

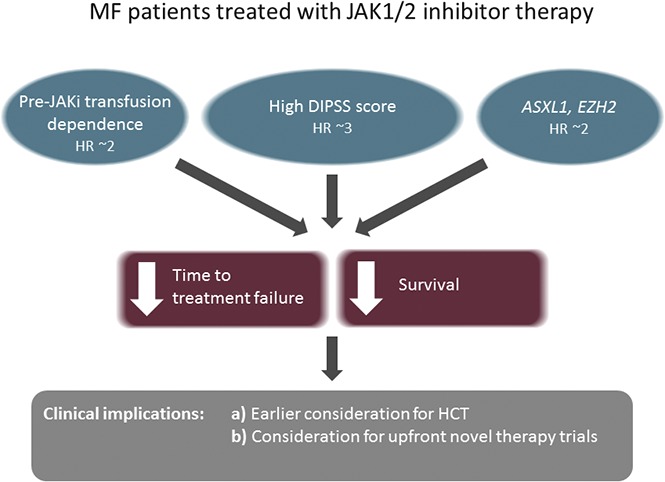
Key Points
ASXL1/EZH2, transfusion dependence, and a high prognostic risk score predict shorter TTF in MF patients on JAK1/2 inhibitors.
These clinical and genetic factors were also associated with decreased overall survival.
Abstract
In myelofibrosis (MF), driver mutations in JAK2, MPL, or CALR impact survival and progression to blast phase, with the greatest risk conferred by triple-negative status. Subclonal mutations, including mutations in high–molecular risk (HMR) genes, such as ASXL1, EZH2, IDH1/2, and SRSF2 have also been associated with inferior prognosis. However, data evaluating the impact of next-generation sequencing in MF patients treated with JAK1/2 inhibitors are lacking. Using a 54-gene myeloid panel, we performed targeted sequencing on 100 MF patients treated with ruxolitinib (n = 77) or momelotinib (n = 23) and correlated mutational profiles with treatment outcomes. Ninety-nine patients had at least 1 mutation identified, 46 (46%) had 2 mutations, and 34 (34%) patients had ≥3 mutations. Seventy-nine patients carried a mutation in JAK2V617F, 14 patients had mutations in CALR, 6 patients had an MPL mutation, and 2 patients were triple negative. No mutation was significantly associated with spleen or anemia response. A high Dynamic International Prognostic Scoring System score and pretreatment transfusion dependence were associated with a shorter time to treatment failure (TTF), and this association retained significance on multivariable analysis. Patients with ASXL1 (hazard ratio [HR], 1.86; P = .03) and EZH2 mutations (HR, 2.94; P = .009) and an HMR profile (HR, 2.06; P = .01) had shorter TTF. On multivariate analysis, ASXL1 or EZH2 mutations were independently associated with shorter TTF and overall survival. These findings help identify patients unlikely to have a durable response with current JAK1/2 inhibitors and provide a framework for future studies.
Visual Abstract
Introduction
The advent of next-generation sequencing (NGS) techniques has brought intense interest to the complex genetic landscape of myeloproliferative neoplasms (MPNs), particularly the ability of mutational data to confer information with respect to prognosis. Driver mutations in JAK2, MPL, or CALR, either alone1 or in conjunction with subclonal mutations in genes, such as ASXL1,2 have been associated with differences in overall survival (OS). Triple-negative patients who lack canonical mutations in JAK2, MPL, or CALR, have an increased risk of leukemic transformation as well as shortened OS.1
There is a growing body of literature on the adverse impact of somatic mutations in ASXL1, EZH2, IDH1/2, and SRSF2 on OS and leukemic transformation in patients with primary myelofibrosis (PMF) treated with supportive therapy.3-5 However, data regarding the impact of this high–molecular risk (HMR) profile on clinical outcomes in the context of novel MPN therapies, such as JAK1/2 inhibitors, are limited. A recent study of 95 patients treated with ruxolitinib using a panel of 29 genes, excluding SRSF2, reported that mutations in any of the HMR genes studied as well as a total of ≥3 mutations were associated with decreased spleen response and a shorter time to discontinuation of therapy.6 A retrospective analysis of the controlled myelofibrosis study with oral JAK inhibitor treatment (COMFORT-II) trial, which compared ruxolitinib to the best available therapy by using a 14-gene panel excluding CALR, found no correlation between mutational pattern and either spleen or hemoglobin response.7 Pardanani and colleagues8 evaluated the clinical response stratified by using a gene panel comprised of driver mutations and ASXL1 in 100 MF patients on momelotinib and found that CALR-positive patients had improved spleen response, whereas mutations in ASXL1 portended an inferior response. Neither of the latter 2 studies evaluated time to treatment failure (TTF) on JAK1/2 inhibitor therapy.7
Identifying patients who may not have durable responses on JAK1/2 inhibitor therapy is of immense clinical interest in guiding the timing of transplant for patients potentially eligible for allogeneic hematopoietic cell transplantation (HCT).9 Given the limited data in this area, we evaluated the effect of mutation profiles on the clinical outcomes of MF patients treated with JAK1/2 inhibitors by sequencing 54 genes commonly described in myeloid malignancies in a cohort of patients treated at our institution.
