations. (C) Cells were treated with 33 nM of IgG1 control Ab, m15-IgG1, m15-IgG4, or rituximab. (D) Cells were treated with 33 nM of IgG1 control Ab, m15-IgG1, PF-06747143, or rituximab.
The CDC activity of m15-IgG1 and m15-IgG4 was assessed in NHL cells, in the presence of human donor complement. m15-IgG1 induced complement-dependent cell lysis (32%), similar to the positive control rituximab (25%), whereas m15-IgG4 had no activity (Figure 2C), consistent with the hypothesis that the human IgG1 Ab has more potent effector function than the human IgG4 Ab. When CDC activities of m15-IgG1, PF-06747143, and rituximab were compared, all 3 Ab’s triggered similar levels of complement fixation-driven cell lysis (Figure 2D).
PF-06747143 induces dose-dependent tumor growth inhibition (TGI) in an NHL xenograft model
We next assessed the in vivo efficacy of PF-06747143 in the subcutaneous NHL Ramos xenograft model, which expresses CXCR4 (99% positive) (supplemental Figure 2; supplemental Table 2). Both PF-06747143 and its parental IgG1 Ab, m15-IgG1, were equally effective at inhibiting tumor growth, compared with IgG1 control Ab (P < .0001) (Figure 3A). The number of animals exhibiting tumor regressions was similar in both parental m15-IgG1 Ab–treated and PF-06747143 Ab–treated groups, with 80% and 70% of animals showing tumor volumes below their initial sizes at the end of the study, respectively. The in vivo exposures of PF-06747143 and m15-IgG1 were similar, with mean serum concentrations of 400 nM and 424 nM 1 week after the first dose, respectively. These results indicate that the parental m15-IgG1 Ab and PF-06747143 Ab have comparable exposure and antitumor activity in vivo.
Figure 3.

Efficacy of PF-06747143 and role of Fc-effector function in the Ramos NHL efficacy model. Ramos cells were implanted subcutaneously (5 × 106 cells), and when mean tumor volume reached 350 to 400 mm3, mice were randomized (n = 10-12 per group). Arrows indicate Ab treatment days. Data points represent the mean tumor volume ± SEM. (A) Animals were treated weekly, subcutaneously, with Ab’s at 10 mg/kg for 2 doses, on days 1 and 8. (B) Animals were treated weekly, subcutaneously, with control IgG1 Ab at 10 mg/kg and PF-06747143 at 0.1, 1, 10, and 30 mg/kg for 4 doses, or until day 21. (C) Animals were treated weekly, subcutaneously, with m15-IgG1 or m15-IgG4 Ab’s at 1 and 10 mg/kg for 3 doses. Study was terminated at day 15. (D) Antibody exposure was evaluated 24 hours after last dose. Serum from 3 animals per group was collected, and human IgG concentration determined by enzyme-linked immunosorbent assay. Bars points represent the mean concentration ± SEM. (E) Cleaved caspase-3 in tumor tissue collected 48 and 72 hours post–Ab treatment at 10 mg/kg was determined by western blot (n = 3 animals per group).
PF-06747143 dose-dependent response was demonstrated in the Ramos subcutaneous model, with similar TGI relative to initial tumor volume observed at the highest doses, 10 mg/kg and 30 mg/kg (Figure 3B). These effects were sustained until the end of the study (day 38), even after Ab treatment was suspended (day 21).
Fc-effector function plays a role in TGI
To evaluate the role of the Ab effector function in TGI, m15-IgG1 and m15-IgG4 Ab’s were compared in the Ramos subcutaneous model, at 1 and 10 mg/kg. The antitumor activity of m15-IgG1 was greater than that of m15-IgG4 at each of the doses tested (Figure 3C). m15-IgG1 at 10 mg/kg exhibited significantly greater TGI than m15-IgG4 Ab at the same dose (P = .014), with complete tumor regressions in 50% of m15-IgG1-treated (10 mg/kg) animals, compared with partial TGI responses observed in the m15-IgG4 group at the same dose. Moreover, TGI achieved by m15-IgG1 at 1 mg/kg was comparable to the TGI seen with m15-IgG4 at a 10-fold higher dose (10 mg/kg) (P = .63). The mean serum concentration of m15-IgG1 and m15-IgG4 were comparable for both doses, suggesting both Ab isoforms have similar exposure in vivo (Figure 3D).