Methods
Patients
All patients with MF seen in the MPN program at the Princess Margaret Cancer Centre between November 2009 and May 2016 and treated with either ruxolitinib or momelotinib were identified by using the MPN database. Patients received JAK1/2 inhibitor therapy via commercial supply or in the context of a clinical trial. Retrospective clinical data were obtained from chart review. All patients consented to the collection of clinical data as well as the collection of molecular samples through the University Health Network (UHN) Hematologic Malignancy Tissue Bank. The study was approved by the UHN Research and Ethics Board (study number 15-9690-CE). Patients were eligible for inclusion in the study if they met the following criteria: (1) diagnosis of PMF, post–essential thrombocythemia (PET-MF), or post–polycythemia vera (PPV-MF) myelofibrosis as per the World Health Organization 2008 or the International Working Group for Myelofibrosis Research and Treatment (IWG-MRT) criteria; (2) in chronic phase (blasts <10%) prior to the initiation of JAK1/2 inhibitor therapy; (3) received ≥1 dose of JAK1/2 inhibitor therapy; and (4) had an available molecular sample for analysis. Patients were excluded if they received a JAK1/2 inhibitor as a bridge to transplant, received treatment as part of an ongoing or blinded clinical trial, or had dual malignancies or a myelodysplastic syndrome/MPN overlap syndrome.
DNA sequencing
DNA was extracted from peripheral blood (n = 88) or bone marrow (n = 12) samples. Targeted sequencing was performed using the TruSight Myeloid Sequencing Panel (Illumina; San Diego, CA) on the MiSeq benchtop genome sequencer (Illumina) by the UHN Advanced Molecular Diagnostics Laboratory. Fifty-four genes were profiled (39 genes with hotspot region coverage; 15 genes with complete coding region coverage; supplemental Table 1A-B) by using amplicon-based library preparation and (2 × 250 bp) paired-end sequencing with 50 ng of input DNA. Sequence data were analyzed by the NextGENe (version 2.3.1, SoftGenetics) and MiSeq Reporter (version 2.4.60, Illumina) software packages. Data files from each sample were uploaded into Cartagenia Bench NGS version 4.2 (Agilent; Santa Clara, CA) for subsequent filtering to prioritize for reporting those variants that passed all MiSeq Reporter quality criteria, including depth of coverage of ≥100× and a variant allele frequency threshold of >5% (>2% for well-documented hotspots). Variants with a global population frequency >1% according to population databases (1000 Genomes, Exome Sequencing Project [ESP6500SI-V2 data set, http://evs.gs.washington.edu/EVS/], Exome Aggregation Consortium)10-12 and/or present in the Advanced Molecular Diagnostics Laboratory internal database of recurring variants were excluded. Variants selected for downstream analysis included exonic frameshift and nonsense mutations, previously reported intronic splice-site variants, missense variants, and in-frame insertions/deletions. Variants were annotated by using established criteria13 and then classified as drivers (ie, canonical MPN-associated JAK2, CALR, and MPL mutations), HMR (ie, involving ASXL1, EZH2, IDH1/2, or SRSF2), oncogenic mutations, or variants of unknown significance, which were not used for downstream analysis. The complete list of annotated variants is reported in supplemental Table 2.
Statistical analysis
The primary endpoint was TTF, defined as the time from the start of JAK1/2 inhibitor therapy to one of the following: treatment discontinuation; progression to accelerated (blast count 10% to 19%) or blast phase (blast count ≥20%); spleen progression; or death. Secondary endpoints included OS and best spleen, anemia, and IWG-MRT response achieved over 48 weeks of treatment. OS was defined as the time from the start of JAK 1/2 inhibitor therapy until death or last follow-up. Treatment responses were assessed as per the 2013 revised IWG-MRT criteria.14 Dynamic International Prognostic Scoring System (DIPSS) scores were calculated for all patients on the first day of JAK1/2 inhibitor therapy.15 Transfusion dependence was defined as any transfusion in the 12 weeks prior to the start of JAK1/2 inhibitor treatment.16
Group comparisons were performed by using the Kruskal-Wallis test and Fisher’s exact test for continuous and categorical variables, respectively. TTF endpoints were estimated by using the Kaplan-Meier method. Group comparisons were performed by using the log-rank test. Patients who were event free at the time of analysis were censored at the date of the last follow-up. Both univariable and multivariable analyses for TTF endpoints were fit by using the Cox proportional hazards model. Assumptions for each model were tested and not found to be in violation. The selection of the final multivariate TTF and OS analysis was performed as follows. First, a univariable analysis was conducted for preliminary screening using P < .05 as a threshold. Based on these results and clinical input, 4 candidate explanatory variables were included in the preliminary multivariable model: DIPSS, pretreatment transfusion dependence, ASXL1/EZH2 mutations, and number of mutations. Bivariate tests of associations were performed based on Fisher’s exact test. The φ correlation coefficient was also calculated. This led to subsequent model fits that did not incorporate both DIPSS and pretreatment transfusion dependence or both ASXL1/EZH2 mutations and number of mutations. All possible combinations were subsequently fit and led to the final models. The fit of each model was assessed by using the likelihood ratio test and the Akaike information criterion. All analyses were performed in SAS software, version 9.4 (SAS Institute, Cary, NC).
Results
Patient characteristics
A total of 159 MF patients at the Princess Margaret Cancer Centre who received JAK1/2 inhibitor therapy were identified from the MPN database. Patients were excluded if they: had no available sample for sequencing (n = 22); were treated with a JAK1/2 inhibitor as a bridge to transplant (n = 9); were part of an ongoing or blinded clinical trial (n = 5); were treated in the accelerated phase (n = 4); or were diagnosed with dual malignancy (n = 2; in both cases, chronic lymphocytic leukemia) or myelodysplastic syndrome/MPN overlap syndrome (n = 2). Other exclusions included treatment with a JAK inhibitor other than ruxolitinib or momelitinib (n = 3), inadequate clinical information (n = 8), and miscellaneous (n = 4). In total, 100 patients were included in the study. There were no differences in the baseline characteristics of included patients vs those who had missing molecular samples or were treated as a part of an ongoing/blinded clinical trial (data not shown). The baseline demographic and clinical data of the study cohort are presented in Table 1.
Table 1.
Baseline clinical and demographic characteristics of patients with myelofibrosis
| Characteristic | All patients (n = 100) | PMF (n = 50) | PPV-MF (n = 27) | PET-MF (n = 23) |
|---|---|---|---|---|
| Age, median (range), y | 68 (43-86) | 66 (43-86) | 68 (54-80) | 70 (48-82) |
| Male, n (%) | 62 (62) | 32 (64) | 18 (67) | 12 (52) |
| Hb concentration, median (range), g/L | 98 (64-158) | 103 (73-158) | 106 (64-148) | 96 (72-121) |
| WCC, median (range), ×109/L | 14.6 (1.6-109.4) | 11.7 (1.6-65) | 19.8 (3.1-109.4) | 14.4 (2.2-50.5) |
| Plt count, median (range), ×109/L | 199 (17-1345) | 225 (17-1072) | 168 (46-647) | 237 (45-1345) |
| PBB ≥1%, n (%) | 56 (56) | 28 (56) | 13 (48) | 15 (65) |
| Baseline palpable spleen size, median (range), cm | 17 (0-38) | 17 (0-38) | 16 (8-32) | 18 (5-31) |
| Transfusion dependence prior to starting JAK inhibitor, n (%) | 39 (39) | 18 (36) | 11 (41) | 10 (43) |
| ECOG scale score, n (%) | ||||
| 0 | 20 (20) | 11 (22) | 6 (22) | 3 (13) |
| 1 | 68 (68) | 34 (68) | 16 (59) | 18 (78) |
| 2 | 10 (10) | 5 (10) | 4 (15) | 1 (4) |
| Missing | 2 (2) | 0 (0) | 1 (4) | 1 (4) |
| DIPSS score, n (%) | ||||
| Low | 1 (1) | 1 (2) | 0 (0) | 0 (0) |
| Intermediate-1 | 27 (27) | 17 (34) | 7 (26) | 3 (13) |
| Intermediate-2 | 43 (43) | 22 (44) | 9 (33) | 12 (52) |
| High | 29 (29) | 10 (20) | 11 (41) | 8 (35) |
| Cytogenetics, n (%) | ||||
| Normal | 47 (47) | 23 (46) | 11 (41) | 13 (57) |
| Abnormal, favorable | 23 (23) | 12 (24) | 7 (26) | 4 (17) |
| Abnormal, not favorable | 5 (5) | 3 (6) | 0 (0) | 2 (9) |
| Failed/not done | 25 (25) | 12 (24) | 9 (33) | 4 (17) |
| JAK inhibitor, n (%) | ||||
| Ruxolitinib | 77 (77) | 35 (70) | 25 (93) | 17 (74) |
| Momelotinib | 23 (23) | 15 (30) | 2 (7) | 6 (26) |
| Received as part of clinical trial, n (%) | 47 (47) | 24 (48) | 12 (44) | 11 (48) |
No P value was found to be statistically significant between MF subtypes.
ECOG, Eastern Cooperative Oncology Group; Hb, hemoglobin; PBB, peripheral blood blast; Plt, platelet; WCC, white blood cell count.
Sequencing results
A total of 301 variants were identified in our cohort, with 51 variants deemed variants of unknown significance and not used in downstream analyses (supplemental Table 2). Ninety-nine (99%) patients had ≥1 mutation identified. Nineteen (19%) patients had 1 mutation, 46 (46%) patients had 2 mutations, and 34 (34%) patients had ≥3 mutations. Seventy-nine patients (79%) carried a JAK2 V617F mutation, 14 (14%) patients had mutations in CALR (12 patients had type 1–like mutations: 2 patients had type II–like mutations), 6 (6%) patients had an MPL mutation, and 2 patients were triple negative (Figure 1). One patient had a cooccurrence of an MPLW515L and a type 1–like CALR mutation. Other genes mutated in ≥5% of the cohort were ASXL1 (33%), TET2 (28%), SF3B1 (13%), U2AF1 (10%), EZH2 (8%), SRSF2 (7%), DNMT3A (6%), CBL (5%), and NRAS (5%) (Figure 1). No oncogenic mutations were found in ABL1, ATRX, BCOR, BCORL1, BRAF, CBLB, CBLC, CDKN2A, CEBPA. FBXW7, FLT3, GATA1, HRAS, IKZF1, JAK3, KDM6A, KIT, KMT2A, KRAS, MYD88, NOTCH1, PDGFRA, RAD21, SMC1A, SMC3, STAG2, and WT1. Cooccurrences of various mutations are shown in a Circos plot (supplemental Figure 1). Forty-three (43%) patients had mutations in HMR genes. An HMR mutation was seen in 24% of patients with 0 to 2 mutations, whereas 79% of patients with ≥3 mutations had an HMR mutation.
Figure 1.

Landscape plot of mutations. Each column represents an individual patient. The bar graph represents the number of mutations per patient.