Treatment-induced cell death was assessed by characterizing caspase-3 activation in tumors collected from Ramos xenograft mice treated with a single dose of m15-IgG1 or m15-IgG4 at 10 mg/kg. Cleaved caspase-3 was observed as early as 48 hours postdose in m15-IgG1–treated animals and increased over time (72 hours), whereas minimal caspase-3 activation was detected in tumors of animals treated with the m15-IgG4 Ab at similar time points (Figure 3E). Taken together, these results suggest that Fc-effector function is an important factor driving in vivo efficacy, contributing to the superior antitumor effect of m15-IgG1 relative to that of m15-IgG4 Ab.
PF-06747143 inhibits tumor growth, increases survival, and synergizes with bortezomib in a disseminated MM model
In hematologic diseases, malignant cells proliferate in protective niches such as the BM. To determine if PF-06747143 is effective in eliminating cells from the BM environment, a disseminated MM model, in which the OPM2-Luc tumor cells are implanted IV and migrated spontaneously to the BM, was used (Figure 4A). The activity of PF-06747143 was compared with that of melphalan, a component of the SOC regimen for MM treatment.35 PF-06747143 significantly inhibited BM tumor growth compared with the IgG1 control Ab and melphalan on day 30 (P < .0001) (Figure 4B-C). Importantly, the strong antitumor response translated into a significant survival benefit. The human IgG1 control Ab and melphalan-treated animals showed median survival of 33.5 and 36 days, respectively, whereas the PF-06747143–treated group showed no deaths by the end of the study (day 50) (P < .0001) (Figure 4D).
Figure 4.

PF-06747143 greatly reduces BM tumor burden as a monotherapy and in combination with SOCs in a disseminated MM model. OPM-2-Luc MM cells were implanted IV (5 × 106 cells) and allowed to spontaneously migrate and home in the BM for 8 days, when animals were randomized based on luciferase activity detected in the large bones (n = 10 per group). Animals showing hind leg paralysis were euthanized, and this was the survival end point of the study. (A) Treatment schematic representation. Animals were treated with 10 mg/kg of IgG1 control and PF-06747143 Ab’s, subcutaneously, weekly, for 5 doses. Melphalan was dosed 1 mg/kg, intraperitoneally (IP), twice a week, for a total of 4 cycles. (B) Tumor burden was determined by bioluminescence imaging and quantification. Data points represent the mean bioluminescence ± SEM. (C) Whole body bioluminescence representative imaging showing tumor burden in n = 5 mice per group over time. (D) Kaplan-Meier survival curve. (E) Treatment schematic representation. Animals were treated with 1 mg/kg of IgG1 control and PF-06747143 Ab’s, subcutaneously, weekly, for a total of 7 doses. Bortezomib was dosed at 0.5 mg/kg, administered IP, 2 times per week, for a total of 4 cycles. (F) Tumor burden was determined by bioluminescence imaging quantification. Data points represent the mean bioluminescence ± SEM. (G) Whole body bioluminescence representative images showing tumor burden in n = 5 mice per group over time. (H) Kaplan-Meier survival curve.
Given the robust single-agent activity of PF-06747143 in the OPM-2 model at the 10 mg/kg dose, we next evaluated efficacy of PF-06747143 at a lower dose (1 mg/kg), both as a monotherapy or in combination with bortezomib (Velcade), a therapeutic agent approved for the treatment of MM36 (Figure 4E). Tumor burden (Figure 4F-G) in the combination group was significantly reduced compared with either agent alone. This translated into a survival benefit in the combination arm, with no deaths observed (P < .0003) at the end of the study (day 51) (Figure 4H). The IgG1 control median survival was 34 days, and single-agent treatments with PF-06747143 or bortezomib showed median survivals of 47 and 44 days, respectively. Taken together, these results show that PF-06747143 reduces BM tumor burden, improves survival as a monotherapy, and has potent synergistic activity with bortezomib in a disseminated MM model.
Efficacy of PF-06747143 in an AML disseminated tumor model
Next, PF-06747143 activity was investigated in a staged, disseminated AML model, using MV4-11 cells. The activity of PF-06747143 at 0.1, 1, and 10 mg/kg was compared with that of daunorubicin, a chemotherapeutic used for the treatment of AML patients,37 and crenolanib, a FLT-3 inhibitor currently in phase 2 clinical trials in AML38 (Figure 5A). Bioluminescence imaging showed that PF-06747143 treatment decreased tumor burden in a dose-dependent manner, compared with human IgG1 control Ab (P < .05) (Figure 5B-C). Daunorubicin was efficacious at reducing tumor burden, with similar activity to PF-06747143 at 10 mg/kg.
Figure 5.