Primary endpoint: TTF
Fifty-two (52%) patients experienced treatment failure, with a median TTF of 116 weeks (Table 2). Eight patients died while on therapy. Twenty-two patients discontinued therapy due to toxicity: 11 patients stopped therapy due to hematologic adverse events (7 patients experienced severe thrombocytopenia; 4 patients experienced transfusion-dependent anemia), whereas 11 patients experienced nonhematologic toxicities. One patient required an emergent splenectomy after splenic rupture and discontinued therapy at that point, whereas 3 (6%) patients discontinued therapy due to a lack of spleen response. The remaining patients (n = 18) failed therapy due to advancement of their disease. Transformation to accelerated or blast phase occurred in 2 and 6 patients, respectively. Five patients met the IWG-MRT criteria for spleen progression, whereas another 5 patients developed progressive MF without meeting IWG-MRT criteria for progression.
Table 2.
Toxicity, response, and outcome data
| Characteristic | All patients (n = 100) | PMF (n = 50) | PPV-MF (n = 27) | PET-MF (n = 23) |
|---|---|---|---|---|
| Grade 3/4 hematologic toxicity | ||||
| Grade 3/4 anemia | 36 (36) | 21 (42) | 6 (22) | 9 (39) |
| Grade 3/4 thrombocytopenia | 23 (23) | 11 (22) | 4 (15) | 8 (35) |
| Grade 3/4 neutropenia | 2 (2) | 2 (4) | 0 (0) | 0 (0) |
| Best IWG-MRT response over 48 wk | ||||
| Clinical improvement | 27 (27) | 14 (28) | 6 (22) | 7 (30) |
| Spleen response | 18 (18) | 8 (16) | 6 (22) | 4 (17) |
| Anemia response | 1 (1) | 0 (0) | 0 (0) | 1 (4) |
| Stable disease | 52 (52) | 28 (56) | 13 (48) | 11 (48) |
| Progressive disease | 1 (1) | 0 (0) | 1 (4) | 0 (0) |
| Not available | 1 (1) | 0 (0) | 1 (4) | 0 (0) |
| IWG-MRT spleen response over 48 wk | 41 (41) | 22 (44) | 9 (35) | 10 (45) |
| IWG-MRT anemia response over 48 wk | 11 (11) | 3 (6) | 5 (19) | 3 (13) |
| Transformation to accelerated/blast phase | 9 (9) | 3 (6) | 3 (11) | 3 (13) |
| Median TTF, wk | 116 | 100 | 149 | 119 |
| Median OS, wk | 182 | 172 | 226 | 143 |
All values expressed as n (%), unless otherwise indicated. No P value was found to be statistically significant between MF subtypes.
The baseline clinical variables associated with shorter TTF included anemia (P = .001), peripheral blood blasts >1% (P = .01), elevated white blood cell count (P = .03), high DIPSS score (P = .001, Figure 2A), and pretreatment transfusion dependence (P = .002, Figure 2B) (Table 3). Of the individual mutations identified in ≥5% of the cohort, ASXL1 (P = .03), EZH2 (P = .009), and CBL (P = .04) mutations were associated with a shorter TTF. ASXL1/EZH2 mutations (P = .002, Figure 2C) as well as HMR mutations (P = .01) were also significantly associated with shorter TTF, whereas there was a trend toward shorter TTF in patients with ≥3 mutations (P = .07). When the number of mutations was stratified by HMR, only ≥3 mutations with HMR reached significance (P = .006), whereas there was a trend toward shorter TTF for 0 to 2 mutations in the presence of HMR (P = .08) (Figure 2D).
Figure 2.

Kaplan-Meier curves for TTF. (A) DIPSS score. (B) Pretreatment transfusion dependence. (C) ASXL1/EZH2 mutations. (D) Number of mutations, stratified by HMR.
Table 3.
Univariate analysis of variables associated with TTF and OS
| TTF | OS | |||||||
|---|---|---|---|---|---|---|---|---|
| Median, y | HR | 95% CI | P | Median, y | HR | 95% CI | P | |
| Median age, y | ||||||||
| >68 | 2.00 | 1.48 | 0.85-2.59 | .17 | 2.90 | 1.94 | 0.96-3.90 | .06 |
| ≤68 | 2.81 | 4.32 | ||||||
| Hb concentration, g/L | ||||||||
| ≥100 | 3.26 | 0.38 | .001 | 4.32 | 0.41 | 0.21-0.83 | .01 | |
| <100 | 1.55 | 0.21-0.69 | 2.75 | |||||
| Median WCC, ×109/L | ||||||||
| >14.6 | 2.85 | 0.55 | 0.32-0.96 | .03 | 4.26 | 0.74 | 0.38-1.43 | .37 |
| ≤14.6 | 1.78 | 3.17 | ||||||
| Median platelet count, ×109/L | ||||||||
| >199 | 2.23 | 0.86 | 0.49-1.50 | .59 | 4.32 | 0.54 | 0.28-1.05 | .07 |
| ≤199 | 2.00 | 2.90 | ||||||
| Median PBB, % | ||||||||
| >1 | 1.55 | 2.10 | 1.20-3.67 | .01 | 2.91 | 2.53 | 1.30-4.94 | .006 |
| ≤1 | 2.76 | 4.32 | ||||||
| Transfusion dependence | ||||||||
| Yes | 1.46 | 2.37 | 1.36-4.12 | .002 | 2.47 | 2.52 | 1.28-4.95 | .007 |
| No | 3.26 | 5.51 | ||||||
| Baseline spleen size, cm | ||||||||
| ≥10 | 2.28 | 0.72 | 0.33-1.55 | .40 | 4.26 | 0.67 | 0.28-1.65 | .39 |
| <10 | 1.71 | 2.37 | ||||||
| JAK inhibitor | ||||||||
| Ruxolitinib | 2.27 | 0.71 | 0.39-1.31 | .27 | 4.26 | 1.23 | 0.60-2.55 | .57 |
| Momelotinib | 1.71 | 4.32 | ||||||
| Diagnosis | ||||||||
| PMF | 1.92 | 0.72 | 0.41-1.25 | .24 | 3.30 | 1.07 | 0.56-2.06 | .84 |
| PPV-/PET-MF | 2.53 | 4.32 | ||||||
| DIPSS | ||||||||
| Low/Intermediate-1 | 4.69 | 1.81 | 0.83-3.96 | .14 | NA | 1.66 | 0.63-4.32 | .30 |
| Intermediate-2 | 2.53 | 3.55 | 1.63-7.73 | .001 | 4.26 | 4.09 | 1.59-10.50 | .004 |
| High | 1.46 | 2.37 | ||||||
| JAK2/MPL (n = 85) | ||||||||
| Yes | 2.23 | 0.75 | 0.40-1.40 | .36 | 4.26 | 0.80 | 0.38-1.71 | .57 |
| No | 1.78 | 2.90 | ||||||
| CALR (n = 14) | ||||||||
| Yes | 1.78 | 0.94 | 0.43-2.10 | .89 | NA | 0.75 | 0.27-2.13 | .59 |
| No | 2.23 | 3.30 | ||||||
| ASXL1 (n = 33) | ||||||||
| Yes | 1.71 | 1.86 | 1.06-3.24 | .03 | 2.37 | 2.27 | 1.17-4.40 | .02 |
| No | 2.76 | 5.51 | ||||||
| SRSF2 (n = 7) | ||||||||
| Yes | 2.52 | 1.00 | 0.40-2.54 | .99 | 2.64 | 1.30 | 0.46-3.70 | .62 |
| No | 2.09 | 4.26 | ||||||
| EZH2 (n = 8) | ||||||||
| Yes | 1.10 | 2.94 | 1.31-6.60 | .009 | 1.83 | 4.17 | 1.69-10.28 | .002 |
| No | 2.28 | 4.32 | ||||||
| IDH1/IDH2 (n = 4) | ||||||||
| Yes | 2.76 | 0.73 | 0.18-3.01 | .67 | 2.90 | 1.34 | 0.32-5.61 | .69 |
| No | 2.09 | 4.26 | ||||||
| TET2 (n = 28) | ||||||||
| Yes | 1.95 | 0.98 | 0.52-1.85 | .95 | 2.45 | 1.57 | 0.77-3.21 | .21 |
| No | 2.28 | 4.32 | ||||||
| U2AF1 (n = 10) | ||||||||
| Yes | 1.95 | 1.60 | 0.68-3.77 | .28 | 2.37 | 1.14 | 0.35-3.76 | .82 |
| No | 2.23 | 4.26 | ||||||
| SF3B1 (n = 13) | ||||||||
| Yes | 1.99 | 1.36 | 0.64-2.91 | .42 | NA | 0.91 | 0.32-2.58 | .86 |
| No | 2.23 | 3.30 | ||||||
| CBL (n = 5) | ||||||||
| Yes | 1.03 | 3.66 | 1.09-12.24 | .04 | 1.20 | 2.92 | 0.69-12.44 | .15 |
| No | 2.28 | 4.26 | ||||||
| DNMT3A (n = 6) | ||||||||
| Yes | 1.13 | 2.06 | 0.74-5.74 | .17 | 2.00 | 2.34 | 0.71-7.72 | .16 |
| No | 2.28 | 4.26 | ||||||
| NRAS (n = 5) | ||||||||
| Yes | 1.38 | 1.57 | 0.57-4.38 | .39 | 4.26 | 1.63 | 0.50-5.35 | .42 |
| No | 2.23 | 4.32 | ||||||
| ASXL1/EZH2 (n = 37) | ||||||||
| Yes | 1.49 | 2.36 | 1.36-4.11 | .002 | 2.37 | 2.90 | 1.48-5.99 | .002 |
| No | 3.26 | 5.51 | ||||||
| HMR profile (n = 43) | ||||||||
| Yes | 1.67 | 2.06 | 1.19-3.58 | .01 | 2.40 | 2.61 | 1.33-5.14 | .005 |
| No | 3.46 | 5.51 | ||||||
| Number of mutations (including driver) | ||||||||
| 1 | NA | 4.32 | ||||||
| 2 | 2.53 | 1.23 | 0.54-2.81 | .63 | NA | 0.99 | 0.37-2.66 | .99 |
| ≥3 | 1.49 | 2.19 | 0.95-5.05 | .07 | 2.37 | 2.65 | 1.04-6.75 | .04 |
| 0-2 mutations | ||||||||
| No HMR | 3.45 | NA | ||||||
| HMR | 1.78 | 1.94 | 0.92-4.08 | .08 | 3.30 | 1.75 | 0.68-4.54 | .25 |
| ≥3 mutations | ||||||||
| No HMR | 2.21 | 1.87 | 0.63-5.52 | .26 | 4.26 | 1.65 | 0.46-5.93 | .45 |
| HMR | 1.49 | 2.50 | 1.30-4.80 | .006 | 2.28 | 4.16 | 1.87-9.27 | .0005 |
NA, event-free rate did not reach 50%.