PF-06747143 reduces BM tumor burden in a dose-response dependent manner in a disseminated AML tumor model. MV4-11-Luc AML cells were implanted IV (1 × 106 cells) and allowed to spontaneously migrate and home in the BM for 11 days, when animals were randomized (n = 5-10 per group). (A) Treatment schematic representation. Animals were treated with IgG1 control or PF-06747143 Ab’s, subcutaneously, weekly, for 4 doses. Daunorubicin was dosed at 2 mg/kg, IV, 3 times (days 1, 3, and 5). Crenolanib was dosed at 7.5 mg/kg, IP, twice a day, on days 11 through 15 and days 25 through 29. (B) Tumor burden was determined by bioluminescence imaging and quantification. Data points represent the mean bioluminescence ± SEM. (C) Whole body bioluminescence representative imaging showing tumor burden in n = 5 mice per group over time. (D) On day 35, 3 days following the final Ab treatment, tumor burden was evaluated in PB and BM cells by flow cytometry, using hCD45 and hCD33 Ab’s as AML markers (n = 5-10 mice per group). Data points represent each individual mouse. (E) Kaplan-Meier survival curve (n = 5 animals per group), using hind leg paralysis as the end point.
To evaluate the effect of PF-06747143 in BM and PB tumor burden, the percentage of human CD45 (hCD45+) and human CD33 (hCD33+) positive cells was determined (Figure 5D). Treatment with PF-06747143, daunorubicin, and crenolanib resulted in a significant reduction in the number of tumor cells in both PB and BM, compared with human IgG1 control Ab (P < .05). PF-06747143 induced dose-dependent reduction in the number of human AML cells (hCD45+ and hCD33+) in both PB and BM. Importantly, the reduction in the number of human AML cells in the BM with PF-06746143 treatment at 10 mg/kg (95.9%) was similar to that achieved by the AML therapeutic agents daunorubicin (84.5%) and crenolanib (80.5%).
A PF-06747143 dose-dependent increase in survival was observed (Figure 5E). PF-06747143 at 10 mg/kg resulted in a median survival of 63 days, and animals treated with PF-06747143 at 1 mg/kg showed median survival of 47 days. PF-06747143 at 0.1 mg/kg and the IgG1 control Ab–treated groups showed similar median survival of 36 and 41 days, respectively. The increase in survival observed for PF-06747143 at 10 mg/kg and 1 mg/kg was statistically significant (P < .05) when compared with the IgG1 control Ab group. Taken together these results demonstrate that treatment with PF-06747143 as a monotherapy translates into dose-dependent BM tumor burden reduction and significant survival benefit in a disseminated AML model.
PF-06747143 has a strong combinatorial effect with daunorubicin and cytarabine (Ara-C) in a chemotherapy-resistant AML PDX model
To investigate the activity of PF-06747143 in an AML chemoresistant model, in combination with the AML SOC agents daunorubicin and Ara-C, we used the disseminated PDX model (BM012407L). This model was derived from an AML patient who developed resistance to induction chemotherapy, consisting of daunorubicin and Ara-C, the main components of AML SOC treatment. Animals were treated according to the dosing scheme in Figure 6A. Of note, CXCR4 expression in this PDX model is lower than in the xenograft cell line models. Only 36% of the BM cells are positive for CXCR4, as indicated by the percentage of CXCR4+ AML (CD45+CD33+) cells observed in the IgG1 control Ab group (Figure 6B). Treatment with PF-06747143 as a monotherapy significantly decreased the total number of CXCR4+ AML cells (CXCR4+CD45+CD33+) in the BM to 2.9%, compared with IgG1 control–treated group (36%). The combination of daunorubicin with Ara-C reduced the BM CXCR4+CD45+CD33+ AML cells to 9.3%. Importantly, a strong combinatorial effect was observed with the triple combination of PF-06747143 added to the SOC agents, daunorubicin and Ara-C, resulting in minimal levels of CXCR4+CD45+CD33+cells in the BM (group average 0.04%) (Figure 6B), with 2 out of 5 animals exhibiting undetectable malignant cells in the BM or no minimal residual disease.
Figure 6.