Secondary endpoints
OS
Over a median follow-up of 109 weeks, 36% of patients died, with a median OS of 182 weeks (Table 2). The primary causes of death included transformation to blast phase (n = 9), progressive MF without AML transformation (n = 19), infectious complications (n = 5), second malignancy (n = 1), and cardiac events (n = 2). The clinical variables associated with a shorter OS included anemia (P = .01), peripheral blood blasts >1% (P = .006), high DIPSS score (P = .004; Figure 3A), and pretreatment transfusion dependence (P = .007; Figure 3B) (Table 3). ASXL1 (P = .02) and EZH2 (P = .002) mutations were the only individual mutations associated with an inferior OS. ASXL1/EZH2 mutations (P = .001; Figure 3C), HMR mutations (P = .005), and ≥3 mutations (P = .04) were also associated with a shorter OS. When the number of mutations was stratified by HMR, having ≥3 mutations was significantly associated with TTF only in the setting of HMR (P = .0005; Figure 3D).
Figure 3.

Kaplan-Meier curves for OS. (A) DIPSS score. (B) Pretreatment transfusion dependency. (C) ASXL1/EZH2 mutations. (D) Number of mutations, stratified by HMR.
IWG-MRT response
The best IWG-MRT 2013 response achieved over 48 weeks of treatment was clinical improvement in 27 patients (Table 2). No patients achieved a complete or partial response. Nineteen patients had a response with respect to either the spleen (n = 18) or anemia (n = 1), but they did not meet criteria for clinical improvement. Fifty-two patients had stable disease, and 1 patient met the criteria for progressive disease. Of 100 patients, 94 were evaluable for spleen response, with 46 (49%) achieving a spleen response at any time point on JAK1/2 inhibitor therapy. The lack of spleen response was correlated with anemia and a high DIPSS score, whereas a trend was noted toward inferior spleen response in patients with HMR (P = .06) (supplemental Table 3). Seventeen (32%) of the 54 patients evaluated met criteria for anemia response at any time point. There was no correlation between anemia response and any clinical or mutational variable (supplemental Table 4). When comparing ruxolitinib with momelotinib, there was no significant difference in any measured outcomes (data not shown).
Grade 3 or 4 hematological toxicity included anemia in 36 (36%) patients, thrombocytopenia in 23 (23%), and neutropenia in 2 (2%) patients (Table 2). Thirty (30%) patients experienced infectious complications of all grades associated with JAK1/2 inhibitor therapy, with the most common infection being herpes zoster. One patient had reactivation of peritoneal tuberculosis. Nonhematological toxicities were primarily grade 1 or 2, seen in 20% of patients (all grades), and included cardiac arrhythmia, acute kidney injury, and Wernicke’s encephalopathy in 1 patient on momelotinib. Two of 23 patients treated with momelotinib developed peripheral neuropathy.
To further evaluate important clinical and genetic variables found to be significant on univariate analysis, we selected DIPSS score, pretreatment transfusion dependence, ASXL1/EZH2 mutations, and number of mutations for an exploratory multivariable analysis of TTF and OS. Our initial 4-variable model demonstrated significant associations between transfusion dependence and DIPSS (Φ = 0.62) and between ASXL1/EZH2 and number of mutations (Φ = 0.53) (supplemental Table 5). We therefore generated models where ASXL1/EZH2 mutations and number of mutations were independently assessed against each of the 2 clinical variables in isolation. A high DIPSS score (HR, 2.79; 95% confidence interval [CI], 1.24-6.31; P = .01) and transfusion dependence (HR, 2.07; 95% CI, 1.18-3.64; P = .01) were independently associated with shorter TTF when combined with ASXL1/EZH2 mutations (Table 4). ASXL1/EZH2 mutations remained independently significant when assessed against both DIPSS score (HR, 1.87; 95% CI, 1.05-3.33; P = .04) and transfusion dependence (HR, 2.05; 95% CI, 1.16-3.61; P = .01). Both clinical variables and ASXL1/EZH2 mutations remained independently significant for OS (Table 4). Models including the number of mutations did not demonstrate significance for either TTF or OS.
Table 4.
Multivariable analysis for TTF and OS.
| Variable | TTF | OS | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P | HR | 95% CI | P | |
| Model 1 | ||||||
| Transfusion dependence: Y vs N | 2.07 | 1.18-3.64 | .01 | 2.04 | 1.02-4.11 | .05 |
| ASXL1/EZH2: Y vs N | 2.05 | 1.16-3.61 | .01 | 2.46 | 1.23-4.92 | .01 |
| Model 2 | ||||||
| DIPSS score: int-2 vs int-1 | 1.57 | 0.71-3.49 | .27 | 1.40 | 0.53-3.69 | .50 |
| DIPSS score: high vs int-1 | 2.79 | 1.24-6.31 | .01 | 3.00 | 1.12-8.03 | .03 |
| ASXL1/EZH2 mutation: Y vs N | 1.87 | 1.05-3.33 | .04 | 2.28 | 1.13-4.61 | .02 |
ASXL1/EZH2 was tested in 2 models against transfusion dependence and DIPSS.
int-1, intermediate-1; int-2, intermediate-2; N, no; Y, yes.