PF-06747143 reduces BM tumor burden in a PDX chemoresistant AML tumor model. (A) NSG mice received 150 cGy whole body irradiation, and within 24 hours, 1.5 × 106 PDX BM cells were implanted IV and allowed to spontaneously migrate and home in the BM for 43 days, when tumor burden in PB reached 0.8% to 1.6%. Animals were then randomized in 5 animals per treatment group and treated with IgG1 control or PF-06747143 Ab’s, subcutaneously, weekly, for 8 doses. Daunorubicin was dosed at 1.5 mg/kg, IV, 3 times per week, for 2 cycles. Ara-C was dosed at 15 mg/kg, IP, once per day, from days 43 through 50 and days 64 through 71. On day 99, n = 5 animals per group were euthanized, and PB and BM samples were analyzed by flow cytometry to detect CXCR4+ malignant AML cells (hCD45+ and hCD33+) (B) or total AML cells (hCD45+ and hCD33+) (C).
When the BM results were analyzed based on the number of AML cells (CD45+CD33+), irrespective of CXCR4 expression (Figure 6C), we observed that treatment with the SOC agents daunorubicin and Ara-C resulted in partial response, with 27% of malignant AML cells remaining in the BM (Figure 6C) (P < .05). This is in line with the chemoresistant nature of this PDX model. Treatment with PF-06747143 as a monotherapy was not very effective, with a group average of 80% CD45+CD33+ AML cells remaining in the BM. This is expected based on the fact that ∼65% of BM AML cells do not express CXCR4 in this PDX model. Importantly, the number of BM CD45+CD33+ AML cells was significantly reduced by the combination of PF-06747143 with the 2 AML SOC agents, with 0.3% residual AML cells in the BM (Figure 6C) of animals in this combination group.
Overall, the levels of CD45+CD33+ AML cells were lower in PB than in the BM, with similar trends to inhibition with PF-06747143 and SOC alone and synergistic effect with the triple combination (Figure 6C). Higher sensitivity to treatment is expected for malignant cells circulating in PB because they are not protected by the growth factors and chemokines present in the BM. Taken together, these data support clinical use of PF-06747143 in combination with SOC in AML, including patients who have developed resistance to chemotherapy.
CXCR4 Ab–induced cell mobilization duration is dependent on Fc-effector function
Mobilization of malignant cells from growth factor–rich BM niches into the PB increases the sensitivity of these cells to therapy. However, prolonged mobilization has the potential to redistribute the malignant cells and may lead to safety-related issues, such as hyperleukocytosis.32 To determine the ability of PF-06747143 to mobilize tumor cells in vivo, we used the MV4-11 AML disseminated mouse model. Animals with high tumor burden (day 12) postimplant were treated with a single dose of PF-06747143 (10 mg/kg). PB samples were evaluated at various time points postdose to determine human AML malignant cell mobilization in PB by flow cytometry (hCD45+hCD33+) and Ab exposure by enzyme-linked immunosorbent assay (Figure 7A). PF-06747143 induced MV4-11 cell mobilization, with a peak observed at 3 hours postdose, followed by a rapid decrease in PB mobilized cells observed at 16 and 24 hours postdose. These results demonstrate that PF-06747143 has the ability to induce malignant cell mobilization, as expected for a CXCR4 antagonist, but the mobilization is followed by a rapid reduction in PB cell number, with levels similar to baseline at 24 hours postdose. Because PF-06747143 serum concentration was high and sustained over the 24-hour study duration (supplemental Figure 5), we hypothesized that malignant AML cell number reduction at 16 and 24 hour time points is likely because of the cytotoxic Fc-effector function of the Ab.
Figure 7.

Role of Fc-Effector function in cell mobilization and normal hematopoiesis. (A) Cell mobilization into the PB was detected by flow cytometry, using hCD45 and hCD33 Ab’s following a single dose of PF-06747143 (10 mg/kg) in MV4-11 AML tumor-bearing mice. Data represent mean hCD45+ and hCD33+ staining in PB relative to baseline (time 0 hours) ± SEM. (B) Cynomolgus monkeys (n = 3 per group) received a single dose of m15-IgG1 or m15-IgG4, at 50 mg/kg, or vehicle (control). Mean absolute PB WBC numbers ± SEM are shown over time. (C) Cynomolgus monkeys (n = 2 per group) received 2 weekly doses of PF-06747143 (10 mg/kg) or vehicle, at 0 and 168 hours. Mean absolute PB WBC numbers ± SEM are shown over time. (D) Cynomolgus monkeys (n = 8-12 per group) received 2 weekly doses of PF-06747143 (10 mg/kg), at 0 and 168 hours. PB number of hematopoietic progenitor CD34+ cells ± SEM, relative to each individual baseline level, are shown over time.
CXCR4 antagonists have been shown to mobilize leukocytes or WBCs and hematopoietic progenitor cells (HPCs) (CD34+) in NHPs or patients.28,29 In order to evaluate the role of Fc-effector function in cell mobilization, we compared m15-IgG1 and m15-IgG4 effects in WBCs in NHPs. After a single dose, mobilization of WBCs in the PB was observed with both Ab’s; however, there were some sharp differences observed. The mean peak concentration of WBCs in m15-IgG1–treated animals (40 × 103 cells per μL) was lower than that for the m15-IgG4 group (54 × 103 cells per μL). Moreover, m15-IgG1 peak mobilization occurred at 1 hour postdose, which was earlier than that for the m15-IgG4 group (6 hours) (Figure 7B). Furthermore, transient mobilization, with a rapid decline to baseline levels, similar to that of vehicle-treated animals, was observed with m15-IgG1 at 24 hours postdose, and m15-IgG4 led to prolonged and sustained mobilization, with high WBC counts 2.6-fold above baseline, at 4 days (96 hours) postdose. In a separate WBC mobilization study in NHPs, the ability of PF-06747143 to induce rapid and transient WBC cell mobilization was evaluated (Figure 7C). Two weekly doses of PF-06747143 Ab (days 1 and 8 [168 hours]) were administered. The PF-06747143 mobilization profile was very similar to its parent Ab (m15-IgG1); WBCs were rapidly mobilized between 1 and 6 hours postdose, and mobilized levels came down to baseline at 24 hours postdose. The HPCs (CD34+) mobilization profile was characterized in NHPs treated with 2 weekly doses of PF-06747143 (Figure 7D). The CD34+ cells followed a similar mobilization pattern to that of the WBCs, with rapid mobilization into PB between 1 and 6 hours postdose, returning to baseline levels after 24 hours, and no significant depletion of the CD34+ cells relative to baseline was observed.
The differences between the IgG1 and IgG4 CXCR4 Ab’s observed in these studies are most likely related to the ability of the IgG1 Ab to induce cell killing through Fc-effector function (ADCC/CDC), whereas such activity is not observed with the IgG4 Ab (Figure 2A,C). PF-06747143 induced cell mobilization followed by rapid reduction of mobilized cell numbers in PB. The WBCs or HPCs numbers did not reach levels significantly below baseline, indicative of an adequate safety profile related to leukocytes.
Discussion
One of the major limitations of chemotherapy-based treatments for AML and other hematologic malignancies has been the failure to clear cancer cells protected by the BM environment. The CXCR4 receptor and its ligand, CXCL12, control processes critical for tumor cell homing and retention in the BM niche. Consistent with the protective role that this pathway plays, increased activity of this signaling axis is correlated with poor prognosis.4,7,8,39,40 CXCR4 inhibitors currently being evaluated in the clinic, including small molecules,23 peptides,24,25 and an IgG4 Ab,27-30 can mobilize tumor cells, sensitizing them to cell killing by chemotherapy.17,41 PF-06747143, a novel humanized IgG1 Ab, binds to CXCR4 and potently inhibits CXCL12/CXCR4-mediated cell signaling pathways such as cAMP and cancer cell migration. These characteristics are in line with those of other CXCR4 inhibitors. However, here we demonstrated that PF-06747143 has an additional potent mechanism of action, related to the ability to induce tumor cytotoxicity via Fc-effector function (ADCC and CDC), which is not present in the other CXCR4 inhibitors currently in clinical trials. Moreover, the Fc-effector activity of PF-06747143 in vitro was comparable to that of rituximab, an anti-CD20 monoclonal Ab, whose main mechanism of action in the clinic relies largely on effector function.34 The importance of effector function for antitumor activity was demonstrated comparing the in vivo activities of PF-06747143 parent Ab, m15-IgG1, to that of its IgG4 version, m15-IgG4, which is deprived of Fc-effector function. The m15-IgG1 Ab had superior TGI, associated with a significant increase in cell death in tumors.
Furthermore, the antitumor effect observed with PF-06747143 across multiple hematologic malignancy in vivo models suggests that the Fc-effector function activity translates into strong efficacy. As a monotherapy, PF-06747143 induced sustained tumor regression, dramatically reduced BM tumor burden, and improved survival in NHL, MM, and AML staged mouse tumor models. Importantly, PF-06747143 had robust combinatorial efficacy with a wide range of SOC agents, including bortezomib in an MM model and daunorubicin plus Ara-C in an AML chemotherapy-resistant PDX model. Overall, these data support a potential for effective use of PF-06747143 in combination regimens and in patients that develop resistance to chemotherapy.