Discussion
There is now substantial literature on the long-term follow-up of patients treated with ruxolitinib, with discontinuation rates of ∼50% at 3 years of treatment.17-20 To date, no clinical factors have consistently been shown to be predictive of spleen response,21,22 and few studies have investigated the factors that determine durability of response to JAK1/2 inhibitors. In our study, we demonstrate that patients with anemia and high DIPSS scores had lower spleen response rates. No individual mutation was associated with spleen response. However, mutations in HMR genes exhibited a trend toward inferior spleen response. Previous post-hoc analysis of the COMFORT-II trial did not suggest a negative impact of mutations regarding the efficacy of ruxolitinib,7 however, other studies have shown inferior spleen response in patients with ASXL1,6,8 EZH2, and IDH1/2 mutations.6
There are limited data on the understanding of factors associated with TTF in MF patients treated with JAK1/2 inhibitors. In the current study, we demonstrated that a high DIPSS score, pretreatment transfusion dependence, and mutations in ASXL1 and EZH2 were associated with shorter TTF. A previous study from the MD Anderson group reported shorter time to treatment discontinuation in patients with ASXL1 and EZH2 mutations.6 This study also showed that patients with ≥3 mutations had shorter time to treatment discontinuation.6 Our data raise the possibility that shorter TTF as well as OS in patients with ≥3 mutations may primarily be driven by the presence of an HMR mutation. However, larger studies are needed to confirm this finding. Our study did not find any impact of SRSF2 and IDH1/2 mutations on treatment outcomes. The data on the impact of SRSF2 and IDH1/2 mutations in patients treated with JAK1/2 inhibitor therapy are limited because these mutations are seen in a small proportion of patients. Moreover, none of the previously reported studies evaluated SRSF2 in the setting of JAK1/2 inhibitor therapy. Taken together, accumulating evidence suggests a shorter duration of treatment response as well as inferior survival in ASXL1- and EZH2-mutated patients treated with JAK1/2 inhibitor therapy.
We were unable to validate the negative impact of mutation number on treatment failure, as previously published, in patients treated with JAK1/2 inhibitor therapy6 or supportive care.3,23 We demonstrated that as the number of mutations increased, the frequency of a HMR mutation increased as well. However, the number of mutations is highly dependent on the number of genes interrogated (which varies between centers due to differing NGS panels) and differing variant annotation criteria. The standardization of an algorithm for identifying pathogenic variants is necessary prior to considering widespread implementation of NGS in routine clinical practice.
Although JAK1/2 inhibitors are important agents in MF patients for alleviating burdensome symptoms and improving quality of life, they are not curative and are associated with significant rates of treatment failure. At present, the only curative therapeutic option for MF is HCT. The expert guidelines recommend the option of HCT in intermediate-2/high-risk disease and selected patients with intermediate-1 disease.24,25 However, physicians as well as patients face a dilemma on the optimal timing of transplant,9,26 especially for patients who are responding well to JAK1/2 inhibitor therapy. There is equipoise among physicians as to whether to proceed to HCT in such patients or consider HCT at the failure of JAK1/2 inhibitor therapy. Our study provides evidence for higher rates of treatment failure in patients with high DIPSS scores, transfusion dependence prior to JAK1/2 inhibitor therapy, and mutations in ASXL1/EZH2. These data suggest that transplant should be considered upfront in MF patients with any of these high-risk features. These findings provide a framework for future prospective studies evaluating the optimal timing of transplant in MF patients. In addition, our study identifies a subgroup of patients for whom the benefit of current JAK1/2 inhibitors is limited, and who therefore potentially could be candidates for future clinical trials using novel agents.
In conclusion, we demonstrate that clinical and molecular variables, including high DIPSS score, pretreatment transfusion dependence, and ASXL1/EZH2 mutations, are associated with a shorter TTF in MF patients treated with JAK1/2 inhibitors. The findings of this study are particularly useful in the identification of patients who are unlikely to have a durable response with current JAK1/2 inhibitors and provide a framework for the design of future studies.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
This work was supported by a grant from the Princess Margaret Cancer Centre Foundation and the Elizabeth and Tony Comper Foundation to the MPN program at the Princess Margaret Cancer Centre.
Authorship
Contribution: D.M., A. Schimmer, A. Schuh, H.S., K.Y., M.D.M., V.G. and J.H. contributed to clinical patient management; A.A. and J.C. collected the samples; J.Y.S., C.M., R.D., J.B., and N.S. collected the clinical data; T.S., M.S., M.T., and S.K.-R. performed the molecular investigations; T.P. performed the statistical analyses; J.S., C.M., and V.G. compiled the clinical and mutational data, analyzed the data, and wrote the manuscript; J.A.K., M.S., and M.T. analyzed and annotated the mutational data; V.G. designed and supervised the study and was responsible for the conduct of the study overall; and all authors reviewed the manuscript and approved it for publication.
Conflict-of-interest disclosure: T.P. provided consultancy services to Celgene. A. Schimmer, D.M., and N.S. received honoraria from Novartis. A. Schuh served on the advisory board of Amgen. K.Y. received research funding and served on the advisory board of Novartis. S.K.-R. received research funding from Bristol-Myers Squibb. V.G. received research funding through his institution and honoraria from Novartis and Incyte and has served on the advisory board of Novartis. The remaining authors declare no competing financial interests.