The ability of CXCR4 antagonists to mobilize tumor cells into the PB and to sensitize them to chemotherapy was previously described in AML and MM patients.18,41,42 However, strategies that simply mobilize malignant cells into circulation, without prompt elimination of these cells, have the potential to lead to serious safety-related issues, such as hyperleukocytosis.32 Our results show that PF-06747143, and its parent Ab m15-IgG1, mobilized cells to the PB in both mice and NHPs, as monotherapies; yet this mobilization was rapid and transient, with the number of mobilized cell levels returning back to baseline at no later than 24 hours postdose. In contrast, treatment with m15-IgG4 induced a sustained level of mobilized WBCs in the PB, lasting for at least 4 days. This is similar to reports of prolonged WBC mobilization observed in NHPs following monotherapy treatment with the IgG4 CXCR4 Ab, ulocuplumab, where the WBC levels returned to baseline only after 15 days postdose.28 Sustained malignant cell mobilization was also reported in MM and AML patients treated with ulocuplumab as a monotherapy.20,28,29 These observations indicate that Fc-effector function may decrease the risk of prolonged cell mobilization in patients.
In summary, in comparison with CXCR4 antagonists currently being evaluated in clinical trials, PF-06747143 exhibits a differentiated product profile because of its unique mechanisms of action. Treatment with PF-06747143 is a one-two punch therapeutic approach, resulting in (1) disruption of CXCR4/CXCL12 axis leading to rapid malignant cell mobilization from protective niches (2) and tumor cell killing via Fc-effector function. Taken together, the robust efficacy and safety margins observed in preclinical models suggest the potential of PF-06747143 as a best-in-class CXCR4-targeted treatment of hematologic cancers. A phase 1 clinical trial evaluation of PF-06747143 is currently underway (registered at www.clinicaltrials.gov as #NCT02954653).
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Arvind Rajpal for project advice, Anirban Adhikari for protein engineering technical assistance, and Karsten Sauer for helpful comments on the manuscript.
Authorship
Contribution: S.-H.L. designed research, analyzed data, and wrote the manuscript; Y.G., B.P., Z.Y., M.H., and R.Y. performed research and analyzed data; C.Z. designed research and analyzed data; C.F. performed research; W.W. performed research and analyzed data; J.L. designed research and analyzed data; M.E.S. analyzed data; E.B. designed research and analyzed data; B.S., N.H., and W.-H.H. performed research and analyzed data; K.L. designed research, performed research, and analyzed data; T.-T.T. performed research and analyzed data; J.K. performed research; J.-T.M. performed research and analyzed data; G.J. wrote the manuscript; I.B. and C.B. performed research; S.M.C. designed research, performed research, and analyzed data; M.J.C. analyzed data; D.S. designed research; T.S. and V.R.F. designed research and wrote the manuscript; and F.P. designed research, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: All authors were employees of Pfizer Inc. at the time the work was performed. None of the authors received grants for this work.
The current affiliation for M.H. is Mirati Therapeutics, San Diego, CA.
The current affiliation for T.S. is Eli Lilly and Company, Indianapolis, IN.
The current affiliation for V.R.F. is ORIC Pharmaceuticals, South San Francisco, CA.
Correspondence: Flavia Pernasetti, 10646 Science Center Dr, San Diego, CA 92121; e-mail: flavia.pernasetti@pfizer.com.
References
- 1.Caruz A, Samsom M, Alonso JM, et al. Genomic organization and promoter characterization of human CXCR4 gene. FEBS Lett. 1998;426(2):271-278. [DOI] [PubMed] [Google Scholar]
- 2.Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 1998;393(6685):595-599. [DOI] [PubMed] [Google Scholar]
- 3.Spoo AC, Lübbert M, Wierda WG, Burger JA. CXCR4 is a prognostic marker in acute myelogenous leukemia. Blood. 2007;109(2):786-791. [DOI] [PubMed] [Google Scholar]
- 4.Burger JA, Peled A. CXCR4 antagonists: targeting the microenvironment in leukemia and other cancers. Leukemia. 2009;23(1):43-52. [DOI] [PubMed] [Google Scholar]
- 5.Konopleva MY, Jordan CT. Leukemia stem cells and microenvironment: biology and therapeutic targeting. J Clin Oncol. 2011;29(5):591-599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mazur G, Butrym A, Kryczek I, et al. Decreased expression of CXCR4 chemokine receptor in bone marrow after chemotherapy in patients with non-Hodgkin lymphomas is a good prognostic factor. PLoS One. 2014;9(5):e98194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Konoplev S, Rassidakis GZ, Estey E, et al. Overexpression of CXCR4 predicts adverse overall and event-free survival in patients with unmutated FLT3 acute myeloid leukemia with normal karyotype. Cancer. 2007;109(6):1152-1156. [DOI] [PubMed] [Google Scholar]
- 8.Orimo A, Gupta PB, Sgroi DC, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121(3):335-348. [DOI] [PubMed] [Google Scholar]
- 9.Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. 2010;16(11):2927-2931. [DOI] [PubMed] [Google Scholar]
- 10.Burger JA, Kipps TJ. CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood. 2006;107(5):1761-1767. [DOI] [PubMed] [Google Scholar]
- 11.Broxmeyer HE, Kohli L, Kim CH, et al. Stromal cell-derived factor-1/CXCL12 directly enhances survival/antiapoptosis of myeloid progenitor cells through CXCR4 and G(alpha)i proteins and enhances engraftment of competitive, repopulating stem cells. J Leukoc Biol. 2003;73(5):630-638. [DOI] [PubMed] [Google Scholar]
- 12.Ratajczak MZ, Serwin K, Schneider G. Innate immunity derived factors as external modulators of the CXCL12-CXCR4 axis and their role in stem cell homing and mobilization. Theranostics. 2013;3(1):3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sison EA, McIntyre E, Magoon D, Brown P. Dynamic chemotherapy-induced upregulation of CXCR4 expression: a mechanism of therapeutic resistance in pediatric AML. Mol Cancer Res. 2013;11(9):1004-1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holm NT, Abreo F, Johnson LW, Li BD, Chu QD. Elevated chemokine receptor CXCR4 expression in primary tumors following neoadjuvant chemotherapy predicts poor outcomes for patients with locally advanced breast cancer (LABC). Breast Cancer Res Treat. 2009;113(2):293-299. [DOI] [PubMed] [Google Scholar]
- 15.Zeng Z, Samudio IJ, Munsell M, et al. Inhibition of CXCR4 with the novel RCP168 peptide overcomes stroma-mediated chemoresistance in chronic and acute leukemias. Mol Cancer Ther. 2006;5(12):3113-3121. [DOI] [PubMed] [Google Scholar]
- 16.Zeng Z, Shi YX, Samudio IJ, et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood. 2009;113(24):6215-6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Domanska UM, Kruizinga RC, Nagengast WB, et al. A review on CXCR4/CXCL12 axis in oncology: no place to hide. Eur J Cancer. 2013;49(1):219-230. [DOI] [PubMed] [Google Scholar]
- 18.Nervi B, Ramirez P, Rettig MP, et al. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood. 2009;113(24):6206-6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dillmann F, Veldwijk MR, Laufs S, et al. Plerixafor inhibits chemotaxis toward SDF-1 and CXCR4-mediated stroma contact in a dose-dependent manner resulting in increased susceptibility of BCR-ABL+ cell to imatinib and nilotinib. Leuk Lymphoma. 2009;50(10):1676-1686. [DOI] [PubMed] [Google Scholar]
- 20.Chien S, Beyerle LE, Wood BL, et al. Mobilization of blasts and leukemia stem cells by anti-CXCR4 antibody BMS-936564 (MDX 1338) in patients with relapsed/refractory acute myeloid leukemia [abstract]. Blood. 2013;122(21). Abstract 3882. [Google Scholar]
- 21.Hendrix CW, Flexner C, MacFarland RT, et al. Pharmacokinetics and safety of AMD-3100, a novel antagonist of the CXCR-4 chemokine receptor, in human volunteers. Antimicrob Agents Chemother. 2000;44(6):1667-1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hendrix CW, Collier AC, Lederman MM, et al. ; AMD3100 HIV Study Group. Safety, pharmacokinetics, and antiviral activity of AMD3100, a selective CXCR4 receptor inhibitor, in HIV-1 infection. J Acquir Immune Defic Syndr. 2004;37(2):1253-1262. [DOI] [PubMed] [Google Scholar]