Correspondence: Vikas Gupta, Department of Medical Oncology and Hematology, Princess Margaret Cancer Centre, 610 University Ave, Toronto, ON M5G 2M9, Canada; e-mail: vikas.gupta@uhn.ca.
References
- 1.Rumi E, Pietra D, Pascutto C, et al. ; Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative Investigators. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood. 2014;124(7):1062-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tefferi A, Guglielmelli P, Lasho TL, et al. . CALR and ASXL1 mutations-based molecular prognostication in primary myelofibrosis: an international study of 570 patients. Leukemia. 2014;28(7):1494-1500. [DOI] [PubMed] [Google Scholar]
- 3.Guglielmelli P, Lasho TL, Rotunno G, et al. . The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: an international study of 797 patients. Leukemia. 2014;28(9):1804-1810. [DOI] [PubMed] [Google Scholar]
- 4.Lundberg P, Karow A, Nienhold R, et al. . Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood. 2014;123(14):2220-2228. [DOI] [PubMed] [Google Scholar]
- 5.Vannucchi AM, Lasho TL, Guglielmelli P, et al. . Mutations and prognosis in primary myelofibrosis. Leukemia. 2013;27(9):1861-1869. [DOI] [PubMed] [Google Scholar]
- 6.Patel KP, Newberry KJ, Luthra R, et al. . Correlation of mutation profile and response in patients with myelofibrosis treated with ruxolitinib. Blood. 2015;126(6):790-797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guglielmelli P, Biamonte F, Rotunno G, et al. ; COMFORT-II Investigators; Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative (AGIMM) Investigators. Impact of mutational status on outcomes in myelofibrosis patients treated with ruxolitinib in the COMFORT-II study. Blood. 2014;123(14):2157-2160. [DOI] [PubMed] [Google Scholar]
- 8.Pardanani A, Abdelrahman RA, Finke C, et al. . Genetic determinants of response and survival in momelotinib-treated patients with myelofibrosis. Leukemia. 2015;29(3):741-744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Devlin R, Gupta V. Myelofibrosis: to transplant or not to transplant? Hematology Am Soc Hematol Educ Program. 2016;2016(1):543-551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.1000 Genomes Project Consortium, Auton A, Brooks LD, et al. . A global reference for human genetic variation. Nature. 2015;526(7571):68-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lek M, Karczewski KJ, Minikel EV, et al. ; Exome Aggregation Consortium. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sherry ST, Ward MH, Kholodov M, et al. . dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lindsley RC, Saber W, Mar BG, et al. . Prognostic Mutations in Myelodysplastic Syndrome after Stem-Cell Transplantation. N Engl J Med. 2017;376(6):536-547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tefferi A, Cervantes F, Mesa R, et al. . Revised response criteria for myelofibrosis: International Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) and European LeukemiaNet (ELN) consensus report. Blood. 2013;122(8):1395-1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Passamonti F, Cervantes F, Vannucchi AM, et al. . Dynamic International Prognostic Scoring System (DIPSS) predicts progression to acute myeloid leukemia in primary myelofibrosis. Blood. 2010;116(15):2857-2858. [DOI] [PubMed] [Google Scholar]
- 16.Bartoszko J, Panzarella T, Lau A, et al. . Effect of red blood cell transfusion dependence on the natural history of myeloproliferative neoplasm-associated myelofibrosis. Clin Lymphoma Myeloma Leuk. 2015;15(11):e151-e156. [DOI] [PubMed] [Google Scholar]
- 17.Harrison C, Kiladjian JJ, Al-Ali HK, et al. . JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787-798. [DOI] [PubMed] [Google Scholar]
- 18.Verstovsek S, Mesa RA, Gotlib J, et al. . A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799-807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verstovsek S, Mesa RA, Gotlib J, et al. ; COMFORT-I investigators. Efficacy, safety, and survival with ruxolitinib in patients with myelofibrosis: results of a median 3-year follow-up of COMFORT-I. Haematologica. 2015;100(4):479-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cervantes F, Vannucchi AM, Kiladjian JJ, et al. ; COMFORT-II investigators. Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood. 2013;122(25):4047-4053. [DOI] [PubMed] [Google Scholar]
- 21.Verstovsek S, Mesa RA, Gotlib J, et al. . The clinical benefit of ruxolitinib across patient subgroups: analysis of a placebo-controlled, Phase III study in patients with myelofibrosis. Br J Haematol. 2013;161(4):508-516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vannucchi AM, Kantarjian HM, Kiladjian JJ, et al. ; COMFORT Investigators. A pooled analysis of overall survival in COMFORT-I and COMFORT-II, 2 randomized phase III trials of ruxolitinib for the treatment of myelofibrosis. Haematologica. 2015;100(9):1139-1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tefferi A, Lasho TL, Finke CM, et al. . Targeted deep sequencing in primary myelofibrosis. Blood Adv. 2016;1(2):105-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mesa R, Jamieson C, Bhatia R, et al. . Myeloproliferative neoplasms, version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2016;14(12):1572-1611. [DOI] [PubMed] [Google Scholar]
- 25.Kröger NM, Deeg JH, Olavarria E, et al. . Indication and management of allogeneic stem cell transplantation in primary myelofibrosis: a consensus process by an EBMT/ELN international working group. Leukemia. 2015;29(11):2126-2133. [DOI] [PubMed] [Google Scholar]
- 26.Shanavas M, Gupta V. Controversies and dilemmas in allogeneic transplantation for myelofibrosis. Best Pract Res Clin Haematol. 2014;27(2):165-174. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.