- 23.Uy GL, Rettig MP, Motabi IH, et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood. 2012;119(17):3917-3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peng SB, Zhang X, Paul D, et al. Identification of LY2510924, a novel cyclic peptide CXCR4 antagonist that exhibits antitumor activities in solid tumor and breast cancer metastatic models. Mol Cancer Ther. 2015;14(2):480-490. [DOI] [PubMed] [Google Scholar]
- 25.Galsky MD, Vogelzang NJ, Conkling P, et al. A phase I trial of LY2510924, a CXCR4 peptide antagonist, in patients with advanced cancer. Clin Cancer Res. 2014;20(13):3581-3588. [DOI] [PubMed] [Google Scholar]
- 26.Peled A, Abraham M, Avivi I, et al. The high-affinity CXCR4 antagonist BKT140 is safe and induces a robust mobilization of human CD34+ cells in patients with multiple myeloma. Clin Cancer Res. 2014;20(2):469-479. [DOI] [PubMed] [Google Scholar]
- 27.Kashyap KM, Amaya-Chanaga CI, Jones H, et al. BMS-936564 (anti-CXCR4 antibody) induces specific leukemia cell mobilization and objective clinical responses in CLL patients treated under a phase I clinical trial [abstract]. Blood. 2013;122(21). Abstract 4190. [Google Scholar]
- 28.Ghobrial I, Perez R, Baz R, et al. Phase 1b study of the novel anti-CXCR4 antibody ulocuplumab (BMS-936564) in combination with lenalidomide plus low-dose dexamethasone, or with bortezomib plus dexamethasone in subjects with relapsed or refractory multiple myeloma [abstract]. Blood. 2014;124(21). Abstract 3483. [Google Scholar]
- 29.Becker PS, Foran JM, Altman JK, et al. Targeting the CXCR4 pathway: safety, tolerability and clinical activity of ulocuplumab (BMS-936564), an anti-CXCR4 antibody, in relapsed/refractory acute myeloid leukemia [abstract]. Blood. 2014;124(21). Abstract 386. [Google Scholar]
- 30.Jiang XR, Song A, Bergelson S, et al. Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat Rev Drug Discov. 2011;10(2):101-111. [DOI] [PubMed] [Google Scholar]
- 31.Kashyap MK, Kumar D, Jones H, et al. Ulocuplumab (BMS-936564/MDX1338): a fully human anti-CXCR4 antibody induces cell death in chronic lymphocytic leukemia mediated through a reactive oxygen species-dependent pathway. Oncotarget. 2016;7(3):2809-2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Porcu P, Cripe LD, Ng EW, et al. Hyperleukocytic leukemias and leukostasis: a review of pathophysiology, clinical presentation and management. Leuk Lymphoma. 2000;39(1-2):1-18. [DOI] [PubMed] [Google Scholar]
- 33.Rozmyslowicz T, Majka M, Kijowski J, et al. Platelet- and megakaryocyte-derived microparticles transfer CXCR4 receptor to CXCR4-null cells and make them susceptible to infection by X4-HIV. AIDS. 2003;17(1):33-42. [DOI] [PubMed] [Google Scholar]
- 34.Harrison AM, Thalji NM, Greenberg AJ, Tapia CJ, Windebank AJ. Rituximab for non-Hodgkin’s lymphoma: a story of rapid success in translation. Clin Transl Sci. 2014;7(1):82-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mateos MV, Ocio EM, Paiva B, et al. Treatment for patients with newly diagnosed multiple myeloma in 2015. Blood Rev. 2015;29(6):387-403. [DOI] [PubMed] [Google Scholar]
- 36.San Miguel JF, Mateos MV, Ocio E, Garcia-Sanz R. Multiple myeloma: treatment evolution. Hematology. 2012;17(suppl 1):S3-S6. [DOI] [PubMed] [Google Scholar]
- 37.Kimby E, Nygren P, Glimelius B; SBU-group. Swedish Council of Technology Assessment in Health Care. A systematic overview of chemotherapy effects in acute myeloid leukaemia. Acta Oncol. 2001;40(2-3):231-252. [DOI] [PubMed] [Google Scholar]
- 38.Galanis A, Ma H, Rajkhowa T, et al. Crenolanib is a potent inhibitor of FLT3 with activity against resistance-conferring point mutants. Blood. 2014;123(1):94-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Azab AK, Hu J, Quang P, et al. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood. 2012;119(24):5782-5794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brault L, Rovó A, Decker S, Dierks C, Tzankov A, Schwaller J. CXCR4-SERINE339 regulates cellular adhesion, retention and mobilization, and is a marker for poor prognosis in acute myeloid leukemia. Leukemia. 2014;28(3):566-576. [DOI] [PubMed] [Google Scholar]
- 41.Azab AK, Runnels JM, Pitsillides C, et al. CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood. 2009;113(18):4341-4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li X, Guo H, Duan H, et al. Improving chemotherapeutic efficiency in acute myeloid leukemia treatments by chemically synthesized peptide interfering with CXCR4/CXCL12 axis. Sci Rep. 2015;5:16228. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.